

Abstract
This study demonstrates the inhibitory effect of 42 pyrimidonic pharmaceuticals (PPs) on the 3-chymotrypsin-like protease of SARS-CoV-2 (3CLpro) through molecular docking, molecular dynamics simulations, and free binding energies by means of molecular mechanics–Poisson Boltzmann surface area (MM-PBSA) and molecular mechanics–generalized Born surface area (MM-GBSA). Of these tested PPs, 11 drugs approved by the US Food and Drug Administration showed an excellent binding affinity to the catalytic residues of 3CLpro of His41 and Cys145: uracil mustard, cytarabine, floxuridine, trifluridine, stavudine, lamivudine, zalcitabine, telbivudine, tipiracil, citicoline, and uridine triacetate. Their percentage of residues involved in binding at the active sites ranged from 56 to 100, and their binding affinities were in the range from −4.6 ± 0.14 to −7.0 ± 0.19 kcal/mol. The molecular dynamics as determined by a 200 ns simulation run of solvated docked complexes confirmed the stability of PP conformations that bound to the catalytic dyad and the active sites of 3CLpro. The free energy of binding also demonstrates the stability of the PP–3CLpro complexes. Citicoline and uridine triacetate showed free binding energies of −25.53 and −7.07 kcal/mol, respectively. Therefore, I recommend that they be repurposed for the fight against COVID-19, following proper experimental and clinical validation.
Keywords: coronavirus SARS-CoV-2, COVID-19, 3-chymotrypsin-like protease, pyrimidonic pharmaceuticals, molecular dynamics simulations, binding free energy
1. Introduction
Over a year has passed since the COVID-19 pandemic began. Some vaccines, such as those by Pfizer and Moderna, and some drugs, such as remdesivir, have been approved for use in therapy. The efforts by governments, health organizations, and other sectors to stem the alarmingly increasing numbers of deaths and cases were unprecedented [1,2,3,4,5,6]. However, SARS-CoV-2 continues to threaten the world, with over four million deaths and 227 million cases as of 16 September 2021 (https://www.worldometers.info/coronavirus/ accessed on 27 November 2021). COVID-19 was declared a pandemic by the World Health Organization on 11 March 2020. Today, the new SARS-CoV-2 virus, the causative agent of COVID-19, has been detected in almost every country on the planet [5,7,8,9].
Coronaviruses are positive-stranded RNA viruses with the largest viral genomes ever known, ranging from 16 to 32 kb. The 3-chymotrypsin-like protease (3CLpro) produced by SARS-CoV-2 is a cysteine protease encoded as nonstructural protein 3 in the polyprotein. 3CLpro is responsible for the cleavage of 11 specific sites of polyproteins (pp1a, pp1ab) produced by the 229E gene. These polyproteins are involved in the production of a functional polypeptide essential for viral replication and transcription. Further, the specificity of 3CLpro is dissimilar to that of human host-cell protease. Thus, 3CLpro has become the focus of drug repurposing and development programs to combat the COVID-19 pandemic [10,11,12,13].
Recent and ongoing research has reported that some pharmaceutical, synthetic, and natural products can act as 3CLpro inhibitors or against SARS-CoV-2 in general. These include selenium-containing heterocyclic compounds, chloroquine phosphate, indinavir, darunavir, lopinavir, eravacycline, naproxen, salix cortex, antioxidants, chiral phytochemicals from Opuntia ficus-indica, elbasvir, valrubicin, favipiravir isoflavone, and myricitrin [6,14,15,16,17,18,19,20,21,22]. Although some of these have entered human clinical trials or were even approved, more studies are still needed. The importance of the pyridone ring was highlighted in synthetic materials and drugs containing pyridone [11,23]. The pyrimidone ring has the exact shape of pyridine but is more functionalized and electron-deficient. Herein, we screened the inhibitory activity of 42 approved pyrimidonic pharmaceuticals (PPs) against 3CLpro using a combination of molecular docking analyses, molecular dynamics simulations, and calculations of the MM-PBSA and MM-GBSA binding free energies. The sites of action of active inhibitors were investigated, discussed, and explored.
2. Materials and Methods
2.1. The Pyrimidonic Pharmaceuticals (PPs)
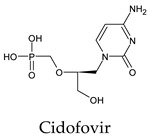
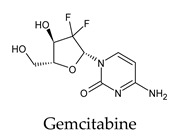
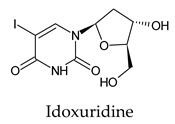
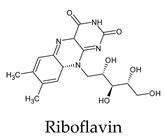
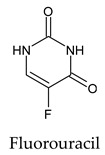
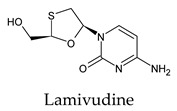
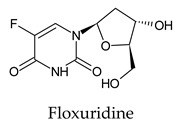
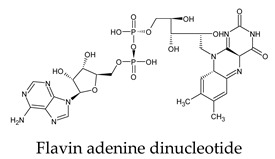
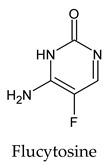
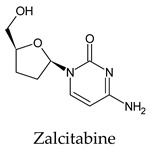
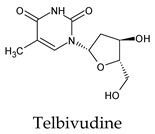
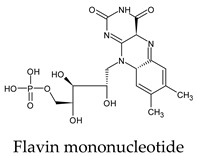
The PPs were selected using the search engine of the drug bank database. The search uncovered 46 PPs; the pharmaceuticals containing caffeine were entirely excluded as all except enprofylline have previously been studied. In addition, the macropolymeric drug mipomersen, drugs composed of a mixture of drugs, and withdrawn drugs were not included in this study. The chosen drugs were classified into four categories according to their structures. 1PPs have only one heterocycle, 2aPPs have two, 2bPPs have two heterocycles with a pyrimidone ring having an extra carbonyl group, and 3PPs have three or more heterocycles (Table 1).
Table 1.
Structures of pyrimidonic pharmaceuticals and their classification according to the number of rings.
| 1PPs | 2aPPs | 2bPPs | 3PPs |
|---|---|---|---|
|
|
|
|
|
|
|
|
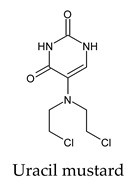
|
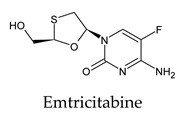
|
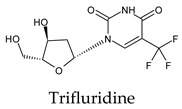
|
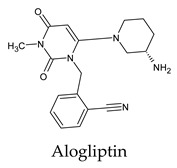
|
|
|
|
|
| - |
|
|
|
| - |
|
|
|
| - |
|
|
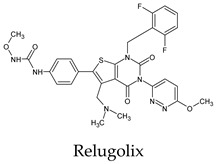
|
| - |
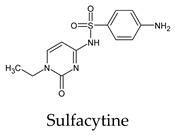
|
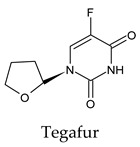
|
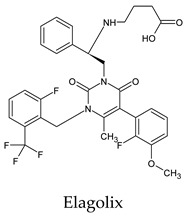
|
| - | - |
|
|
| - | - |
|
- |
| - | - |
|
- |
2.2. Generation and Energy Minimization of the PPs and 3CLpro
The 3D structures of the selected PPs were downloaded from the PubChem website as SDF files; their energy was minimized for 10,000 steepest descent steps at 5000 conjugate gradient steps using antechamber plugin UCSF Chimera [24,25]. For alogliptin, the 3D structure was obtained by utilizing OpenBabel converter tools and ChemSkech [26]. The crystal structure of SARS-CoV-2 3CLpro was obtained from the Protein Data Bank database website (PDB ID: 6Y2E). For analysis, water was removed from the 3CLpro structure, and the energy was then minimized for 1000 steepest descent steps at 20 conjugate gradient steps.
2.3. Molecular Docking
Blind molecular docking experiments were performed using the AutoDock Vina tool implemented with the interactive visualization and analysis program UCSF Chimera. The default parameter values were adopted with a grid box (−15 × −25 × 15) Å, centered at (35, 65, 65) Å. The predicted affinity values of the score were observed using the View Dock tool. The binding between ligands and 3CLpro active sites and the images were processed and visualized using UCSF Chimera [24,25,26,27,28].
2.4. Molecular Dynamics Simulations
MD simulations were performed as previously described [29]. The PP ligands were separated from the docked complexes using UCSF Chimera. The missed hydrogens were added and saved as PDB files using AMBER’s large-structure serial numbering. Topology files and parameters of the receptor and the ligands were made using leap and antechamber of Amber Tools 21 [30,30], utilizing Amber force fields of GAFF2 [31] and ff14SB [32] to assign inhibitors and 3CLpro structure, respectively. The systems were solvated with TIP3P water molecules [33] and were neutralized via sodium ions. Subsequently, molecular dynamics (MD) simulations were performed by means of the Nanoscale Molecular Dynamics (NAMD) Simulation 2.6 program [34]. Each system was minimized for 1 ps at 273.15 K using the NVE ensemble. The temperature was gradually increased to 310 K using the NVT ensemble in a protocol consisting of 1600 minimization steps. Then, each system was minimized for 10 ps at 310 K followed by 200 ns of MD simulation control using the NVT ensemble at 310 K and a time step of 2 fs. In order to calculate electrostatic interactions, the particle mesh Ewald process and periodic boundary conditions were applied [35,36]. The root-mean-square fluctuation (RMSF) and the root-mean-square deviation (RMSD) for each system were obtained by analyzing the trajectory using the VMD 1.8 program [37].
2.5. The Binding Free Energies
The binding free energies of the PP-3CLpro complexes were calculated by means of molecular mechanics–Poisson Boltzmann surface area (MM-PBSA) and molecular mechanics–generalized Born surface area (MM-GBSA) using the MMPBSA.py module of Amber Tools 21 [38]. The MD simulation over 200 ns provided several conformations sampled after equilibrium, using the last frames to lessen the computational cost. CPPTRAJ was used to obtain the snapshots [39]. The conformational changes were evaluated through quasi-harmonic entropy approximation [40]. The free energy of the binding interaction between inhibitors and 3CLpro complexes can be obtained via the following equations:
| ΔG = ΔH − TΔS | (1) |
| ΔH = ΔGgas + ΔGsol | (2) |
| ΔGgas = Evdw + Eelec | (3) |
| ΔGsol = Epb/gb + Enp | (4) |
where ΔH represents enthalpy change, TΔS represents the entropic contribution, Evdw represents the van der Waals interaction energy, Eele represents the electrostatic interaction energy, ΔGsol represents the polar solvation energy, and Enp represents the nonpolar solvation energy.
3. Results and Discussion
The results of the molecular docking are tabulated in Tables S1–S4. Figure 1 shows the catalytic dyad and the active sites of 3CLpro. The crucial residues HIS 41, GLY 143, SER 144, and CYS 145 forming the S1′ site are shown in black. Then, PHE 140, LUE 141, ASN 142, HIS 163, GLU 166 (magenta), and the N-terminal amino acid residues (blue) are involved in the formation of the S1 subsite of the substrate-binding pocket. The MET 49, TYR 54, HIS 164, ASP 187, and ARG R188 residues form the S2 site (green). MET 165, LEU 167, GLN 189, THR 190, and GLN 192 comprise the S4 site (cyan). The SER 284, ALA 285, and LEU 286 residues (yellow) are a result of genetic mutation leading to an increase in the SASR-CoV-2 3CLpro activity of 3.6 fold over that of the 3CLpro predecessor of SARS-CoV [12,41].
Figure 1.

The crystal structure of chymotrypsin-like protease of SARS-CoV-2 (PDB ID: 6Y2E) and its active residues. (a) Color indicates the residues involved in the formation of the S1 site (shown in magenta), S1 site from the other promotor (blue), S2 site (green), S4 site (cyan), and S1’ site (black), in addition to SER 284, ALA 285, and LEU 286 (yellow). (b) Only the catalytic dyad and GLU 166 residues.
3.1. Molecular Docking
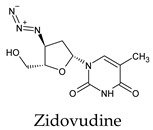
The docked complexes of the top 11 candidates are depicted in Figure 2. Their binding affinities to the active sites of 3CLpro are shown in Table 2. The 3PPs showed significant interactions with the residues LEU 286, SER 284, and ALA 285, and a relatively lower interaction ratio to the catalytic dyad, in contrast to the other groups. Of the 3PPs, alogliptin and flavin mononucleotide were found to have the highest binding percentage with the catalytic dyad and to form hydrogen bonds with the S1 and S’1 sites. These were followed by riboflavin and sofosbuvir with an advantage in binding to the LEU 286 residue (Table S1). Flavin adenine dinucleotide showed excellent binding affinity to LEU 286 but not with the catalytic dyad. Zidovudine and gemcitabine demonstrated similar activity to alogliptin and flavin mononucleotide (Tables S2 and S3).
Figure 2.

The PPs docked with 3CLpro, focusing on contacts with HIS 41, CYS 145, and GLU 166. (a) uracil mustard, (b) cytarabine, (c) floxuridine, (d) trifluridine, (e) stavudine, (f) lamivudine, (g) zalcitabine, (h) telbivudine, (i) tipiracil, (j) citicoline, (k) uridine triacetate. The hydrocarbon skeleton is shown in cyan, nitrogen atoms are blue, and oxygens are red. Hydrogen bonds are represented by blue lines; van der Waals forces are represented in yellow.
Table 2.
The binding affinities of the potential pyrimidonic pharmaceuticals with 3-chymotrypsin-like protease (3CLpro).
| Pharmaceutical Name | Binding Percentage a | Score ± SD (kcal/mol) b |
RMSD | Hydrogen Bond (Number of Bonds/Number of Conformations) | Van Der Waals (Distance) (Number of Bonds/Number of Conformations) |
|---|---|---|---|---|---|
| Uracil mustard | a. 100 * c. 89 d. 100 e. 100 All. 100 |
a. −4.6 ± 0.14 c. −4.6 ± 0.14 d. −4.6 ± 0.14 e. −4.6 ± 0.14 |
a. 0.00–7.13 c. 0.00–7.13 d. 0.00–7.13 e. 0.00–7.13 |
a. HIS 163 (5/5), GLU 166 (3/3), LEU 141 (3/3), ASN 142 (1/1) | a. HIS 163 (31/5), GLU 166 (40/8), LEU 141 (19/5), ASN 142 (50/7), PHE 140 (8/5) c. MET 49 (52/7), HIS 164 (12/4) d. GLN 189 (32/9), MET 165 (28/8) e. HIS 41 (53/9), SER 144 (10/5), GLY 143 (4/1), CYS 145 (28/9) |
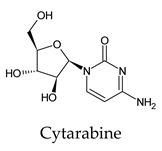
| Cytarabine | a. 67 c. 67 d. 67 e. 67 All. 67 |
a. −5.4 ± 0.26 c. −5.4 ± 0.26 d. −5.4 ± 0.26 e. −5.4 ± 0.26 |
a. 0.00–6.07 c. 0.00–6.07 d. 0.00–6.07 e. 0.00–6.07 |
a. GLU 166 (3/3), LEU 141 (2/2), HIS 163 (1/1), PHE 140 (2/2), ASN 142 (1/1) c. HIS 164 (2/2) d. GLN 189 (1/1) e. GLY 143 (1/1) |
a. GLU 166 (44/5), LEU 141 (18/3), HIS 163 (18/4), PHE 140 (19/3), ASN 142 (27/6) c. HIS 164 (10/4), MET 49 (20/6) d. MET 165 (28/4), GLN 189 (4/2) e. GLY 143 (11/2), SER 144 (8/2), CYS 145 (15/6), HIS 41 (21/5) |
| Floxuridine | a. 44 c. 44 d. 44 e. 44 All. 56 |
a. 5.5 ± 0.13 c. 5.6 ± 0.25 d. 5.6 ± 0.25 e. 5.6 ± 0.19 |
a. 3.24–8.28 c. 0.00–8.28 d. 0.00–8.28 e. 0.00–8.28 |
a. ASN 142 (1/1), HIS 163 (2/2), GLU 166 (2/2), PHE 140 (1/1), LEU 141 (1/1) e. HIS 41 (1/1) |
a. GLU 166 (23/4), LEU 141 (6/2), PHE 140 (8/2), HIS 163 (5/2), ASN 142 (19/4) c. MET 49 (9/2), HIS 164 (3/3) d. MET 165 (5/3), GLN 189 (4/1) e. GLY 143 (6/1), SER 144 (4/1), CYS 145 (5/3), HIS 41 (16/3) |
| Trifluridine | a. 44 c. 44 d. 44 e. 44 f. 11 All. 56 |
a. −6.03 ± 0.17 c. −6.03 ± 0.17 d. −6.03 ± 0.17 e. −6.03 ± 0.17 f. −5.7 |
a. 0.00–5.58 c. 0.00–5.58 d. 0.00–5.58 e. 0.00–5.58 f. 28.34–30.21 |
a. GLU 166 (2/2), ASN 142 (1/1). c. HIS 164 (2/2) e. GLY 143 (1/1) |
a. GLU 166 (18/4), ASN 142 (15/3), HIS 163 (2/1), LEU 141 (2/1) b. c. HIS 164 (10/3), MET 49 (17/4) d. MET 165 (19/4), GLN 189 (1/1). e. CYS 145 (13/4), GLY 143 (10/2), HIS 41 (10/4) f. SER 284 (9/1) |
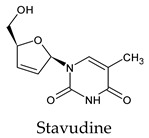
| Stavudine | a. 44 c. 56 d. 44 e. 56 All. 56 |
a. −5.6 ± 0.28 c. −5.6 ± 0.28 d. −5.6 ± 0.28 e. −5.6 ± 0.28 |
a. 27.12–32.34 c. 27.12–35.03 d. 27.12–32.34 e. 27.12–35.03 |
a. GLU 166 (1/1) | a. ASN 142 (13/3), GLU 166 (14/4), HIS 163 (2/1), LEU 141 (1/1) c. HIS 164 (6/3), MET 49 (20/5) d. MET 165 (12/4), GLN 189 (1/1) e. HIS 41 (39/5), GLY 143 (9/2), CYS 145 (7/3) |
| Lamivudine | a. 56 c. 44 d. 44 e. 56 f. 11 All. 67 |
a. −5.4 ± 0.24 c. −5.4 ± 0.28 d. −5.4 ± 0.25 e. −5.4 ± 0.24 f. −5.2 |
a. 0.00–4.77 c. 0.00–3.38 d. 0.00–4.77 e. 0.00–4.77 f. 26.28–28.62 |
a. HIS 163 (3/3), ASN 142 (1/1), PHE 140 (3/3), LEU 141 (2/2), GLU 166 (2/2) d. GLN 189 (1/1) e. SER 144 (2/2) |
a. HIS 163 (26/5), ASN 142 (19/2), PHE 140 (31/5), LEU 141 (15/5), GLU 166 (46/5) c. MET 49 (12/4), HIS 164 (1/1) d. GLN 189 (12/2), MET 165 (10/4), LEU 167 (1/1) e. SER 144 (18/4), HIS 41 (5/2), CYS 145 (7/3) f. LEU 286 (1/1) |
| Zalcitabine | a. 44 b. 11 c. 33 d. 44 e. 44 f. 22 All. 67 |
a. −5.5 ± 0.29 b. −5.1 c. −5.4 ± 0.35 d. −5.5 ± 0.29 e. −5.5 ± 0.29 f. −5.1 ± 0.00 |
a. 0.00–6.39 b. 28.02–29.48 c. 0.00–6.39 d. 0.00–6.39 e. 0.00–6.39 f. 28.02–31.88 |
a. PHE 140 (3/3), LEU 141 (1/1), GLU 166 (3/3), ASN 142 (1/1) c. GLN 189 (1/1) |
a. HIS 163 (15/3), PHE 140 (16/3), LEU 141 (9/3), GLU 166 (25/4), ASN 142 (11/3) b. LYS 5 (6/1), ARG 4 (12/1), PHE 3 (4/1) c. MET 49 (16/2), HIS 164 (5/3) d. GLN 189 (6/2), MET 165 (15/4) e. SER 144 (7/2), HIS 41 (13/2), CYS 145 (6/3) f. SER 284 (4/1), LEU 286 (4/1) |
| Telbivudine | a. 56 c. 44 d. 44 e. 56 f. 11 All. 67 |
a. −5.6 ± 0.38 c. −5.7 ± 0.42 d. −5.7 ± 0.42 e. −5.6 ± 0.38 f. −5.3 |
a. 0.00–7.50 c. 0.00–7.50 d. 0.00–7.50 e. 0.00–7.50 f. 22.85–24.17 |
a. ASN 142 (2/1), HIS 163 (3/3), GLU 166 (1/1), PHE 140 (2/2), LEU 141 (1/1) c. HIS 164 (1/1) d. GLN 189 (1/1) e. HIS 41 (1/1) |
a. GLU 166 (36/4), HIS 163 (11/3), PHE 140 (11/3), ASN 142 (27/5), LEU 141 (9/3) c. HIS 164 (6/3), MET 49 (27/3) d. GLN 189 (10/3), MET 165 (14/4), e. CYS 145 (11/4), SER 144 (8/2), GLY 143 (3/1), HIS 41 (12/5) f. LEU 286 (5/1) |
| Tipiracil | a. 56 b. 11 c. 44 d. 44 e. 44 f. 11 All. 67 |
a. −5.8 ± 0.16 b. −5.7 c. −5.9 ± 0.17 d. −5.8 ± 0.08 e. −5.9 ± 0.17 f. −5.7 |
a. 26.28–29.69 b. 19.12–20.08 c. 26.28–29.69 d. 26.67–29.69 e. 26.28–29.69 f. 19.12–20.08 |
a. HIS 163 (2/2), GLU 166 (2/1), PHE 140 (2/2), ASN 142 (1/1) b. LYS 5 (1/1) c. HIS 164 (1/1) e. GLY 143 (1/1) |
a. HIS 163 (14/3), GLU 166 (40/4), PHE 140 (8/3), ASN 142 (22/4), LEU 141 (10/3) b. PHE 3 (6/1), LYS 5 (9/1), ARG 4 (5/1) c. HIS 164 (8/4), MET 49 (22/3) d. MET 165 (9/3), GLN 189 (13/3) e. GLY 143 (7/2), HIS 41 (26/4), SER 144(2/1), CYS 145 (13/4) f. SER 284 (7/1), LEU 286 (2/1) |
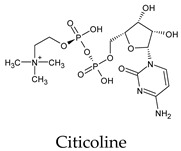
| Citicoline | a. 56 * c. 56 d. 56 e. 56 All. 56 |
a. −7.0 ± 0.19 c. −7.0 ± 0.19 d. −7.0 ± 0.19 e. −7.0 ± 0.19 |
a. 0.00–8.01 c. 0.00–8.01 d. 0.00–8.01 e. 0.00–8.01 |
a. PHE 140 (2/2), GLU 166 (4/4), HIS 163 (3/3), ASN 142 (2/1), LEU 141 (1/1) e. SER 144 (1/1) |
a. PHE 140 (24/5), GLU 166 (57/5), HIS 163 (21/5), ASN 142 (34/5), LEU 141 (21/5) c. MET 49 (23/5), HIS 164 (1/1) d. MET 165 (29/5), GLN 189 (6/3) e. SER 144 (12/3), GLY 143 (4/1), CYS 145 (9/5), HIS 41 (21/5) |
| Uridine triacetate | a. 56 c. 56 d. 56 e. 56 f. 11 All. 67 |
a. −6.2 ± 0.26 c. −6.2 ± 0.26 d. −6.2 ± 0.26 e. −6.2 ± 0.26 f. −6.4 |
a. 0.00–6.79 c. 0.00–6.79 d. 0.00–6.79 e. 0.00–6.79 f. 24.97–28.01 |
a. HIS 163 (2/2), GLU 166 (3/2) e. HIS 41 (1/1) |
a. HIS 163 (5/2), GLU 166 (31/5), ASN 142 (30/5), PHE 140 (4/2), LEU 141 (6/2) c. MET 49 (18/5), HIS 164 (5/2) d. GLN 189 (15/4), MET 165 (18/4) e. CYS 145 (16/5), HIS 41 (35/5), SER 144 (4/2), GLY 143 (2/1) f. LEU 286 (1/1) |
a Binding percentage was calculated based on the number of conformations attached to the active sites of the CLpro (nine conformations in total). b SD based on the other score energies of conformations. * Alphabetical order indicates the type of active site involved in bonding: a. S1 site, b. S1 site from the other promotor, c. S2 site, d. S4 site, e. S’1 site, and f. SER 284, ALA 285, and LEU 286 residues. When letters are missing, this means no interactions were observed at that site.
Among the 2bPPs, anti-hepatitis B infection telbivudine, anti-orotic aciduria uridine triacetate, and anticancer tipiracil were found to have the highest binding to 3CLpro active sites, followed by antimetabolite floxuridine, anti-herpesvirus trifluridine, and anti-HIV stavudine. Here, it is worth noting the importance of the molecular structure, as this set differed from the previous one by its increased ability to bind to the 3CLpro catalytic dyad. The 2aPPs showed similar activity to that of the 2bPPs. Anticancer cytarabine, anti-glaucoma citicoline, and anti-HIV drugs lamivudine and zalcitabine showed promising inhibitory activity (Table S3).
Finally, but very importantly, of the 1PPs, the chemotherapy drug uracil mustard showed binding to the catalytic dyad with all of its simulated conformations, followed by anti-cytomegalovirus cidofovir (Table S4).
3.2. Molecular Dynamics Simulations
MD was performed on the hole complexes of the top 11 PPs candidates. Based on the conformer score energy from docking, the complex with the conformer with the lowest value and interacting with the 3CLpro active site was selected. The RMSDs were computed along the trajectories using the initial structure as a reference. Figure 3 shows that the binding of PPs significantly affected the equilibration states of 3CLpro, as the majority of the tested systems reached their equilibrium at around 100 ns. The PP-3CLpro complexes revealed relatively lower average values for the RMSDs, between 0.41 and 0.52 Å, throughout the simulation, clarifying their good behavior in forming stable complexes. Moreover, the fluctuations in the 3CLpro backbone residues were analyzed by means of the RMSF (Figure 4). The 3CLpro/PPs exhibited lower fluctuations, particularly at the active site. The fluctuations at the catalytic dyad and GLU 166 were minor, demonstrating the loss of flexibility at these regions upon binding to the PPs. Table 3 shows the superior stability of the PP-3CLpro complexes formed throughout the production runs; these results support the use of these PPs as 3CLpro inhibitors.
Figure 3.


The RMSD values of the simulated PP-3CLpro complexes throughout the 200 ns production runs.
Figure 4.

The RMSF values of the simulated PP-3CLpro complexes throughout the 200 ns production runs.
Table 3.
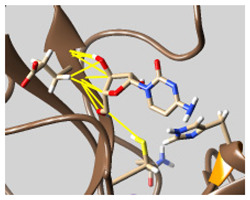
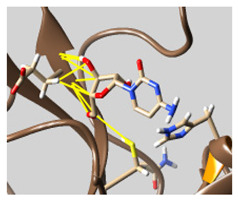
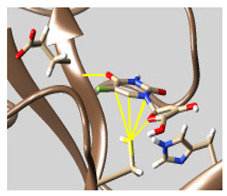
The binding interactions of the potential pyrimidonic pharmaceuticals/3-chymotrypsin-like protease 3CLpro complexes at different times throughout the production runs.
| Pharmaceutical Name | 100 ns | 150 ns | 200 ns |
|---|---|---|---|
| Uracil mustard |
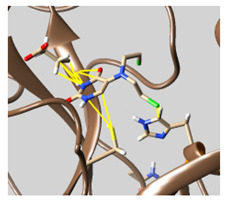
|
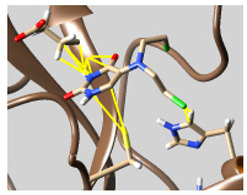
|
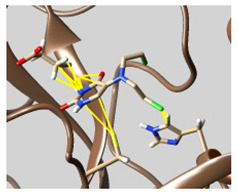
|
| Cytarabine |
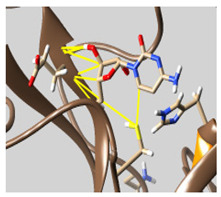
|
|
|
| Floxuridine |
|
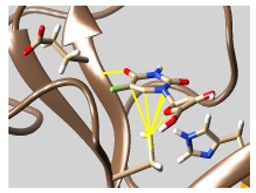
|
|
| Trifluridine |
|
|
|
| Stavudine |
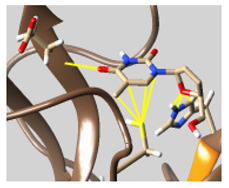
|
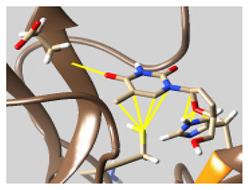
|
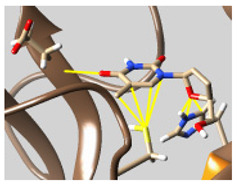
|
| Lamivudine |
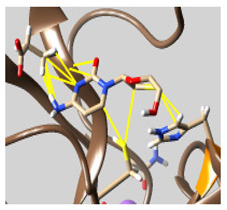
|
|
|
| Zalcitabine |
|
|
|
| Telbivudine |
|
|
|
| Tipiracil |
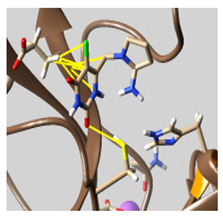
|
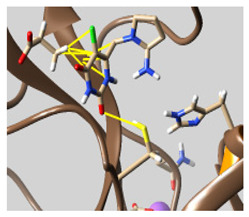
|
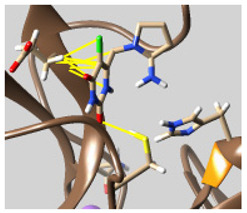
|
| Citicoline |
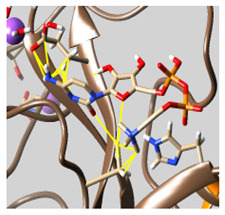
|
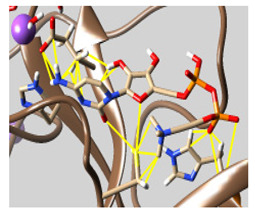
|
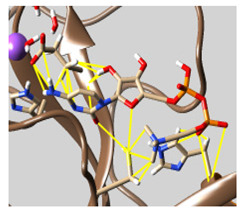
|
| Uridine triacetate |
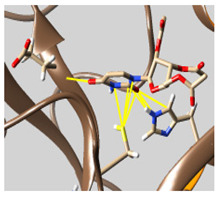
|
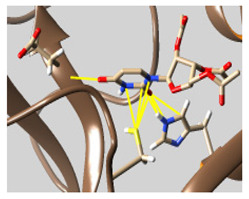
|
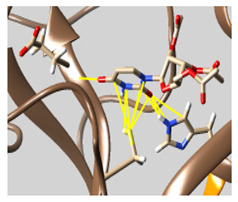
|
3.3. The Binding Free Energies
The data on the binding free energies of the PP-3CLpro complexes are tabulated in Table 4. In both MM-GBSA and MM-PBSA, van der Waals and electrostatic interactions acted as driving forces for the PP ligands to bind to 3CLpro, contrasting the solvation energies. The MM-GBSA and MM-PBSA results suggest that the PPs have an excellent ability to inhibit 3CLpro. Of these PPs, citicoline revealed the most promising inhibitory activity, followed by uridine triacetate (Table 4).
Table 4.
The MMPBSA and MMGBSA data for the binding of pyrimidone containing-pharmaceuticals to 3CLPro of SARS-CoV-2.
| 3CLpro Complex Type | −TΔS | Evdw | MMGBSA | MMPBSA | ||||
|---|---|---|---|---|---|---|---|---|
| Ealac | Esol | Δg (kcal/mol) | Ealac | Esol | Δg (kcal/mol) | |||
| Uracil mustard | 22.58 | −23.76 | −16.08 | 20.46 | 3.20 | −0.80 | 0.94 | −1.05 |
| Cytarabine | 22.39 | −19.25 | −33.50 | 28.39 | −1.96 | −1.67 | 1.09 | 2.55 |
| Floxuridine | 22.42 | −24.54 | 0.00 | 3.70 | 1.58 | 0.00 | 0.39 | −1.73 |
| Trifluridine | 22.96 | −29.48 | 0.00 | 4.20 | −2.32 | 0.00 | 0.41 | −6.11 |
| Stavudine | 22.25 | −24.41 | −17.67 | 17.90 | −1.94 | −0.88 | 0.93 | −2.12 |
| Lamivudine | 22.26 | −20.58 | −35.11 | 28.94 | −4.50 | −1.76 | 1.06 | 0.98 |
| Zalcitabine | 22.12 | −24.34 | −30.10 | 26.50 | −5.81 | −1.50 | 1.19 | 2.53 |
| Telbivudine | 22.48 | −25.29 | −46.88 | 42.53 | −7.16 | −2.34 | 1.61 | −3.54 |
| Tipiracil | 22.54 | −28.79 | 0.00 | 5.07 | −1.18 | 0.00 | 0.44 | −5.81 |
| Citicoline | 24.17 | −54.50 | 0.00 | 4.80 | −25.53 | 0.00 | 0.64 | −29.69 |
| Uridine triacetate | 23.60 | −32.98 | −31.09 | 33.40 | −7.07 | −1.55 | 1.43 | −9.51 |
Among the challenges of discovering 3CLpro inhibitors for COVID-19 treatment, these inhibitors must be highly bioavailable inside the cytosol [13]. The 2aPP and 2bPP structures contain a primidone heterocycle and ribose ring. These heterocycles increase their hydrophilicity and solubility in plasma. Thus, they can satisfy the requirement of bioavailability. Citicoline, the most promising inhibitor among the PPs investigated, has high hydrophilicity and good ADME properties [42,43]. The 2aPPs and 2bPPs also have an intermediate structure size among the PP groups. This sheds light on the importance of the size and general structural features of PPs acting as 3CLpro inhibitors.
Recent reports suggest a general hypothesis that 3CLpro inhibitors comprise electrophilic sites such as Michael acceptors [12]. That the pyrimidone ring is highly electron-deficient clarifies and confirms this hypothesis. The pyrimidone ring plays an essential role in PPs’ inhibitory activity and has a high tendency to form hydrogen bonds, particularly with the GLU 166 residue. This may preclude the formation of the S1 pocket. Contacts between the pyrimidone ring and HIS 41 were observed in floxuridine, stavudine, and telbivudine. In all these cases, HIS 41 interacts with the oxygen of the pyrimidone group. Further, the electrophilic carbon, nitrogen, and oxygen in the pyrimidone ring were attracted to bind with the sulfur of the CYS 145 residue.
Interestingly, most PPs investigated were previously studied against SARS-CoV-2. For example, flavin mononucleotide and flavin adenine dinucleotide have been suggested as good 3CLpro and RNA-dependent RNA polymerase inhibitors, respectively [22,44]. Riboflavin and sofosbuvir were shown to be suitable inhibitors of the spike protein S1 domain/ACE2 and RNA-dependent RNA polymerase [45,46,47]. Alogliptin was also suggested as a 3CLpro inhibitor; however, the enzymatic assay demonstrated its inactivity against 3CLpro [48]. Compelling clues have been found regarding the use of zidovudine and gemcitabine against spike protein/human ACE2 and in the inhibition SARS-CoV-2 in cell culture [49,50,51,52,53]. Gemcitabine and cidofovir were reported to inhibit SARS-CoV and SARS-CoV-2 proteins with IC50 values of 4.95 μM and 36 μM [54,55]. Further, telbivudine, tipiracil, cytarabine, and citicoline were recommended as 3CLpro inhibitors [56,57,58,59,60,61]. This transitory literature scanning confirms these pharmaceuticals’ activity against 3CLpro of SARS-CoV-2, as demonstrated in the present study.
To conclude, the inhibitory effect on 3CLpro by PPs was investigated based on their ability to form hydrogen bonds and van der Waals interactions with the 3CLpro active side through molecular docking, MD simulations, and the calculation of binding free energy. The overall analysis revealed 11 candidates from the initial set of 42 investigated PPs are promising 3CLpro inhibitors. These include citicoline and uridine triacetate as the best choices, followed by telbivudine, trifluridine, lamivudine, cytarabine, stavudine, zalcitabine, tipiracil, floxuridine, and flavin mononucleotide. The interactions of PPs with the catalytic dyad and the active sides of 3CLpro of SARS-CoV-2 were comprehensively and thoroughly investigated. The pyrimidone ring was found to play an essential role in the PPs’ inhibitory activity.
Supplementary Materials
The following are available online, Table S1: The binding affinities of the pyrimidonic pharmaceuticals (group 3PPs) with 3-chymotrypsin-like protease (3CLpro), Table S2: The binding affinities of the pyrimidonic pharmaceuticals (group 2bPPs) with 3-chymotrypsin-like protease (3CLpro), Table S3: The binding affinities of the pyrimidonic pharmaceuticals (group 2aPPs) with 3-chymotrypsin-like protease (3CLpro), Table S4: The binding affinity of the pyrimidone containing-pharmaceuticals (group 1PCPs) with 3-chymotrypsinlike protease (3CLpro).
Funding
This research and the APC were funded by the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, grant number [21-13-18-040].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data will be available upon request.
Conflicts of Interest
There is no conflict of interest to be reported.
Sample Availability
Not applicable.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ledford H., Cyranoski D., Van Noorden R. The UK has approved a COVID vaccine-here’s what scientists now want to know. Nature. 2020;588:205–206. doi: 10.1038/d41586-020-03441-8. [DOI] [PubMed] [Google Scholar]
- 2.Mahase E. Covid-19: UK approves Moderna vaccine to be given as two doses 28 days apart. BMJ. 2021;372 doi: 10.1136/bmj.n74. [DOI] [PubMed] [Google Scholar]
- 3.Mahase E. Covid-19: Moderna applies for US and EU approval as vaccine trial reports 94.1% efficacy. BMJ. 2020;371 doi: 10.1136/bmj.m4709. [DOI] [PubMed] [Google Scholar]
- 4.Lamb Y.N. Remdesivir: First approval. Drugs. 2020;80:1355–1363. doi: 10.1007/s40265-020-01378-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubin D., Chan-Tack K., Farley J., Sherwat A. FDA approval of remdesivir—A step in the right direction. N. Engl. J. Med. 2020;383:2598–2600. doi: 10.1056/NEJMp2032369. [DOI] [PubMed] [Google Scholar]
- 6.Rivero-Segura N.A., Gomez-Verjan J.C. In Silico Screening of Natural Products Isolated from Mexican Herbal Medicines against COVID-19. Biomolecules. 2021;11:216. doi: 10.3390/biom11020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization Coronavirus Disease 2019 (COVID-19): Situation Report, 94. 2020. [(accessed on 27 November 2021)]. Available online: https://apps.who.int/iris/handle/10665/331865.
- 8.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization COVID-19 Weekly Epidemiological Update. 2020. [(accessed on 2 December 2021)]. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
- 10.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020;30:127377. doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amin S.A., Banerjee S., Gayen S., Jha T. Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem. 2021:113294. doi: 10.1016/j.ejmech.2021.113294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steuten K., Kim H., Widen J.C., Babin B.M., Onguka O., Lovell S., Bolgi O., Cerikan B., Neufeldt C.J., Cortese M. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis. 2021 doi: 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck B.R., Shin B., Choi Y., Park S., Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020 doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qamar M.T.U., Alqahtani S.M., Alamri M.A., Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020;10:313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomez C.R., Espinoza I., Faruke F.S., Hasan M., Rahman K.M., Walker L.A., Muhammad I. Therapeutic intervention of COVID-19 by natural products: A population-specific survey directed approach. Molecules. 2021;26:1191. doi: 10.3390/molecules26041191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020;60:3277–3286. doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang Y.-C., Tung Y.-A., Lee K.-H., Chen T.-F., Hsiao Y.-C., Chang H.-C., Hsieh T.-T., Su C.-H., Wang S.-S., Yu J.-Y. Potential therapeutic agents for COVID-19 based on the analysis of protease and RNA polymerase docking. Preprints. 2020 doi: 10.20944/preprints202002.0242.v2. [DOI] [Google Scholar]
- 19.Terrier O., Dilly S., Pizzorno A., Chalupska D., Humpolickova J., Bouřa E., Berenbaum F., Quideau S., Lina B., Fève B. Antiviral Properties of the NSAID Drug Naproxen Targeting the Nucleoprotein of SARS-CoV-2 Coronavirus. Molecules. 2021;26:2593. doi: 10.3390/molecules26092593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le N.P.K., Herz C., Gomes J.V.D., Förster N., Antoniadou K., Mittermeier-Kleßinger V.K., Mewis I., Dawid C., Ulrichs C., Lamy E. Comparative Anti-Inflammatory Effects of Salix Cortex Extracts and Acetylsalicylic Acid in SARS-CoV-2 Peptide and LPS-Activated Human In Vitro Systems. Int. J. Mol. Sci. 2021;22:6766. doi: 10.3390/ijms22136766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhter S., Batool A.I., Selamoglu Z., Sevindik M., Eman R., Mustaqeem M., Akram M.S., Kanwal F., Lu C., Aslam M. Effectiveness of Natural Antioxidants against SARS-CoV-2? Insights from the In-Silico World. Antibiotics. 2021;10:1011. doi: 10.3390/antibiotics10081011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elzupir A.O. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020;1222:128878. doi: 10.1016/j.molstruc.2020.128878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 25.Wang J., Wang W., Kollman P.A., Case D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 26.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011;3:1–14. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shapovalov M.V., Dunbrack Jr R.L. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19:844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elzupir A.O. Caffeine and caffeine-containing pharmaceuticals as promising inhibitors for 3-chymotrypsin-like protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020:1–8. doi: 10.1080/07391102.2020.1835732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Da Silva A.W.S., Vranken W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes. 2012;5:1–8. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 32.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 34.Nelson M.T., Humphrey W., Gursoy A., Dalke A., Kalé L.V., Skeel R.D., Schulten K. NAMD: A parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. Comput. 1996;10:251–268. doi: 10.1177/109434209601000401. [DOI] [Google Scholar]
- 35.De Leeuw S.W., Perram J.W., Smith E.R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. Lond. A Math. Phys. Sci. 1980;373:27–56. [Google Scholar]
- 36.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 37.Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 38.Miller B.R., III, McGee T.D., Jr., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 39.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 40.Numata J., Wan M., Knapp E.-W. Conformational entropy of biomolecules: Beyond the quasi-harmonic approximation. Genome Inform. 2007;18:192–205. [PubMed] [Google Scholar]
- 41.Lim L., Shi J., Mu Y., Song J. Dynamically-driven enhancement of the catalytic machinery of the SARS 3C-like protease by the S284-T285-I286/A mutations on the extra domain. PLoS ONE. 2014;9:e101941. doi: 10.1371/journal.pone.0101941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saver J.L. Citicoline: Update on a promising and widely available agent for neuroprotection and neurorepair. Rev. Neurol. Dis. 2008;5:167–177. [PubMed] [Google Scholar]
- 43.Dinsdale J., Griffiths G., Rowlands C., Castelló J., Ortiz J., Maddock J., Aylward M. Pharmacokinetics of 14C CDP-choline. Arzneim. Forsch. 1983;33:1066–1070. [PubMed] [Google Scholar]
- 44.Anwar M.U., Adnan F., Abro A., Khan M.R., Rehman A.U., Osama M., Javed S., Baig A., Shabbir M.R., Assir M.Z. Combined Deep Learning and Molecular Docking Simulations Approach Identifies Potentially Effective FDA Approved Drugs for Repurposing Against SARS-CoV-2. Comput. Biol. Med. 2021 doi: 10.1016/j.compbiomed.2021.105049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prajapat M., Shekhar N., Sarma P., Avti P., Singh S., Kaur H., Bhattacharyya A., Kumar S., Sharma S., Prakash A. Virtual screening and molecular dynamics study of approved drugs as inhibitors of spike protein S1 domain and ACE2 interaction in SARS-CoV-2. J. Mol. Graph. Model. 2020;101:107716. doi: 10.1016/j.jmgm.2020.107716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elfiky A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020;253:117592. doi: 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chien M., Anderson T.K., Jockusch S., Tao C., Li X., Kumar S., Russo J.J., Kirchdoerfer R.N., Ju J. Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res. 2020;19:4690–4697. doi: 10.1021/acs.jproteome.0c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nar H., Schnapp G., Hucke O., Hardman T.C., Klein T. Action of dipeptidyl peptidase-4 inhibitors on SARS-CoV-2 main protease. ChemMedChem. 2021;16:1425–1426. doi: 10.1002/cmdc.202000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou L., Wang J., Liu G., Lu Q., Dong R., Tian G., Yang J., Peng L. Probing antiviral drugs against SARS-CoV-2 through virus-drug association prediction based on the KATZ method. Genomics. 2020;112:4427–4434. doi: 10.1016/j.ygeno.2020.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y.-N., Zhang Q.-Y., Li X.-D., Xiong J., Xiao S.-Q., Wang Z., Zhang Z.-R., Deng C.-L., Yang X.-L., Wei H.-P. Gemcitabine, lycorine and oxysophoridine inhibit novel coronavirus (SARS-CoV-2) in cell culture. Emerg. Microbes Infect. 2020;9:1170–1173. doi: 10.1080/22221751.2020.1772676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng Z., Groaz E., Snoeck R., De Jonghe S., Herdewijn P., Andrei G. Influence of 4′-Substitution on the Activity of Gemcitabine and Its ProTide against VZV and SARS-CoV-2. ACS Med. Chem. Lett. 2020;12:88–92. doi: 10.1021/acsmedchemlett.0c00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verma A.K., Aggarwal R. Repurposing potential of FDA-approved and investigational drugs for COVID-19 targeting SARS-CoV-2 spike and main protease and validation by machine learning algorithm. Chem. Biol. Drug Des. 2021;97:836–853. doi: 10.1111/cbdd.13812. [DOI] [PubMed] [Google Scholar]
- 53.Borbone N., Piccialli G., Roviello G.N., Oliviero G. Nucleoside analogs and nucleoside precursors as drugs in the fight against SARS-CoV-2 and other coronaviruses. Molecules. 2021;26:986. doi: 10.3390/molecules26040986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jo S., Kim S., Yoo J., Kim M.-S., Shin D.H. A Study of 3CLpros as Promising Targets against SARS-CoV and SARS-CoV-2. Microorganisms. 2021;9:756. doi: 10.3390/microorganisms9040756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pillaiyar T., Meenakshisundaram S., Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today. 2020;25:668–688. doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maurya S.K., Maurya A.K., Mishra N., Siddique H.R. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J. Recept. Signal Transduct. 2020;40:605–612. doi: 10.1080/10799893.2020.1772298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kandeel M., Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. doi: 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi R., Zhou M., Shek R., Wilson J.W., Tillery L., Craig J.K., Salukhe I.A., Hickson S.E., Kumar N., James R.M. High-throughput screening of the ReFRAME, Pandemic Box, and COVID Box drug repurposing libraries against SARS-CoV-2 nsp15 endoribonuclease to identify small-molecule inhibitors of viral activity. PLoS ONE. 2021;16:e0250019. doi: 10.1371/journal.pone.0250019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharanya C., Abhithaj J., Sadasivan C. Drug repurposing to identify therapeutics against COVID 19 with SARS-Cov-2 spike glycoprotein and main protease as targets: An in silico study. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12090408.v1. [DOI] [Google Scholar]
- 60.Fadlalla M. COVID19 Approved Drug Repurposing: Pocket Similarity Approach. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12722483.v1. [DOI] [Google Scholar]
- 61.Rahman N., Basharat Z., Yousuf M., Castaldo G., Rastrelli L., Khan H. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2) Molecules. 2020;25:2271. doi: 10.3390/molecules25102271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data will be available upon request.