

Abstract
Triple negative breast cancer (TNBC) cells lack expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor‐2 (HER‐2). Thus, TNBC does not respond to hormone‐based therapy. TNBC is also an aggressive subtype associated with poorer prognoses compared to other breast cancers. Conventional chemotherapeutics are used to manage TNBC although systemic relapse is common with limited benefits being reported as well as adverse events being documented. Here, we discuss current therapies for TNBC in the neo‐ and adjuvant settings, as well as recent advancements in the targeting of PD‐L1‐positive tumors and inclusion of PARP inhibitors for TNBC patients with BRCA mutations. The recent development of cyclin‐dependent kinase (CDK) 4/6 inhibitors in ER‐positive breast cancers has demonstrated significant improvements in progression free survival in patients. Here, we review preclinical data of CDK 4/6 inhibitors and describe current clinical trials assessing these in TNBC disease.
Keywords: CDK/4/6 inhibitors, metastatic, triple negative breast cancer
Triple negative breast cancer (TNBC) is an aggressive subtype associated with poorer prognoses compared to other breast cancers. Conventional chemotherapeutics are used to manage TNBC although systemic relapse is common with limited benefits being reported. The identification of alternative pathways important for the survival of cancer cells has led to the development of CDK4/6 inhibitors that have shown great promise in preliminary clinical trials in TNBC patients, in the presence of chemotherapy and alone as monotherapy. With early preclinical studies further novel agents have been identified that may lead to new strategies in treating TNBC and providing improved prognoses for patients.
1. INTRODUCTION
Breast cancer (BC) remains the most common cause of death in women, resulting in over half a million deaths annually worldwide. 1 BC falls into four main subtypes, and the most common are luminal A (LBC‐A) and luminal B (LBC‐B) that represent 60% (40% and 20%, respectively) of cases. The more aggressive human epidermal growth factor receptor‐2 (HER‐2)‐positive and triple negative (lacking ER/PR and HER‐2) BC (TNBC) subtypes account for approximately 25% and 15% of cases, respectively. Each subtype is characterized by prognostic markers that can be identified by molecular profiling. 2 LBC‐A is positive for the estrogen receptor (ER) and/or progesterone receptor (PR) receptors but negative for HER‐2. Similarly, LBC‐B is ER and/or PR positive but can also be HER‐2 positive or negative. Thus, LBC‐B has a higher rate of recurrence and poorer prognosis compared to LBC‐A. Growth of ER‐/PR‐positive BCs is heavily dependent on the expression of these receptors as they stimulate signaling pathways involved in cellular processes such as proliferation, apoptosis, and angiogenesis. 3 This dependence on ER/PR signaling has allowed for the development of endocrine therapies that target these receptors and their downstream pathways, such as ER blockers (often known as selective ER modulators [SERMs]), aromatase inhibitors (AIs), and ER downregulators (selective ER downregulators [SERDs]). Tamoxifen (SERM), letrozole (AI), and fulvestrant (SERD) are examples of endocrine therapies that are widely used in ER‐positive BC, often recommended to patients for a 5‐ or 10‐year period after breast surgery with significant improved outcomes. 4 , 5 In HER‐2‐positive BC, overexpression of the HER‐2 receptor has served as a suitable target for novel agents. For example, the monoclonal antibodies, trastuzumab and pertuzumab, cause downregulation and inhibition of the HER‐2 and HER‐1 receptors, respectively, resulting in improved survival outcomes. 6 Additionally, in HER‐2‐positive BC, the antibody‐conjugated agents TDM1 (trastuzumab conjugated to the cytotoxic agent DM1) and T‐Dxd (trastuzumab deruxtecan) have shown significant antitumor activity and are used as standard therapy in early and advanced disease, respectively. 7 , 8 , 9 , 10 In contrast, TNBC accounts for 170,000 cases annually worldwide in which patients have poor prognosis and overall survival (OS) as well as higher distant recurrence rates. 11 , 12 Although initially responsive to chemotherapy, TNBC patients have relatively short disease‐free survival (DFS) rates compared to those with hormone‐receptor‐positive disease. 13 Coupled with the lack of targetable receptors, effective treatment remains a challenge for TNBC patients with novel agents urgently needed to improve outcome.
2. TNBC PATHOLOGY AND METASTASIS
TNBC falls into four main subtypes identified by early gene profiling studies and histological analyses; basal‐like, luminal (androgen receptor [AR]) like (LAR), mesenchymal (MES), and mesenchymal stem like (MSL). Although the terms basal‐like BC and TNBC are often used interchangeably, they are not the same. TNBC is a description of the immunophenotype of BC that is negative for ER, PR, and HER‐2, whereas basal‐like BC describes the molecular phenotype, with approximately 50% of TNBC falling within this subtype (Figure 1). 14 Basal‐like BC, first discovered by first generation cDNA microarrays, 15 is characterized by enriched cell cycle and cell division pathways, as well as elevated DNA damage response pathways. 14 , 16 Additionally, immune suppressed basal‐like (IM) subtypes, identified by gene profiling, are enriched for factors involved in immune cell signaling (basal‐like immune activated [BLIA]), resulting in a favorable prognosis despite its association with a high‐grade histology. 17 Burstein et al. also described an IM subtype (basal‐like immune suppressed [BLIS]) that exhibited low expression of immune cell differentiation and immune signaling and is associated with poor prognosis as demonstrated by low DFS rates. 17 The LAR subtype is characterized by its overexpression of the AR, 10‐fold greater than the other subtypes. 18 Although ER negative, LAR BC cells may express ESR1 (gene encoding ER) and other estrogen‐regulated genes, as well as pathways that regulate steroid synthesis, porphyrin metabolism, and androgen/estrogen metabolism. 14 The MES and MSL subtypes are heavily enriched in mechanisms involved in cell motility and extracellular receptor interaction and cell differentiation. 14 , 16 More recent analyses of breast tumors obtained from the molecular taxonomy of BC international consortium (METABRIC) identified 10 subtypes that were associated with histological type, tumor grade, receptor status, and lymphocytic infiltration. 19 Further analysis of the METABRIC and The Cancer Genome Atlas (TCGA) databases using copy number variants (CNVs) of the different subtypes proved to be an accurate method for the diagnosis of BCs compared to mRNA biomarkers. 20 These biomarkers, and their associated signaling pathways, provide targetable opportunities for therapeutic agents in TNBC disease.
FIGURE 1.
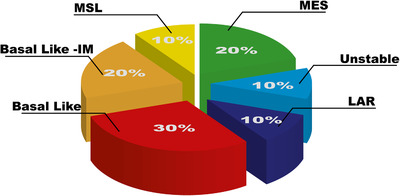
Proportion of TNBC subtypes. The unstable subtype is characterized by its cellular proliferation and responses to DNA damage
Abbreviations: IM, immunomodulatory; LAR, luminal androgen receptor; MES, mesenchymal; MSL, mesenchymal stem like.
Identification of these various subtypes is important for determining the most suitable therapeutic approaches for TNBC patients. In a retrospective study of early BC, investigators revalidated existing gene expression microarray data from 146 TNBC patients, 130 of which had received standard neoadjuvant chemotherapy with evaluable pathologic response data. The authors were then able to classify the TNBC subtypes and correlate them to the pathological complete response (pCR) status. 21 They found that TNBC subtype was significantly associated with pCR status. For example, the basal‐like1 (BL1) subtype was associated with the highest pCR rate (52%), whereas the basal‐like2 (BL2) and LAR had the lowest pCRs (0% and 10%, respectively). 21 Though specific tumor subtypes can be a predictor of pCR, further classification and understanding of these subtypes may help direct personalized therapeutic strategies for TNBC patients who have currently incurable metastatic disease.
2.1. Metastatic TNBC
Metastasis in BC is a complex multistep process that involves the infiltration of tumor cells into the surrounding tissue followed by transendothelial migration into blood vessels (intravasation) and subsequently extravasation into distant sites. 22 Metastasis may also occur through the lymphatic system into the lymph nodes; thus, tumor‐positive lymph nodes are important predictors of tumor aggressiveness for most BCs. 23 More specifically, in metastatic TNBC (mTNBC), a higher rate of node positivity is observed with visceral metastasis more likely to occur in the lungs and brain. 24 , 25 In addition, TNBC patients with visceral metastases demonstrate shorter median survival rates compared to non‐TNBC with limited response to chemotherapy. 26 , 27
3. CURRENT TREATMENTS FOR TNBC
Due to the biologically aggressive nature of TNBC, prognosis is very poor (median OS being 10.2 months with a 5‐year survival rate of 65% for localized tumors and 11% that have spread to distal organs 28 , 29 ) despite patients sometimes responding better to chemotherapy than non‐TNBC. 11 , 30 The lack of target receptors (e.g., ER/PR or HER‐2), means that TNBC patients do not benefit from endocrine or targeted therapies. Therefore, surgery and chemotherapy (alone or in combination) are the modalities available for TNBC. Anthracyclines (A) and taxanes (T) are the mainstay of chemotherapy regimens with the recent addition of platinum‐based agents.
3.1. Neoadjuvant therapeutic agents
Neoadjuvant chemotherapy is commonly used to treat patients with early TNBC, aiming to target DNA repair and cell proliferation mechanisms (Figure 2). A study looking at the relationship between anthracycline‐based neoadjuvant chemotherapy (doxorubicin plus cyclophosphamide, AC) and long‐term endpoints in different subtypes of BC showed that TNBC patients (n = 34) demonstrated the highest clinical response rates compared to HER‐2 (n = 11) and ER‐positive (n = 62) BCs (85% vs 70% vs 47%, respectively). 31 Moreover, pCR rates, defined as the absence of residual invasive tumor in breast and regional nodes at the time of surgery, were significantly higher in TNBC (27%) than ER‐positive patients (7%). A larger study described similar findings where higher pCR rates are observed in TNBC (n = 255) than in non‐TNBC (n = 893) in response to neoadjuvant AC chemotherapy (30% vs 6.7%, respectively). 30 However, in both of the above studies, TNBC OS and distant‐free survival were significantly lower than for non‐TNBC. 31 , 32 Following these findings, the NSABP B‐27 trial evaluated long‐term outcomes in response to AC therapy with the addition of docetaxel (T). 33 Here, 2411 women were assigned into one of three arms; the first arm received AC followed by surgery, the second received AC followed by T and then surgery, and the third arm received AC followed by surgery then T. The addition of T to AC preoperatively resulted in an increase in pCR rates compared to AC alone in TNBC patients (22.8% vs 13.6%, respectively); however, an updated analysis of the study showed that the addition of T did not improve DFS or OS in these patients. 34
FIGURE 2.
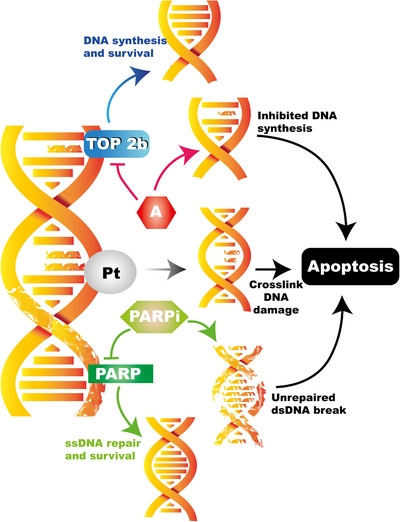
Current TNBC therapeutics aim to disrupt DNA structure thereby leading to DNA damage and consequent cell death. Chemotherapeutic agents such as anthracyclines (A) inhibit molecules required for DNA synthesis. Platinum‐based compounds (Pt) target DNA cross‐linking resulting in cell apoptosis. PARP inhibitors prevent the repair of single strand DNA (ssDNA) damage. The accumulation of ssDNA damage results in unrepairable double strand DNA (dsDNA) breaks leading to cell death
Platinum‐based compounds (such as cisplatin and carboplatin) have also been considered as treatment in TNBC due to their DNA cross‐linking properties (Figure 2), consequently resulting in tumor cell apoptosis. 35 The Alliance phase 2 trial (Cancer and Leukemia Group B 40603) studied pCR rates in 443 stage 2–3 TNBC patients in response to carboplatin treatment with addition of paclitaxel (followed by AC). 36 pCR rates were significantly higher in patients treated with carboplatin, compared to those that received paclitaxel and doxorubicin alone (60% vs 44%; p = 0.0018). 36 Similar results were observed in the parallel GeparSixto trial, also in stage 2–3 TNBC patients receiving paclitaxel and doxorubicin neoadjuvant chemotherapy with the addition of weekly carboplatin treatment. 37 Here, carboplatin‐treated patients demonstrated 53.2% pCR (compared to 36.9%, p = 0.005) without platinum‐based therapy, but significantly more toxic effects were observed in the carboplatin group than the no‐carboplatin group (neutropenia: 65% vs 27%, anemia 15% vs 1%, diarrhea 17% vs 11%). 37 However, DFS and OS were not assessed in these studies; thus, the hypothesis that OS benefit can be predicted by increased rates of pCR, as proposed by Cortazar et al., 38 remains controversial.
3.2. Addition of immunotherapy
Approximately 20% of TNBC cases are highly enriched in immune cell markers, and these are classified as immunomodulatory. Tumors that possess more than 50% tumor‐infiltrating lymphocytes (TILs) are associated with better prognosis with improved OS, increased metastasis‐free survival, and decreased distant recurrence. 39 The immune‐checkpoint receptor, PD1, and its ligand, PD‐L1, are correlated with high levels of TILs in the BC microenvironment and is one of the most common subtypes in TNBC. 40 , 41 The overexpressed PD‐L1 (on tumor cells) binds to the PD1 receptor of activated T‐cells, thereby inhibiting their cytotoxic activities on the tumor cell. Thus, targeting the PD1–PD‐L1 axis has become an attractive approach in TNBC because PD‐L1 is expressed in 20% of all TNBC cases. 42 PD1 inhibitors such as pembrolizumab have proven to be effective in the treatment of lung, melanoma, and bladder cancers. 43 , 44 , 45 In the assessment of safety and antitumor activity of pembrolizumab in TNBC, a phase‐1b (KEYNOTE‐012) trial enrolled 111 TNBC patients, 58.6% of which expressed PD‐L1‐positive tumors. 46 From these, mild toxicities were noted such as arthralgia, fatigue, and nausea with only five patients exhibiting grade ≥ 3 toxicity and the overall response rate was 18.5% (ClinicalTrials.gov identifier: NCT02447003). Currently, an ongoing open‐label, adaptively randomized phase‐2 trial will assess pembrolizumab plus neoadjuvant chemotherapy in stage 2/3 BC patients (ClinicalTrials.gov identifier: NCT01042379). The latest results from this study (obtained in 2017) demonstrated pCR rates of 44% versus 17% (HER2‐negative), 30% versus 13% (HR‐positive), and 60% versus 22% (TNBC) for pembrolizumab versus control, respectively. 47 In a similar phase‐3 trial (IMpassion031), atezolizumab plus chemotherapy was assessed in the neoadjuvant setting compared to placebo plus chemotherapy (ClinicalTrials.gov identifier: NCT03197935). It has been recently reported that pCR rates improved with atezolizumab plus chemotherapy compared to the placebo plus chemotherapy arm (57.6% vs 41.1%, respectively), particularly in PD‐L1‐positive patients with pCR rates reaching 68.8%. 48 Further follow‐up of patients on these studies will establish whether the improved pCR translates into increased DFS or OS.
3.3. Adjuvant chemotherapy
Optimizing early‐stage chemotherapy in TNBC is imperative for reducing the risk of recurrence, distant metastases, and eventual death. Recent guidelines set by the European Society for Medical Oncology (ESMO) do not recommend further adjuvant therapy for patients with TNBC if residual disease is present after completion of neoadjuvant chemotherapy. 49 Despite this, a number of trials have shown that statistically significant improvements were observed in DFS and OS when patients also received adjuvant chemotherapy. 50 , 51 For example, a randomized study assessing the effects of capecitabine following neoadjuvant chemotherapy in 910 patients with HER2‐negative residual invasive BC described longer DFS and OS compared to a noncapecitabine‐treated control group (74.1% vs 67.6% and 89.2% vs 83.5%, respectively). 51 For the patients with TNBC (32.2% of the study population), DFS was 69.8% in the capecitabine group and 56.1% in the control group and OS was also significantly improved (78% vs 70.3%). Adjuvant immunotherapy has also been considered postneoadjuvant chemotherapy in TNBC. The current multicenter phase 2 c‐TRAK‐TN trial utilizes circulating tumor DNA (ctDNA) screening to detect residual disease following primary treatment for TNBC (ClinicalTrials.gov identifier: NCT03145961). Originating from tumor cells, ctDNA are extracellular DNA molecules found in the plasma or serum of cancer patients. ctDNA screening allows for early cancer detection and is able to determine the tissue of origin, prognosis, and detection of minimal residual disease. 52 In the instance of a positive ctDNA result, patients will be randomized into a pembrolizumab treatment arm or an observation arm. Although focused on the utility of ctDNA in monitoring disease progression, this study also includes descriptive differences in time between ctDNA detection and disease recurrence, and DFS, between patients in the pembrolizumab and the observation groups, among the outcome measures.
Conversely, a number of studies have reported no differences in prognosis or survival rates between TNBC patients who received only adjuvant anthracycline‐ and nonanthracycline‐based chemotherapy. 53 , 54 , 55 , 56 However, dose dense adjuvant anthracycline‐based chemotherapy has demonstrated good survival advantages. Increasing the dose intensity of chemotherapy by shortening the intervals between cycles or by administering individual drug sequentially at full dose may improve efficacy. A recent meta‐analysis conducted by the Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) studied the benefits of dose‐intense chemotherapy comparing 2‐weekly versus 3‐weekly schedules reported from 26 trials. 57 Patients receiving dose‐intense chemotherapy exhibited lower recurrences compared to standard‐schedule chemotherapy (10‐year recurrence risk 28.0% vs 31.4%). Ten‐year BC mortality and death without recurrence were also lower in response to dose‐intense chemotherapy than standard schedule (18.9% vs 21.3% and 4.1% vs 4.6%, respectively). Similar reductions in recurrence were observed in 2‐weekly chemotherapy compared to the same treatment given 3‐weekly (10‐year risk; 24% vs 28.3%). This was also observed when anthracycline plus taxanes chemotherapy was administered sequentially as opposed to concurrently (28.1 vs 31.3%). These differences may arise due to heterogeneity of TNBC disease. The success of anthracycline‐based therapies depends on the TNBC subtype and/or the underlying genetic factor (for example, BRCA1 mutations), as it is even suggested that BRCA1‐associated TNBC may be less sensitive to anthracycline‐based therapies compared to sporadic TNBC. 58 BRCA1/2 are tumor suppressor genes that play important roles in DNA damage repair, cell cycle checkpoint control, apoptosis, and transcriptional regulation. 59 Thus, mutations in BRCA1/2 induce defects in the DNA damage repair processes that are associated with the risk of development of BC. 60 The OlympiA phase‐3 trial assessed olaparib, a poly(adenosine diphosphate‐ribose) polymerase inhibitor, in patients with TNBC (and ER+HER2‐) germline BRCA1/2 mutations (ClinicalTrials.gov identifier: NCT02032823). In this study, 1836 participants were enrolled and randomized (1:1) into an olaparib arm or a placebo arm. Patients underwent 12 months of treatment and recent interim results have shown that olaparib resulted in significant improvements in 3 year invasive DFS (85.9% vs 77.1% in the placebo group), 3 year distant DFS (87.5% vs 80.4% in the placebo group), as well as fewer deaths (59 compared to 86 in the placebo group). 61 However contradictory results are still being reported in a number of studies relating to prognosis of neo‐ and adjuvant chemotherapy 62 , 63 , 64 ; thus, the search for alternative therapies has become an imperative avenue for exploration.
3.4. Treatment of metastatic TNBC
Once metastases develop, biopsy assessment is conducted when clinically achievable, to confirm hormone receptor and HER‐2 status, as 8% of tumors that were ER negative convert to ER positive at the metastatic site. 65 , 66 Consequently, modification of the therapeutic approach is necessary upon reevaluation of metastatic disease. 67 Cytotoxic chemotherapy is currently the backbone of first‐line treatment options for mTNBC and is aimed to prolong survival, palliate symptoms, and delay disease progression. Current guidelines recommend that systemic chemotherapy should be individualized based on tumor burden, rate of disease progression, previous chemotherapy treatments, and patient preferences. 68 More recently, the addition of platinum‐based agents to first‐line chemotherapy has been suggested as a more effective approach in mTNBC. In a retrospective cohort study (n = 379), patients treated with platinum‐based chemotherapy demonstrated longer PFS compared to nonplatinum‐based chemotherapy (7.8 vs 4.9 months). 69 Additionally, the phase‐3 Triple Negative Breast Cancer Trial (TNT) compared carboplatin with docetaxel in 400 patients with either mTNBC or with known BRCA1/2 mutations. 70 A 2014 snapshot analysis showed that there were no statistically significant differences in PFS (carboplatin 3.1 months vs docetaxel 4.5 months) and OS (carboplatin 12.3 months vs docetaxel 12.4 months) in the TNBC group. On the other hand, PFS and objective response rates were improved in response to carboplatin in the BRCA1/2 carriers (6.8 months vs 3.1 months and 68% vs 33%, respectively). 71 With these data, a platinum‐based chemotherapy regimen has been recommended by the European Society of Medical Oncology for patients with BRCA‐associated TNBC, if not previously administered. 49
The phase‐3 study, KEYNOTE‐355, with pembrolizumab in mTNBC or inoperable TNBC is being undertaken in two parts (ClinicalTrials.gov identifier: NCT02819518); the first aims to assess the safety of pembrolizumab in combination with chemotherapy on tumors that have not been previously treated with chemotherapy. The second part of the study will compare the safety and efficacy of pembrolizumab plus chemotherapy to placebo plus chemotherapy in the aim that pembrolizumab, in combination with chemotherapy, will prolong progression free survival (PFS) and OS. In parallel, the KEYNOTE‐522 study will assess the safety and efficacy of pembrolizumab plus chemotherapy as neoadjuvant therapy (ClinicalTrials.gov identifier: NCT03036488). Here, after screening and randomization, patients with locally advanced TNBC will receive pembrolizumab plus chemotherapy or placebo plus chemotherapy for 24 weeks (eight cycles). Each patient will then undergo definitive surgery in which adjuvant treatment of pembrolizumab or placebo will be administered for a further 27 weeks (nine cycles). Patients will be monitored for safety, survival, and disease recurrence. A phase‐3 trial (Impassion130) assessed atezolizumab (anti – PD‐L1 antibody) plus nab‐paclitaxel versus placebo plus nab‐paclitaxel in mTNBC. 72 Here, 451 patients were assigned to each group in which PFS and OS were improved in the atezolizumab plus nab‐paclitaxel treatment arm (PFS: 7.2 months vs 5.5 months, OS: 21.3 months vs 17.6 months, respectively). In contrast, the IMpassion131 (phase‐3) trial reported no differences in the treatment arms when atezolizumab and paclitaxel were combined compared to placebo plus paclitaxel (ClinicalTrials.gov identifier: NCT03125902). Nab‐paclitaxel and paclitaxel are drugs from the same class though differ in delivery method. Nab‐paclitaxel is albumin‐bound, whereas paclitaxel is delivered in a solvent and requires pretreatment with steroids. Although yet not clear, this difference maybe a contributing factor to the conflicting results. From these results, all TNBC patients are now PD‐L1 checked at diagnosis of metastatic disease and, following the approval by the FDA, atezolizumab plus nab‐paclitaxel is used as first‐line therapy in PD‐L1‐positive TNBC.
In addition to targeting the PD‐L1 axis, DNA‐targeting molecules such as PARP inhibitors have shown efficacy in mTNBC. The BRCA1 and BRCA2 tumor suppressor genes code for proteins involved in the DNA damage‐sensing process and double‐stranded DNA break repair mechanisms. BRCA1 mutations are present in 50–87% of TNBC patients. 73 , 74 , 75 In addition to BRCA1, the PARP1 and PARP2 enzymes are also activated by DNA single‐strand breaks that subsequently facilitate DNA repair, essential processes for cancer cell survival (Figure 2). In the absence of PARP, the accumulation of single‐strand breaks results in cytotoxic double‐strand breaks that would normally be rectified by BRCA. 76 However, BRCA1‐mutated BC lacks this mechanism; thus, inhibiting PARP poses as an attractive therapeutic approach in BRCA1‐associated TNBC. Studies using in vitro and in vivo models of BRCA1/2 TNBC have shown sensitivity to PARP inhibitors, demonstrating significant tumor regression and longer DFS and OS in mice. 77 , 78 , 79 , 80 The recent olympiAD trial (ClinicalTrials.gov identifier NCT02000622) studied the PARP inhibitor olaparib in 302 mBC patients with known BRCA mutations. 81 Patients were randomized to single‐agent chemotherapy (n = 97) or olaparib (n = 205); 49.8% of patients in the olaparib group and 49.5% of the chemotherapy group exhibited TNBC disease. Response rates and PFS were significantly higher in the olaparib compared to chemotherapy group (59.9% vs 28.8% and 7 vs 4.2 months, respectively), and hence, olaparib was approved for the treatment of metastatic HER‐2‐negative BC patients with BRCA mutations by the FDA in January 2018. A trial of Talazoparib (EMBRACA; ClinicalTrial.gov identifier: NCT01945775), another PARP inhibitor, also resulted in significantly improved PFS and ORR (objective response rate) rates compared to standard therapy (DFS; 8.6 vs 5.6 months, ORR; 62.6% vs 27.2, respectively). 82 Talozoparib is now also FDA approved in patients pretreated with chemotherapy with mTNBC. In addition to BRCA1/2 mutations, a number of (mutated) genes have been identified as biomarkers in TNBC. For example, an inactivating mutation of TP53 that is also involved in DNA damage repair and genome integrity is associated with poor prognosis due to poor responses to chemotherapy. 83 Other biomarkers such as PTEN, PIK3CA, and EGFR have been described in playing roles in TNBC and reviewed in detail by Sporikova et al. 84 More recently, a phase‐3 trial (ASCENT) assessed the trophoblast cell‐surface antigen 2 (Trop‐2) as a potential biomarker for the treatment of mTNBC using the Trop‐2 directed antibody‐drug conjugate sacituzumab govitecan (SG). 85 It is understood that elevated levels of Trop‐2 are associated with tumor growth in TNBC, poor prognoses, and decreased survival. 86 , 87 Biopsy samples of patients with mTNBC were therefore taken to determine Trop‐2 expression levels and were randomized to receive SG (10 mg/kg) or TPC (apecitabine, eribulin, vinorelbine, or gemcitabine). It was found that patients with high and medium expressions of Trop‐2 benefited from SG treatment compared to those with low expression. For example, PFS in SG‐treated and TPC‐treated patients was 6.9 versus 2.5 (high Trop‐2) 5.6 versus 2.2 (medium Trop‐2) and 2.7 versus 1.6 (low Trop‐2). 85 This was also the case for OS (14.2, 14.9, and 9.3 months versus 6.9, 6.9, and 7.6 months) and ORR (44%, 38%, and 22% versus 1%, 11%, and 6%) for SG‐ and TPC‐treated patients with high, medium, and low Trop‐2 expression levels, respectively. Interestingly, it was also found that BRCA1/2 status did not impact the effects of SG as PFS and OS were improved in BRCA1/2‐positive (PFS: 4.6 vs 2.5 months, OS: 15.6 vs 4.4 months SG and TPC, respectively) and BRCA1/2‐negative patients (PFS: 4.9 vs 1.6 months, OS: 10.9 vs 7 months SG and TPC, respectively). This study illustrates the importance of identifying suitable biomarkers, in this case to target DNA repair factors, which allow for successful targeted therapies.
As well as targeting PARP directly, findings from a recent study suggest that targeting PARP‐associated molecules could also prove to be interesting therapeutic approaches. For example, the transcription factor KLF4 (involved in a number of cellular processes such as cell cycle control and genome stability) undergoes PARP‐mediated parylation. 88 More specifically, parylation of KLF4 on the YYR motif by PARP is essential for the survival of cancer cells in which both proteins have been found to be overexpressed in TNBC cell lines. 88 Disruption of the KLF4‐PARP axis by knocking out KLF4 (KLF4−/−) resulted in a dysfunctional DNA damage response in TNBC cell lines. Furthermore, when exposed to the PARP inhibitor, olaparib, KLF4−/− cells significantly enhanced olaparib‐induced cell death in BRCA1‐proficient TNBC cell lines. In contrast, BRCA1‐defient TNBC cell lines did not respond to olaparib regardless of KLF4 status. The sensitizing efficacy for olaparib in BRCA1‐proficeint tumors was also demonstrated in vivo where KLF4−/‐ xenograft tumor growth was significantly perturbed compared to control KLF4−/− tumors. Analysis of these tumors showed that KLF4−/‐ resulted in decreased proliferation and increased cell death in response to olaparib treatment. With such findings from preclinical studies, and clinical outcomes that have provided patients with small successes, the identification of alternative therapeutic targets in TNBC remains a high clinical priority while taking major challenges such as tumor heterogeneity and biomarkers into consideration. 89
4. CYCLIN‐DEPENDENT KINASE INHIBITORS IN METASTATIC TNBC
4.1. Preclinical studies of CDK4/6 inhibitors in TNBC
Cyclin‐dependent kinase (CDK) 4/6 is a key regulator of the transition from the G1 phase of the cell cycle and initiates cell cycle progression. In this pathway, cyclin D1 forms an activating complex with CDK 4 and CDK6 that go on to phosphorylate the retinoblastoma protein (Rb). Once phosphorylated, phosphor‐Rb (pRb) binds to the E2F transcription factors that subsequently regulate the expression of a series of genes that initiate progression through the cell cycle (Figure 3). 90 In cancer, this pathway is dysregulated, resulting in aberrant cell proliferation. 91 Almost 50% of BC patients exhibit cyclin D1 overexpression, and this, in turn, results in the phosphorylation of Rb and progression of the cell cycle. 92 This has led to the development of CDK4/6 inhibitors that have proved successful, alone and in combination with endocrine therapy, in ER‐positive BC. Importantly, not only were direct anticancer effects observed, but sensitization of endocrine therapy‐resistant BCs was induced. 4 , 93
FIGURE 3.

CDK 4/6 mediated cell cycle progression. Under normal conditions, the phosphorylation of the retinoblastoma protein (Rb) by the CDK 4/6—cyclin D complex results in its dissociation from E2F thereby allowing for the transcription of genes for cell cycle progression. CDK4/6 inhibitors such as Palbociclib aim to prevent the progression of the cell cycle and maintain cell cycle arrest
In TNBC, Rb dysfunction occurs in approximately 30% of cases. 94 Additionally, from 180 TNBC patient samples, 51% were found to be Rb positive, thus representing a relevant target for therapy. 95 Early in vitro studies aimed to investigate of the effects of CDK4/6 inhibition to identify potential targetable pathways in TNBC. 93 , 96 Using the CDK4/6 inhibitor palbociclib, Finn et al. showed that growth of a panel of TNBC cell lines was inhibited, although less sensitive to CDK4/6 inhibition than ER‐positive cell lines. 93 A more detailed analysis, using a panel of 12 TNBC cell lines, showed that LAR TNBC cells demonstrated higher sensitivity compared to basal‐like and MES lines that were found to be resistant to the CDK4/6 inhibitors palbociclib and ribociclib. 96 This was also the case for tumors grown in vivo; LAR xenograft tumors were significantly reduced in size when treated with palbociclib compared to the vehicle‐treated group. 96 This limited evidence suggests that particular TNBC subgroups may be sensitive to CDK4/6 inhibition and further preclinical studies are needed to establish how these can be identified.
The mechanisms responsible for CDK4/6 resistance are poorly understood; however, the loss of Rb (observed in 7–20% of TNBC) and overexpression of cyclin E have been shown to confer resistance to CDK4/6 inhibitors. 97 , 98 , 99 Asghar et al. describe that when exiting mitosis, palbociclib‐sensitive TNBC cells exhibit lower levels of CDK2 compared to palbociclib resistant cells. 96 The cyclin E–CDK2 axis also plays an important role in the regulation of the cell cycle 100 ; therefore, the Rb and CDK2 status may act as biomarkers for CDK4/6‐targeted inhibition. In a more recent study, investigators assessed palbociclib in combination with the chemotherapeutic agent paclitaxel in vitro. 101 When MDA‐MB‐231 cells were exposed to a combination of palbociclib and paclitaxel, an antagonistic effect was observed, where the inhibition of cell proliferation caused by palbociclib impeded the cytotoxic effects of paclitaxel. However, sequential treatment with palbociclib followed by paclitaxel resulted in an additive inhibitory effect of cell proliferation. 101 Cell death was also significantly higher following exposure to palbociclib then paclitaxel, compared to that caused by palbociclib or paclitaxel alone. These data demonstrate the importance of palbociclib being administered first when given in sequence with antiproliferative drugs, as this allows tumor cells to reenter the cell cycle synchronously once palbociclib is removed and thus sensitizing cells to the effects of chemotherapeutic agents.
Outside the CDK4/6 axis, it has also been shown that palbociclib reduces glucose metabolism by downregulating the GLUT‐1 glucose transporter in the TNBC MDA‐MB‐231 cell line. 102 Glucose uptake and consumption were further inhibited when cells underwent sequential exposure to palbociclib followed by paclitaxel. 101 Enzalutamide, an AR antagonist that is commonly used in the treatment of prostate cancer, has also shown antitumor effects in TNBC because 30% of TNBC patients demonstrate AR‐positive disease. 103 , 104 , 105 The combination of palbociclib and enzalutamide resulted in an enhanced cytostatic effect compared to palbociclib and enzalutamide alone, with no cell death observed in the TNBC cell lines. 106 In the metastatic setting, an in vivo study investigating the effects of palbociclib on distal site invasion found a significant decrease in liver (12% vs 75%) and lung (25% vs 75%) metastases (compared to saline treated mice) in TNBC xenograft models. 107 Treatment with palbociclib, initiated after the resection of the primary tumor, inhibited lung colonization resulting in a significantly lower number of lung nodules compared to the saline treated control group. In the mechanism of invasion and EMT, SNAIL1 is a key regulator of this process by repressing the expression of CDH1 (the gene encoding E‐cadherin) and activating the expression of invasion‐associated genes. 108 , 109 Upon treatment with palbociclib, a decrease in SNAIL1 protein stability and increased ubiquitination (marking proteins for degradation) was observed in TNBC cells. It was also shown that palbociclib did not directly interact with SNAIL1, but instead acted through the phosphorylation of deubiquitinating enzyme 3 (DUB3), which, in turn, downregulates SNAIL1, 107 suggesting a new target for palbociclib. Separately, a recent study investigated the effects of palbociclib treatment preceding cisplatin chemotherapy. 110 In the MDA‐MB‐231 and MDA‐MB‐468 cell lines, palbociclib alone resulted in cell cycle arrest as expected, but when cells were treated in combination with cisplatin, cell apoptosis was unaffected. This is due to the inability of the chemotherapeutic agent to act on already arrested cells. However, when cells were treated with palbociclib for 48 h followed by its removal for 48 h, then treated with cisplatin for 48 h, significantly increased apoptosis was observed compared to monotherapy. Additionally, sequential treatment resulted in increased DNA damage and lower cell viability compared to single drug treatments. The MDA‐MB‐231 xenograft model was used to study the effects of sequential treatment in vivo. Mice were treated with vehicle (PBS), palbociclib only, cisplatin only, or palbociclib followed by cisplatin 48 h later. Palbociclib alone did not affect tumor volume and weight, whereas cisplatin alone caused a significant decrease compared to vehicle control. 110 Further inhibition in tumor growth was observed in the sequential treatment group, with lower tumor Ki‐67 expression than that seen in single treatments and vehicle controls. Thus, pretreatment with palbociclib sensitizes cells to cisplatin and further increases its antitumor effect. Taken together, these preclinical studies suggest that targeting the CDK4/6 signaling pathway, sequentially with current chemotherapy agents, provides an alternative therapeutic approach in TNBC. In addition to providing alternative approaches, the safety of CDK4/6 inhibitors in combination with current therapies must be vigorously assessed. CDK4/6 inhibitor use in ER‐positive BC has proved to be a safe option when combined with hormone therapies 4 ; however, little data of the safety of CDK4/6 inhibitors exist and are discussed below
4.2. CDK4/6 inhibitors in treatment of TNBC—Clinical studies
In ER‐positive BC, CDK4/6 inhibitors have made major advancements in improving DFS and OS, particularly in combination with endocrine therapies as recently reviewed. 4 The success of CDK4/6 inhibitors such as abemaciclib (MONARCH studies), ribociclib (MONALEESA studies), as well as palbociclib (PALOMA studies), in combination with endocrine therapy, has been demonstrated in a number of trials in ER‐positive BC. 111 , 112 , 113 , 114 , 115 , 116 Recently, the safety of ribociclib plus tamoxifen (or letrozole) was assessed in 672 ER‐positive, HER‐2 negative BC patients in the MONALEESA‐7 trial. 117 A total of 335 patients were assigned to the ribociclib group and 337 to the placebo group. Here, median PFS was significantly higher in the ribociclib group compared to the placebo group (23.8 months vs 13 months, respectively). Follow‐up analysis found that 24.8% of deaths occurred in the ribociclib group compared to 32.3% in the placebo group; thus, OS was significantly higher in the ribociclib group than the placebo group (42 months vs 46 months, respectively). 118 In addition to these findings, a study found that ribociclib plus letrozole resulted in greater cost‐savings than other CDK4/6 inhibitor‐letrozole combination making it a cost‐effective treatment for ER‐positive BC. 119
In contrast, little data exist for CDK4/6 inhibitors in clinical trials in TNBC (Table 1), despite preclinical studies demonstrating that TNBCs express CDK4/6 and their growth is inhibited by CDK4/6 inhibitors, as described above. In 2015, a phase‐2 clinical trial assessing the safety of palbociclib, where 11% of the 37 patients enrolled had TNBC disease. 120 Unfortunately, no further TNBC patients were recruited due to the rapid disease progression observed in all four TNBC patients. These patients also showed a significantly lower PFS compared to ER‐positive patients (1.5 months vs 4.5 months) in response to palbociclib treatment. At the same time, a nonrandomized phase‐1/2 open‐label, single‐arm trial studied the effects of palbociclib and bicalutamide (anti‐AR agent) (ClinicalTrials.gov identifier: NCT02605486). Thus far, the combination of palbociclib and bicalutamide has proven to be safe and well tolerated by AR‐positive TNBC patients, with no dose‐limiting toxicities recorded or grade 4 or 5 adverse events. 121 The study aims to establish the recommended phase‐2 dose of combination as well as overall response rates and 1‐year PFS, though as of yet no clinical data have been recorded. Additionally, the phase‐1 PAveMenT trial (still recruiting at the time of writing) will test the safety of using palbociclib and avelumab in combination, as it is hypothesized that this may be an effective approach compared to sequential treatment in AR‐positive TNBC patients (ClinicalTrials.gov identifier: NCT04360941). Similarly to palbociclib, ribociclib has been investigated in an open‐label phase‐1/2 trial in combination with bicalutamide in AR‐positive TNBC disease to assess toxicity and clinical benefit rate (ClinicalTrials.gov identifier: NCT03090165—though patient recruitment is yet to commence at the time of writing this review). A third CDK4/6 inhibitor, abemaciclib, has also been investigated in TNBC. A phase‐1 trial assessing the efficacy and safety of abemaciclib reported nine (out of 225) patients had TNBC demonstrated only 1.1 months of median PFS and a clinical benefit rate of 11%. 122 Although results from this trial were more promising for ER‐positive patients, other trials have been established to identify better strategies for TNBC. One such approach is the single‐arm phase‐2 trial that is currently recruiting an estimated 37 metastatic Rb‐positive TNBC patients (invasive tumor has > 50% of Rb‐positive cells) that are to receive abemaciclib as a single agent (ClinicalTrials.gov identifier: NCT03130439). Results from these trials will help pave the way for CDK4/6 inhibitors to be incorporated into existing therapeutic strategies for TNBC.
TABLE 1.
Clinical trials with CDK4/6 inhibitors in TNBC obtained from ClinicalTrials.gov
| Trial ID | Phase | Arms | Treatment plan | Disease | Primary outcome | Results | Status | REF |
|---|---|---|---|---|---|---|---|---|
| Abemaciclib | ||||||||
| NCT03130439 | II | Single arm: Abemaciclib | 150 mg twice daily for 28 days | Rb‐positive metastatic TNBC | ORR | Awaiting | Recruiting | |
| Patnaik et al. | I | Single arm: Abemaciclib |
once daily: 50 mg (n = 4), 100 mg (n = 3), 150 mg (n = 3), or 225 mg (n = 3) Twive daily: 75 mg (n = 3), 100 mg (n = 4), 150 mg (n = 3), 200 mg (n = 7), and 275 mg (n = 3) |
TNBC | Safety and tolerability | Median PFS 1.1 months (vs HR+ 8.8 months) | 122 | |
| Palbociclib | ||||||||
| NCT02605486 | I/II | Palbociclib + Bicalutamide |
Phase I: to be determined Phase II: bicalutamide orally once daily. Palbociclib will be given orally daily for 3 weeks on followed by 1 week off at the doses determined in phase I |
AR‐positive metastatic TNBC |
Phase II dose Phase II PFS |
Awaiting | Active: not recruiting | |
| DeMichele et al. | II | Single arm: Palbociclib | 125 mg for 3 weeks on followed by 1 week off | Rb‐positive metastatic TNBC | Median PFS 1.5 months (vs HR+/Her2−: 3.8 months vs HR+/Her2+: 5.1 months) | 120 | ||
| NCT04360941 | I | Palbociclib + Avelumab |
Part A: palbociclib dose escalation + fixed dose of avelumab (n = 18) Part B: MTD and schedule determined by plan A. |
AR‐positive metastatic TNBC |
MTD ORR |
Recruiting | ||
| Ribociclib | ||||||||
| NCT03090165 | I/II | Ribociclib + Bicalutamide |
Phase I: cohort 1: bicalutamide 150 mg daily on days 1–28 + ribociclib 400mg daily on days 1–21 of a 28 day cycle cohort 2: bicalutamide 150 mg daily on days 1–28 + ribociclib 400 mg daily on days 1–28 of a 28 day cycle cohort 3: bicalutamide 150 mg daily on days 1–28 + ribociclib 600 mg daily on days 1–21 of a 28 day cycle Phase II: maximum safe dose of ribociclib in combination with bicalutamide (n = 25) |
Rb‐positive metastatic TNBC |
Phase I MTD Phase II CBR |
Awaiting | Recruiting | |
Abbreviations: ORR, objective response rate; PFS, progression free survival; CBR, clinical benefit rate; MTD, maximum tolerated dose.
5. ALTERNATIVE PATHWAY INHIBITORS IN COMBINATION WITH CDK4/6 INHIBITION
5.1. mTOR/PI3K inhibitors
Alternative pathways that also regulate cell proliferation, survival, and growth, such as the mTOR/PI3K and PARP pathways, have been shown to be deregulated in BC. 123 , 124 , 125 These pathways have therefore been targeted in ER‐positive BC through agents such as the PIK3 inhibitor, BYL719, in combination with ribociclib and letrozole (AI) (ClinicalTrials.gov : NCT 01872260), or the mTOR inhibitor, everolimus plus ribociclib and exemestane (AI) (ClinicalTrials.gov : NCT 01857193). 4 , 126 In TNBC disease, the combination of ribociclib and alpelisib (a PI3K inhibitor) demonstrated significantly increased apoptosis and cell cycle arrest in Rb‐positive TNBC cell lines in vitro. 127 In addition, in vivo xenograft models of TNBC demonstrated improved disease control in response to combined CDK4/6 and PI3K inhibition. Combined inhibition of CDK4/6, PI3K, and immune checkpoint pathways resulted in complete regression of established TNBC tumors. 127 The mTOR and PI3K pathway are closely linked and often considered as a single pathway. The mTOR kinase inhibitor, MLN0128, has been tested in combination with palbociclib in TNBC cell lines. 128 In this study, cell proliferation was significantly inhibited through cell cycle arrest and western blot analysis showed inhibition of the CDK4/6‐Rb and mTOR pathways in response to combination treatment. 128 Moreover, in TNBC patient‐derived xenograft models, palbociclib or MLN0128 monotherapy, resulted in significantly inhibited tumor growth. However, when used in combination, further significant tumor growth inhibition was observed compared to monotherapy or the saline control group. As previously mentioned, palbociclib has inhibitory effects on glucose metabolism outside the CDK4/6‐Rb axis. When palbociclib is combined with alpelisib or BEZ235 (a dual PI3K and mTORC1‐2 inhibitor), an additive inhibitory effect is observed on cell proliferation in the TNBC MDA‐MB‐231 and MDA‐MB‐468 cell lines. 102 Moreover, preincubation with palbociclib for 24 h, followed by simultaneous PI3K inhibition, resulted in a synergistic inhibition of cell proliferation in TNBC cell lines. However, once palbociclib is removed during combined treatment with the PI3K inhibitors, this synergistic inhibition is lost, suggesting that continuous palbociclib treatment is required to maintain the inhibitory effects. In addition to cell cycle arrest (increased proportion in the G0 phase of the cell cycle), sequential treatment gave rise to greater inhibition of the Rb and PI3K/AKT/mTOR pathways that result in the induction of cell death compared to single and combination treatments. 102 Furthermore, combination with alpelisib resulted in significantly reduced expression of GLUT‐1 and glucose uptake and consumption (25%) compared to palbociclib alone (15%). 102 The inhibition of CDK4/6 by palbociclib has been implicated in the activation of AKT and the mTOR pathway resulting in activation of cell proliferation, conferring resistance in Rb‐positive BC. 129 , 130 Thus, further investigations in preclinical models of TNBC disease are required to pave the way for the clinical utility of dual inhibition of the CDK4/6 and mTOR/PI3K pathways for TNBC patients.
5.2. Novel CDK7 inhibitors—Overcoming resistance to CDK4/6 inhibitors
As part of the mechanism of resistance to CDK4/6 inhibition, other cell cycle regulators may be activated to compensate for CDK4/6 activity. One of these is CDK7, which acts as a CDK‐activating kinase (CAK) to regulate the G2/M phase by establishing CDK1 and CDK2 activity. In addition, CDK7 has been shown to maintain CDK4/6 in an active state. 131 In addition to cell cycle regulation, CDK7 is a component of the basal transcriptional factor TFIIH that0 phosphorylates RNA polymerase 2 (PolII), resulting in transcription of genes required for progression through the cell cycle. 132 CDK7 thus makes an interesting potential target for anti‐cancer therapies and the agent ICEC0942 is shown to be a potent CDK7‐specific inhibitor in both ER‐positive and TNBC cell lines. 133 ICEC0942 elicited strong inhibitory effects on cell proliferation and promoted apoptosis through reduced levels of PolII in both the ER‐positive (MCF7 and T47D) and TNBC (MDA‐MB‐231 and MDA‐MB‐468) cells. A study investigated that the CDK7 inhibitor THZ1 reported that TNBC cell lines exhibited greater sensitivity to CDK7 inhibition than ER‐positive cells, as seen by potent inhibition of cell proliferation; however, phospho‐PolII status was equally affected in both BC subtypes. 134 In contrast, cell death was elicited in TNBC cells by the induction of PARP and caspase 3 cleavage, but not in ER‐positive BC cells. In vivo studies showed that THZ2 (an improved analogue of THZ1) significantly reduced the rate of tumor growth in a TNBC xenograft mouse model compared to the vehicle‐treated mice. 134 Interestingly, the knock‐down of CDK7 by CRISPR/Cas9 preferentially suppressed TNBC cell growth, whereas ER‐positive BC cells were unaffected, demonstrating the high dependency of CDK7 in TNBC. These studies provide an interesting foundation for the testing the combination of CDK4/6 and CDK7 inhibitors in for patients with TNBC, where otherwise limited options are available. The combination of such inhibitors will likely give rise to various adverse effects and toxicities, because a number of pathways are targeted. It is therefore imperative to assess the off‐target effects (for example, the impact on hematological cells) and safety of the drugs in combination or sequence.
6. SUMMARY
Cytotoxic chemotherapy continues to serve as the backbone therapy for TNBC; however, prognosis remains poor for TNBC patients. Neoadjuvant chemotherapy results in greater pCR in TNBC than non‐TNBC, with recent trials showing improvements in OS and DFS. Furthermore, incremental benefits are being observed in the neoadjuvant TNBC setting with the addition of platinum‐based chemotherapy and more recently PD‐L1 inhibitors. Additionally, although utilizing polychemotherapy in a dose dense manner seems to be the most effective approach in the adjuvant setting, optimized regimens are yet to be determined particularly for mTNBC. Additionally, following the success of CDK4/6 inhibitors in ER‐positive disease, the efforts to identify novel therapeutic approaches to TNBC continue. Preclinical studies support CDK4/6 inhibition as a plausible approach in TNBC in combination with current chemotherapies, particularly in sequential treatments playing a role in sensitizing cells providing further benefit compared to chemotherapy alone. With results from ongoing CDK4/6 inhibitor trials in TNBC, it may be possible to identify timings for the administration of these inhibitors in parallel with chemotherapeutic or novel agents in (neo)adjuvant treatments. With more recent cases of resistance to CDK4/6 inhibitors, further data from these trials will also provide valuable resources to identify potential biomarkers to help determine subgroups of patients with TNBC most likely to benefit from CDK4/6 inhibitors. Importantly, TNBC is a heterogeneous disease characterized by molecular phenotypes and signaling pathways. Thus, the profiling of these different subtypes is imperative for the success of personalized therapies for TNBC patients while avoiding exposure to toxicities for those who may not benefit.
CONFLICT OF INTEREST
All authors declare no conflicts of interests.
FUNDING
Not applicable.
ETHICS APPROVAL
Not applicable.
AVAILABILITY OF DATA
Not applicable.
CONTRIBUTIONS
All authors contributed to the writing, review and revision of the manuscript.
ACKNOWLEDGMENTS
Not applicable.
Saleh L, Wilson C, Holen I CDK4/6 inhibitors: a potential therapeutic approach for triple negative breast cancer. MedComm. 2021;2:514–530. 10.1002/mco2.97.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: gLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3). [DOI] [PubMed] [Google Scholar]
- 2. Kittaneh M, Montero AJ, Glück S. Molecular profiling for breast cancer: a comprehensive review. Biomark Cancer. 2013; 2013:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saleh L, Wilson C, Holen I, CDK4/6 inhibitors in breast cancer ‐ from in vitro models to clinical trials. Acta Oncol. 2020;59(2):219‐232. [DOI] [PubMed] [Google Scholar]
- 5. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase‐inhibitor adjuvant therapy to 10 years. N Engl J Med. 2016;375(3):209‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pernas S, Tolaney SM. HER2‐positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol. 2019;11:1758835919833519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2‐positive breast cancer. N Engl J Med. 2020;382(7):610‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krop IE, Beeram M, Modi S, et al. Phase I study of trastuzumab‐DM1, an HER2 antibody‐drug conjugate, given every 3 weeks to patients with HER2‐positive metastatic breast cancer. J Clin Oncol. 2010;28(16):2698‐2704. [DOI] [PubMed] [Google Scholar]
- 9. Baselga JLPG, Verma S, Jungsil R, Pegram M. Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of trastuzumab emtansine in HER2‐positive metastatic breast cancer. Clin Cancer Res. 2016;22:3755‐3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Indini A, Rijavec E, Grossi F. Changing the destiny of HER2 expressing solid tumors. Int J Mol Sci. 2021;22(9):4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ismail‐Khan R, Bui MM, A review of triple‐negative breast cancer. Cancer Control. 2010;17(3):173‐176. [DOI] [PubMed] [Google Scholar]
- 12. Yin L, Duan JJ, Bian XW, Yu SC. Triple‐negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020;22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaplan HG, Malmgren JA, Atwood M. T1N0 triple negative breast cancer: risk of recurrence and adjuvant chemotherapy. Breast J. 2009;15(5):454‐460. [DOI] [PubMed] [Google Scholar]
- 14. Hubalek M, Czech T, Muller H. Biological subtypes of triple‐negative breast cancer. Breast Care (Basel). 2017;12(1):8‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747‐752. [DOI] [PubMed] [Google Scholar]
- 16. Mehanna J, Haddad FG, Eid R, Lambertini M, Kourie HR. Triple‐negative breast cancer: current perspective on the evolving therapeutic landscape. Int J Womens Health. 2019;11:431‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burstein MD, Tsimelzon A, Poage GM, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple‐negative breast cancer. Clin Cancer Res. 2015;21(7):1688‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple‐negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750‐2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukherjee A, Russell R, Chin S‐F, et al. Associations between genomic stratification of breast cancer and centrally reviewed tumour pathology in the METABRIC cohort. Breast Cancer. 2018;4(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan X, Hu X, Zhang YH, et al. Identification of the copy number variant biomarkers for breast cancer subtypes. Mol Genet Genom. 2019;294(1):95‐110. [DOI] [PubMed] [Google Scholar]
- 21. Masuda H, Baggerly KA, Wang Y, et al. Differential response to neoadjuvant chemotherapy among 7 triple‐negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19(19):5533‐5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;1‐2:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGF‐C promotes breast cancer metastasis. Nat Med. 2001;7(2):192‐198. [DOI] [PubMed] [Google Scholar]
- 24. Dent R, Trudeau M, Pritchard KI, et al. Triple‐negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429‐4434. [DOI] [PubMed] [Google Scholar]
- 25. Criscitiello C, Azim HA Jr, Schouten PC, Linn SC, Sotiriou C. Understanding the biology of triple‐negative breast cancer. Ann Oncol. 2012;23(Suppl 6):vi13‐8. [DOI] [PubMed] [Google Scholar]
- 26. Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple‐negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638‐2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kassam F, Enright K, Dent R, et al. Survival outcomes for patients with metastatic triple‐negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer. 2009;9(1):29‐33. [DOI] [PubMed] [Google Scholar]
- 28. Bonotto M, Gerratana L, Poletto E, et al. Measures of outcome in metastatic breast cancer: insights from a real‐world scenario. Oncologist. 2014;19(6):608‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kohler BA, Sherman RL, Howlader N, et al. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst. 2015;107(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von Minckwitz G, Untch M, Blohmer JU, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012;30(15):1796‐1804. [DOI] [PubMed] [Google Scholar]
- 31. Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329‐2334. [DOI] [PubMed] [Google Scholar]
- 32. Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long‐term survival in patients with triple‐negative breast cancer. J Clin Oncol. 2008;26(8):1275‐1281. [DOI] [PubMed] [Google Scholar]
- 33. Bear HD, Anderson S, Brown A, et al. The effect on tumor response of adding sequential preoperative docetaxel to preoperative doxorubicin and cyclophosphamide: preliminary results from National Surgical Adjuvant Breast and Bowel Project Protocol B‐27. J Clin Oncol. 2003;21(22):4165‐4174. [DOI] [PubMed] [Google Scholar]
- 34. Rastogi P, Anderson SJ, Bear HD, et al. Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project Protocols B‐18 and B‐27. J Clin Oncol. 2008;26(5):778‐785. [DOI] [PubMed] [Google Scholar]
- 35. Silver DP, Richardson AL, Eklund AC, et al. Efficacy of neoadjuvant cisplatin in triple‐negative breast cancer. J Clin Oncol. 2010;7:1145‐1153.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sikov WM, Berry DA, Perou CM, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once‐per‐week paclitaxel followed by dose‐dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple‐negative breast cancer: cALGB 40603 (Alliance). J Clin Oncol. 2015;33(1):13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. von Minckwitz G, Schneeweiss A, Loibl S, et al. Neoadjuvant carboplatin in patients with triple‐negative and HER2‐positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 2014;15(7):747‐756. [DOI] [PubMed] [Google Scholar]
- 38. Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long‐term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet (London, England). 2014;384(9938):164‐172. [DOI] [PubMed] [Google Scholar]
- 39. Stanton SE, Adams S, Disis ML. Variation in the incidence and magnitude of tumor‐infiltrating lymphocytes in breast cancer subtypes: a systematic review. JAMA Oncol. 2016;2(10):1354‐1360. [DOI] [PubMed] [Google Scholar]
- 40. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor‐infiltrating lymphocytes predict response to anthracycline‐based chemotherapy in estrogen receptor‐negative breast cancer. Breast Cancer Res. 2011;13(6):R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mittendorf EA, Philips AV, Meric‐Bernstam F, et al. PD‐L1 expression in triple‐negative breast cancer. Cancer Immunol Res. 2014;2:361‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med. 2015;372(21):2018‐2028. [DOI] [PubMed] [Google Scholar]
- 44. Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second‐line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521‐2532. [DOI] [PubMed] [Google Scholar]
- 46. Nanda R, Chow LQ, Dees EC, et al. Pembrolizumab in patients with advanced triple‐negative breast cancer: phase Ib KEYNOTE‐012 study. J Clin Oncol. 2016;34(21):2460‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nada R, Liu MC, Yau C, et al. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early‐stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I‐SPY2 trial. JAMA Oncol. 2020;6(5):676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harbeck N, Zhang H, Barrios CH, et al. IMpassion031: results from a phase III study of neoadjuvant (neoadj) atezolizumab + chemotherapy in early triple‐negative breast cancer (TNBC) | OncologyPRO. Ann Oncol. 2020;31(Suppl_4):S1142‐S1215. [Google Scholar]
- 49. Senkus E, Kyriakides S, Ohno S, et al. Primary breast cancer: eSMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2015;26(Suppl 5):v8‐30. [DOI] [PubMed] [Google Scholar]
- 50. Gluz O, Nitz UA, Harbeck N, et al. Triple‐negative high‐risk breast cancer derives particular benefit from dose intensification of adjuvant chemotherapy: results of WSG AM‐01 trial. Ann Oncol. 2008;19(5):861‐870. [DOI] [PubMed] [Google Scholar]
- 51. Masuda N, Lee SJ, Ohtani S, et al. Adjuvant capecitabine for breast cancer after preoperative chemotherapy. N Engl J Med. 2017;376(22):2147‐2159. [DOI] [PubMed] [Google Scholar]
- 52. Keller L, Belloum Y, Wikman H, Pantel K. Clinical relevance of blood‐based ctDNA analysis: mutation detection and beyond. Br J Cancer. 2021;124(2):345‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steponaviciene L, Lachej‐Mikeroviene N, Smailyte G, Aleknavicius E, Meskauskas R, Didziapetriene J. Triple negative breast cancer: adjuvant chemotherapy effect on survival. Adv Med Sci. 2011;56(2):285‐290. [DOI] [PubMed] [Google Scholar]
- 54. Kashiwagi S, Yashiro M, Takashima T, et al. Advantages of adjuvant chemotherapy for patients with triple‐negative breast cancer at Stage II: usefulness of prognostic markers E‐cadherin and Ki67. Breast Cancer Res. 2011;13(6):R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gennari A, Sormani MP, Pronzato P, et al. HER2 status and efficacy of adjuvant anthracyclines in early breast cancer: a pooled analysis of randomized trials. J Natl Cancer Inst. 2008;100(1):14‐20. [DOI] [PubMed] [Google Scholar]
- 56. Peto R, Davies C, Godwin J, et al. Comparisons between different polychemotherapy regimens for early breast cancer: meta‐analyses of long‐term outcome among 100,000 women in 123 randomised trials. Lancet. 2012;379(9814):432‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Early Breast Cancer Trialists' Collaborative Group (EBCTCG) . Increasing the dose intensity of chemotherapy by more frequent administration or sequential scheduling: a patient‐level meta‐analysis of 37 298 women with early breast cancer in 26 randomised trials. Lancet (London, England). 2019;393(10179):1440‐1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wahba HA, El‐Hadaad HA. Current approaches in treatment of triple‐negative breast cancer. Cancer Biol Med. 2015;2:106‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014;343(6178):1470‐1475. [DOI] [PubMed] [Google Scholar]
- 60. Chen H, Wu J, Zhang Z, et al. Association between BRCA status and triple‐negative breast cancer: a meta‐analysis. Front Pharmacol. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tutt AN, Garber JE, Kaufman B, et al. Adjuvant Olaparib for Patients with BRCA1‐ or BRCA2‐Mutated Breast Cancer | NEJM. 2021. [DOI] [PMC free article] [PubMed]
- 62. Fisher CS, Ma CX, Gillanders WE, et al. Neoadjuvant chemotherapy is associated with improved survival compared with adjuvant chemotherapy in patients with triple‐negative breast cancer only after complete pathologic response. Ann Surg Oncol. 2012;19(1):253‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Clifton K, Gutierrez‐Barrera A, Ma J, et al. Adjuvant versus neoadjuvant chemotherapy in triple‐negative breast cancer patients with BRCA mutations. Breast Cancer Res Treat. 2018;170(1):101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kennedy CR, Gao F, Margenthaler JA. Neoadjuvant versus adjuvant chemotherapy for triple negative breast cancer. J Surg Res. 2010;163(1):52‐57. [DOI] [PubMed] [Google Scholar]
- 65. Van Poznak C, Somerfield MR, Bast RC, et al. Use of biomarkers to guide decisions on systemic therapy for women with metastatic breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2015;33(24):2695‐2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Broom RJ, Tang PA, Simmons C, et al. Changes in estrogen receptor, progesterone receptor and Her‐2/neu status with time: discordance rates between primary and metastatic breast cancer. Anticancer Res. 2009;29(5):1557‐1562. [PubMed] [Google Scholar]
- 67. Aurilio G, Monfardini L, Rizzo S, et al. Discordant hormone receptor and human epidermal growth factor receptor 2 status in bone metastases compared to primary breast cancer. Acta Oncol (Stockholm, Sweden). 2013;52(8):1649‐1656. [DOI] [PubMed] [Google Scholar]
- 68. Cardoso F, Costa A, Senkus E, et al. 3rd ESO‐ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 3). Ann Oncol. 2017;28(1):16‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang J, Fan M, Xie J, et al. Chemotherapy of metastatic triple negative breast cancer: experience of using platinum‐based chemotherapy. Oncotarget. 2015;6(40):43135‐43143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kilburn LS, Group TNTTM . ‘Triple negative’ breast cancer: a new area for phase III breast cancer clinical trials. Clin Oncol (R Coll Radiol). 2008;1:35‐39. [DOI] [PubMed] [Google Scholar]
- 71. Tutt A, Tovey H, Cheang MCU, et al. A randomised phase III trial of carboplatin compared with docetaxel in BRCA1/2 mutated and pre‐specified triple negative breast cancer “BRCAness” subgroups: the TNT Trial. Natl Med. 2018:628‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379(22):2108‐2121. [DOI] [PubMed] [Google Scholar]
- 73. Atchley DP, Albarracin CT, Lopez A, et al. Clinical and pathologic characteristics of patients with BRCA‐positive and BRCA‐negative breast cancer. J Clin Oncol. 2008;26(26):4282‐4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haffty BG, Yang Q, Reiss M, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early‐stage breast cancer. J Clin Oncol. 2006;24(36):5652‐5657. [DOI] [PubMed] [Google Scholar]
- 75. Byrski T, Gronwald J, Huzarski T, et al. Response to neo‐adjuvant chemotherapy in women with BRCA1‐positive breast cancers. Breast Cancer Res Treat. 2008;108(2):289‐296. [DOI] [PubMed] [Google Scholar]
- 76. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;4:387‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105(44):17079‐17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐917. [DOI] [PubMed] [Google Scholar]
- 79. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 80. Ibrahim YH, Garcia‐Garcia C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA‐proficient triple‐negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523‐533. [DOI] [PubMed] [Google Scholar]
- 82. Litton JK, Rugo HS, Ettle J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chae BJ, Bae JS, Lee A, et al. p53 as a specific prognostic factor in triple‐negative breast cancer. Jpn J Clin Oncol. 2009;39(4):217‐224. [DOI] [PubMed] [Google Scholar]
- 84.Sporikova Z, Koudelakova V, Trojanec R, Hajduch M. Genetic markers in triple‐negative breast cancer. Clin Breast Cancer. 2018;18(5):e841‐e850. [DOI] [PubMed] [Google Scholar]
- 85. Bardia A, Tolaney SM, Punie K, et al. Biomarker analyses in the phase III ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple‐negative breast cancer. Ann Oncol. 2021;32(9):1148‐1156. [DOI] [PubMed] [Google Scholar]
- 86. Goldenberg DM, Stein R, Sharkey RM. The emergence of trophoblast cell‐surface antigen 2 (TROP‐2) as a novel cancer target. Oncotarget. 2018;9(48):28989‐29006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ambrogi F, Fornili M, Boracchi P, et al. Trop‐2 is a determinant of breast cancer survival. PLoS One. 2014;9(5):e96993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhou Z, Huang F, Shrivastava I, et al. New insight into the significance of KLF4 PARylation in genome stability, carcinogenesis, and therapy. EMBO Mol Med. 2020;12:e12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jhan JR, Andrechek ER. Triple‐negative breast cancer and the potential for targeted therapy. Pharmacogenomics. 2017;18(17):1595‐1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672‐1677. [DOI] [PubMed] [Google Scholar]
- 92. Ishii Y, Pirkmaier A, Alvarez JV, et al. Cyclin D1 overexpression and response to bortezomib treatment in a breast cancer model. J Natl Cancer Inst. 2006;98(17):1238‐1247. [DOI] [PubMed] [Google Scholar]
- 93. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor‐positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;5:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Witkiewicz AK, Cox D, Knudsen ES. CDK4/6 inhibition provides a potent adjunct to Her2‐targeted therapies in preclinical breast cancer models. Genes Cancer. 2014;5(7‐8):261‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Patel JM, Goss A, Garber JE, et al. Retinoblastoma protein expression and its predictors in triple‐negative breast cancer. Breast Cancer. 2020;6(1):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Asghar US, Barr AR, Cutts R, et al. Single‐cell dynamics determines response to CDK4/6 inhibition in triple negative breast cancer. Clin Cancer Res. 2017;23(18):5561‐5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Herrera‐Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor‐positive breast cancer. Cancer Res. 2016;76(8):2301‐2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal‐like and luminal B breast carcinomas. Breast Cancer Res. 2008;10(5):R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Guarducci C, Bonechi M, Benelli M, et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor‐positive breast cancer. NPJ Breast Cancer. 2018;4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Neganova I, Vilella F, Atkinson SP, et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells. 2011;29(4):651‐659. [DOI] [PubMed] [Google Scholar]
- 101. Cretella D, Fumarola C, Bonelli M, et al. Pre‐treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci Rep. 2019;9(1):13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cretella D, Ravelli A, Fumarola C, et al. The anti‐tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J Exp Clin Cancer Res. 2018;37(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cochrane DR, Bernales S, Jacobsen BM, et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014;16(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Caiazza F, Murray A, Madden SF, et al. Preclinical evaluation of the AR inhibitor enzalutamide in triple‐negative breast cancer cells. Endocr Relat Cancer. 2016;23(4):323‐334. [DOI] [PubMed] [Google Scholar]
- 105. Barton VN, D'Amato NC, Gordon MA, et al. Multiple molecular subtypes of triple‐negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015;14(3):769‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Liu CY, Lau KY, Hsu CC, et al. Combination of palbociclib with enzalutamide shows in vitro activity in RB proficient and androgen receptor positive triple negative breast cancer cells. PLoS One. 2017;12(12):e0189007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu T, Yu J, Deng M, et al. CDK4/6‐dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat Commun. 2017;8(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2(2):84‐89. [DOI] [PubMed] [Google Scholar]
- 109. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Huang Y, Wu H, Li X. Novel sequential treatment with palbociclib enhances the effect of cisplatin in RB‐proficient triple‐negative breast cancer. Cancer Cell Int. 2020;20:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638‐3646. [DOI] [PubMed] [Google Scholar]
- 112. Sledge GW Jr, Toi M, Neven P. et al. MONARCH 2: abemaciclib in Combination With Fulvestrant in Women With HR+/HER2‐ advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875‐2884. [DOI] [PubMed] [Google Scholar]
- 113. Slamon DJ, Neven P, Chia S, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor‐positive, human epidermal growth factor receptor 2‐negative advanced breast cancer: mONALEESA‐3. J Clin Oncol. 2018;36(24):2465‐2472. [DOI] [PubMed] [Google Scholar]
- 114. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first‐line therapy for HR‐positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738‐1748. [DOI] [PubMed] [Google Scholar]
- 115. Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926‐1936. [DOI] [PubMed] [Google Scholar]
- 116. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425‐439. [DOI] [PubMed] [Google Scholar]
- 117. Tripathy D, Im SA, Colleoni M, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone‐receptor‐positive, advanced breast cancer (MONALEESA‐7): a randomised phase 3 trial. Lancet Oncol. 2018;19(7):904‐915. [DOI] [PubMed] [Google Scholar]
- 118. Im SA, Lu YS, Bardia A, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. 2019;381(4):307‐316. [DOI] [PubMed] [Google Scholar]
- 119. Suri G, Chandiwana D, Lee A, Mistry R. Cost‐effectiveness analysis of ribociclib plus letrozole versus palbociclib plus letrozole in the United Kingdom. J Health Econ Outcomes Res. 2019;6(2):20‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. DeMichele A, Clark AS, Tan KS, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. 2015;21(5):995‐1001. [DOI] [PubMed] [Google Scholar]
- 121. Gucalp A, Edelweiss M, Patil S, et al. Abstract P3‐11‐04: Phase I/II trial of palbociclib in combination with bicalutamide for the treatment of androgen receptor (AR)+ metastatic breast cancer (MBC). Cancer Res. 2018;78(4 Suppl):Abstract nr P3‐11‐04. [Google Scholar]
- 122. Patnaik A, Rosen LS, Tolaney SM, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non‐small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6(7):740‐753. [DOI] [PubMed] [Google Scholar]
- 123. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: erbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100(15):8933‐8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nur Husna SM, Tan HTT, Mohamud R, Dyhl‐Polk A, Wong KK. Inhibitors targeting CDK4/6, PARP and PI3K in breast cancer: a review. Ther Adv Med Oncol. 2018;10:1758835918808509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dey N, De P, Leyland‐Jones B. PI3K‐AKT‐mTOR inhibitors in breast cancers: from tumor cell signaling to clinical trials. Pharmacol Ther. 2017;175:91‐106. [DOI] [PubMed] [Google Scholar]
- 126. Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;8:671‐688. [DOI] [PubMed] [Google Scholar]
- 127. Teo ZL, Versaci S, Dushyanthen S, et al. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple‐negative breast cancer. Cancer Res. 2017;77(22):6340‐6352. [DOI] [PubMed] [Google Scholar]
- 128. Yamamoto T, Kanaya N, Somlo G, Chen S. Synergistic anti‐cancer activity of CDK4/6 inhibitor palbociclib and dual mTOR kinase inhibitor MLN0128 in pRb‐expressing ER‐negative breast cancer. Breast Cancer Res Treat. 2019;174(3):615‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang J, Xu K, Liu P, et al. Inhibition of Rb phosphorylation leads to mTORC2‐mediated activation of Akt. Mol Cell. 2016;62(6):929‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bonelli MA, Digiacomo G, Fumarola C, et al. Combined inhibition of CDK4/6 and PI3K/AKT/mTOR pathways induces a synergistic anti‐tumor effect in malignant pleural mesothelioma cells. Neoplasia. 2017;19(8):637‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Schachter MM, Merrick KA, Larochelle S, et al. A Cdk7‐Cdk4 T‐loop phosphorylation cascade promotes G1 progression. Mol Cell. 2013;50(2):250‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Fisher RP. The CDK network: linking cycles of cell division and gene expression. Genes Cancer. 2012;3(11‐12):731‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Patel H, Periyasamy M, Sava GP, et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol Cancer Ther. 2018;17(6):1156‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wang Y, Zhang T, Kwiatkowski N, et al. CDK7‐dependent transcriptional addiction in triple‐negative breast cancer. Cell. 2015;163(1):174‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.