

Abstract
Pheochromocytomas are rare in children. The diagnosis is usually established from a raised urinary or plasma catecholamine or their metabolites. We present a girl aged 11 years who manifested with a hypertensive crisis secondary to an adrenal tumour but with unexpectedly normal urinary metanephrine and catecholamine results. She improved spontaneously following the crisis and underwent surgery later. The histopathological study confirmed a pheochromocytoma with large central necrosis. Her genetic screening reported a pathogenic von Hippel-Lindau gene mutation. Surveillance scan postsurgery detected no other tumours. Following the catecholamine crisis, an acute infarct occurred, resulting in extensive tumour necrosis and subsequent rapid remission of symptoms and paradoxically normal biochemical markers. Although not unheard of in adults, we believe this is the first reported case of an extensive spontaneous necrosis resulting in a biochemically normal pheochromocytoma in a child.
Keywords: adrenal disorders, radiology
Background
Pheochromocytomas are catecholamine-secreting tumours arising from the chromaffin cells of the adrenal medulla with variable clinical presentation. The disease can range from asymptomatic ‘incidentaloma’ to paroxysmal hypertensive crises. The most common signs and symptoms include sustained hypertension, headaches, tachycardia and diaphoresis.1
These tumours are uncommon in children, accounting for about 10% of all pheochromocytoma cases.1 2 When they occur, a large majority of the paediatric cases are associated with genetic syndromes such as familial paraganglioma, multiple endocrine neoplasia (MEN) type 2 and von Hippel-Lindau (VHL) disease.3 The latter is an autosomal dominant disorder associated with a germline mutation in the VHL suppressor gene. It is associated with an increased risk of neoplasms such as haemangioblastoma, renal cell carcinoma, cystadenomas and pheochromocytoma. About 10%–20% of individuals with VHL will develop pheochromocytoma and the mean age at presentation is about 30 years.4
The diagnosis of pheochromocytoma relies on the biochemical documentation of excess catecholamine secretion, coupled with imaging studies for localisation and staging. Up to 90% of paediatric pheochromocytomas are secretory.5 Therefore, it can be challenging to make a diagnosis in the context of a clinically functioning adrenal tumour but with a biochemically normal profile. While imaging studies may provide clues in assisting the diagnostic process, it requires careful interpretation and experience to identify the radiological features that differentiate the adrenal tumours.
Here, we describe a child with VHL-associated left adrenal pheochromocytoma who presented with hypertensive crisis but with normal urinary fractionated metanephrines and catecholamine levels following an extensive necrosis of the tumour.
Case presentation
A girl aged 11 years who had a history of bronchial asthma presented to the hospital with an acute onset of breathlessness, chest discomfort, pallor and sweatiness. Her weight had faltered for the past 6 months, crossing two major percentiles (figure 1). She did not have polydipsia, polyuria, recurrent headache or blurring of vision. There was no family history of malignancy or hypertension.
Figure 1.
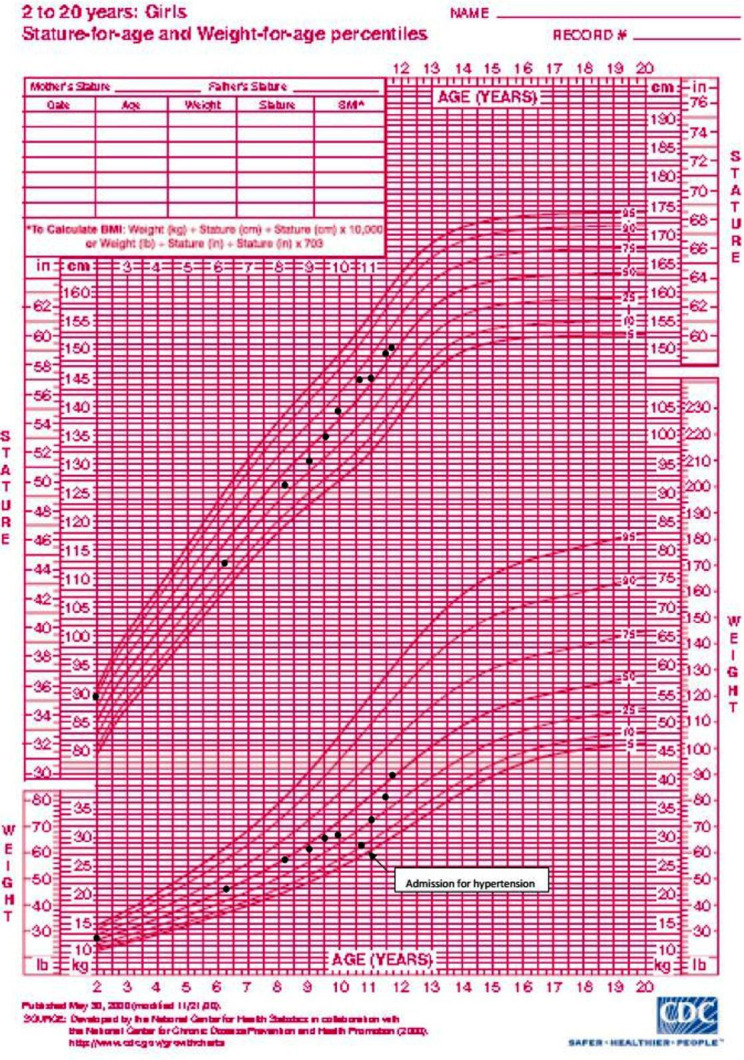
Growth chart indicating faltered weight, crossing two major percentiles over 6 months before presentation.
On physical examination at presentation, vital signs showed an elevated blood pressure up to 168/105 mmHg (>95th percentile) and tachycardia of 140 beats per minute. Apart from rhonchi on lung auscultation, there were no other cardiopulmonary abnormalities or abdominal masses detected. Her weight was 28 kg (10th percentile), height 145 cm (50th percentile) and a body mass index of 13.3 kg/m2 (<3rd percentile).
She was given nebulised salbutamol at the emergency department and then referred to the paediatric team for hypertension. Initial blood investigation showed mild hypokalaemia that presumably resulted from salbutamol administered twice in the emergency department. Repeated potassium level the following day was normal without the need for any replacement. Serum sodium, glucose, thyroid, full blood count, liver and renal functions were normal. Serum direct renin was 13.00 mU/L (3–66 mU/L) and aldosterone <103 pmol/L (<582.6 pmol/L). Lung fields were clear with no cardiomegaly on the chest radiograph. An ECG showed features of left ventricular hypertrophy. In addition, an echocardiogram revealed a hypertrophic left ventricular wall with apical hypokinesia. The finding was attributed to a ‘stunned’ myocardium secondary to a sudden high afterload. There was no feature of coarctation of the aorta.
Given her presentation, we conducted further investigations. Ultrasonography of the abdomen detected a well-defined hypoechoic solid lesion at the left renal hilar region with no internal vascularity or calcification, measuring 2.6 cm×3.2 cm×3.9 cm (figure 2A, B). A concurrent renal Doppler study showed no evidence of renal artery stenosis. An abdominal CT was done the day after the acute presentation. It confirmed an encapsulated non-enhancing left adrenal soft tissue lesion with Hounsfield Unit (HU) 20–30. It had minimal internal fat attenuation and no intralesional calcification. The size measured 2.4 cm×2.9 cm×2.3 cm (APxWxCC) (figure 2C).
Figure 2.

(A) Initial ultrasound shows a well-defined hypoechoic solid lesion at the left renal hilar region measuring 2.6 cm×3.2 cm×3.9 cm. (B) The lack of internal vascularity is demonstrated by the lack of signal on the Doppler flow study. (C) Abdominal CT shows a well-defined encapsulated non-enhancing hypodense soft tissue (*) with Hounsfield Unit 20–30 and a pronounced enhancement of the peripheral rim of the viable tumour tissue described as the ‘ring sign’ arising from the lateral limb of the left adrenal gland. (D) Repeated ultrasound 6 weeks later shows a regressed size of the lesion.
She was started on enalapril and prazosin for initial control of her blood pressure. However, when pheochromocytoma was suspected, enalapril was weaned off after 4 days, given the clinical presentation and imaging features. Her blood pressure was well-controlled with just prazosin and cardiac function returned to normal on subsequent echocardiogram 2 weeks later.
Investigations
A 24-hour urine catecholamine taken 3 days after stopping enalapril and fractionated metanephrine, normetanephrine and 3-methoxytyramine 2 weeks after were all not elevated (table 1). As most clinically functioning tumours are biochemically active, the normal urinary results had caused some confusion to the initial suspicion of pheochromocytoma.
Table 1.
24-hour urine catecholamines and 24-hour urine fractionated metanephrines taken shortly after the first presentation
| Investigation | Results | Reference range |
| 24-hour urinary catecholamines | ||
| Norepinephrine | 48 nmol/day | 89–473 nmol/day |
| Dopamine | 392 nmol/day | 424–2611 nmol/day |
| Epinephrine | 5.5 nmol/day | 2.7–109.2 nmol/day |
| 24-hour urinary fractionated metanephrine, normetanephrine and 3-MT | ||
| Metanephrine | 0.41 µmol/L | 0.33–1.53 µmol/L |
| Normetanephrine | 1.18 µmol/L | 0.88–2.88 µmol/L |
| 3-MT | 0.50 µmol/L | 0.60–2.60 µmol/L |
3-MT, 3-methoxytyramine.
She was discharged well after 2 weeks of admission. An abdominal ultrasound repeated in 6 weeks showed a reduction in the size of the lesion with no further intervention (figure 2D). A repeated 24-hour urine biochemistry 2 months later on single antihypertensive agent (prazosin) was again normal. The child remained asymptomatic and normotensive on follow-up. A functional scan was not done because it was not available in our hospital.
Differential diagnosis
The clinical presentation of a hypertensive crisis, complicated with stunned myocardium, is consistent with a catecholamine crisis. However, the 24-hour urine fractionated metanephrines and catecholamines were repeatedly within normal ranges for age. Therefore, while pheochromocytoma should still be suspected, other causes of adrenal mass associated with hypertension such as aldosterone or cortisol-producing tumours should be considered. Our patient did not have features of Cushing syndrome. Biochemical workup did not demonstrate hyperaldosteronism or hypercortisolism. On CT imaging, adrenal adenomas are typically smaller in size (<3 cm) and tend to have a lower attenuation index of <10 HU. They also lack internal vascularity on colour Doppler ultrasound, which was seen in this case.
Preoperatively making the right diagnosis for pheochromocytoma is imperative, because of the risk of acute catecholamine secretion during surgery. This will result in life-threatening complications if the patient’s care is not optimised preoperatively.
Treatment
The child underwent a diagnostic laparoscopy 4 months after the initial presentation. As a small pheochromocytoma remained a possibility given the clinical presentation, she was admitted 1 week prior to ensure her blood pressure was optimal with prazosin. Intraoperatively, a left adrenal tumour was densely adhered to the left renal vein measuring 4 cm×2 cm with a 1 cm×1 cm nodule. Left anterior adrenalectomy was done and the tumour was excised completely. The surgery was uneventful with stable vital signs perioperatively. The histopathological report confirmed the diagnosis of pheochromocytoma with extensive central necrosis rimmed by granulomatous inflammation. The necrosis and inflammation were bordered by some viable tumour cells. Mitotic activity was rare and the Ki67 proliferative index was about 5%. The surgical margin was clear, and no periadrenal adipose tissue invasion or vascular tissue invasion was seen. The Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) was 4 due to the presence of necrosis and cellular monotony (figure 3A–F).
Figure 3.
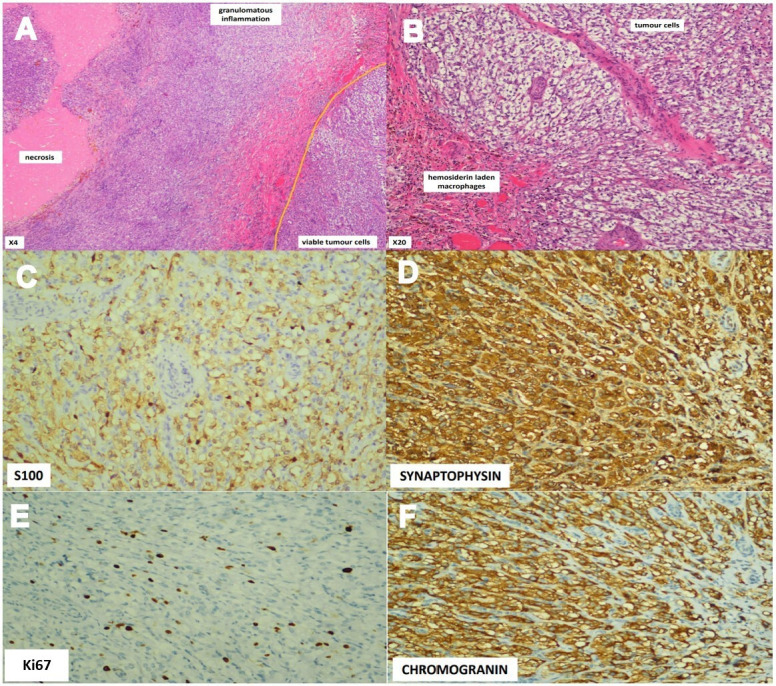
(A and B) Histopathological study shows a large area of necrosis rimmed by granulomatous inflammation (admixture of histiocytes and small lymphocytes). The necrosis and inflammation are bordered by some viable tumour cells. (C) S100 protein immunostain is positive for sustentacular cells. (D) The tumour cells stained positive for synaptophysin. (E) Ki67 proliferative index is about 5% with rare mitotic activity. (F) Chromogranin stain was positive for the tumour cells.
Outcome and follow-up
Postoperatively, prazosin was successfully weaned off and the child was discharged well with normal blood pressure. Her genetic study later showed the presence of pathogenic variant VHL (c.467A>G(p.Tyr156Cys)) heterozygous mutation. Her father has the same gene mutation and was referred to the adult physicians for tumour screening and subsequent follow-up. The histopathological assessment showed a PASS of 4, which may suggest malignant potential. However, her tumour was removed completely and on follow-up over a year, she had remained well and normotensive without medication. Repeated 24-hour urine fractionated metanephrines and catecholamines were normal, and her full body MRI scan did not show evidence of other tumours or metastatic lesions.
Discussion
The detection of adrenal pheochromocytoma largely depends on biochemical evidence of raised urine catecholamines and urine or plasma fractionated metanephrines which are highly sensitive and specific. In contrast to patients with symptomatic pheochromocytomas who have clearly abnormal values for any of these tests, biochemically normal pheochromocytomas are mostly small, measuring <1 cm and asymptomatic.6 While the presence of an acute hypertensive crisis with a moderate-sized adrenal lesion on the CT scan was suggestive of a hyperfunctioning pheochromocytoma, the normal biochemical results in our case had caused a diagnostic dilemma.
Necrotic, cystic and haemorrhagic changes are common among pheochromocytomas that are vascular in nature. However, spontaneous, extensive necrosis within the tumour and associated with acute catecholamine crisis is infrequent. Although there have been reports in the adults on spontaneous necrosis resulting in a biochemically normal tumour,7 such presentation in children and adolescents remains unclear. Prompt resolution of symptoms, hypertension and cardiac function following the acute crisis presentation, coupled with the regressed size of the tumour prior to surgery, suggested that the necrosis occurred acutely, similar to a case reported by Ohara et al in an adult male patient.7
The extensive necrosis with minimal residual viable tumour cells could likely explain the normal biochemical results sampled after the acute event. The mechanism causing the necrosis is obscure, but a possible explanation would be due to the catecholamine-induced vasoconstriction of the vessels within the tumour. This would elucidate the hypertensive emergency followed by rapid improvement in symptoms and blood pressure. Other presumed causal factors for necrosis of pheochromocytoma include haemorrhage into the tumour, high intracapsular pressure due to malignant growth and hypotension.7 These features were not present in our patient.
Necrosis is a commonly described radiological feature in pheochromocytoma, especially in larger tumours.8–10 Typically, pheochromocytoma is described to have variable echogenicity and high internal vascularity on colour Doppler. Our patient’s initial sonographic finding showed a hypoechoic lesion with a lack of internal vascularity. The latter could represent the early onset necrotic changes. The CT finding of a ring-enhanced lesion with a central hypodensity of 20–30 HU further suggests the possibility of central necrosis as described in previous studies.9 10 Ctvrtlik et al described the presence of ‘ring sign’ in up to 40% of CT images of cases of pheochromocytoma in their study,10 as also observed in our case. In a biochemically normal lesion, the various appearances on ultrasound and CT can be complementary and assist in differentiating pheochromocytoma from other adrenal lesions. Functional scans (Iodine-123 metaiodobenzylguanidine (I-123 MIBG) or integrated PET/CT) could be useful but are often not readily available.
Pheochromocytoma is rare in children. When diagnosed, one should seek the associations with hereditary syndromes such as VHL disease, familial paragangliomas and MEN type 2A. Genetic testing should be considered for all cases of pheochromocytoma. Cases that involve bilateral adrenal glands, a family history of pheochromocytoma or those who present at the age of <20 years are associated with a higher likelihood of hereditary causes; hence, genetic testing should be prioritised.11 In our patient, a pathogenic mutation of (c.467A>G (p.Tyr156Cys)) was detected in the VHL gene.
VHL disease is an autosomal dominant disorder associated with a germline mutation in the VHL gene. Besides pheochromocytoma, individuals with this disorder are at higher risk of developing other neoplasms such as haemangioblastoma, retinal angioma, clear cell renal cell carcinoma, pancreatic neuroendocrine tumours and papillary cystadenomas of the epididymis or broad ligament. These neoplasms were not detected in our patient during surveillance scan. The pathogenic variant seen in our patient is reported to be associated with VHL syndrome type 2C, which is more commonly associated with pheochromocytoma or paraganglioma alone.12 Nevertheless, regular tumour surveillance with imaging and biochemical markers in addition to a life-long follow-up would be needed.
In conclusion, an acute infarct within an adrenal pheochromocytoma causing extensive necrosis may manifest with a catecholamine crisis, followed by subsequent improvement of symptoms and normal biochemistry. As this paradox may cause confusion, it is essential to correlate the clinical picture with careful assessment of the lesion from imaging studies. In confirmed cases of pheochromocytoma, genetic study and evaluation for hereditary syndromes should be performed.
Patient’s perspective.
My child has asthma which was diagnosed when she was a young toddler. Otherwise, she has been well. I still remember when I had to rush her to the hospital as she was short of breath, lethargic and had excessive sweating. After being managed in the emergency department and told that it was one of her asthma attacks, she was transferred to the general ward for observation. I noticed that she was still sweating profusely even at sleep, which is not her usual self. I recalled the doctor telling me that her blood pressure has been persistently high since admission. It was a very long night battling many questions about what was wrong with my girl.
The next morning, the doctors did a battery of tests and a scan later in the day showed a tumour on the left side of her body. That came as shocking news to both my husband and I. What a turn of event! She had more scans after that to try to identify what the tumour was. We were told that it could be a tumour called pheochromocytoma, but her investigation was not consistent with the suspicion. She had her blood pressure monitored very closely the next few days in the ward. Fortunately, her blood pressure was under control and we were allowed home. However, the managing doctor reminded us that we still had to watch her blood pressure closely at home. Both my husband and I were anxious, fearful of the uncertainties. What if she had a similar episode at home? What if we failed to detect her being ill? The weeks to follow were hectic, with frequent clinic visits but fortunately my girl remained well.
On follow-up, we were told the tumour was still present on the repeated scan, but now smaller in size. She had her surgery 4 months later. Her surgery was uneventful and we were told the tumour was successfully removed. Her blood pressure had normalised without any medications. I was thankful. We met the doctor 2 weeks later in the clinic. The biopsy confirmed the tumour to be a pheochromocytoma. The doctor explained that the initial tests for this tumour were negative because a large portion of the tumour cells had ‘died’, causing the lesion to not produce the chemicals it was supposed to.
Unfortunately, with the diagnosis, she needed to have more tests done; this time involving a genetic test that was costly for us. After sending off the test, we had to wait anxiously for the result. The COVID-19 pandemic worsened during this time, and that created even more delay. After 3 months of wait, I received a call from the doctor to meet at his office to discuss the results. We were told that my daughter was tested positive for von Hippel-Lindau (VHL). Honestly, that term was foreign to me, even till this day though I have learnt to understand a bit more of the disease. With that genetic mutation, it means my girl would have a future risk of recurrence and risk of other tumours. Both my husband and I, along with my youngest daughter were screened for the same disease. It hit hard when my husband was also found to be positive. We were both dumbfounded by the news. However, we are thankful for the attending doctors who had arranged for further workup for my girl and husband—their scans are clear, for now. Nevertheless, we understand they will need regular follow-up.
Looking back at the whole sequence of events, I am thankful for the doctors’ dedication in managing my child up till this very day. I did not anticipate my child to have a tumour and how that spiralled off to a genetic disease that puts her at risk of getting tumours. Worse, I have my husband to worry about also. The genetic result also means my husband’s siblings and children may also carry the gene, which has caused considerable anxiety to their families. I have learnt to live with their diagnosis but I would be lying if I said I have no worries. Having said that, I feel reassured how this had led to the detection of VHL. At least now, they can be placed on regular surveillance and earlier treatment if needed. My experience with my child allows me to now share my understanding of the disease and encourage my in-laws to get genetically screened, if financially feasible. Even if they do not, at least we can create awareness for regular health monitoring.
Learning points.
Normal urinary biochemistry does not exclude pheochromocytoma in a child presented with hypertensive crisis; the diagnosis should be considered and further investigated if clinically indicated.
Spontaneous, extensive necrosis can result in an acute catecholamine crisis with prompt resolution of symptoms and subsequently normal biochemistry.
Catecholamine-induced vasoconstriction of the vessels may explain the infarction and necrosis of pheochromocytoma.
Imaging studies are vital in localising the tumour and provide clues to differentiate pheochromocytoma from other adrenal tumours.
Genetic testing for familial disease should be considered for all patients with pheochromocytoma, particularly those who present at a young age.
Acknowledgments
The authors would like to thank the Director General of Health, Malaysia for his permission to publish this case report. The authors would also like to thank Dr Noraini Binti Mohamad from the Pathology Department, Sarawak General Hospital for her assistance in providing the histopathology images and the Radiology Department in Sibu Hospital for the scan images.
Footnotes
Contributors: BW-LN: conceptualised, designed and drafted the initial draft of the manuscript and approved the final manuscript. T-HT and JS-LW conceptualised and designed the manuscript, critically revised the manuscript as submitted and also approved the final manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Consent obtained from parent(s)/guardian(s)
References
- 1.Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer 2014;38:7–41. 10.1016/j.currproblcancer.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dağdeviren Çakır A, Turan H, Aykut A, et al. Two childhood pheochromocytoma cases due to von Hippel-Lindau disease, one associated with pancreatic neuroendocrine tumor: a very rare manifestation. J Clin Res Pediatr Endocrinol 2018;10:179–82. 10.4274/jcrpe.5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Igaki J, Nishi A, Sato T, et al. A pediatric case of pheochromocytoma without apparent hypertension associated with von Hippel-Lindau disease. Clin Pediatr Endocrinol 2018;27:87–93. 10.1297/cpe.27.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenders JWM, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. The Lancet 2005;366:665–75. 10.1016/S0140-6736(05)67139-5 [DOI] [PubMed] [Google Scholar]
- 5.Sarathi V. Characteristics of pediatric pheochromocytoma/paraganglioma. Indian J Endocrinol Metab 2017;21:470–4. 10.4103/ijem.IJEM_558_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kota SK, Kota SK, Panda SSk K, Sandip P, et al. Pheochromocytoma: an uncommon presentation of an asymptomatic and biochemically silent adrenal incidentaloma. Malays J Med Sci 2012;19:86–91. [PMC free article] [PubMed] [Google Scholar]
- 7.Ohara N, Uemura Y, Mezaki N, et al. Histopathological analysis of spontaneous large necrosis of adrenal pheochromocytoma manifested as acute attacks of alternating hypertension and hypotension: a case report. J Med Case Rep 2016;10:279. 10.1186/s13256-016-1068-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersenn S, Richter P-A, Broemel T, et al. Computed tomography criteria for discrimination of adrenal adenomas and adrenocortical carcinomas: analysis of the German ACC registry. Eur J Endocrinol 2015;172:415–22. 10.1530/EJE-14-0916 [DOI] [PubMed] [Google Scholar]
- 9.Clifton-Bligh R. Diagnosis of silent pheochromocytoma and paraganglioma. Expert Rev Endocrinol Metab 2013;8:47–57. 10.1586/eem.12.76 [DOI] [PubMed] [Google Scholar]
- 10.Ctvrtlik F, Tudos Z, Szasz P, et al. Characteristic CT features of pheochromocytomas - probability model calculation tool based on a multicentric study. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2019;163:212–9. 10.5507/bp.2019.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenders JWM, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915–42. 10.1210/jc.2014-1498 [DOI] [PubMed] [Google Scholar]
- 12.Bholah R, Bunchman TE. Review of pediatric pheochromocytoma and paraganglioma. Front Pediatr 2017;5:155. 10.3389/fped.2017.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]