

Abstract
Helicobacter pylori is a pathogen that confers the highest known risk for gastric cancer. Research directed at understanding the pathogenesis of H. pylori is crucial to identify colonized persons that may subsequently develop neoplasia. Imai et al. describe how H. pylori elicits BRCAness and endows epithelial cells with the ability to evade apoptosis.
Gastric adenocarcinoma is the fourth leading cause of cancer-related death in the world [1]. Adenocarcinoma is the most common type of cancer that affects the stomach, and two distinct variants of gastric adenocarcinoma can be differentiated histologically: diffuse-type gastric cancer, which consists of individually infiltrating neoplastic cells that do not form glandular structures, and intestinal-type adenocarcinoma, which progresses through a series of well-defined histological steps [2]. Intestinal-type adenocarcinoma is initiated by the transition from normal mucosa to chronic superficial gastritis; this is followed by the development of atrophic gastritis and intestinal metaplasia, finally leading to dysplasia and adenocarcinoma [3]. Typically, gastric cancer is not diagnosed until late in the disease course and 5-year survival rates for gastric cancer in the USA are less than 30% [4]. It is therefore of great importance to gain a comprehensive understanding of the factors that contribute to this malignancy.
Helicobacter pylori infection is usually acquired in childhood and can persist for the lifetime of the host. Factors that influence pathologic disease outcomes of H. pylori infection include strain-specific bacterial constituents, host genetic factors, alterations of the stem cell niche and host microbiota, and environmental influences, including diet [5]. One strain-specific bacterial determinant that augments cancer risk is the cag type IV secretion system (T4SS), which translocates the oncoprotein CagA from adherent H. pylori into host epithelial cells. Intracellular CagA undergoes tyrosine phosphorylation and activates a eukaryotic phosphatase (SHP-2), leading to carcinogenic cellular responses [5]. In normal cells, aberrant activation of oncogenes is typically counteracted by tumor suppressor mechanisms. The p53 protein, encoded by the human gene TP53, plays a key role in this process by limiting aberrant cellular proliferation and eliminating transformed cells that otherwise may cause tumors to develop [6]. p53 is inactivated by mutations in TP53 in approximately 50% of patients with gastric cancer and CagA has been shown to induce degradation of p53 [7], providing a strong rationale for investigating tumor suppressor pathways within the context of H. pylori-induced gastric cancer.
In a recent study published in Cell Host and Microbe, Imai et al. demonstrate that CagA induced BRCAness, a defect in DNA double-strand break (DSB) repair by homologous recombination-mediated repair to induce genome instability and gastric carcinogenesis [8]. The authors revealed that CagA expression in gastric epithelial cells, either via coculture with viable CagA positive H. pylori or plasmid-mediated transfection, induced DSBs through direct inhibition of polarity-regulating serine/threonine kinase PAR1b kinase activity (Figure 1). Coexpression of CagA with wild type PAR1b, but not kinase-inactive PAR1b, prevented DSBs. The tumor suppressors, BRCA1 and BRCA2, are critical for DNA repair by homologous recombination, which is largely involved in the repair of DNA lesions that stall DNA replication forks; replication stress is one factor that generates DSBs by inducing fork stalling or collapse [9]. The tumor suppressors BRCA1 and BRCA2 specifically protect stalled forks from being degraded by cellular nucleases. The caveat is that these tumor suppressors can only exert their protective role in the nucleus. Using a combination of in vitro experiments utilizing gastric cell lines, transfection, inducible CagA systems, and cocultures with H. pylori, Imai et al. demonstrated that CagA inhibited translocation of the tumor suppressor BRCA1, but not BRCA2, to the nucleus and this was mediated through a mechanism of inhibition of PAR1b kinase-mediated phosphorylation of BRCA1 (Figure 1).
Figure 1. Schematic representation of Helicobacter pylori CagA-induced BRCAness.
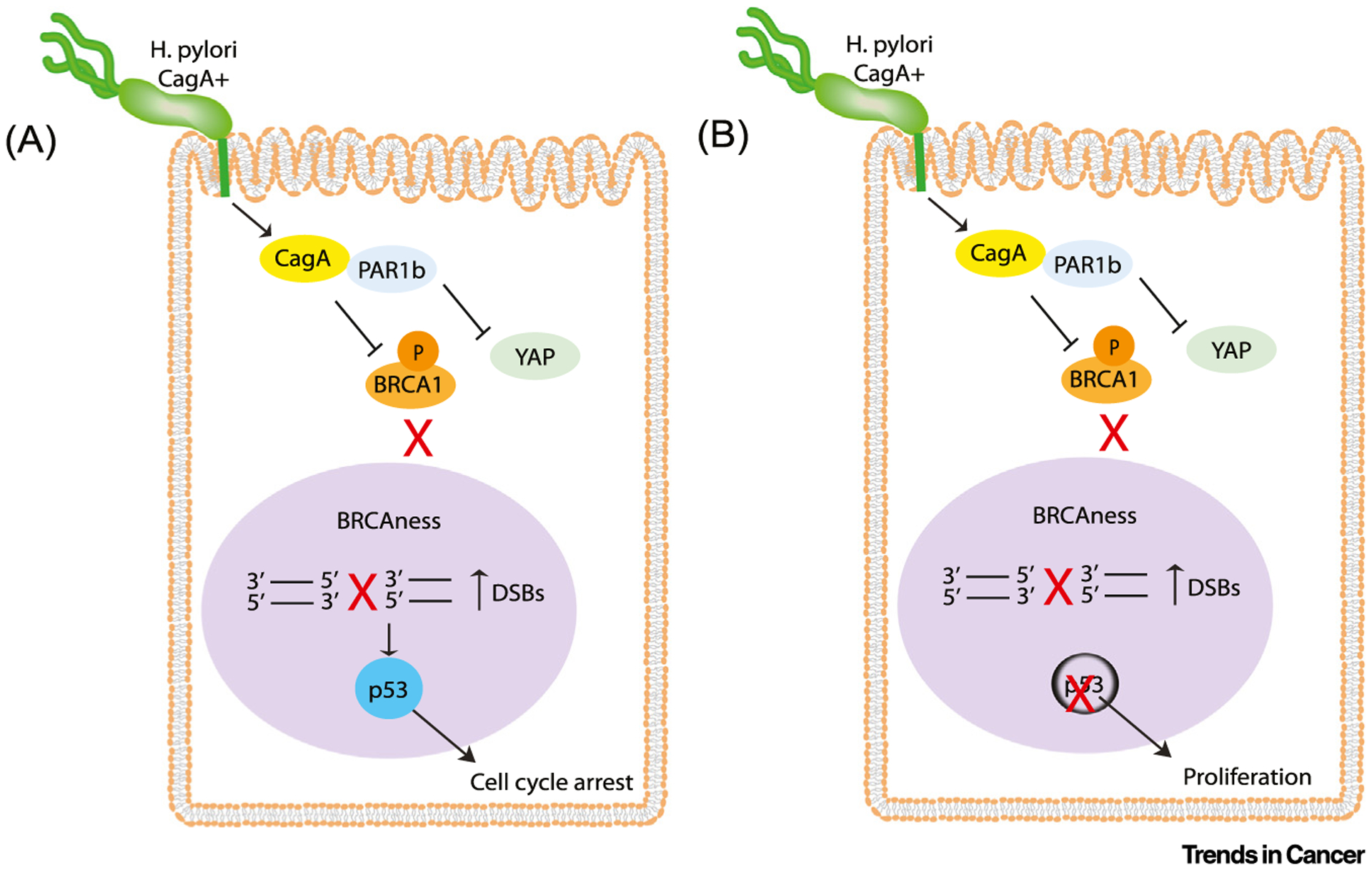
H. pylori translocates CagA into host epithelial cells where it inactivates PAR1b kinase activity, leading to decreased nuclear translocation of BRCA1 and, as a result, increased double strand breaks (DSBs). In parallel, CagA-mediated inhibition of PAR1b activates Hippo signaling and prevents nuclear translocation of Yes-associated protein (YAP), thus inhibiting apoptosis. (A) In the presence of functional p53, p21 is activated and cells enter cell cycle arrest. (B) When cellular p53 is lost or mutated, CagA-expressing cells with BRCAness continue to proliferate.
Traditionally, in vivo experiments with H. pylori involve oral gavage of mice with the pathogen to infect the stomach; however, Imai et al. utilized a recently established floxed cagA-transgenic mouse model where the cagA transgene is conditionally induced by tamoxifen treatment. Concordant with their in vitro findings, Imai et al. observed that 5 days following tamoxifen treatment and the subsequent induction of CagA, levels of nuclear BRCA1 were reduced and DSBs were increased within mouse gastric mucosa. Using a quantitative reporter assay that evaluates the ability of DSB repair by error-free homologous recombination or error-prone non-homologous end-joining, CagA was found to inhibit homologous recombination-mediated DSB repair. Similar to CagA expression, knockdown of PAR1b also prevented homologous recombination-mediated DSB repair, suggesting that through inhibition of PAR1b kinase activity, CagA is able to inhibit homologous recombination-mediated DSB repair and elicit a state of transient BRCAness in gastric epithelial cells (Figure 1).
It was somewhat of an enigma that CagA-expressing cells, with enriched DSBs and BRCAness, were able to evade apoptosis and persist in a seemingly healthy state. To address this observation, the authors demonstrated that apoptosis was suppressed through transient activation of Hippo signaling via CagA-mediated PAR1b inhibition, which prevented nuclear translocation of the proapoptotic Yes-associated protein (YAP)/p73 complex (Figure 1). However, CagA-induced DSBs also activated the p53 tumor suppressor and, as a result, its direct transcriptional target, p21, was activated, which promotes cell cycle arrest. However, in the absence of p53, CagA-expressing cells did not accumulate p21 and were able to continue proliferating. To recapitulate what happens in chronic H. pylori infection in humans, Imai et al. used a tet-off system to induce CagA expression in epithelial cells on and off over 20 cycles. Under these conditions, repeated cycles of BRCAness resulted in accumulation of genomic alterations that constitute the BRCAness-specific genome mutational signature.
In human gastric mucosa infected with cagA-positive H. pylori, BRCA1 was decreased in the nucleus of gastric epithelial cells and DSBs were increased compared with uninfected gastric mucosa. Of note is that in the isthmus of the corpus glands, where stem cells reside, decreased BRCA1 and increased DSBs were also observed in H. pylori-infected individuals, but not in uninfected persons.
In summary, Imai et al. have provided a mechanism through which H. pylori CagA may elicit the development of gastric cancer through repeated exposure of gastric epithelial cells to CagA over many years. The interaction of CagA with PAR1b prevents the translocation of BRCA1 to the nucleus and, in doing so, elicits BRCAness, promoting DSBs and preventing DNA repair by homologous recombination. In parallel, the interaction of CagA with PAR1b activates Hippo signaling and cells with DNA damage escape apoptosis and repair DCBs through error-prone mechanisms. In the presence of functional p53, proliferation of CagA-expressing cells is inhibited by p21. However, in the absence of functional p53, such as occurs in approximately 50% of gastric cancer cases, CagA-expressing cells displaying BRCAness can proliferate. Since loss of cellular p53 usually occurs because of aging-associated somatic TP53 mutations [10], this finding may explain why H. pylori infection in young individuals leads to primarily mild disease compared with elderly individuals where gastric cancer is more prevalent.
Footnotes
Declaration of interests
No interests are declared.
References
- 1.Bray F et al. (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 68, 394–424 [DOI] [PubMed] [Google Scholar]
- 2.Correa P (1992) Human gastric carcinogenesis: a multistep and multifactorial process – First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52, 6735–6740 [PubMed] [Google Scholar]
- 3.Sipponen P et al. (2000) Gastritis and gastric cancer. Western countries. Gastroenterol. Clin. N. Am 29, 579–592 [DOI] [PubMed] [Google Scholar]
- 4.Allemani C et al. (2018) Global surveillance of trends in cancer survival: analysis of individual records for 37,513,025 patients diagnosed with one of 18 cancers during 2000–2014 from 322 population-based registries in 71 countries (CONCORD-3). Lancet 391, 1023–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amieva M et al. (2016) Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology 150, 64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivlin N (2011) Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumor-igenesis. Genes Cancer 2, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei J et al. (2010) Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139, 1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imai S et al. (2021) Helicobacter pylori CagA elicits BRCAness to induce genome instability that may underlie bacterial gastric carcinogenesis. Cell Host Microbe 29, 941–958 [DOI] [PubMed] [Google Scholar]
- 9.Venkitaraman AR et al. (2014) Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 343, 1470–1475 [DOI] [PubMed] [Google Scholar]
- 10.Richardson R (2013) p53 mutations associated with aging-related rise in cancer incidence rates. Cell Cycle 12, 2468–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]