

Abstract
Microsatellite instability (MSI) is the landmark feature of DNA mismatch repair deficiency, which can be found in 15–20% of all colorectal cancers (CRC). This specific set of tumors has been initially perceived as a niche for geneticists or gastroenterologists focused on inherited predispositions. However, over the years, MSI has established itself as a key biomarker for the diagnosis, then extending to forecasting the disease behavior and prognostication, including the prediction of responsiveness to immunotherapy and eventually to kinase inhibitors, and possibly even to specific biological drugs. Thanks to the contribution of the characterization of MSI tumors, researchers have first acknowledged that a strong lymphocytic reaction is associated with a good prognosis. This understanding supported the prognostic implications in terms of the low metastatic potential of MSI-CRC and has led to modifications in the indications for adjuvant treatment. Furthermore, with the emergence of immunotherapy, this strong biomarker of responsiveness has exemplified the capability of re-activating an effective immune control by removing the brakes of immune evasion. Lately, a subset of MSI-CRC emerged as the ideal target for kinase inhibitors. This therapeutic scenario implies a paradox in which appropriate treatments for advanced disease are effective in a set of tumors that seldom evolve towards metastases.
Keywords: microsatellite instability, colorectal cancer, immunotherapy, targeted therapy
1. Introduction
Colorectal cancer (CRC) is the third most common malignancy and cause of cancer mortality in Europe and the United States, accounting for nearly 900.000 deaths every year worldwide [1]. Among the newly diagnosed CRC, approximately 20% of patients still present with a metastatic disease, and a further 25% of those with an initially localized disease will eventually develop distant metastases [2,3]. Despite the fact that staging has traditionally represented the backbone of the prognostic factors in oncology, the growing knowledge of the molecular mechanisms of CRC has revolutionized the traditional or “old school” methods of managing tumor conditions. Indeed, CRC is a highly heterogeneous disease in regard to molecular expression and genetic abnormalities. It is known that a small subset of CRCs, approximately 15% of the cases, demonstrate microsatellite instability (MSI) due to an impaired DNA mismatch repair (MMR) system, though the vast majority of CRCs belong to the microsatellite stable (MSS) biomarker list [4]. MSI-CRCs are mostly sporadic, while approximately 3% of all CRCs harbor a germline mutation of mismatch repair genes (i.e., MLH1, MSH2, MSH6, PMS2, and EpCAM) identifying the Lynch syndrome [5]. The understanding of the carcinogenesis of MMR deficient tumors and subsequent clinical research has had an enormous therapeutic impact in the field of gastrointestinal oncology. In particular, the MSI status defines the largest group of inherited predispositions to gastrointestinal cancers and impacts the prognosis of CRC, giving better stage-adjusted survival rates compared to MSS tumors [6,7]. Moreover, MSI colorectal tumors are more frequently seen at early stages (i.e., stage II–III), and only 3.5% of the cases present with a metastatic disease [8], in accordance with a reduced distant metastasis, which is intrinsic to MSI status. MMR/MSI testing is increasingly being incorporated as a standard of care for all CRC patients and is collectively recommended by the most important scientific societies involved in the field, such as AGA, ASGE, ASCRS, ASCO, and ESMO [9]. This review summarizes the evidence demonstrating the value of MSI as a diagnostic and prognostic tool and eventually also a predictive biomarker in the personalized approach to CRC.
2. Discovery of MSI, Its Relevance in Lynch Syndrome and Understanding the Different Molecular Pathogenesis of CRC
2.1. Parallel Discovery
The discovery of DNA mismatch repair (MMR) defects is an interesting outcome, which testifies how the contemporary efforts of different teams have helped to elucidate the molecular basis of Lynch syndrome (LS) in a relatively short period of time. However, in addition to contributing to the development of a new era in molecular medicine, it has also raised other lessons in LS management that are worth recalling. The reason for this is chiefly that different methodological approaches were used by the groups involved in the research. To be precise, finding the mechanism behind LS was not the shared aim of these teams. The study led by Perucho was involved in identifying a particular mechanism of carcinogenesis through an unbiased molecular approach, defined as an “arbitrarily primed polymerase chain reaction” (PCR) [10,11]. In doing so, his group found that a fraction of CRCs harbored un-corrected frame-shifted DNA tracts, and they referred to such changes as ubiquitous somatic mutations. The team led by Thibodeau [12] was looking for allelic losses (and gains) by PCR and noted that there was “instability” at the amplified microsatellite sequences (hence microsatellite instability or MSI), in some proportion of the CRCs. Neither study was familiar with or looking for familial cancer or Lynch syndrome genes. Meanwhile, an international consortium with a strong membership from Finland, including Aaltonen, was trying to identify the loci associated with Lynch syndrome by employing an allelotyping approach to search for loss of heterozygosity [13,14]. With the exploration of dinucleotide repeats in tumor DNA compared to normal subjects, CRC patients were found to have frame-shifted sequences, which they described as replication errors (RER). Subsequently, the term MSI was used to describe the same phenomenon that these groups identified and described, although the degree of competition was very high. Perucho’s reference to a probable inherited syndrome was incorporated within the manuscript after Aaltonen and Vogelstein’s group had mapped and reported a Lynch syndrome locus on 2p, a finding already detected by Perucho. In a timely editorial, it was noted that “the cancers whose cells carry shortened repeats are differently distributed in the colon from others and metastasize less frequently. If Perucho is right in believing that the underlying fault may be a mutation of a DNA repair gene, the ramifications of that may be exceedingly important” [15]. These words summarized the relevant biological and clinical implications of the discovery of DNA MMR defects.
These inherent differences led to a dual development of research efforts in the field. On one side, the genes involved in DNA mismatch repair in humans were targeted, being first identified by Kolodner [16] and subsequently largely addressed in their relevance by various teams, including that led by Bert Vogelstein [17], as part of his landmark work unravelling the molecular bases of CRC, before and after the discovery of MMR defects.
On the other side, the research focused on the molecular pathogenesis of MMR deficient CRC and addressed the role of these types of mutations in the peculiar behavior of MSI tumors. It soon became evident that these cancers remain in a class of their own among tumors [18], as compared to other known genetic pathways to CRC, mainly driven by APC gene damage both in inherited (i.e., Familial Adenomatous Polyposis) and sporadic carcinogenesis. In this respect, MMR-deficient tumors appear mainly a disease marked by accelerated tumor progression rather than by an accelerated tumor initiation. It was appreciated that the burden of unrepaired mutations in these tumors contributes to their indolent behavior [19,20] and to the amount of immune response that they elicit [21,22]. Surprisingly, these areas of investigation took years to generate translational research aimed at systematically identifying prognostic markers for CRC and then influencing clinical practice. It should be mentioned that for the first time since the discovery of MMR defects and MSI, a molecular phenotype has recently been proposed for the molecular screening of a specific disease subtype [23]. This long journey led to the exclusion from adjuvant therapy of patients with stage IIA MSI CRC, even though they displayed high-risk hallmarks and contributed to defining the role of tumor-infiltrating lymphocytes (TILs) as a prognostic marker in CRC staging (see below).
2.2. Unraveling the Pool of Genes Involved in DNA MMR and Deranged in Lynch Syndrome
MMR is a mechanism whereby proteins identify and repair mismatched bases occurring mostly by statistical chance during DNA replication or genetic recombination, a mechanism that is present among many species. DNA mismatching, however, is also enhanced by chemical or physical damage. The high conservation rate among species accounts for its importance, as does the discovery of its involvement in human disease by a basic scientist [16]. He was able to cross its defects with the by-then emerging phenotype of MSI in human CRC, thus developing a strategy to identify one of its components (namely, MSH2) as the culprit for a fraction of the cases of Lynch syndrome, moving from the similarities of molecular signatures in yeasts. That is why the human genes were initially labelled as homologues of their counterpart in yeasts.
As the result of a plurality of efforts, we now know that this system is constituted by multiple proteins, including MLH1 (MutL homologue), PMS2 (post-meiotic segregation protein), MSH2 (MutS homologue), MSH6, MLH3, MSH3, and PMS1, which form heterodimers with different roles: MSH2/MSH6 and MSH2/MSH3 heterodimers recognize and bind base–base mismatches and insertion/deletion loops, and subsequently, they recruit MLH1/PMS2 heterodimers to excise and allow the resynthesis of corrected strands [4,24,25]. Later, deletions of the 3′ distal portion of the EPCAM gene, containing the termination codon, have been demonstrated to influence the MMR system by leading to the methylation of the promoter of the downstream neighbor MSH2 and therein to its silencing [26]. Genetic or epigenetic events leading to the silencing of one of the genes of the MMR system ensues in the appearance of the mutator phenotype. Irrespective of the underlying molecular mechanisms, the inactivation of any of the members of the MMR genes leads to the disappearance of the encoded protein. However, the loss of MSH2 or MLH1 also leads to the loss of expression of that protein itself and its heterodimer partner, whereas the loss of MSH6 or PMS2 results in the loss of expression only of the specific protein. Accordingly, germline inactivating mutations of the genes encoding for one among the MMR proteins stay at the basis of MSI as the first pathogenetic damage of the Lynch syndrome and should be followed by a second somatic inactivation hit according to the Knudson hypothesis turning off the second allele [24,27].
In the seminal phase of the late 1990s, addressing MSI in clinical practice was mostly based on clinical criteria, namely the Bethesda ones [28,29]. In other words, the clinical criteria used to define Lynch syndrome (by then referred to as Hereditary Non-Polyposis CRC, HNPCC) or Amsterdam criteria [30,31] were loosened and expanded to identify those patients suitable for the analysis of the MS-status of their CRC and then to germline sequencing if the results of the somatic analysis revealed MSI. Initially, the characterizations of tumor samples based on MS-status comprised the classification into microsatellite instability high (MSI-H) if two or more of the microsatellite markers show instability (or >30% of unstable markers if a larger panel is used) and microsatellite instability low (MSI-L) if only one marker shows instability, as opposed to MSS cancers [24,32]. However, such a classification has been variably criticized, and the distinction in MSI-H and MSI-L progressively lost relevance, and the latter group is cumulated with MSS tumors [33,34].
The systematization of the characterization of the MS status in CRC has confirmed the initial findings by Perucho et al. that most MSI tumors are not the epiphenomenon of LS but are instead sporadic. In fact, considering that MSI cancers account for 15% of all CRCs, only 3% of the total (or 20% among MSI cases) are attributable to Lynch syndrome [35]. It is also now clear that hereditary MSI cancers differ from sporadic ones by means of the type of underlying alteration causing the impairment of the MMR system (as well as in their clinical behavior).
2.3. Sporadic MSI Cancers and Hypermethylation
Patients with sporadic MSI CRC are significantly older than those affected by Lynch syndrome, and most of them lack any significant familial clustering, nevertheless maintaining a better prognosis than those with MSS tumors [36]. The molecular features of sporadic MSI tumors, instead of germline pathogenic variants of MMR genes plus second hit on the other allele, are the methylation of MLH1 promoter frequently coupled with the mutation BRAF(V600E) [4,37].
Understanding the molecular pathogenesis of sporadic MSI CRC was the sequel of the discovery of germline MMR defects, which has helped clarify the mechanism for a portion of otherwise unexplained cases, as well as introducing one additional cancer phenotype [38,39,40]. In fact, the main mechanism for a sporadic MSI CRC going through the inactivation of the promoter region of the DNA mismatch repair gene MLH1 by hypermethylation [41] mostly occurs in the context of the CpG island methylator phenotype (CIMP) [42]. CpG islands are genomic regions rich in cytosine and guanine repeats present in about 40–50% of human genes, usually located at the promoter region and crucial for the epigenetic inactivation of gene transcription by hypermethylation [42].
Although the methylator phenotype can be intended as the main molecular biomarker of sporadic MSI tumors, CIMP can also be found in a group of patients who present no anomalies of the MMR system. Further studies by Ogino et al. [43] and Samowitz et al. [44] demonstrated that not all sporadic MSI tumors with MLH1 hypermethylation have a methylator phenotype. The scenario of CRC molecular characterization has become more and more complex over the years, adding the CIMP status as a separate parameter of classification [41,45]. CIMP+ (or CIMP-high) CRCs are reported to be more frequent in the elderly and in women, are often located in the proximal location, show poor differentiation, and have a high frequency of MSI and BRAF mutation [41,46,47], largely overlapping with sporadic MSI cases. CIMP was originally described as the de novo methylation of the 5′ CpG island of p16 (now CDNK2A) detectable in approximately 1/5 of different tumor types and acting as an alternative mechanism for the silencing of tumor suppressor genes [48].
Although the value of CIMP is not well known, CIMP+ CRC seems to have a better outcome than CIMP-low (particularly if showing wild-type BRAF) and appears to respond more efficiently to adjuvant treatments [41].
2.4. Lynch Syndrome versus Lynch-Like Syndrome
The seminal report on what will be later referred to as HNPCC and Lynch syndrome dates to the end of the XIX century by Aldred S. Warthin, who reported the pedigree of “family G” with a cluster of uterine, gastric, and abdominal cancer, which led him to suspect the existence of a form of predisposition [49]. Years later, Henry Lynch reported similar familial clusters of cancer and reviewed the history of family G, with a predominance of cancers of the colon, uterus and stomach [50]. Notably, Lynch concluded the culprit was an autosomal dominant inheritance of this otherwise unrecognized syndromic cluster, referred to as “Cancer Family Syndrome” (for an exhaustive perspective on the historical development of the medical perspective on the topic, see Boland, 2013) [50]. Later, the term HNPCC was used to refer to the lack of a phenotypic hallmark compared to polyposis syndromes [51]. However, once a molecular phenotype had been identified and its basis clarified, the term Lynch syndrome was encouraged and adopted for those cases with a defined MMR defect and a germline mutation in the MMR genes. Alternately, the lack of a pathogenic germline mutation in a patient with an MSI CRC and features suggestive of an underlying predisposition is called “Lynch-like” syndrome [52,53]. The two syndromes have the development of MSI CRCs at a young age and the presence of extracolonic cancers in common. However, although in patients affected by Lynch-like syndrome, the onset of cancer is in the fifth decade (mean age, 54.9 years) [53], the standardized incidence ratios of CRC and extracolonic cancers is lower (2.12 vs. 6.04 and 1.69 vs. 2.81, respectively) [54].
Although Lynch-like syndrome patients lack germline mutations of the MMR system, they exhibit in almost half of all cases the biallelic somatic inactivation of DNA MMR genes within the tumor [54,55]; moreover, they might harbor germline mutations of unknown genes other than MMR ones. Nevertheless, due to the increased cancer risk for the proband and his or her relatives, a careful follow-up remains advisable from a clinical perspective [54,55].
3. Prognostic Value of MSI in CRC
3.1. Lower Metastatic Potential and Better Survival of MSI CRC
MSI is undoubtedly a positive prognostic factor in CRC patients, which is promptly explained by the low prevalence of MSI tumors among metastatic CRCs, corresponding to 2–4% of stage IV cases [4,25], as compared to their prevalence in earlier stages [56,57]. MSI CRCs typically present a dense immune cell infiltration, particularly rich in TILs, which has been associated with a better prognosis and a reduced tendency to metastasize [8]. Substantial evidence supports that MSI is a strong prognostic marker in early-stage CRCs with a favorable impact on survival, beyond the TNM staging system also from pooled retrospective analyses [58]. With respect to stage II CRC patients, in the ACCENT database analysis, the MSI profile significantly improved the disease-free survival and the overall survival [59].
Compared to stage II, the prognostic value of MSI in stage III CRC is less defined, and contradictory data have emerged from randomized clinical trials (RCTs) and meta-analysis [60,61,62] (see below).
Summarizing the available data, MSI confers a favorable prognosis in stage II CRC, and this effect seems to be progressively reduced with advancing stage (i.e., stage III) [60,61,62]. A speculative explanation of this phenomenon lies in the evasion of immune surveillance that is possibly acquired in more advanced stages of the disease. In accordance with the above statement, in stage IV CRCs, MSI no longer provides an advantage in terms of prognosis [63,64], though interactions with chemotherapy, as the standard adjuvant treatment for stage III CRC, should not be disregarded despite being difficult to disentangle.
3.2. Adaptive Immune Response and the Relevance of Immune Parameters
MSI-CRCs attract a dense lymphocytic infiltrate [21,22], parallelly driving the infiltration of specific subsets of immune cells (i.e., cytotoxic and helper T-lymphocytes) that are associated with an improved prognosis and reduced recurrence rates after surgery [63], especially in patients with early, node-negative CRC, largely contributing to the prognostic advantage of high densities of infiltrating lymphocytes [65].
Among the immune subpopulations recruited by MSI-CRC, dendritic cells and T cells activate the immune antitumoral response, which is downstream accomplished by activated memory CD4 + T cells, NK cells, M1 macrophages, and neutrophils [66]. The attempt to measure the immune infiltrate in the primary tumor and to assess its prognostic value has been pursued by trying to build a reliable “immuno-score” that quantifies the amount of infiltrating T-lymphocytes and allows inferences on CRC outcomes [67]. The immuno-score has been suggested to be superior to the conventional TNM classification in CRC, given its ability to differentiate patients with a better or worse prognosis in MSS and MSI disease, as across the various stages according to AJCC/UICC [68]. The measure of CD8+ cells and CD45RO+ memory cells in specific tumor regions (i.e., at the invasive front) has been, in fact, linked to longer overall survival in MSI-CRC patients [69]. This parameter is likely to be included in the TNM staging, similarly to its use for MSI, although some refinement is necessary in order to better define its reliability in stage III disease [36,44].
4. Predictive Value of MSI
4.1. Implication for the Adjuvant Treatment: Stage 2 vs. Stage 3
In stage II CRC, MSI has been endorsed as a reliable predictive indicator associated with a lack of benefit from adjuvant chemotherapy (5-fluorouracil-based (5FU)). This clinical endorsement first moved from the better prognosis and lower metastatic potential of MSI CRCs [56,57].
The initial report on non-responsiveness came from a study by Ribic et al. in which patients with MSI CRC were found to have a better overall 5-year survival, especially when not receiving adjuvant chemotherapy [70]. Subsequently, Sargent, in a collaborative study, confirmed this finding by showing that MSI interacted significantly with chemotherapy and that there was no improvement in patients with stage II MSI CRC who had received 5-FU [71]. Sinicrope et al. shortly after confirmed that patients with MSI CRC have lower rates of tumor recurrence, delayed time to relapse, and improved survival rates, with respect to MSS CRC patients [72]. Adjuvant treatment also reduced the rate of distant recurrences in patients with stage III CRC, which could be significant in patients with germline pathogenic variants compared to those with sporadic tumors [73].
A milestone in modern oncology was placed in the phase III Quick and Simple and Reliable (QUASAR) trial that randomized more than 2000 patients affected by stage II CRC to either receive adjuvant chemotherapy with 5-FU or for observation [74]. The study showed a significantly reduced risk of recurrence for MMR-deficient CRC (risk ratio, 0.53, 95% C.I., 0.40–0.70; p < 0.001) as compared to proficient ones, and the subanalysis for MMR status demonstrated no benefit from adjuvant chemotherapy [74]. This evidence has been confirmed by several meta-analyses that established MSI status as a predictive factor for both therapy response and relapse rates as concerns in stage II CRC [75,76,77]. Overall, data support MSI as the leading molecular marker with clinical value in early-stage CRC; no further molecular stigma has been incorporated in the management algorithms of CRC yet.
The situation in stage III appears more complex. In a study on patients included in a randomized trial on adjuvant 5-FU plus Oxaliplatin and folinic acid (FOLFOX) after resection of stage III CRC, Sinicrope et al. found that KRAS and BRAF mutations had a negative prognostic effect on disease-free survival, while MSI was not prognostic in all patients but significantly interacted with the tumor site and nodal status [78]. Accordingly, only patients with right-sided MSI CRC had a better outcome, and such an advantage was lost in those with N2 tumors [78].
In an interesting study assessing the value of lymphocyte infiltration in patients included in the PETACC8 phase III study [79], the authors found that MSI was not a predictive factor for overall survival in treated patients [79]. However, a larger study adding patients from the NCCTG N0147 trial [80] found that patients with MMR-deficient CRCs had significantly longer disease-free survival than those with proficient tumors at multivariate analyses (HR, 0.73; 95% CI, 0.54–0.97; p = 0.03), although such advantage may become evident only after 18 months at Kaplan–Meier survival curves. One issue involves the benefit of oxaliplatin added to 5-flurouracil [80]. Interestingly, it had been shown earlier that in an MSH2-deficient mouse model developing CRC, FOLFOX treatment led to a reduction in tumor volume, and MMR status was found not to modify responsiveness to oxaliplatin in previous studies [81,82].
Other studies further clarified that KRAS and BRAF mutations act as negative prognostic factors in MSS CRC patients treated with adjuvant FOLFOX, but not in MSI patients [83].
4.2. Removing the Breaks from the Immune Response: Immunotherapy
In the last decade, translational research in oncology has been focusing on the molecular mechanisms driving the interaction between MSI CRC and the immune system. The MSI status influences the tumoral microenvironment and the interactions with the immune system through multiple aspects, therefore impacting the efficacy of immunotherapy. A defective MMR leads to a high tumor mutational burden (TMB) [11,19,20], which means that tumoral cells profusely generate highly immunogenic soluble and surface neoantigens able to attract cytotoxic and helper T-lymphocytes [22,84]. The higher somatic mutational load that increases the presentation of neoepitopes has been epitomized as one of the mediators of the observed augmented response to immunotherapy as well in MSI tumors [85,86]. The immunogenicity of these neoantigens, structurally frame-shifted peptides, lies in their ability to bind with major histocompatibility complex class I (MHC-I) alleles [87]. Moreover, the neo-antigen load was directly associated with the T-cell memory tumoral infiltration [87].
Secondly, as demonstrated by Llosa et al., neoplastic cells with MMR defect overexpress several immune checkpoint proteins (e.g., PD-1, PD-L1, CTLA-4, LAG-3, and IDO), compared to MSS cancers [88].
These findings, together with evidence stemming from clinical trials, initially led immune checkpoint inhibitors (i.e., anti-PD1) to be approved by the regulatory authorities exclusively according to the MSI status, regardless of cancer type [8].
Recent studies investigating anti-programmed death-1 (PD-1) checkpoint inhibitors have identified and demonstrated MS status as a biomarker predictive of therapy response [89,90]. MMR-deficient cancers are now acknowledged to be sensitive to anti-PD1 (nivolumab, pembrolizumab) with or without anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibodies [89,90].
4.3. Silencing Map Kinases in Sporadic MSI
In the current landscape, it has become clear that BRAF-mutant CRC represents a distinct biologic entity, typically refractory to the traditional chemotherapy regimens [91]. BRAF is a serine/threonine kinase that acts downstream of KRAS in the mitogen-activated protein kinase (MAPK) cellular signaling pathway. BRAF-mutant CRC commonly exhibits a valine to glutamic-acid variation, specifically at codon 600 (V600E; or 1799T>A). The effect of this change is a constitutively activated protein. The BRAF V600E mutation overlaps with sporadic MSI-CRC in up to 33% of the cases [92].
Historically, BRAF-mutated CRCs have been associated with a significantly worse prognosis [93]. The therapeutic implications of targeting this mutation came as a lesson from the management of BRAF-mutated melanomas, and currently, several ongoing clinical trials are investigating the efficacy of BRAF-inhibitors (i.e., dabrafenib, vemurafenib, or encorafenib) alone or in combination in patients with metastatic BRAF (V600E)-mutated CRC [94].
In terms of personalized medicine, the inhibition of MAPK signaling in sporadic MSI-CRCs has been explored with promising results. In a pivotal, single-arm study that included 43 patients with BRAF-V600E metastatic CRC treated with the adjunct of a MEK inhibitor (Trametinib), the results showed improved response rates compared with BRAF inhibition alone [95]. A further phase II study, comparing dabrafenib, trametinib, and panitumumab triple therapy with double therapies (either dabrafenib plus panitumumab or trametinib plus panitumumab), assessed a disease control rate (response and stable disease together) in 86% of patients [96]. The median progression-free survival (PFS) and the duration of response were 4.2 and 7.6 months, respectively [96].
Based on these preliminary data, the combination therapies of BRAF/MEK have not been approved by the Food and Drug Administration (FDA) for the treatment of metastatic BRAF V600E CRC yet. Lastly, clinical trials examining immunotherapy in combination with inhibitors of the MAPK pathway are expected.
5. Discussion and Concluding Remarks
This review illustrates the current evidence on the prognostic and predictive value of MSI as a trail maker of the personalized medicine approach to CRC. Compared to MSS CRC, MSI status is associated with a more favorable prognosis in early-stage CRCs [58]. Furthermore, based on the evidence that adjuvant chemotherapy does not add any advantage for the prognosis in stage II, knowledge of MSI status drives clinical decisions for these patients [59]. Conversely, the prognostic value of MSI with respect to stage III disease appears attenuated, and these patients are, so far, recommended to receive standard adjuvant chemotherapy.
Regarding the predictive value of MSI status, it has been extensively demonstrated to be a robust biomarker for a good response to immune checkpoint inhibitors in patients with metastatic disease [89,90]. However, the precise role of immunotherapy in earlier-stage CRCs needs to be clarified by ongoing randomized studies. The studies on the molecular heterogeneity and tumoral microenvironment surrounding MSI tumors have led to an increased understanding of possible innovative therapeutic targets.
Figure 1 summarizes the timeline of the gradual achievement of a progressively wider clinical usefulness of MSI status in the field of CRC.
Figure 1.
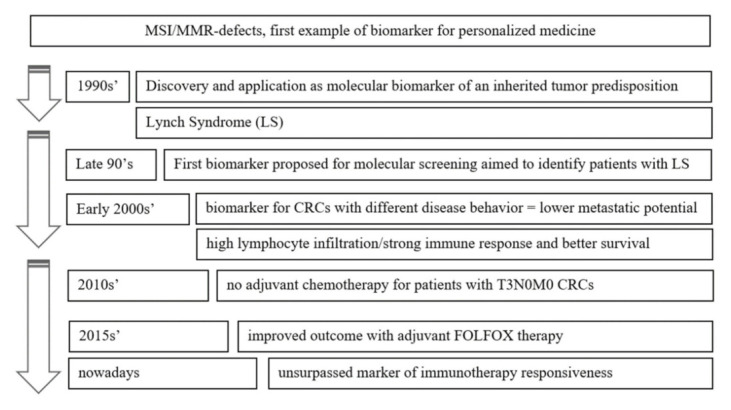
MMR story: lessons from a long-lasting biomarker. Timeline of its gradual achievement of wider clinical usefulness.
Finally, research has recently been focusing on the relationship between gut microbiota and CRC tumorigenesis, with a particular interest in the induced molecular profile, such as MSI. What is emerging is that, among the different microbiological species, Fusobacterium nucleatum is linked to the development of MSI tumors [97,98]. Indeed, tumors with high levels of Fusobacterium nucleatum tend to occur in the proximal colon and have a higher incidence of MSI with rather poor survival, as reported in a prospective cohort study [98]. This seems somehow counterintuitive, and it has been associated with the capability of Fusobacterium nucleatum to suppress the adaptive immune response in MSI-CRCs [99].
In the foreseeable future, gut bacterial modulation or a fecal microbiota transplant could stimulate the immune response in patients with MSI-CRCs that have developed a secondary resistance to immunotherapy. Thus, the modulation of the microbiota and increased antigen presentation appear to be two possible therapeutic targets for new and personalized strategies aimed, for example, at restoring a competent immune response and immunotherapy efficacy in MSI tumors. As we have gleaned much more than we would have expected from the MSI tumor subtype, we should be confident there is yet more to learn.
T3N0M0 CRCs, or stage IIA, invade through the muscolaris propria into the subserosa but have not reached nearby organs and lymph nodes and have not spread to distant organs [100]. FOLFOX, comprising of 5-FU, Oxaliplatin, and Folinic acid, is administered after surgery as adjuvant treatment.
Abbreviations
| CRC | colorectal carcinoma |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| LS | Lynch syndrome |
| TILs | tumor-infiltrating lymphocytes |
Author Contributions
Conceptualization, L.L.; methodology, A.D.B., F.G.; writing—original draft preparation, A.D.B., F.G.; writing—review and editing, L.P. and C.C.; supervision, L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Biller L.H., Schrag D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA. 2021;325:669–685. doi: 10.1001/jama.2021.0106. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Goding Sauer A., Fedewa S.A., Butterly L.F., Anderson J.C., Cercek A., Smith R.A., Jemal A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020;70:145–164. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 3.National Cancer Institute Surveillance, Epidemiology, and End Results Program Cancer Stat Facts: Colorectal Cancer. [(accessed on 9 September 2021)]; Available online: https://seer.cancer.gov/statfacts/html/colorect.html.
- 4.Boland C.R., Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–2087. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward R., Meagher A., Tomlinson I., O’Connor T., Norrie M., Wu R., Hawkins N. Microsatellite instability and the clinico-pathological features of sporadic colorectal cancer. Gut. 2001;48:821–882. doi: 10.1136/gut.48.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taieb J., Shi Q., Pederson L., Alberts S., Wolmark N., Van Cutsem E., de Gramont A., Kerr R., Grothey A., Lonardi S., et al. Prognosis of microsatellite instability and/or mismatch repair deficiency stage III colon cancer patients after disease recurrence following adjuvant treatment: Results of an ACCENT pooled analysis of seven studies. Ann. Oncol. 2019;30:1466–1471. doi: 10.1093/annonc/mdz208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Z., Sinicrope F.A. Prognostic and Predictive Values of Mismatch Repair Deficiency in Non-Metastatic Colorectal Cancer. Cancers. 2021;13:300. doi: 10.3390/cancers13020300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganesh K., Stadler Z.K., Cercek A., Mendelsohn R.B., Shia J., Segal N.H., Diaz L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019;16:361–375. doi: 10.1038/s41575-019-0126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Messersmith W.A. NCCN Guidelines Updates: Management of Metastatic Colorectal Cancer. J. Natl. Compr. Canc. Net. 2019;17:599–601. doi: 10.6004/jnccn.2019.5014. [DOI] [PubMed] [Google Scholar]
- 10.Peinado M.A., Malkhosyan S., Velazquez A., Perucho M. Isolation and characterization of allelic losses and gains in colorectal tumors by arbitrarily primed polymerase chain reaction. Proc. Natl. Acad. Sci. USA. 1992;89:10065–10069. doi: 10.1073/pnas.89.21.10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ionov Y., Peinado M.A., Malkhosyan S., Shibata D., Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 12.Thibodeau S.N., Bren G., Schaid D. Microsatellite Instability in Cancer of the Proximal Colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 13.Aaltonen L.A., Peltomäki P., Leach F.S., Sistonen P., Pylkkänen L., Mecklin J.-P., Järvinen H., Powell S.M., Jen J., Hamilton S.R., et al. Clues to the Pathogenesis of Familial Colorectal Cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 14.Peltomäki P., Aaltonen L.A., Sistonen P., Pylkkänen L., Mecklin J.-P., Järvinen H., Green J.S., Jass J.R., Weber J.L., Leach F.S., et al. Genetic Mapping of a Locus Predisposing to Human Colorectal Cancer. Science. 1993;260:810–812. doi: 10.1126/science.8484120. [DOI] [PubMed] [Google Scholar]
- 15.Maddox J. Competition and the death of science. Nature. 1993;363:667. doi: 10.1038/363667a0. [DOI] [PubMed] [Google Scholar]
- 16.Fishel R., Lescoe M.K., Rao M.R., Copeland N.G., Jenkins N.A., Garber J., Kane M., Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 17.Leach F.S., Nicolaides N.C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomäki P., Sistonen P., Aaltonen L., Nyström-Lahti M., et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-S. [DOI] [PubMed] [Google Scholar]
- 18.Kinzler K.W., Vogelstein B. Lessons from Hereditary Colorectal Cancer. Cell. 1996;87:159–170. doi: 10.1016/S0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 19.Malkhosyan S., Rampino N., Yamamoto H., Perucho M. Frameshift mutator mutations. Nature. 1996;382:499–500. doi: 10.1038/382499a0. [DOI] [PubMed] [Google Scholar]
- 20.Perucho M. Microsatellite instability: The mutator that mutates the other mutator. Nat. Med. 1996;2:630–631. doi: 10.1038/nm0696-630. [DOI] [PubMed] [Google Scholar]
- 21.Kim H., Jen J., Vogelstein B., Hamilton S.R. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am. J. Pathol. 1994;145:148–156. [PMC free article] [PubMed] [Google Scholar]
- 22.Guidoboni M., Gafà R., Viel A., Doglioni C., Russo A., Santini A., Del Tin L., Macrì E., Lanza G., Boiocchi M., et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am. J. Pathol. 2001;159:297–304. doi: 10.1016/S0002-9440(10)61695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aaltonen L.A., Salovaara R., Kristo P., Canzian F., Hemminki A., Peltomäki P., Chadwick R.B., Kääriäinen H., Eskelinen M., Järvinen H., et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N. Engl. J. Med. 1998;338:1481–1487. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 24.De’ Angelis G.L., Bottarelli L., Azzoni C., De’Angelis N., Leandro G., Di Mario F., Gaiani F., Negri F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018;89:97–101. doi: 10.23750/abm.v89i9-S.7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peltomäki P. Update on Lynch syndrome genomics. Fam. Cancer. 2016;15:385–393. doi: 10.1007/s10689-016-9882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tutlewska K., Lubinski J., Kurzawski G. Germline deletions in the EPCAM gene as a cause of Lynch syndrome—Literature review. Hered. Cancer Clin. Pr. 2013;11:9. doi: 10.1186/1897-4287-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 28.Boland C.R., Thibodeau S.N., Hamilton S.R., Sidransky D., Eshleman J.R., Burt R.W., Meltzer S.J., Rodriguez-Bigas M.A., Fodde R., Ranzani G.N., et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 29.Umar A., Boland C.R., Terdiman J.P., Syngal S., Chapelle A.D.L., Rüschoff J., Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasen H.F., Mecklin J.P., Khan P.M., Lynch H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis. Colon Rectum. 1991;34:424–425. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 31.Vasen H.F., Watson P., Mecklin J.P., Lynch H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/S0016-5085(99)70510-X. [DOI] [PubMed] [Google Scholar]
- 32.Jass J.R. HNPCC and sporadic MSI-H colorectal cancer: A review of the morphological similarities and differences. Fam Cancer. 2004;3:93–100. doi: 10.1023/B:FAME.0000039849.86008.b7. [DOI] [PubMed] [Google Scholar]
- 33.Laiho P., Launonen V., Lahermo P., Esteller M., Guo M., Herman J.G., Mecklin J.P., Järvinen H., Sistonen P., Kim K.M., et al. Low-level microsatellite instability in most colorectal carcinomas. Cancer Res. 2002;62:1166–1170. [PubMed] [Google Scholar]
- 34.Tomlinson I., Halford S., Aaltonen L., Hawkins N., Ward R. Does MSI-low exist? J. Pathol. 2002;197:6–13. doi: 10.1002/path.1071. [DOI] [PubMed] [Google Scholar]
- 35.Hampel H., Frankel W.L., Martin E., Arnold M., Khanduja K., Kuebler P., Nakagawa H., Sotamaa K., Prior T.W., Westman J., et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N. Engl. J. Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 36.Laghi L., Negri F., Gaiani F., Cavalleri T., Grizzi F., De’ Angelis G.L., Malesci A. Prognostic and Predictive Cross-Roads of Microsatellite Instability and Immune Response to Colon Cancer. Int. J. Mol. Sci. 2020;21:9680. doi: 10.3390/ijms21249680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domingo E., Laiho P., Ollikainen M., Pinto M., Wang L., French A.J., Westra J., Frebourg T., Espín E., Armengol M., et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J. Med. Genet. 2004;41:664–668. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kane M.F., Loda M., Gaida G.M., Lipman J., Mishra R., Goldman H., Jessup J.M., Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 39.Cunningham J.M., Christensen E.R., Tester D.J., Kim C.Y., Roche P.C., Burgart L.J., Thibodeau S.N. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–3460. [PubMed] [Google Scholar]
- 40.Herman J.G., Umar A., Polyak K., Graff J.R., Ahuja N., Issa J.P., Markowitz S., Willson J.K., Hamilton S.R., Kinzler K.W., et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gallois C., Laurent-Puig P., Taieb J. Methylator phenotype in colorectal cancer: A prognostic factor or not? Crit. Rev. Oncol. Hematol. 2016;99:74–80. doi: 10.1016/j.critrevonc.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Toyota M., Ahuja N., Ohe-Toyota M., Herman J.G., Baylin S.B., Issa J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogino S., Kawasaki T., Kirkner G.J., Kraft P., Loda M., Fuchs C.S. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J. Mol. Diagn. 2007;9:305–314. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samowitz W.S., Albertsen H., Herrick J., Levin T.R., Sweeney C., Murtaugh M.A., Wolff R.K., Slattery M.L. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 45.Gaiani F., Marchesi F., Negri F., Greco L., Malesci A., de’Angelis G.L., Laghi L. Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes. Int. J. Mol. Sci. 2021;22:5246. doi: 10.3390/ijms22105246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kambara T., Simms L.A., Whitehall V.L., Spring K.J., Wynter C.V., Walsh M.D., Barker M.A., Arnold S., McGivern A., Matsubara N., et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53:1137–1144. doi: 10.1136/gut.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nosho K., Irahara N., Shima K., Kure S., Kirkner G.J., Schernhammer E.S., Hazra A., Hunter D.J., Quackenbush J., Spiegelman D., et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS ONE. 2008;3:e3698. doi: 10.1371/journal.pone.0003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ref Merlo A., Herman J.G., Mao L., Lee D.J., Gabrielson E., Burger P.C., Baylin S.B., Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 49.Warthin A.S. Heredity with reference to carcinoma as shown by the study of the cases examined in the Pathological Laboratory of the University of Michigan, 1895−1912. Arch. Int. Med. 1913;12:546–555. doi: 10.1001/archinte.1913.00070050063006. [DOI] [Google Scholar]
- 50.Boland C.R., Lynch H.T. The history of Lynch syndrome. Fam. Cancer. 2013;12:145–157. doi: 10.1007/s10689-013-9637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lynch H.T., Kimberling W., Albano W.A., Lynch J.F., Biscone K., Schuelke G.S., Sandberg A.A., Lipkin M., Deschner E.E., Mikol Y.B., et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). I. Clinical description of resource. Cancer. 1985;56:934–938. doi: 10.1002/1097-0142(19850815)56:4<934::AID-CNCR2820560439>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 52.Carethers J.M., Stoffel E.M. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J. Gastroenterol. 2015;21:9253–9261. doi: 10.3748/wjg.v21.i31.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pico M.D., Castillejo A., Murcia O., Giner-Calabuig M., Alustiza M., Sanchez A., Moreira L., Pellise M., Castells A., Carrillo-Palau M., et al. Clinical and Pathological Characterization of Lynch-Like Syndrome. Clin. Gastroenterol. Hepatol. 2020;18:368–374. doi: 10.1016/j.cgh.2019.06.012. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez-Soler M., Perez-Carbonell L., Guarinos C., Zapater P., Castillejo A., Barbera V.M., Juarez M., Bessa X., Xicola R.M., Clofent J., et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013;144:926.e921–932.e921. doi: 10.1053/j.gastro.2013.01.044. quiz e913–e924. [DOI] [PubMed] [Google Scholar]
- 55.Mensenkamp A.R., Vogelaar I.P., van Zelst-Stams W.A., Goossens M., Ouchene H., Hendriks-Cornelissen S.J., Kwint M.P., Hoogerbrugge N., Nagtegaal I.D., Ligtenberg M.J. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146:643.e648–646.e648. doi: 10.1053/j.gastro.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 56.Sankila R., Aaltonen L.A., Järvinen H.J., Mecklin J.P. Better survival rates in patients with MLH1-associated hereditary colorectal cancer. Gastroenterology. 1996;110:682–687. doi: 10.1053/gast.1996.v110.pm8608876. [DOI] [PubMed] [Google Scholar]
- 57.Gryfe R., Gallinger S. Microsatellite instability, mismatch repair deficiency, and colorectal cancer. Surgery. 2001;130:17–20. doi: 10.1067/msy.2001.112738. [DOI] [PubMed] [Google Scholar]
- 58.Dienstmann R., Mason M.J., Sinicrope F.A., Phipps A.I., Tejpar S., Nesbakken A., Danielsen S.A., Sveen A., Buchanan D.D., Clendenning M., et al. Prediction of overall survival in stage II and III colon cancer beyond TNM system: A retrospective, pooled biomarker study. Ann. Oncol. 2017;28:1023–1031. doi: 10.1093/annonc/mdx052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sargent D.J., Shi Q., Yothers G., Tejpar S., Bertagnolli M.M., Thibodeau S.N., Andre T., Labianca R., Gallinger S., Hamilton S.R., et al. Prognostic impact of deficient mismatch repair (dMMR) in 7,803 stage II/III colon cancer (CC) patients (pts): A pooled individual pt data analysis of 17 adjuvant trials in the ACCENT database. J. Clin. Oncol. 2014;32:3507. doi: 10.1200/jco.2014.32.15_suppl.3507. [DOI] [Google Scholar]
- 60.Klingbiel D., Saridaki Z., Roth A.D., Bosman F.T., Delorenzi M., Tejpar S. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: Results of the PETACC−3 trial. Ann. Oncol. 2015;26:126–132. doi: 10.1093/annonc/mdu499. [DOI] [PubMed] [Google Scholar]
- 61.Wang B., Li F., Zhou X., Ma Y., Fu W. Is microsatellite instability-high really a favorable prognostic factor for advanced colorectal cancer? A meta-analysis. World J. Surg. Oncol. 2019;17:169. doi: 10.1186/s12957-019-1706-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buckowitz A., Knaebel H.P., Benner A., Blaker H., Gebert J., Kienle P., von Doeberitz K.M., Kloor M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br. J. Cancer. 2005;92:1746–1753. doi: 10.1038/sj.bjc.6602534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Venderbosch S., Nagtegaal I.D., Maughan T.S., Smith C.G., Cheadle J.P., Fisher D., Kaplan R., Quirke P., Seymour M.T., Richman S.D., et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin. Cancer Res. 2014;20:5322–5330. doi: 10.1158/1078-0432.CCR-14-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laghi L., Bianchi P., Miranda E., Balladore E., Pacetti V., Grizzi F., Allavena P., Torri V., Repici A., Santoro A., et al. CD3+ cells at the invasive margin of deeply invading (pT3-T4) colorectal cancer and risk of post-surgical metastasis: A longitudinal study. Lancet Oncol. 2009;10:877–884. doi: 10.1016/S1470-2045(09)70186-X. [DOI] [PubMed] [Google Scholar]
- 65.Pagès F., Kirilovsky A., Mlecnik B., Asslaber M., Tosolini M., Bindea G., Galon J. In situ cytotoxic and memory T cells predict outcome in patients with early stage colorectal cancer. J. Clin. Oncol. 2009;27:5944–5951. doi: 10.1200/JCO.2008.19.6147. [DOI] [PubMed] [Google Scholar]
- 66.Lin A., Zhang J., Luo P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front Immunol. 2020;11:2039. doi: 10.3389/fimmu.2020.02039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galon J., Mlecnik B., Bindea G., Angell H.K., Berger A., Lagorce C., Pagès F. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirta E.V., Seppälä T., Friman M., Väyrynen J., Ahtiainen M., Kautiainen H., Kuopio T., Kellokumpu I., Mecklin J.P., Böhm J. Immunoscore in mismatch repair-proficient and -deficient colon cancer. J. Pathol. Clin. Res. 2017;3:203–213. doi: 10.1002/cjp2.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Noepel-Duennebacke S., Juette H., Schulmann K., Graeven U., Porschen R., Stoehlmacher J., Hegewisch-Becker S., Raulf A., Arnold D., Reinacher-Schick A., et al. Microsatellite instability (MSI-H) is associated with a high immunoscore but not with PD-L1 expression or increased survival in patients (pts.) with metastatic colorectal cancer (mCRC) treated with oxaliplatin (ox) and fluoropyrimidine (FP) with and without bevacizumab (bev): A pooled analysis of the AIO KRK 0207 and RO91 trials. J. Cancer Res. Clin. Oncol. 2021;147:3063–3072. doi: 10.1007/s00432-021-03559-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ribic C.M., Sargent D.J., Moore M.J., Thibodeau S.N., French A.J., Goldberg R.M., Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sargent D.J., Marsoni S., Monges G., Thibodeau S.N., Labianca R., Hamilton S.R., French A.J., Kabat B., Foster N.R., Torri V., et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sinicrope F.A., Foster N.R., Thibodeau S.N., Marsoni S., Monges G., Labianca R., Kim G.P., Yothers G., Allegra C., Moore M.J., et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J. Natl. Cancer Inst. 2011;103:863–875. doi: 10.1093/jnci/djr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hutchins G., Southward K., Handley K., Magill L., Beaumont C., Stahlschmidt J., Richman S., Chambers P., Seymour M., Kerr D., et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J. Clin. Oncol. 2011;29:1261–1270. doi: 10.1200/JCO.2010.30.1366. [DOI] [PubMed] [Google Scholar]
- 74.Petrelli F., Ghidini M., Cabiddu M., Pezzica E., Corti D., Turati L., Costanzo A., Varricchio A., Ghidini A., Barni S., et al. Microsatellite Instability and Survival in Stage II Colorectal Cancer: A Systematic Review and Meta-analysis. Anticancer Res. 2019;39:6431–6441. doi: 10.21873/anticanres.13857. [DOI] [PubMed] [Google Scholar]
- 75.Fomiti A., Rulli E., Pilozzi E., Gerardi C., Roberto M., Legramandi L., Falcone R., Pacchetti I., Marchetti P., Floriani I. Exploring the prognostic role of microsatellite instability in patients with stage II colorectal cancer: A systematic review and meta-analysis. Clin. Colorectal Cancer. 2017;16:e55–e59. doi: 10.1016/j.clcc.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 76.Laghi L., Malesci A. Microsatellite instability and therapeutic consequences in colorectal cancer. Dig. Dis. 2012;30:304–309. doi: 10.1159/000337003. [DOI] [PubMed] [Google Scholar]
- 77.Koopman M., Kortman G.A., Mekenkamp L., Ligtenberg M.J., Hoogerbrugge N., Antonini N.F., Punt C.J., van Krieken J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer. 2009;100:266–273. doi: 10.1038/sj.bjc.6604867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sinicrope F.A., Mahoney M.R., Smyrk T.C., Thibodeau S.N., Warren R.S., Bertagnolli M.M., Nelson G.D., Goldberg R.M., Sargent D.J., Alberts S.R. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J. Clin. Oncol. 2013;31:3664–3672. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Emile J.F., Julié C., Le Malicot K., Lepage C., Tabernero J., Mini E., Budnik T.M. Prospective validation of a lymphocyte infiltration prognostic test in stage III colon cancer patients treated with adjuvant FOLFOX. Eur. J. Cancer. 2017;82:16–24. doi: 10.1016/j.ejca.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 80.Zaanan A., Shi Q., Taieb J., Alberts S.R., Meyers J.P., Smyrk T.C., Julie C., Zawadi A., Tabernero J., Mini E., et al. Role of Deficient DNA Mismatch Repair Status in Patients with Stage III Colon Cancer Treated with FOLFOX Adjuvant Chemotherapy: A Pooled Analysis From 2 Randomized Clinical Trials. JAMA Oncol. 2018;4:379–383. doi: 10.1001/jamaoncol.2017.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kucherlapati M.H., Lee K., Nguyen A.A., Clark A.B., Hou H., Jr., Rosulek A., Li H., Yang K., Fan K., Lipkin M., et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology. 2010;138:993.e1–1002.e1. doi: 10.1053/j.gastro.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gavin P.G., Colangelo L.H., Fumagalli D., Tanaka N., Remillard M.Y., Yothers G., Kim C., Taniyama Y., Kim S.I., Choi H.J., et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: An assessment of their prognostic and oxaliplatin predictive value. Clin. Cancer Res. 2012;18:6531–6541. doi: 10.1158/1078-0432.CCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taieb J., Le Malicot K., Shi Q., Penault-Llorca F., Bouché O., Tabernero J., Mini E., Goldberg R.M., Folprecht G., Luc Van Laethem J., et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J. Natl. Cancer Inst. 2016;109:272. doi: 10.1093/jnci/djw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S., et al. Cancer immunology. Mutational landscape determines sensitivity to PD−1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giannakis M., Mu X.J., Shukla S.A., Qian Z.R., Cohen O., Nishihara R., Garraway L.A. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15:857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Le D.T., Durham J.N., Smith K.N., Wang H., Bartlett B.R., Aulakh L.K., Diaz L.A. Mismatch repair deficiency predicts response of solid tumors to PD−1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roudko V., Bozkus C.C., Orfanelli T., McClain C.B., Carr C., O’Donnell T., Chakraborty L., Samstein R., Huang K.L., Blank S.V., et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell. 2020;183:1634–1649.e17. doi: 10.1016/j.cell.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Llosa N.J., Cruise M., Tam A., Wicks E.C., Hechenbleikner E.M., Taube J.M., Blosser R.L., Fan H., Wang H., Luber B.S., et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Diaz L.A., Jr. PD−1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Overman M.J., Lonardi S., Wong K.Y.M., Lenz H.J., Gelsomino F., Aglietta M., André T. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018;36:773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 91.Ursem C., Atreya C.E., Van Loon K. Emerging treatment options for BRAF-mutant colorectal cancer. Gastrointest. Cancer. 2018;8:13–23. doi: 10.2147/GICTT.S125940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tran B., Kopetz S., Tie J., Gibbs P., Jiang Z.Q., Lieu C.H., Desai J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hyman D.M., Puzanov I., Subbiah V., Faris J.E., Chau I., Blay J.Y., Baselga J. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Corcoran R.B., Atreya C.E., Falchook G.S., Kwak E.L., Ryan D.P., Bendell J.C., Hamid O., Messersmith W.A., Daud A., Kurzrock R., et al. Combined BRAF and MEK Inhibition with Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015;33:4023–4031. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corcoran R.B., André T., Atreya C.E., Schellens J.H.M., Yoshino T., Bendell J.C., Hollebecque A., McRee A.J., Siena S., Middleton G., et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018;8:428–443. doi: 10.1158/2159-8290.CD-17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tahara T., Yamamoto E., Suzuki H., Maruyama R., Chung W., Garriga J., Issa J.P.J. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014;74:1311–1318. doi: 10.1158/0008-5472.CAN-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mima K., Cao Y., Chan A.T., Qian Z.R., Nowak J.A., Masugi Y., Ogino S. Fusobacterium nucleatum in colorectal carcinoma tissue according to tumor location. Gut. 2016;65:1973–1980. doi: 10.1136/gutjnl-2015-310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hamada T., Zhang X., Mima K., Bullman S., Sukawa Y., Nowak J.A., Kosumi K., Masugi Y., Twombly T.S., Cao Y., et al. Fusobacterium nucleatum in Colorectal Cancer Relates to Immune Response Differentially by Tumor Microsatellite Instability Status. Cancer Immunol. Res. 2018;6:1327–1336. doi: 10.1158/2326-6066.CIR-18-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Compton C.C., Sullivan D.C., Byrd D.R., Greene F.L., Gershenwald J.E., Hess K.R., Gaspar L.E., Washington M.K., Schilsky R.L., Edge S. In: AJCC Cancer Staging Manual. 8th ed. Amin M.B., Edge S.B., Greene F.L., Byrd D.R., Brookland R.K., Washington M.K., Gershenwald J.E., Compton C.C., Hess K.R., Sullivan D.C., Jessup J.M., Brierley J.D., Gaspar L.E., Schilsky R.L., Balch C.M., Winchester D.P., Asare E.A., Madera M., Gress D.M., Meyer L.R., editors. Springer International Publishing; New York, NY, USA: 2017. [Google Scholar]