

Abstract
The retinoblastoma tumor suppressor protein (RB) is a negative regulator of the cell cycle that inhibits both G1 and S-phase progression. While RB-mediated G1 inhibition has been extensively studied, the mechanism utilized for S-phase inhibition is unknown. To delineate the mechanism through which RB inhibits DNA replication, we generated cells which inducibly express a constitutively active allele of RB (PSM-RB). We show that RB-mediated S-phase inhibition does not inhibit the chromatin binding function of MCM2 or RPA, suggesting that RB does not regulate the prereplication complex or disrupt early initiation events. However, activation of RB in S-phase cells disrupts the chromatin tethering of PCNA, a requisite component of the DNA replication machinery. The action of RB was S phase specific and did not inhibit the DNA damage-mediated association of PCNA with chromatin. We also show that RB-mediated PCNA inhibition was dependent on downregulation of CDK2 activity, which was achieved through the downregulation of cyclin A. Importantly, restoration of cyclin-dependent kinase 2 (CDK2)–cyclin A and thus PCNA activity partially restored S-phase progression in the presence of active RB. Therefore, the data presented identify RB-mediated regulation of PCNA activity via CDK2 attenuation as a mechanism through which RB regulates S-phase progression. Together, these findings identify a novel pathway of RB-mediated replication inhibition.
The retinoblastoma tumor suppressor protein RB is a critical negative regulator of cell cycle progression (1, 24, 54, 6 2, 63). Extensive analysis has shown that RB functions as a protein-binding protein, assembling multiprotein complexes that regulate gene transcription (24, 62, 63). For example, RB binds to the E2F transcription factor and recruits histone deacetylase through an independent protein-binding module to repress specific E2F target genes (4, 42). Similarly, RB interacts with Brg-1 to mediate transcriptional repression of discrete target genes (57, 65). Naturally occurring mutant alleles of RB found in tumors invariably disrupt RB-mediated transcriptional repression function, lending credence to the hypothesis that this function is required to block inappropriate proliferation.
Cell cycle progression is a highly ordered process driven by the activity of cyclin-dependent kinase (CDK)-cyclin complexes. Mitogenic signaling interfaces with the cell cycle machinery through the activation of D-type cyclins and their associated catalytic subunits (CDK4 or CDK6) (1, 52, 54). The CDK-cyclin D complexes initiate the phosphorylation of RB in mid-G1, with complete RB hyperphosphorylation mediated by CDK2-associated kinase activity (43). RB is the critical substrate for cyclin D-associated kinase activity, as cyclin D and CDK4 are not required in the absence of functional RB (38, 40). In contrast, CDK2-cyclin E and -cyclin A complexes have additional substrates, as RB-deficient cells are readily arrested by attenuation of CDK2 activity (46). CDK-mediated phosphorylation of RB disrupts its protein-binding activity, transcriptional repression activity, and growth-inhibitory function (5, 21, 27, 43, 62).
To study RB function, we previously generated mutants of RB that are resistant to CDK phosphorylation (PSM-RB), since wild-type RB is rapidly phosphorylated by endogenous CDK activity (26, 28). PSM-RB proteins are potent inhibitors of cell cycle progression (29). We and others have previously shown that tumor cells must bypass RB-mediated transcriptional repression to escape PSM-RB-mediated cell cycle arrest (9, 26, 32, 39, 57, 65). In general, RB-mediated cell cycle inhibition is dependent on the downregulation of transcriptional targets that stimulate CDK2 activity (26, 37, 41, 65).
It has recently been demonstrated that RB is also an important regulator of S-phase progression (9, 26, 41, 65). Initially, it was found that the introduction of PSM-RB alleles into S-phase cells inhibits DNA replication (9, 26). Subsequently, it has been shown that endogenous RB is required to inhibit cellular DNA replication in response to specific stresses (31, 53). For example, DNA damage promotes RB dephosphorylation and activation in S-phase cells. This dephosphorylation-activation of RB precedes the inhibition of DNA synthesis induced by genotoxic damage and is required for S-phase inhibition, as RB-deficient cells continue DNA replication in the presence of DNA damage that blocks replication in matched RB-positive cells (31). Similarly, the expression of adeno-associated virus replication protein 78 arrests DNA replication via the activation of endogenous RB (53). Together, these studies indicate that RB regulates both G1 and S-phase progression and is required for the appropriate response to specific environmental stresses. The mechanism underlying RB regulation of DNA synthesis has not been described.
DNA replication is regulated to ensure the accurate duplication of genetic material only once per cell division cycle (12, 25, 56, 61). In early G1, multiprotein complexes containing ORC, MCM, and cdc6 proteins assemble at origins of replication (pre-replication complex [preRC]) (10, 11, 12, 18, 25, 56, 61). As cells enter S phase, discrete origins of replication fire, leading to the displacement of MCM and cdc6 from the origin (10, 11, 23, 25, 48, 58). Upon DNA unwinding, RPA and additional factors bind to the single-stranded DNA (61). Subsequently, replication factor C (RFC) loads PCNA onto chromatin; PCNA in turn recruits DNA polymerase δ and serves as a processivity factor during DNA synthesis (61). Discontinuous origin activation and DNA polymerase-mediated elongation events continue until all origins have been replicated and MCM proteins are no longer associated with the chromatin (10, 11, 25, 56, 61). Disruption of any of these discrete processes leads to the inhibition of DNA replication.
Here we sought to identify the mechanism by which RB regulates DNA synthesis. Cells which harbor inducible expression of PSM-RB were generated. Using these cells, we show that active RB inhibits DNA replication in both S-phase-synchronized and asynchronously proliferating cells. We show that RB activation does not disrupt MCM2 or RPA chromatin tethering, suggesting that RB does not affect the establishment or maintenance of the preRC or early initiation events; however, induction of PSM-RB specifically disrupted PCNA activity. The ability of RB to regulate PCNA was dependent on the inhibition of CDK2 activity, as achieved through downregulation of cyclin A expression. Although the chromatin association of PCNA was dependent on CDK2 activity, we show that this regulation can be dissociated from the presence of cyclin A. Lastly, restoration of CDK2-cyclin A activity restored PCNA tethering and partially reversed PSM-RB-mediated inhibition of DNA replication. Together, these data demonstrate that RB inhibits DNA synthesis through CDK2-mediated regulation of PCNA tethering. These data are the first to delineate a pathway of RB action upon the DNA replication machinery.
MATERIALS AND METHODS
Cloning, cell culture, and synchronization.
PSM-RB (PSM.7-LP) was subcloned into the unique BamHI site of the expression pTRE plasmid (Clontech). This plasmid was cotransfected with the pTK-Hyg plasmid into Rat 16 cells. The Rat 16 cells were engineered to express the Tet-VP16 fusion protein and provide tight regulation of Tet-regulated gene expression. These cells were kindly provided by S. Salama and E. Harlow (Massachusetts General Hospital). Several clones expressing PSM-RB were isolated, and one (A2-4: PSM.7-LP) was used for the present study. Consistent S-phase inhibition is observed with independent clones and other PSM-RB alleles.
The cell lines harboring inducible expression of PSM-RB were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 1 μg of doxycycline (Dox) per ml to keep the expression of active RB off. Culture media also contained 10% fetal bovine serum (FBS), hygromycin B (200 μg/ml), and G418 (400 μg/ml). To induce PSM-RB expression, cells were washed three times in phosphate-buffered saline (PBS) and subsequently placed into media lacking Dox.
In order to synchronize cells in quiescence, asynchronously growing cells were cultured in 0.1% FBS for 72 h in the presence of Dox. To induce PSM-RB expression in these cultures, cells were washed with PBS after 48 h in 0.1% FBS and then placed in medium containing 0.1% FBS in either the presence or absence of Dox for an additional 24 h. Following synchronization in quiescence, cells were serum stimulated by adding fresh medium containing 10% FBS either with or without Dox.
To synchronize cells in early S phase, proliferating cells with Dox were first made quiescent with 0.1% FBS, placed into 10% FBS for 16 h, and then incubated in the presence of aphidicolin (APH; 2 μg/ml; Sigma) for 10 h. This concentration of aphidicolin does not prevent initiation but impedes chain elongation (6, 11, 36). Thus, PCNA is tethered to chromatin under these conditions (3, 7). To induce active RB expression in these S-phase cultures, the APH-synchronized cells were washed with PBS and placed in medium containing aphidicolin in either the presence (PSM-RB expression off) or absence (PSM-RB expression induced) of Dox. These matched APH-synchronized cell populations were used for reverse transcription (RT)-PCR analysis, analysis of replication proteins, and analysis of CDK-cyclin expression and activity.
To monitor S-phase progression, APH-synchronized cells that were cultured either with or without Dox were released from the aphidicolin block by washing as previously described (26) and propagated in medium containing or lacking Dox, respectively.
Reporter assays.
Approximately 3 × 105 cells were plated in 60-mm dishes. Cells were transfected with 8 μg of total plasmid DNA using FuGENE 6 (Roche) according to the manufacturer's instructions. Sixteen hours posttransfection, APH was added to synchronize cells in S-phase. After sixteen hours, cells were switched to media with or without Dox but containing APH. Sixteen hours after PSM-RB induction, cells were harvested and processed for relative luciferase activity as described previously (32). The plasmids used have been previously described (32).
RT-PCR.
Total cellular RNA was isolated with Trizol reagent (Gibco-BRL) according to the manufacturer's instructions. First-strand cDNAs were synthesized from 1 μg of RNA by use of ThermoScript RT-PCR system (Gibco-BRL) following the manufacturer's protocol. cDNAs were subjected to PCR amplifications using the PCR Core System I (Promega) with two sets of primers specific for glyceraldheyde-3-phosphate dehydrogenase (GAPDH) (5′-GGTCATCAATGGGAAACCCATCAC-3′ and 3′-TGATGGCATGGACTGTGGTCATGA-5′) and cyclin A (5′-AGACCCTGCATTTGGCTGTG-3′ and 3′-ACAAACTCTGCTACTTCTGG-5′). PCR was performed with the following conditions: 94°C for 4 min, 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s for 26 cycles. Approximately 10 μl of the PCR products was then separated by electrophoresis on a 2% agarose gel. The PCR products were visualized by ethidium bromide staining and then photographed (26).
Immunofluorescence microscopy.
Approximately 105 cells were seeded on glass coverslips in six-well dishes. For bromodeoxyuridine (BrdU) incorporation, cells were labeled with BrdU for the indicated period of time and stained for BrdU incorporation as previously described (30).
For PCNA, MCM2, RPA, and lamin B immunofluorescence, cells were grown on coverslips which were washed and incubated in cold PBS and fixed with methanol (unextracted) for 5 min at room temperature. For staining of chromatin-tethered proteins, coverslips were washed three times in cold PBS and extracted with a modification of cold CSK buffer (10 mM PIPES [piperazine N,N′-bis(2-ethanesulfonic acid), pH 6.8], 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) supplemented with 0.1% Triton X-100 and protease inhibitors for 20 min at 4°C before fixing in methanol. The following primary antibodies were used: PCNA (PC10; Santa Cruz), lamin B (sc-6217; Santa Cruz), MCM2 (46), and RPA (Ab-3; Oncogene Research Products).
Nuclear fractionation and immunoblotting.
A modification of a previously described method by Fujita et al. (19) was used to isolate chromatin-bound proteins. Briefly, cells were cultured in 100-mm plates, washed three times with ice-cold PBS, collected in 1 ml of PBS by scraping, and pelleted by quick spinning at 1,000 rpm for 1 to 2 min. Soluble proteins were then extracted with ice-cold 0.1% Triton X-100 in CSK buffer for 20 min at 4°C. The insoluble, chromatin-bound fraction was then pelleted by low-speed centrifugation at 3,000 rpm for 5 min at 4°C. These pellets were then reextracted by incubating in CSK buffer and collected by centrifugation at 3,000 rpm for 10 min at 4°C. The final pellet fraction (containing chromatin-bound proteins) or total cell pellets were solubilized in radioimmunoprecipitation assay (RIPA) buffer and equal protein was resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). Immunoblotting and kinase assays were performed using standard procedures and the following antibodies: cyclin A (C-19 or H432; Santa Cruz), cyclin E (M20; Santa Cruz), and CDK2 (M2-G; Santa Cruz), and PSM-RB was detected using 851 antibody (64).
Flow cytometry analysis.
Following cell synchronization and drug treatments, cells were harvested by trypsinization, fixed with ethanol, and processed for propidium iodide staining as described (26). For bivariate flow analyses, cells were pulse labeled with BrdU for 1 h and then processed for BrdU staining and propidium iodide staining (31).
Adenoviral infections.
Cyclin A-green fluorescent protein (GFP) and GFP adenoviruses were a kind gift from Gustavo Leone (Ohio State University). Approximately 105 cells were seeded on coverslips in six-well dishes and synchronized in S-phase as described above. Just before infection, cells were washed with PBS and switched to media with and without Dox in the presence of APH. The infections were performed at a calculated multiplicity of infection (MOI) of 50 to 100 (the actual infection efficiency was approximately 95 to 100%, as determined by GFP fluorescence) for 16 h.
RESULTS
PSM-RB induction inhibits S-phase progression.
To study the mechanism by which RB regulates S phase, Rat-1 cells harboring inducible expression of active RB (PSM-RB) were generated (A2-4 cells). PSM-RB migrates at a lower molecular weight than the endogenous rat RB, enabling unambiguous detection of the induced PSM-RB protein (26). As shown in Fig. 1A, expression of PSM-RB was absent in the nontransduced parental line (lanes 1 and 2), minimally detectable in the presence of Dox (lane 3), but readily apparent following removal of Dox from the medium for 16 h (lane 4). To examine the effect of PSM-RB expression on cellular proliferation, growth curves were performed through counting and trypan blue exclusion. In the presence of Dox, A2-4 cells doubled approximately every 24 h, while in the absence of Dox, proliferation ceased (Fig. 1B). After 6 days in culture, cells formed tight colonies of proliferating cells in Dox medium, whereas single cells exhibiting a large flattened morphology were observed in the absence of Dox (not shown). Analysis of BrdU incorporation revealed that induction of PSM-RB virtually halted DNA replication (Fig. 1C). To specifically analyze the G1/S transition, A2-4 cells were synchronized by culture in medium containing 0.1% FBS for 72 h. Cells were then cultured for 24 h in fresh 0.1% FBS medium either containing or lacking Dox to induce PSM-RB expression (Fig. 1D, hour 0). These quiescent cells were then stimulated with medium containing 10% FBS, harvested at 12, 18, 24, or 32 h post-serum stimulation, and monitored for cell cycle entry by flow cytometry. With PSM-RB expression inhibited (with Dox), cells entered the cell cycle within 18 h, as indicated by the accumulation of cells with a DNA content greater than 2N (Fig. 1D, upper panel). In contrast, cells expressing PSM-RB failed to exhibit cell cycle advancement out of G1 over a 32-h period (Fig. 1D, lower panel). Thus, PSM-RB expression retards the transition from G1 to S phase.
FIG. 1.

Inhibition of proliferation and G1/S by inducible PSM-RB expression. (A) Asynchronously proliferating Rat-16 cells (lanes 1 and 2) or A2-4 cells (lanes 3 and 4) were cultured in medium containing (+Dox) or lacking (−Dox) Dox for 16 h. Whole-cell lysates were prepared, equal protein was resolved by SDS-PAGE, and PSM-RB was detected by immunoblotting. (B) A2-4 cells (2 × 104) were cultured in the presence or absence of Dox. Cells were harvested at 72 and 144 h and counted. Average cell number from duplicate plates is shown as a function of time. (C) Asynchronously proliferating A2-4 cells were seeded onto coverslips and cultured in the presence or absence of Dox for 16 h. BrdU was added for a total labeling time of 3 h. Cells were fixed and stained for BrdU incorporation. The percentage of BrdU-positive cells was determined by indirect immunofluorescence. Data shown are from two independent experiments with at least 100 cells counted per experiment. Representative photomicrographs at ×60 magnification are also shown. (D) A2-4 cells were synchronized in quiescence by culture in medium containing Dox and 0.1% FBS for 72 h. In half of the cultures PSM-RB expression was induced by changing to 0.1% FBS medium lacking Dox for 24 h. Cells were then serum stimulated (10% FBS) in the presence or absence of Dox and harvested at the indicated time points. Cells were fixed, stained with propidium iodide, and analyzed by flow cytometry. Shown are representative histograms of 10,000 gated events from two independent experiments.
However, the G1-retarded cells eventually entered S phase and ceased proliferation, consistent with the observation that RB is also known to inhibit S-phase progression. Demonstrating this, quiescent cells which had been serum stimulated in the presence or absence of PSM-RB were analyzed for cell cycle distribution over a 4-day period. Cells stimulated with 10% FBS in the absence of PSM-RB expression (plus Dox) entered the cell cycle and continued proliferating until confluency halted cell cycle progression at 96 h (Fig. 2A, upper panel). In the presence of PSM-RB, virtually no cells stimulated with serum entered S phase before 24 h; by 72 h, the majority of cells expressing PSM-RB had entered S phase but were blocked for progression through S phase (Fig. 2A, lower panel). The ability of RB to inhibit S phase was verified using bivariate flow cytometry (31). In the absence of PSM-RB expression (plus Dox), A2-4 cells presented in all phases of the cell cycle (x axis). Approximately 90% of cells with an S-phase DNA content incorporated BrdU (y axis) (Fig. 2B, left panel). After PSM-RB induction, cells were present in both G1 and S phases of the cell cycle, but less than 15% of S-phase cells demonstrated active DNA replication (Fig. 2B, right panel).
FIG. 2.

Active RB inhibits S-phase progression. (A) A2-4 cells were synchronized in quiescence by culture in medium containing Dox and 0.1% FBS for 72 h. In half of the cultures PSM-RB expression was induced by changing to 0.1% FBS medium lacking Dox for 24 h. Cells were then serum stimulated (10% FBS) in the presence or absence of Dox and harvested at the indicated time points. Cells were fixed and stained with propidium iodide and analyzed by flow cytometry. Shown are representative histograms of 10,000 gated events from two independent experiments. (B) Asynchronously growing A2-4 cells were cultured in the presence or absence of Dox for 16 h. Cells were pulse-labeled with BrdU for 1 h and then processed for bivariate flow cytometry. Two-dimensional contour maps are shown, with BrdU incorporation on the y axis and DNA content (propidium iodide [PI]) on the x axis. Data shown are representative of two independent experiments. (C) A2-4 cells were synchronized in early S phase with APH. PSM-RB expression was then induced by culture in the absence of Dox (see Materials and Methods). Cells were released from APH (see Materials and Methods) and labeled with BrdU for 4 h. Cells were fixed and immunostained for BrdU incorporation to monitor S-phase progression. Data shown are from two independent experiments with at least 150 cells counted per experiment. (D) A2-4 cells were synchronized in S phase with APH and then cultured for an additional 16 h in APH with or without Dox. Cells were then released from the APH block and pulse labeled with BrdU for 1 h. Cells were harvested and processed for bivariate flow cytometry. Shown are representative contour maps with BrdU (y axis) and propidium iodide (PI, x axis) staining.
To directly assess the influence of RB on S-phase progression, A2-4 cells were synchronized in S phase with the DNA polymerase inhibitor APH. We have previously shown that APH arrests Rat-1 cells reversibly in S phase (26). To monitor the influence of PSM-RB expression in S-phase cells, we first synchronized cultures in S phase with APH (see Materials and Methods). The synchronized cultures were washed and then propagated in the presence of both Dox and APH (PSM-RB off) or in the absence of Dox but presence of APH (PSM-RB expressed). After 16 h to allow the accumulation of PSM-RB, the cells were released from APH arrest and S-phase progression was monitored by BrdU incorporation.
As shown in Fig. 2C, cells synchronized with APH and then released in the absence of PSM-RB expression (plus Dox) readily incorporated BrdU. However, S-phase-synchronized cells that were cultured with PSM-RB expression induced (no Dox) failed to incorporate BrdU following APH release (Fig. 2C). Analysis of the cell cycle and DNA replication simultaneously by bivariate flow cytometry illustrated that cells synchronized with APH in early S phase were inhibited for DNA replication. Release of these cells from APH in the absence of PSM-RB resulted in rapid incorporation of BrdU in approximately 50% of the cells (Fig. 2D, upper right panel). In the presence of PSM-RB, less than 5% of cells incorporated BrdU following release (Fig. 2D, lower right panel). Collectively, these data show that the PSM-RB-inducible cells recapitulate the ability of RB to inhibit DNA synthesis and provide the means necessary to probe the underlying mechanism.
Active RB disrupts association of PCNA with chromatin.
We hypothesized that RB must influence the activity of the replication machinery to inhibit S-phase progression. During early G1, the preRC is sequentially assembled at origins of replication and contains ORC, cdc6, and MCM proteins (10, 11, 18, 25). Following activation of replication origins, PCNA is recruited to the origin in S phase, where it functions as a homotrimeric clamp facilitating the loading and processivity of DNA polymerase δ. Both PCNA and MCM2 are E2F-regulated genes, and as such would be candidates for transcriptional regulation by RB.
To analyze the action of PSM-RB on replication proteins in S-phase cells, we first synchronized cultures in S phase with APH. These synchronized cultures were washed and then cultured either in the presence of both Dox and APH (PSM-RB off) or in the absence of Dox but presence of APH (PSM-RB expressed). After 16 h to allow the accumulation of PSM-RB in the S-phase cultures, these cell populations were analyzed to determine how active RB influences the expression or activity of proteins involved in DNA replication.
Surprisingly, PCNA protein levels were unchanged and MCM2 protein levels were only minimally attenuated (approximately two fold) following PSM-RB induction in APH-synchronized cells (Fig. 3A). Similar results were observed in asynchronously proliferating cells (not shown). Since PCNA and MCM2 expression was maintained during RB-mediated S-phase arrest, their respective activation states could be assessed. When active, MCM (a preRC component) and PCNA (a replisome component) proteins are tethered to the chromatin and cannot be extracted with low concentrations of nonionic detergents (10, 11, 19, 58). Thus, the fraction retained on chromatin appears to represent the active forms of these proteins. To determine the influence of PSM-RB on replication protein activity, the association of MCM2 and PCNA with chromatin was assessed using two distinct approaches (Fig. 3B to D). First, in situ extraction was used, which determines the percentage of cells with tethered replication proteins (19, 58). In these studies, lamin-B was used as a positive control (top panel). As shown in Fig. 3B, virtually 100% of cells stain positively for lamin-B, and lamin-B was not extracted following exposure to nonionic detergent (compare gray and black bars plus Dox). Under identical conditions, soluble proteins (e.g., GFP) were completely extracted from the cell (data not shown). PSM-RB expression had no influence on the tethering of lamin-B (compare black bars with and without Dox). MCM2, as part of the preRC, is known to be tethered to chromatin in both G1 and S phase. In unextracted cells, MCM2 staining was present irrespective of PSM-RB expression (Fig. 3B, middle panel, compare gray bars). This finding is consistent with the immunoblotting data indicating that PSM-RB does not significantly alter MCM2 protein levels (Fig. 3A). In extracted cells, PSM-RB expression did not inhibit MCM2 chromatin tethering (compare black bars), suggesting that PSM-RB does not disrupt the preformed preRC complexes in APH-synchronized cells.
FIG. 3.

PSM-RB expression specifically disrupts PCNA activity. (A) Cell lysates were collected from APH-synchronized cells that were cultured in the presence or absence of Dox for 16 h. Total cellular protein was resolved by SDS-PAGE, and the levels of lamin-B, MCM2, and PCNA were detected by immunoblotting. (B) Cells were seeded on coverslips, synchronized in early S-phase with APH, and then cultured in the presence or absence of Dox for 16 h. Coverslips were either extracted (black bars) or mock extracted (gray bars). Cells were then fixed in methanol and processed for immunofluorescence staining with lamin-B, MCM2, or PCNA antibodies. Values shown are the averages from three independent experiments with at least 150 cells counted per experiment. Representative photomicrographs at ×60 magnification are also shown. (C) Cells were synchronized in early S-phase with APH and then cultured in the presence or absence of Dox for 16 h. Cells were harvested and extracted with CSK buffer to separate soluble proteins from the chromatin-bound pellet fraction. The pellet fractions were resolved on SDS–10% PAGE and immunoblotted with lamin-B, MCM2, or PCNA antibody. Data shown are representative of several independent experiments. (D) Asynchronously growing cells were cultured in the presence or absence of Dox for 16 h. Cells were processed as described for panel B (black bars, extracted; gray bars, unextracted). Values shown are averages from two independent experiments with at least 100 cells counted per experiment.
In agreement with the data in Fig. 3A, PCNA staining was unchanged in unextracted cells following expression of PSM-RB (Fig. 3B, bottom panel, gray bars). However, unlike lamin-B or MCM2 tethering, PSM-RB expression abrogated the PCNA-chromatin association in APH-synchronized cells (Fig. 3B, black bars). To confirm that PCNA was no longer tethered to chromatin, fractionation was performed (Fig. 3C). Cells were subjected to extraction in situ, wherein chromatin-tethered proteins are retained in the pellet fraction (as described in Materials and Methods). Cell pellets retained lamin-B and MCM2 in the presence of PSM-RB expression, as determined by immunoblotting (Fig. 3C, compare lanes 1 and 2). In contrast, PCNA was absent from the pellet fraction following PSM-RB induction (Fig. 3C, compare lanes 1 and 2).
To determine if PSM-RB regulation of PCNA also applied to nonsynchronized cells, asynchronously proliferating cells were employed. As shown in Fig. 3D, expression of PSM-RB did not disrupt the chromatin tethering of MCM2 or lamin-B (black bars, compare with and without Dox). Since PCNA specifically tethers to chromatin during S phase, only 25 to 35% of cells in an asynchronous culture stained positively. Importantly, PCNA tethering to chromatin was specifically disrupted in the presence of PSM-RB in the asynchronous cultures (black bars). This effect was not due to a reduction in PCNA protein levels, as PSM-RB expression had no influence on PCNA staining in unextracted cells (gray bars). Together, these data indicate that PSM-RB expression specifically disrupts the tethering of PCNA to chromatin and provides a link between RB and the DNA replication machinery.
PSM-RB does not inhibit RPA-chromatin association.
To further refine the mechanism through which RB impacts the DNA replication machinery, we analyzed the chromatin association of RPA. The RPA heterotrimer (p32, p70, and p14) stabilizes single-stranded DNA as origins unwind during initiation and is required at the replication fork (61). Therefore, RPA tethering can be used as a marker for early initiation events. Quiescent (as a control) and S-phase-synchronized cells were subjected to in situ extraction and immunostaining for RPA (p32 subunit). Consistent with its role in S-phase, less than 5% of extracted quiescent cells retained detectable RPA staining (Fig. 4A, top graph). However, in S-phase-synchronized populations, RPA tethered to chromatin was detectable in greater than 50% of the extracted cells, and this association was not significantly inhibited by the expression of PSM-RB (Fig. 4A, top graph, compare with and without Dox, and Fig. 4B). As a control for these studies, we monitored PCNA staining (Fig. 4A, bottom graph). PCNA fails to significantly tether to chromatin in quiescent cells (Fig. 4A, bottom graph), and PCNA tethering was again diminished by the induction of PSM-RB (Fig. 4A, bottom graph, compare with and without Dox). Results consistent with the immunostaining were also observed by fractionation (not shown). Thus, PSM-RB does not perturb the chromatin association of RPA while disrupting the tethering of PCNA.
FIG. 4.
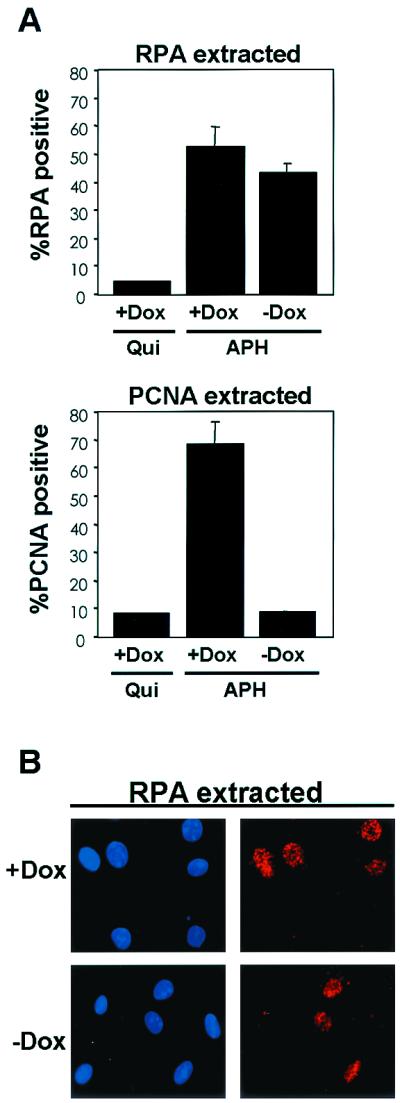
PSM-RB does not disrupt RPA tethering. (A) Cells were seeded on coverslips and synchronized in quiescence (Qui) or in early S phase with APH. Cells were cultured for an additional 16 h in APH medium in either the presence or absence of Dox. Coverslips were extracted, fixed in methanol, and processed for immunofluorescence using RPA p32 (top graph) or PCNA (bottom graph) antibodies. Values shown for APH with and without Dox are the averages of four independent experiments with at least 200 cells counted per experiment; that for the Qui plus Dox control is from duplicate samples with at least 200 cells counted. (B) Representative photomicrographs of RPA (APH with and without Dox) staining as described in panel A. Magnification, ×60.
RB-mediated regulation of PCNA correlates with inhibition of CDK2-cyclin A activity.
To determine if the effect of RB on PCNA was direct, we examined the kinetics of RB induction and the dissociation of PCNA from chromatin (Fig. 5A to C). Cells were synchronized in S phase, and PSM-RB expression was induced by removal of Dox (in the presence of APH). PSM-RB expression was first detected within 4 h of removal of Dox in APH-synchronized cultures (Fig. 5A). Analysis of PCNA tethering in these APH-synchronized cells by fractionation showed that greater than 80% of tethered PCNA was dissociated from chromatin 16 h after removal of Dox (Fig. 5B). Similar results were observed by immunostaining analysis (Fig. 5C). Since PSM-RB expression significantly preceded the dissociation of PCNA from chromatin, this suggested that RB does not act directly upon PCNA but that an intermediate step is involved.
FIG. 5.

RB-mediated inhibition of CDK2-cyclin A correlates with the disruption of PCNA function. (A) Cells were synchronized in early S phase with APH. PSM-RB expression was then induced by culturing cells in the absence of Dox. Cells were harvested at the indicated times following removal of Dox. Total cellular protein was resolved by SDS-PAGE, and PSM-RB was detected by immunoblotting. (B) S-phase-synchronized cells were cultured in the presence of APH and Dox for an additional 16 h (lane 1) or without Dox (lanes 2, 3, and 4) for the indicated times. Either whole-cell lysates (total) or chromatin-bound proteins (pellet) were resolved by SDS-PAGE. Lamin-B and PCNA were detected by immunoblotting. (C) Cells were seeded on coverslips and synchronized with APH. PSM-RB expression was induced by the removal of Dox. Coverslips were washed with PBS, either extracted with CSK or mock extracted, and then fixed at the indicated times. PCNA was detected by immunostaining. Values shown are the averages from four experiments with at least 150 cells counted per experiment. (D) Cells were synchronized in S phase with APH and then cultured for an additional 16 h with APH in the presence or absence of Dox. (Left) Equal total protein from cells cultured in Dox (lane 1) or in the absence of Dox (lane 2) was resolved by SDS-PAGE, and CDK2, cyclin E, and cyclin A protein was detected by immunoblotting. (Right) Total protein (200 μg) was subjected to immunoprecipitation with E1A (control, lane 1) or CDK2 (2 to 5) antibodies. Resulting immune complexes were used for in vitro kinase reactions with histone H1 substrate. Shown are kinase reactions from independent lysates of cells propagated in the presence (lanes 2 and 3) or absence (lanes 4 and 5) of Dox. Data are representative of several independent experiments. (E) (Left) Cells were transfected with 1 μg of CMV-β-gal, 2 μg of cyclin A promoter (human −608CycALuc reporter), and 5 μg of empty vector. Transfected cells were then synchronized with APH. Subsequently, PSM-RB was induced (−Dox) or not (+Dox) for 16 h in the presence of APH. Relative luciferase activity was set to 100% for cells cultured in the presence of Dox. Data shown are from two independent experiments. (Right) Cells were synchronized in S phase with APH and then cultured for 16 h in the presence or absence of Dox. Total RNA was prepared, and RT-PCR was performed with primers specific to cyclin A and GAPDH. Resultant products were resolved by agarose gel electrophoresis and visualized by ethidium bromide staining. The results are representative of three independent experiments. (F) Cell lysates were prepared from APH-synchronized cells that were cultured for an additional 16 h in medium containing or lacking Dox for the indicated times. Total protein was resolved by SDS–10% PAGE and immunoblotted with cyclin A antibody.
RB is implicated in transcriptional repression of cell cycle target genes, such as cyclin E and cyclin A. However, the efficacy of RB signaling to these targets has not been extensively studied in S-phase cultures. Therefore, we initially analyzed the influence of PSM-RB expression on the levels of CDK2, cyclin E, and cyclin A proteins in APH-synchronized cells (Fig. 5D, left panel). CDK2 and cyclin E expression was not significantly attenuated in the presence of PSM-RB (Fig. 5D, left panel, compare lanes 1 and 2). In contrast, cyclin A expression was dramatically inhibited following PSM-RB (no Dox) induction in the S-phase-synchronized cells (Fig. 5D, left panel, lanes 1 and 2). Cyclin A downregulation occurred at the level of RNA, as the endogenous transcript is downregulated (Fig. 5E, right panel). Additionally, the cyclin A promoter was repressed upon PSM-RB induction in S-phase cells (Fig. 5E, left panel). Since cyclin A is the principal activating cyclin for CDK2 in S-phase cells, the influence of PSM-RB expression on CDK2 activity was assessed. We found that the expression of PSM-RB inhibited CDK2 activity in S-phase cells (Fig. 5D, right panel, compare duplicate experiments in lanes 2 and 3 versus 4 and 5). Because cyclin A protein levels were attenuated following PSM-RB expression in S phase, we determined if attenuation of cyclin A expression correlated with loss of PCNA tethering. Downregulation of cyclin A occurred approximately 12 h after removal of Dox (Fig. 5F), just preceding the dissociation of PCNA from chromatin (Fig. 5B and C). Since the kinetics of cyclin A attenuation correlated with the disruption of PCNA-chromatin association, this suggested that RB signaling to CDK2-cyclin A may represent a pathway which regulates PCNA activity.
PCNA chromatin tethering is dependent on CDK2 activity and can be dissociated from the presence of cyclin A.
Cyclin A is known to bind PCNA as part of a ternary complex, suggesting that PCNA tethering to chromatin may be dependent on cyclin A as a binding partner (51), as opposed to the action of cyclin A as an activator of CDK2. To differentiate between these two distinct activities of cyclin A, we determined whether CDK2 activity is required for PCNA-chromatin tethering. A pharmacological inhibitor of CDK2 activity, olomucine, was used to inhibit CDK2 activity independent of PSM-RB expression. As shown in Fig. 6A, 100 μM olomucine significantly inhibited CDK2 activity (left panel, compare duplicate experiments in lanes 3 and 4 versus 5 and 6). Cyclin A expression was retained following 16 h of olomucine treatment in the absence of PSM-RB expression, which contrasts with the ability of PSM-RB to downregulate cyclin A over the same time period (right panel, lanes 1 to 3). Analysis of PCNA activity in APH-synchronized cultures showed that PCNA tethering was disrupted by olomucine (Fig. 6B, black bars), while olomucine had no effect on total PCNA (Fig. 6B, gray bars). Therefore, PCNA tethering in these S-phase cultures is dependent on CDK2 activity and can be dissociated from the presence of cyclin A.
FIG. 6.

PCNA activity can be dissociated from presence of cyclin A: requirement of CDK2 and bypass by DNA damage. (A) (Left) Duplicate E1A (lane 1 and 2) or CDK2 (lanes 3 to 6) immunocomplexes incubated with vehicle (lanes 1 to 4) or 100 μM olomucine (lanes 5 and 6) were used in in vitro kinase reactions with histone H1. (Right) Cells were synchronized with APH and then incubated an additional 16 h in APH medium supplemented with Dox and vehicle (lane 1); Dox and 100 μM olomucine (lane 2); or no Dox with vehicle (lane 3). Total cellular protein was resolved by SDS-PAGE, and PSM-RB and cyclin A proteins were detected by immunoblotting. (B) Cells were seeded on coverslips and synchronized with APH. Cells were cultured either in the presence of Dox, in the presence of Dox and 100 μM olomucine, or in the absence of Dox. Coverslips were washed with PBS and either extracted with CSK (black bars) or mock extracted (gray bars) and then fixed. PCNA was detected by immunostaining. Values are averages from three experiments with at least 150 cells counted per experiment. (C) Asynchronous cells were seeded on coverslips and cultured in the presence or absence of Dox for 16 h. During incubation, cisplatin CDDP (16 μM) was added to cultures for the indicated times. Coverslips were extracted with CSK, and PCNA was detected by immunostaining. Values shown are the averages from two experiments with at least 150 cells counted per experiment. (D) Asynchronous cells which were cultured in either the presence (lane 1) or absence (lanes 2 to 4) of Dox for 16 h were exposed to 16 μM CDDP for the indicated times. Equal total protein was resolved by SDS-PAGE, and PSM-RB and cyclin A were detected by immunoblotting.
DNA damage facilitates the recruitment of PCNA to chromatin, which is not dependent on S-phase positioning. Since RB is also implicated in DNA damage checkpoints (31), we assessed the action of PSM-RB on DNA damage-mediated tethering of PCNA. To perform these experiments, asynchronously growing cells were treated with cisplatin (CDDP) in the presence of PSM-RB (Fig. 6C and D). Active RB inhibited cyclin A expression in the presence of cisplatin (Fig. 6D). However, RB-mediated inhibition of PCNA tethering was overcome by cisplatin damage (Fig. 6C). This result indicates that RB only abrogates the S-phase recruitment of PCNA and that the DNA damage-mediated tethering of PCNA is independent of CDK2-cyclin A activity.
Restoration of CDK2-cyclin A facilitates PCNA tethering in presence of active RB.
To assess the functional role of active CDK2-cyclin A kinase in the disruption of PCNA-chromatin association by PSM-RB, we attempted to restore cyclin A protein in PSM-RB-expressing cells. Previous attempts to produce cyclin A in rat fibroblasts by transfection were unsuccessful and coupled with cell death (26). Therefore, we initially analyzed whether recombinant human cyclin A-encoding adenovirus would be tolerated and induce cyclin A expression in our cell cultures. APH-synchronized cells in the presence or absence of PSM-RB expression were either mock infected or infected with an adenovirus encoding both human cyclin A and GFP. Sixteen hours postinfection, cyclin A protein levels were determined by immunoblotting with antibodies which react preferentially against rodent cyclin A (C19) (Fig. 7A, top panel) or against human cyclin A (H432) (Fig. 7A, bottom panel). Induction of PSM-RB reduced the expression of the endogenous rat cyclin A protein in uninfected cells (Fig. 7A, compare lanes 1 and 2). Expression of endogenous cyclin A was not significantly rescued by infection with the adenovirus encoding human cyclin A (top panel, lane 3), indicating that RB signaling to endogenous cyclin A was intact following infection. However, the virus-encoded human cyclin A was readily detected by immunoblotting (Fig. 7A, bottom panel, lane 3). These cells showed no significant cell death after infection, and the infections did not influence the level of PSM-RB in the cells (not shown). Thus, cyclin A expression can be restored in the presence of PSM-RB in this cell system. To confirm that infected cyclin A was sufficient to restore CDK2 activity, CDK2 activity was monitored after PSM-RB induction and cyclin A infection. As shown in Fig. 7B, cyclin A infection restored CDK2 activity in the presence of PSM-RB (no Dox), similar to levels observed in the absence of PSM-RB (with Dox) (compare duplicate experiments in lanes 2 and 3 versus 4 and 5 versus 6 and 7).
FIG. 7.

Ectopic expression of cyclin A restores PCNA chromatin association. (A) Cells were synchronized in S phase with APH, then cultured in the presence of APH in medium containing Dox (lanes 1) or shifted to APH medium lacking Dox and then immediately infected with adenoviruses encoding GFP or cyclin A-GFP (lanes 2 and 3, respectively). Cells were harvested after 16 h, and total protein was resolved by SDS-PAGE and immunoblotted with either C-19 or H432 cyclin A antibodies. (B) Cell were synchronized in S phase with APH and then cultured in the presence (lanes 1 to 3) or absence (lanes 4 to 7) of Dox and either mock infected (lanes 1 to 5) or infected with the cyclin A-GFP adenovirus (lanes 6 and 7). CDK2 was immunoprecipitated, and resulting immune complexes were used in in vitro kinase reactions against histone H1 (duplicate experiments are shown). (C) Cells were seeded on coverslips and synchronized in S-phase with APH. These cells were then subsequently cultured with and without Dox (with APH) and infected with adenoviruses bearing GFP or cyclin A-GFP. Soluble proteins were extracted, and cells were fixed and processed for PCNA immunofluorescence. Values shown are the averages of three independent experiments with at least 150 cells counted per experiment. Representative photomicrogaphs taken at ×60 magnification are shown. (D) Asynchronously growing cells were noninfected or infected with adenovirus constructs expressing GFP or cyclin A-GFP. Cells were extracted, fixed, and processed for PCNA immunofluorescence. At least 150 cells were counted in the experiment.
To determine the influence of restored CDK2-cyclin A activity on PCNA tethering, infected cells were subjected to in situ extraction and stained for PCNA. As expected, infection with the GFP virus did not influence PCNA-chromatin tethering, whereas PSM-RB induction abrogated this interaction (Fig. 7C, compare with and without Dox). In contrast, restored CDK2-cyclin A activity preempted the ability of PSM-RB to disrupt PCNA tethering (Fig. 7C). These results provide a link between RB-mediated inhibition of CDK2-cyclin A and subsequent disruption of a specific component of the replication machinery. To expand this finding, asynchronous cells were used (Fig. 7D). In such a population, only a limited number of cells will be in S-phase; this approach allowed us to determine whether the overproduction of cyclin A may force PCNA tethering in non-S-phase cells. Approximately 23% of asynchronous cells showed PCNA tethering, consistent with the percentage of cells in S phase, and PCNA tethering was reduced to approximately 5% upon PSM-RB induction (Fig. 7D). In cyclin A-infected cells, tethering was observed in 27% of cells, which was only reduced to 23% by PSM-RB induction (Fig. 7D). Together, these results demonstrate that CDK2-cyclin A-mediated tethering will only occur in primed S-phase cells and that CDK2-cyclin A rescue of the PCNA-chromatin interaction occurs in S-phase cells present in an asynchronous population.
Restoration of CDK2-cyclin A attenuates replication block elicited by active RB.
Since PCNA tethering is required for efficient DNA replication, we evaluated the ability of cells with restored CDK2-cyclin A activity (via cyclin A infection) to progress through S phase in the presence of PSM-RB expression (no Dox). Initially, cells were synchronized with APH, PSM-RB was induced by removal of Dox in the presence of APH, and cells were immediately infected with either GFP or GFP-cyclin A adenovirus. After 16 h, the cells were released from APH and subjected to BrdU labeling for 4 h. Cells were fixed and stained for BrdU incorporation. As shown in Fig. 8A, viral infection had no influence on BrdU incorporation in the absence of PSM-RB expression, as GFP or GFP-cyclin A-infected cells incorporated roughly equivalent amounts of BrdU (60 to 70%). In the presence of PSM-RB, less than 10% of GFP-infected cells incorporated BrdU following APH release (Fig. 8A, GFP), similar to the inhibition observed in uninfected cells (Fig. 2C). In cells infected with cyclin A, 27% of cells incorporated BrdU in the presence of PSM-RB expression (Fig. 8A, GFP plus cyclin A, no Dox). Therefore, ectopic cyclin A expression significantly restored the capacity for DNA replication in the presence of PSM-RB. Similar results were observed with asynchronously proliferating cells (Fig. 8B). Less than 5% of uninfected or GFP-infected cells incorporated BrdU upon PSM-RB expression. Cyclin A expression led to 20% of cells incorporating BrdU; however, this was significantly less than the levels observed in cells which do not express PSM-RB (40 to 50%). Although restoration of the CDK2-cyclin A-PCNA pathway was not sufficient to completely bypass RB-mediated inhibition of DNA replication, these results demonstrate that the ability of RB to regulate PCNA activity is CDK2-cyclin A dependent and plays a critical role in the regulation of DNA replication (Fig. 8C).
FIG. 8.

Partial restoration of DNA synthesis by ectopic cyclin A expression. (A) S-phase-synchronized cells were switched to medium containing APH with or without Dox. These cells were then immediately infected with adenovirus constructs expressing GFP or cyclin A-GFP. Following 16 h, cells were released from APH block and pulse labeled with BrdU. Cells were fixed and processed for BrdU immunofluorescence. Average values from three independent experiments are shown, with at least 150 cells counted per experiment. Photomicrographs are shown, taken at ×60 magnification. (B) Asynchronously proliferating cells were infected immediately following removal of Dox from the medium. These cells were then labeled with BrdU 16 h post infection, fixed, and stained for BrdU incorporation. Data shown are from two independent experiments with at least 100 cells counted per experiment. (C) Model of PSM-RB-mediated S-phase inhibition. In S-phase cells, PSM-RB specifically downregulates cyclin A protein levels and CDK2 kinase activity. This leads to the disruption of PCNA activity without influencing RPA or MCM2 function. Restoration of cyclin A protein leads to a restoration of PCNA tethering and a partial recovery of DNA replication in the presence of PSM-RB. This finding argues for an additional pathway through which RB can inhibit DNA replication.
DISCUSSION
Recent studies have shown that RB inhibits cell cycle progression in S phase and that this function of RB is distinct from its role in G1. The S-phase inhibitory action of RB is invoked by specific types of stress, underscoring the importance of this function in controlling inappropriate cell cycle progression. However, the mechanism by which RB halts DNA replication has remained elusive. Here we report that RB regulates PCNA activity. Using cells harboring inducible expression of active PSM-RB, we showed that S-phase inhibition by RB coincided with the specific dissociation of PCNA from chromatin. This effect on PCNA was dependent on the downregulation of CDK2 kinase activity, as achieved (in the case of RB) through downregulation of cyclin A, and that RB-mediated PCNA regulation can be dissociated from the presence of cyclin A alone. Restoration of CDK2-cyclin A activity restored PCNA chromatin tethering even in the presence of activated RB. The critical nature of the RB-CDK2-PCNA pathway was indicated by the partial restoration of S-phase progression by ectopic cyclin A expression in the presence of PSM-RB. These studies delineate a signaling pathway from RB through CDK2 to the replication machinery in S phase (Fig. 8C).
RB is believed to mediate cell cycle inhibition by repressing the expression of numerous proteins whose promoters contain E2F binding sites (24). This mechanism of RB function is believed to act principally in G1, as the E2F partner DP-1 is inactive in S-phase cells (34, 35). However, we find that in S-phase cells RB does have the capacity to inhibit E2F-dependent transcription (not shown). Additionally, active RB expression in S-phase cells downregulates the cyclin A promoter and leads to attenuation of cyclin A RNA and protein levels. These results, coupled with studies on the RB-mediated DNA-damage response (31), indicate that the transcriptional repression function of RB contributes to S-phase inhibition.
The activity of CDK2-cyclin A complexes is important for DNA replication. CDK2-cyclin A has been shown to promote DNA replication in vitro (16), while ablation of CDK2 activity has been shown to inhibit DNA replication in vivo (45). Several DNA replication proteins are substrates for cyclin A-associated kinase activity; however, the role of their phosphorylation in the promotion of DNA replication is not clear (2, 12, 14, 17). For example, cdc6 is phosphorylated by CDK2-cyclin A and subsequently translocated to the cytoplasm and degraded (23, 48). However, the phosphorylation of cdc6 is apparently not required for DNA replication (47). CDK2-cyclin A complexes phosphorylate MCM proteins, but this phosphorylation inhibits their helicase activity, which would be contrary to a positive effect of cyclin A on DNA replication (22). RPA and the large subunit of DNA polymerase delta are substrates of CDK-cyclin complexes; however, phosphorylation has not been linked to changes in activity or DNA replication (61). For example, elimination of all CDK-cyclin sites in the RPA p32 subunit fails to influence RPA function (20), which is consistent with our finding that expression of PSM-RB and resultant inhibition of CDK2 activity does not inhibit RPA-chromatin association. During DNA replication, PCNA functions as a homotrimeric clamp, which facilitates the assembly and processivity of the DNA polymerase holoenzyme (61). In this active functional state, PCNA is tightly tethered to chromatin and resistant to extraction with low concentrations of Triton X-100. We find that in the absence of PSM-RB expression (and in the presence of CDK2-cyclin A), APH-synchronized cells exhibit PCNA tethering. This result is consistent with previous results that tethered PCNA is detected in APH-synchronized cells (3, 7). Following PSM-RB induction, PCNA association with chromatin was disrupted with a significant delay. This result argues against a direct influence of the RB protein on PCNA and for an intermediary step. We found that during this lag period, cyclin A protein levels were specifically attenuated, implying that RB leads to the dissociation of PCNA from chromatin via regulation of CDK2-cyclin A activity. The causal relationship between cyclin A and PCNA tethering was illustrated by restoring PCNA tethering through the ectopic expression of cyclin A in the APH-synchronized cells expressing PSM-RB. Interestingly, we failed to detect disruption of MCM2 or RPA tethering by PSM-RB. This finding indicates that PSM-RB does not target the establishment or maintenance of the preRC complex or early initiation events; rather, RB is targeting the recruitment of specific machinery involved in replication.
A variety of studies have shown that disruption of PCNA activity inhibits DNA replication. In Saccharomyces cerevisiae, mutation of PCNA stalls DNA replication (61). In mammalian cell extracts, disruption of PCNA function inhibits in vitro DNA replication (8, 49, 60). In the presence of PSM-RB, we find that although PCNA protein levels are not altered, the PCNA protein is no longer active, as determined by chromatin binding. Since efficient DNA replication does not occur in the absence of PCNA function, this clearly represents a mechanism through which RB inhibits DNA replication. Restoration of PCNA tethering through restored CDK2-cyclin A activity enabled a significant induction of DNA replication in the presence of active RB.
The RB-CDK2-cyclin A pathway is independent of the DNA damage-induced recruitment of PCNA, since DNA damage can actually rescue PCNA tethering in the presence of active RB. It is believed that PCNA is recruited to sites of DNA damage to facilitate repair processes. This role of PCNA in the DNA damage response has been previously dissociated from DNA replication. For example, in in vitro systems, it is possible to inhibit PCNA-dependent DNA replication without inhibiting nucleotide excision repair (55). Additionally, DNA damage promotes the chromatin association of PCNA in non-S-phase (quiescent) cells (59). How PCNA is recruited to damaged chromatin remains unclear and perhaps lesion specific. A number of PCNA-associated proteins are implicated in DNA damage response and repair (MSH2, MLH1, GADD45, and hRad9), suggesting that these factors could mediate chromatin association (15, 33, 50).
The mechanism through which CDK2-cyclin A acts to stimulate or maintain PCNA tethering is at present unknown. The requirement for cyclin A lies in its ability to activate CDK2 and not apparently the simple association of cyclin A with PCNA. Ectopic overproduction of cyclin E also promotes PCNA tethering in the presence of PSM-RB (not shown), further supporting the idea that CDK2 activity is the key determinant of PCNA activity. PCNA has a single conserved site (Thr-209) for CDK phosphorylation, and it has been shown that phosphorylated PCNA is specifically tethered to chromatin (51). However, the Thr-209 site has never been described as being phosphorylated or having functional consequence. While direct phosphorylation of PCNA is appealing, CDK2-cyclin A may stimulate phosphorylation of other components of the replication machinery, such as RFC, which are required to assemble PCNA complexes on chromatin (61). Our experiments demonstrate that in populations of APH-synchronized cells, where PCNA is largely tethered to chromatin, induction of PSM-RB disrupts this existing association. This finding suggests two possible mechanisms: first, that CDK2-cyclin A is required for maintaining PCNA on chromatin, and second, that PCNA is continuously dissociating and associating with chromatin and CDK2-cyclin A is required for PCNA loading. Interestingly, PCNA tethering is dependent on both cell cycle phase and the presence of cyclin A. In asynchronously proliferating cells, PCNA tethering was only observed in approximately 25% of cells, and after ectopic expression of cyclin A, there was no increase in PCNA tethering above this threshold, despite the observation that greater than 95% of cells were infected with cyclin A-producing adenovirus. Together, these results indicate that the inactivation of PCNA tethering by RB is an S-phase event which can be specifically restored through CDK2 activity.
Although ectopic cyclin A expression completely restored the tethering of PCNA to chromatin, it did not fully restore DNA replication in the presence of PSM-RB expression. It could be argued that high-level expression of cyclin A may have a deleterious effect on DNA replication; however, we failed to detect any influence of cyclin A on DNA replication in the absence of PSM-RB expression. Thus, PSM-RB likely has additional targets which are involved in DNA replication (Fig. 8C). In the absence of efficient RB-mediated transcriptional repression, RB fails to elicit cell cycle inhibition in both G1 and S phase (57, 65), suggesting that additional transcriptional targets of RB may facilitate the inhibition of S-phase progression. Clearly the requirement for these targets is not strictly essential for replication, since the ectopic expression of cyclin A does facilitate DNA replication in approximately half of the PSM-RB arrested cells. Since RB represses the expression of numerous proteins involved in nucleotide biosynthesis (13, 44), an intriguing possibility would be that the levels of these enzymes and/or pools of deoxyribonucleotides might dictate whether replication is capable of proceeding in the presence of ectopic cyclin A expression.
In summary, we demonstrate that active RB inhibits CDK2-cyclin A activity in S phase, resulting in the loss of PCNA-chromatin association. These data reveal a new pathway of RB function that contributes to its efficacy as an inhibitor of cellular proliferation.
ACKNOWLEDGMENTS
We thank Shelley Barton, Kenji Fukasawa, Jean Wang, and Peter Stambrook for thought provoking discussion and critical reading of the manuscript. We are grateful to Sofie Salama and Ed Harlow for the provision of Rat-16 cells. RPA antibodies were kindly provided by Thomas Melendy and Marc Wold. George Babcock and Jim Cornelius provided exquisite flow cytometric analysis. Special thanks to Gustavo Leone, who provided the recombinant adenoviruses.
This work was supported by grant CA82525 to E.S.K. from the NIH/NCI. E.S.K. is a Kimmel Scholar.
The first two authors contributed equally.
REFERENCES
- 1.Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway in cell cycle control and cancer. Exp Cell Res. 1997;237:1–6. doi: 10.1006/excr.1997.3776. [DOI] [PubMed] [Google Scholar]
- 2.Bashir T, Horlein R, Rommelaere J, Willwand K. Cyclin A activates the DNA polymerase delta-dependent elongation machinery in vitro: a parvovirus DNA replication model. Proc Natl Acad Sci USA. 2000;97:5522–5527. doi: 10.1073/pnas.090485297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bravo R. Synthesis of the nuclear protein cyclin (PCNA) and its relationship with DNA replication. Exp Cell Res. 1986;163:287–293. doi: 10.1016/0014-4827(86)90059-5. [DOI] [PubMed] [Google Scholar]
- 4.Brehm A, Miska E A, McCance D J, Reid J L, Bannister A J, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 5.Bremner R, Cohen B L, Sopta M, Hamel P A, Ingles C J, Gallie B L, Phillips R A. Direct transcriptional repression by pRB and its reversal by specific cyclins. Mol Cell Biol. 1995;15:3256–3265. doi: 10.1128/mcb.15.6.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brylawski B P, Cohen S M, Longmire J L, Doggett N A, Cordeiro-Stone M, Kaufman D G. Construction of a cosmid library of DNA replicated early in the S phase of normal human fibroblasts. J Cell Biochem. 2000;78:509–517. doi: 10.1002/1097-4644(20000901)78:3<509::aid-jcb15>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 7.Bunch R T, Eastman A. 7-Hydroxystaurosporine (UCN-01) causes redistribution of proliferating cell nuclear antigen and abrogates cisplatin-induced S-phase arrest in Chinese hamster ovary cells. Cell Growth Differ. 1997;8:779–788. [PubMed] [Google Scholar]
- 8.Chen J, Peters R, Saha P, Lee P, Theodoras A, Pagano M, Wagner G, Dutta A. A 39 amino acid fragment of the cell cycle regulator p21 is sufficient to bind PCNA and partially inhibit DNA replication in vivo. Nucleic Acids Res. 1996;24:1727–1733. doi: 10.1093/nar/24.9.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chew Y P, Ellis M, Wilkie S, Mittnacht S. pRB phosphorylation mutants reveal role of pRB in regulating S phase completion by a mechanism independent of E2F. Oncogene. 1998;17:2177–2186. doi: 10.1038/sj.onc.1202443. [DOI] [PubMed] [Google Scholar]
- 10.Dimitrova D S, Todorov I T, Melendy T, Gilbert D M. Mcm2, but not RPA, is a component of the mammalian early G1-phase prereplication complex. J Cell Biol. 1999;146:709–722. doi: 10.1083/jcb.146.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dimitrova D S, Gilbert D M. Temporally coordientated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol. 2000;2:686–694. doi: 10.1038/35036309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donovan S, Diffley J F. Replication origins in eukaroytes. Curr Opin Genet Dev. 1996;6:203–207. doi: 10.1016/s0959-437x(96)80051-7. [DOI] [PubMed] [Google Scholar]
- 13.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 14.Findeisen M, El-Denary M, Kapitza T, Graf R, Strausfeld U. Cyclin A-dependent kinase activity affects chromatin binding of ORC, Cdc6, and MCM in egg extracts of Xenopus laevis. Eur J Biochem. 1999;264:415–426. doi: 10.1046/j.1432-1327.1999.00613.x. [DOI] [PubMed] [Google Scholar]
- 15.Flores-Rozas H, Clark D, Kolodner R D. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- 16.Fotedar A, Cannella D, Fitzgerald P, Rousselle T, Gupta S, Doree M, Fotedar R. Role for cyclin A-dependent kinase in DNA replication in human S phase cell extracts. J Biol Chem. 1996;271:31627–31637. doi: 10.1074/jbc.271.49.31627. [DOI] [PubMed] [Google Scholar]
- 17.Fotedar R, Fotedar A. Cell cycle control of DNA replication. Prog Cell Cycle Res. 1995;1:73–89. doi: 10.1007/978-1-4615-1809-9_6. [DOI] [PubMed] [Google Scholar]
- 18.Fujita M. Cell cycle regulation of DNA replication initiation proteins in mammalian cells. Front Biosci. 1999;4:D816–D823. doi: 10.2741/fujita. [DOI] [PubMed] [Google Scholar]
- 19.Fujita M, Kiyono T, Hayashi Y, Ishibashi M. hCDC47, a human member of the MCM family: dissociation of the nucleus-bound form during S phase. J Biol Chem. 1996;271:4349–4354. doi: 10.1074/jbc.271.8.4349. [DOI] [PubMed] [Google Scholar]
- 20.Henricksen L A, Wold M S. Replication protein A mutants lacking phosphorylation sites for p34cdc2 kinase support DNA replication J. Biol Chem. 1994;269:24203–24208. [PubMed] [Google Scholar]
- 21.Hinds P W, Mittnacht S, Dulic V, Arnold A, Reed S I, Weinberg R A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 22.Ishimi Y, Komamura-Kohno Y, You Z, Omori A, Kitagawa M. Inhibition of Mcm4,6,7 helicase activity by phosphorylation with cyclin A/Cdk2. J Biol Chem. 2000;275:16235–16241. doi: 10.1074/jbc.M909040199. [DOI] [PubMed] [Google Scholar]
- 23.Jiang W, Wells N J, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci USA. 1999;96:6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaelin W G., Jr Recent insights into the functions of the retinoblastoma susceptibility gene product. Cancer Investig. 1997;15:243–254. doi: 10.3109/07357909709039722. [DOI] [PubMed] [Google Scholar]
- 25.Kearsey S E, Maiorano D, Holmes E C, Todorov I T. The role of MCM proteins in the cell cycle control of genome duplication. Bioessays. 1996;18:183–190. doi: 10.1002/bies.950180305. [DOI] [PubMed] [Google Scholar]
- 26.Knudsen E S, Buckmaster C, Chen T T, Feramisco J R, Wang J Y. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 1998;12:2278–2292. doi: 10.1101/gad.12.15.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knudsen E S, Wang J Y. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 28.Knudsen E S, Wang J Y. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knudsen K E, Weber E, Arden K C, Cavenee W K, Feramisco J R, Knudsen E S. RB inhibits cellular proliferation through two distinct mechanisms: inhibition of cell cycle progression and induction of cell death. Oncogene. 1999;20:345–349. doi: 10.1038/sj.onc.1202910. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen K E, Arden K C, Cavenee W K. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998;273:20213–20222. doi: 10.1074/jbc.273.32.20213. [DOI] [PubMed] [Google Scholar]
- 31.Knudsen K E, Booth D, Naderi S, Sever-Chroneos Z, Fribourg A F, Hunton I, Feramisco J R, Wang J Y J, Knudsen E S. RB-dependent S-phase response to DNA-damage. Mol Cell Biol. 2000;20:7751–7763. doi: 10.1128/mcb.20.20.7751-7763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knudsen K E, Fribourg A F, Strobeck M W, Blanchard J M, Knudsen E S. Cyclin A is a functional target of retinoblastoma tumor suppressor protein-mediated cell cycle arrest. J Biol Chem. 1999;274:27632–27641. doi: 10.1074/jbc.274.39.27632. [DOI] [PubMed] [Google Scholar]
- 33.Komatsu K, Wharton W, Hang H, Wu C, Singh S, Lieberman H B, Pledger W J, Wang H G. PCNA interacts with hHus1/hRad9 in response to DNA damage and replication inhibition. Oncogene. 2000;19:5291–5297. doi: 10.1038/sj.onc.1203901. [DOI] [PubMed] [Google Scholar]
- 34.Krek W, Ewen M E, Shirodkar S, Arany Z, Kaelin W G, Jr, Livingston D M. Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell. 1994;78:161–172. doi: 10.1016/0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 35.Krek W, Xu G, Livingston D M. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell. 1995;83:1149–1158. doi: 10.1016/0092-8674(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 36.Levenson V, Hamlin J L. A general protocol for evaluating the specific effects of DNA replication inhibitors. Nucleic Acids Res. 1993;21:3997–4004. doi: 10.1093/nar/21.17.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukas C, Sorensen C S, Kramer E, Santoni-Rugiu E, Lindeneg C, Peters J M, Bartek J, Lukas J. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature. 1999;401:815–818. doi: 10.1038/44611. [DOI] [PubMed] [Google Scholar]
- 38.Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol Cell Biol. 1995;15:2600–2611. doi: 10.1128/mcb.15.5.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukas J, Herzinger T, Hansen K, Moroni M C, Resnitzky D, Helin K, Reed S I, Bartek J. Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- 40.Lukas J, Parry D, Aagaard L, Mann D J, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- 41.Lukas J, Sorensen C S, Lukas C, Santoni-Rugiu E, Bartek J. p16INK4a, but not constitutively active pRb, can impose a sustained G1 arrest: molecular mechanisms and implications for oncogenesis. Oncogene. 1999;18:3930–3935. doi: 10.1038/sj.onc.1202777. [DOI] [PubMed] [Google Scholar]
- 42.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain J P, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 43.Mittnacht S. Control of pRB phosphorylation. Curr Opin Genet Dev. 1998;8:21–27. doi: 10.1016/s0959-437x(98)80057-9. [DOI] [PubMed] [Google Scholar]
- 44.Nevins J R. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9:585–593. [PubMed] [Google Scholar]
- 45.Ogryzko V V, Wong P, Howard B H. WAF1 retards S-phase progression primarily by inhibition of cyclin-dependent kinases. Mol Cell Biol. 1997;17:4877–4882. doi: 10.1128/mcb.17.8.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohtsubo M, Theodoras A M, Schumacher J, Roberts J M, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pelizon C, Madine M A, Romanowski P, Laskey R A. Unphosphorylatable mutants of cdc6 disrupt its nuclear export but still support DNA replication once per cell cycle. Genes Dev. 2000;14:2526–2533. doi: 10.1101/gad.176300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen B O, Lukas J, Sorensen C S, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999;18:396–410. doi: 10.1093/emboj/18.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prelich G, Kostura M, Marshak D R, Mathews M B, Stillman B. The cell-cycle regulated proliferating cell nuclear antigen is required for SV40 DNA replication in vitro. Nature. 1987;326:471–475. doi: 10.1038/326471a0. [DOI] [PubMed] [Google Scholar]
- 50.Prosperi E. Multiple roles of the proliferating cell nuclear antigen: DNA replication, repair and cell cycle control. Prog Cell Cycle Res. 1997;3:193–210. doi: 10.1007/978-1-4615-5371-7_15. [DOI] [PubMed] [Google Scholar]
- 51.Prosperi E, Scovassi A I, Stivala L A, Bianchi L. Proliferating cell nuclear antigen bound to DNA synthesis sites: phosphorylation and association with cyclin D1 and cyclin A. Exp Cell Res. 1994;215:257–262. doi: 10.1006/excr.1994.1341. [DOI] [PubMed] [Google Scholar]
- 52.Roussel M F. Key effectors of signal transduction and G1 progression. Advances in Cancer Res. 1998;74:1–24. doi: 10.1016/s0065-230x(08)60763-0. [DOI] [PubMed] [Google Scholar]
- 53.Saudan P, Vlach J, Beard P. Inhibition of S-phase progression by adeno-associated virus rep78 protein is mediated by hypophosphorylated pRb. EMBO J. 2000;19:4351–4361. doi: 10.1093/emboj/19.16.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sherr C J. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 55.Shivji M K, Grey S J, Strausfeld U P, Wood R D, Blow J J. Cip1 inhibits DNA replication but not PCNA-dependent nucleotide excision-repair. Curr Biol. 1994;4:1062–1068. doi: 10.1016/s0960-9822(00)00244-x. [DOI] [PubMed] [Google Scholar]
- 56.Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- 57.Strobeck M W, DeCristafaro M, Fribourg A F, Knudsen K E, Weissman B, Imbalzano A N, Knudsen E S. Brg-1 is required for RB-mediated arrest. Proc Natl Acad Sci USA. 2000;57:3475–3480. doi: 10.1073/pnas.97.14.7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Todorov I T, Attaran A, Kearsey S E. BM28, a human member of the MCM2–3-5 family, is displaced from chromatin during DNA replication. J Cell Biol. 1995;129:1433–1445. doi: 10.1083/jcb.129.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toschi L, Bravo R. Changes in cyclin/proliferating cell nuclear antigen distribution during DNA repair synthesis. J Cell Biol. 1988;107:1623–1628. doi: 10.1083/jcb.107.5.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waga S, Hannon G J, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 61.Waga S, Stillman B. The DNA replication fork in eukaryotic cells. Annu Rev Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 62.Wang J Y, Knudsen E S, Welch P J. The retinoblastoma tumor suppressor protein. Adv Cancer Res. 1994;64:25–85. doi: 10.1016/s0065-230x(08)60834-9. [DOI] [PubMed] [Google Scholar]
- 63.Weinberg R A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 64.Welch P J, Wang J Y. A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Ab1 tyrosine kinase in the cell cycle. Cell. 1993;75:779–790. doi: 10.1016/0092-8674(93)90497-e. [DOI] [PubMed] [Google Scholar]
- 65.Zhang H S, Gavin M, Dahiya A, Postigo A A, Ma D, Luo R X, Harbour J W, Dean D C. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]