

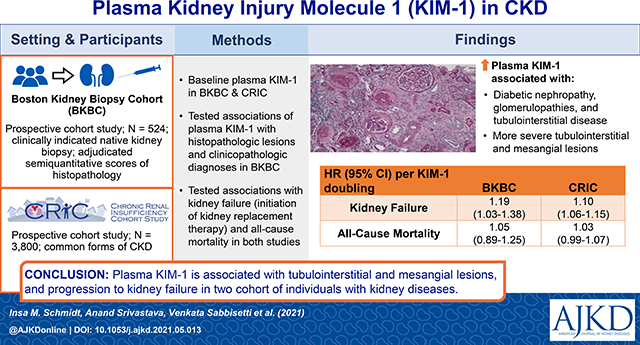
Abstract
Rationale & Objective:
Plasma kidney injury molecule-1 (KIM-1) is a sensitive marker of proximal tubule injury, but its association with risks of adverse clinical outcomes across a spectrum of kidney diseases is unknown.
Study Design:
Prospective, observational cohort study.
Setting & Participants:
524 individuals undergoing clinically indicated native kidney biopsy with biopsy specimens adjudicated for semiquantitative scores of histopathology by two kidney pathologists enrolled into the Boston Kidney Biopsy Cohort (BKBC) Study and 3,800 individuals with common forms of chronic kidney disease (CKD) enrolled into the Chronic Renal Insufficiency Cohort (CRIC) Study.
Exposure:
Histopathologic lesions and clinicopathologic diagnosis in cross-sectional analyses, baseline plasma KIM-1 in prospective analyses.
Outcomes:
Baseline plasma KIM-1 in cross-sectional analyses, kidney failure (defined as initiation of kidney replacement therapy) and death in prospective analyses.
Analytical Approach:
Multivariable-adjusted linear regression models tested associations of plasma KIM-1 with histopathologic lesions and clinicopathologic diagnoses. Cox proportional hazards models tested associations of plasma KIM-1 with future kidney failure and death.
Results:
In the BKBC Study, higher plasma KIM-levels were associated with more severe acute tubular injury, tubulointerstitial inflammation, and more severe mesangial expansion after multivariable adjustment. Participants with diabetic nephropathy, glomerulopathies, and tubulointerstitial disease had significantly higher plasma KIM-1 levels after multivariable adjustment. In the BKBC Study, 124 participants progressed to kidney failure and 85 participants died during a median follow-up time of 5 years. In the CRIC Study, 1153 participants progressed to kidney failure and 1356 participants died during a median follow-up time of 11.5 years. In both cohorts, each doubling of plasma KIM-1 was associated with an increased risk of kidney failure after multivariable adjustment (BKBC: HR 1.19, 95% CI 1.03 to 1.38 and CRIC: HR 1.10, 95% CI 1.06 to 1.15). There was no statistically significant association of plasma KIM-1 with death in either cohort.
Limitations:
Generalizability and unmeasured confounding.
Conclusions:
Plasma KIM-1 is associated with underlying tubulointerstitial and mesangial lesions and progression to kidney failure in two cohort studies of individuals with kidney diseases.
Plain-language Summary:
Kidney tubular injury may lead to the development or progression of chronic kidney disease (CKD). Plasma KIM-1 is a sensitive marker of tubular injury, but its association with adverse clinical outcomes across a spectrum of kidney diseases is not known. In two prospective cohort studies of individuals with common and diverse forms of CKD, higher plasma KIM-1 levels were independently associated with progression to kidney failure. In individuals who underwent a native kidney biopsy with adjudicated histopathology, higher plasma KIM-1 levels were associated with more severe acute tubular injury, tubulointerstitial inflammation, and mesangial expansion. Collectively, the findings suggest that plasma KIM-1 may serve as a non-invasive tool to assess histopathologic lesions and has prognostic value across a variety of kidney diseases.
Graphical Abstract
Introduction
Kidney injury molecule-1 (KIM-1), originally identified as hepatitis A virus receptor (HAVCR1, also known as TIM-1), is a type 1 transmembrane glycoprotein strongly induced by ischemic and toxic insults to the kidney.1–3 In healthy kidneys, KIM-1 is virtually undetectable. In both acute kidney injury (AKI) and chronic kidney disease (CKD), KIM-1 expression is markedly upregulated in proximal tubules.4–7 Animal models suggest that acute upregulation of KIM-1 appears to be protective after kidney insults, whereas chronic upregulation of KIM-1 tubular expression may lead to progressive kidney fibrosis.8, 9
KIM-1 expression has been demonstrated in urine samples and kidney biopsy specimens of patients with various etiologies of kidney disease.10 In animal and human studies, kidney tissue expression of KIM-1 was present in areas of inflammation and fibrosis and associated with the presence of interstitial macrophages.1, 10, 11 Since the ectodomain of KIM-1 is cleaved by metalloproteinases and detectable in the urine after kidney damage,6 the majority of the literature has focused on the investigation of urinary KIM-1.2, 12–24 While more recent studies suggest that plasma KIM-1 is a risk factor for CKD progression,25–29 less is known about the associations of plasma KIM-1 with histopathologic lesions and the risks of adverse clinical outcomes across a spectrum of kidney diseases. To address this, we measured plasma KIM-1 levels in participants from two independent CKD cohort studies, the Boston Kidney Biopsy Cohort (BKBC) and the Chronic Renal Insufficiency Cohort (CRIC). We hypothesized that higher levels of plasma KIM-1 are associated with underlying histopathologic lesions, clinicopathologic diagnoses, and increased risks of subsequent progression to kidney failure and death.
Methods
Study Design and Oversight
The institutional review boards at the participating hospitals of each study approved the study protocol, which is in accordance with the principles of the Declaration of Helsinki. All participants provided written informed consent. The design and methods of both studies have been described previously.30, 31
BKBC Study
Study Population
The BKBC Study is a prospective, observational cohort study of individuals undergoing native kidney biopsy at three tertiary care hospitals in Boston, MA. The study includes adults (≥18 years of age) who underwent a clinically indicated native kidney biopsy between September 2006 and June 2016. Exclusion criteria included the inability to provide written consent, severe anemia, pregnancy, and enrollment in competing studies. Measurements of KIM-1 were made in baseline plasma samples of 524 participants collected on the day of native biopsy.
Evaluation of Histopathology
Methods to evaluate and score histopathologic lesions in the BKBC Study were previously described.31 Briefly, kidney biopsies were adjudicated under light microscopy by two experienced kidney pathologists who provided semiquantitative scores of kidney inflammation, fibrosis, vascular sclerosis, and tubular injury (Supplemental Table 1). The kidney pathologists reviewed the light microscopy slides in joint pathology review sessions and verbally suggested their score for each individual histopathologic lesion severity in the presence of A.S. and/or S.S.W. If they did not initially agree, the case was discussed in more detail to obtain consensus on each individual histopathologic lesion severity. Of the 13 histopathologic lesions adjudicated, all were scored during study sessions except for grades of global or segmental glomerulosclerosis, which were taken from the biopsy report, because they were each calculated as a percentage of the total number of glomeruli. We dichotomized endocapillary glomerular inflammation, extracapillary cellular crescents, focal glomerular necrosis, and fibrocellular crescents into present/absent and then further created a single dichotomous variable named ‘glomerular inflammation’ due to the relatively low prevalence and limited range of severity for each lesion in this cohort. Glomerular inflammation was considered present if any of these lesions were present. All participants’ charts were reviewed alongside the histopathologic evaluations to provide the final primary clinicopathologic diagnosis.
Outcomes
The primary outcome was progression to kidney failure, defined as initiation of kidney replacement therapy (dialysis or kidney transplantation). The secondary outcome was death. Kidney replacement therapy status was confirmed by reviewing the electronic medical record (EMR) and linkage with the United States Renal Data System (USRDS).31 Mortality status was confirmed with the Social Security Death Index. Participants were followed up until the occurrence of death, voluntary study withdrawal, loss to follow-up, or February 1, 2020.
Ascertainment of Covariates
We collected participants’ information at the biopsy visit, including demographics, medical history, medication lists, and pertinent laboratory data. All data were stored at the REDCap electronic data capture tools hosted at Partners Health Care.32 We used the creatinine-based Chronic Kidney Disease Epidemiology Collaboration equation to calculate eGFR.33 We obtained serum creatinine from the EMR on the day of biopsy. In participants for whom this was unavailable, we measured serum creatinine in blood samples collected on the day of biopsy. We obtained spot urine protein-to-creatinine ratio or urine albumin-creatinine ratio (UACR) from the date of kidney biopsy up to 3 months before biopsy from the EMR. If a participant did not have either of these values, we measured UACR from urine collected on the day of kidney biopsy. Serum and urine creatinine were measured using a Jaffe-based method, and urine albumin was measured by an immunoturbidometric method.
CRIC Study
Study Population
The CRIC Study is a multicenter, prospective, observational cohort study of individuals with CKD that was designed to investigate risk factors for progression of CKD, development of cardiovascular disease, and mortality. During Phase 1, the CRIC Study enrolled 3939 men and women aged 21 to 74 years between June 2003 and September 2008 across seven clinical centers in the United States. Individuals were included if they met specific age-defined criteria for estimated glomerular filtration rate (eGFR) of 20 to 70 ml/min/1.73m2. Black and Hispanic participants were oversampled. Exclusion criteria included inability to provide consent, institutionalization, New York Heart Association class III or IV congestive heart failure, human immunodeficiency virus infection, cirrhosis, multiple myeloma, kidney cancer, recent chemotherapy or immunosuppressive therapy, polycystic kidney disease, organ transplantation, or prior treatment with dialysis for at least 1 month.30 CRIC Study participants were followed annually by clinic visits, with interim phone contact at 6 months. Measurements of KIM-1 were made in baseline plasma samples of 3,800 study participants.
Outcomes
The primary outcome was progression to kidney failure, defined as initiation of kidney replacement therapy (dialysis or kidney transplantation). The secondary outcome was death. Ascertainment of kidney replacement therapy status within CRIC was supplemented by cross-linkage with the USRDS.30 Participants were followed up until the occurrence of the event of interest, voluntary study withdrawal, loss to follow-up, or end of the follow-up period on January 30, 2018.
Ascertainment of Covariates
Data obtained at the baseline visit included sociodemographic characteristics, medical history, lifestyle behaviors, medications, standardized blood pressure measurements, anthropometric measures, and laboratory measurements. eGFR was calculated using the CRIC-derived estimating equation based on serum creatinine and cystatin C.34 UACR was calculated as spot urine albumin concentration divided by spot urine creatinine concentration from urine samples. Urine creatinine concentration was measured using a Jaffe-based method and urine albumin concentration was measured using an immunoturbidometric method.
Sample Collection and Biomarker Assays
In the BKBC Study, blood samples were collected from participants on the day of kidney biopsy and stored at −80°C. In the CRIC Study, blood samples were collected from participants at baseline and stored at −80°C. Plasma KIM-1 was measured in BKBC and CRIC samples using multiplex microbead-based enzyme-linked immunosorbent assays on a Luminex platform as previously described.25 We measured plasma KIM-1 levels using monoclonal antibody (ADI-905–906-0100, Enzo life sciences) and polyclonal antibody (AF1750, R&D systems) against KIM-1 in the BKBC and the CRIC Study, respectively. Plasma samples were diluted 1:5 in diluent buffer and 30 μl of sample or recombinant human KIM-1 protein (1750-TM, R&D systems) was incubated with microbeads coupled with KIM-1 capture antibody for one hour, washed three times with PBST, and incubated with detection antibody (BAF 1750, R&D Systems) for 45 min. Beads were washed again with PBST and incubated for 15 min with streptavidin-PE solution (Invitrogen). The signal from the fluorochrome, which is directly proportional to the amount of antigen bound at the micro-bead surface, was captured using the Bio-Plex 200 system (Bio-Rad). Data were generated and interpreted using parametric logistic regression analysis and the amount of KIM-1 in plasma was determined using Bio-Plex 200. Additional information about the plasma KIM-1 intra- and inter-assay coefficients of variation are in the Supplemental Methods.
Statistical Analysis
Continuous variables were expressed as means with standard deviation (SD) or medians with interquartile range (IQR) and categorical variables were presented as count with percentages. For skewed data distributions, we performed natural logarithmic transformation as appropriate. We assessed plasma KIM-1 levels in two-group comparisons using the Wilcoxon rank sum test and in multiple-group comparisons using the Kruskal-Wallis test. We used Spearman correlations to assess the association between baseline plasma KIM-1 and continuous variables. To assess differences in frequency distributions of categorical variables, normally distributed continuous variables, and non-normally distributed continuous variables by tertiles (in BKBC) or quintiles (in CRIC) of plasma KIM-1, we used chi-squared, ANOVA, and Kruskal-Wallis tests, respectively. We limited statistical analyses on histopathologic lesions to participants with adjudicated histopathology by both kidney pathologists (n=458, 87%) except for analyses of global or segmental glomerulosclerosis since they were taken from the biopsy report (n=524). Missing data on histopathologic lesions are specified in the legend of Figure 1. To calculate percent differences of plasma KIM-1 by histopathologic lesion and clinicopathologic diagnosis, we used multivariable linear regression models. Percent differences in plasma KIM-1 were calculated by raising 2 to the power of the beta-coefficient, subtracting 1, and multiplying by 100 [(2β – 1) * 100)] for each respective histopathologic lesion or clinicopathologic diagnosis from the linear regression model. Recognizing that proteinuria may be a collider variable35 in analyses of histopathologic lesions and clinicopathologic diagnosis with plasma KIM-1, our primary models omitted proteinuria as an adjustment variable. Exploratory analyses were conducted with further adjustment for proteinuria.
Figure 1. Differences in plasma KIM-1 by histopathologic lesion.

Boxplots show differences between grades of adjudicated histopathologic lesions and plasma KIM-1 on a log scale. Boxplots show median and interquartile range (IQR) of plasma KIM-1 measured using a monoclonal detection antibody. Whiskers represent the IQR of the lower and upper quartile (25th and 75th percentile, respectively). P-values obtained from Wilcoxon or Kruskal-Wallis tests: p<0.001 for interstitial fibrosis/tubular atrophy, acute tubular injury, inflammation in the fibrosed and non-fibrosed interstitium, global glomerulosclerosis, mesangial expansion, arterial and arteriolar sclerosis; p=0.7 for glomerular inflammation; and p=0.4 for segmental sclerosis. 3 individuals had KIM-1 levels below the lower limit of quantification (LLOQ) which were imputed as LLOQ/2. Data on histopathologic lesion scores were missing for n=7 (global glomerulosclerosis), n=13 (segmental sclerosis), n=10 (interstitial fibrosis/tubular atrophy), n=12 (acute tubular injury), n=12 (inflammation in the non-fibrosed interstitium), n=10 (inflammation in the fibrosed interstitium), n=14 (mesangial expansion), n=16 (arterial sclerosis), and n=12 (arteriolar sclerosis).
We performed time-to-event analyses to examine the risk of the outcomes, evaluating plasma KIM-1 as a continuous variable (log base 2) in both cohorts and as tertiles (lowest tertile as the reference group) and quintiles (lowest quintile as the reference group) in the BKBC Study and the CRIC Study, respectively. We used cause-specific Cox proportional hazards regression to investigate multivariable-adjusted associations between plasma KIM-1 and outcomes. Subdistribution hazard competing risk models were not used because the goal was to understand the biologic associations of plasma KIM-1 with each outcome. Our multivariable adjustment strategy was hierarchical and based on biological and clinical plausibility of covariates as potential confounders of the association between plasma KIM-1 and outcomes. In the BKBC Study, model 1 was stratified by site and adjusted for age, sex, and race; model 2 included model 1 and further adjusted for natural log transformed proteinuria and primary clinicopathologic diagnosis, angiotensin-converting enzyme inhibitor (ACEi)/angiotensin II receptor blocker (ARB) and immunosuppression/corticosteroids use; model 3 included model 2 and further adjusted for eGFR. We tested for statistical interaction between plasma KIM-1 and primary clinicopathologic diagnosis (glomerulopathy vs. other diagnoses) for both outcomes through multiplicative interaction terms. In the CRIC Study, model 1 was stratified by site and adjusted for age, sex, race, diabetes, any cardiovascular disease (CVD), smoking status, systolic blood pressure (SBP), and body mass index (BMI); model 2 included model 1 and further adjusted for hemoglobin, natural log transformed UACR, and medications including ACEi/ARBs, statins, and antiplatelets drugs; model 3 included model 2 and further adjusted for eGFR. There were less than 5% missing data in both cohorts. Based on the Little Test,36 missing data were considered as missing completely at random and complete case analysis was used for the analyses. All analyses were performed with SAS version 9.4 (SAS Institute, Cary, NC) and STATA 15.0 (STATACorp, College Station, TX). All statistical tests were 2-sided, and P < 0.05 was considered statistically significant.
Results
BKBC Study
Baseline Characteristics
Table 1 summarizes the baseline characteristics of the BKBC participants by tertiles of plasma KIM-1. The median (IQR) plasma KIM-1 concentration using the monoclonal detection antibody from Enzo Life Sciences was 210.2 (102.2 – 452.4) pg/ml. The mean age was 53 ± 17 years and the mean eGFR was 56 ± 36 ml/min/1.73m2. The most common primary clinicopathologic diagnoses were proliferative glomerulonephritis (29.1%), non-proliferative glomerulopathy (18.3%), advanced glomerulosclerosis (11.3%), and diabetic nephropathy (11.1%). Plasma KIM-1 correlated negatively with eGFR (rs = −0.6, p < 0.001) and positively with proteinuria (rs = 0.3, p < 0.001) (Supplemental Table 2).
Table 1.
Baseline characteristics of the BKBC Study participants by tertiles of plasma KIM-1.
| Plasma KIM-1 (pg/mL)* | All participants 4.6 – 17305.3 (n = 524) |
Tertile 14.6 – 125.2 (n = 174) |
Tertile 2125.4 – 342.3 (n = 175) |
Tertile 3343.3 – 17305.3 (n = 175) |
p-value |
|---|---|---|---|---|---|
|
| |||||
| Clinical Characteristics | |||||
|
| |||||
| Age, years | 52.8 ± 16.6 | 46.4 ± 15.3 | 55.7 ± 16.0 | 56.4 ± 16.4 | <0.001 |
| Female | 267 (51.0) | 104 (59.8) | 83 (47.4) | 80 (45.7) | 0.02 |
| Race | 0.2 | ||||
| White | 334 (63.7) | 104 (59.8) | 118 (67.4) | 112 (64.0) | |
| Black | 104 (19.8) | 32 (18.4) | 32 (18.3) | 40 (22.9) | |
| Other | 86 (16.4) | 38 (21.8) | 25 (14.3) | 23 (13.1) | |
| eGFR, ml/min/1.73m2 | 56.4 ± 36.0 | 81.8 ± 34.1 | 54.2 ± 31.6 | 33.4 ± 23.5 | <0.001 |
| Proteinuria, g/g creatinine | 1.6 [0.4 – 4.0] | 1.0 [0.3 – 2.6] | 1.5 [0.3 – 3.7] | 2.6 [0.7 – 6.7] | <0.001 |
|
| |||||
| Reason for Biopsy ** | |||||
|
| |||||
| Proteinuria | 306 (58.4) | 110 (63.2) | 113 (64.6) | 83 (47.4) | 0.001 |
| Hematuria | 127 (24.2) | 57 (32.8) | 38 (21.7) | 32 (18.3) | 0.004 |
| Nephrotic syndrome | 70 (13.4) | 24 (13.8) | 18 (10.3) | 28 (16.0) | 0.3 |
| Nephritic syndrome | 12 (2.3) | 7 (4.0) | 3 (1.7) | 2 (1.1) | 0.2 |
| Abnormal eGFR | 271 (51.7) | 56 (32.2) | 93 (53.1) | 122 (69.7) | <0.001 |
|
| |||||
| Primary Clinicopathologic Diagnosis | <0.001 | ||||
|
| |||||
| Proliferative glomerulonephritis | 150 (29.1) | 64 (37.0) | 45 (26.3) | 41 (24.0) | |
| Non-proliferative glomerulopathy | 94 (18.3) | 34 (19.7) | 36 (21.1) | 24 (14.0) | |
| Advanced glomerulosclerosis | 58 (11.3) | 18 (10.4) | 25 (14.6) | 15 (8.8) | |
| Diabetic nephropathy | 57 (11.1) | 4 (2.3) | 14 (8.2) | 39 (22.8) | |
| Vascular disease | 44 (8.5) | 12 (6.9) | 23 (13.5) | 9 (5.3) | |
| Paraprotein-related disease | 42 (8.2) | 17 (9.8) | 11 (6.4) | 14 (8.2) | |
| Tubulointerstitial disease | 39 (7.6) | 3 (1.7) | 11 (6.4) | 25 (14.6) | |
| Other | 31 (6.0) | 21 (12.1) | 6 (3.5) | 4 (2.3) | |
|
| |||||
| Comorbidities | |||||
|
| |||||
| Diabetes mellitus | 120 (22.9) | 13 (7.5) | 42 (24.0) | 65 (37.1) | <0.001 |
| Hypertension | 279 (53.2) | 65 (37.4) | 98 (56.0) | 116 (66.3) | <0.001 |
| Systemic lupus erythematosus | 77 (14.7) | 49 (28.2) | 16 (9.1) | 12 (6.9) | <0.001 |
| Hepatitis C | 10 (1.9) | 2 (1.1) | 0 (0.0) | 8 (4.6) | 0.005 |
| Malignancy | 82 (15.6) | 18 (10.3) | 35 (20.0) | 29 (16.6) | 0.04 |
|
| |||||
| Medications | |||||
|
| |||||
| ACEi/ARB | 243 (46.4) | 77 (44.3) | 92 (52.6) | 74 (42.3) | 0.1 |
| MRA | 12 (2.3) | 1 (0.6) | 5 (2.9) | 6 (3.4) | 0.2 |
| Calcium channel blockers | 138 (26.3) | 38 (21.8) | 45 (25.7) | 55 (31.4) | 0.1 |
| Beta-blockers | 168 (32.1) | 30 (17.2) | 58 (33.1) | 80 (45.7) | <0.001 |
| Immunosuppression | 94 (17.9) | 34 (19.5) | 31 (17.7) | 29 (16.6) | 0.8 |
| Corticosteroids | 97 (18.5) | 42 (24.1) | 32 (18.3) | 23 (13.1) | 0.03 |
Data presented as mean ± standard deviation, median [IQR], and count with frequencies (%) for binary and categorical variables. ACEi, angiotensin converting enzyme inhibitor; ARB, angiotensin II receptor blocker; MRA, mineralocorticoid receptor blocker; eGFR, estimated glomerular filtration rate
Monoclonal detection antibody; 3 individuals had KIM-1 levels below the lower limit of quantification (LLOQ) which were imputed as LLOQ/2.
Reasons for biopsy were based on the referring nephrologist’s judgement. Percentages do not add to 100 as there may have been more than one reason for kidney biopsy. Percentages do not add to 100 as there may have been more than one reason for kidney biopsy
Nine individuals had insufficient tissue to make a clinicopathologic diagnosis
Associations of Plasma KIM-1 with Histopathologic Lesions and Clinicopathologic Diagnoses
Figure 1 shows the differences in plasma KIM-1 concentrations across severity scores of histopathologic lesions. Compared to participants with less severe lesions, plasma KIM-1 levels were significantly higher in participants with more severe interstitial fibrosis/tubular atrophy, acute tubular injury, inflammation in the non-fibrosed and fibrosed interstitium, global glomerulosclerosis, mesangial expansion, and arterial and arteriolar sclerosis. Figure 2 shows adjusted differences in plasma KIM-1 concentration across histopathologic lesions and clinicopathologic diagnoses using multivariable linear regression models adjusted for age, sex, race, and eGFR. Plasma KIM-1 levels were significantly higher in participants with more severe acute tubular injury, more severe mesangial expansion, and the presence of inflammation in the non-fibrosed interstitium compared to participants with less severe lesions (Figure 2A). Plasma KIM-1 levels remained significantly higher in participants with more severe acute tubular injury and the presence of inflammation in the non-fibrosed interstitium even after adjustment for proteinuria (Supplemental Table 3). Compared to participants with minor abnormalities or relatively preserved kidney parenchyma, plasma KIM-1 levels were significantly higher in participants with diabetic nephropathy, tubulointerstitial disease, non-proliferative glomerulopathy, and proliferative glomerulonephritis (Figure 2B). Plasma KIM-1 levels remained significantly higher in participants with diabetic nephropathy and tubulointerstitial disease compared to participants with minor abnormalities or relatively preserved kidney parenchyma even after adjustment for proteinuria (Supplemental Table 4).
Figure 2. Adjusted differences in plasma KIM-1 by histopathologic lesion and clinicopathologic diagnosis.

Models were fit using log-transformed plasma KIM-1 as the outcome and histopathologic lesion (A) and clinicopathologic diagnosis (B) as the predictor variable. Percent differences are derived from linear regression models of log base 2 plasma KIM-1. Percent differences in plasma KIM-1 were calculated by raising 2 to the power of the beta-coefficient, subtracting 1, and multiplying by 100, [(2β – 1) * 100)], for each respective histopathologic lesion or clinicopathologic diagnosis from the linear regression model. Each individual model was adjusted for age, sex, race, and eGFR.
A. Reference is absence of lesion for glomerular inflammation (p=0.2), segmental sclerosis (p=0.2), inflammation in the non-fibrosed interstitium (p=0.01);
Reference is none/mild lesion severity for mesangial expansion (p=0.006), acute tubular injury (p<0.001), arterial sclerosis (p=0.8), arteriolar sclerosis (p=0.7);
Reference is 0-25% of cortical volume affected for global glomerulosclerosis (p=0.8), inflammation in the fibrosed interstitium (p=0.6), interstitial fibrosis/tubular atrophy (p=0.1).
B. Reference is near-normal tissue (comprised of individuals with minor abnormalities or relatively preserved parenchyma). Proliferative glomerulonephritis, p=0.01; non-proliferative glomerulopathy, p<0.001; paraprotein disease, p=0.07; diabetic nephropathy, p<0.001; vascular disease, p=0.5; tubulointerstitial disease, p<0.001; advanced glomerulosclerosis, p=0.3.
Associations between Plasma KIM-1 and Risks of Adverse Clinical Outcomes
During a median follow-up time of 5 years, 124 individuals progressed to kidney failure and 85 participants died. Table 2 shows the multivariable-adjusted associations of plasma KIM-1 with subsequent kidney failure and death in the BKBC Study. In the fully adjusted model, each doubling of plasma KIM-1 was associated with a 1.19-fold increased risk of progression to kidney failure. Participants in the highest tertile of plasma KIM-1 had a higher risk of kidney failure, but the confidence interval crossed 1.0 in the fully adjusted model. There was no evidence of statistical interaction between plasma KIM-1 and primary clinicopathologic diagnosis for progression to kidney failure (P for interaction: 0.3) or death (P for interaction: 0.7) (Supplemental Table 5). Higher levels of plasma KIM-1 were associated with a higher risk of death, but these associations were not statistically significant after multivariable adjustment including eGFR (Table 2).
Table 2.
Plasma KIM-1 and the risk of adverse clinical outcomes in the BKBC Study.
| Plasma KIM-1 | Events | Events per 100 person-years | Model 1 | p-value | Model 2 | p-value | Model 3 | p-value |
|---|---|---|---|---|---|---|---|---|
| Kidney Failure | ||||||||
| Tertile 1 | 19 | 2.1 | Reference | Reference | Reference | |||
| Tertile 2 | 37 | 5.4 | 2.39 (1.35 – 4.22) |
0.003 | 2.35 (1.30 – 4.27) |
0.005 | 1.40 (0.77 – 2.56) |
0.3 |
| Tertile 3 | 68 | 12.3 | 5.12 (3.00 – 8.73) |
<0.001 | 4.62 (2.57 – 8.32) |
<0.001 | 1.46 (0.80 – 2.64) |
0.2 |
| Continuous, per doubling KIM-1 | 124 | 5.8 | 1.66 (1.48 – 1.87) |
<0.001 | 1.65 (1.43 – 1.90) |
<0.001 | 1.19 (1.03 – 1.38) |
0.02 |
| Mortality | ||||||||
| Tertile 1 | 12 | 1.2 | Reference | Reference | Reference | |||
| Tertile 2 | 31 | 3.6 | 2.17 (1.09 – 4.29) |
0.03 | 1.93 (0.96 – 3.90) |
0.07 | 1.46 (0.72 – 2.96) |
0.3 |
| Tertile 3 | 42 | 5.0 | 2.92 (1.51 – 5.68) |
0.002 | 2.63 (1.30 – 5.34) |
0.007 | 1.48 (0.69 – 3.18) |
0.3 |
| Continuous, per doubling KIM-1 | 85 | 3.1 | 1.25 (1.09 – 1.43) |
0.001 | 1.23 (1.06 – 1.44) |
0.008 | 1.05 (0.89 – 1.25) |
0.6 |
Results are expressed as hazard ratios (95% CI); Kidney failure, defined as initiation of kidney replacement therapy.
Model 1 is stratified by site and adjusted for age, sex, and race.
Model 2 is model 1 and further adjusts natural log transformed proteinuria, primary clinicopathologic diagnosis, ACEi/ARB use, and immunosuppression/corticosteroids.
Model 3 is model 2 and further adjusts for eGFR
Abbreviations: ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin II receptor blocker; eGFR, estimated glomerular filtration rate
CRIC Study
Baseline Characteristics
Table 3 summarizes baseline characteristics of 3,800 CRIC Study participants by quintiles of plasma KIM-1 concentration. The median (IQR) plasma KIM-1 concentration using the polyclonal detection antibody from R&D was 1073.0 (557.5 – 2052.4) pg/ml. The mean age was 58 ± 11 years, and the mean eGFR was 45 ± 17 ml/min/1.73m2. Participants who had higher levels of plasma KIM-1 were more likely to be non-white, had a greater prevalence of diabetes mellitus and CVD, had higher SBP, and had lower hemoglobin. Plasma KIM-1 had a negative correlation with eGFR (rs = −0.3, p < 0.001) and a positive correlation with UACR (rs = 0.4, p < 0.001) (Supplemental Table 2).
Table 3.
Baseline characteristics of the CRIC Study participants by quintiles of plasma KIM-1.
| Plasma KIM-1 (pg/mL)* | All Participants 16.5 – 965023.4 (n = 3800) |
Quintile 1 12.6 – 484.5 (n = 760) |
Quintile 2 485.6 – 830.0 (n = 760) |
Quintile 3 831.7 – 1374.0 (n = 760) |
Quintile 4 1374.8 – 2444.4 (n = 759) |
Quintile 5 2446.1 – 965023.4 (n = 761) |
p-value |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Clinical characteristics | |||||||
|
| |||||||
| Age, years | 57.7 ± 11.0 | 56.4 ± 11.3 | 58.5 ± 10.6 | 58.4 ± 11.0 | 58.1 ± 11.1 | 56.8 ± 10.9 | <0.001 |
| Female | 1694 (44.6) | 336 (44.2) | 341 (44.9) | 347 (45.7) | 344 (45.3) | 326 (42.8) | 0.8 |
| Race | |||||||
| White | 1587 (41.8) | 360 (47.4) | 371 (48.8) | 339 (44.6) | 283 (37.3) | 234 (30.7) | <0.001 |
| Black | 1570 (41.3) | 310 (40.8) | 287 (37.8) | 312 (41.1) | 317 (41.8) | 344 (45.2) | |
| Hispanic | 489 (12.9) | 55 (7.2) | 82 (10.8) | 78 (10.3) | 129 (17.0) | 145 (19.1) | |
| Other | 154 (4.1) | 35 (4.6) | 20 (2.6) | 31 (4.1) | 30 (4.0) | 38 (5.0) | |
| eGFR (ml/min/1.73m2) | 44.9 ± 16.9 | 53.0 ± 18.4 | 47.7 ± 16.1 | 43.5 ± 15.7 | 41.1 ± 15.0 | 39.1 ± 15.4 | <0.001 |
| UACR, mg/g | 51.6 [8.6 – 453.5] | 12.4 [5.1 – 62. 1] | 25.7 [6.6 – 161.5] | 53.8 [9.5 – 335.5] | 153.2 [18.3 – 873.3] | 433.8 [34.0 – 2227.0] | <0.001 |
| Hemoglobin, g/dL | 12.6 ± 1.8 | 13.2 ± 1.6 | 12.8 ± 1.8 | 12.6 ± 1.7 | 12.3 ± 1.7 | 12.1 ± 1.8 | <0.001 |
| Total cholesterol | 183.6 ± 45.5 | 177.5 ± 38.9 | 179.0 ± 40.7) | 180.8 ± 43.5 | 186.0 ± 46.6 | 194.6 ± 54.1 | <0.001 |
|
| |||||||
| Comorbidities | |||||||
|
| |||||||
| Hypertension | 3271 (86.1) | 600 (78.9) | 652 (85.8) | 667 (87.8) | 654 (86.2) | 698 (91.7) | <0.001 |
| Diabetes mellitus | 1842 (48.5) | 236 (31.1) | 320 (42.1) | 352 (46.3) | 444 (58.5) | 490 (64.4) | <0.001 |
| SBP, mm Hg | 128.6 ± 22.2 | 122.3 ± 20.2 | 125.0 ± 19.4 | 126.9 ± 21.0 | 131.5 ± 22.2 | 137.1 ± 25.0 | <0.001 |
| BMI, kg/m2 | 32.1 ± 7.8 | 31.5 ± 7.2 | 32.3 ± 7.7 | 32.3 ± 8.2 | 32.3 ± 7.9 | 32.2 ± 7.8 | 0.2 |
| Any CVD | 1269 (33.4) | 189 (24.9) | 270 (35.5) | 254 (33.4) | 277 (36.5) | 279 (36.7) | <0.001 |
| Current smoking | 496 (13.1) | 83 (10.9) | 98 (12.9) | 88 (11.6) | 113 (14.9) | 114 (15.0) | 0.06 |
|
| |||||||
| Medications ** | |||||||
|
| |||||||
| ACEI/ARB use | 2594 (68.8) | 495 (65.4) | 506 (66.9) | 541 (71.9) | 513 (68.2) | 539 (71.4) | 0.02 |
| Statins | 2087 (55.3) | 368 (48.6) | 401 (53.0) | 430 (57.2) | 443 (58.9) | 445 (58.9) | <0.001 |
| Antiplatelets | 1731 (45.9) | 327 (43.2) | 369 (48.8) | 338 (44.9) | 338 (44.9) | 359 (47.5) | 0.2 |
Data presented as mean ± standard deviation, median [IQR], and count with frequencies (%) for binary and categorical variables. ACEi, angiotensin converting enzyme inhibitor; ARB, angiotensin II receptor blocker; SBP, systolic blood pressure; CVD, cardiovascular disease; BMI, body mass index; eGFR, estimated glomerular filtration rate; UACR, urine albumin-to-creatinine ratio
Polyclonal detection antibody; 2 individuals had KIM-1 levels below the lower limit of quantification (LLOQ) which were imputed as LLOQ/2.
Data on medications were missing for 28 individuals.
Associations between Plasma KIM-1 and Risks of Adverse Clinical Outcomes
During a median follow-up time of 11.5 years, 1153 participants progressed to kidney failure and 1356 participants died. Table 4 shows multivariable-adjusted associations of plasma KIM-1 with subsequent kidney failure and death in the CRIC Study. In the fully adjusted model, each doubling of plasma KIM-1 was associated with a 1.10-fold increased risk for progression to kidney failure. Participants in the highest quintile of plasma KIM-1 had a 1.58-fold increased risk of progression to kidney failure compared to participants in the lowest quintile. Higher levels of plasma KIM-1 were associated with a higher risk of death, but this association was attenuated and no longer statistically significant after further adjustment for eGFR (Table 4).
Table 4.
Plasma KIM-1 and the risks of adverse clinical outcomes in the CRIC Study.
| Plasma KIM-1 | Events | Events per 100 person-years | Model 1 | p-value | Model 2 | p-value | Model 3 | p-value |
|---|---|---|---|---|---|---|---|---|
| Kidney Failure | ||||||||
| Quintile 1 | 100 | 1.3 | Reference | Reference | Reference | |||
| Quintile 2 | 140 | 1.9 | 1.35 (1.05 – 1.75) |
0.02 | 1.03 (0.79 – 1.34) |
p=0.8 | 1.03 (0.79 – 1.35) |
0.8 |
| Quintile 3 | 211 | 3.2 | 2.15 (1.69 – 2.73) |
<0.001 | 1.29 (1.01 – 1.66) |
p=0.04 | 1.14 (0.89 – 1.46) |
0.3 |
| Quintile 4 | 292 | 5.1 | 2.81 (2.23 – 3.55) |
<0.001 | 1.40 (1.10 – 1.79) |
p=0.01 | 1.22 (0.95 – 1.55) |
0.1 |
| Quintile 5 | 410 | 8.8 | 4.64 (3.70 – 5.81) |
<0.001 | 1.86 (1.45 – 2.37) |
p<0.001 | 1.58 (1.24 – 2.02) |
<0.001 |
| Continuous, per doubling KIM-1 | 1153 | 3.6 | 1.24 (1.21 – 1.28) |
<0.001 | 1.13 (1.09 – 1.17) |
p<0.001 | 1.10 (1.06 – 1.15) |
<0.001 |
| Mortality | ||||||||
| Quintile 1 | 182 | 2.2 | Reference | Reference | Reference | |||
| Quintile 2 | 235 | 3.0 | 1.08 (0.89 – 1.31) |
0.4 | 1.04 (0.85 – 1.27) |
0.7 | 1.01 (0.83 – 1.24) |
0.9 |
| Quintile 3 | 269 | 3.5 | 1.28 (1.06 – 1.55) |
0.01 | 1.14 (0.93 – 1.39) |
0.2 | 1.04 (0.85 – 1.27) |
0.7 |
| Quintile 4 | 317 | 4.5 | 1.45 (1.20 – 1.75) |
<0.001 | 1.23 (1.01 – 1.50) |
0.04 | 1.14 (0.93 – 1.39) |
0.2 |
| Quintile 5 | 353 | 5.0 | 1.63 (1.35 – 1.97) |
<0.001 | 1.23 (1.00 – 1.51) |
0.05 | 1.13 (0.92 – 1.39) |
0.2 |
| Continuous, per doubling KIM-1 | 1356 | 3.6 | 1.09 (1.06 – 1.13) |
<0.001 | 1.04 (1.00 – 1.08) |
0.03 | 1.03 (0.99 – 1.07) |
0.1 |
Results are expressed as hazard ratios (95% CI); Kidney failure, defined as initiation of kidney replacement therapy
Model 1 is stratified by site and adjusted for age, sex, race, diabetes, any CVD, smoking status, SBP, and BMI.
Model 2 is model 1 and further adjusts for ACEi/ARB use, statins, antiplatelet drugs, hemoglobin, and natural log transformed UACR.
Model 3 is model 2 and further adjusts for eGFR
Abbreviations: ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin II receptor blocker; SBP, systolic blood pressure; CVD, cardiovascular disease; BMI, body mass index; eGFR, estimated glomerular filtration rate; UACR, urine albumin-to-creatinine ratio
Discussion
Kidney tubular injury leads to a decrease in kidney function, most commonly associated with the clinical syndrome of AKI, but tubular injury may be interconnected to the development and progression of CKD.37 In two prospective cohort studies that included common forms of CKD as well as more rare glomerular, interstitial, and vascular diseases, we found a consistent association between higher levels of plasma KIM-1, a sensitive marker of tubular injury, and an increased risk of progression to kidney failure. In individuals who underwent a native kidney biopsy with adjudicated histopathology, we confirmed that higher plasma KIM-1 levels were associated with more severe acute tubular injury, and we further identified associations between higher levels of plasma KIM-1 with tubulointerstitial inflammation and more severe mesangial expansion independent of kidney function. The results provide additional evidence for the importance of kidney tubular injury in glomerulopathies and more common forms of CKD, such as diabetic kidney disease. Our findings demonstrate that plasma KIM-1 has prognostic value across a spectrum of kidney diseases and may serve as a tool in the non-invasive assessment of kidney tubular injury.
KIM-1 is strongly expressed in proximal tubules in response to hypoxic or nephrotoxic events.1, 6, 38 While tubular KIM-1 expression is nearly undetectable in healthy kidney tissue, multiple human studies have demonstrated an elevated expression not only in AKI but also in the setting of chronic kidney damage.4, 6, 7, 39 Rodent models of CKD showed that sustained KIM-1 upregulation triggers the development of fibrosis with associated monocyte chemotactic protein 1 (MCP-1)-mediated macrophage chemotaxis.8 Our finding of an association between higher levels of plasma KIM-1 and more severe acute tubular injury and tubulointerstitial inflammation confirms similar observations in animal and smaller cross-sectional human studies, which identify the tubules as the primary site for KIM-1 expression.6, 11 Our findings of higher plasma KIM-1 levels in those with more severe mesangial expansion could reflect downstream tubular ischemia from abnormal glomerular capillaries and post-glomerular blood flow.10 The observed associations of plasma KIM-1 with tubulointerstitial inflammation and mesangial expansion provide additional evidence that tubular injury and sustained KIM-1 expression may be implicated in the progressive cycle of interstitial inflammation and fibrosis, glomerular ischemia, and microvascular rarefaction.8, 40
Urinary and plasma KIM-1 are correlated but each may reflect separate processes of injury, which could lead to differences in the prognostic value for kidney disease progression.25 Urinary KIM-1 was associated with lower eGFR and greater proteinuria across 5 cohorts of individuals with CKD,2 which was similar in magnitude to the associations of plasma KIM-1 with eGFR and proteinuria in the present study. Some, but not all, studies of urinary KIM-1 in kidney diseases have shown that KIM-1 was a risk factor for future kidney disease progression after adjustment for eGFR and proteinuria.13, 14, 16, 20–24 Studies that examined plasma KIM-1 in individuals with diabetes have demonstrated prognostic value for the development or progression of CKD.26–28 Two studies that measured both plasma and urinary KIM-1 showed that higher plasma, but not urinary, KIM-1 was associated with an increased risk of early kidney function decline in individuals with type 1 and type 2 diabetes without CKD.26, 28 In participants with early and advanced diabetic kidney disease from the ACCORD and VA NEPHRON-D clinical trials, respectively, higher levels of plasma KIM-1 were associated with worsening kidney function over time.27 Similar associations have been reported between higher levels of plasma KIM-1 and the development of CKD in healthy individuals29 and progression to kidney failure in individuals with moderate to severe CKD.41 One potential explanation for the discordance in prognostic value for urinary and plasma KIM-1 is that urinary KIM-1 may reflect the extent of acute tubular damage and KIM-1 production in response to injury, whereas plasma KIM-1 may better mirror the integral of injury over time and continuing production, being released into systemic circulation in response to a loss of tubular cell polarity.25, 29, 42, 43
We extend the findings in the prior studies of plasma KIM-1 by demonstrating a consistent association of higher plasma KIM-1 levels with progression to kidney failure in cohorts of individuals with both common and more rare forms of kidney disease. Notably, our findings were similar in individuals with and without glomerulopathies, which is consistent with results from the Chronic Kidney Disease in Children (CKID) Study that included both glomerular diseases as well as congenital anomalies of the kidney and urinary tract.44 While we found a nominally higher risk of death in individuals with higher levels of plasma KIM-1, these associations were attenuated by eGFR and no longer statistically significant after adjustment.
Our prospective findings confirm that plasma KIM-1 may serve as a tool to enrich selection of high-risk individuals with kidney disease for enrollment in clinical trials.27, 45, 46 Although there was overlap of plasma KIM-1 levels across histopathologic severity grades, our findings suggest that plasma KIM-1 may be able to provide a non-invasive estimate of more severe acute tubular injury independent of kidney function, which warrants further investigation. While we are unable to demonstrate a causal association of plasma KIM-1 with histopathology or CKD progression, confirmation of our findings may show that plasma KIM-1 is able to serve as a tool to assess for kidney tubular injury6, 47 or to monitor response to treatment in the setting of a clinical trial48 for the development of therapies across a variety of chronic kidney diseases.49–51
Strengths of our study are the inclusion of two large cohorts of individuals with established kidney disease. Both studies have a long duration of follow-up and low rates of missing outcome data. Specific strengths of the BKBC Study include biopsy-confirmation of the underlying cause of kidney disease, adjudicated histopathologic scores on lesion severity, and the inclusion of individuals with a diverse range of kidney diseases. Specific strengths of the CRIC Study include the adjustment for a large number of potential confounders. Our study has limitations that warrant consideration. The ascertainment of variables differed across the two cohorts, but we were able to adjust for major confounders, including eGFR and proteinuria in both cohorts. Plasma KIM-1 was measured using different platforms, so direct comparisons of absolute levels cannot be done across the two cohorts, which may limit generalizability of the findings. Although we did not find a statistically significant association between the amount of time in storage and plasma KIM-1 concentration using multivariable regression analyses, we cannot rule out that the duration of storage time may have impacted the stability of plasma KIM-1 which could have altered plasma KIM-1 levels. Given the heterogeneity of the BKBC Study, the number of individuals within each clinicopathologic diagnostic category was small, particularly for rare diseases. We did not measure urinary KIM-1 in the BKBC Study and therefore we are unable to compare plasma KIM-1 with urinary KIM-1 as a biomarker of histopathologic lesions. While the effect sizes between plasma KIM-1 and adverse clinical outcomes were relatively modest, additional research is warranted to determine if multi-marker panels that include plasma KIM-1 are able to improve risk estimation for CKD progression. Although we adjusted for a number of clinical predictors, there may be residual confounding from unmeasured confounders.
In conclusion, we found that higher levels of plasma KIM-1 are associated with more severe tubulointerstitial and mesangial lesions and progression to kidney failure in two cohort studies of individuals with kidney diseases. The strong associations with histopathologic lesions and progression to kidney failure suggest that plasma KIM-1 may potentially serve as a kidney-specific marker to enhance the estimation of the risk of progression to kidney failure across a diverse spectrum of kidney diseases.
Supplementary Material
Item S1: Supplementary methods.
Table S1. Histopathologic scoring system for light microscopy in the BKBC Study.
Table S2. Spearman correlation coefficients between plasma KIM-1, kidney function, and proteinuria.
Table S3. Adjusted differences in plasma KIM-1 by histopathologic lesions in the BKBC Study.
Table S4. Adjusted differences in plasma KIM-1 by clinicopathologic diagnoses in the BKBC Study.
Table S5. Associations of plasma KIM-1 with adverse clinical outcomes by primary clinicopathologic diagnosis in the BKBC Study.
Acknowledgements:
The authors thank the staff and participants of the studies for their important contributions and invaluable assistance.
Support: This work was supported by the CKD Biomarker Consortium by National Institutes of Health (NIH) grants U01DK85649, U01DK085673, U01DK085660, U01DK085688, U01DK085651, and U01DK085689, DK072381, R37DK39773, the National Center for Advancing Translational Sciences of the NIH (UL1TR000003), and the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). I.M.S. is supported by the American Philosophical Society Daland Fellowship in Clinical Investigation. A.S. is supported by NIH grant K23DK120811, NIDDK Kidney Precision Medicine Project Opportunity Pool grant under U2CDK114886, and core resources from the George M. O’Brien Kidney Research Center at Northwestern University (NU-GoKIDNEY) P30DK114857. S.S.W. is also supported by NIH grants UH3DK114915, U01DK085660, U01DK104308, R01DK103784, and R21DK119751. Funding for the CRIC Study was obtained under a cooperative agreement from NIDDK (U01DK060990, U01DK060984, U01DK061022, U01DK061021, U01DK061028, U01DK060980, U01DK060963, and U01DK060902). In addition, this work was supported in part by: the Perelman School of Medicine at the University of Pennsylvania Clinical and Translational Science Award NIH/NCATS UL1TR000003, Johns Hopkins University UL1 TR-000424, University of Maryland GCRC M01 RR-16500, Clinical and Translational Science Collaborative of Cleveland, UL1TR000439 from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research, Michigan Institute for Clinical and Health Research (MICHR) UL1TR000433, University of Illinois at Chicago CTSA UL1RR029879, Tulane University Translational Research in Hypertension and Renal Biology P30GM103337, Kaiser Permanente NIH/NCRR UCSF-CTSI UL1 RR-024131. Funding for the BKBC was supported by the NIH grant R01DK093574 (S.S.W.). This work was conducted with support from Harvard Catalyst. The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH award UL1TR001102) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the NIH. The funders of the studies had no role in study design; data collection, analysis, or reporting; or the decision to submit for publication.
Financial Disclosure: A.S. reports personal fees from Horizon Therapeutics, PLC, AstraZeneca, CVS Caremark, and Tate & Latham (medicolegal consulting). J.V.B. is cofounder and holds equity in Goldfinch Bio, is co-inventor on KIM-1 patents assigned to Partners Healthcare, received grant funding from Boehringer Ingelheim and has received consulting income related to biomarkers from Biomarin, Aldeyra, Angion, PTC, Praxis, and Sarepta. S.S.W. reports personal fees from Public Health Advocacy Institute, CVS, Roth Capital Partners, Kantum Pharma, Mallinckrodt, Wolters Kluewer, GE Health Care, GSK, Mass Medical International, Barron and Budd (vs. Fresenius), JNJ, Venbio, Strataca, Takeda, Cerus, Pfizer, Bunch and James, Harvard Clinical Research Institute (aka Baim), and grants and personal fees from Allena Pharmaceuticals. The remaining authors declare that they have no relevant financial interests.
Article Information
Chronic Kidney Disease Biomarkers Consortium Investigators: Aside from authors Ramachandran (Chair, Steering Committee), Bonventre (co-Principal Investigator [PI]), Waikar (co-PI), Sabbisetti, Hsu (PI), Liu (PI), Feldman (PI), Xie, Zhang, Mifflin (Laboratory), Nelson, Kimmel, and Kusek, the CKD Biomarkers Consortium Investigators comprise: Cedars Sinai Medical Center: Jennifer Van Eyk, PhD, Dawn Chen, PhD, Qin Fu, PhD; Cincinnati Children’s Hospital Medical Center: Hermine Brunner, MD; Columbia University College of Physicians and Surgeons: Vivette D’Agati, MD, Jonathan Barasch, MD; Johns Hopkins University: Josef Coresh, MD, PhD (PI), Casey Rebholz, PhD; Division of Research, Kaiser Permanente Northern California: Alan S. Go, MD; Icahn School of Medicine at Mount Sinai: Erwin Bottinger, MD (PI), Avelino Teixeira, PhD, Ilse Daehn, PhD; Northwestern University: Mark Molitch, MD (PI), Daniel Batlle, MD; Ohio State University: Brad Rovin, MD (PI), Haifeng Wu, MD; Tufts Medical Center: Andrew S. Levey, MD, Lesley A. Inker, MD, MS, Meredith Foster, PhD; University of Louisville: Jon Klein, MD, PhD; University of Minnesota: Michael Mauer, MD (PI), Paola Fioretto, MD, PhD, Gary Nelsestuen, PhD, John H. Eckfeldt, MD, PhD, Amy Karger, MD, PhD; University of Padova: Paola Fioretto, MD, PhD; University of Pennsylvania (Coordinating Center): Shawn Ballard, MS, Krista Whitehead, MS, Phyllis Gimotty, PhD, Haochang Shou, PhD, Kellie Ryan; University of Utah: Tom Greene, PhD.
CRIC Study Investigators: Aside from authors Feldman, Chen, Nelson, the CRIC Study Investigators comprise Lawrence J. Appel, MD, MPH, Alan S. Go, MD, James P. Lash, MD, Mahboob Rahman, MD, Panduranga S. Rao, MD, Vallabh O Shah, PhD, MS, Raymond R. Townsend, MD, and Mark L. Unruh, MD, MS
Footnotes
Peer Review: Received Oct 09, 2020. Evaluated by 4 external peer reviewers and a statistician, with editorial input from an Acting Editor-in-Chief (Editorial Board Member Emmanuel Burdmann, MD, PhD). Accepted in revised form May 3, 2021. The involvement of an Acting Editor-in-Chief to handle the peer-review and decision-making processes was to comply with AJKD’s procedures for potential conflicts of interest for editors, described in the Information for Authors & Journal Policies.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ichimura T, Bonventre JV, Bailly V, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is upregulated in renal cells after injury. J Biol Chem. 1998;273(7): 4135–4142. [DOI] [PubMed] [Google Scholar]
- 2.Waikar SS, Sabbisetti V, Arnlov J, et al. Relationship of proximal tubular injury to chronic kidney disease as assessed by urinary kidney injury molecule-1 in five cohort studies. Nephrol Dial Transplant. 2016;31(9): 1460–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song J, Yu J, Prayogo G, et al. Understanding kidney injury molecule 1: a novel immune factor in kidney pathophysiology. Am J Transl Res. 2019;11: 1219–1229. [PMC free article] [PubMed] [Google Scholar]
- 4.Nauta FL, Boertien WE, Bakker SJ, et al. Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care. 2011;34(4): 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a specific and sensitive biomarker of kidney injury. Scand J Clin Lab Invest Suppl. 2008;241: 78–83. [DOI] [PubMed] [Google Scholar]
- 6.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62(1): 237–244. [DOI] [PubMed] [Google Scholar]
- 7.Schroppel B, Kruger B, Walsh L, et al. Tubular expression of KIM-1 does not predict delayed function after transplantation. J Am Soc Nephrol. 2010;21(3): 536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humphreys BD, Xu F, Sabbisetti V, et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest. 2013;123(9): 4023–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuehn E, Park K, Somlo S, Bonventre J. Kidney injury molecule-1 expression in murine polycystic kidney disease. Am J Physiol Renal Physiol. 2002;283: F1326–1336. [DOI] [PubMed] [Google Scholar]
- 10.van Timmeren MM, van den Heuvel MC, Bailly V, Bakker SJ, van Goor H, Stegeman CA. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol. 2007;212(2): 209–217. [DOI] [PubMed] [Google Scholar]
- 11.Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV. Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Renal Physiol. 2004;286(3): F552–563. [DOI] [PubMed] [Google Scholar]
- 12.Vaidya VS, Niewczas MA, Ficociello LH, et al. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-beta-D-glucosaminidase. Kidney Int. 2011;79(4): 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen SE, Andersen S, Zdunek D, Hess G, Parving HH, Rossing P. Tubular markers do not predict the decline in glomerular filtration rate in type 1 diabetic patients with overt nephropathy. Kidney Int. 2011;79(10): 1113–1118. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen SE, Reinhard H, Zdunek D, et al. Tubular markers are associated with decline in kidney function in proteinuric type 2 diabetic patients. Diabetes Res Clin Pract. 2012;97(1): 71–76. [DOI] [PubMed] [Google Scholar]
- 15.Fu WJ, Xiong SL, Fang YG, et al. Urinary tubular biomarkers in short-term type 2 diabetes mellitus patients: a cross-sectional study. Endocrine. 2012;41(1): 82–88. [DOI] [PubMed] [Google Scholar]
- 16.Peralta CA, Katz R, Bonventre JV, et al. Associations of urinary levels of kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) with kidney function decline in the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2012;60(6): 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Driver TH, Katz R, Ix JH, et al. Urinary kidney injury molecule 1 (KIM-1) and interleukin 18 (IL-18) as risk markers for heart failure in older adults: the Health, Aging, and Body Composition (Health ABC) Study. Am J Kidney Dis. 2014;64(1): 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlsson AC, Calamia M, Riserus U, et al. Kidney injury molecule (KIM)-1 is associated with insulin resistance: results from two community-based studies of elderly individuals. Diabetes Res Clin Pract. 2014;103(3): 516–521. [DOI] [PubMed] [Google Scholar]
- 19.Sarnak MJ, Katz R, Newman A, et al. Association of urinary injury biomarkers with mortality and cardiovascular events. J Am Soc Nephrol. 2014;25(7): 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panduru NM, Sandholm N, Forsblom C, et al. Kidney injury molecule-1 and the loss of kidney function in diabetic nephropathy: a likely causal link in patients with type 1 diabetes. Diabetes Care. 2015;38(6): 1130–1137. [DOI] [PubMed] [Google Scholar]
- 21.Foster MC, Coresh J, Bonventre JV, et al. Urinary Biomarkers and Risk of ESRD in the Atherosclerosis Risk in Communities Study. Clin J Am Soc Nephrol. 2015;10(11): 1956–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fufaa GD, Weil EJ, Nelson RG, et al. Association of urinary KIM-1, L-FABP, NAG and NGAL with incident end-stage renal disease and mortality in American Indians with type 2 diabetes mellitus. Diabetologia. 2015;58(1): 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu CY, Xie D, Waikar SS, et al. Urine biomarkers of tubular injury do not improve on the clinical model predicting chronic kidney disease progression. Kidney Int. 2017;91(1): 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhotra R, Katz R, Jotwani V, et al. Urine Markers of Kidney Tubule Cell Injury and Kidney Function Decline in SPRINT Trial Participants with CKD. Clin J Am Soc Nephrol. 2020;15(3): 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabbisetti VS, Waikar SS, Antoine DJ, et al. Blood Kidney Injury Molecule-1 Is a Biomarker of Acute and Chronic Kidney Injury and Predicts Progression to ESRD in Type I Diabetes. J Am Soc Nephrol. 2014; 25(10): 2177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowak N, Skupien J, Niewczas MA, et al. Increased plasma kidney injury molecule-1 suggests early progressive renal decline in non-proteinuric patients with type 1 diabetes. Kidney Int. 2016;89(2): 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coca SG, Nadkarni GN, Huang Y, et al. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J Am Soc Nephrol. 2017;28(9): 2786–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak N, Skupien J, Smiles AM, et al. Markers of early progressive renal decline in type 2 diabetes suggest different implications for etiological studies and prognostic tests development. Kidney Int. 2018;93(5): 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulz CA, Engstrom G, Nilsson J, et al. Plasma kidney injury molecule-1 (p-KIM-1) levels and deterioration of kidney function over 16 years. Nephrol Dial Transplant. 2020;35(2): 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldman H, Appel L, Chertow G, et al. The Chronic Renal Insufficiency Cohort (CRIC) Study: Design and Methods. J Am Soc Nephrol. 2003;14(7 Suppl 2): S148–153. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava A, Palsson R, Kaze A, et al. The Prognostic Value of Histopathologic Lesions in Native Kidney Biopsy Specimens: Results from the Boston Kidney Biopsy Cohort Study. J Am Soc Nephrol. 2018;29(8): 2213–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2): 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9): 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson AH, Yang W, Hsu CY, et al. Estimating GFR among participants in the Chronic Renal Insufficiency Cohort (CRIC) Study. Am J Kidney Dis. 2012;60(2): 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Staplin N, Haynes R, Herrington WG, et al. Smoking and Adverse Outcomes in Patients With CKD: The Study of Heart and Renal Protection (SHARP). Am J Kidney Dis. 2016;68(3): 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Little RJ. A test of missing completely at random for multivariate data with missing values. J Am Stat Assoc. 1988;83(404): 1198–1202. [Google Scholar]
- 37.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371(1): 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song J, Yu J, Prayogo GW, et al. Understanding kidney injury molecule 1: a novel immune factor in kidney pathophysiology. Am J Transl Res. 2019;11(3): 1219–1229. [PMC free article] [PubMed] [Google Scholar]
- 39.Han WK, Alinani A, Wu CL, et al. Human kidney injury molecule-1 is a tissue and urinary tumor marker of renal cell carcinoma. J Am Soc Nephrol. 2005;16(4): 1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82(2): 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alderson HV, Ritchie JP, Pagano S, et al. The Associations of Blood Kidney Injury Molecule-1 and Neutrophil Gelatinase-Associated Lipocalin with Progression from CKD to ESRD. Clin J Am Soc Nephrol. 2016;11(12): 2141–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sutton TA. Alteration of microvascular permeability in acute kidney injury. Microvasc Res. 2009;77(1): 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaidya VS, Waikar SS, Ferguson MA, et al. Urinary biomarkers for sensitive and specific detection of acute kidney injury in humans. Clin Transl Sci. 2008;1(3): 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenberg JH, Abraham AG, Xu Y, et al. Plasma Biomarkers of Tubular Injury and Inflammation Are Associated with CKD Progression in Children. J Am Soc Nephrol. 2020;31(5): 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamanouchi M, Skupien J, Niewczas MA, et al. Improved clinical trial enrollment criterion to identify patients with diabetes at risk of end-stage renal disease. Kidney Int. 2017;92(1): 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parikh CR, Liu C, Mor MK, et al. Kidney Biomarkers of Injury and Repair as Predictors of Contrast-Associated AKI: A Substudy of the PRESERVE Trial. Am J Kidney Dis. 2019;75: 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaidya VS, Ozer JS, Dieterle F, et al. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28(5): 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dekkers CCJ, Petrykiv S, Laverman GD, Cherney DZ, Gansevoort RT, Heerspink HJL. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes Metab. 2018;20(8): 1988–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakris GL, Agarwal R, Chan JC, et al. Effect of Finerenone on Albuminuria in Patients With Diabetic Nephropathy: A Randomized Clinical Trial. JAMA. 2015;314(9): 884–894. [DOI] [PubMed] [Google Scholar]
- 50.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, et al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J Am Soc Nephrol. 2015;26(1): 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuttle KR, Brosius FC 3rd, Adler SG, et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol Dial Transplant. 2018;33(11): 1950–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Item S1: Supplementary methods.
Table S1. Histopathologic scoring system for light microscopy in the BKBC Study.
Table S2. Spearman correlation coefficients between plasma KIM-1, kidney function, and proteinuria.
Table S3. Adjusted differences in plasma KIM-1 by histopathologic lesions in the BKBC Study.
Table S4. Adjusted differences in plasma KIM-1 by clinicopathologic diagnoses in the BKBC Study.
Table S5. Associations of plasma KIM-1 with adverse clinical outcomes by primary clinicopathologic diagnosis in the BKBC Study.