

Abstract
Over the last decades, the growing understanding on DNA damage response (DDR) pathways has broadened the therapeutic landscape in oncology. It is becoming increasingly clear that the genomic instability of cells resulted from deficient DNA damage response contributes to the occurrence of cancer. One the other hand, these defects could also be exploited as a therapeutic opportunity, which is preferentially more deleterious in tumor cells than in normal cells. An expanding repertoire of DDR‐targeting agents has rapidly expanded to inhibitors of multiple members involved in DDR pathways, including PARP, ATM, ATR, CHK1, WEE1, and DNA‐PK. In this review, we sought to summarize the complex network of DNA repair machinery in cancer cells and discuss the underlying mechanism for the application of DDR inhibitors in cancer. With the past preclinical evidence and ongoing clinical trials, we also provide an overview of the history and current landscape of DDR inhibitors in cancer treatment, with special focus on the combination of DDR‐targeted therapies with other cancer treatment strategies.
Keywords: cancer, combination therapy, DNA damage response, PARP
In this review, we sought to summarize the complex network of DNA repair machinery in cancer cells and discuss the underlying mechanism for the application of DDR inhibitors in cancer. With the past preclinical evidence and ongoing clinical trials, we also provide an overview of the history and current landscape of DDR inhibitors in cancer treatment, with especial focus on the combination of DDR‐targeted therapies with other cancer treatment strategies.
1. INTRODUCTION
As early as 1914, a German scientist Theodor Boveri published his work on the origin of malignant tumors, which suggested the “specific and abnormal chromosome constitution” could attribute to the onset of cancer. 1 Through out the century, compelling data are emerging to support the role of genomic instability in cancer, including the alteration in chromosome number and structure, and moreover, in DNA compositions. These changes may lead to oncogenic transformation and confer resistance to anticancer therapies. Alongside direct damage caused by genetic alterations, some mutations have been characterized as collateral damage from the loss of genome integrity caused by carcinogens. Common oncogenic factors that result in genomic instability include chemical carcinogens in the environment, genotoxic anticancer drugs, 2 and endogeneous carcinogens such as microbial metabolism products 3 and free radicals produced by ionizing radiation. 4
To limit the progression of DNA lesions, cells have evolved complex DNA repair machinery, which triggers cell‐cycle checkpoints and allows DNA damage repair before it further interferes with the replication process. Excessive DNA damage or deficient DNA repair would thus result in accumulating genomic disorders that ultimately contribute to cell death. Thus, the fate of a cell following critical DNA damage is largely decided by the amount of DNA damage and its repair capacity. On the other hand, the misrepair of single‐strand breaks (SSBs) and double‐strand breaks (DSBs) of DNA may result in genome rearrangement. The DNA repair capacity varies among different cell types, with some tumor cells exhibit significantly enhanced DNA repair following replication and genotoxic stress. 5
In parallel with the advances in tumor biology that introduce DDR as potential therapeutic targets, a range of inhibitors targeting DDR components have emerged, some of which are now under clinical investigation. Moreover, emerging evidence suggests the sensitization effect of DDR inhibitors to conventional cancer therapies, and the correlation between DDR pathways and immune checkpoint inhibitor (ICI) response, which together encourages the design DDR inhibitor‐based combination treatments. In this review, we sought to summarize the complex network of DNA repair machinery in cancer cells and to discuss the underlying mechanism for the application of DDR inhibitors in cancer. With the past preclinical evidence and ongoing clinical trials, we especially summarized the ongoing clinicals that involve DDR inhibitors, with special focus on the combination therapy of DDR inhibitors including chemotherapy, radiotherapy, immunotherapies, and combinations DDR inhibitors, hopefully providing an overview of the history and current landscape of DDR inhibitors.
2. DNA DAMAGE AND THE DNA DAMAGE RESPONSE
To maintain genomic integrity, an intricate DNA repair system is evolved to counteract various forms of DNA lesions, and these mechanisms are referred to as the DNA damage response (DDR). Here we classified DDR pathways into three functionally interwoven parts: the sensor that detects DNA damage, signal transducer that triggers signaling cascades, and effector that impedes DNA repair. Numerous efforts have been undertaken to elucidate the machinery for the repair of genotoxic lesions in mammalian cells. These pathways are not mutually exclusive processes, but rather coordinated with each other to form a precise regulation network of DNA repair. Figure 1 presents an overview of major pathways for the repair of different DNA damage.
FIGURE 1.
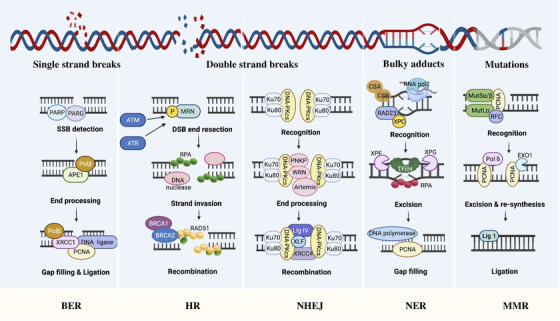
Overview of major pathways for the repair of different DNA damage. Single‐strand breaks (SSBs) are repaired by direct and indirect base excision repair (BER) and double‐strand breaks (DSBs) are repaired by homologous recombination (HR) and nonhomologous end joining (NHEJ). Replication error is repaired by mismatch repair (MMR) and DNA adducts by nucleotide excision repair (NER). Figure was created with Biorender
2.1. Base excision repair (BER) and nucleotide excision repair (NER)
The genome of all organisms are continuously experiencing subtle changes due to various genotoxicants generated endogenously such as reactive oxygen species (ROS), or environmental insults such as ionizing radiation and alkylating agents. The majority of these subtle changes in DNA such as SSBs are repaired through the base excision repair (BER) pathway. BER is initiated with damaged bases, which are then excised and replaced with newly synthesized DNA. 6 In the next step, the apurinic/apyrimidinic (AP)‐endonuclease (APE) cleaves the AP site to form 3′ OH terminus at the damage site. 7 Finally, the DNA polymerase and DNA ligase are recruited at the nucleotide gap produced by lesion base removal, thereby sealing the nick. Whereas BER is responsible for the repair of small lesions, the nucleotide excision repair (NER) is needed for bulkier SSBs that deform the DNA helical structure. 8 The NER machinery involves a crucial protein, the excision repair cross‐complementing protein 1 (ERCC1), which takes an active part in the excision of DNA surrounding the lesion followed by replacement with normal DNA replication. 9
2.2. Homologous recombination (HR) and nonhomologous end joining (NHEJ)
In mammalian cells, HR and NHEJ represent the two major pathways for repairing DSBs. 10 , 11 Since a homologous sister chromatid is required as a template for new DNA synthesis, HR pathways arguably repair DSBs during the S/G2 cell‐cycle phase, whereas NHEJ are active through all cell‐cycle phases except M phase. The HR analyses the homologous sequences from other parts of genome and thus collects the lost information at break sites. The HR pathway is initiated with the resection of break ends, followed by the formation of Rad51 nucleoprotein filament by Brca2 and Rad51, which retrieves the homologous sequence and promotes the formation of a joint molecule between the broken DNA and the homologous template. 12 With minimal processing on DNA break ends, NHEJ is believed to be mechanistically simpler than HR, which directly rejoins the break ends together. The fundamental factor required for NHEJ is the heterodimer composed of Ku70/Ku80 and the catalytic subunit of the DNA‐dependent protein kinase (DNA‐PKcs) which recognize DSBs and facilitates downstream signaling factors for NHEJ, such as XRCC4, XLF, and DNA ligase IV. 13 Although simpler among these repair mechanisms, NHEJ sometimes leads to rearrangements, especially the slow resection‐dependent NHEJ process, whereas HR is believed to be error free. However, in some cases, cross‐overs are formed in HR pathways, resulting in potential chromosomal rearrangements. 10 , 14 , 15 These scenarios contribute to the preference of cells to NHEJ over HR in the absence of sister chromatid.
In addition to HR and NHEJ, a group of DSB repair pathways that share similar mechanisms to the two major DSB repair pathways, but are genetically distinct, are collectively known as alternative end‐joining (a‐EJ) pathways. The a‐EJ pathway can either share similar initiation process or constitute factors with HR, 16 , 17 but also with NHEJ in terms of DNA ends joining without homologous templates. Growing body of literature has reported that a‐EJ can cause gene deletions, translocations, and rearrangements in cancer cells. 18 , 19 Growing interest has been attached to a‐EJ pathways as potential therapeutic targets in cancer cells with compromised NHEJ or HR activities. 20 , 21 , 22
2.3. Mismatch repair (MMR)
Apart from those produced by cells exposed to genotoxins, DNA damage can also derive from aberrant DNA processing. A DNA repair pathway targeting replication‐associated errors is known as MMR. During DNA synthesis, MMR corrects nucleotide misincorporation and thereby prevents permanent DNA change in dividing cells. 23 , 24 , 25 Thus, defects in MMR either by gene mutation or epigenetic silencing may contribute to increased incidence of spontaneous mutation, which is typically associated with inherited and sporadic cancers. 26 , 27
2.4. Translesion synthesis and template switching
As an essential bypass mechanism for the repair of replication‐stalling DNA lesions, DNA damage tolerance (DDT) allows DNA replication across the obstructing element. 28 The translesion synthesis (TLS) is one of the two distinct DDT modes that depends on the function of a special TLS polymerase, rather than replicative DNA polymerases, and directly replicates across the lesions. 29 The TLS mechanism has been characterized as error‐prone due to the deficient proofreading activity of the TLS polymerase, which increases the risk of mutation. Not surprisingly, TLS is a major source of cellular mutagenesis. 30 In contrast, another mode of DDT, the template switching (TS), involves recombination to a homologous DNA template on a sister chromatid, which is similar to the HR process and is believed to be more accurate in the outcome than TLS 21539841. The repair activities of TLS and TS start behind the replication fork, suggesting that they could occur during or after DNA replication, with TS beginning earlier in the S cell‐cycle phase and TLS in the late S phase. 31 , 32 , 33
2.5. The Fanconi anemia (FA) pathway
Fanconi anemia is a rare genetic disease resulting from biallelic mutations of FANC genes, and affected patients are companied by deficient response to DNA damage. 34 , 35 , 36 , 37 , 38 Affected patients have deficient ICL repair. The Fanconi anemia (FA) has been identified as a DNA repair pathway for its removal of a barrier that impedes DNA replication and transcription, the DNA interstrand crosslink (ICL). 39 ICLs can be formed by aldehydes during multiple metabolic reactions such as lipid peroxidation and alcohol metabolism, and chemotherapies such as platinum. 40 , 41 Whereas intrastrand crosslinks are repaired by NER pathway as described above, 42 , 43 the highly toxic ICL is primarily repaired by the FA pathway. 44 Following the detection of ICL by UHRF1 protein and the FANCM–MHF1–MHF2 complex, the FA core complex is recruited to chromatin and monoubiquitylates FANCD2‐I incorporation with UBE2T/FANCT E2 conjugating enzyme. Ubiquitylated FANCD2‐I recruits scaffolding protein for various DNA endonucleases, which split the strands near the ICL and facilitate the production of ICL‐derived double‐strand breaks. Given the considerable role that the FA pathway plays in DNA repair, it is not surprising that the FA pathway is also extensively studied in the context of cancer and that targeting the FA pathway is a potential cancer intervention strategy. 45 , 46
2.6. O6‐methylguanine‐DNA methyltransferase pathway
DNA methylating agents are known for their ability to inhibit DNA methylation and produce a wide range of DNA adducts, such as O6‐methylguanine (O6MeG) and O4‐methylthymine, which may result in base mispairing and subsequent point mutations. 47 Given the smaller incidence of O4‐methylthymine production by methylating agents (< 0.3% compared with 8% of O6MeG), 48 O6MeG is referred to as major source of methylating agents‐induced DNA adducts that cause mutagenesis and carcinogenesis. O6MeG can be repaired by O6‐methylguanine‐DNA methyltransferase, also known as MGMT, in a single‐step suicide reaction. 49 MGMT transfers the methyl at O6 site of damaged guanine to its cysteine residues, and thus prevents gene mutation. It is conceivable that MGMT reduces the efficacy of alkylating agents in cancer cells, potentially contributing to chemoresistance. Because DNA methylation can inhibit transcription, the methylation of MGMT promoter, which hampers its transcription, could be used to increase cell sensitivity to alkylating agents. 50 A wide breadth of recent literature has identified the methylation of MGMT promoter as a response predictor for alkylating agents in gliomas. 51 , 52 , 53 , 54 , 55
3. MECHANISMS UNDERLYING THE THERAPEUTIC APPLICATION OF DDR
As DNA‐damaging chemotherapies and ionizing radiation are used as the backbone of many therapeutic regimens in cancer, it is intriguing to speculate whether DNA repair deficiency represents a good source of anticancer therapeutic targets. Moreover, in some cases, the DDR deficiency is characterized as predicting biomarkers both for prognosis and treatment responses. A typical example has been discussed earlier in the review that MGMT promoter methylation can be used to predict the response to temozolomide in glioblastoma multiforme. 52 , 56 The underlying mechanisms for increased sensitivity of tumor cells to DNA‐damaging agents relative to normal cells lie in the three differentiating aspects: loss of at least one DDR pathways, elevated replication stress, and increased endogenous DNA damage.
3.1. DDR defects
Although DDR defects are implicated in the initiation and progression of cancers, 57 defects in DDR pathways also provide therapeutic opportunities to target tumor cells with minimum impact on normal cells. 58 Tumor cells carrying DDR deficiency leads to enhanced genomic instability and its dependency on remaining DDR pathways for survival. The combinational targeting of the remaining DNA repair pathways as a therapeutic approach reflects a concept known as synthetic lethality. 59 The concept of synthetic lethality was based on two concurrent loss‐of‐function genetic events, either of which alone does not cause lethality but collectively contribute to cell death. 60 As one genetic alteration on DDR pathways that are unique to cancer cells occurs, the second loss‐of‐function event caused by pharmacological inhibition with DDR inhibitor then becomes synthetic lethal to a cancer cells without affecting normal cells. 58 , 61 , 62 , 63
DNA‐damaging agents such as chemotherapies and radiotherapies have been used for years as the keystone of many anticancer therapeutics. Although these agents have demonstrated potent activity in a wide range of cancers, treatment resistance occurs through a variety of mechanisms and presents ongoing challenges including the upregulation of DDR components. 64 DDR inhibitors were first developed as a combination partner for with platinum compounds, but later presented difficulty in application due to overlapping toxicities. 65 Targeting DDR components as monotherapies is largely based on the concept of synthetic lethality. 66 This approach would deliver considerable benefit to cancer patients compared with conventional treatments such as cytotoxic chemotherapies. Small‐molecule inhibitors targeting DDR are often DDR components that demonstrate enzymatic activities, including the PIKK family kinases, ChK1/2 and PARP‐1.
3.2. Replication stress
The intricate DNA replication system of Eukaryotic cells is tightly regulated during cell division by various proteins in cell cycles. 66 , 67 This is issue is a particularly prominent in the early S‐phase due to the fact that replication stress can be induced by untimely entry into S cell‐cycle phase before necessary molecules required for replication are generated. 68 Numerous DNA nucleotides need to be accurately polymerized to ensure cellular homeostasis. Endogenous or exogenous obstacles that retard or terminate the progression of replication forks activate conserved cellular response pathways, which is referred to as replication stress. The molecular mechanism for replication stress is the stalled progression of DNA polymerase and the subsequent uncoupling of DNA polymerization from DNA helicases. 69 One example of replication stress inducers are deficient G1/S cell‐cycle checkpoints, either caused by the loss of retinoblastoma tumor suppressor (pRb) function, deletion of the CDKN2A, 70 or amplification of Cyclin D1 or Cyclin E. 71 , 72
Early stages of tumorigenesis is characterized with chronic replication stress and the subsequent collision of replication forks. 73 , 74 Some of collapsed replication forks are resolved by DDR pathways such as HR 75 or mitotic DNA synthesis. 76 However, increased genomic instability and mutagenesis can not be rescued in regions where the DNA replication process is not resumed. In order to accomplish bulk genome replication, cells often recruit error‐prone DNA polymerases. On the other hand, the replication failures and the subsequent presence of incompletely‐replicated DNA in mitosis would further lead to chromosomal entanglements between sister chromatids 77 or the generation of micronuclei. 78 Finally, if replication stress is not eliminated after mitosis, nuclear bodies, characterized by the DNA damage response protein p53 binding protein 1 (53BP1), are formed in daughter cells as protective machinery. 79 Recent evidence has revealed an important role of RNA in DDR, particularly in human cells. Two substes of RNA were identified, damage‐induced long noncoding RNAs (dilncRNAs) and small DDR RNAs (DDRNAs). 80 , 81 The dilncRNAs potentially forms DNA–RNA hybrids and attracts DNA repair‐associated proteins such as BRCA1, BRCA2, RAD51, and MRE11 to the DNA damage sites and thus promotes DNA repair. 82
Apart from being a crucial etiologic factor for cancer, 71 , 83 , 84 elevated replication stress has also been observed during cancer therapies. Nucleoside analogues are widely used as chemotherapies such as acute myeloid leukemia (AML) induction therapy, which decrease the amount of dNTPs and delay DNA synthesis, and thus promote replication stress. For example, fluorouracil (5‐FU) is a pyrimidine analogue, which is incorporated into RNA following its conversion to 5‐fluoro‐deoxyuridine monophosphate (5‐FdUMP). 85 In addition to RNA metabolism, 5‐FU has also been found to hamper DNA metabolism according to reported genetic screening results, which suggested increased 5‐FU sensitivity in cells deficient in the ATR‐Chk1 signaling pathway and homologous recombinational repair. 86 Oxaliplatin, a platinum‐type chemotherapeutic drugs, inhibits DNA replication and G2/M cell‐cycle progression independent of ATM and ATR. 87 , 88 The underlying mechanism for the independence of oxaliplatin on DDR pathway lies in its ability to induce ribosome biogenesis stress by suppressing the transcription of deoxyuridine triphosphatase and the enzymes required for thymidylate biosynthesis. 89 , 90 Similar inhibitory effect on DNA synthesis can also be observed on TFTD (TAS‐102), a novel anticancer drug that suppresses dTTP biosynthesis 91 and accelerates its incorporation into DNA. 92
4. INHIBITORS TARGETING DNA REPAIR PATHWAYS
The current anticancer strategies that exploit DDR defects have largely been addressed by the development of targeted agents that inhibit molecules involved in DNA repair process. We herein summarized single‐agent DDR inhibitors currently under clinical trial development (Table 1).
TABLE 1.
Single‐agent DDR inhibitors currently under clinical trial development
| Target | Conditions | Interventions | Phase | Clinical trial* |
|---|---|---|---|---|
| PARP | ||||
| Metastatic breast cancer | Drug: PARP inhibitor 2X‐121 | Phase II | NCT03562832 | |
| Breast cancer | Talazoparib | Phase II | NCT03990896 | |
| Ovarian cancer | AK112 | Phase I/II | NCT04999605 | |
| Breast cancer | Rucaparib | Phase I | NCT03911453 | |
| BRCA‐positive advanced breast cancer | KU‐0059436 (AZD2281) | Phase II | NCT00494234 | |
| Ovarian cancer | EP0057 olaparib | Phase II | NCT04669002 | |
| Pancreatic cancer | Niraparib | Phase II | NCT03601923 | |
| Neoplasms | Talazoparib | Phase I | NCT03343054 | |
| Ovarian carcinoma, breast cancer | AZD2281 | Phase II | NCT00679783 | |
| Advanced breast cancer | Talazoparib tosylate | Phase II | NCT02401347 | |
| Advanced malignant solid neoplasm | Talazoparib | Phase II | NCT04550494 | |
| HRR mutated solid tumors (VASTUS) | IDX‐1197 | Phase I/II | NCT04174716 | |
| Ovarian cancer | Niraparib | Phase II | NCT02354586 | |
| Advanced tumors with ATM/BRCA1/2 gene mutation | Talazoparib | Phase II | NCT02286687 | |
| Ovarian neoplasms | Niraparib | Phase III | NCT01847274 | |
| Advanced/metastatic solid tumors | NMS‐03305293 | Phase I | NCT04182516 | |
| Solid tumor | RP12146 | Phase I | NCT05002868 | |
| Platinum sensitive BRCAm Serous ovarian cancer | Olaparib, Cediranib,AZD2281 | Phase I | NCT02855697 | |
| Ovarian neoplasms | KU‐0059436 (AZD2281) | Phase I | NCT00516373 | |
| Ovarian cancer (neoadjuvant setting) | Niraparib | Phase II | NCT04284852 | |
| Advanced tumors with HRR gene mutations | Olaparib oral capsule | Phase II | NCT03967938 | |
| Ovarian cancer | Fluzoparib capsules | Phase III | NCT03863860 | |
| Advanced malignant solid neoplasm | Olaparib | Phase II | NCT03212274 | |
| Ovarian cancer | IMP4927 | Phase III | NCT04169997 | |
| Ovarian cancer | ZL‐2306 (nirapairb) | Phase III | NCT03709316 | |
| Ovarian, breast cancer | Lynparza (olaparib) | Phase I | NCT04041128 | |
| Ovarian cancer | ZL‐2306 (niraparib) | Phase II | NCT04392102 | |
| Ovarian cancer | Talazoparib oral capsule | Phase I | NCT04598321 | |
| Digestive cancers | Individualized PARP inhibitor | Not applicable | NCT04584008 | |
| gBRCA mutated pancreatic cancer | Olaparib | Phase III | NCT02184195 | |
| BRCAm pancreatic cancer | Olaparib | Phase II | NCT04858334 | |
| Pancreatic cancer | RUCAPARIB | Phase II | NCT03140670 | |
| Metastatic breast cancer | Olaparib | |||
| Relapsed ovarian cancer | Olaparib tablets | Phase III | NCT03534453 | |
| Metastatic bladder urothelial carcinoma | Olaparib | Phase II | NCT03375307 | |
| Advanced solid tumors | TALZENNA capsule | Phase I | NCT04672460 | |
| Relapsed ovarian cancer | Olaparib tablets | Phase III | NCT01874353 | |
| Stage IV pancreatic cancer | Olaparib | Phase II | NCT02677038 | |
| HER2‐negative, germline BRCA mutation‐positive breast cancer | Niraparib | Phase III | NCT01905592 | |
| Ovarian, fallopian tube, primary peritoneal cancer | Niraparib | Phase II | NCT03891576 | |
| Metastatic castration‐resistant prostate cancer | Rucaparib | Phase III | NCT02975934 | |
| Ovarian, fallopian tube, primary peritoneal cancer | Rucaparib | Phase III | NCT01968213 | |
| Ovarian, fallopian tube, primary peritoneal cancer | Rucaparib | NCT04539327 | ||
| Prostatic neoplasms | Niraparib | Phase II | NCT02854436 | |
| Breast cancer patients with chest wall recurrences | Olaparib | Phase I | NCT03955640 | |
| gBRCAm breast cancer | Olaparib | Phase III | NCT02000622 | |
| Biliary tract cancer with aberrant DNA repair gene mutations | Olaparib | Phase II | NCT04042831 | |
| Solid tumors and with deleterious mutations in HRR genes | Rucaparib | Phase II | NCT04171700 | |
| Ovarian, fallopian tube, or primary peritoneal cancer | Oral rucaparib | Phase II | NCT01891344 | |
| Advanced malignant solid neoplasm | Olaparib | Phase II | NCT03233204 | |
| Castration‐resistant prostate carcinoma | Olaparib | Phase II | NCT03516812 | |
| Advanced malignant neoplasm | AMXI‐5001 | Phase I/II | NCT04503265 | |
| Metastatic carcinoma of the cervix | Nirapaib | Phase I/II | NCT03644342 | |
| Solid tumor, adult | RBN‐2397 | Phase I | NCT04053673 | |
| Recurrent solid tumor | Olaparib | Phase II | NCT01078662 | |
| Prostate, ovarian cancer | Rucaparib | Phase III | NCT04676334 | |
| IDH1/2‐mutant Grade I–IV gliomas | Drug: PARP Inhibitor BGB‐290 | Phase I | NCT03749187 | |
| Advanced gastric adenocarcinoma | Olaparib | Phase II | NCT04209686 | |
| Malignant mesothelioma | Rucaparib | Phase II | NCT03654833 | |
| Acute myeloid leukemia | Olaparib | Phase II | NCT03953898 | |
| Advanced or inoperable gastric cancer | Pamiparib (BGB‐290) | Phase II | NCT03427814 | |
| Endometrial serous carcinoma | Niraparib | Phase II | NCT04716686 | |
| Small cell lung carcinoma | IDX‐1197 | Phase II | NCT03672773 | |
| Urothelial carcinoma | Olaparib+EP0057 | Phase I/II | NCT02769962 | |
| Neoplasms | Niraparib tablet/capsule | Phase I | NCT03329001 | |
| Advanced ovarian cancer | Olaparib tablets | Phase III | NCT01844986 | |
| Head and neck squamous cell carcinoma | Niraparib | Phase II | NCT04681469 | |
| Advanced solid tumors | JPI‐547 | Phase I | NCT04335604 | |
| Metastatic melanoma with HR mutation | Niraparib | Phase II | NCT03925350 | |
| ATM | ||||
| Advanced solid tumors | M4076 | Phase I | NCT04882917 | |
| Neoplasms | BAY1895344 | Phase I | NCT03188965 | |
| NSCLC | VX‐970 (M6620) | Phase I/II | NCT02487095 | |
| Cancers of the stomach and intestines | BAY 1895344 | Phase I | NCT04535401 | |
| SCLC, neuroendocrine cancer, pancreatic cancer | BAY 1895344 | Phase I | NCT04514497 | |
| Urothelial cancer | BAY 1895344 | Phase I | NCT04491942 | |
| Advanced cancers | LY2606368 (Prexasertib) | Phase II | NCT02873975 | |
| Unresectable solid tumors | M1774 | Phase I | NCT04170153 | |
| Advanced stage solid tumors | M6620 | Phase I | NCT03309150 | |
| ATR | ||||
| Advanced solid tumor | RP‐3500 | Phase I/II | NCT04497116 | |
| Advanced solid tumors and lymphomas | BAY1895344 | Phase I | NCT03188965 | |
| Cancers of the stomach and intestines | BAY 1895344 | Phase I | NCT04535401 | |
| Advanced cancer | ART0380 | Phase I/II | NCT04657068 | |
| Unresectable solid tumors | M1774 | Phase I | NCT04170153 | |
| Pancreatic and ovarian cancer | BAY 1895344 | Phase I | NCT04616534 | |
| CHK1 | ||||
| Advanced cancers | LY2606368 | Phase II | NCT02873975 | |
| WEE1 | ||||
| Advanced solid tumors | IMP7068 | Phase I | NCT04768868 | |
| Uterine cancer | AZD1775 | Phase II | NCT03668340 | |
| Prostate cancer | Adavosertib | Phase II | NCT03385655 | |
| DNA‐PK | ||||
| Advanced solid tumors, non‐Hodgkin's lymphoma, or multiple myeloma | CC‐122 | Phase I | NCT01421524 | |
Data from https://clinicaltrials.gov.
4.1. Poly (ADP‐ribose) polymerase (PARP)
4.1.1. Mechanisms underlying the application of PARP inhibitors
The development of PARP inhibitors represents the paradigm of the concept discussed earlier, known as synthetic lethality. 93 PARP1 and PARP2 are key DDR enzymes that sense DNA damage and pass on signals by modifying target proteins with negatively charged poly(ADP‐ribose) (PAR) chains, known as PARylation. 94 The structural changes of PARP1 following its binding to damaged DNA activate its catalytic function, 95 , 96 which facilitates the recruitment of DNA repair effector molecules and the structural remodeling of chromatins around DNA damage sites. In this way, PARP1 PARylates itself, a process known as autoPARylation, which potentially contributes to its release from repaired DNA. 97 Recent advances in epigenetics have revealed the correlation of specific chromatin remodeling factors with DDR. 98 One such example is PARP1, which PARylates MORC2 and increases its ability to induce chromatin remodeling. Since eukaryotic DNA is surrounded by condensed chromatin, the dynamic remodeling of chromatin would largely affect the efficiency of DNA repair. 99 , 100 More studies are thus warranted to shed light on the collaborative interplay between chromatin‐associated enzymes and DDR. Given the pivotal role of PARP in promoting the effective repair of DNA, PARP inhibitors selectively kill tumor cells with homologous recombination deficiency. Conflicting results were reported regarding whether PARP is required for BER, 101 with some evidence suggesting the increased sensitivity of PARP1‐deficient cells to base‐damaging agents, 102 , 103 , 104 whereas some studies found that PARP was not necessary for the repair of base. 105
Alongside the inhibition on enzymatic activities of PARP, the process referred to as PARP trapping provides an additional mechanism for PARP inhibitors, where PARP1 and PARP2 are trapped at the site of DNA damage and block the recruitment of proteins involved in DNA repair. Since a complete set of repair‐associated proteins is the prerequisite for accurate DNA repair, PARP‐inhibited cells lost the capacity to properly repair their DNA during replication, eventually inducing mitotic catastrophe and subsequent cell death. 94 Multiple PARP inhibitors have demonstrated comparable antitumor efficacy and selective inhibition on PARP1 and PARP2, but their abilities to induce PARP trapping vary, which contributes to the difference of recommended doses among PARP inhibitors. 106 , 107
PARPi is a promising therapeutic strategy for BRCA‐mutant tumors, which is a typical setting of synthetic lethality. 108 BRCA gene has long been identified as crucial components of the HR pathway. 109 In cells harboring BRCA mutation, alternate DNA repair mechanisms such as the PARP pathway are initiated to fix the damage. Thus, PARP inhibition in a BRCA‐deficient setting likely causes the accumulation of DNA damage and thereby leads to cell death. However, as cells with BRCA1 or BRCA2 germline mutation are unable to fix treatment‐induced DSBs, toxicity caused by PARP inhibitor has received considerable attention. Previous studies investigated the association between myelosuppression occurrence and BRCA1 or BRCA2 mutation status in patients receiving platinum‐based chemotherapy and revealed no significant correlation between BRCA mutation status and hematological toxicities. 110 However, it remains unclear whether PARPi toxicity could also be used as a predictive biomarker for PARPi treatment response.
4.1.2. PARP inhibitors as the first‐line therapy
Ovarian cancer is the leading cause of gynecologic cancer‐related deaths in women worldwide, 111 and the standard care for the newly diagnosed advanced ovarian cancer (NADOC) patients in the last two decades is the surgical debulking followed by platinum–taxanes‐based systemic chemotherapy. Unfortunately, an estimated number of 70% of patients with advanced ovarian cancer experience relapsed disease within 3 years posttreatment. 112 The concurrent and maintenance anti‐VEGF bevacizumab was later recommended for the standard first‐line systemic treatment of epithelial ovarian cancer, which improves PFS in patients with higher risk of recurrence (International Federation of Gynecology and Obstetrics FIGO stage IV or suboptimally debulked stage III ovarian cancer—OC). 113 However, the efficacy of the combinational treatment diminishes over time with a 5‐year survival rate being around 35%, and adverse effects accumulate as chemotherapy cycles proceed. 114 , 115 , 116 Thus, recent research of this field aims to identify more efficient drug combinations to aid the systemic treatment of ovarian cancer patients.
In a recent European Society for Medical Oncology (ESMO) Congress, research teams reported preliminary results from clinical trials of three different PARP inhibitors in patients with ovarian cancer, including the PAOLA‐1/ENGOT‐OV25 Phase III trial where the combination of PARP‐inhibitor olaparib and bevacizumab was assessed for the first time as maintenance therapy following platinum‐based chemotherapy in the overall population regardless of the BRCA status. 114 , 115 , 116 The mechanism underlying the application of PARP inhibitors in patients with advanced ovarian cancer is illustrated in Figure 2. Following the promising results from these trials, the oncology community starts to review the practice regime of PARP inhibitors in first‐line treatment of NADOC and the selection criterion for patients that would receive the maximum benefits. The defined subset of patients based on their molecular diagnosis include those with BRCA‐mutation, HR‐deficiency, and HR‐proficiency. 117 Here, we discuss the updated data from the ongoing as well as previous clinical trials regarding the application of PARP inhibitors.
FIGURE 2.
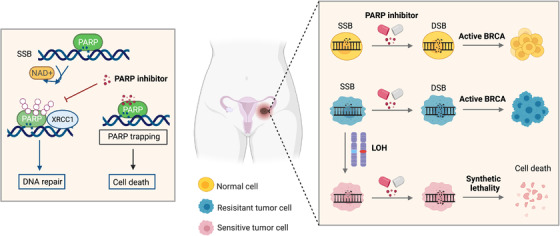
The mechanism underlying the application of PARP inhibitors in patients with advanced ovarian cancer. SSBs, single‐strand breaks; DSBs, double‐strand breaks. Figure was created with Biorender
Olaparib
The first human clinical trials of PARPi evaluated the chemopotentiation effect of low‐dose rucaparib in patients with metastatic melanoma. 118 Currently, four PARP inhibitors, olaparib, rucaparib, niraparib, and talazoparib, have been approved by the US Food and Drug Administration (FDA). Based on accumulating research results on synthetic lethality observed between PARP inhibition and BRCA mutation status, 119 , 120 a clinical evaluation of olaparib was initiated in 2005, where 63% of patients cancer with germline BRCA1 or BRCA2 mutations (gBRCAm) exhibited durable clinical benefit. 121 The evaluation of olaparib later extended to patients with gynecological malignancies and reported a favorable response to olaparib in patients who response better to prior platinum chemotherapies. This finding accorded with the hypothesis that platinum‐based therapies and PARPi shared similar molecular targets. 122 Phase II trials further supported significant clinical benefit in multiple gBRCAm cancer types including breast, ovarian, pancreatic, or prostate cancers. 123 , 124 , 125 In 2014, olaparib was approved as maintenance therapy for platinum‐sensitive advanced ovarian cancer with germline BRCA1 or BRCA2 mutations (gBRCAm). 126 More recently, a randomized Phase III trial reported improved survival outcomes in gBRCAm/HER2‐negative breast cancer patients receiving olaparib than those with standard chemotherapy. 127
A growing number of clinical trials have been conducted since 2009 to investigate the efficacy and safety of PARP inhibitors in multiple cancer types irrespective of the BRCA status. 128 , 129 , 130 , 131 , 132 A Phase II trial metastatic investigated the treatment response to olaparib in patients with castrate‐resistant prostate cancer (mCRPC) by evaluating clinical parameters including PSA decline and radiologic responses. 133 Notably, the overall response rate in unselected CRPC population to PARP inhibitors was only 33%, possibly attributed to the observed tumor mutations in other DDR members. 134 The team then conducted next‐generation sequencing on enrolled patients and the genetic map of these patients revealed homozygous deletions or mutations in DRR‐associated genes including ATM, PALB2, CHEK2, FANCA, and HDAC2. This trial not only granted olaparib approval for the treatment of BRCA1/2‐ or ATM‐mutant mCRPC patients, but also provided additional application of PARPi in DDR‐defective patients beyond BRCA mutations. Thus, it may be insufficient only to use BRCA1 or BRCA2 mutations as predictive biomarker for PARPi responders. Based on the observation that ATM gene alteration resulted in increased sensitivity of cells to PARP inhibition, ATM gene mutation was included as a predictive biomarker for PARPi response in the FDA breakthrough therapy designation. 135 , 136 It has to be addressed that the ideal predicting factor for PARPi response would be recombination deficiency, which does not exist in practice.
Rucaparib
The combination of rucaparib and temozolomide were the first clinical trial containing PARPi treatment regimens. 118 Rucaparib was first indicated for the treatment of advanced ovarian cancer with either germline or somatic BRCA1/2 mutations, and was then approved in 2018 for the maintenance treatment of platinum‐sensitive ovarian, fallopian tubal, and peritoneal cancer regardless of the BRCA status. 137 In the maintenance setting (ARIEL 2, NCT01891344), advanced ovarian cancer patients were divided into three groups based on the genomic features of their tumors including the germline or somatic BRCA status and chromosomal loss of heterozygosity (LOH). The longest progression‐free survival (PFS) was observed in the BRCA mutant group, followed by the high LOH group. 138 BRCA status appeared to be a significant predictor in the maintenance setting of rucaparib, given that the proportion of BRCA wild‐type patients displaying durable responses was smaller than that of patients receiving standard platinum‐based chemotherapies. 139 Thus, the following Phase III trial (NCT01968213) aimed to investigate the potential of the genome‐wide LOH to be transformed into a clinically applicable biomarker for patients’ responses to rucaparib 27908593. Along with the promising results from an additional Phase II trial HGSOvCa (NCT01482715), 140 rucaparib was approved for chemotherapy‐pretreated patients with gBRCAm or sBRCAm advanced ovarian cancer. However, rucaparib has been reported as the least selective clinical PARP1 inhibitor with simultaneous inhibition on multiple PARPs ranging from PARP1, PARP2 to mono(ADP‐ribosyl) transferases PARP3, PARP4, PARP10, PARP15, and PARP16. 141 , 142
Veliparib and niraparib
Some PARP1/2 inhibitors are not highly selective such as rucaparib discussed earlier. 142 For example, niraparib has been reported to interact with non‐PARP targets such as deoxycytidine kinase (DCK). 143 The cross‐inhibition on DCK, which is fundamental for the activation of nucleoside analogs, would decrease the efficacy of niraparib/gemcitabine synergy. 143 On the other hand, due to its formation of a PARP1/2‐unique water‐mediated hydrogen bond that interacts with a highly conservative subdomain D766, veliparib has been identified as the most selective clinical inhibitors targeting PARP1/2, with 100‐fold higher affinities to PARP1/2 relative to olaparib and talazoparib. 144 In a Phase III clinical trial, the median duration of PFS was significantly increased in ovarian cancer patients receiving niraparib, irrespective of gBRCA status (NCT01847274). 145 Though non‐gBRCA mutant, these tumors were identified with a unique mutational profile similar to the genome of gBRCAm tumors, which is referred to as BRCAness DNA scar. 146 Though BRCAness DNA‐scar positive patients appeared to have improved prognosis compared to BRCAness‐scar negative patients, the prognostic value of BRCAness‐scar as a predictive biomarker remains incompletely defined and requires further clarification in larger cohorts. 139 , 145
Though effective in the clinical practice, PARP inhibitors have also demonstrated certain limitations like any other novel development in history. Predominantly, the varying PARP trapping ability by different PARP inhibitors potentially lead to the off‐target PARP trapping on the DNA of normal cells. 147 Besides, the emerging resistance to PARP inhibitors also poses challenges to their clinical application, the underlying mechanisms of which include loss of PARP trapping, 148 , 149 upregulated drug efflux protein expression, 150 , 151 stabilized replication fork stabilization, 152 , 153 , 154 and the restoration of HR pathway. 155 , 156 , 157 , 158 , 159 , 160 , 161 , 162 , 163
4.2. Poly(ADP‐ribose) glycohydrolase (PARG)
The above limitations of PARP inhibitors motivated the design of additional therapeutic targets for BRCA‐proficient and deficient tumors, or PARPi‐resistant tumors. PARG reverses the action of PARP enzymes by hydrolyzing the ribose–ribose bonds in PAR following DNA damage. 164 , 165 , 166 Likewise, the active role of PARG in DNA replication and repair leads to increased sensitivity to DNA damaging agents in PARG‐deficient cells. Though extensive studies have suggested the correlation between PARP inhibitors and synthetic lethality, research on therapeutic mechanisms of PARG inhibition has lagged behind. It has been reported that depletion of the HR proteins such as BRCA1/2 in breast cancer cells could stimulate synthetic lethality in PARG‐inhibited cells, 167 , 168 and that COH34, a PARG inhibitor, is able to induce cell death of ovarian and breast cancers with BRCA mutations or resistance to olaparib. 169 However, conflicting results were reported in other cancer cells. 170 Of the six tested breast cancer lines, only one BRCA‐proficient cell line was sensitive to PARG inhibitor PDD00017273, whereas five cell lines failed to respond to PDD00017273 including those with BRCA mutations. 171
PDD00017273 is a quinazolinedione‐type PARG inhibitor with improved specificity, efficiency, and cell permeability, but lacks bioavailability. 172 Unlike cytotoxic PARP inhibitors, the major effect by PDD00017273 is cytostasis where the replication catastrophe does not progress into mitosis but rather remains static in interphase. 171 However, the exposure to ionizing radiation enhanced centrosome amplification and the subsequent multipolar spindle formation and chromosome missegregation caused by PARG deficiency. 173 , 174 Thus, it is intriguing to speculate that under some circumstances such as PARG inhibition coupled with cell‐cycle checkpoint blockades or DNA‐damaging agents, mitotic abnormalities would occur. 175 , 176 , 177
Neither of the first‐generation PARG inhibitor (GPI 16552 and gallotannin) demonstrates sufficient activity in vitro and its frequent off‐target effects in cells makes it a less than ideal strategy. 178 , 179 Another early PARG inhibitor, rhodanine‐based PARG inhibitor (RBPI) is more selective than previous generation PARGi, with limited cell permeability. 180 , 181 The recently reported COH34 is a novel small‐molecule PARG inhibitor with nanomolar potency both in vitro and in vivo, and notably, with efficiently killing effect on PARP inhibitor‐resistant cancer cells, which makes it a good candidate for clinical studies. 169 Chemical library screening identified methylxanthine derivatives JA2–4 and JA2131 as selective bioavailable PARG inhibitors, which showed comparable killing on PARP inhibitor‐resistant A172 glioblastoma cells. 182
4.3. Ataxia telangiectasia mutated (ATM)
The DDR signaling cascades are driven by serial protein phosphorylation. ATM, ATR, and DNA‐PKs are the key kinases involved in this process and are similar in molecular structure, the C‐terminus of which is responsible for phosphorylation activity especially on serine or threonine residue (Ser/Thr). 183 , 184 , 185 Activated by DNA double‐strand breaks, ATM is recruited to DSB sites by the MRE11‐RAD50‐NBS1 (MRN) complex. 186 Substrates of ATM include p53, CHK1, and CHK2, the phosphorylation of which would lead to intra‐S or G2/M cell‐cycle arrest. 187 , 188 Despite its canonical role in a wide variety of molecular processes such as DNA repair, ATM has also been characterized with noncanonical functions including spliceosome displacement. 189 As ATM is rightly considered as a tumor suppressor, ATM deficiency or deleterious alterations are commonly seen in solid tumors and B‐cell lymphoma. 190 Germline ATM mutation likely contributes to Ataxia Telangiectasia (A‐T), a neural degeneration disorder characterized by increased predisposition to cancer. 191
The main reason for ATM deficiency in cancer cells is hypermethylation of the ATM promoter, 192 with multiple cancer types including brain cancer, breast cancers, lung cancer, and head and neck squamous cell carcinoma exhibiting hypermethylated ATM promoter region. 193 , 194 , 195 , 196 However, ATM signaling can also be advantageous to tumors, increasing their risks of therapeutic resistance to radiation and chemotherapies. 197 Several ATM inhibitors are now under investigation for cancer therapy. 198 , 199 The loss of ATM occurs in prostate cancer and was recently suggested to increase cell sensitivity to ATR inhibition. 200
The first reported selective ATM inhibitor, 2‐morpholin‐4‐yl‐6‐thianthren‐1‐yl‐pyran‐4‐one, (KU‐55933), was developed by screening the PIKK family‐targeting compound library and exhibited 100‐fold higher selectivity for ATM over ATR, DNA‐PK, and PI3K. 199 , 201 Exposure to KU‐55933 sensitizes cells to cytotoxic agents that cause DSB, by blocking HR repair signals and thereby increasing γ‐H2AX and RAD51 foci accumulation. 202 In response to chemotherapy, KU‐55933 inhibits ATM‐mediated repair signals in the presence of inositol polyphosphate‐4‐phosphatase type II (INPP4B), which has contradictory roles in cancer progression. 203 In colon cancer cells, INPP4B acts as an oncogenic factor that positively regultates AKT 26411369, whereas INPP4B suppresses cancer progression in prostate cancer cells by reducing tumor migration, invasion, and angiogenesis. 204
KU‐60019 is an analogue of KU‐55933 with improved pharmacokinetics and bioavailability and is reported to interrupt radiation‐induced ATM phosphorylation in glioma cells. 205 Given that PTEN is an active participant of DNA repair process, it is not surprising that KU‐60019 was specifically toxic to PTEN mutant cancer cells. 206 Besides, the combination of KU‐60019 and cisplatin would induce synthetic lethality in PTEN‐deficient cells, 207 , 208 the underlying mechanism of which involves increased PARP cleavage and γ‐H2AX formation. 209 Thus, PTEN‐deficiency is a potential biomarker for predicting repines to DDR‐targeting agents. KU59403 is the first ATM inhibitor tested in preclinical trials with improved solubility, bioavailability, and selectivity. 210 KU‐59403 potentiates the efficacy of chemotherapies and IR at low doses in cancer cells irrespective of TP53 mutation status. 210 However, KU‐59403 monotherapy failed to demonstrate antitumor effects either in vitro or in vivo, which largely limited its clinical application and was not widely used thereafter. CP466722 was identified as a ATM kinase inhibitor by screening targeted compound library, which does not display inhibitory activities on PI3K family members. Noteworthy, even transient inhibition of ATM by CP466722 is sufficient to induce radiosensitization in cells and suggests that therapeutic radiosensitization, indicating that ATM is required for early stage of the DDR process. 211
The major limitations of earlier developed ATM inhibitors are their bioavailability in central nervous system via the blood brain barrier (BBB). Modified ATM inhibitors AZ31 and AZ32 have higher free brain concentrations and their radiosensitization effects were more prominent in p53 mutant cells than p53 wild‐type glioma cells. 212 In contrary to this finding, previous evidence suggested increased sensitivity of wild‐type p53 glioblastoma cells to radiation than p53 mutant cells. 213 AZD0156 has been reported to enhance the efficacy of DSBs in mouse xenograft models but lack BBB penetration. 214 The further optimized compound, AZD1390, is now under investigation as a radiosensitizer for nervous system malignancies. 215
4.4. Ataxia telangiectasia and Rad3‐related protein (ATR)
In contrast to ATM, which is triggered by DSBs, ATR is activated by and recruited to replication protein A (RPA)‐coated single‐strand DNA (ssDNA). 216 , 217 Single‐strand DNA can be produced by nucleolytic processing of DSBs as well as the uncoupling of the replicative DNA helicase from the DNA polymerase machinery. The intracellular ATR signaling involves the phosphorylation of a series of downstream molecules, triggering a wide array of responses including blocking cell‐cycle checkpoints, DDR, and cell apoptosis. 218 In response to genotoxic stress, Chk1 is phosphorylated on serines 317 (S317) and 345 (S345) by ATR, thereby activating WEE1, 219 , 220 which in turn phosphorylates CDK1 on tyrosine 15 and suppresses mitotic entry. 221 CDC25A is responsible for removing the inactivated phosphates on CDK2. Once CDC25A is phosphorylated by CHK1, the activation of intra‐S phase checkpoints impairs the rate of CDC25‐mediated replication, allowing cells to repair DNA damage. In addiction, as CDK1 is fundamental for the progression via G2/M checkpoints, ATR has been described colloquially as the apex of DDR signaling that acts on both S and G2/M cell‐cycle checkpoints, preventing the entry of damaged DNA into replication process before it has been properly repaired. 187 , 222 , 223 , 224
Cancer‐associated inflammation and cytotoxic treatments such as chemotherapies and radiotherapies are known to cause replication stress, which increases cell reliance on the ATR‐mediated S and G2/M checkpoints activation as countermeasures. Thus, it is intriguing to speculate whether inhibition of ATR would sensitize cells to DNA damaging agents such as chemotherapy, encouraging the development of selective ATR inhibitors. However, compared with other DDR proteins such as PARP, development of ATR inhibitors has lagged behind. Contributing factors may include the large size of the ATR molecule and the lack of knowledge on its crystal structure. In addition, its highly homologous active sites in all PIKKs and the demand for coactivating proteins further restrict its drug design.
The first chemicals reported to inhibit ATR were natural molecules caffeine and schisandrin B, the inhibition of which was nonspecific and only worked at high concentrations. 225 , 226 This finding further confirmed the potential of natural compound for future synthesis of DDR‐regulating drugs. 227 Several approaches were used to identify potentially potent ATR inhibitors. One such example is the cell‐based high‐throughput microscopy that enables the screening of compounds, investigating their specific activity on ATR, 228 , 229 where they identified a highly selective compound, ETP‐46464 with specific action on ATR, rather than ATM or DNA‐PKcs. 229 Recent advancement in gene editing suggests that CRISPR DDR screens can also be used to identify drug candidates. 230
Another identification strategy is the in vitro use of recombinant ATR to test its kinase reactions, through which researchers were able to characterize compounds that directly and specifically targeted ATR, such as VE‐82. 66 , 231 With further modification on pharmacological properties, VE‐821 was later named VE‐822 and is now under clinical investigation as VX‐970 (M6620) (NCT03309150, NCT03022409, NCT02723864, etc.). 232 Interestingly, some ATR inhibitors were discovered during research on inhibitors developed for other targets. NU6027 was originally selected for CDK2 inhibition and was later found to impair HR pathway, thereby sensitizing cells to DNA‐damaging agents and PARP inhibitors. 233 The new‐generation ATR inhibitors include AZD6738, an derivate of the compound AZ20, which is currently under clinical investigations (NCT02567422, NCT03022409, NCT02157792), BAY1895344 (NCT03188965), 234 , 235 , 236 berzosertib (NCT02157792), 237 and a recently reported pyrazolopyrimidine‐containing inhibitor of ATR. 238
4.5. CHK1
As described earlier, CHK1 is actively involved in the ATR‐ and ATM‐initiated DNA damage response by phosphorylating and recruiting a series of regulatory proteins. CHK1 regulates the intra‐S checkpoint by phosphorylating CDC25A, leading to the degradation of CDC25A and the subsequent decrease of cyclin‐dependent kinase 2 (CDK2) activity in S cell‐cycle phase, 239 , 240 and the phosphorylation of CDC25C and WEE1 by CHK1 regulates mitotic entry and G2/M checkpoints. 241 Moreover, CHK1 also phosphorylates RAD51 on Thr‐309 promoting its interaction with BRCA2 during HR. 242 , 243 , 244 , 245 Noteworthy, CHK1 also acts on a number of physiological processes that are critical to cell survival. For example, the suppression of CHK1 leads to p53‐induced death domain (PIDD) signaling and the associated caspase 2‐mediated cell death. 246 It has been recently reported that the phosphorylation of nucleophosmin (NPM) by CHK1, a chaperone protein involved in various cellular functions including, disrupts its interaction with PIDD, thus protecting cells from caspase 2‐mediated cell death. 247 Further studies are warranted to clarify the importance, yet poorly defined role, of CHK1 in other cellular processes independent of DDR.
Though CHK1 deficiency has been reported to induce early embryonic lethality in vivo, 219 the knockdown of which is preferentially more deleterious in tumor cells than in normal cells, suggesting the potential of Chk1 as a therapeutic target in cancer treatments. On the other hand, increased CHK1 levels have been reported to correlate to worse prognosis, disease recurrence, and therapeutic resistance, 248 , 249 , 250 , 251 , 252 further supporting the therapeutic potential of Chk1 inhibition. In circumstances where cells harbor certain genetic alterations, such as c‐MYC, CHK1 inhibitors are able to induce synthetic lethality in malignancies driven by oncogene c‐MYC. 253 , 254 , 255 Likewise, CHK1 inhibitor PF‐00477736 exhibited cytotoxic effects on mantle cell lymphoma (MCL) and myeloma with translocation t(11;14)‐mediated Cyclin D1 overexpression. 256 , 257 Cells with acquired PF‐00477736‐resistant cells displayed enriched prosurvival and proliferation‐associated gene patterns, suggesting that inhibition of prosurvival signaling pathways could potentially sensitize cells to CHK1 inhibitors.
The first‐generation CHK1 inhibitors were used as chemosensitizing agents, the majority of which were nonspecific due to their high affinity to plasma protein 1‐acid glycoprotein, with a long half‐life and low bioavailability. 258 The early CHK1 inhibitors were mostly used as combinational partners with cytotoxic agents in cancer, 187 , 259 the clinical development of which was largely restricted by their unacceptable toxicities and suboptimal pharmacological profiles. 260 With significantly improved selectivity toward CHK1, the second‐generation CHK1 inhibitors such as LY2606368, LY2880070, SRA737, and GDC‐0575 are now under intense clinical studies. These CHK1‐targeting agents potently synergize with drugs that produce DNA damage including cytotoxic chemotherapies and antimetabolites. 261 , 262 One such example is the combinational treatment of low‐dose gemcitabine with GDC‐0575, which induced promising objective response rates in patients with advanced sarcoma. 263
Recently, clinical trials (NCT02797977, NCT02797964) reported promising results that the combination of a novel CHK1 inhibitor SRA737 with low‐dose gemcitabine led to partial responses in 6 patients and stable disease for at least 4 months in 32 patients. SRA737 has also demonstrated synergistic effect with PARP1 inhibitors in cancer both in vitro and in vivo. 264 Despite intense interest in CHK1 inhibitors, no known agents have reached Phase III clinical trial or received FDA approval. According to preclinical results, though the single use of CHK1 inhibitors did not usually cause significant toxicities, the unacceptable cytotoxic effects on normal cells caused by the combination therapy with DNA damaging agents outweighed the modest gains.
4.6. WEE1
In response to DNA damage, the activated ATR phosphorylates Chk1, which in turn phosphorylates WEE1 and CDC25. 265 , 266 , 267 In contrast to CDC25 whose activity is suppressed by the phosphorylation, WEE1 is activated and then phosphorylates downstream CDK1 on Tyr15 and Thr14 to inhibit its activity, leading to G2/M cycle arrest and allowing time for DNA damage repair. In addition, by phosphorylating CDK1 on Tyr15, WEE1 also prevents the progression of S phase to G2 phase before DNA replication is completed. 268 Moreover, WEE1 has also been reported to phosphorylate histone H2B on Tyr37, thereby blocking the transcription of certain histone genes that reduce the burden of the histone mRNA turnover machinery. 269
G1/S and G2/M checkpoints are regulated by p53 gene, which is frequently absent or deficient in cancer cells. Under this circumstance, cancer cells become highly dependent on WEE1‐mediated G2/M checkpoint control for DNA repair. 270 , 271 It is thus not surprising that some cancers are accompanied by WEE1 overexpression, which decreases their sensitivity to radiotherapy and chemotherapy. 272 , 273 Besides, results from whole‐genome characterization of chemoresistant ovarian cancer suggested the feasibility of WEE1 inhibition in multiple tumor‐related pathways. 21 , 274 These evidence support the early therapeutic rationale of WEE1 inhibitors in p53‐deficient tumors. It is becoming increasingly clear that neither p53 deletion nor the loss of G1 checkpoint is a predictor for WEE1 sensitivity. 275 , 276 , 277 Currently, most clinical studies focus on the combinational use of WEE1 inhibition with chemotherapeutic drugs, which will be discussed further in the review.
The first generation of small‐molecule WEE1 inhibitors, represented by PD0166285, was rather unspecific with an inhibitory activity against multiple kinases such as EGFR, CHK1, and c‐Src. 278 , 279 , 280 The first selective WEE1 inhibitor, adavosertib (AZD1775), was obtained from screening a small‐molecule compound library. 281 Though more selective than previous‐generation WEE1 inhibitors, from kinase profiling results, AZD1775 was found to target other kinases as well with reduced potency. 282 , 283 For example, the unspecific targets of AZD1775 include PLK, the role of which in cell‐cycle progression has been described as antagonistic to WEE1. This multiple binding may contribute to the difficulty in interpreting experimental results, but it was recently suggested that therapeutic concentrations of AZD1775 were not sufficient to suppress PLK1 activities. 284 Noteworthy, AZD1775 exhibits potent antitumor activity even as monotherapy. 285 Given that single‐agent therapy is believed to be almost equally toxic to normal and cancer cells, the antitumor activity of WEE1 inhibitors monotherapy potentially arises from the increased replication stress in cancer cells. 286 , 287 , 288
Whereas the rationale for WEE1 inhibitors is clear, its clinical application is restricted by its demand for appropriate therapeutic windows. The substantial >grade 3 adverse effects caused by AZD1775 are often a concern (NCT02341456, NCT02666950, NCT01357161, NCT00648648). As WEE1 is required for a number of physiological processes in normal cells, adverse events are usually expected to impact cells undergoing frequent divisions such as the hematopoietic system and intestinal epithelium. 289 For this reason, numerous efforts have been undertaken to optimize dosing and therapeutic schedule of AZD1775, 290 with its analogues being developed, which remained effective but brought lower toxicity. 291 Another research attempt is to identify additional biomarkers for AZD1775 to reduce the off‐target effects. AZD1775 is able to induce synthetic lethality in cells with defects in the Fanconi Anemia or HR pathways, 285 , 292 suggesting that the efficacy of AZD1775 may be enhanced by further inhibiting additional factors that downregulate DNA replication.
4.7. DNA‐PK inhibitors
DNA‐dependent protein kinase was initially discovered by chance in 1985 when scientists added double‐stranded DNA (dsDNA) into the cell extracts and identified this protein with enhanced phosphorylation. 293 Later in 1990, the DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) was identified. 294 , 295 Encoded by the PRKDC/XRCC7 gene, DNA‐PKcs is abundantly present in human cells with no fewer than 50,000 molecules per cell and the largest PIKK family member. 296 , 297 , 298 , 299 , 300 DNA‐PKcs shares similar domain compositions with two other PIKK family members involved in DDR, ATM, and ATR, such as the kinase domain and the conserved FRAP‐ATM‐TRRAP (FAT) domain. 301
Loss of the key factors in the NHEJ pathway has long been considered as a hallmark for tumor progression and increased sensitivity to DSB‐inducing agents, possibly due to increased genomic instability. 298 , 302 , 303 The upregulation of DNA‐PK expression was observed in various tumor types including the gastrointestinal cancer, lung cancer, and hepatocellular carcinoma and was associated with higher tumor grades and poor prognosis. 304 , 305 , 306 In melanoma, increased DNA‐PKcs expression was related to a progressed phenotype with tumor microenvironment favoring metastasis. 307 In addition, DNA‐PKcs upregulation has been reported to promote resistance to radiotherapy and chemotherapy in thyroid, 308 nasopharynx, 309 cervix cancers, 310 and leukemia. 310 , 311 Moreover, DNA‐PK has been reported to transcriptionally regulate protumorigenic pathways, leading to tumor progression and metastasis. 312 , 313 These findings have encouraged the design of multiple DNA‐PK inhibitory strategies.
Giving the structural similarity between DNA‐PK and PI3K, early attempts to block DNA‐PK were based on pharmacological approaches that directly targeted PI3K or its derivatives. Development of DNA‐PK inhibitors mainly focuses on the catalytic activity of DNA‐PKcs, whereas novel anti‐DNA‐PK approaches such as DNA‐PKcs‐inhibiting microRNAs 314 , 315 or inhibitors targeting the Ku heterodimers were based on the homology model of the ATP‐binding site. 316 , 317 The first reported DNA‐PK inhibiting compound was caffeine, which was identified with in vitro kinase activities on two other DDR master kinases ATM and ATR, and later with inhibition on DNA‐PK. 318 Further application of these early DNA‐PK inhibitors such as wortmannin 226 and vanillin 319 was limited due to poor selectivity and complexed structure. With the advent of a lead compound LY294002, more specific and potent derivate compounds were later developed such as NU7441, NU7427, NU7026, and NU7163. 320 , 321 , 322 , 323
In preclinical studies, NU7427 and NU7026 potentiated the therapeutic effect of IR and topoisomerase II inhibitor chemotherapy in cancer cells, 321 , 324 whereas NU7441 substantially delayed the repair of IR‐ and chemotherapy‐induced DSBs both in vitro and in vivo. 325 There were compelling preclinical data studies suggesting NU7441 as a potent DNA‐PK inhibitor in cancer models. 326 , 327 , 328 , 329 , 330 Another class of DNA‐PK‐targeting compounds studied in preclinical studies are a series of arylmorpholine‐containing compounds derived from IC60211, 331 which include IC86621, IC486154, IC87102, and the most intensively used IC87361. 332 , 333 Despite extensive research, clinical evaluation and application of these inhibitors could not be achieved due to their undesirable pharmacokinetics. 334
VX‐984 and M3814 are the new‐generation DNA‐PK selective inhibitors, which have already progressed into clinical trials in combination with IR or chemotherapy. VX‐984 is known for its potential to cross the blood brain barrier based on the observation that VX‐984 enhanced the response to radiotherapy in glioblastoma mouse models. 335 M3814 has been reported to suppress NHEJ repair induced by chemotherapies and radiation, and to enhance the treatment efficacy in multiple cancer types. 336 , 337 In addition, clinical studies supported the use of peposertib (formerly M3814) with desirable safety profile as monotherapy, 338 but most ongoing clinical trials investigate its effects in combination with chemo‐ or radiotherapy in cancer. LY3023414 and CC‐115 are dual inhibitors that simultaneously target DNA‐PK and the mammalian target of rapamycin (mTOR), selectively blocking class I PI3K isoforms at low nanomolar concentrations. 339 , 340 CC‐115 was initially designed for mTOR, but was later reported to inhibit DNA repair and become particularly active in ATM‐deficient tumors. 341 Recently Phase I trial on LY3023414 reported that LY3023414 was well tolerated as single agent in advanced cancers. 342
5. DDR INHIBITOR‐BASED COMBINATION THERAPY
The combined treatment of DDR inhibitors with other treatment modalities including chemotherapy, radiotherapy, immunotherapy, or other targeted therapies. Moreover, recent data also supported the therapeutic value of concomitant targeting against nonredundant DDR components. 343 , 344 Here we summarized the ongoing combination trials on DDR inhibitors with chemotherapy, radiotherapy, target therapy (Table 2), with other DDR inhibitors (Table 3), and with immunotherapy (Table 4).
TABLE 2.
Ongoing combination trials of DDR inhibitors with chemotherapy, radiotherapy, and target therapy
| Conditions | Interventions | Phase | Clinical trial* | |
|---|---|---|---|---|
| Chemotherapy | ||||
| PARP | ||||
| Cancer | Veliparib + VX‐970 + cisplatin | I | NCT02723864 | |
| Metastatic breast cancer | Veliparib + carboplatin/paclitaxel | III | NCT02163694 | |
| Ovarian, breast, pancreatic, prostate cancer | AZD5305 + Carboplatin/paclitaxe | I /II | NCT04644068 | |
| Ovarian cancer | Veliparib + carboplatin/paclitaxel | III | NCT02470585 | |
| Metastatic pancreatic adenocarcinoma | Veliparib + fluorouracil/irinotecan hydrochloride | II | NCT02890355 | |
| SCLC | Veliparib + topotecan | I | NCT03227016 | |
| Advanced solid tumors | IMP4297 + temozolomide | I | NCT04434482 | |
| Triple negative breast cancer, ovarian cancer | KU‐0059436 (AZD2281) + carboplatin/paclitaxel | I | NCT00516724 | |
| Breast cancer | ABT‐888 + temozolomide | II | NCT01009788 | |
| Metastatic BRCA‐associated breast cancer | Veliparib + cisplatin | II | NCT02595905 | |
| HR deficient advanced solid tumor malignancies | Niraparib + carboplatin | I | NCT03209401 | |
| Prostate carcinoma | Niraparib + chemotherapy | II | NCT04592237 | |
| Breast cancer | Olaparib + paclitaxel/carboplatin | II/III | NCT03150576 | |
| Adrenal gland pheochromocytoma, paraganglioma | Olaparib + temozolomide | II | NCT04394858 | |
| Advanced (stage IIIB‐C‐IV) ovarian, primary peritoneal and fallopian tube cancer | Rucaparib + paclitaxel/carboplatin | I /II | NCT03462212 | |
| BRCA‐mutated ovarian carcinoma | Olaparib + chemotherapy | I | NCT03943173 | |
| Gastric cancer | Olaparib + paclitaxel | II | NCT01063517 | |
| Ovarian cancer | Olaparib + carboplatin/paclitaxel | II | NCT01081951 | |
| Ovarian, fallopian tube, or primary peritoneal cancer | Rucaparib + chemotherapy | III | NCT02855944 | |
| Recurrent solid tumors and ewing sarcoma | Talazoparib + onivyde | I /II | NCT04901702 | |
| Uterine leiomyosarcoma | Olaparib + temozolomide | II | NCT03880019 | |
| Ovarian cancer | Talazoparib + chemotherapy | III | NCT03642132 | |
| Acute leukemia | Veliparib + temozolomide | I | NCT01139970 | |
| Recurrent ovarian carcinoma | Niraparib + chemotherapy + atezolizumab | III | NCT03598270 | |
| Metastatic malignant solid neoplasm | Veliparib + topotecan hydrochloride | I | NCT01012817 | |
| IDH1 mutation | BGB‐290 + temozolomide | I/II | NCT03914742 | |
| Recurrent glioma | Talazoparib + carboplatin | II | NCT04740190 | |
| Refractory lymphomas undergoing stem cell transplant | Olaparib + chemotherapy | I | NCT03259503 | |
| ATM | ||||
| Refractory cancer | AZD6738 + paclitaxel | I | NCT02630199 | |
| Advanced cancer | ART0380 + gemcitabine | I/II | NCT04657068 | |
| ATR | ||||
| Esophageal cancer | M6620 + cisplatin | I | NCT03641547 | |
| Advanced stage solid tumors | BAY 1895344 + chemotherapy | I | NCT04514497 | |
| Ovarian serous tumor | M6620 + gemcitabine | I | NCT02595892 | |
| NSCLC, SCLC | VX‐970 (M6620) + topotecan | I/II | NCT02487095 | |
| Cancer | AZD6738 + gemcitabine | I | NCT03669601 | |
| Metastatic malignant solid neoplasm | M6620 + irinotecan hydrochloride | I | NCT02595931 | |
| Refractory cancer | AZD6738 + paclitaxel | I | NCT02630199 | |
| Advanced solid tumors | BAY 1895344 + cisplatin | I | NCT04491942 | |
| Small cell cancers outside of the lungs | M6620 + topotecan | II | NCT03896503 | |
| CHK1 | ||||
| Brain tumor | LY2606368 + cyclophosphamide/gemcitabine | I | NCT04023669 | |
| WEE1 | ||||
| Metastatic pancreatic adenocarcinoma | MK‐1775 + paclitaxel/gemcitabine hydrochloride | I/II | NCT02194829 | |
| Ovarian, primary peritoneal, or fallopian tube cancer | MK‐1775 + paclitaxel/gemcitabine hydrochloride | II | NCT02101775 | |
| Radiotherapy | ||||
| PARP | ||||
| Triple negative breast cancer | Niraparib + radiation therapy/dostarlimab | II | NCT04837209 | |
| Triple negative breast cancer | Niraparib + radiation therapy | I | NCT03945721 | |
| Breast inflammatory carcinoma | Olaparib + radiation therapy | II | NCT03598257 | |
| Malignant glioma without H3 K27M or BRAFV600 mutations | Veliparib + radiation therapy + temozolomide | II | NCT03581292 | |
| Head and neck neoplasms | Olaparib + radiotherapy | I | NCT02229656 | |
| Malignant gliomas | Temozolomide (TMZ) + radiotherapy | I/II | NCT03212742 | |
| ATM | ||||
| Brain cancer | AZD1390 + radiation therapy | I | NCT03423628 | |
| Advanced cancer | XRD‐0394 + palliative radiotherapy | I | NCT05002140 | |
| WEE1 | ||||
| Esophageal adenocarcinoma | Adavosertib + radiation therapy | I | NCT04460937 | |
| Cervical carcinoma | Adavosertib + cisplatin/radiation therapy | I | NCT03345784 | |
| DNA‐PK | ||||
| Rectal cancer | Peposertib + capecitabine/radiotherapy | I/II | NCT03770689 | |
| Solid tumors | M3814 + radiotherapy | I | NCT03724890 | |
| Advanced solid tumors | M3814 + fractionated RT/cisplatin | I | NCT02516813 | |
| Glioblastoma, gliosarcoma | Nedisertib + radiation therapy/ temozolomide | I | NCT04555577 | |
| Advanced solid tumor | XRD‐0394 + palliative radiotherapy | I | NCT05002140 | |
| Other target therapy | ||||
| PARP | ||||
| BRCA1/2 gene mutated tumors | Niraparib + copanlisib (PI3Ki) | I | NCT03586661 | |
| HER2 positive breast carcinoma | Niraparib + trastuzumab | I/II | NCT03368729 | |
| Ovarian cancer | Olaparib + cediranib (VEGFR inhibitor) | N/A | NCT02681237 | |
| Ovarian cancer patients | Niraparib + bevacizumab | II | NCT04734665 | |
| Advanced solid tumors | Olaparib + CYH33 (PI3Kα inhibitor) | II | NCT04586335 | |
| Breast cancer | Talazoparib + sacituzumab goviteca | I/II | NCT04039230 | |
| Advanced breast carcinoma | Olaparib + cediranib(VEGFRi) | II | NCT04090567 | |
| Metastatic breast cancer | Talazoparib + belinostat (HDACi) | I | NCT04703920 | |
| Metastatic malignant solid neoplasm | Olaparib + onalespib (Hsp90 inhibitor) | I | NCT02898207 | |
| Ovarian cancer | Niraparib + bevacizumab | I/II | NCT02354131 | |
| High‐grade serous ovarian cancer | Olaparib + paclitaxel | II | NCT04261465 | |
| Ovarian cancer | Olaparib + anlotinib (VEGFRi) | II | NCT04566952 | |
| Breast cancer metastatic | Olaparib + vorinostat (HDACi) | I | NCT03742245 | |
| Endometrial and ovarian cancer | Olaparib + AZD5363 (AKTi) | I/II | NCT02208375 | |
| Metastatic prostate carcinoma, malignant neoplasm in the bone | Olaparib + cediranib (AZD‐2171) (VEGFRi) | II | NCT02893917 | |
| EGFR‐mutated advanced lung cancer | Niraparib + osimertinib (EGFRi) | I | NCT03891615 | |
| Ovarian cancer | Olaparib + cediranib | III | NCT03278717 | |
| Advanced malignant solid neoplasm | Talazoparib tosylate + axitinib/ crizotinib (VEGFRi) | I | NCT04693468 | |
| Endometrial serous adenocarcinoma | Olaparib + DS‐8201a (HER2i) | I | NCT04585958 | |
| Ovarian cancer with no germline BRCA mutation | Olaparib + alpelisib (PIK3i) | III | NCT04729387 | |
| Pancreatic cancer | Olaparib + cobimetinib (MEK/ERK inhibition) | I | NCT04005690 | |
| Recurrent ovarian, primary peritoneal, or fallopian tube cancer | Olaparib + cediranib maleate | II | NCT02345265 | |
| Recurrent ovarian, fallopian tube, or peritoneal cancer | Olaparib + cediranib maleate | I/II | NCT01116648 | |
| Gastric or gastroesophageal junction cancer | Olaparib + ramucirumab (VEGFRi) | I/II | NCT03008278 | |
| Metastatic NSCLC | Olaparib + cediranib | I | NCT02498613 | |
| Ovarian, fallopian tube, or primary peritoneal cancer | Olaparib + cediranib maleate | II/II | NCT02502266 | |
| ATR | ||||
| Chronic lymphocytic leukemia | AZD6738 + acalabrutinib (BTK inhibitor) | I/I | NCT03328273 | |
| Other treatments | ||||
| PARP | ||||
| Neuroendocrine tumors | Talazoparib + 177Lu‐DOTA‐octreotate PRRT | I | NCT05053854 | |
| Prostate cancer with ATM/BRCA1/2 gene mutation | Niraparib + radical prostatectomy | II | NCT04030559 | |
| Prostate cancer | Olaparib + radium Ra223 dichloride | I | NCT03317392 | |
| Neuroendocrine tumors, thymoma, mesothelioma | Olaparib + 177Lu‐DOTA‐TATE | I | NCT04375267 | |
| Prostate carcinoma | Talazoparib + androgen deprivation therapy | II | NCT04734730 | |
| Metastatic castration‐resistant prostate cancer | Rucaparib + Enzalutamide/zbiraterone | I | NCT04179396 | |
| Prostate cancer | Talazoparib + enzalutamide | III | NCT04821622 | |
| ATR | ||||
| SCLC, neuroendocrine cancers | Berzosertib + lurbinectedin | I/II | NCT04802174 | |
Data from https://clinicaltrials.gov.
TABLE 3.
Ongoing combination trials of concomitant targeting against nonredundant DDR components
| Combination | Conditions | Interventions | Phase | Clinical trial* |
|---|---|---|---|---|
| PARPi + ATRi | ||||
| Advanced solid tumor | Talazoparib + RP‐3500 | I | NCT04497116 | |
| Advanced solid tumors (excluding prostate cancer) | Niraparib + BAY1895344 | I | NCT04267939 | |
| High‐grade serous carcinoma | Olaparib pill + AZD6738 | II | NCT03462342 | |
| Advanced solid tumor | Niraparib/Olaparib + RP‐3500 | I/I | NCT04972110 | |
| Gynaecological cancers | Olaparib + AZD6738 | II | NCT04065269 | |
| Cancer | AZD2281 + AZD5363 + AZD1775 + AZD6738 | II | NCT02576444 | |
| Advanced solid tumors | Niraparib + M1774 | I | NCT04170153 | |
| Malignant solid neoplasm | Olaparib + Ceralasertib | II | NCT03878095 | |
| Recurrent ovarian, primary peritoneal, or fallopian tube cancer | Olaparib + Adavosertib | II | NCT03579316 | |
| Prostate cancer | Olaparib + AZD6738 | II | NCT03787680 | |
| Clear cell renal cell carcinoma | AZD6738 + Olaparib | II | NCT03682289 | |
| Advanced solid tumor | RP‐3500 + Niraparib/Olaparib | I/II | NCT04972110 | |
| PARPi + BETi | ||||
| Advanced malignant solid neoplasm | Olaparib + Adavosertib | I | NCT04197713 | |
| Ovarian cancer | Olaparib + Adavosertib | I | NCT04633239 | |
| Triple negative breast cancer | Talazoparib + ZEN003694 | II | NCT03901469 | |
| PARPi + CDK4/6i | ||||
| Breast cancer | Niraparib + Abemaciclib | I | NCT04481113 | |
| PARPi + ATMi | ||||
| Advanced solid tumours | Olaparib + AZD0156 | I | NCT02588105 | |
| Other | ||||
| Ovarian cancer | Olaparib + AsiDNATM | I/II | NCT04826198 | |
Data from https://clinicaltrials.gov.
TABLE 4.
Ongoing combination trials of DDR inhibitors with immunotherapy
| DDR | Conditions | Interventions | Phase | Clinical trial* |
|---|---|---|---|---|
| PARP | ||||
| Endometrial neoplasms | Olaparib + durvaluma | II | NCT03951415 | |
| Solid tumor | Rucaparib + atezolizumab | II | NCT04276376 | |
| Biliary tract cancer | Rucaparib + nivolumab | II | NCT03639935 | |
| Lung small cell carcinoma, neuroendocrine carcinoma | Niraparib + dostarlimab | II | NCT04701307 | |
| Cervical cancer | Olaparib + pembrolizumab | II | NCT04483544 | |
| Breast cancer | Olaparib + pembrolizumab | II | NCT03025035 | |
| Ovarian, breast, gastric cancer, SCLC | Olaparib + durvalumab | I/II | NCT02734004 | |
| Ovarian neoplasms | Niraparib + TSR‐042 | II | NCT03574779 | |
| Ovarian, fallopian tube, peritoneal cancer | Olaparib + tremelimumab | I/II | NCT02571725 | |
| Metastatic pancreatic adenocarcinoma | Olaparib + pembrolizumab | II | NCT04548752 | |
| Advanced malignant solid neoplasm | Niraparib + atezolizumab | I | NCT03830918 | |
| Advanced malignant solid neoplasm | Olaparib + durvalumab/copanlisib | I | NCT03842228 | |
| Metastatic breast carcinoma | Olaparib + atezolizumab | II | NCT02849496 | |
| LSCL | Olaparib + durvalumab | I | NCT04728230 | |
| Platinum‐sensitive ovarian cancer | OSE2101 + pembrolizumab | II | NCT04713514 | |
| Advanced malignant solid neoplasm | Talazoparib + paclitaxel | I | NCT02317874 | |
| Colorectal, breast neoplasms | Olaparib + durvalumab | I/II | NCT02484404 | |
| Prostate carcinoma | Olaparib + durvalumab | II | NCT04336943 | |
| Breast cancer | Niraparib + TSR‐042 (dostarlimab) | I | NCT04673448 | |
| Triple negative breast cancer | Olaparib + durvalumab | II | NCT03167619 | |
| Extensive SLSC | Talazoparib + atezolizumab | II | NCT04334941 | |
| Fallopian tube mucinous adenocarcinoma | Olaparib + cediranib + durvalumab | II | NCT04739800 | |
| Metastatic triple negative breast cancer | Olaparib + durvalumab | II | NCT03801369 | |
| Breast, ovarian cancer | Niraparib + pembrolizumab | I/II | NCT02657889 | |
| BRCAm ovarian, fallopian tube or primary peritoneal cancer | Olaparib + durvalumab/tremelimumab | II | NCT02953457 | |
| Ovarian, fallopian tube, or primary peritoneal cancer | Rucaparib + nivolumab | III | NCT03522246 | |
| Ovarian carcinosarcoma | Niraparib + TSR‐042 (dostarlimab) | II/III | NCT03651206 | |
| Pancreatic adenocarcinoma | Niraparib + nivolumab/ipilimumab | I/II | NCT03404960 | |
| Endometrial cancer | Olaparib + durvalumab | II | NCT03660826 | |
| Metastatic solid tumors | Talazoparib + avelumab | II | NCT03330405 | |
| BRCA1/2 and PALB2 mutated metastatic pancreatic cancer | Niraparib + dostarlimab | II | NCT04493060 | |
| Advanced solid neoplasm | Veliparib + nivolumab | I | NCT03061188 | |
| Metastatic melanoma with HR mutation | Olaparib + pembrolizumab | II | NCT04633902 | |
| ATM | ||||
| Advanced solid tumors | Drug: BAY1895344 + pembrolizumab | I | NCT04095273 | |
| ATR | ||||
| Advanced solid tumors | BAY1895344 + pembrolizumab | I | NCT04095273 | |
Data from https://clinicaltrials.gov.
5.1. Combinations with DNA‐damaging agents
5.1.1. DDR inhibitor–chemotherapy combinations
As discussed, synergistic treatment of DDR inhibitors with cytotoxic chemotherapy has been performed, with schedules based on sequential chemotherapy administration followed by DDR inhibitor being proved clinically more beneficial and more tolerable. 145 , 345 , 346 , 347 The underlying mechanism for the synergy is that the rapidly dividing cancer cells are more likely to be affected by DNA damage directly caused by chemotherapy or indirectly from reactive oxygen species. 348 For example, platinum derivatives (carboplatin, cisplatin, and oxaliplatin) produce intrastrand DNA cross‐links repaired by NER or the Fanconi anemia pathway. 349 Antimetabolites result in stalling of the replication fork, whereas alkylating agents such as temozolomide lead to both single‐ and double‐ DNA strand breaks. Topoisomerase (Top) inhibitors include Top 1 inhibitors that generate SSBs, and Top 2 inhibitors that result in DSBs. 350 , 351 Meanwhile, epigenetics regulation also plays an important role in DDR, with the hypomethylation of DDR genes significantly associated with worse prognosis in glioblastoma patients. 352 The epigenetics silencing of PRPF19 and TERT genes in glioblastoma cells overcomes their resistance to temozolomide treatment. 352
Combining cytotoxic chemotherapies and PARP inhibitors has long been proposed based on the capability of PARP inhibitors to eliminate DNA lesions caused by chemotherapy. An early study suggested that a PARP inhibitor 3‐AB reversed tumor resistance to temozolomide (TMZ) in glioma models. 353 , 354 The combination of TMZ and PARP inhibitor NU1025 was later found to suppress tumor growth and improve overall survival of central nervous system lymphoma. 355 These successful preclinical results urged the clinical evaluation of the TMZ/PARPi combination in patients with advanced gliomas, where the combination regimen demonstrated modest antitumor efficacy and overall tolerability. 356 A randomized Phase II/III study (NCT02152982) investigated the combination of PARPi veliparib and TMZ, which improved disease outcome in tumors with MGMT promoter hypermethylation. 357 Interestingly, the combination was previously found to be ineffective in MGMT‐unmethylated cell lines, suggesting the predicting value of MGMT promoter methylation status in tumor response to TMZ/veliparib combination therapy. 357 The combination was further tested in other cancer types (NCT01009788, NCT01638546), but failed to induce significant survival benefits in patients with small cell lung cancer. 358 Early PARP inhibitors 3‐AB and PJ34 were shown to overcome tumor resistance to cisplatin in several cancer types, 359 , 360 , 361 and olaparib was later suggested to enhance the therapeutic effect of cisplatin in lung cancer cells. 362 , 363 These preclinical success allowed the initiation of clinical studies on olaparib in patients with platinum‐sensitive ovarian cancer (NCT01081951), where olaparib increased PFS in patients receiving platinum/paclitaxel monotherapy, but failed to improve overall survival. 65 , 347 The combination of PARP inhibitor veliparib with carboplatin and paclitaxel was tested in patients with triple‐negative breast cancer patients (NCT02032277) but did not bring survival benefits. 364
The ATR inhibitor M6620 demonstrated strong efficacy in combination with cisplatin, which later entered clinical trial and resulted in objective responses in clinical trial either as single agent or cotherapy with carboplatin. 365 , 366 Other DDR inhibitors used along with definitive chemotherapy are underway, including DNA‐PK inhibitor M9831, the Phase I evaluation of which was completed in 2019 to determine the maximum tolerated dose of M9831 and its efficacy with or without doxorubucin in advanced cancer patients (NCT02644278).
5.1.2. DDR inhibitor–radiotherapy combinations
The systematic delivery of chemotherapy poses a challenge to its the combinatorial therapy with DDR inhibitors. The overlapping toxicities, predominantly myelosuppression, have led to the termination of many clinical trials. 367 , 368 To date, DNA‐damaging agents still remain the mainstay of nonoperative cancer treatment, and besides chemotherapy, radiation therapy is an optional treatment. The ionization effect of radiation producing oxygen free radicals causes DNA damage in cells with 1 Gy of ionizing radiation being able to generate 1000 SSBs and 35 DSBs. 369 While radiation has been proved effective by accumulating evidence in combating tumors, an important question is how to reduce the amount of radiation delivered to normal tissues and thus prevent the acute and chronic toxicities. A strategy to intensify the efficacy and at the same time reduce toxicity of radiotherapy is the combination with novel targeted therapies, which increases the radiosensitivity of cancer cells to a greater extent than normal cells. 370 Given that radiation causes different DNA lesions including base damaging, SSBs, and DSBs, the simultaneous inhibition of key DDR enzymes thus becomes a promising strategy. 371 Furthermore, the clear correlation between radioresistance and increased DNA repair capacities further justify the combinational use of radiotherapy and DDR inhibition. 372 However, early efforts on DDR blockade such as PARP inhibitors failed to achieve consistent results. 373 , 374 , 375 The suboptimal synergistic effect might be attributed to the fact that DSBs caused by conventional radiation are repaired predominately through the NHEJ pathway, rather than PARP‐regulated BER pathway. Moreover, compared with conventional photon‐based radiation, HR repair pathway is more engaged in the repair of heavy ion (carbon and iron)‐induced DNA damage. 375 , 376 The radiosensitization approaches include inhibitors that prevent S and G2/M cell‐cycle arrest that allows DNA damage repair, such such as PARP, CHK1, WEE1, ATR, and DNA‐PK inhibitors.
VE‐821 is a ATR inhibitor with potent inhibitory activities on the phosphorylation of H2AX and CHK1 by ATR, and sensitizing effect on cancer cells to radiotherapy and genotoxic chemotherapeutics. 66 , 231 , 377 , 378 , 379 Notably, the radiosensitization of VE‐821 was even more profound in hypoxic cells. 377 M6620 (VX‐970) is the improved analogue of VE‐821 and its synergistic potential with radiotherapy has been widely studied in preclinical settings. 232 In esophageal cancer, M6620 was shown to enhance radiation‐induced tumor growth arrest both in vitro and in vivo. 380 , 381 The concurrent treatment of M6620 and radiation was recently reported to improve the overall survival in mouse models, supporting the ongoing clinical trial (NCT02589522) assessing the sensitizing effects of M6620 to whole brain irradiation in NSCLC patients with brain metastases. 382 AZD6738 was intensively investigated in various cancers, especially ATM‐deficient cancers as a monotherapy; recent attempt has converged on its combination therapies. 286 , 383 , 384 , 385 The multiparametric cell‐based assays measuring DNA damage and cell‐cycle transition are induced by the treatment of AZD6738, and the in vivo mouse xenograft studies provide strong rationale for the design of Phase I clinical trials. 386 The accumulating promising results from preclinical studies encouraged the assessment of AZD6738 in more than 25 clinical trials including monotherapies in hematological malignancies (NCT01955668, NCT03770429) and in refractory solid tumors (NCT02223923, NCT03022409), and in combination with radiotherapy (NCT02223923).
WEE1 is involved in the initiation of G2 checkpoint, and the inhibition of Wee1 would subsequently cause unscheduled mitotic entry and increased replication stress. 281 It has been reported that increased sensitivity to WEE1 inhibition through mechanisms outside of cell‐cycle checkpoint defects, such as DDR aberrations and nucleotide resource starvation, with single‐agent activity observed even in TP53‐wild‐type cancer cells. 387 , 388 , 389 , 390 The critical role of p53 in the regulation of G1 checkpoints provides a strong rationale for the use of WEE1 inhibitors in p53‐deficient cells. 391 A WEE1 inhibitor, adavosertib (AZD1775 or MK‐1775), was shown to sensitize p53‐deficient cells to DNA‐damaging radiotherapy via the induction of mitotic lethality. 281 , 392 Thus, recent clinical development has focused to the concurrent treatment of WEE1 inhibitors and DNA‐damaging treatments such as radiation therapy in TP53 mutant tumors. Following the evaluation of Phase I study as single agent, 393 AZD1775 has demonstrated overall survival benefits when combining radiation in patients with advanced pancreatic cancer. 290
As NHEJ is the predominant pathway for the repair of traditional radiotherapy, 394 the specific targeting of NHEJ by DNA‐PK inhibitors is thus considered as a potential combination partner for radiation. Currently, three DNA‐PK inhibitors are under clinical trials: M9831 (VX‐984), nedisertib (M3814), and CC‐115. In addition to monotherapy, CC‐115 is now being investigating in combination with androgen‐deprivation therapy (ADT) in castrate‐resistant prostate cancer patients (NCT02833883) and with radiation in glioblastoma patients (NCT02977780). Inspired by results from a Phase I trial involving patients with tumors in the head and neck or thorax, 395 a growing number of trials are underway to assess the efficacy of nedisertib monotherapy or with radiation.
5.2. DDR inhibitor–DDR inhibitor combinations
The initial purpose of cotargeting key DDR elements was to overcome the acquired resistance to a single DDR inhibitor, predominantly PARP inhibitors. In the light of the variety of DNA repair mechanisms, the combination of one or more of DDR inhibitors to induce synthetic lethality is biologically applicable, even in HR‐proficient cells. 396 An exciting example was the coinhibition of PARP and WEE1 inhibitor. The combination of adavosertib and olaparib synergistically promoted radiosensitivity of pancreatic cancer cells by impairing their HR repair capacity to achieve synthetic lethality, which led to the initiation of multiple clinical trials (NCT02723864, NCT02576444, and NCT02511795). 397 In PARPi‐resistant cells with SLFN11 deficiency, the additional ATR inhibition would overcome the resistance due to the fact that SLFN11‐inactive cells were more reliant on the ATR pathway for DNA repair. 398 Likewise, ATR blockade further disrupted HR repair pathway in BRCA‐deficient cancer cells. 399 In lymphoma models, ATR inhibitor AZD6738 displayed a strong synergistic cytotoxic effect when combined with Chk1 inhibtor or WEE1 inhibitor, further expanding the repertoire of DDR–DDR therapeutic combinations. 400
In addition to ATR, HR‐deficient tumor cells are also increasingly reliant on other alternative repair pathways such as a type of a‐EJ, named microhomology‐mediated end joining (MMEJ) for survival, suggesting the potential of cotargeting PARP and key members of MMEJ. 21 Other combination partners for PARP inhibitors include the antagonists of PI3K‐AKT pathway 401 and BRD4 protein, which has been shown to downregulate several DDR genes and increase the sensitivity of HR‐proficient tumors to PARP inhibition. 402 , 403 , 404 Previous work shows that recently, the combined inhibition of PARP1 and DNA‐PK was found to suppress HNSCC tumor growth in vitro and in vivo compared to either agent used alone. 405 The underlying mechanism may be the cooperation between PARP1 and DNA‐PKcs to recruit XRCC1 to mediate DNA repair. 406 , 407 , 408
5.3. DDR inhibitor–immunotherapy combinations
The alteration in immune environment caused by DDR deficiency may be used to facilitate the sensitization of immunotherapies. 409 Deficient DDR results in accumulated DNA damage in cells and increases their mutational burden, particularly in tumor cells that normally experience high level of endogenous or exogenous DNA damage. It is becoming increasingly clear that DNA damage could induce the production of immune‐regulatory cytokines such as type I IFNs. 410 , 411 , 412 DNA normally resides in the nucleus or mitochondria, and once it is released to the cytoplasm, it triggers a series of immune response. DNA binds to cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase (cGAS), which leads to the conformational change of the catalytic subunit of cGAS allowing the formation of the second messenger cyclic GMP–AMP (cGAMP). 413 cGAMP then activates STING and its downstream transcription factors IRF3 and NF‐κB via kinases TBK1 and IKK, respectively. As shown in Figure 3, IRF3 and NF‐κB then translocate into the nucleus and induce the expression of multiple cytokines such as IFNs. DDR dysfunction or the combination therapy with DDR inhibitors further enhances DNA damage, which when transfers into cytosolic DNA and triggers the stimulator of interferon genes (STING) pathway to activate innate immune responses. 414 , 415
FIGURE 3.
The interaction between DNA damage with immune responses. The activated STING pathway leads to upregulation of type I IFNs, which enhances the cross‐presentation of dendritic cells (DCs) and T‐cell activation. Unrepaired DNA damage may generate tumor neoantigens and thereby improving tumor recognition by T cells. However, DNA damage or DDR deficiencies have also been shown to upregulate PD‐L1 expression. cGAS, cyclic GMP–AMP synthase; DDR, DNA damage response; DC, dendritic cell; DSB, double‐strand break; ER, endoplasmic reticulum; IRF3, interferon regulatory factor 3, IFN, interferon; NF‐κB, nuclear factor kappa‐B; STING, stimulator of interferon genes; TBK1, TANK‐binding kinase 1. Figure was created with bioRender
Tumors harboring mutations in BRCA1/2 or ATM were identified with high level of cytosolic DNA, which stimulated the innate immune activities and correlated with a durable response to ICIs. 416 , 417 In addition, the induced neoantigens of tumor cells could stimulate the host immune response including the intratumoral infiltration of CD8+ T cells, which have long been characterized as a predictive marker for cell response to ICIs. 418 , 419 , 420 Recent evidence suggested that deleterious DDR‐related gene mutations are a frequent event in NSCLC, which indicates improved clinical outcomes in NSCLC patients with PD‐(L)1 antibody treatment. 421 Thus, it is conceivable that DDR inhibitors may be able to convert immunologically “cold” into “hot” tumors and sensitize tumor cells to ICIs. 422 , 423 A growing number of clinical trials evaluating this drug combination in cancer patients are underway. 93 Figure 3 presents a simplified scheme of the interaction between DNA damage with immune responses.
PARP inhibitors are one of the most extensively studied DDR inhibitors in clinical development and in the context of synthetic lethality such as cells with BRCA1/2 mutations, PARP inhibition is considered proinflammatory. 424 Cells treated with PARP inhibitors exhibited an increased level of PD‐L1 expression, supporting the concomitant use of PARP inhibitors and ICIs. 423 Interestingly, cancer stem cells (CSCs) displayed higher expression of PD‐L1 compared to nonstem cell cancer cells, which might contribute to the long‐term survival improvement by immunotherapy 425 and make ICIs a potential strategy to overcome resistance of CSCs to PARP inhibitors. 426 However, PARP inhibition has recently been shown to attenuate immune response in mice by suppressing thymocyte maturation. 427 It is thus intriguing to speculate whether toxicity of ICIs could be reduced when used in combination with PARP inhibitors.
CDK4/6 inhibitors could convert HR into NHEJ mechanism in cells treated with ionizing radiation in several tumor models, 428 , 429 , 430 which was likely attributed to the active involvement of cyclin D‐CDK4/6‐RB pathway in DDR. 431 Besides their radiosensitization effects, CDK4/6 inhibitors were also reported to reduce the T‐cell exclusion and immune evasion in ICI‐resistant melanoma cells. 432 It is therefore not surprising that the combination of CDK4/6 inhibitors and anti‐PD‐L1 therapy led to substantial tumor regression in xenograft mouse models. 433 , 434 Clinical trials sought to determine the efficacy of FDA‐approved CDK4/6 inhibitors such as palbociclib and abemaciclib combined with pembrolizumab in patients with HR‐positive breast cancer (NCT02779751, NCT02778685), where the drug combination induced a higher objective response rate than either monotherapy and later entered clinical trials on other cancer types. 435
Other combination partner for ICI includes the CHK1 inhibitor prexasertib (LY2606368), which potently activated the STING/TBK1/IRF3 innate immune pathway and upregulated tumor expression of PD‐L1, suggesting its synergistical potential with ICIs. 436 , 437 Several action mechanisms of the combination therapy have been proposed. For example, ATR inhibitor (BAY1895433) targeting the ATR‐CHK1 signaling could activate CDK1‐SPOP axis, wchich results in the destabilization of PD‐L1, proving a strong rationale for the concomitant use of ATRi with anti‐PD‐L1 therapy. 438 Adavosertib is currently the only WEE1 inhibitor under clinical trials and its combination with anti‐PD‐L1 monoclonal antibody durvalumab is under assessment in a Phase I trial (NCT02617277). 439
6. REMAINING CHALLENGES AND FUTURE PERSPECTIVES
Cell response to DNA damage is a complex process involving various signal networks and proteins, which are differentially activated or inactivated in specific cancer types. For instance, breast, ovarian, and bladder cancers are likely accompanied with alterations in HR genes, whereas some gastric and colorectal tumor subgroups present a hypermutator phenotype lacking aneuploidy. Furthermore, the DNA repair capacity also varies among different cell types. For example, the repair efficiency of human embryonic stem cells is the higher than differentiated cell types, 440 and some tumor cells present upregulated damage repair such as the high level of MGMT repair activity in gliomas. 441 , 442 Thus, characterization of every single type of tumor to identify its specific profile of deregulated DDR components will facilitate personalized treatment of cancer patients. Next‐generation sequencing provides an opportunity for precision medicine by analyzing the whole‐genome alterations associated with DNA repair across different cancer types. It is recently found through next‐generation sequencing that epigenetic regulators also appear to play a particularly important role in cancer events. 443 For example, epigenetic silencing of genes leads to loss‐of‐function events of DDR proteins.
The initial idea for the DDR inhibitor‐based combination therapy was to enhance the efficacy of conventional treatments. Although DDR inhibitors have been widely conducted on unselected patients, recent research interest tends to use these drug combinations in tumors with specific genetic backgrounds such as p53 mutation and BRCA alterations, which make cells more susceptible to DDR inhibitors. Emerging clinical trials are ongoing to explore the potential predictive markers for patients’ response to combinational therapy, including alterations in genes such as ATM, BRCA1, BRCA2, CDK12, CHEK1, MYC, PARP1, PIK3CA, and PTEN (NCT03842228, NCT02546661).
The early knowledge that DNA repair deficiency leading to increased neoantigen and tumor mutational load makes ICI a potential combination partner for DDR inhibitors. However, high mutational burden is a not a guarantee for efficient ICI response, given the varying level of immunogenicity induced by different DNA repair–deficient backgrounds. The immune score and mutational signature have been proved feasible in evaluating the response of ovarian cancer patients to niraparib and pembrolizumab. 444 Reliable predictive biomarkers are needed to identify the specific subset of patients responsive to ICI and DDR inhibitor combinations. 445 One such strategy is to integrate indexes from multiple platforms, such as combining tumor mutational burden with immune activity marker. The immune activity can be reflected by intratumor immune infiltrations and STING pathway. 446
Targeting methylation pathways is a promising anticancer strategy. 447 Accumulating evidence has suggested the epigenetics regulation on DDR. Multiple histone methyltransferases and demethylases have been described to facilitate chromatin remodeling and chromatin‐based DDR activities. However, mechanisms of how histone methylation is involved in DDR remains to be elucidated. Given the correlation between PARP and histone methylation, identifying the involvement of methylation signaling in DDR would bring new therapeutic approaches for cancer treatment.
Finally, the increased replication stress and DNA repair defects in tumors provide a therapeutic opportunity that makes cancer cells more vulnerable to DDR inhibition than normal cells. However, the rapid development of clinical DDR inhibitors has raised a concern on toxicity, which is frequently accompanied with other anticancer therapies. It is rather imperative to identify optimal doses, combinations, and schedules of DDR inhibitors to minimize their adverse effects and more ideally, enhance the efficacy. It has to be addressed that DDR proteins initially possess essential physiological functions that recognize and fix DNA damage in normal cells, the repression of which may be deleterious due to the increased mutagenic load in normal tissues. Surveillance on long‐term toxicity of DDR inhibition may thus be added into clinical trial design.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
Wang Manni offered the main direction and significant guidance of this manuscript. Wang Manni, Siyuan Chen and Danyi Ao drafted the manuscript and illustrated the figures for the manuscript.
ETHICS APPROVAL
Not Applicable
ACKNOWLEDGMENTS
This work is supported by The National Postdoctoral Science Foundation of China (No. 2021M702347).
Wang M, Chen S, Ao D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm. 2021;2:654–691. 10.1002/mco2.103
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the review.
REFERENCES
- 1. Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sc. 2008;121(Supplement_1):1‐84. [DOI] [PubMed] [Google Scholar]
- 2. Roos WP, Kaina B. DNA damage‐induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237‐248. [DOI] [PubMed] [Google Scholar]
- 3. Voulgaridou GP, Anestopoulos I, Franco R, Panayiotidis MI, Pappa A. DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat Res. 2011;711(1‐2):13‐27. [DOI] [PubMed] [Google Scholar]
- 4. Cadet J, Ravanat JL, TavernaPorro M, Menoni H, Angelov D. Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett. 2012;327(1‐2):5‐15. [DOI] [PubMed] [Google Scholar]
- 5. Kauffmann A, Rosselli F, Lazar V, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27(5):565‐573. [DOI] [PubMed] [Google Scholar]
- 6. David SS, O'Shea VL, Kundu S. Base‐excision repair of oxidative DNA damage. Nature. 2007;447(7147):941‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doetsch PW, Cunningham RP. The enzymology of apurinic/apyrimidinic endonucleases. Mutat Res. 1990;236(2‐3):173‐201. [DOI] [PubMed] [Google Scholar]
- 8. Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10(11):756‐768. [DOI] [PubMed] [Google Scholar]
- 9. McNeil EM, Melton DW. DNA repair endonuclease ERCC1‐XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012;40(20):9990‐10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lieber MR. NHEJ and its backup pathways in chromosomal translocations. Nat Struct Mol Biol. 2010;17(4):393‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forget AL, Kowalczykowski SC. Single‐molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends Cell Biol. 2010;20(5):269‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shibata A, Jeggo P, Lobrich M. The pendulum of the Ku‐Ku clock. DNA Repair (Amst). 2018;71:164‐171. [DOI] [PubMed] [Google Scholar]
- 14. Paques F, Haber JE. Multiple pathways of recombination induced by double‐strand breaks in Saccharomyces cerevisiae . Microbiol Mol Biol Rev. 1999;63(2):349‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orr‐Weaver TL, Szostak JW. Yeast recombination: the association between double‐strand gap repair and crossing‐over. Proc Natl Acad Sci U S A. 1983;80(14):4417‐4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhargava R, Onyango DO, Stark JM. Regulation of single‐strand annealing and its role in genome maintenance. Trends Genet. 2016;32(9):566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boboila C, Jankovic M, Yan CT, et al. Alternative end‐joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc Natl Acad Sci U S A. 2010;107(7):3034‐3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simsek D, Jasin M. Alternative end‐joining is suppressed by the canonical NHEJ component Xrcc4‐ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17(4):410‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mateos‐Gomez PA, Gong F, Nair N, Miller KM, Lazzerini‐Denchi E, Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518(7538):254‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ceccaldi R, Liu JC, Amunugama R, et al. Homologous‐recombination‐deficient tumours are dependent on Poltheta‐mediated repair. Nature. 2015;518(7538):258‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tobin LA, Robert C, Rapoport AP, et al. Targeting abnormal DNA double‐strand break repair in tyrosine kinase inhibitor‐resistant chronic myeloid leukemias. Oncogene. 2013;32(14):1784‐1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9(1):89‐96. [DOI] [PubMed] [Google Scholar]
- 24. Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681‐710. [DOI] [PubMed] [Google Scholar]
- 25. Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101‐133. [DOI] [PubMed] [Google Scholar]
- 26. Tiraby JG, Fox MS. Marker discrimination in transformation and mutation of pneumococcus. Proc Natl Acad Sci U S A. 1973;70(12):3541‐3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Espanol T, Caragol I, Bertran JM. Postnatal transmission of HIV infection. N Engl J Med. 1992;326(9):642. author reply 643–4. [DOI] [PubMed] [Google Scholar]
- 28. Branzei D, Szakal B. DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair (Amst). 2016;44:68‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73(1):134‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bi X. Mechanism of DNA damage tolerance. World J Biol Chem. 2015;6(3):48‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci U S A. 2006;103(24):8971‐8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lang GI, Murray AW. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol. 2011;3:799‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Daigaku Y, Davies AA, Ulrich HD. Ubiquitin‐dependent DNA damage bypass is separable from genome replication. Nature. 2010;465(7300):951‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sawyer SL, Tian L, Kahkonen M, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics. 2015;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337‐349. [DOI] [PubMed] [Google Scholar]
- 37. Bluteau D, Masliah‐Planchon J, Clairmont C, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;126(9):3580‐3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knies K, Inano S, Ramirez MJ, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. 2017;127(8):3013‐3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim H, D'Andrea AD. Regulation of DNA cross‐link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26(13):1393‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stone MP, Cho YJ, Huang H, et al. Interstrand DNA cross‐links induced by alpha,beta‐unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res. 2008;41(7):793‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475(7354):53‐58. [DOI] [PubMed] [Google Scholar]
- 42. O'Donovan A, Davies AA, Moggs JG, West SC, Wood RD. XPG endonuclease makes the 3' incision in human DNA nucleotide excision repair. Nature. 1994;371(6496):432‐435. [DOI] [PubMed] [Google Scholar]
- 43. Liu W, Palovcak A, Li F, Zafar A, Yuan F, Zhang Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross‐link repair. Cold Spring Harb Perspect Biol. 2013;5(10):a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niraj J, Farkkila A, D'Andrea AD. The Fanconi anemia pathway in cancer. Annu Rev Cancer Biol. 2019;3:457‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rodriguez A, D'Andrea A. Fanconi anemia pathway. Curr Biol. 2017;27(18):R986. [DOI] [PubMed] [Google Scholar]
- 47. Swann PF. Why do O6‐alkylguanine and O4‐alkylthymine miscode? The relationship between the structure of DNA containing O6‐alkylguanine and O4‐alkylthymine and the mutagenic properties of these bases. Mutat Res. 1990;233(1‐2):81‐94. [DOI] [PubMed] [Google Scholar]
- 48. Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231(1):11‐30. [DOI] [PubMed] [Google Scholar]
- 49. Fang Q, Kanugula S, Pegg AE. Function of domains of human O6‐alkylguanine‐DNA alkyltransferase. Biochemistry. 2005;44(46):15396‐15405. [DOI] [PubMed] [Google Scholar]
- 50. Kaina B, Christmann M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair (Amst). 2019;78:128‐141. [DOI] [PubMed] [Google Scholar]
- 51. Criniere E, Kaloshi G, Laigle‐Donadey F, et al. MGMT prognostic impact on glioblastoma is dependent on therapeutic modalities. J Neurooncol. 2007;83(2):173‐179. [DOI] [PubMed] [Google Scholar]
- 52. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997‐1003. [DOI] [PubMed] [Google Scholar]
- 53. Reifenberger G, Hentschel B, Felsberg J, et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int J Cancer. 2012;131(6):1342‐1350. [DOI] [PubMed] [Google Scholar]
- 54. Esteller M, Garcia‐Foncillas J, Andion E, et al. Inactivation of the DNA‐repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350‐1354. [DOI] [PubMed] [Google Scholar]
- 55. Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O‐6‐methylguanine‐DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10(6):1871‐1874. [DOI] [PubMed] [Google Scholar]
- 56. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987‐996. [DOI] [PubMed] [Google Scholar]
- 57. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338‐345. [DOI] [PubMed] [Google Scholar]
- 58. O'Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene. 2007;26(56):7816‐7824. [DOI] [PubMed] [Google Scholar]
- 59. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double‐strand break repair. J Clin Oncol. 2008;26(22):3785‐3790. [DOI] [PubMed] [Google Scholar]
- 60. Lucchesi JC. Synthetic lethality and semi‐lethality among functionally related mutants of Drosophila melanogaster . Genetics. 1968;59(1):37‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801‐817. [DOI] [PubMed] [Google Scholar]
- 62. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689‐698. [DOI] [PubMed] [Google Scholar]
- 63. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287‐294. [DOI] [PubMed] [Google Scholar]
- 64. Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33(1):9‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum‐sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87‐97. [DOI] [PubMed] [Google Scholar]
- 66. Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428‐430. [DOI] [PubMed] [Google Scholar]
- 67. Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739‐750. [DOI] [PubMed] [Google Scholar]
- 68. Buisson R, Boisvert JL, Benes CH, Zou L. Distinct but concerted roles of ATR, DNA‐PK, and Chk1 in countering replication stress during S phase. Mol Cell. 2015;59(6):1011‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145(3):435‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425‐448. [DOI] [PubMed] [Google Scholar]
- 72. Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR‐dependent checkpoint. Genes Dev. 2005;19(9):1040‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell. 2010;37(4):492‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene‐induced DNA replication stress. Nature. 2018;555(7694):112‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Costantino L, Sotiriou SK, Rantala JK, et al. Break‐induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343(6166):88‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Minocherhomji S, Ying S, Bjerregaard VA, et al. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528(7581):286‐290. [DOI] [PubMed] [Google Scholar]
- 77. Gisselsson D, Pettersson L, Hoglund M, et al. Chromosomal breakage‐fusion‐bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A. 2000;97(10):5357‐5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Crasta K, Ganem NJ, Dagher R, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482(7383):53‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lukas C, Savic V, Bekker‐Jensen S, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13(3):243‐253. [DOI] [PubMed] [Google Scholar]
- 80. Michelini F, Pitchiaya S, Vitelli V, et al. Damage‐induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double‐strand breaks. Nat Cell Biol. 2017;19(12):1400‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Michelini F, Jalihal AP, Francia S, et al. From “cellular” RNA to “smart” RNA: multiple roles of RNA in genome stability and beyond. Chem Rev. 2018;118(8):4365‐4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. D'Alessandro G, Whelan DR, Howard SM, et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat Commun. 2018;9(1):5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bartkova J, Hamerlik P, Stockhausen MT, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010;29(36):5095‐5102. [DOI] [PubMed] [Google Scholar]
- 84. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907‐913. [DOI] [PubMed] [Google Scholar]
- 85. Longley DB, Harkin DP, Johnston PG. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330‐338. [DOI] [PubMed] [Google Scholar]
- 86. Fujinaka Y, Matsuoka K, Iimori M, et al. ATR‐Chk1 signaling pathway and homologous recombinational repair protect cells from 5‐fluorouracil cytotoxicity. DNA Repair (Amst). 2012;11(3):247‐258. [DOI] [PubMed] [Google Scholar]
- 87. Voland C, Bord A, Peleraux A, et al. Repression of cell cycle‐related proteins by oxaliplatin but not cisplatin in human colon cancer cells. Mol Cancer Ther. 2006;5(9):2149‐2157. [DOI] [PubMed] [Google Scholar]
- 88. Kiyonari S, Iimori M, Matsuoka K, et al. The 1,2‐diaminocyclohexane carrier ligand in oxaliplatin induces p53‐dependent transcriptional repression of factors involved in thymidylate biosynthesis. Mol Cancer Ther. 2015;14(10):2332‐2342. [DOI] [PubMed] [Google Scholar]
- 89. Wilson PM, Fazzone W, LaBonte MJ, Lenz HJ, Ladner RD. Regulation of human dUTPase gene expression and p53‐mediated transcriptional repression in response to oxaliplatin‐induced DNA damage. Nucleic Acids Res. 2009;37(1):78‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bruno PM, Liu Y, Park GY, et al. A subset of platinum‐containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med. 2017;23(4):461‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tanaka N, Sakamoto K, Okabe H, et al. Repeated oral dosing of TAS‐102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep. 2014;32(6):2319‐2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Matsuoka K, Iimori M, Niimi S, et al. Trifluridine induces p53‐dependent sustained G2 phase arrest with its massive misincorporation into DNA and few DNA strand breaks. Mol Cancer Ther. 2015;14(4):1004‐1013. [DOI] [PubMed] [Google Scholar]
- 93. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Satoh MS, Lindahl T. Role of poly(ADP‐ribose) formation in DNA repair. Nature. 1992;356(6367):356‐358. [DOI] [PubMed] [Google Scholar]
- 95. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84(2):137‐146. [DOI] [PubMed] [Google Scholar]
- 96. Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Eustermann S, Wu WF, Langelier MF, et al. Structural basis of detection and signaling of dna single‐strand breaks by human PARP‐1. Mol Cell. 2015;60(5):742‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012;46(6):722‐734. [DOI] [PubMed] [Google Scholar]
- 99. Ziv Y, Bielopolski D, Galanty Y, et al. Chromatin relaxation in response to DNA double‐strand breaks is modulated by a novel ATM‐ and KAP‐1 dependent pathway. Nat Cell Biol. 2006;8(8):870‐876. [DOI] [PubMed] [Google Scholar]
- 100. Murga M, Jaco I, Fan Y, et al. Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol. 2007;178(7):1101‐1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dantzer F, Schreiber V, Niedergang C, et al. Involvement of poly(ADP‐ribose) polymerase in base excision repair. Biochimie. 1999;81(1‐2):69‐75. [DOI] [PubMed] [Google Scholar]
- 102. de Murcia JM, Niedergang C, Trucco C, et al. Requirement of poly(ADP‐ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94(14):7303‐7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Vodenicharov MD, Sallmann FR, Satoh MS, Poirier GG. Base excision repair is efficient in cells lacking poly(ADP‐ribose) polymerase 1. Nucleic Acids Res. 2000;28(20):3887‐3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Pachkowski BF, Tano K, Afonin V, et al. Cells deficient in PARP‐1 show an accelerated accumulation of DNA single strand breaks, but not AP sites, over the PARP‐1‐proficient cells exposed to MMS. Mutat Res. 2009;671(1‐2):93‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Poly HelledayT. ADP‐ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single‐strand intermediate. Nucleic Acids Res. 2011;39(8):3166‐3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588‐5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pommier Y, O'Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17. [DOI] [PubMed] [Google Scholar]
- 109. Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage‐dependent poly(ADP‐ribosyl)ation by human PARP‐1. Science. 2012;336(6082):728‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kotsopoulos J, Willows K, Trat S, et al. BRCA mutation status is not associated with increased hematologic toxicity among patients undergoing platinum‐based chemotherapy for ovarian cancer. Int J Gynecol Cancer. 2018;28(1):69‐76. [DOI] [PubMed] [Google Scholar]
- 111. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 112. Ledermann JA, Raja FA, Fotopoulou C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2013;24(6):vi24‐vi32. Suppl. [DOI] [PubMed] [Google Scholar]
- 113. Franzese E, Diana A, Centonze S, et al. PARP inhibitors in first‐line therapy of ovarian cancer: are there any doubts? Front Oncol. 2020;10:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ray‐Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first‐line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416‐2428. [DOI] [PubMed] [Google Scholar]
- 115. Gonzalez‐Martin A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391‐2402. [DOI] [PubMed] [Google Scholar]
- 116. Coleman RL, Fleming GF, Brady MF, et al. Veliparib with first‐line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403‐2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Banerjee S, Gonzalez‐Martin A, Harter P, et al. First‐line PARP inhibitors in ovarian cancer: summary of an ESMO Open—Cancer Horizons round‐table discussion. ESMO Open. 2020;5(6):e001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Plummer R, Lorigan P, Steven N, et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF‐01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71(5):1191‐1199. [DOI] [PubMed] [Google Scholar]
- 119. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 120. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐917. [DOI] [PubMed] [Google Scholar]
- 121. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123‐134. [DOI] [PubMed] [Google Scholar]
- 122. Fong PC, Yap TA, Boss DS, et al. Poly(ADP)‐ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum‐free interval. J Clin Oncol. 2010;28(15):2512‐2519. [DOI] [PubMed] [Google Scholar]
- 123. Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof‐of‐concept trial. Lancet. 2010;376(9737):245‐251. [DOI] [PubMed] [Google Scholar]
- 124. Kaufman B, Shapira‐Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration‐resistant prostate cancer with DNA repair gene aberrations (TOPARP‐B): a multicentre, open‐label, randomised, phase 2 trial. Lancet Oncol. 2020;21(1):162‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kim G, Ison G, McKee AE, et al. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA‐mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257‐4261. [DOI] [PubMed] [Google Scholar]
- 127. Robson M, Goessl C, Domchek S. Olaparib for metastatic germline BRCA‐mutated breast cancer. N Engl J Med. 2017;377(18):1792‐1793. [DOI] [PubMed] [Google Scholar]
- 128. Sonnenblick A, de Azambuja E, Azim HA, Piccart M. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12(1):27‐41. [DOI] [PubMed] [Google Scholar]
- 129. Mirza MR, Pignata S, Ledermann JA. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol. 2018;29(6):1366‐1376. [DOI] [PubMed] [Google Scholar]
- 130. Franzese E, Centonze S, Diana A, et al. PARP inhibitors in ovarian cancer. Cancer Treat Rev. 2019;73:1‐9. [DOI] [PubMed] [Google Scholar]
- 131. Mateo J, Lord CJ, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30(9):1437‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pilie PG, Gay CM, Byers LA, O'Connor MJ, YapTA. PARP inhibitors: extending benefit beyond BRCA‐mutant cancers. Clin Cancer Res. 2019;25(13):3759‐3771. [DOI] [PubMed] [Google Scholar]
- 133. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162(2):454. [DOI] [PubMed] [Google Scholar]
- 134. Helleday T. PARP inhibitor receives FDA breakthrough therapy designation in castration resistant prostate cancer: beyond germline BRCA mutations. Ann Oncol. 2016;27(5):755‐757. [DOI] [PubMed] [Google Scholar]
- 135. Bryant HE, Helleday T. Inhibition of poly (ADP‐ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34(6):1685‐1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP‐ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109‐8115. [DOI] [PubMed] [Google Scholar]
- 137. Pujade‐Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum‐resistant recurrent ovarian cancer: the AURELIA open‐label randomized phase III trial. J Clin Oncol. 2014;32(13):1302‐1308. [DOI] [PubMed] [Google Scholar]
- 138. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum‐sensitive high‐grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open‐label, phase 2 trial. Lancet Oncol. 2017;18(1):75‐87. [DOI] [PubMed] [Google Scholar]
- 139. Gonzalez Martin A. Progress in PARP inhibitors beyond BRCA mutant recurrent ovarian cancer? Lancet Oncol. 2017;18(1):8‐9. [DOI] [PubMed] [Google Scholar]
- 140. Shapira‐Frommer R, Oza AM, Domchek SM, et al. A phase II open‐label, multicenter study of single‐agent rucaparib in the treatment of patients with relapsed ovarian cancer and a deleterious BRCA mutation. J Clin Oncol. 2015;33(15):5513‐5513. suppl. [Google Scholar]
- 141. Thomas HD, Calabrese CR, Batey MA, et al. Preclinical selection of a novel poly(ADP‐ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007;6(3):945‐956. [DOI] [PubMed] [Google Scholar]
- 142. Wahlberg E, Karlberg T, Kouznetsova E, et al. Family‐wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol. 2012;30(3):283‐288. [DOI] [PubMed] [Google Scholar]
- 143. Knezevic CE, Wright G, Rix LLR, et al. Proteome‐wide profiling of clinical PARP inhibitors reveals compound‐specific secondary targets. Cell Chem Biol. 2016;23(12):1490‐1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Thorsell AG, Ekblad T, Karlberg T, et al. Structural basis for potency and promiscuity in poly(ADP‐ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem. 2017;60(4):1262‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum‐sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154‐2164. [DOI] [PubMed] [Google Scholar]
- 146. Telli ML, Timms KM, Reid J, et al. Homologous recombination deficiency (HRD) score predicts response to platinum‐containing neoadjuvant chemotherapy in patients with triple‐negative breast cancer. Clin Cancer Res. 2016;22(15):3764‐3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hopkins TA, Ainsworth WB, Ellis PA, et al. PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res. 2019;17(2):409‐419. [DOI] [PubMed] [Google Scholar]
- 148. Gogola E, Duarte AA, de Ruiter JR, et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor‐mediated synthetic lethality. Cancer Cell. 2018;33(6):1078‐1093. e12. [DOI] [PubMed] [Google Scholar]
- 149. Pettitt SJ, Krastev DB, Brandsma I, et al. Genome‐wide and high‐density CRISPR‐Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9(1):1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079‐17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Vaidyanathan A, Sawers L, Gannon AL, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel‐ and olaparib‐resistant ovarian cancer cells. Br J Cancer. 2016;115(4):431‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Ray Chaudhuri A, Callen E, Ding X, et al. Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature. 2016;535(7612):382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Taglialatela A, Alvarez S, Leuzzi G, et al. Restoration of replication fork stability in BRCA1‐ and BRCA2‐deficient cells by inactivation of SNF2‐family fork remodelers. Mol Cell. 2017;68(2):414‐430. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Murai J, Tang SW, Leo E, et al. SLFN11 blocks stressed replication forks independently of ATR. Mol Cell. 2018;69(3):371‐384. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple‐negative and BRCA‐mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1‐mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Goodall J, Mateo J, Yuan W, et al. Circulating cell‐free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7(9):1006‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dev H, Chiang TW, Lescale C, et al. Shieldin complex promotes DNA end‐joining and counters homologous recombination in BRCA1‐null cells. Nat Cell Biol. 2018;20(8):954‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. He YJ, Meghani K, Caron MC, et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1‐deficient cells. Nature. 2018;563(7732):522‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mirman Z, Lottersberger F, Takai H, et al. 53BP1‐RIF1‐shieldin counteracts DSB resection through CST‐ and Polalpha‐dependent fill‐in. Nature. 2018;560(7716):112‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ter Brugge P, Kristel P, van der Burg E, et al. Mechanisms of therapy resistance in patient‐derived xenograft models of BRCA1‐deficient breast cancer. J Natl Cancer Inst. 2016;108(11). [DOI] [PubMed] [Google Scholar]
- 163. Noordermeer SM, Adam S, Setiaputra D, et al. The shieldin complex mediates 53BP1‐dependent DNA repair. Nature. 2018;560(7716):117‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hatakeyama K, Nemoto Y, Ueda K, Hayaishi O. Purification and characterization of poly(ADP‐ribose) glycohydrolase. Different modes of action on large and small poly(ADP‐ribose). J Biol Chem. 1986;261(32):14902‐14911. [PubMed] [Google Scholar]
- 165. Wielckens K, Schmidt A, George E, Bredehorst R, Hilz H. DNA fragmentation and NAD depletion. Their relation to the turnover of endogenous mono(ADP‐ribosyl) and poly(ADP‐ribosyl) proteins. J Biol Chem. 1982;257(21):12872‐12877. [PubMed] [Google Scholar]
- 166. Alvarez‐Gonzalez R, Althaus FR. Poly(ADP‐ribose) catabolism in mammalian cells exposed to DNA‐damaging agents. Mutat Res. 1989;218(2):67‐74. [DOI] [PubMed] [Google Scholar]
- 167. Gravells P, Grant E, Smith KM, James DI, Bryant HE. Specific killing of DNA damage‐response deficient cells with inhibitors of poly(ADP‐ribose) glycohydrolase. DNA Repair (Amst). 2017;52:81‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Fathers C, Drayton RM, Solovieva S, Bryant HE. Inhibition of poly(ADP‐ribose) glycohydrolase (PARG) specifically kills BRCA2‐deficient tumor cells. Cell Cycle. 2012;11(5):990‐997. [DOI] [PubMed] [Google Scholar]
- 169. Chen SH, Yu X. Targeting dePARylation selectively suppresses DNA repair‐defective and PARP inhibitor‐resistant malignancies. Sci Adv. 2019;5(4):eaav4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Noll A, Illuzzi G, Ame JC, Dantzer F, Schreiber V. PARG deficiency is neither synthetic lethal with BRCA1 nor PTEN deficiency. Cancer Cell Int. 2016;16:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Pillay N, Tighe A, Nelson L, et al. DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP‐ribose) glycohydrolase inhibitors. Cancer Cell. 2019;35(3):519‐533. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. James DI, Smith KM, Jordan AM, et al. First‐in‐class chemical probes against poly(ADP‐ribose) glycohydrolase (PARG) inhibit DNA repair with differential pharmacology to olaparib. ACS Chem Biol. 2016;11(11):3179‐3190. [DOI] [PubMed] [Google Scholar]
- 173. Min W, Cortes U, Herceg Z, Tong WM, Wang ZQ. Deletion of the nuclear isoform of poly(ADP‐ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis. Carcinogenesis. 2010;31(12):2058‐2065. [DOI] [PubMed] [Google Scholar]
- 174. Gravells P, Neale J, Grant E, et al. Radiosensitization with an inhibitor of poly(ADP‐ribose) glycohydrolase: a comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair (Amst). 2018;61:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Koh DW, Lawler AM, Poitras MF, et al. Failure to degrade poly(ADP‐ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101(51):17699‐17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Ame JC, Fouquerel E, Gauthier LR, et al. Radiation‐induced mitotic catastrophe in PARG‐deficient cells. J Cell Sci. 2009;122(12):1990‐2002. Pt. [DOI] [PubMed] [Google Scholar]
- 177. Slade D. Mitotic functions of poly(ADP‐ribose) polymerases. Biochem Pharmacol. 2019;167:33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Falsig J, Christiansen SH, Feuerhahn S, et al. Poly(ADP‐ribose) glycohydrolase as a target for neuroprotective intervention: assessment of currently available pharmacological tools. Eur J Pharmacol. 2004;497(1):7‐16. [DOI] [PubMed] [Google Scholar]
- 179. Erdelyi K, Kiss A, Bakondi E, et al. Gallotannin inhibits the expression of chemokines and inflammatory cytokines in A549 cells. Mol Pharmacol. 2005;68(3):895‐904. [DOI] [PubMed] [Google Scholar]
- 180. Slama JT, Aboul‐Ela N, Goli DM, Cheesman BV, Simmons AM, Jacobson MK. Specific inhibition of poly(ADP‐ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J Med Chem. 1995;38(2):389‐393. [DOI] [PubMed] [Google Scholar]
- 181. Finch KE, Knezevic CE, Nottbohm AC, Partlow KC, Hergenrother PJ. Selective small molecule inhibition of poly(ADP‐ribose) glycohydrolase (PARG). ACS Chem Biol. 2012;7(3):563‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Houl JH, Ye Z, Brosey CA, et al. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat Commun. 2019;10(1):5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Bannister AJ, Gottlieb TM, Kouzarides T, Jackson SP. c‐Jun is phosphorylated by the DNA‐dependent protein kinase in vitro; definition of the minimal kinase recognition motif. Nucleic Acids Res. 1993;21(5):1289‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Bockstahler LE, Lytle CD, Hellman KB. A review of photodynamic therapy for herpes simplex: benefits and potential risks. N Y J Dent. 1975;45(5):148‐157. [PubMed] [Google Scholar]
- 185. Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274(53):37538‐37543. [DOI] [PubMed] [Google Scholar]
- 186. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA‐PKcs to sites of DNA damage. Nature. 2005;434(7033):605‐611. [DOI] [PubMed] [Google Scholar]
- 187. Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 2014;6(6):442‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Tresini M, Warmerdam DO, Kolovos P, et al. The core spliceosome as target and effector of non‐canonical ATM signalling. Nature. 2015;523(7558):53‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Bea S, Valdes‐Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(45):18250‐18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197‐210. [PubMed] [Google Scholar]
- 192. Begam N, Jamil K, Raju SG. Promoter hypermethylation of the ATM gene as a novel biomarker for breast cancer. Asian Pac J Cancer Prev. 2017;18(11):3003‐3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Bolt J, Vo QN, Kim WJ, et al. The ATM/p53 pathway is commonly targeted for inactivation in squamous cell carcinoma of the head and neck (SCCHN) by multiple molecular mechanisms. Oral Oncol. 2005;41(10):1013‐1020. [DOI] [PubMed] [Google Scholar]
- 194. Mehdipour P, Karami F, Javan F, Mehrazin M. Linking ATM oromoter methylation to cell cycle protein expression in brain tumor patients: cellular molecular triangle correlation in ATM territory. Mol Neurobiol. 2015;52(1):293‐302. [DOI] [PubMed] [Google Scholar]
- 195. Safar AM, Spencer H, Su X, et al. Methylation profiling of archived non‐small cell lung cancer: a promising prognostic system. Clin Cancer Res. 2005;11(12):4400‐4405. [DOI] [PubMed] [Google Scholar]
- 196. Vo QN, Kim WJ, Cvitanovic L, Boudreau DA, Ginzinger DG, Brown KD. The ATM gene is a target for epigenetic silencing in locally advanced breast cancer. Oncogene. 2004;23(58):9432‐9437. [DOI] [PubMed] [Google Scholar]
- 197. Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2014;33(26):3351‐3360. [DOI] [PubMed] [Google Scholar]
- 198. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15(8):1781‐1791. [DOI] [PubMed] [Google Scholar]
- 199. Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM signaling network in development and disease. Front Genet. 2013;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Rafiei S, Fitzpatrick K, Liu D, et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res. 2020;80(11):2094‐2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele‐specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Herrero AB, Gutierrez NC. Targeting ongoing DNA damage in multiple myeloma: effects of DNA damage response inhibitors on plasma cell survival. Front Oncol. 2017;7:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Wang P, Ma D, Wang J, et al. INPP4B‐mediated DNA repair pathway confers resistance to chemotherapy in acute myeloid leukemia. Tumour Biol. 2016;37(9):12513‐12523. [DOI] [PubMed] [Google Scholar]
- 204. Chen H, Li H, Chen Q. INPP4B overexpression suppresses migration, invasion and angiogenesis of human prostate cancer cells. Clin Exp Pharmacol Physiol. 2017;44(6):700‐708. [DOI] [PubMed] [Google Scholar]
- 205. Golding SE, Rosenberg E, Valerie N, et al. Improved ATM kinase inhibitor KU‐60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8(10):2894‐2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. McCabe N, Hanna C, Walker SM, et al. Mechanistic rationale to target PTEN‐deficient tumor cells with inhibitors of the DNA damage response kinase ATM. Cancer Res. 2015;75(11):2159‐2165. [DOI] [PubMed] [Google Scholar]
- 207. Konig G, Remberger K, Hofling B, Erdmann E, Fruhmann G. A 45‐year‐old patient with a progressive, currently therapy‐resistant disease of the lung. Internist (Berl). 1986;27(1):65‐69. [PubMed] [Google Scholar]
- 208. Mansour WY, Tennstedt P, Volquardsen J, et al. Loss of PTEN‐assisted G2/M checkpoint impedes homologous recombination repair and enhances radio‐curability and PARP inhibitor treatment response in prostate cancer. Sci Rep. 2018;8(1):3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Li K, Yan H, Guo W, et al. ATM inhibition induces synthetic lethality and enhances sensitivity of PTEN‐deficient breast cancer cells to cisplatin. Exp Cell Res. 2018;366(1):24‐33. [DOI] [PubMed] [Google Scholar]
- 210. Batey MA, Zhao Y, Kyle S, et al. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol Cancer Ther. 2013;12(6):959‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008;68(18):7466‐7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Karlin J, Allen J, Ahmad SF, et al. Orally bioavailable and blood‐brain barrier‐penetrating ATM inhibitor (AZ32) radiosensitizes intracranial gliomas in mice. Mol Cancer Ther. 2018;17(8):1637‐1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Williams JR, Zhang Y, Zhou H, et al. A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int J Radiat Biol. 2008;84(4):253‐264. [DOI] [PubMed] [Google Scholar]
- 214. Pike KG, Barlaam B, Cadogan E, et al. The identification of potent, selective, and orally available inhibitors of ataxia telangiectasia mutated (ATM) kinase: the discovery of AZD0156 (8‐{6‐[3‐(dimethylamino)propoxy]pyridin‐3‐yl}‐3‐methyl‐1‐(tetrahydro‐2 H‐pyran‐4‐yl)‐1,3‐dihydro‐2 H‐imidazo[4,5‐ c]quinolin‐2‐one). J Med Chem. 2018;61(9):3823‐3841. [DOI] [PubMed] [Google Scholar]
- 215. Durant ST, Zheng L, Wang Y, et al. The brain‐penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv. 2018;4(6):eaat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science. 2003;300(5625):1542‐1548. [DOI] [PubMed] [Google Scholar]
- 217. Lecona E, Fernandez‐Capetillo O. Replication stress and cancer: it takes two to tango. Exp Cell Res. 2014;329(1):26‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet. 2016;50:155‐173. [DOI] [PubMed] [Google Scholar]
- 219. Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14(12):1448‐1459. [PMC free article] [PubMed] [Google Scholar]
- 220. Zhao H, Piwnica‐Worms H. ATR‐mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21(13):4129‐4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Parker LL, Piwnica‐Worms H. Inactivation of the p34cdc2‐cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257(5078):1955‐1957. [DOI] [PubMed] [Google Scholar]
- 222. Sorensen CS, Syljuasen RG, Falck J, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation‐induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3(3):247‐258. [DOI] [PubMed] [Google Scholar]
- 223. Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14‐3‐3 proteins. Mol Biol Cell. 2001;12(3):551‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Chen MS, Ryan CE. Piwnica‐Worms H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14‐3‐3 binding. Mol Cell Biol. 2003;23(21):7488‐7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Nishida H, Tatewaki N, Nakajima Y, et al. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009;37(17):5678‐5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Sarkaria JN, Busby EC, Tibbetts RS, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59(17):4375‐4382. [PubMed] [Google Scholar]
- 227. van Stuijvenberg J, Proksch P, Fritz G. Targeting the DNA damage response (DDR) by natural compounds. Bioorg Med Chem. 2020;28(4):115279. [DOI] [PubMed] [Google Scholar]
- 228. Toledo LI, Murga M, Gutierrez‐Martinez P, Soria R, Fernandez‐Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22(3):297‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Toledo LI, Murga M, Zur R, et al. A cell‐based screen identifies ATR inhibitors with synthetic lethal properties for cancer‐associated mutations. Nat Struct Mol Biol. 2011;18(6):721‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Su D, Feng X, Colic M, et al. CRISPR/CAS9‐based DNA damage response screens reveal gene‐drug interactions. DNA Repair (Amst). 2020;87:102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Charrier JD, Durrant SJ, Golec JM, et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem. 2011;54(7):2320‐2330. [DOI] [PubMed] [Google Scholar]
- 232. Fokas E, Prevo R, Pollard JR, et al. Targeting ATR in vivo using the novel inhibitor VE‐822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Peasland A, Wang LZ, Rowling E, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105(3):372‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Foote KM, Blades K, Cronin A, et al. Discovery of 4‐{4‐[(3R)‐3‐Methylmorpholin‐4‐yl]‐6‐[1‐(methylsulfonyl)cyclopropyl]pyrimidin‐2‐y l}‐1H‐indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J Med Chem. 2013;56(5):2125‐2138. [DOI] [PubMed] [Google Scholar]
- 235. Vendetti FP, Lau A, Schamus S, Conrads TP, O'Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti‐tumor effects of cisplatin to resolve ATM‐deficient non‐small cell lung cancer in vivo. Oncotarget. 2015;6(42):44289‐44305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Wengner AM, Siemeister G, Lucking U, et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage‐inducing or repair‐compromising therapies in preclinical cancer models. Mol Cancer Ther. 2020;19(1):26‐38. [DOI] [PubMed] [Google Scholar]
- 237. Terranova N, Jansen M, Falk M, Hendriks BS. Population pharmacokinetics of ATR inhibitor berzosertib in phase I studies for different cancer types. Cancer Chemother Pharmacol. 2021;87(2):185‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Ramachandran SA, Jadhavar PS, Singh MP, et al. Discovery of pyrazolopyrimidine derivatives as novel inhibitors of ataxia telangiectasia and rad3 related protein (ATR). Bioorg Med Chem Lett. 2017;27(4):750‐754. [DOI] [PubMed] [Google Scholar]
- 239. Gatei M, Sloper K, Sorensen C, et al. Ataxia‐telangiectasia‐mutated (ATM) and NBS1‐dependent phosphorylation of Chk1 on Ser‐317 in response to ionizing radiation. J Biol Chem. 2003;278(17):14806‐14811. [DOI] [PubMed] [Google Scholar]
- 240. Busino L, Donzelli M, Chiesa M, et al. Degradation of Cdc25A by beta‐TrCP during S phase and in response to DNA damage. Nature. 2003;426(6962):87‐91. [DOI] [PubMed] [Google Scholar]
- 241. Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277(5331):1497‐1501. [DOI] [PubMed] [Google Scholar]
- 242. Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27(28):3977‐3985. [DOI] [PubMed] [Google Scholar]
- 243. Morgan MA, Parsels LA, Zhao L, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70(12):4972‐4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Parsels LA, Morgan MA, Tanska DM, et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol Cancer Ther. 2009;8(1):45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell‐cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195‐201. [DOI] [PubMed] [Google Scholar]
- 246. Pan Y, Ren KH, He HW, Shao RG. Knockdown of Chk1 sensitizes human colon carcinoma HCT116 cells in a p53‐dependent manner to lidamycin through abrogation of a G2/M checkpoint and induction of apoptosis. Cancer Biol Ther. 2009;8(16):1559‐1566. [DOI] [PubMed] [Google Scholar]
- 247. Hiregange D, Naick H, Rao BJ. ATR signalling mediates the prosurvival function of phospho‐NPM against PIDDosome mediated cell death. Cell Signal. 2020;71:109602. [DOI] [PubMed] [Google Scholar]
- 248. David L, Fernandez‐Vidal A, Bertoli S, et al. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signal. 2016;9(445):ra90. [DOI] [PubMed] [Google Scholar]
- 249. Wang WJ, Wu SP, Liu JB, et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell‐like population of nasopharyngeal carcinoma cells. Cancer Res. 2013;73(3):1219‐1231. [DOI] [PubMed] [Google Scholar]
- 250. Zhang P, Wei Y, Wang L, et al. ATM‐mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16(9):864‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756‐760. [DOI] [PubMed] [Google Scholar]
- 252. Morton NE. Gene maps and location databases. Ann Hum Genet. 1991;55(3):235‐241. [DOI] [PubMed] [Google Scholar]
- 253. Cole KA, Huggins J, Laquaglia M, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108(8):3336‐3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC‐driven lymphoma cells. Oncogene. 2012;31(13):1661‐1672. [DOI] [PubMed] [Google Scholar]
- 255. Sowden M, Harrison S, Ashfield R, Kingsman AJ, Kingsman SM. Multiple cooperative interactions constrain BPV‐1 E2 dependent activation of transcription. Nucleic Acids Res. 1989;17(8):2959‐2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Barker HE, Patel R, McLaughlin M, et al. CHK1 inhibition radiosensitizes head and neck cancers to paclitaxel‐based chemoradiotherapy. Mol Cancer Ther. 2016;15(9):2042‐2054. [DOI] [PubMed] [Google Scholar]
- 257. Walton MI, Eve PD, Hayes A, et al. The clinical development candidate CCT245737 is an orally active CHK1 inhibitor with preclinical activity in RAS mutant NSCLC and Emicro‐MYC driven B‐cell lymphoma. Oncotarget. 2016;7(3):2329‐2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Fuse E, Tanii H, Kurata N, et al. Unpredicted clinical pharmacology of UCN‐01 caused by specific binding to human alpha1‐acid glycoprotein. Cancer Res. 1998;58(15):3248‐3253. [PubMed] [Google Scholar]
- 259. McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142(1):1‐10. [DOI] [PubMed] [Google Scholar]
- 260. Sakurikar N, Eastman A. Will targeting Chk1 have a role in the future of cancer therapy? J Clin Oncol. 2015;33(9):1075‐1077. [DOI] [PubMed] [Google Scholar]
- 261. Daud AI, Ashworth MT, Strosberg J, et al. Phase I dose‐escalation trial of checkpoint kinase 1 inhibitor MK‐8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33(9):1060‐1066. [DOI] [PubMed] [Google Scholar]
- 262. Infante JR, Hollebecque A, Postel‐Vinay S, et al. Phase I study of GDC‐0425, a checkpoint kinase 1 inhibitor, in combination with gemcitabine in patients with refractory solid tumors. Clin Cancer Res. 2017;23(10):2423‐2432. [DOI] [PubMed] [Google Scholar]
- 263. Laroche‐Clary A, Lucchesi C, Rey C, et al. CHK1 inhibition in soft‐tissue sarcomas: biological and clinical implications. Ann Oncol. 2018;29(4):1023‐1029. [DOI] [PubMed] [Google Scholar]
- 264. Booth L, Roberts J, Poklepovic A, Dent P. The CHK1 inhibitor SRA737 synergizes with PARP1 inhibitors to kill carcinoma cells. Cancer Biol Ther. 2018;19(9):786‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Ma M, Rodriguez A, Sugimoto K. Activation of ATR‐related protein kinase upon DNA damage recognition. Curr Genet. 2020;66(2):327‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40(2):477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Mahajan K, Mahajan NP. WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet. 2013;29(7):394‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Mahajan K, Fang B, Koomen JM, Mahajan NP. H2B Tyr37 phosphorylation suppresses expression of replication‐dependent core histone genes. Nat Struct Mol Biol. 2012;19(9):930‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Mir SE, De Witt Hamer PC, Krawczyk PM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18(3):244‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53‐deficient cells rely on ATM‐ and ATR‐mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11(2):175‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Wang F, Zhu Y, Huang Y, et al. Transcriptional repression of WEE1 by Kruppel‐like factor 2 is involved in DNA damage‐induced apoptosis. Oncogene. 2005;24(24):3875‐3885. [DOI] [PubMed] [Google Scholar]
- 273. Mak JP, Man WY, Chow JP, Ma HT, Poon RY. Pharmacological inactivation of CHK1 and WEE1 induces mitotic catastrophe in nasopharyngeal carcinoma cells. Oncotarget. 2015;6(25):21074‐21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole‐genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489‐494. [DOI] [PubMed] [Google Scholar]
- 275. Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single‐agent antitumor activity against sarcoma cells. Mol Cancer Ther. 2012;11(1):174‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK‐1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53‐deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17(9):2799‐2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Van Linden AA, Baturin D, Ford JB, et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013;12(12):2675‐2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Li C, Andrake M, Dunbrack R, Enders GH. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1‐dependent nuclear export. Mol Cell Biol. 2010;30(1):116‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Palmer BD, Smaill JB, Rewcastle GW, et al. Structure‐activity relationships for 2‐anilino‐6‐phenylpyrido[2,3‐d]pyrimidin‐7(8H)‐ones as inhibitors of the cellular checkpoint kinase Wee1. Bioorg Med Chem Lett. 2005;15(7):1931‐1935. [DOI] [PubMed] [Google Scholar]
- 280. Panek RL, Lu GH, Klutchko SR, et al. In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J Pharmacol Exp Ther. 1997;283(3):1433‐1444. [PubMed] [Google Scholar]
- 281. Hirai H, Iwasawa Y, Okada M, et al. Small‐molecule inhibition of Wee1 kinase by MK‐1775 selectively sensitizes p53‐deficient tumor cells to DNA‐damaging agents. Mol Cancer Ther. 2009;8(11):2992‐3000. [DOI] [PubMed] [Google Scholar]
- 282. Zhu JY, Cuellar RA, Berndt N, et al. Structural basis of Wee kinases functionality and inactivation by diverse small molecule inhibitors. J Med Chem. 2017;60(18):7863‐7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Wright G, Golubeva V, Remsing Rix LL, et al. Dual targeting of WEE1 and PLK1 by AZD1775 elicits single agent cellular anticancer activity. ACS Chem Biol. 2017;12(7):1883‐1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Serpico AF, D'Alterio G, Vetrei C, et al. Wee1 rather than Plk1 is inhibited by AZD1775 at therapeutically relevant concentrations. Cancers (Basel). 2019;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Leijen S, van Geel RM, Pavlick AC, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34(36):4371‐4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR kinases produces tumor‐selective synthetic lethality and suppresses metastasis. J Clin Invest. 2019;129(3):1329‐1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Hauge S, Naucke C, Hasvold G, et al. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S‐phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget. 2017;8(7):10966‐10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Aarts M, Bajrami I, Herrera‐Abreu MT, et al. Functional genetic screen identifies increased sensitivity to WEE1 inhibition in cells with defects in fanconi anemia and HR pathways. Mol Cancer Ther. 2015;14(4):865‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Elbaek CR, Petrosius V, Sorensen CS. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat Res. 2020;819‐820:111694. [DOI] [PubMed] [Google Scholar]
- 290. Cuneo KC, Morgan MA, Sahai V, et al. Dose escalation trial of the wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019;37(29):2643‐2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Matheson CJ, Venkataraman S, Amani V, et al. A WEE1 inhibitor analog of AZD1775 maintains synergy with cisplatin and demonstrates reduced single‐agent cytotoxicity in medulloblastoma cells. ACS Chem Biol. 2016;11(4):921‐930. [DOI] [PubMed] [Google Scholar]
- 292. Kausar T, Schreiber JS, Karnak D, et al. Sensitization of pancreatic cancers to gemcitabine chemoradiation by WEE1 kinase inhibition depends on homologous recombination repair. Neoplasia. 2015;17(10):757‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Walker AI, Hunt T, Jackson RJ, Anderson CW. Double‐stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat‐shock protein in animal cell extracts. EMBO J. 1985;4(1):139‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Carter T, Vancurova I, Sun I, Lou W, DeLeon S. A DNA‐activated protein kinase from HeLa cell nuclei. Mol Cell Biol. 1990;10(12):6460‐6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Lees‐Miller SP, Chen YR, Anderson CW. Human cells contain a DNA‐activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol. 1990;10(12):6472‐6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Blunt T, Finnie NJ, Taccioli GE, et al. Defective DNA‐dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. 1995;80(5):813‐823. [DOI] [PubMed] [Google Scholar]
- 297. van der Burg M, Ijspeert H, Verkaik NS, et al. A DNA‐PKcs mutation in a radiosensitive T‐B‐ SCID patient inhibits Artemis activation and nonhomologous end‐joining. J Clin Invest. 2009;119(1):91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Woodbine L, Neal JA, Sasi NK, et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J Clin Invest. 2013;123(7):2969‐2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Miller RD, Hogg J, Ozaki JH, Gell D, Jackson SP, Riblet R. Gene for the catalytic subunit of mouse DNA‐dependent protein kinase maps to the scid locus. Proc Natl Acad Sci U S A. 1995;92(23):10792‐10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Chang HH, Lieber MR. Structure‐specific nuclease activities of Artemis and the Artemis: DNA‐PKcs complex. Nucleic Acids Res. 2016;44(11):4991‐4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Blackford AN, Jackson SP. ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801‐817. [DOI] [PubMed] [Google Scholar]
- 302. van der Burg M, van Dongen JJ, van Gent DC. DNA‐PKcs deficiency in human: long predicted, finally found. Curr Opin Allergy Clin Immunol. 2009;9(6):503‐509. [DOI] [PubMed] [Google Scholar]
- 303. Schwartz C, Rohr O, Wallet C. Targeting the DNA‐PK complex: its rationale use in cancer and HIV‐1 infection. Biochem Pharmacol. 2019;160:80‐91. [DOI] [PubMed] [Google Scholar]
- 304. Cornell L, Munck JM, Alsinet C, et al. DNA‐PK‐A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin Cancer Res. 2015;21(4):925‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Hosoi Y, Watanabe T, Nakagawa K, et al. Up‐regulation of DNA‐dependent protein kinase activity and Sp1 in colorectal cancer. Int J Oncol. 2004;25(2):461‐468. [PubMed] [Google Scholar]
- 306. Abdel‐Fatah TM, Arora A, Moseley P, et al. ATM, ATR and DNA‐PKcs expressions correlate to adverse clinical outcomes in epithelial ovarian cancers. BBA Clin. 2014;2:10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Kotula E, Berthault N, Agrario C, et al. DNA‐PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle. 2015;14(12):1961‐1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Ihara M, Ashizawa K, Shichijo K, Kudo T. Expression of the DNA‐dependent protein kinase catalytic subunit is associated with the radiosensitivity of human thyroid cancer cell lines. J Radiat Res. 2019;60(2):171‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Shintani S, Mihara M, Li C, et al. Up‐regulation of DNA‐dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003;94(10):894‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Beskow C, Skikuniene J, Holgersson A, et al. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA‐PKcs, Ku70 and Ku86. Br J Cancer. 2009;101(5):816‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Willmore E, Elliott SL, Mainou‐Fowler T, et al. DNA‐dependent protein kinase is a therapeutic target and an indicator of poor prognosis in B‐cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14(12):3984‐3992. [DOI] [PubMed] [Google Scholar]
- 312. George VC, Ansari SA, Chelakkot VS, et al. DNA‐dependent protein kinase: epigenetic alterations and the role in genomic stability of cancer. Mutat Res Rev Mutat Res. 2019;780:92‐105. [DOI] [PubMed] [Google Scholar]
- 313. Um JH, Kang CD, Bae JH, et al. Association of DNA‐dependent protein kinase with hypoxia inducible factor‐1 and its implication in resistance to anticancer drugs in hypoxic tumor cells. Exp Mol Med. 2004;36(3):233‐242. [DOI] [PubMed] [Google Scholar]
- 314. Piotto C, Biscontin A, Millino C, Mognato M. Functional validation of miRNAs targeting genes of DNA double‐strand break repair to radiosensitize non‐small lung cancer cells. Biochim Biophys Acta Gene Regul Mech. 2018;1861(12):1102‐1118. [DOI] [PubMed] [Google Scholar]
- 315. Yan D, Ng WL, Zhang X, et al. Targeting DNA‐PKcs and ATM with miR‐101 sensitizes tumors to radiation. PLoS One. 2010;5(7):e11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Weterings E, Gallegos AC, Dominick LN, et al. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair (Amst). 2016;43:98‐106. [DOI] [PubMed] [Google Scholar]
- 317. Xiong H, Lee RJ, Haura EB, Edwards JG, Dynan WS, Li S. Intranuclear delivery of a novel antibody‐derived radiosensitizer targeting the DNA‐dependent protein kinase catalytic subunit. Int J Radiat Oncol Biol Phys. 2012;83(3):1023‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Block WD, Merkle D, Meek K, Lees‐Miller SP. Selective inhibition of the DNA‐dependent protein kinase (DNA‐PK) by the radiosensitizing agent caffeine. Nucleic Acids Res. 2004;32(6):1967‐1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Durant S, Karran P. Vanillins—a novel family of DNA‐PK inhibitors. Nucleic Acids Res. 2003;31(19):5501‐5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Griffin RJ, Fontana G, Golding BT, et al. Selective benzopyranone and pyrimido[2,1‐a]isoquinolin‐4‐one inhibitors of DNA‐dependent protein kinase: synthesis, structure‐activity studies, and radiosensitization of a human tumor cell line in vitro. J Med Chem. 2005;48(2):569‐585. [DOI] [PubMed] [Google Scholar]
- 321. Hardcastle IR, Cockcroft X, Curtin NJ, et al. Discovery of potent chromen‐4‐one inhibitors of the DNA‐dependent protein kinase (DNA‐PK) using a small‐molecule library approach. J Med Chem. 2005;48(24):7829‐7846. [DOI] [PubMed] [Google Scholar]
- 322. Hollick JJ, Golding BT, Hardcastle IR, et al. 2,6‐disubstituted pyran‐4‐one and thiopyran‐4‐one inhibitors of DNA‐dependent protein kinase (DNA‐PK). Bioorg Med Chem Lett. 2003;13(18):3083‐3086. [DOI] [PubMed] [Google Scholar]
- 323. Leahy JJ, Golding BT, Griffin RJ, et al. Identification of a highly potent and selective DNA‐dependent protein kinase (DNA‐PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14(24):6083‐6087. [DOI] [PubMed] [Google Scholar]
- 324. Willmore E, de Caux S, Sunter NJ, et al. A novel DNA‐dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103(12):4659‐4665. [DOI] [PubMed] [Google Scholar]
- 325. Zhao Y, Thomas HD, Batey MA, et al. Preclinical evaluation of a potent novel DNA‐dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66(10):5354‐5362. [DOI] [PubMed] [Google Scholar]
- 326. Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ. DNA‐PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat. 2014;143(1):47‐55. [DOI] [PubMed] [Google Scholar]
- 327. Cowell IG, Durkacz BW, Tilby MJ. Sensitization of breast carcinoma cells to ionizing radiation by small molecule inhibitors of DNA‐dependent protein kinase and ataxia telangiectsia mutated. Biochem Pharmacol. 2005;71(1‐2):13‐20. [DOI] [PubMed] [Google Scholar]
- 328. Yanai M, Makino H, Ping B, et al. DNA‐PK inhibition by NU7441 enhances chemosensitivity to topoisomerase inhibitor in non‐small cell lung carcinoma cells by blocking DNA damage repair. Yonago Acta Med. 2017;60(1):9‐15. [PMC free article] [PubMed] [Google Scholar]
- 329. Tichy A, Durisova K, Salovska B, et al. Radio‐sensitization of human leukaemic MOLT‐4 cells by DNA‐dependent protein kinase inhibitor, NU7441. Radiat Environ Biophys. 2014;53(1):83‐92. [DOI] [PubMed] [Google Scholar]
- 330. Yang C, Wang Q, Liu X, et al. NU7441 enhances the radiosensitivity of liver cancer cells. Cell Physiol Biochem. 2016;38(5):1897‐1905. [DOI] [PubMed] [Google Scholar]
- 331. Kashishian A, Douangpanya H, Clark D, et al. DNA‐dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Mol Cancer Ther. 2003;2(12):1257‐1264. [PubMed] [Google Scholar]
- 332. Knight ZA, Chiang GG, Alaimo PJ, et al. Isoform‐specific phosphoinositide 3‐kinase inhibitors from an arylmorpholine scaffold. Bioorg Med Chem. 2004;12(17):4749‐4759. [DOI] [PubMed] [Google Scholar]
- 333. Shinohara ET, Geng L, Tan J, et al. DNA‐dependent protein kinase is a molecular target for the development of noncytotoxic radiation‐sensitizing drugs. Cancer Res. 2005;65(12):4987‐4992. [DOI] [PubMed] [Google Scholar]
- 334. Nutley BP, Smith NF, Hayes A, et al. Preclinical pharmacokinetics and metabolism of a novel prototype DNA‐PK inhibitor NU7026. Br J Cancer. 2005;93(9):1011‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Timme CR, Rath BH, O'Neill JW, Camphausen K, Tofilon PJ. The DNA‐PK inhibitor VX‐984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol Cancer Ther. 2018;17(6):1207‐1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Wang M, Chen S, Wei Y, Wei X. DNA‐PK inhibition by M3814 enhances chemosensitivity in non‐small cell lung cancer. Acta Pharmaceutica Sinica B. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Zenke FT, Zimmermann A, Sirrenberg C, et al. Abstract 1658: m3814, a novel investigational DNA‐PK inhibitor: enhancing the effect of fractionated radiotherapy leading to complete regression of tumors in mice. Cancer Res. 2016;76:1658. Supplement. [Google Scholar]
- 338. van Bussel MTJ, Awada A, de Jonge MJA, et al. A first‐in‐man phase 1 study of the DNA‐dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br J Cancer. 2021;124(4):728‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Smith MC, Mader MM, Cook JA, et al. Characterization of LY3023414, a novel PI3K/mTOR dual inhibitor eliciting transient target modulation to impede tumor growth. Mol Cancer Ther. 2016;15(10):2344‐2356. [DOI] [PubMed] [Google Scholar]
- 340. Mortensen DS, Perrin‐Ninkovic SM, Shevlin G, et al. Optimization of a series of triazole containing mammalian target of rapamycin (mTOR) kinase inhibitors and the discovery of CC‐115. J Med Chem. 2015;58(14):5599‐5608. [DOI] [PubMed] [Google Scholar]
- 341. Tsuji T, Sapinoso LM, Tran T, et al. CC‐115, a dual inhibitor of mTOR kinase and DNA‐PK, blocks DNA damage repair pathways and selectively inhibits ATM‐deficient cell growth in vitro. Oncotarget. 2017;8(43):74688‐74702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Bendell JC, Varghese AM, Hyman DM, et al. A first‐in‐human phase 1 study of LY3023414, an oral PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin Cancer Res. 2018;24(14):3253‐3262. [DOI] [PubMed] [Google Scholar]
- 343. Carrassa L, Chila R, Lupi M, et al. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11(13):2507‐2517. [DOI] [PubMed] [Google Scholar]
- 344. Sanjiv K, Hagenkort A, Calderon‐Montano JM, et al. Cancer‐specific synthetic lethality between ATR and CHK1 kinase activities. Cell Rep. 2016;14(2):298‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852‐861. [DOI] [PubMed] [Google Scholar]
- 346. Pujade‐Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum‐sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT‐Ov21): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274‐1284. [DOI] [PubMed] [Google Scholar]
- 347. Lheureux S, Lai Z, Dougherty BA, et al. Long‐term responders on olaparib maintenance in high‐grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res. 2017;23(15):4086‐4094. [DOI] [PubMed] [Google Scholar]
- 348. Biau J, Chautard E, Verrelle P, Dutreix M. Altering DNA repair to improve radiation therapy: specific and multiple pathway targeting. Front Oncol. 2019;9:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Jung Y, Lippard SJ. Direct cellular responses to platinum‐induced DNA damage. Chem Rev. 2007;107(5):1387‐1407. [DOI] [PubMed] [Google Scholar]
- 350. Kamchatnov RA, Shul'gin VS. Raising the qualifications of physicians and of paramedical personnel in the medical service troop team. Voen Med Zh. 1979(12):19‐21. [PubMed] [Google Scholar]
- 351. Caldecott KW. Single‐strand break repair and genetic disease. Nat Rev Genet. 2008;9(8):619‐631. [DOI] [PubMed] [Google Scholar]
- 352. Kessler T, Berberich A, Sadik A, et al. Methylome analyses of three glioblastoma cohorts reveal chemotherapy sensitivity markers within DDR genes. Cancer Med. 2020;9(22):8373‐8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Cheng CL, Johnson SP, Keir ST, et al. ADP‐ribose) polymerase‐1 inhibition reverses temozolomide resistance in a DNA mismatch repair‐deficient malignant glioma xenograft. Mol Cancer Ther. 2005;4(9):1364‐1368. [DOI] [PubMed] [Google Scholar]
- 354. Wedge SR, Porteous JK, Newlands ES. 3‐aminobenzamide and/or O6‐benzylguanine evaluated as an adjuvant to temozolomide or BCNU treatment in cell lines of variable mismatch repair status and O6‐alkylguanine‐DNA alkyltransferase activity. Br J Cancer. 1996;74(7):1030‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Tentori L, Leonetti C, Scarsella M, et al. Combined treatment with temozolomide and poly(ADP‐ribose) polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site. Blood. 2002;99(6):2241‐2244. [DOI] [PubMed] [Google Scholar]
- 356. Hussain M, Carducci MA, Slovin S, et al. Targeting DNA repair with combination veliparib (ABT‐888) and temozolomide in patients with metastatic castration‐resistant prostate cancer. Invest New Drugs. 2014;32(5):904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Gupta SK, Kizilbash SH, Carlson BL, et al. Delineation of MGMT hypermethylation as a biomarker for veliparib‐mediated temozolomide‐sensitizing therapy of glioblastoma. J Natl Cancer Inst. 2016;108(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Pietanza MC, Krug LM, Waqar SN, et al. A multi‐center, randomized, double‐blind phase II study comparing temozolomide (TMZ) plus either veliparib (ABT‐888), a PARP inhibitor, or placebo as 2nd or 3rd‐line therapy for patients (Pts) with relapsed small cell lung cancers (SCLCs). J Clin Oncol. 2016;34(5):8512‐8512. suppl. [Google Scholar]
- 359. Nguewa PA, Fuertes MA, Cepeda V, et al. ADP‐ribose) polymerase‐1 inhibitor 3‐aminobenzamide enhances apoptosis induction by platinum complexes in cisplatin‐resistant tumor cells. Med Chem. 2006;2(1):47‐53. [DOI] [PubMed] [Google Scholar]
- 360. Hastak K, Alli E, Ford JM. Synergistic chemosensitivity of triple‐negative breast cancer cell lines to poly(ADP‐Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res. 2010;70(20):7970‐7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Huang SH, Xiong M, Chen XP, Xiao ZY, Zhao YF, Huang ZY. PJ34, an inhibitor of PARP‐1, suppresses cell growth and enhances the suppressive effects of cisplatin in liver cancer cells. Oncol Rep. 2008;20(3):567‐572. [PubMed] [Google Scholar]
- 362. Cheng H, Zhang Z, Borczuk A, et al. PARP inhibition selectively increases sensitivity to cisplatin in ERCC1‐low non‐small cell lung cancer cells. Carcinogenesis. 2013;34(4):739‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Michels J, Vitale I, Galluzzi L, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73(7):2271‐2280. [DOI] [PubMed] [Google Scholar]
- 364. Loibl S, O'Shaughnessy J, Untch M, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple‐negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19(4):497‐509. [DOI] [PubMed] [Google Scholar]
- 365. Yap TA, O'Carrigan B, Penney MS, et al. Phase I trial of first‐in‐class ATR inhibitor M6620 (VX‐970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38(27):3195‐3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Gorecki L, Andrs M, Rezacova M, Korabecny J. Discovery of ATR kinase inhibitor berzosertib (VX‐970, M6620): clinical candidate for cancer therapy. Pharmacol Ther. 2020;210:107518. [DOI] [PubMed] [Google Scholar]
- 367. Bendell J, O'Reilly EM, Middleton MR, et al. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol. 2015;26(4):804‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Rajan A, Carter CA, Kelly RJ, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18(8):2344‐2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Rothkamm K, Lobrich M. Evidence for a lack of DNA double‐strand break repair in human cells exposed to very low x‐ray doses. Proc Natl Acad Sci U S A. 2003;100(9):5057‐5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Berbis P, Hesse S, Privat Y. Essential fatty acids and the skin. Allerg Immunol (Paris). 1990;22(6):225‐231. [PubMed] [Google Scholar]
- 371. Reuvers TGA, Kanaar R, Nonnekens J. DNA damage‐inducing anticancer therapies: from global to precision damage. Cancers (Basel). 2020;12(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11(4):239‐253. [DOI] [PubMed] [Google Scholar]
- 373. Chabot P, Hsia TC, Ryu JS, et al. Veliparib in combination with whole‐brain radiation therapy for patients with brain metastases from non‐small cell lung cancer: results of a randomized, global, placebo‐controlled study. J Neurooncol. 2017;131(1):105‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Reiss KA, Herman JM, Armstrong D, et al. A final report of a phase I study of veliparib (ABT‐888) in combination with low‐dose fractionated whole abdominal radiation therapy (LDFWAR) in patients with advanced solid malignancies and peritoneal carcinomatosis with a dose escalation in ovarian and fallopian tube cancers. Gynecol Oncol. 2017;144(3):486‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Blumenthal DT, Rankin C, Stelzer KJ, et al. A Phase III study of radiation therapy (RT) and O(6)‐benzylguanine + BCNU versus RT and BCNU alone and methylation status in newly diagnosed glioblastoma and gliosarcoma: Southwest Oncology Group (SWOG) study S0001. Int J Clin Oncol. 2015;20(4):650‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Bhattacharya S, Srinivasan K, Abdisalaam S, et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017;45(8):4590‐4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Prevo R, Fokas E, Reaper PM, et al. The novel ATR inhibitor VE‐821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13(11):1072‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Huntoon CJ, Flatten KS, Wahner Hendrickson AE, et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73(12):3683‐3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Pires IM, Olcina MM, Anbalagan S, et al. Targeting radiation‐resistant hypoxic tumour cells through ATR inhibition. Br J Cancer. 2012;107(2):291‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Leszczynska KB, Dobrynin G, Leslie RE, et al. Preclinical testing of an Atr inhibitor demonstrates improved response to standard therapies for esophageal cancer. Radiother Oncol. 2016;121(2):232‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Shi Q, Shen LY, Dong B, et al. The identification of the ATR inhibitor VE‐822 as a therapeutic strategy for enhancing cisplatin chemosensitivity in esophageal squamous cell carcinoma. Cancer Lett. 2018;432:56‐68. [DOI] [PubMed] [Google Scholar]
- 382. Baschnagel AM, Elnaggar JH, VanBeek HJ, et al. ATR inhibitor M6620 (VX‐970) enhances the effect of radiation in non‐small cell lung cancer brain metastasis patient‐derived xenografts. Mol Cancer Ther. 2021;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Dillon MT, Barker HE, Pedersen M, et al. Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Mol Cancer Ther. 2017;16(1):25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Wallez Y, Dunlop CR, Johnson TI, et al. The ATR inhibitor AZD6738 synergizes with gemcitabine in vitro and in vivo to induce pancreatic ductal adenocarcinoma regression. Mol Cancer Ther. 2018;17(8):1670‐1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Jin J, Fang H, Yang F, et al. Combined inhibition of ATR and WEE1 as a novel therapeutic strategy in triple‐negative breast cancer. Neoplasia. 2018;20(5):478‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Checkley S, MacCallum L, Yates J, et al. Bridging the gap between in vitro and in vivo: dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2015;5:13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Pfister SX, Markkanen E, Jiang Y, et al. Inhibiting Wee1 selectively kills histone H3K36me3‐deficient cancers by dNTP starvation. Cancer Cell. 2015;28(5):557‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Mizuarai S, Yamanaka K, Itadani H, et al. Discovery of gene expression‐based pharmacodynamic biomarker for a p53 context‐specific anti‐tumor drug Wee1 inhibitor. Mol Cancer. 2009;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Pfister SX, Ahrabi S, Zalmas LP, et al. SETD2‐dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7(6):2006‐2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Guertin AD, Li J, Liu Y, et al. Preclinical evaluation of the WEE1 inhibitor MK‐1775 as single‐agent anticancer therapy. Mol Cancer Ther. 2013;12(8):1442‐1452. [DOI] [PubMed] [Google Scholar]
- 391. Aarts M, Sharpe R, Garcia‐Murillas I, et al. Forced mitotic entry of S‐phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2(6):524‐539. [DOI] [PubMed] [Google Scholar]
- 392. Bridges KA, Hirai H, Buser CA, et al. MK‐1775, a novel Wee1 kinase inhibitor, radiosensitizes p53‐defective human tumor cells. Clin Cancer Res. 2011;17(17):5638‐5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Do K, Wilsker D, Ji J, et al. Phase I study of single‐agent AZD1775 (MK‐1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2015;33(30):3409‐3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Willers H, Dahm‐Daphi J, Powell SN. Repair of radiation damage to DNA. Br J Cancer. 2004;90(7):1297‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Van Triest B, Damstrup L, Falkenius J, et al. A phase Ia/Ib trial of the DNA‐dependent protein kinase inhibitor (DNA‐PKi) M3814 in combination with radiotherapy in patients with advanced solid tumors. Journal of Clinical Oncology. 2017;35(15):e14048‐e14048. suppl. [Google Scholar]
- 396. O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547‐560. [DOI] [PubMed] [Google Scholar]
- 397. Karnak D, Engelke CG, Parsels LA, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20(19):5085‐5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534‐76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Yazinski SA, Comaills V, Buisson R, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor‐resistant BRCA‐deficient cancer cells. Genes Dev. 2017;31(3):318‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Restelli V, Lupi M, Chila R, et al. DNA damage response inhibitor combinations exert synergistic antitumor activity in aggressive B‐cell lymphomas. Mol Cancer Ther. 2019;18(7):1255‐1264. [DOI] [PubMed] [Google Scholar]
- 401. Yap TA, Kristeleit R, Michalarea V, et al. Phase I trial of the PARP inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2‐ and non‐BRCA1/2‐mutant cancers. Cancer Discov. 2020;10(10):1528‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Karakashev S, Zhu H, Yokoyama Y, et al. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017;21(12):3398‐3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Yang L, Zhang Y, Shan W, et al. Repression of BET activity sensitizes homologous recombination‐proficient cancers to PARP inhibition. Sci Transl Med. 2017;9(400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Sun C, Yin J, Fang Y, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(3):401‐416. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Fok JHL, Ramos‐Montoya A, Vazquez‐Chantada M, et al. AZD7648 is a potent and selective DNA‐PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun. 2019;10(1):5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Spagnolo L, Barbeau J, Curtin NJ, Morris EP, Pearl LH. Visualization of a DNA‐PK/PARP1 complex. Nucleic Acids Res. 2012;40(9):4168‐4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Azad A, Bukczynska P, Jackson S, et al. Co‐targeting deoxyribonucleic acid‐dependent protein kinase and poly(adenosine diphosphate‐ribose) polymerase‐1 promotes accelerated senescence of irradiated cancer cells. Int J Radiat Oncol Biol Phys. 2014;88(2):385‐394. [DOI] [PubMed] [Google Scholar]
- 408. Ying S, Chen Z, Medhurst AL, et al. DNA‐PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016;76(5):1078‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Mouw KW, Goldberg MS, Konstantinopoulos PA, D'Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ. A prosurvival DNA damage‐induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017;31(4):353‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Gluck S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Wu J, Sun L, Chen X, et al. Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Corrales L, Gajewski TF. Molecular pathways: targeting the stimulator of interferon genes (STING) in the immunotherapy of cancer. Clin Cancer Res. 2015;21(21):4774‐4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Corrales L, McWhirter SM, Dubensky TW, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126(7):2404‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Hartlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity. 2015;42(2):332‐343. [DOI] [PubMed] [Google Scholar]
- 417. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69‐74. [DOI] [PubMed] [Google Scholar]
- 419. Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non‐small cell lung cancer. Cancer Discov. 2017;7(3):264‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Ricciuti B, Recondo G, Spurr LF, et al. Impact of DNA damage response and repair (DDR) gene mutations on efficacy of PD‐(L)1 immune checkpoint inhibition in non‐small cell lung cancer. Clin Cancer Res. 2020;26(15):4135‐4142. [DOI] [PubMed] [Google Scholar]
- 422. Prasanna T, Wu F, Khanna KK, et al. Optimizing poly (ADP‐ribose) polymerase inhibition through combined epigenetic and immunotherapy. Cancer Sci. 2018;109(11):3383‐3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP inhibitor efficacy depends on CD8(+) T‐cell recruitment via intratumoral STING pathway activation in BRCA‐deficient models of triple‐negative breast cancer. Cancer Discov. 2019;9(6):722‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer. 2018;118(3):312‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Wu Y, Chen M, Wu P, Chen C, Xu ZP, Gu W. Increased PD‐L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol. 2017;44(5):602‐604. [DOI] [PubMed] [Google Scholar]
- 426. Friedlander M, Meniawy T, Markman B, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose‐escalation stage of a multicentre, open‐label, phase 1a/b trial. Lancet Oncol. 2019;20(9):1306‐1315. [DOI] [PubMed] [Google Scholar]
- 427. Navarro J, Gozalbo‐Lopez B, Mendez AC, et al. PARP‐1/PARP‐2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci Rep. 2017;7:41962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287(34):29075‐29087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Salvador‐Barbero B, Alvarez‐Fernandez M, Zapatero‐Solana E, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;37(3):340‐353. e6. [DOI] [PubMed] [Google Scholar]
- 430. Pesch AM, Hirsh NH, Chandler BC, et al. Short‐term CDK4/6 inhibition radiosensitizes estrogen receptor‐positive breast cancers. Clin Cancer Res. 2020;26(24):6568‐6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Pestell RG. New roles of cyclin D1. Am J Pathol. 2013;183(1):3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Jerby‐Arnon L, Shah P, Cuoco MS, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984‐997. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Schaer DA, Beckmann RP, Dempsey JA, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD‐L1 checkpoint blockade. Cell Rep. 2018;22(11):2978‐2994. [DOI] [PubMed] [Google Scholar]
- 434. Teo ZL, Versaci S, Dushyanthen S, et al. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple‐negative breast cancer. Cancer Res. 2017;77(22):6340‐6352. [DOI] [PubMed] [Google Scholar]
- 435. Comorosan S. On a possible biological spectroscopy. Bull Math Biol. 1975;37(4):419‐425. [DOI] [PubMed] [Google Scholar]
- 436. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING‐mediated T‐cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Sen T, Della Corte CM, Milutinovic S, et al. Combination treatment of the oral CHK1 inhibitor, SRA737, and low‐dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. J Thorac Oncol. 2019;14(12):2152‐2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Tang Z, Pilie PG, Geng C, et al. ATR inhibition induces CDK1‐SPOP signaling and enhances anti‐PD‐L1 cytotoxicity in prostate cancer. Clin Cancer Res. 2021;27(17):4898‐4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Patel MR, Falchook GS, Wang JS‐Z, et al. Open‐label, multicenter, phase I study to assess safety and tolerability of adavosertib plus durvalumab in patients with advanced solid tumors. J Clin Oncol. 2019;37(15):2562‐2562. _suppl. [Google Scholar]
- 440. Maynard S, Swistowska AM, Lee JW, et al. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells. 2008;26(9):2266‐2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Christmann M, Verbeek B, Roos WP, Kaina B. O(6)‐Methylguanine‐DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta. 2011;1816(2):179‐190. [DOI] [PubMed] [Google Scholar]
- 442. Weller M, Stupp R, Reifenberger G, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6(1):39‐51. [DOI] [PubMed] [Google Scholar]
- 443. Klinakis A, Karagiannis D, Rampias T. Targeting DNA repair in cancer: current state and novel approaches. Cell Mol Life Sci. 2020;77(4):677‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Farkkila A, Gulhan DC, Casado J, et al. Immunogenomic profiling determines responses to combined PARP and PD‐1 inhibition in ovarian cancer. Nat Commun. 2020;11(1):1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Cleary JM, Aguirre AJ, Shapiro GI, D'Andrea AD. Biomarker‐guided development of DNA repair inhibitors. Mol Cell. 2020;78(6):1070‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol. 2014;232(2):199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12(12):917‐930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the review.