

Abstract
EphB3 is a major key player in a variety of cellular activities, including cell migration, proliferation, and apoptosis. However, the exact role of EphB3 in cancer remains ambiguous. Accordingly, new EphB3 inhibitors can increase the understanding of the exact roles of the receptor and may act as promising therapeutic candidates. Herein, a hybrid approach of structure-based design and virtual combinatorial library generated 34 quinazoline sulfonamides as potential selective EphB3 inhibitors. A molecular docking study over EphB3 predicted the binding affinities of the generated library, and the top seven hit compounds (3a and 4a–f), with GlideScore ≥ −6.20 Kcal/mol, were chosen for further MM-GBSA calculations. Out of the seven top hits, compound 4c showed the highest MM-GBSA binding free energy (−74.13 Kcal/mol). To validate these predicted results, compounds 3a and 4a–f were synthesized and characterized using NMR, HRMS, and HPLC. The biological evaluation revealed compound 4c as a potent EphB3 inhibitory lead (IC50 = 1.04 µM). The screening of 4c over a mini-panel of kinases consisting of EGFR, Aurora A, Aurora B, CDK2/cyclin A, EphB1, EphB2, EphB4, ERBB2/HER2, and KDR/VEGFR2, showed a promising selective profile against EphB3 isoform. A dose-dependent assay of compound 4c and a molecular docking study over the different forms of EphB provided insights into the elicited biological activities and highlighted reasonable explanations of the selectivity.
Keywords: EphB3, kinase inhibitors, quinazoline sulfonamide, MM-GBSA calculations, selectivity profile, virtual combinatorial library
1. Introduction
Erythropoietin-producing hepatocellular carcinoma type-B receptor 3 (EphB3) belongs to the Eph receptor tyrosine kinase family, which is divided into classes A and B according to their sequence homology and ligand-binding affinity [1,2]. EphB3 and its ligands were found to be key players in a variety of cellular activities including cell migration, proliferation, and apoptosis. The EphB3 receptor subtype is expressed during embryonic development and in discrete areas of the adult brain, including the cerebellum and hippocampus. It co-localizes to brain regions with high levels of ephrin B ligand expression [3,4]. Several EphB3-related physiological and pathological processes are involved with the development and disease of many organ systems. Overexpression of EphB3 in non-small-cell lung cancer was reported to increase metastasis via increasing the tumor migratory capabilities, which in turn increases the tumor survival [5]. A recent study by Lee et al. reported EphB3 upregulation in gastric cancer with acquired resistance to AZD4547 (a selective inhibitor of a fibroblast growth factor receptor) [6]. In the colorectal cancer mice model, EphB3 expression was also found to be increased along with tumor development [7] and coamplified with PIK3CA in head and neck squamous cell carcinoma (HNSCC) [8]. In addition, Zhao et al. recently reported a potential function of EphB3 in carcinogenesis of ovarian serous carcinoma [9]. Li et al. also reported EphB3 as a tumor promoter in papillary thyroid cancer (PTC) via enhancing the in vitro migration and the in vivo metastasis of PTC cells through regulating the activities of Vav2 and Rho GTPases in a kinase-dependent manner [10]. Moreover, silencing of EphB3 significantly affected cycle progression distribution and increased apoptosis in CRC cell which revealed that knockdown of EphB3 inhibited tumor growth [11,12]. Despite these finding, the exact role of EphB3 in cancer remains controversial since some reports have documented conflicting roles of EphB3 in carcinogenesis [13,14,15,16,17]. Accordingly, EphB3 inhibitors can help to understand the exact roles of the receptor and may act as promising therapeutic candidates. Even though numerous Eph kinase inhibitors have been described in the literature (Figure 1) [18,19,20,21,22,23,24,25,26], almost all suffer from a lack of selectivity for the different Eph isoforms and/or other kinases due to the great conservation within the kinase domains. Unfortunately, the absence of a well-diversified library of isoform-selective inhibitors has hampered the pharmacological mapping of individual Eph kinase activities [27].
Figure 1.

Reported small molecules as Eph inhibitors [18,19,20,21,22,23,24,25,26].
The investigation of the exact role of EphB3 isoforms has been hampered by the unavailability of many identified selective EphB3 inhibitors. Searching the literature, only two studies successfully reported small molecules as selective EphB3 inhibitors (Figure 2) [15,28]. Accordingly, identification of new small molecules able to reversibly inhibit EphB3 would be a step toward the development of new selective inhibitory candidates. This in turn would further enhance the understanding of the exact functions of the receptor and offer potential therapeutic agents for several cancer disorders related to EphB3.
Figure 2.

Reported small molecules as selective EphB3 inhibitors.
Consequently, our aim in this study was to design and synthesize new small molecules as potential EphB3 selective inhibitors with a reversible mode of action. Thus, the quinazoline ring of compound II was used as a main scaffold of our target compounds since compound II showed a significant selectivity among Eph subtypes. The 2-chloroacetamide moiety in compound II, which is responsible for the covalent binding with Cys717 in EphB3 was replaced with several aromatic moieties aiming at development of alternative hydrophobic interactions (Van der Waals interactions) and/or hydrogen bond(s). Moreover, since the phenyl sulfonamide moiety was found in compound III (a potent EphB4 inhibitor [29], Figure 3) to interact with either backbone or hydrophilic residues in the binding pocket to improve the ligand-solubility properties, a structural hybridization-based strategy was applied via incorporating the phenyl sulfonamide moiety of compound III at position 4 of compound II (Figure 3). Herein, we report a new series of small molecules was designed, synthesized, and biologically evaluated as potential selective and reversible EphB3 inhibitors.
Figure 3.

Design of the target small molecules via applying a structural hybridization strategy of compounds II and III.
2. Results and Discussion
2.1. Validation of the Crystal Structures and Generation of a Virtual Library
2.1.1. Validation of the Protein Structure
The crystal structures of the EphB isoforms have not been frequently utilized in previous studies; therefore, validation of their crystal structure was necessary. The PROCHECK server was utilized to produce Ramachandran plots for the four downloaded crystal structures, displaying allowed and disallowed regions (https://servicesn.mbi.ucla.edu/PROCHECK/, accessed on 21 May 2021) [30]. More than 90% residues should be in the most favored regions for the crystal structure to act as a standard model of good quality and reliability. The protein structures of EphB1, 2, 3 and 4 utilized in this study had 90.3%, 88.0%, 88.8%, and 89.0% residues, respectively, in the most favored region and 8.4%, 11.1%, 10.8%, and 9.8% residues, respectively, in the additional allowed regions (Figure 4). Hence, the structures selected for conducting the molecular docking study were predicted to be reliable, as more than 99% of the residues were found to be in the allowed regions.
Figure 4.

Ramachandran plots of EphB1 (A), EphB2 (B), EphB3 (C) and EphB4 (D); residues present in red-colored region signify the residues present in most favored region, additional allowed residues are found in the dark yellow-colored region, additional allowed residues are found in the light-yellow area and the disallowed residues are represented on white background color.
2.1.2. Generation of Virtual Combinatorial Library and Compounds Selection
To speed up the identification process and to decrease the cost, an in silico virtual combinatorial library was enumerated. This library was based on the hybridization-based strategy of incorporating the benzenesulfonamide moiety of compound III at position 4 of compound II to generate various derivatives. Additionally, to avoid any irreversible inhibition via the interaction with Cys717, a C-C linker was chosen to avoid any flexibility in the “tail” moiety which would prevent any possible interaction with Cys717. A virtual library was constructed using all aromatic and cyclic boronic acid derivatives available in our laboratory chemicals resulting in a virtual library of 34 hit compounds (Figure S1). The constructed library was subjected to a molecular docking simulation, using Maestro Schrodinger software, as a structure-based approach to facilitate and support the selection of compounds for synthesis. The virtual library was docked into the active site of the EphB3 crystal structure (PDB: 5L6P). GlideScore was used to rank the compounds based on their affinity to the target protein. The more negative the GlideScore the higher the affinity of the docked ligand to the target protein [31]. The top seven hits (1–7, Table 1) generated from docking of the virtual combinatorial library were chosen for further in-depth molecular docking study, MM-GBSA calculations, chemical synthesis, and biological evaluation. Each selected hit was given a synthetic code as follows: 1 (4e), 2 (4c), 3 (4d), 4 (4a), 5 (4f), 6 (4b), and 7 (3a).
Table 1.
The hits generated from the virtual combinatorial library and their respective GlideScores.
| Hit Cpd | GlideScore (kcal/mol) | Hit Cpd | GlideScore (kcal/mol) |
|---|---|---|---|
| 1 | −7.30 | 18 | −5.78 |
| 2 | −7.16 | 19 | −5.63 |
| 3 | −6.65 | 20 | −5.50 |
| 4 | −6.47 | 21 | −5.49 |
| 5 | −6.23 | 22 | −5.47 |
| 6 | −6.21 | 23 | −5.46 |
| 7 | −6.20 | 24 | −5.35 |
| 8 | −6.18 | 25 | −5.35 |
| 9 | −6.13 | 26 | −5.32 |
| 10 | −6.12 | 27 | −5.31 |
| 11 | −6.11 | 28 | −5.16 |
| 12 | −6.10 | 29 | −5.09 |
| 13 | −6.07 | 30 | −4.80 |
| 14 | −6.06 | 31 | −4.59 |
| 15 | −5.90 | 32 | −4.49 |
| 16 | −5.84 | 33 | −4.24 |
| 17 | −5.84 | 34 | −4.22 |
2.2. Molecular Docking
Using the molecular docking to clarify the main protein-ligand interactions, the binding modes of the top seven generated hits from the combining virtual library was predicted in the EphB3 substrate-binding cavity. Docking of the active and selective co-crystallized inhibitor 6P8 (PDB: 5L6P [15]) demonstrated the ability of the modelling system to reproduce the reported crystal poses within 0.3 Å accuracy (data not shown). This, combined with the validation performed employing the PROCHECK server (Figure 4), offered assurance about the reliability of the chosen crystal structure in identifying potential EphB3 inhibitors.
Although each of the seven hits investigated by the molecular docking displayed a unique pattern of interaction with the active site of EphB3 active site, the sulfonamide and quinazoline moieties in all seven hit compounds formed an essential interaction, giving additional reassurance to the chosen design. Furthermore, all seven compounds interacted with Leu779, with the top two compounds (4e and 4c) forming a hydrogen bond with Leu779 via the oxygen of the sulfonamide group (Table 1). Conversely, compounds 3a, 4a, 4b, 4d and 4f formed π-Alkyl interactions with Leu779, implying that compounds 4e and 4c possess a higher affinity for EphB3. Compound 4e, GlideScore of −7.30 kcal/mol, was predicted to interact with EphB3 through four typical hydrogen bonds, one carbon-hydrogen bond and three π-Alkyl interaction (Table 2). Although, the predicted binding mode of compound 4c exhibited one less hydrogen bond when compared to compound 4e, compound 4c was able to interact with the active site residue through both; “head” and “tail” moieties indicating a better fit and predicting a higher biological activity. In summary, the predicted binding modes of compounds 3a and 4a–4f indicate their potential activity as EphB3 reversible inhibitors, marking them as good candidates for chemical synthesis and biological assay.
Table 2.
Molecular docking study results of the top seven hits generated from the virtual combinatorial library.
| Cpd | GlideScore (kcal/mol) |
2D Diagram | Amino Acids |
Binding Group | Molecular Interactions |
|---|---|---|---|---|---|
| 3a | −6.20 |
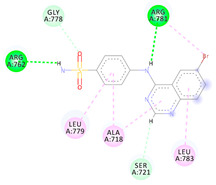
|
Arg762 Arg781 Ser721 Leu779 Ala718 Leu783 |
NH (SO2NH2) NH (amide) Benzene Benzene Benzene Benzene |
H-bond H-bond π-donor H-bond π-Alkyl π-Alkyl π-Alkyl |
| 4a | −6.47 |
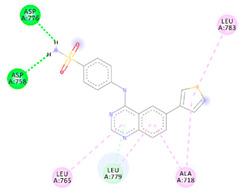
|
Asp776 Asp758 Leu779 Leu765 Ala718 Leu783 |
NH (SO2NH2) NH (SO2NH2) Benzene Benzene Benzene Benzene |
H-bond H-bond C-H-bond π-Alkyl π-Alkyl π-Alkyl |
| 4b | −6.21 |
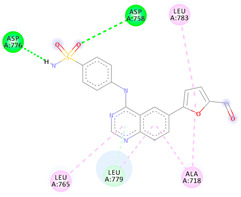
|
Asp776 Asp758 Leu779 Leu765 Ala718 Leu783 |
NH (SO2NH2) O (SO2NH2) Benzene Benzene Benzene Benzene |
H-bond H-bond π-donor H-bond π-Alkyl π-Alkyl π-Alkyl |
| 4c | −7.16 |
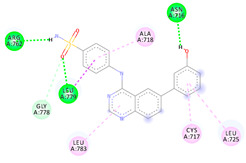
|
Arg762 Asn716 Leu779 Gly778 Ala718 Leu783 Cys717 Leu725 |
NH (SO2NH2) OH O (SO2NH2) O (SO2NH2) Benzene Benzene Benzene Benzene |
H-bond H-bond H-bond C-H-bond π-Alkyl π-Alkyl π-Alkyl π-Alkyl |
| 4d | −6.65 |
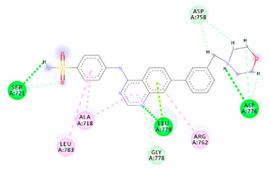
|
Ser721 Leu779 Asp776 Gly778 Asp758 Leu783 Arg762 |
NH (SO2NH2) H (morpholine) H (morpholine) N (quinazoline) H (morpholine) Benzene Benzene |
H-bond H-bond H-bond C-H-bond C-H-bond π-Alkyl π-Alkyl |
| 4e | −7.30 |
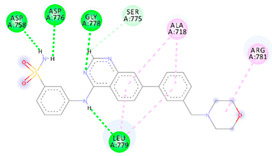
|
Asp758 Asp776 Gly778 Leu779 Ser775 Ala718 Arg781 |
NH (SO2NH2) NH (SO2NH2) N (quinazoline) NH (amide) H (quinazoline) Benzene Benzene |
H-bond H-bond H-bond H-bond C-H-bond π-Alkyl π-Alkyl |
| 4f | −6.23 |
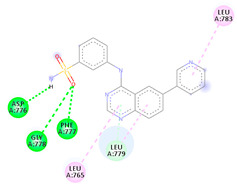
|
Asp776 Gly778 Phe777 Leu779 Leu765 Leu783 |
NH (SO2NH2) O (SO2NH2) O (SO2NH2) Benzene Benzene Benzene |
H-bond H-bond H-bond π-donor H-bond π-Alkyl π-Alkyl |
2.3. MM-GBSA Calculations
The free energy estimated using the MM-GBSA method is a useful tool to confirm the predicted potential affinity of a compound with its target. This is due to MM-GBSA calculations accounting for the effect of solvents on the complex’s stability. The lower the calculated binding free energy of a ligand protein complex, the more stable it is projected to be, as well as the higher the predicted activity and potency. As such, the MM-GBSA was calculated for compounds 3a and 4a–f as demonstrated in Table 3. Out of the top seven generated hits, compound 4c displayed the highest free energy of −74.13 kcal/mol indicating its high stability when complexed with EphB3. Compound 4e exhibited a slightly lowered free binding energy of −63.69 kcal/mol, while compounds 3a, 4a, 4b, 4d and 4f displayed moderate to poor free binding energy of −53.14, −52.09, −35.79, −57.47 and −40.08 kcal/mol, respectively. Therefore, compounds 4e and 4c were predicted to possess the highest stability and predicted biological activity.
Table 3.
MM-GBSA calculations for compounds 3a and 4a–f.
| Cpd | MM-GBSA (kcal/mol) |
|---|---|
| 3a | −53.14 |
| 4a | −52.09 |
| 4b | −35.79 |
| 4c | −74.13 |
| 4d | −57.47 |
| 4e | −63.69 |
| 4f | −40.08 |
2.4. Chemical Synthesis
The synthetic route used to obtain the desired target compounds 3a and 4a–f is outlined in Scheme 1. Refluxing of the commercially available 2-amino-5-bromobenzonitrile (1a) or 2-amino-4-bromobenzonitrile (1b) with dimethylformamide dimethyl acetal afforded compounds 2a–b. Reacting compounds 2a–b with the appropriate sulfonamide in glacial acetic acid under reflux resulted in the formation of compounds 3a–d. The intermediates 3a–d were then reacted with the appropriate boronic acid derivative via Suzuki cross-coupling using Bis(di-tert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II) as a coupling catalyst and the inorganic base, propylphosphonic anhydride, dissolved in dioxane and water at 80 °C under pressure in the presence of nitrogen to yield compounds 4a–f.
Scheme 1.

Reagents and conditions: (i) Dimethylformamide dimethyl acetal, 100 °C, 1.5 h; (ii) appropriately substituted aniline, glacial acetic acid, reflux, 2 h; (iii) appropriate boronic acid, Bis(di-tert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II), propylphosphonic anhydride, dioxane, 80 °C, 2 h.
Different spectroscopic techniques including NMR, HRMS and HPLC were utilized to elucidate the chemical structures and the purity of the synthesized target compounds. The 1H NMR spectra of compounds 3a–d are characterized by the appearance of two peaks at 7.20–7.50 ppm attributable to the sulfonamide NH2 and another peak at 10.10–10.30 ppm attributable to the NH of the amide group confirming the formation of compounds 3a–d from their parent compounds 2a–b. The success of the Suzuki cross-coupling reaction for compounds 4a–f was confirmed by the appearance of characteristic peaks in the 1H NMR and 13C NMR spectra of the synthesized compounds. In particular, the appearance of a peak at 9.66 ppm in compound 4b attributable to the carbonyl group or the appearance of a peak at 9.64 ppm in compound 4c attributable to the hydroxy group confirmed the formation of these products. Similarly, compounds 4d and 4e displayed three characteristic peaks in the range of 2.30–2.40 and 2.40–3.50 ppm, respectively, validating the presence of the aliphatic hydrogens.
2.5. Biological Studies
2.5.1. Assessment of the EphB3 Inhibitory Activity
The synthesized compounds were subjected to in vitro screening over EphB3 kinase. A single dose of 10 µM concentration of each target compound was used in EphB3 kinase inhibition assay, at 10 µM ATP, applying HotSpotSM technology. Among the synthesized compounds, compound 4c exhibited a potent EphB3 inhibitory activity of 93.27%. Consequently, it was selected for a further dose dependent assay to determine its IC50 value over EphB3 kinase. It exhibited a potent IC50 value of 1.04 µM (Figure 5). The results of the EphB3 inhibitory activity assay are demonstrated in Table 4.
Figure 5.

IC50 values of compound 4c (A) and staurosporine (B) over EphB3 kinase.
Table 4.
% Inhibition values of the synthesized compounds 3a and 4a–f over EphB3 kinase at 10 µM concentration.
| Cpd | Structure | Average % Enzyme Inhibition at 10 µM (Relative to DMSO Control) |
|---|---|---|
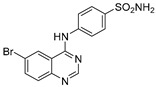
| 3a |
|
26.97 ± 0.24 |
| 4a |
|
25.81 ± 0.24 |
| 4b |
|
26.71 ± 0.45 |
| 4c |
|
93.27 ± 0.06 |
| 4d |
|
18.86 ± 1.09 |
| 4e |
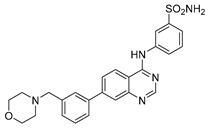
|
−21.35 ± 0.09 |
| 4f |
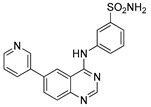
|
23.48 ± 5.36 |
2.5.2. Assessment of the EGFR Inhibitory Activity
Many small-molecule inhibitors of EGFR kinase, epidermal growth factor receptor, are based on a quinazoline core, indicating that quinazolines have a high affinity for EGFR [32,33,34,35]. Accordingly, compounds possessing a quinazoline skeleton may have a potential to interact with the EGFR. Therefore, the synthesized compounds were tested over EGFR kinase at 10 µM concentrations in the presence of 10 M ATP (Table 5). Only compounds 4c, 4d, and 4e displayed inhibition > 50%. Accordingly, the three compounds were selected for a further dose dependent assay to determine their IC50 values over EGFR. As shown in Figure 6, compounds 4c, 4d, and 4e exhibited a modest EGFR inhibitory with IC50 values of 8.04, 2.76 and 1.06 µM, respectively, compared to the standard staurosporine (IC50 = 0.084 µM).
Table 5.
% Inhibition values of the synthesized compounds 3a and 4a–f over EGFR kinase at a single dose concentration of 10 µM.
| Cpd | Average % Enzyme Inhibition (Relative to DMSO Controls) at 10 µM |
|---|---|
| 3a | −13.16 ± 4.31 |
| 4a | 0.56 ± 4.32 |
| 4b | 25.25 ± 1.25 |
| 4c | 46.78 ± 0.54 |
| 4d | 65.76 ± 0.98 |
| 4e | 63.28 ± 0.21 |
| 4f | 4.57 ± 2.16 |
| Staurosporine | - |
Figure 6.

The IC50 values of compounds 4c, 4e, 4d (A) and staurosporine over EGFR (B).
2.5.3. Selectivity Assay of Compound 4c over a Kinase Panel
To evaluate the selectivity and the kinase inhibition profile of the most potent EphB3 inhibitor of this new series (4c), an in vitro screening was carried out over a panel of cancer-related kinases and different isoforms of EphB kinase (Aurora A, Aurora B, CDK2/cyclin A, EphB1, EphB2, EphB4, ERBB2/HER2, and KDR/VEGFR2) in 10-dose IC50 mode with a 3-fold serial dilution starting at a concentration of 10 µM. The results of the selectivity study are demonstrated in Table 6. The results of the mini-kinase panel [36,37,38,39,40] indicate that among the four isoforms of EphB kinase, compound 4c is selective toward EphB3 with more than 93% inhibition, while it exhibited a moderate activity against the other three isoforms EphB1, EphB2 and EphB4 with average inhibition of 58.89, 76.32, and 70.15%, respectively. Similarly, compound 4c displayed poor to moderate inhibitory activity against the tested kinases with 67.86, 74.96, 67.75, 47.75, and 46.78% inhibition over Aurora A, CDK2, HER2, VEGFR2 and EGFR, respectively.
Table 6.
% Inhibition values of compound 4c over a panel of different kinases.
| Kinase | Average % Enzyme Inhibition (Relative to DMSO Controls) at 10 µM |
|---|---|
| Aurora A | 67.86 ± 4.93 |
| CDK2/cyclin A | 74.96 ± 2.07 |
| EphB1 | 58.89 ± 1.98 |
| EphB2 | 76.32 ± 0.61 |
| EphB3 | 93.27 ± 0.06 |
| EphB4 | 70.15 ± 3.64 |
| ERBB2/HER2 | 67.75 ± 0.99 |
| KDR/VEGFR2 | ±0.66 |
A further dose-dependent assay was conducted to validate the selectivity of compound 4c over the EphB isoforms. The assay assessed the IC50 values of 4c over the EphB isoforms 1, 2 and 4 to be 5.86, 3.95 and 3.98 µM, respectively (Table 7). These values were found to be higher than the standard staurosporine which showed IC50 values of 30, 80 and 220 nM over EphB isoforms 1, 2 and 4, respectively. However, compound 4c exhibited a 4-fold selectivity for EphB3 over EphB 2 and 4 isoforms and six-fold selectivity over the EphB1 isoform. Accordingly, it could be considered as a promising selective inhibitor for EphB3 isoform.
Table 7.
IC50 values of compound 4c over the EphB isoforms.
| Kinase | 4c IC50 (µM) |
Staurosporine IC50 (µM) |
|---|---|---|
| EphB1 | 5.86 | 0.03 |
| EphB2 | 3.95 | 0.08 |
| EphB3 | 1.04 | 1.20 |
| EphB4 | 3.98 | 0.22 |
2.6. Molecular Docking Study
2.6.1. Molecular Docking Study for Compound 4c over the EphB Isoforms
A molecular docking study was employed to elucidate the key protein-ligand interactions which may contribute to future structural modifications and SAR development. Docking of compound 4c over EphB four isoforms revealed that the selectivity for EphB3 was due to two factors (the number of interactions formed with the active site and the conformation of 4c inside the active site). The docking study demonstrated that compound 4c was in a good conformation inside the active site of both EphB3 and 4 crystal structures, with both the “head” and “tail” regions of the compound participating in favorable interactions. Conversely, when compound 4c was docked over EphB1 and 2 isoforms, only the sulfonamide of the “head” region was able to interact with the active site, leaving a large part of the compound outside the active site cavity, unable to form any favorable interactions (Figure 7). Compound 4c formed four hydrogen bonds with the EphB1 isoform, involving Asp762, Ser761, and Met700 amino acids. However, the poor fit of the compound inside the active site may have contributed to its modest inhibition (58.89%). Compound 4c was able to establish two hydrogen bonds with the EphB2 isoform. Conversely, compound 4c established four hydrogen bonds with the amino acids Arg762, Gly778, Leu779, and Asn716 (Figure 8) when docked over the EphB3 isoform, which combined with a good conformation inside the active site explains its potent inhibition. In summary, out of the four isoforms of EphB, compound 4c showed both; a good fit with the active site and several favorable interactions over the EphB3 isoform, which help to explain its potent IC50 value of 1.04 µM. On the contrary, the lack of a good fit or the presence of favorable interactions led to weaker binding profile over the EphB1, 2, and 4 isoforms.
Figure 7.

Docked complexes of compounds 4c with EphB1 and EphB2 isoforms: (A) 3D model of the crystal structure of EphB1 with the compound 4c; (B) 3D docking model of compound 4c into EphB1 binding pocket; (C) 2D interaction diagram of compound 4c with EphB1 active site; (D) 3D model of the crystal structure of EphB2 with compound 4c; (E) 3D docking model of compound 4c into EphB2 binding pocket; (F) 2D interaction diagram of compound 4c with EphB2 active site.
Figure 8.

Docked complexes of compounds 4c with EphB3 and EphB4 isoforms: (A) 3D model of the crystal structure of EphB3 with compound 4c; (B) 3D docking model of compound 4c into EphB3 binding pocket; (C) 2D interaction diagram of compound 4c with EphB3 active site; (D) 3D model of the crystal structure of EphB4 with compound 4c; (E) 3D docking model of compound 4c into EphB4 binding pocket; (F) 2D interaction diagram of compound 4c with EphB4 active site.
2.6.2. Molecular Docking Study for Compound 4c over the Tested Kinase Panel
A molecular docking study was carried out for compound 4c over the tested kinases to clarify the key protein-ligand interactions and get molecular insights for its modest biological activity over these kinases. Compound 4c established two hydrogen bonds with ALA220 and GLN 234 amino acid residues of the Aurora A binding site (Figure 9A). It was able to form three further weak interactions with the active site of Aurora A which explains its moderate inhibitory activity (67.86%). Similarly, it formed two hydrogen bonds with VEGFR2 and HER2 (Figure 9B and Figure 9C, respectively) with inhibitory activity of 47.94% and 67.75%, respectively. Conversely, it established three hydrogen bonds with CDK2 (Figure 9D), which may clarify the increase in its inhibitory activity against CDK2 (74.96%).
Figure 9.

The 2D interaction pattern of compound 4c in the binding sites of Aurora A (A), VEGFR2 (B), HER2 (C) and CDK2 (D). Favorable interactions are color coded as follows: green, hydrogen bonds; dark pink, π–π stacking interactions; purple, π–sigma interactions; light green, unconventional carbon–hydrogen bonds.
3. Materials and Methods
3.1. Protein Preparation
The crystal structures of EphB1 (PDB code: 6UMW, Resolution: 1.98 Å) [41], EphB2 (PDB code: 2HEN, Resolution: 2.60 Å) [42], EphB3 (PDB ID: 5L6P, Resolution: 2.26 Å) [15], EphB4 (PDB ID: 3ZEW, Resolution: 2.50 Å) [43], VEGFR2 (PDB code: 3VHE, Resolution: 1.55 Å) [44], Aurora A (PDB code: 2X6E, Resolution: 3.35 Å) [45], HER2 (PDB code: 3PP0, Resolution: 2.25 Å) [46] and CDK2 (PDB code3PY0, Resolution: 1.75 Å) [47] were downloaded from protein data bank (www.pdb.org, accessed on 18 April 2021). To ensure the validity of the downloaded crystal structures, PROCHECK server was employed to generate a Ramachandran plot for all four downloaded crystal structures to ensure their validity [30]. The preparation wizard of Schrodinger 2021 suite package was used to prepare the downloaded Protein structures at the default setting with pH = 7.4. The ligands were sketched employing ChemDraw Professional 17.0 (PerkinElmer, Inc., Waltham, MA, USA) and saved in the structure data file format which was then uploaded to the Ligprep module. Subsequently, the Ligprep module of Schrodinger was applied in the ligand preparation step to further optimizing the geometry. The generated docking models were validated by re-docking of the X-ray ligand, thus confirming the reproducibility of the results (data not shown). The minimized ligands were docked into the appropriate crystal structure binding site using Glide’s standard precision module to generate ten poses for the docked ligand. Discovery Studio visualizer Client 2021 package (Dassault Systèmes, Vélizy-Villacoublay, France) was used for the visualization and illustration of the generated ligand-protein complexes.
3.2. Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) Calculations
The binding free energy of the examined protein-ligand complexes was calculated using the MM-GBSA approach, which combined molecular mechanics (MM) force fields with a generalized Born and surface area continuum (implicit) solvation solvent model. OPLS-2005 force field, VSGB solvent model, and rotamer search algorithms were applied in the MM-GBSA computation [48]. The MM-GBSA was calculated using the prime module of the Schrodinger suite (Prime, Schrödinger, LLC, New York, NY, USA) according to the equation below:
| ΔG = E_complex (minimized) − [E_ligand (minimized) + E_receptor (minimized)] |
The MM-GBSA was calculated using the default configuration, which rendered all protein atoms rigid while allowing ligand atoms to be in a relaxed state.
3.3. Chemistry
The purchased solvents and reagents were used without further purification in the conducted experiments. Varian 400 MHz spectrometer (Varian Medical Systems, Inc., Palo Alto, CA, USA) was used to record the 1H NMR spectra with chemical shifts being recorded in parts per million (ppm) and coupling constants in Hz, as reported previously [49]. The high-resolution electrospray ionization mass spectrometry (HR-ESIMS) data were recorded on a JMS-700 mass spectrometer (Jeol, Japan) or by HR-ESIMS data recorded on a G2 QTOF mass spectrometer (Thermo Scientific™, Waltham, MA, USA). The reactions were monitored using TLC on 0.25 mm silica plates (E. Merck, Darmstadt, Germany; silica gel 60 F254). The purity of the products was determined by reversed-phase high performance liquid chromatography (RP-HPLC) utilizing Waters Corp. HPLC system equipped with a UV detector set at 254 nm (Milford, MA, USA). The mobile phases used were: (A) H2O containing 0.05% TFA and (B) CH3CN. HPLC employed a YMC Hydrosphere C18 (HS-302) column (5 μM particle size, 12 nm pore size) that was 4.6 mm in diameter × 150 mm in size, with a flow rate of 1.0 mL/min. The final compounds purity was assessed using either a gradient of 75% B or 100% B in 30 min. A Fisherbrand digital melting point apparatus was used to measure the melting points.
3.3.1. General Procedure for Synthesis of Compounds 2a–b
A mixture containing 1 mmol of 2-amino-5-bromobenzonitrile (1a) or 2-amino-4-bromobenzonitrile (1b) and dimethylformamide dimethyl acetal (10 mL) was stirred at reflux for 1.5 h. The mixture was cooled to room temperature and refrigerated. The solid was filtered, washed with several portions of ether, and dried to yield the desired compound which was used in the next step without further purification [50].
3.3.2. General Procedure for Synthesis of Compounds 3a–d
A mixture of (E)-N′-(5-bromo-2-cyanophenyl)-N,N-dimethylformimidamide (2a) or (E)-N′-(4-bromo-2-cyanophenyl)-N,N-dimethylformimidamide (2b) and the appropriate aniline derivative (1 eq.) in glacial acetic acid (15.0 mL) was refluxed for 2 h. The acetic acid was evaporated and the solid was washed with water and diethyl ether and dried to afford the titled compounds which were used in the next step without further purification [50].
4-((6-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3a)
Off-white solid, yield: 76%, mp: 322.2–324.5 °C, HPLC purity: 15 min, 99.45%, 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.88 (s, 1H), 8.69 (s, 1H), 8.09–8.03 (d, J = 8.4 Hz, 2H), 8.03–7.98 (d, J = 8.8 Hz, 1H), 7.85–7.79 (d, J = 8.4 Hz, 2H), 7.78–7.74 (d, J = 8.8 Hz, 1H), 7.27 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.10, 155.03, 149.04, 142.44, 139.05, 136.75, 130.57, 126.77, 125.91, 121.93, 119.58, 116.93. HRMS (ESI) m/z calculated for C14H11BrN4O2S: [M + H]+: 378.9859. Found: 378.9856.
3-((6-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3b)
Off-white solid, yield: 90%, mp: 325.0–326.4 °C, HPLC purity: 12 min, 95.11%, 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 8.92 (d, J = 2.0 Hz, 1H), 8.68 (s, 1H), 8.38 (s, 1H), 8.20 (m, 1H), 8.02 (dd, J = 8.0, 2.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.63–7.58 (m, 2H), 7.42 (s, 2H). HRMS (ESI) m/z calculated for C14H11BrN4O2S: [M + H]+: 378.9859. Found: 378.9850.
4-((7-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3c)
Off white solid, yield: 18%, mp: 320.3–322.1 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.68 (s, 1H), 8.56 (d, J = 9.2 Hz, 1H), 8.11–8.02 (m, 3H), 7.85 (m 3H), 7.29 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 158.15, 155.58, 150.68, 142.39, 139.29, 130.13, 129.90, 127.51, 126.76, 125.84, 122.21.
3-((7-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3d)
White solid, yield: 97%, mp: 318.6–320.5 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 8.66 (s, 1H), 8.56 (d, J = 8.8 Hz, 1H), 8.37 (s, 1H), 8.20–8.13 (m, 1H), 8.03 (d, J = 2.0 Hz, 1H), 7.86 (dd, J = 8.8, 2.0 Hz, 1H), 7.61 (t, J = 6.6 Hz, 2H), 7.42 (s, 2H). 13C NMR (100 MHz, DMSO-d6) 158.15, 155.82, 151.27, 144.89, 139.85, 130.07, 127.34, 125.74, 119.53, 114.51.
3.3.3. General Procedure for Synthesis of Compounds 4a–f
Compounds 3a–d were allowed to react with the appropriate boronic acid derivative via the Suzuki cross-coupling reaction in the presence of the coupling catalyst bis(di-tert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II) and an inorganic base propylphosphonic anhydride. The reaction mixture was dissolved in dioxane and water and then heated to 80 °C for 2 h under pressure in the presence of nitrogen to produce compounds 4a–f [51].
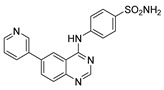
4-((6-(Pyridin-3-yl)quinazolin-4-yl)amino)benzenesulfonamide (4a)
Off-white solid, yield: 22%, mp: 315.1–316.2 °C, HPLC purity: 8 min, 97.84%, 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.13 (s, 1H), 8.93 (s, 1H), 8.71–8.59 (m, 2H), 8.27 (d, J = 8.0 Hz, 2H), 8.07 (d, J = 8.0 Hz, 2H), 7.92 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.61–7.53 (m, 1H), 7.29 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 158.10, 154.95, 149.93, 149.48, 149.38, 148.71, 148.49, 142.66, 139.09, 135.66, 134.97, 132.37, 129.33, 126.90, 126.68, 124.54, 122.17, 121.55, 115.89. HRMS (ESI) m/z calculated for C19H15N5O2S: [M + H]+: 378.1019. Found: 378.1012.
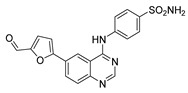
4-((6-(5-Formylfuran-2-yl)quinazolin-4-yl)amino)benzenesulfonamide (4b)
Off-white solid, yield: 22%, mp: 263.4–236.9 °C, HPLC purity: 13 min, 95.58%, 1H (400 MHz, DMSO-d6) δ10.36 (s, 1H), 9.66 (s, 1H), 9.03 (s, 1H), 8.67 (s, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 4.0 Hz, 1H), 7.44 (d, J = 4.0 Hz, 1H), 7.30 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 178.41, 158.26, 157.97, 155.49, 152.60, 150.76, 139.23, 130.37, 129.39, 127.06, 126.73, 122.45, 120.01, 115.97, 110.50, 40.36, 40.09, 39.95, 39.67. HRMS (ESI) m/z calculated for C19H14N4O4S: [M + H]+: 395.0809. Found: 395.0805.
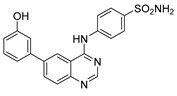
4-((6-(3-Hydroxyphenyl)quinazolin-4-yl)amino)benzenesulfonamide (4c)
Off-white solid, yield: 45%, mp: 313.2–315.1 °C, HPLC purity: 13 min, 95.10%, 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.64 (s, 1H), 8.80 (s, 1H), 8.65 (s, 1H), 8.13 (dd, J = 8.0, 1.6 Hz, 1H), 8.07 (d, J = 8.0 Hz, 2H), 7.85 (m, 3H), 7.29 (m, 5H), 6.84 (dd, J = 8.0, 1.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 158.37, 158.10, 154.58, 149.62, 142.75, 140.99, 139.01, 138.94, 132.49, 130.49, 128.89, 126.72, 122.07, 120.81, 118.46, 115.84, 115.37, 114.53. HRMS (ESI) m/z calculated for C20H16N4O3S: [M + H]+: 393.1016. Found: 393.1007.
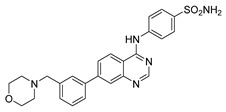
4-((7-(4-(Morpholinomethyl)phenyl)quinazolin-4-yl)amino)benzenesulfonamide (4d)
Off-white crystals, yield: 24%, mp: 228.1–229.8 °C, HPLC purity: 10 min, 96.63%, 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 8.72–8.66 (m, 2H), 8.13 (d, J = 8.8 Hz, 2H), 8.08 (s, 1H), 8.04 (d, J = 8 Hz, 1H), 7.86 (t, J = 8 Hz, 4H), 7.49 (d, J = 8 Hz, 2H), 7.29 (s, 2H) 3.61–3.56 (m, 4H), 3.58 (s, 1H), 3.55 (s, 1H), 2.38 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 157.87, 155.21, 155.18, 150.79, 144.89, 142.85, 138.96, 138.80, 137.68, 130.22, 130.03, 127.64, 127.49, 126.83, 125.87, 125.22, 125.08, 124.33, 121.83, 114.65, 66.67, 62.46, 53.66. HRMS (ESI) m/z calculated for C25H25N5O3S: [M + H]+: 476.1751. Found: 476.1745.
3-((7-(3-(Morpholinomethyl)phenyl)quinazolin-4-yl)amino)benzenesulfonamide (4e)
Off-white solid, yield: 32%, mp: 267.5–269.0 °C, HPLC purity: 10 min, 99.15%, 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 8.72–8.64 (m, 2H), 8.43 (s, 1H), 8.22 (m, 1H), 8.08–8.01 (m, 2H), 7.87 (d, J = 8.0 Hz, 2H), 7.60 (m, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.41 (s, 2H), 3.62–3.58 (m, 4H), 3.55 (s, 2H), 2.40 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 157.89, 155.27, 150.79, 144.90, 142.85, 138.83, 137.69, 130.30, 127.69, 127.42, 126.90, 126.61, 125.70, 125.25, 125.05, 124.38, 124.13, 121.88, 114.66, 66.68, 62.46, 53.66, 49.06. HRMS (ESI) m/z calculated for C25H25N5O3S: [M + H]+: 476.1751. Found: 476.1744.
3-((6-(Pyridin-3-yl)quinazolin-4-yl)amino)benzenesulfonamide (4f)
Off-white solid, yield: 26%, mp: 320.0–321.5 °C, HPLC purity: 8 min, 98.93%, 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.15 (s, 1H), 8.97 (s, 1H), 8.70–8.64 (m, 2H), 8.37 (s, 1H), 8.33–8.26 (m, 2H), 8.24–8.16 (m, 1H), 7.94 (d, J = 8.2 Hz, 1H), 7.65–7.57 (m, 3H), 7.42 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 158.18, 155.06, 149.87, 149.40, 148.55, 144.91, 139.94, 135.59, 134.90, 132.32, 129.62, 129.19, 125.68, 124.39, 121.44, 121.19, 119.48, 115.79. HRMS (ESI) m/z calculated for C19H15N5O2S: [M + H]+: 379.1019. Found: 378.1010.
3.4. In Vitro Kinase Assay
The screening of the tested compounds was performed using the Kinase HotSpotSM service from Reaction Biology Co. (http://www.reactionbiology.com, accessed on 25 September 2021). The HotSpot assay platform contains specific kinase/substrate pairs along with required cofactors. Base reaction buffer: 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO. The tested compounds were dissolved in 100% DMSO to specific concentration. The serial dilution was conducted by Integra Viaflo Assist in DMSO (Sigma-Aldrich, Burlington, MA, USA). The reaction mixture containing the examined compound and 33P-ATP was incubated at room temperature for 2 h, and radioactivity was detected by filter-binding method. Kinase activity data were expressed as the percent remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software, San Diego, CA, USA).
4. Conclusions
As an efficient approach to speed up the identification process of new EphB3 inhibitors, a virtual library was constructed based on a structure-based design and was subjected to an in-depth molecular docking study. The docking study was used to select the most promising compounds for MM-GBSA calculations, chemical synthesis, and biological evaluation. The MM-GBSA calculations predicted compound 4c to possess the highest stability with a binding free energy (−74.13 Kcal/mol). The kinase assays confirmed the potency of compound 4c as a potent EphB3 inhibitor (IC50 = 1.04 µM). Furthermore, a kinase panel assay composed of EGFR, Aurora A, Aurora B, CDK2/cyclin A, EphB1, EphB2, EphB4, ERBB2/HER2, and KDR/VEGFR2 revealed that compound 4c has a promising selectivity profile, with a four-fold selectivity for EphB3 over EphB2 and four isoforms and six-fold selectivity over the EphB1 isoform. These results indicate that compound 4c is a promising selective EphB3 inhibitor. Accordingly, it may be a useful tool to enhance the understanding of EphB3 biological role and pave the way toward identification of more potent and selective inhibitors. Additionally, the results show the validity of the constructed experimental model to predict novel EphB3 inhibitors.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ph14121247/s1, Figure S1: The chemical structures of the generated virtual library, 1H NMR, 13C NMR, HPLC, and HRMS data of compounds 3a–d and 4a–f.
Author Contributions
Conceptualization, A.E. and K.L.; methodology, H.N.; software, H.N.; validation, H.N.; formal analysis, H.J.B. and C.H.L.; investigation, H.J.B. and C.H.L.; resources, K.L.; data curation, A.E.; writing—original draft preparation, H.N. and A.E.; writing—review and editing, all authors; visualization, H.N.; project administration, A.E.; supervision, K.L.; funding acquisition, K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the national Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. NRF-2018R1A5A2023127).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pasquale E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaught D., Brantley-Sieders D.M., Chen J. Eph receptors in breast cancer: Roles in tumor promotion and tumor suppression. Breast Cancer Res. 2008;10:217. doi: 10.1186/bcr2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willson C.A., Foster R.D., Onifer S.M., Whittemore S.R., Miranda J.D. EphB3 receptor and ligand expression in the adult rat brain. J. Mol. Histol. 2006;37:369–380. doi: 10.1007/s10735-006-9067-0. [DOI] [PubMed] [Google Scholar]
- 4.Darling T.K., Lamb T.J. Emerging Roles for Eph Receptors and Ephrin Ligands in Immunity. Front. Immunol. 2019;10:1473. doi: 10.3389/fimmu.2019.01473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji X.D., Li G., Feng Y.X., Zhao J.S., Li J.J., Sun Z.J., Shi S., Deng Y.Z., Xu J.F., Zhu Y.Q., et al. EphB3 is overexpressed in non-small-cell lung cancer and promotes tumor metastasis by enhancing cell survival and migration. Cancer Res. 2011;71:1156–1166. doi: 10.1158/0008-5472.CAN-10-0717. [DOI] [PubMed] [Google Scholar]
- 6.Lee S.Y., Na Y.J., Jeong Y.A., Kim J.L., Oh S.C., Lee D.H. Upregulation of EphB3 in gastric cancer with acquired resistance to a FGFR inhibitor. Int. J. Biochem. Cell Biol. 2018;102:128–137. doi: 10.1016/j.biocel.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 7.Jang B.G., Kim H.S., Bae J.M., Kim W.H., Hyun C.L., Kang G.H. Expression Profile and Prognostic Significance of EPHB3 in Colorectal Cancer. Biomolecules. 2020;10:602. doi: 10.3390/biom10040602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatia S., Griego A., Lennon S., Oweida A., Sharma J., Rohmer C., Uyanga N., Bukkapatnam S., Van Court B., Raben D., et al. Role of EphB3 Receptor in Mediating Head and Neck Tumor Growth, Cell Migration, and Response to PI3K Inhibitor. Mol. Cancer Ther. 2018;17:2049–2059. doi: 10.1158/1535-7163.MCT-17-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao K., He J., Wang Y.F., Jin S.D., Fan Y., Fang N., Qian J., Xu T.P., Guo R.H. EZH2-mediated epigenetic suppression of EphB3 inhibits gastric cancer proliferation and metastasis by affecting E-cadherin and vimentin expression. Gene. 2019;686:118–124. doi: 10.1016/j.gene.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Li J.J., Sun Z.J., Yuan Y.M., Yin F.F., Bian Y.G., Long L.Y., Zhang X.L., Xie D. EphB3 Stimulates Cell Migration and Metastasis in a Kinase-dependent Manner through Vav2-Rho GTPase Axis in Papillary Thyroid Cancer. J. Biol. Chem. 2017;292:1112–1121. doi: 10.1074/jbc.M116.750349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang G., Liu X., Li Y., Wang Y., Liang H., Li K., Li L., Chen C., Sun W., Ren S., et al. EphB3-targeted regulation of miR-149 in the migration and invasion of human colonic carcinoma HCT116 and SW620 cells. Cancer Sci. 2017;108:408–418. doi: 10.1111/cas.13161. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Herath N.I., Boyd A.W. The role of Eph receptors and ephrin ligands in colorectal cancer. Int. J. Cancer. 2010;126:2003–2011. doi: 10.1002/ijc.25147. [DOI] [PubMed] [Google Scholar]
- 13.Chiu S.-T., Chang K.-J., Ting C.-H., Shen H.-C., Li H., Hsieh F.-J. Over-expression of EphB3 enhances cell–cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis. 2009;30:1475–1486. doi: 10.1093/carcin/bgp133. [DOI] [PubMed] [Google Scholar]
- 14.Li G., Ji X.-D., Gao H., Zhao J.-S., Xu J.-F., Sun Z.-J., Deng Y.-Z., Shi S., Feng Y.-X., Zhu Y.-Q., et al. EphB3 suppresses non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt signalling complex. Nat. Commun. 2012;3:667. doi: 10.1038/ncomms1675. [DOI] [PubMed] [Google Scholar]
- 15.Kung A., Chen Y.-C., Schimpl M., Ni F., Zhu J., Turner M., Molina H., Overman R., Zhang C. Development of Specific, Irreversible Inhibitors for a Receptor Tyrosine Kinase EphB3. J. Am. Chem. Soc. 2016;138:10554–10560. doi: 10.1021/jacs.6b05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montero-Herradón S., García-Ceca J., Zapata A.G. EphB receptors, mainly EphB3, contribute to the proper development of cortical thymic epithelial cells. Organogenesis. 2017;13:192–211. doi: 10.1080/15476278.2017.1389368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Z., Liu R., Xiong L., Miao X., Li D., Zou Q., Yuan Y., Yang Z. Prognostic and Clinicopathological Significance of EphB3 and Dysadherin Expression in Extrahepatic Cholangiocarcinoma. Cancer Manag. Res. 2020;12:221–232. doi: 10.2147/CMAR.S232278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martiny-Baron G., Holzer P., Billy E., Schnell C., Brueggen J., Ferretti M., Schmiedeberg N., Wood J.M., Furet P., Imbach P. The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis. 2010;13:259–267. doi: 10.1007/s10456-010-9183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L., Shan Y., Ji X., Zhu M., Li C., Sun Y., Si R., Pan X., Wang J., Ma W., et al. Discovery and evaluation of triple inhibitors of VEGFR-2, TIE-2 and EphB4 as anti-angiogenic and anti-cancer agents. Oncotarget. 2017;8:104745–104760. doi: 10.18632/oncotarget.20065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Werner T.L., Wade M.L., Agarwal N., Boucher K., Patel J., Luebke A., Sharma S. A pilot study of JI-101, an inhibitor of VEGFR-2, PDGFR-β, and EphB4 receptors, in combination with everolimus and as a single agent in an ovarian cancer expansion cohort. Investig. New Drugs. 2015;33:1217–1224. doi: 10.1007/s10637-015-0288-5. [DOI] [PubMed] [Google Scholar]
- 21.Pietanza M.C., Lynch T.J., Jr., Lara P.N., Jr., Cho J., Yanagihara R.H., Vrindavanam N., Chowhan N.M., Gadgeel S.M., Pennell N.A., Funke R., et al. XL647—A multitargeted tyrosine kinase inhibitor: Results of a phase II study in subjects with non-small cell lung cancer who have progressed after responding to treatment with either gefitinib or erlotinib. J. Thorac. Oncol. 2012;7:219–226. doi: 10.1097/JTO.0b013e31822eebf9. [DOI] [PubMed] [Google Scholar]
- 22.Du E., Li X., He S., Li X., He S. The critical role of the interplays of EphrinB2/EphB4 and VEGF in the induction of angiogenesis. Mol. Biol. Rep. 2020;47:4681–4690. doi: 10.1007/s11033-020-05470-y. [DOI] [PubMed] [Google Scholar]
- 23.Burkholder T.P., Clayton J.R., Rempala M.E., Henry J.R., Knobeloch J.M., Mendel D., McLean J.A., Hao Y., Barda D.A., Considine E.L., et al. Discovery of LY2457546: A multi-targeted anti-angiogenic kinase inhibitor with a novel spectrum of activity and exquisite potency in the acute myelogenous leukemia-Flt-3-internal tandem duplication mutant human tumor xenograft model. Investig. New Drugs. 2012;30:936–949. doi: 10.1007/s10637-011-9640-6. [DOI] [PubMed] [Google Scholar]
- 24.Caligiuri M., Molz L., Liu Q., Kaplan F., Xu J.P., Majeti J.Z., Ramos-Kelsey R., Murthi K., Lievens S., Tavernier J., et al. MASPIT: Three-hybrid trap for quantitative proteome fingerprinting of small molecule-protein interactions in mammalian cells. Chem. Biol. 2006;13:711–722. doi: 10.1016/j.chembiol.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Choi Y., Syeda F., Walker J.R., Finerty P.J., Jr., Cuerrier D., Wojciechowski A., Liu Q., Dhe-Paganon S., Gray N.S. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:4467–4470. doi: 10.1016/j.bmcl.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noberini R., Lamberto I., Pasquale E.B. Targeting Eph receptors with peptides and small molecules: Progress and challenges. Semin. Cell Dev. Biol. 2012;23:51–57. doi: 10.1016/j.semcdb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyd A.W., Bartlett P.F., Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014;13:39–62. doi: 10.1038/nrd4175. [DOI] [PubMed] [Google Scholar]
- 28.Qiao L., Choi S., Case A., Gainer T.G., Seyb K., Glicksman M.A., Lo D.C., Stein R.L., Cuny G.D. Structure–activity relationship study of EphB3 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:6122–6126. doi: 10.1016/j.bmcl.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazaki Y., Nakano M., Sato H., Truesdale A.T., Stuart J.D., Nartey E.N., Hightower K.E., Kane-Carson L. Design and effective synthesis of novel templates, 3,7-diphenyl-4-amino-thieno and furo-[3,2-c]pyridines as protein kinase inhibitors and in vitro evaluation targeting angiogenetic kinases. Bioorg. Med. Chem. Lett. 2007;17:250–254. doi: 10.1016/j.bmcl.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 31.Sirous H., Chemi G., Campiani G., Brogi S. An integrated in silico screening strategy for identifying promising disruptors of p53-MDM2 interaction. Comput. Biol. Chem. 2019;83:107105. doi: 10.1016/j.compbiolchem.2019.107105. [DOI] [PubMed] [Google Scholar]
- 32.Ravez S., Castillo-Aguilera O., Depreux P., Goossens L. Quinazoline derivatives as anticancer drugs: A patent review (2011–present) Expert Opin. Ther. Pat. 2015;25:789–804. doi: 10.1517/13543776.2015.1039512. [DOI] [PubMed] [Google Scholar]
- 33.Li H.-Q., Li D.-D., Lu X., Xu Y.-Y., Zhu H.-L. Design and synthesis of 4,6-substituted-(diaphenylamino)quinazolines as potent EGFR inhibitors with antitumor activity. Bioorg. Med. Chem. 2012;20:317–323. doi: 10.1016/j.bmc.2011.10.085. [DOI] [PubMed] [Google Scholar]
- 34.Cruz-Lopez O., Conejo-Garcia A., Nunez M.C., Kimatrai M., Garcia-Rubino M.E., Morales F., Gomez-Perez V., Campos J.M. Novel Substituted Quinazolines for Potent EGFR Tyrosine Kinase Inhibitors. Curr. Med. Chem. 2011;18:943–963. doi: 10.2174/092986711794940824. [DOI] [PubMed] [Google Scholar]
- 35.Elkamhawy A., Hassan A.H.E., Paik S., Sup Lee Y., Lee H.H., Shin J.S., Lee K.T., Roh E.J. EGFR inhibitors from cancer to inflammation: Discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorg. Chem. 2019;86:112–118. doi: 10.1016/j.bioorg.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 36.Karaman M., Herrgard S., Treiber D., Gallant P., Atteridge C., Campbell B., Chan K., Ciceri P., Davis M., Edeen P.J.N.B., et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 37.Bembenek S.D., Hirst G., Mirzadegan T. Determination of a Focused Mini Kinase Panel for Early Identification of Selective Kinase Inhibitors. J. Chem. Inf. Model. 2018;58:1434–1440. doi: 10.1021/acs.jcim.8b00222. [DOI] [PubMed] [Google Scholar]
- 38.Asquith C.R.M., Treiber D.K., Zuercher W.J. Utilizing comprehensive and mini-kinome panels to optimize the selectivity of quinoline inhibitors for cyclin G associated kinase (GAK) Bioorg. Med. Chem. Lett. 2019;29:1727–1731. doi: 10.1016/j.bmcl.2019.05.025. [DOI] [PubMed] [Google Scholar]
- 39.Brandt P., Jensen A.J., Nilsson J. Small kinase assay panels can provide a measure of selectivity. Bioorg. Med. Chem. Lett. 2009;19:5861–5863. doi: 10.1016/j.bmcl.2009.08.083. [DOI] [PubMed] [Google Scholar]
- 40.Fabian M., Biggs W., Treiber D., Atteridge C.E., Azimioara M.D., Benedetti M.G., Carter T.A., Ciceri P., Edeen P.T., Floyd M., et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed M.S., Wang P., Nguyen N.U.N., Nakada Y., Menendez-Montes I., Ismail M., Bachoo R., Henkemeyer M., Sadek H.A., Kandil E.S. Identification of tetracycline combinations as EphB1 tyrosine kinase inhibitors for treatment of neuropathic pain. Proc. Natl. Acad. Sci. USA. 2021;118:e2016265118. doi: 10.1073/pnas.2016265118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiesner S., Wybenga-Groot L.E., Warner N., Lin H., Pawson T., Forman-Kay J.D., Sicheri F. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006;25:4686–4696. doi: 10.1038/sj.emboj.7601315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Overman R.C., Debreczeni J.E., Truman C.M., McAlister M.S., Attwood T.K. Completing the structural family portrait of the human EphB tyrosine kinase domains. Protein Sci. 2014;23:627–638. doi: 10.1002/pro.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oguro Y., Miyamoto N., Okada K., Takagi T., Iwata H., Awazu Y., Miki H., Hori A., Kamiyama K., Imamura S. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo [3,2-d]pyrimidine derivatives: Novel VEGFR2 kinase inhibitors binding to inactive kinase conformation. Bioorg. Med. Chem. 2010;18:7260–7273. doi: 10.1016/j.bmc.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 45.Bavetsias V., Large J.M., Sun C., Bouloc N., Kosmopoulou M., Matteucci M., Wilsher N.E., Martins V., Reynisson J., Atrash B., et al. Imidazo[4,5-b]pyridine Derivatives As Inhibitors of Aurora Kinases: Lead Optimization Studies toward the Identification of an Orally Bioavailable Preclinical Development Candidate. J. Med. Chem. 2010;53:5213–5228. doi: 10.1021/jm100262j. [DOI] [PubMed] [Google Scholar]
- 46.Du Y., Shi W.-W., He Y.-X., Yang Y.-H., Zhou C.-Z., Chen Y. Structures of the substrate-binding protein provide insights into the multiple compatible solute binding specificities of the Bacillus subtilis ABC transporter OpuC. Biochem. J. 2011;436:283–289. doi: 10.1042/BJ20102097. [DOI] [PubMed] [Google Scholar]
- 47.De Azevedo W.F., Leclerc S., Meijer L., Havlicek L., Strnad M., Kim S.-H. Inhibition of Cyclin-Dependent Kinases by Purine Analogues. Eur. J. Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- 48.Greenidge P.A., Kramer C., Mozziconacci J.-C., Wolf R.M. MM/GBSA Binding Energy Prediction on the PDBbind Data Set: Successes, Failures, and Directions for Further Improvement. J. Chem. Inf. Model. 2013;53:201–209. doi: 10.1021/ci300425v. [DOI] [PubMed] [Google Scholar]
- 49.Elkamhawy A., Kim N.Y., Hassan A.H.E., Park J.E., Yang J.E., Oh K.S., Lee B.H., Lee M.Y., Shin K.J., Lee K.T., et al. Design, synthesis and biological evaluation of novel thiazolidinedione derivatives as irreversible allosteric IKK-β modulators. Eur. J. Med. Chem. 2018;157:691–704. doi: 10.1016/j.ejmech.2018.08.020. [DOI] [PubMed] [Google Scholar]
- 50.Elkamhawy A., Farag A.K., Viswanath A.N.I., Bedair T.M., Leem D.G., Lee K.-T., Pae A.N., Roh E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg. Med. Chem. Lett. 2015;25:5147–5154. doi: 10.1016/j.bmcl.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida S., Morita T., Hosoya T. Synthesis of Diverse Benzotriazoles from Aryne Precursors Bearing an Azido Group via Inter- and Intramolecular Cycloadditions. Chem. Lett. 2016;45:726–728. doi: 10.1246/cl.160349. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article.