

Abstract
Cell-free nucleic acids (cfNAs) such as short non-coding microRNA (miRNA) and circulating tumor DNA (ctDNA) that reside in bodily fluids have emerged as potential cancer biomarkers. Methods for the rapid, highly specific, and sensitive monitoring of cfNAs in biofluids have, therefore, become increasingly attractive as clinical diagnosis tools. As a next generation technology, we provide a practical guide for an amplification-free, single molecule Förster resonance energy transfer (smFRET)-based kinetic fingerprinting approach termed intramolecular single molecule recognition through equilibrium Poisson sampling, or iSiMREPS, for the rapid detection and counting of miRNA and mutant ctDNA with virtually unlimited specificity and single molecule sensitivity. iSiMREPS utilizes a pair of fluorescent detection probes, wherein one probe immobilizes the target molecules on the surface, and the other probe transiently and reversibly binds to the target to generate characteristic time-resolved fingerprints as smFRET signal that are detected in a total internal reflection fluorescence microscope. Analysis of these kinetic fingerprints enables near-perfect discrimination between specific binding to target molecules and nonspecific background binding. By accelerating kinetic fingerprinting using the denaturant formamide and reducing background signals by removing target-less probes from the surface via toehold-mediated strand displacement, iSiMREPS has been demonstrated to count miR-141 and EGFR exon 19 deletion ctDNA molecules with a limit of detection (LOD) of ~1 and 3 fM, respectively, as well as mutant allele fractions as low as 0.0001%, during a standard acquisition time of only ~10 s per field of view. In this review, we provide a detailed roadmap for implementing iSiMREPS more broadly in research and clinical diagnostics, combining rapid analysis, high specificity, and high sensitivity.
Keywords: TIRF microscopy, intramolecular kinetic fingerprinting, smFRET, rapid diagnostics, amplification-free detection, nucleic acid biomarkers
Graphical Abstract
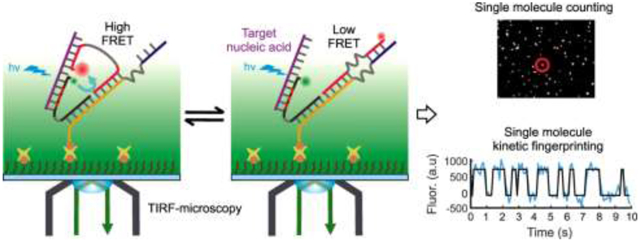
1. Introduction
Analysis of nucleic acids is expanding in significance because of their deep involvement in all biological processes and many disease states. In recent years, cell-free nucleic acids (cfNAs) such as circulating tumor-derived DNA (ctDNA), mRNA, and non-coding RNA found in biofluids (e.g., blood, urine or saliva) have emerged as potential diagnostic and prognostic biomarkers for many different types of human cancers [1]. The concentrations of these nucleic acids in biofluids of cancer patients can differ significantly from those of healthy people. Detecting abnormal sequences and expression levels of these cfNA biomarkers by so-called “liquid biopsy” has been a topic of rising interest for clinical development and shows promise in both the early detection of cancers and monitoring of treatment efficacy [2].
To gain clinical utility, “liquid biopsy” methods should have high sensitivity and specificity, as well as fast analysis speed. However, detection and quantification of cfNAs in biofluids pose several technical challenges. For example, mutant DNA often has allele frequencies that are quite low (e.g., 0.1%–1%) within a large background of wild-type DNA from normal cells [3]. Advanced techniques such as digital PCR [4] and next generation sequencing (NGS) [5] have become the gold standard methods to detect mutant alleles at the lowest allele frequencies, since they routinely detect point mutations present at <1%. However, such amplification-based techniques suffer from several drawbacks, including heat induced chemical damage [6, 7], amplification errors and bias [8], and inefficient amplification of short nucleic acids such as miRNAs [8] that often handicap their specificity and sensitivity. While several amplification- free methods for detecting cfNAs have been reported [9–11], these techniques suffer from limited specificity due to the upper limit of thermodynamics of hybridization that precludes detection of rare alleles.
To address the shortcomings of conventional methods, we previously reported an amplification-free single molecule kinetic fingerprinting approach named single molecule recognition through equilibrium Poisson sampling (SiMREPS) for the highly sensitive and ultraspecific detection of diverse nucleic acid biomarkers [7, 12]. SiMREPS utilizes a surface-tethered high affinity capture probe that is complementary to part of the target molecule and binds it strongly, while a short (8–10 nt), weakly binding fluorescent probe complementary to another part of the target that transiently binds and dissociates to generate blinking fluorescent signals (i.e., kinetic fingerprints) that are recorded by a total internal reflection fluorescence (TIRF) microscope. Continuous recording of the kinetic fingerprints over a time window of 10 min or more showed near-perfect specificity (> 99.99999%) and high sensitivity (limit of detection, or LOD, of ~1 fM) for detecting ctDNAs as well various synthetic and endogenous miRNAs [7, 12]. Despite its high specificity and sensitivity, SiMREPS has an upper speed limit for analyzing a sample because it relies on the binding of free fluorescent probe from solution to the surface-bound target. Improving SiMREPS’s speed in principle simply requires a higher fluorescent probe concentration to reduce the time that a target spends without a probe bound. However, this approach increases diffuse background signal from unbound probe molecules that approach the surface, thereby degrading the signal-to-noise ratio (S/N) until single probe binding events are no longer detectable against background.
To improve SiMREPS for use as a next generation liquid biopsy tool, we increased the speed of kinetic fingerprinting by developing intramolecular SiMREPS, or iSiMREPS, utilizing single molecule Förster resonance energy transfer (smFRET) for detecting single nucleic acid biomarkers (Fig. 1). iSiMREPS is based on a dynamic DNA nanoassembly of a surface tethered anchor and a pair of fluorescent detection probes (Fig. 1). The sensor is immobilized on a surface suitable for TIRF microscopy using an affinity tag (e.g., biotin) at one end of the anchor. One of the fluorescent probes, named the capture probe (CP), strongly and stably surface immobilizes a target molecule. Another fluorescent probe, named the query probe (QP), transiently interacts with the target and a competitor (C) to generate alternating on and off FRET signals (kinetic fingerprint) (Fig. 1). The intramolecular design of iSiMREPS makes detection faster because it engenders high local effective concentrations of target, QP and C, thereby both accelerating the association kinetics of the QP and removing the long unbound dwell times seen in conventional SiMREPS [7, 12]. By optimizing the design of iSiMREPS components using a pair of invaders to remove target-less sensors from the surface, and denaturant (formamide) in the imaging buffer to accelerate dissociation, we achieved a 60-fold faster standard acquisition time (10 s) than conventional (intermolecular) SiMREPS without any compromise in sensitivity or specificity [13]. In this article, we enumerate practical considerations for the use of iSiMREPS to rapidly detect short cfNAs, with both miR-141 and an EGFR exon 19 deletion mutant DNA serving as proof-of-principle examples.
Fig. 1.
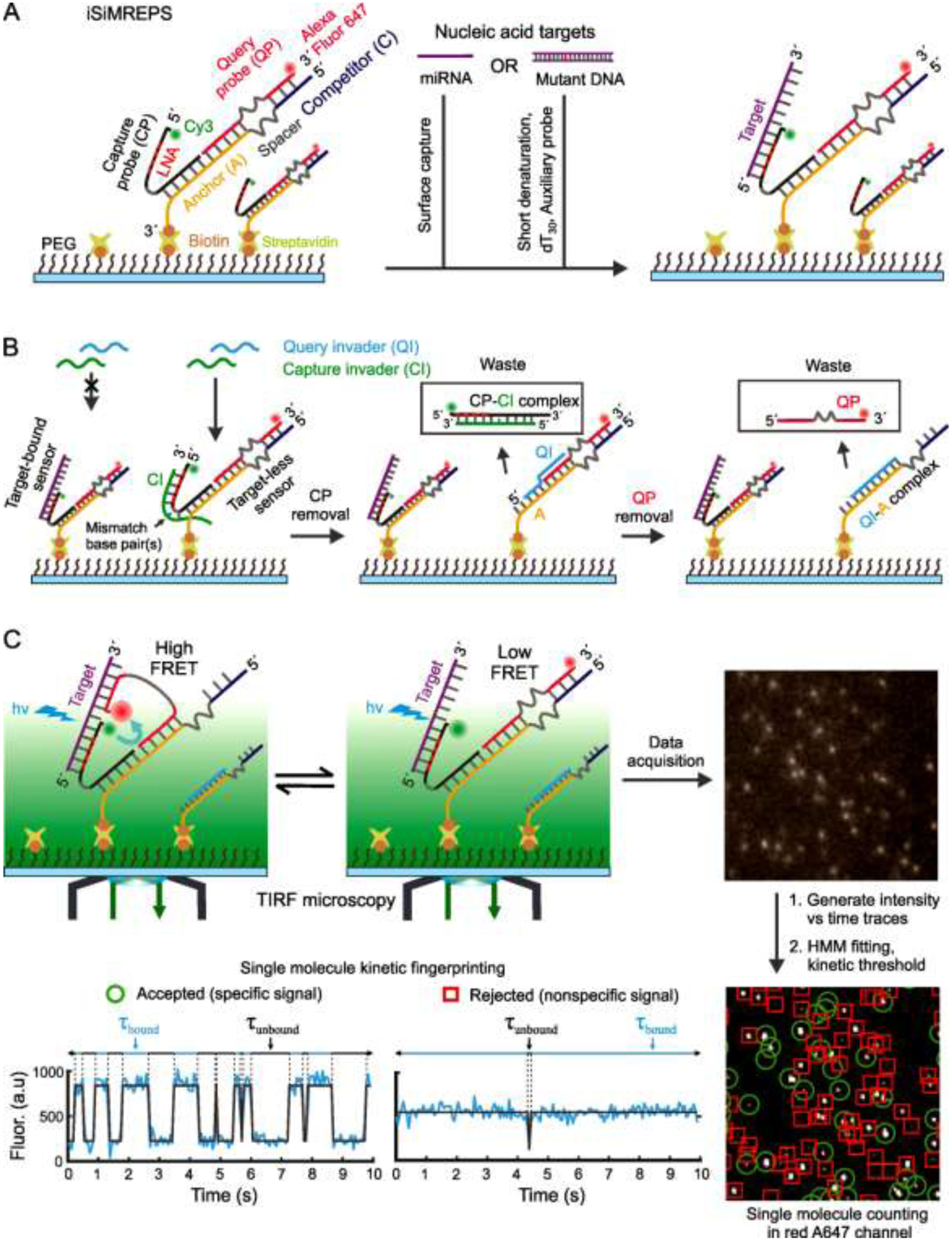
Schematic of smFRET-based intramolecular SiMREPS for digital counting of single nucleic acid molecules. (A) Schematic of iSiMREPS sensor assembly and target capture at the surface. iSiMREPS uses a surface immobilized and intramolecularly assembled FRET pair of fluorescent probes for surface tethering and imaging of single nucleic acid molecules. The 5´ Cy3-labeled capture probe contains several LNA residues for high-affinity, stable capturing of target miRNA or mutant DNA molecules. (B) Mechanism of background improvement by removing non-target bound fluorescent probes from the surface using a pair of capture and query invaders. The capture invader (green) binds with the toehold of non-target-bound capture probe (CP) and invades through the CP-Anchor (A) duplex to remove CP, while query invader (cyan) binds with the exposed toehold on A and displaces QP from A. (C) Mechanism of smFRET signal generation and schematic of data acquisition and processing to obtain single molecule kinetic fingerprints. The A647-labeled query probe interacts transiently and reversibly with the target and competitor sequence and generates a single molecule kinetic fingerprint that represents donor (Cy3) or FRET-mediated-acceptor (A647) emission recorded by a TIRF microscope. A representative field of view (top right corner, not in scale) from TIRF microscopy analyzed with MATLAB programs to identify spots with potential smFRET signals in a field of view (bottom right corner, not to scale) and generate intensity vs time traces (bottom left, cyan). HMM idealization (grey lines) for each intensity vs. time trace and application of kinetic thresholds like the Nb+d, dwell times, S/N, and signal intensity distinguishes target-specific signals (green circle) from nonspecific background signals (red rectangle) (see section 2.7 and 2.8 for details).
2. Materials and Methods
2.1. Single-molecule fluorescence microscopy
iSiMREPS as described here requires a microscope capable of TIRF illumination to record signals only from surface-bound fluorescent probes. In our studies, all experiments are carried out using either a prism-type or objective-type TIRF microscope. Detailed instrumentation for SiMREPS has been reported previously [7, 12, 14, 15]. Briefly, prism-type TIRF (P-TIRF) measurements were made with a microscope based on an Olympus IX-71 frame equipped with a 60× water-immersion objective (Olympus Uplanapo, 1.2NA) with an ICCD (I-Pentamax, Princeton Instruments) or sCMOS (Hamamatsu C13440-20CU) camera for movie recording. A 640 nm red laser (Coherent CUBE 640-100C, 100 mW) and 532 nm green laser (CrystaLaser CL532-150mW-L) is used for excitation of Alexa Fluor 647 (A647) and Cy3 fluorophores, respectively, with an illumination intensity of ~100W/cm2. A dichroic mirror with a cut-off wavelength of 610 nm (Chroma) was used to separate the Cy3 and A647 emission signals with a full-frame acquisition rate of 10Hz. The Cy3 channel image was passed through a bandpass filter (HQ580/60m, Chroma) and the A647 channel was passed through a long-pass filter (HQ655LP, Chroma).
Objective-type TIRF (O-TIRF) measurements were performed using an Olympus IX-81 microscope equipped with a 60× oil-immersion objective (APON 60XOTIRF, 1.49 NA) with both cell^TIRF™ and Z-drift control modules. The O-TIRF microscope uses an EMCCD camera (Evolve 512, Photometrics) to record movies. The smFRET signals were obtained by exciting the Cy3 fluorophore with the green laser at 532 nm at an output power of typically 20–30 mW. For reliably detecting FRET signals with satisfactory signal-to-noise, an illumination intensity of ~50 W/cm2 was typically used, the TIRF angle was adjusted to achieve a calculated evanescent field penetration depth of ~70–85 nm, and a Cy3-A647 FRET dichroic mirror (zt640drc-UF2, Chroma) and emission filter (ET655LP-TRF filter, Chroma) were utilized to permit direct excitation of the donor and selective detection of the acceptor.
2.2. Preparation of slides, coverslips, and sample cell
SiMREPS experiments require a sample cell designed to hold an imaging solution comprising the fluorescent probe and an oxygen scavenger system that limits exposure of the fluorophores to oxygen (which accelerates photobleaching) during imaging. The sample cells typically used for SiMREPS assays vary depending on the microscope used, which is typically a prism-type TIRF (P-TIRF) or objective-type TIRF (O-TIRF) [14, 15]. For P-TIRF experiments, the flow sample cells are commonly sandwiched between a passivated microscope slide (fused silica) and glass coverslip (VWR Micro Cover Glasses 22 × 30 mm, catalog no. 48393-026) for easy set up between a prism and an objective lens. Each microscope slide has a hole drilled on each of two ends that together allow facile exchange of sample solutions and buffers using Tygon tubing (U.S. plastic corporation, 0.020” ID × .060” OD Tygon® ND 100-80, catalog no. 56515) connected to the holes. In contrast, O-TIRF permits an open-top sample geometry that requires only a single passivated glass coverslip (VWR No. 1.5, 24×50 mm, catalog no. 48393-241) for placement of sample wells. The sample wells are constructed from cut pipette tips or 3D printed wells by attaching them to passivated coverslips using epoxy (Ellsworth adhesives, hardman double, catalog no. 4001). Since the smaller surface/volume ratio of these wells focuses the immobilization of analyte to the imaging surface at higher density, O-TIRF reaches greater sensitivity than the thin flow cell used for P-TIRF and is recommended for more sensitive detection of the target.
Prior to assembly of the sample cell, the microscope slide or glass coverslip is cleaned using a previously reported “base piranha” protocol, after which the surface is passivated [7, 15]. Briefly, the slide or coverslip is first thoroughly washed with water and then sonicated in acetone for 10 min followed by 1 M KOH for 20 min to remove aqueous and organic residues from the surface. Next, the slide or coverslip is treated with “base piranha” solution consists of 14.3% v/v of 28–30 wt% NH4OH, and 14.3% v/v of 30–35 wt% H2O2 that is heated to 70–80 °C. In an alternative cleaning procedure, the coverslip is cleaned by applying air plasma for 3 min in a plasma cleaner and is subsequently washed twice with acetone. The clean glass coverslip is then surface functionalized with an aminosilane using a 2% v/v solution of (3-aminopropyl) triethoxysilane (APTES) (Sigma-Aldrich, catalog no. A3648-100ML) in acetone for 20 min. This amine-modified surface is functionalized with a mixture of succinimide esters of biotin-PEG and methoxy-PEG (Laysan Bio, Inc. catalog no. BIO-PEG-SVA-5K-100MG & MPEG-SVA-5K-1g) at a certain ratio (e.g., 1:100 ratio or final concentration of 0.0025 and 0.25 mg/μL in 0.1 M NaHCO3), and residual amines quenched by 30 mg/mL disulfosuccinimidyl tartrate (Soltec Ventures, catalog no. CL107) in 1 M NaHCO3, pH 8.5, to quench any unreacted amine groups. Finally, the coverslips are dried completely under nitrogen flow and stored for up to 2–3 weeks in the dark in a nitrogen-flushed cabinet until further use.
2.3. Single molecule FRET-based intramolecular kinetic fingerprinting for detecting nucleic acids
2.3.1. Basic architecture and working principle of iSiMREPS
The smFRET-based iSiMREPS technique utilizes an intramolecular assembly of a pair of fluorescent probes (i.e., a FRET pair) for digital counting of single nucleic acid molecules. The basic architecture of an iSiMREPS sensor features three DNA oligonucleotides: a biotinylated anchor (A) that hybridizes to the unlabeled ends of a 5′ donor fluorophore (Cy3)-labeled capture probe (CP) and a 3′ acceptor fluorophore (Alexa Fluor 647, A647)-labeled query probe (QP) (Fig. 1A). The two ends of both the CP and QP are separated by poly-thymidine oligonucleotide (poly-dT) spacers that allow for the flexibility necessary for sensor assembly and to generate kinetic fingerprints. The pre-hybridized A, CP and QP are immobilized on a passivated coverslip or slide via a streptavidin-biotin affinity linkage on the 3′ end of the anchor strand A, upon which the nucleic acid target is introduced. The fluorophore-labeled end of the CP is partially modified with locked nucleic acid (LNA) residues, and thus captures target molecules with high affinity. Subsequently, any target-less CP and QP are removed from the surface via toehold mediated strand displacement (TMSD)[16] by a pair of capture and query invaders that reduce background signals before imaging under a TIRF microscope (Fig. 1B). The fluorophore-labeled end of the QP alternates between a target-bound state and a competitor (C)-bound state, where C is a single-stranded DNA (ssDNA) segment that extends from the anchor. When the QP is target-bound, the donor and acceptor fluorophores are in close proximity and a high-FRET signal is generated (Fig. 1C). In contrast, the fluorophores are far apart when the QP is competitor-bound or dissociated from the target and low-FRET signal is observed (Fig. 1C). The repeated alternation between high- and low-FRET signals generates a characteristic kinetic fingerprint that readily and with high confidence distinguishes target-bound sensor molecules from non-target molecules or background signals (Fig. 1C). We demonstrate here that the intramolecular arrangement of iSiMREPS allows ~60-fold faster identification [13] and counting of single nucleic acid molecules than intermolecular SiMREPS probing [7, 12].
2.4. Design of the oligonucleotide components for iSiMREPS
2.4.1. Anchor A
The role of the anchor in iSiMREPS is to bind both the CP and QP while surface-tethering the sensor through a streptavidin-biotin affinity linkage to a passivated sample cell. Effective detection and signal generation requires that (1) the anchor maintain stable surface-tethering and (2) the anchor-CP and anchor-QP binding be stable. In general, the streptavidin-biotin interaction is stable enough to maintain strong surface tethering for the assay. The most common methods for increasing the thermodynamic stability of the duplex DNA (dsDNA) include either locked nucleic acid (LNA) modification or an increase in GC content. The anchor in our current iSiMREPS sensor design consists of two GC-rich (≥75%) 12-nt segments with melting temperatures (Tm) of ~70 °C designed to not form DNA secondary structures and instead assemble stably with the CP and QP. The anchor strand also contains an extended segment separated from the QP/CP binding region by a poly-thymidine DNA oligonucleotide spacer of empirically optimized length (dT3–18). The extended segment is partially or fully complementary to the query sequence that binds with the target and acts as a competitor to the query-target interaction to allow for modulation of the high- and low-FRET dwell times (Fig. 1). Its design is described in more detail below.
2.4.2. Capture probe CP
The CP serves two functions in iSiMREPS: (1) target capture and (2) acting as a detection probe. Strong, high-affinity target capture is necessary for both basic sensor function and high sensitivity. This can be achieved by using a relatively long CP-target sequence (~20 bp). While a long CP is amenable for detecting long nucleic acid targets like ctDNA [1], it is unsuitable for short nucleic acids like miRNAs (~22 nt) where it is difficult to accommodate both a long CP the and QP (Fig. 2A and 3A). Therefore, our iSiMREPS sensor design uses an 11–12 nt CP with 4 LNA residues for stable, high affinity capture of the target. Since CPs sometimes form secondary structures, it is critical to carefully choose which residues are LNA-modified to avoid creating stable structures that compromise CP binding to the anchor or target. This is done with the assistance of NUPACK [17, 18] and the vendor Qiagen’s oligo optimizer tool, which allow for visualization of intrinsic secondary structures and assessment of their stability, as well as indicating the potential for self-binding with a self-structure score. In general, LNA placement should avoid residues involved in secondary structures, and should also avoid GC residues that typically pose a higher risk for self-complementarity. The target-bound 5′ end of the CP is Cy3-labeled so that Cy3 acts as the FRET donor in the target-bound state (Fig. 1). The CPs with LNA residues were purchased from either Integrated DNA Technologies (IDT, https://www.idtdna.com) with a 5′ Cy3 modification or from Qiagen (https://www.qiagen.com/us) with a 5′ amino modification for subsequent labeling with Cy3 monoreactive dye (GE life sciences, catalog no. PA23001), followed by purification via ethanol precipitation (https://www.qiagen.com/us/resources).
Fig. 2.
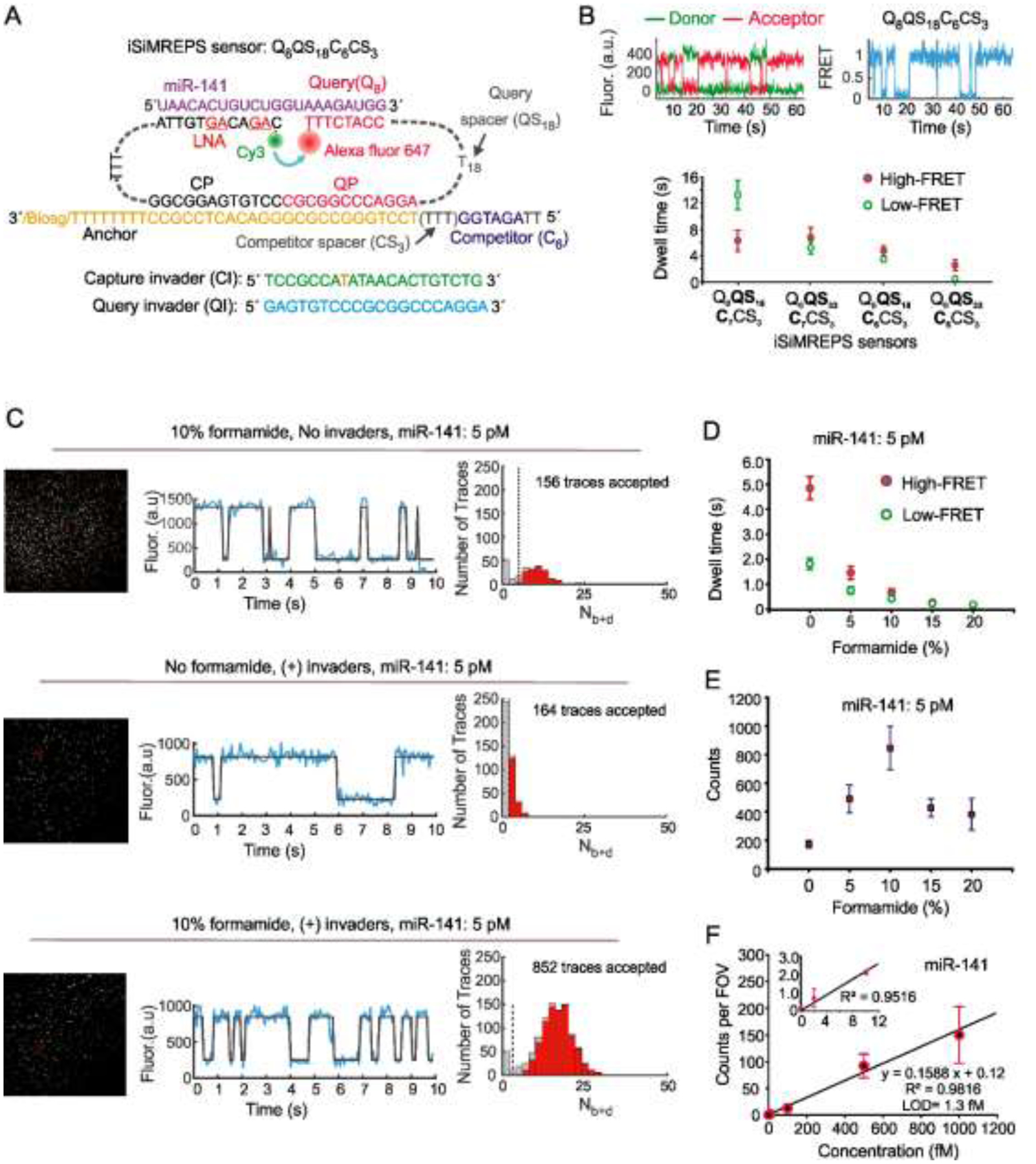
Development of smFRET based intramolecular SiMREPS approach for detection of miR-141. (A) Schematic of the optimal iSiMREPS sensor design for detecting miR-141 detection. (B) A representative single molecule kinetic trace and average dwell times of high- and low-FRET states for different sensor designs. Dwell time averages were calculated using an exponential decay fitting model. (C) A representative field of view, single-molecule kinetic trace, and Nb+d histogram with and without application of invaders and in presence of 0 and 10% formamide for detecting miR-141. (D, E) The average dwell times of high- and low-FRET states and the average number of candidate miR-141 bound molecules per FOV for different formamide v/v% conditions with a standard data acquisition of 10 s after applying optimized kinetic filtering parameters. (F) Standard curve for miR-141 showing an LOD of ~1.3 fM. Linear fits were constrained to a y-intercept of accepted counts at 0 fM and R2 = 0.9816. All data are presented as the mean ± s.d of n = ≥ 3 independent measurements. For each independent measurement, 10 FOVs were collected. Panels A, B, D and E were adapted with permission from reference 13.
Fig. 3.
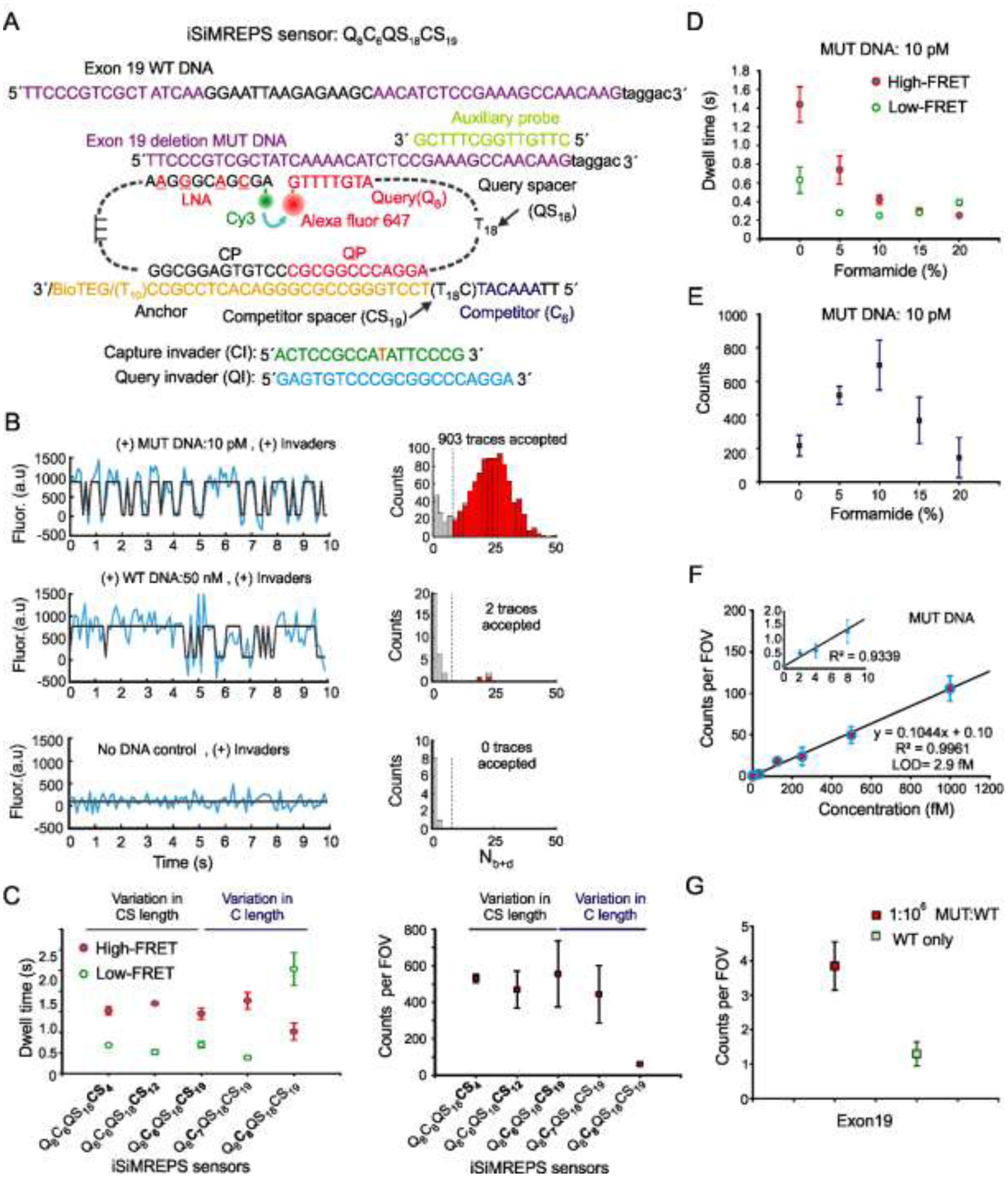
Development of smFRET-based iSiMREPS for detection of EGFR exon 19 deletion mutant DNA. (A) Schematic of the sensor design of exon 19 deletion mutant and WT DNA detection. (B) Representative single-molecule kinetic fingerprints and histograms of the number of candidate molecules per field-of-view (FOV) showing a given number of binding and dissociation events (Nb+d) after applying thresholds for FRET intensity, signal-to-noise, and dwell times of target bound and non-target-bound states in presence of 10 pM exon 19 deletion mutant DNA (top), 50 nM WT DNA (middle), and no DNA control (bottom). (C) The average dwell times spent in the high-FRET and low-FRET states for different iSiMREPS sensors designs and accepted counts per FOV at a standard acquisition of 10 s. Lifetime averages were calculated using an exponential decay fitting model. (D, E) The average dwell times of high- and low-FRET states and the number of candidate mutant DNA bound molecules per FOV for each formamide v/v% condition with a standard data acquisition of 10 s after applying kinetic thresholds. (F) Standard curve for exon 19 deletion mutant DNA showing an LOD of ~2.9 fM. Linear fits were constrained to a y-intercept of accepted counts at 0 fM, yielding R2 values = 0.9961. (G) Comparison of counts from low MUT allelic fraction and WT only conditions for determining specificity. The specificity obtained was 99.9999% over the MUT fraction of 0.0001%. All data are presented as the mean ± s.d of n = ≥ 3 independent measurements. For each independent measurement, 10 FOVs were collected. Panels A, D, E and G were adapted with permission from reference 13.
2.4.3. Query probe QP
Single-molecule kinetic fingerprinting approaches require a fluorescent probe that can undergo transient and reversible binding and dissociation with the target. In general, SiMREPS uses an 8–10-nt fluorescent probe to generate kinetic fingerprints in 4× PBS (Phosphate-buffered saline) (40 mM Na2HPO4, 7.2 mM KH2PO4, pH 7.4, 548 mM NaCl, 10.8 mM KCl) at ambient room temperature [7, 12]. For iSiMREPS, the QPs used typically have an 8-nt segment (Tm = ~25 °C) for transient binding and dissociation with their target (miRNA or mutant DNA). The Tm of QPs were calculated using IDT oligo analyzer (https://www.idtdna.com/calc/analyzer) at 25°C and at concentrations of 1μM oligo and 600 mM Na+ ions, using the complementary segments that hybridize with the target. The QP for detecting mutant DNA is selected based on its ability to discriminate mutant (MUT) from wild-type (WT) DNA based on the following equation 1[19]:
| (1) |
where Qmax ,therm is the maximum theoretical discrimination, ΔΔG0 is the difference in the Gibbs free energy of the QP hybridizing with either MUT or WT DNA target. The free energy of hybridization is calculated using the web software NUPACK [17, 18]. More detailed guidelines for designing SiMREPS query or fluorescent probes have been discussed elsewhere [15]. The A647-labeled QPs were purchased with HPLC purification from IDT (https://www.idtdna.com). The two working ends of a QP are separated by a poly-thymidine spacer of length dT18–33 to allow sufficient flexibility [20] for switching between high- and low-FRET states (Fig. 2A and 3A).
2.4.4. Competitor C as part of the Anchor A
Due to the intramolecular arrangement of the iSiMREPS sensor, the effective local concentration of probes is estimated to be ~50 μM based on a previous study of proximity-induced intramolecular DNA strand displacement assay[21], which renders the target-bound (high-FRET) state very favorable. To reduce this bias without losing target specificity, iSiMREPS sensors use a short 6–8-nt competitor (with a sequence analogous to the target) extended from the anchor A that transiently and reversibly binds with the 8-nt QP, thus allowing fine tuning of smFRET state dwell times (Fig. 3). To accelerate kinetic fingerprinting, it is best to avoid a fully complementary competitor-query pair that often favors the competitor-bound (and thus not target-bound) state because of the antiparallel arrangement of QP and C in the same DNA duplex (Fig. 3). A 6-nt competitor was found to work best with an 8-nt QP for detecting both miRNA and mutant DNA. The anchor strand A includes an ssDNA spacer separating the competitor and CP/QP binding regions to allow sufficient flexibility for the competitor-query interaction, as described under the anchor design.
2.4.5. Auxiliary probes
The term auxiliary probe is used here for a short ssDNA (~14 nt, Tm = ~50 °C) that can be employed in SiMREPS assays to promote efficient capture of target and suppress unwanted secondary structure in unbound regions on long target strands to maximize sensitivity and specificity [7, 15]. An example for a relatively long nucleic acid biomarker is EGFR exon 19 deletion mutant DNA that is found in biofluids predominantly as ~70–200 bp fragments [1, 3] (Fig. 3A). The use of an auxiliary probe for long duplex DNA targets has a couple of advantages: Firstly, it binds with the melted ssDNA strand originating from dsDNA during sample preparation and helps to maintain the DNA in single-stranded form in solution, rendering it more easily captured by the CP. Secondly, the binding of an auxiliary probe to the target can minimize unwanted secondary structure and maximize target capture and by extension, specificity, and sensitivity.
2.4.6. Invaders
Since the iSiMREPS approach uses two immobilized fluorescent probes, crosstalk from the donor fluorophore into the acceptor detection channel and a small amount of direct excitation of the acceptor fluorophore can result in high background. This is especially problematic when a high surface density of sensors is required for efficient target capture and high sensitivity. However, the iSiMREPS sensor design allows us to remove most non-target-bound fluorescent probes from the surface before imaging under a TIRF microscope by utilizing toehold mediated strand displacement (TMSD)[16] executed by a pair of capture and query invader DNA oligonucleotides (Fig. 1B). Firstly, the capture invader (CI) uses a 9–15-nt complementary region (Fig. 2A and 3A) to bind a toehold on the CP exposed only on a non-target bound sensor. It then invades 6–8-nt into the 12-bp CP-anchor duplex to displace the CP from the anchor and reveal an 8-nt toehold that the query invader (QI) can bind. Secondly, the QI binds to this newly exposed toehold and fully invades the 12-bp QP-anchor duplex to remove the QP from the anchor. The CI typically has a single or full mismatch with the poly-dT spacer of the CP to prevent target removal using that spacer as a toehold. Once completed, the removed probes are washed out so that background is reduced for imaging.
2.5. Analyte scope
In principle, single molecule kinetic fingerprinting can detect any analyte molecules that can be surface captured in a manner that leaves a region of the analyte free to interact with a (short) fluorescent probe. Conventional (intermolecular) SiMREPS uses a single fluorescent probe to generate kinetic fingerprinting and has been successfully applied to detect diverse analytes including miRNAs, ctDNAs, proteins, and small molecules as well as to measure the activity of enzymes [22]. The intramolecular nature of iSiMREPS, on the other hand, requires the analyte to accommodate a FRET pair of fluorescent probes to generate a kinetic fingerprint from the continuously surface-immobilized sensor. Specific detection is achieved via an intramolecular conformational change rather than a binding event of an external fluorescent probe. So far, iSiMREPS has been implemented for the rapid, sensitive, and highly specific detection of nucleic acids including a miRNA (miR-141), and a ctDNA (an EGFR exon 19 deletion mutant). The scope of iSiMREPS analytes can potentially be further expanded to any analyte that can accommodate a pair of fluorescent probes that repeatedly change their relative positioning in a detectable way.
2.6. iSiMREPS assay preparation
2.6.1. O-TIRF assay
The O-TIRF experiment uses a sample cell made, e.g., from cut pipette tips that are glued onto a biotin-PEG and m-PEG passivated glass coverslip (see Section 2.2) on demand. Each cell or well can optionally be washed first with ~ 200 μL of T50 (10 mM Tris-HCl pH 8.0, 50 mM NaCl) to remove potential contaminants and rehydrate the surface. Next, approximately 40–50 μL of streptavidin (Thermo Fisher Scientific, catalog no. S-888) is added at a concentration of 0.1–0.5 mg/mL for a 10–20 min incubation; this creates stably surface-linked multivalent streptavidin affinity tags with binding sites available for capturing the biotinylated sensor. Excess streptavidin is removed by washing the cell 3 times with 4× PBS. The concentration and incubation time with streptavidin need to be optimized for desired density of affinity tags on the surface, and thus sensors for superior sensitivity. The time and concentration mentioned above should work well for most assays.
The next step is the preparation and introduction of sensor into the sample cell. The anchor, CP and QP are typically assembled in a PCR tube at ~400 nM each in 4× PBS and subjected to thermocycler heating at 95 °C for 3 min, 72 °C for 7 min, and then cooling to 25 °C for 20 min, followed by 4 °C until ready for use. The concentrated sensor is then diluted to the desired concentration of ~10–25 nM and 100 μL of the diluted sensor is added to the sample cell and incubated for about 30–45 min. The surface density of sensors and subsequent capture of the target is dictated by concentration and incubation time of the sensor and it thus critical to be optimized. The sample cell is washed typically 3 times using 4× PBS to remove excess materials in between any successive steps in the assay. Overall, the O-TIRF iSiMREPS assay requires approximately 2.5–3 hours to prepare a sample ready for data acquisition.
The iSiMREPS sensors described in this study detect miR-141 and EFGR exon 19 deletion mutant DNA in buffer. The miR-141 target sample is prepared in 4× PBS at the desired concentrations (2 fM-10 pM), optionally with 2 μM of the carrier dT30. 100 μL of target is then added to the sample cell and incubated for 90 min for efficient capture by the surface-immobilized sensors. In an alternative assay strategy, miR-141 target is preassembled with the sensor (A, CP and QP) following a very similar procedure described in section 2.6.1 with ~200 pM and 5 pM as the final sensor and miR-141 concentrations, respectively. By contrast, mutant DNA exists as dsDNA in biofluids and therefore requires a few additional target preparation steps. Firstly, to provide a more authentic context, the synthetic double-stranded exon 19 deletion mutant and wild-type DNA substrates are prepared by annealing complementary single-stranded oligonucleotides (of 44 nt length) at 1 μM final concentration in annealing buffer (10 mM Tris-HCl, pH 8.0, at 25 °C supplemented with 50 mM NaCl and 1 mM EDTA), heating at 95 °C for 5 min, cooling to room temperature for 25 min, and finally keeping at 4 °C for 10 min before storage at −20 °C for further use. Secondly, the EGFR exon 19 mutant and wild-type DNA samples are prepared in a 100 μL solution at their desired concentrations (1.96 fM–100 pM) along with 100 nM of auxiliary probe and 2 μM of the carrier dT30. The 100 μL target sample is added to the sample cell and incubated for 90 min to maximize capturing of targets on surface. We have found a 90 min target incubation sufficient to obtain maximal sensitivity [13]; however, for new targets, this variable may be subject to optimization to obtain the best performance.
After flushing the target solutions from the cell and washing with 4× PBS buffer, the sample cell is treated with capture and query invaders at a concentration of in large (at least 100-fold) excess (e.g., 1–2 μM invaders for 10 nM sensor) over the sensor for 20 min before imaging under a TIRF microscope. This step ensures the removal of non-target-bound sensors from the surface via TMSD[16] (see Section 2.4.6), which reduces the background signals significantly. The final step of the assay before imaging is to prepare the imaging buffer with an oxygen scavenger system (OSS) and denaturant (formamide, 0–20% v/v) in 4× PBS. The OSS consists of 1 mM Trolox (ACROS Organics, catalog no. AC218940050), 5 mM 3,4-dihydroxybenzoate, and 50 nM protocatechuate dioxygenase (Sigma Aldrich, catalog no. P8279-25UN). This OSS serves to protect fluorophores from oxidation, photobleaching and blinking as well as ensure accurate FRET signal measurements. Approximately 200 μL of imaging buffer is added to a sample cell just prior to imaging with an O-TIRF microscope.
2.6.2. P-TIRF assay
P-TIRF assays use the slide sandwich sample cell outlined in Section 2.2 to exchange sample materials and buffers while providing space to seat a prism for imaging. This cell is first incubated with 150 μL of 1 mg/mL biotinylated Bovine Serum Albumin (bBSA) (Thermo Fischer, 25mg ImmunoPure) for 10 min to passivate the surface with bBSA as the source of biotin for surface tethering. Unbound bBSA is washed out with T50 buffer (10 mM Tris-HCl pH 8.0 at 25 °C, 50 mM NaCl), 1 mg/mL streptavidin is then injected into the chamber and incubated for 10 min to establish the biotin-streptavidin linkage, upon which unbound streptavidin is washed out with 4× PBS (Phosphate-buffered saline, pH 7.4 at 25 °C). iSiMREPS sensors were then assembled at ~200 nM initial concentration in 4× PBS buffer with miR-141 target at a 1.000:1.125:1.125:1.250 ratio for A, CP, QP, and target respectively. This solution was heated at 70 °C for 7 min in a metal bath and then cooled by holding at ambient room temperature for 20–25 min. This solution was then diluted to ~100 pM final concentration and injected into the chamber at 150 μL volume for 10 min incubation to allow surface capture by binding to open streptavidin sites established by prior steps. Finally, an imaging buffer containing an OSS outlined in the previous section is filled into the cell prior to imaging with a P-TIRF microscope.
2.7. Microscope Imaging
2.7.1. O-type TIRF Imaging
When performing O-TIRF imaging, the laser power, camera settings, and acquisition time are especially important variables to control. Since iSiMREPS neither requires nor allows probe exchange, photobleaching of either fluorophore in a sensor will render it non-functional. The laser power must thus be chosen carefully to permit sufficient S/N without significant photobleaching during the observation window. An appropriate camera exposure time is also necessary because this dictates how much light is available per frame and how accurately transitions and short dwell times can be captured. The chosen exposure time must be long enough to provide sufficient S/N to identify a target and also short enough to accurately record any transitions so that the FRET states are clearly distinguishable. Finally, a sufficiently long acquisition time must be set so that any target molecule signal collected is readily discriminated from background signal. A shorter acquisition time can also be set when aiming to optimize sensor performance for a specific analysis speed. iSiMREPS experiments typically use 20–30 mW of laser power, a 60–100 ms exposure time, and a 10 s acquisition time window for imaging. Once appropriate parameters are chosen, the sample cell can be focused, the laser turned on, and movies collected with the help of the software to set TIRF mode (e.g., using the cell^TIRF™ Illuminator by Olympus), focus the sample and collect movies (Metamorph), and control the laser output (Coherent Connection). For each experiment, a total of 10 FOVs were collected to improve the chance of detecting a statistically significant set of molecules by running a journal in Metamorph which follows a predefined stage-scanning protocol where each pair of consecutive FOVs is separated by 134 nm (equivalent to the width or height of one FOV) and a 2s delay time is imposed when changing the FOV to allow the autofocus system to re-establish focus.
2.7.2. P-type TIRF Imaging
As with O-TIRF imaging, it is critical to control the laser power, camera settings, and acquisition time in P-TIRF imaging. Since the P-TIRF microscope outlined in Section 2.1 has 2 channel recording, more detailed kinetic information can be obtained as both donor and acceptor signals can be observed and inspected for FRET-based anti-correlation. A longer acquisition time can thus be beneficial as it can allow for thorough and accurate information on FRET states and kinetic behavior of an iSiMREPS sensor. Brief direct excitation of the acceptor at low power (~3 mW) is also beneficial for selecting high-quality traces as outlined in Section 2.8.2. Additionally, a bead slide must be imaged prior to the sample because a short movie with easily identified spots in both channels is needed to pair signals from the same molecule in both channels when processing FRET movies. When ready, the cell can be placed on the objective with the prism on top, focused, and imaged. In P-TIRF imaging, the signal integration time (exposure time) per frame is typically 100 ms, and up to 9000 movie frames are acquired per field-of -view (FOV). Since this P-TIRF imaging setup allows visualization of both the donor and acceptor signals, detailed information about the sensor FRET states and kinetic behavior over the observation time window can be obtained. It is thus recommended to use such a two-channel setup when more detailed information is desired, such as estimating FRET efficiency or distinguishing between low-FRET states and fluorophore blinking or bleaching.
2.8. Data analysis
2.8.1. O-TIRF Data Analysis
O-TIRF analysis is performed with two MATLAB programs called SiMREPS analysis suite (SAS) and SiMREPS optimizer. The former identifies spots in a field of view, extracts traces from those spots, fits them with a hidden Markov model (HMM), and uses filtering criteria to separate traces containing target from those that lack it. The latter program uses data from experiments with and without the target as training sets for a Monte Carlo optimization that outputs an initial list of optimized filtering parameters that can be further refined by hand, if desired.
The SAS uses intensity averaging to identify regions with higher intensity than background and generates intensity-time traces for each such region. It then runs a two-state HMM [23] on all traces to evaluate the FRET state in each frame and when transitions occur. It creates idealized, noiseless traces that display the FRET behavior and applies kinetic filtering to distinguish target-bound molecule traces from non-target traces and background. Finally, it saves an Excel sheet with detailed trace and kinetic data and creates an Nb+d distribution histogram for each field of view and the whole dataset where red bars are accepted molecules and grey ones rejected molecules. The filtering criteria, as detailed in Table 1, are based on metrics like the Nb+d, dwell times in the high- and low-FRET states, S/N, and signal intensity, and are critical for proper distinction of the target, maximizing counts of genuine molecules and minimizing false positives (Fig. 1C).
Table 1.
Acquisition parameters and default kinetic filtering criteria for iSiMREPS sensors for detecting miR-141 and EGFR exon 19 deletion mutant DNA in 10% v/v formamide.
| iSMREPS sensor | Q8C6 QS18CS3 for detecting miR-141 | Q8C6 QS18CS19 for detecting exon 19 deletion mutant DNA |
|---|---|---|
| Movie frames start-to-end | 1–166 | 1–100 |
| Exposure time per frame (s) | 0.06 | 0.1 |
| Acquisition time (s) | 10 | 10 |
| Intensity threshold per trace | 200 | 500 |
| S/N threshold per event | 2 | 1.5 |
| S/N threshold per trace | 3.5 | 1.5 |
| Minimum Nb+d | 5 | 6 |
| Maximum Nb+d | Inf | Inf |
| Minimum τon, median (s) | 0.06 | 0.1 |
| Maximum τon, median (s) | 10 | 6 |
| Minimum τoff, median (s) | 0.06 | 0.1 |
| Maximum τoff, median (s) | 0.9 | 6 |
| Maximum τon, event (s) | 5 | 8 |
| Maximum τoff, event (s) | 4 | 8 |
Note: τon and τoff indicate target-bound (high-FRET) and non-target-bound (low-FRET) states, respectively. These criteria were obtained with the help of SiMREPS optimizer using several independent datasets with ≥ 10 FOVs both with and without target. Optimizer parameters were further tweaked manually to obtain a default that generally maximized true positives and minimized false positives. Each formamide variation experiment was individually optimized to allow each condition to be tested for its best possible performance.
2.8.2. P-TIRF Data Analysis
Two-channel P-TIRF data analysis is like that for single-channel O-TIRF, although it utilizes slightly different software and processing scripts and a manual selection of traces by the user. Firstly, a MATLAB script is used to find regions with intensity higher than background in one or both detection channels, to use the bead map movies to pair these regions with the corresponding regions of the other channel, and then generate two-channel (donor-acceptor) time-intensity traces. Manual trace viewing and selection is then done. Establishing selection criteria that are clear, consistent and result in the picking of traces with clear, readily interpretable kinetic behavior for the target is critical. The criteria used for the manual trace picking with their respective rationales are listed in Table 2. Once the traces are selected, several MATLAB scripts are used to clean up and organize the selected traces’ data. This data is placed into the software QuB [24]where each trace is subjected to a 2-state HMM fitting and idealized traces with detailed trace data is saved. These idealized traces are then further processed with additional MALTAB scripts to generate transition occupancy density plots (TODPs) that give a detailed picture of FRET transitions across all traces, and to extract the dwell times for each event that occurred for both FRET states[25].
Table 2.
Selection criteria for P-TIRF traces with rationales
| Criterion | Rationale |
|---|---|
| Clear anti-correlation in donor and acceptor signal when FRET states switch | In genuine FRET, only one signal is active at a time |
| Acceptor signal is present | Filters out traces with a bleached acceptor or no QP. |
| No multistep transitions | It is hard to distinguish genuine FRET transitions from noise in such traces |
| No signal drifting into the baseline | Worsening S/N renders some FRET states indistinguishable from noise |
| Very low high-FRET or very high low-FRET values and very weak S/N | These traces are susceptible to incorrect FRET assignments in HMM modeling |
| Only the longest segment or the one with the best S/N is chosen | Prevents data bias by a few traces with a large number of transitions |
| Low-FRET last events in segment accepted only if acceptor is present after that event. | Prevents acceptor photobleaching from tainting kinetic data |
| Traces must have distinction between signal and baseline | A static signal and an unusually intense baseline cannot be distinguished |
2.8.3. Obtaining dwell times
A MATLAB script (version 2019a or later) is used to first place the data into bins ranging from the minimum to maximum dwell time that are incremented by the length of the camera exposure. It then calculates the cumulative frequency of the range of dwell times in the data and fit it with a single- (Equation 1) or double-exponential (Equation 2) function.
| (1) |
| (2) |
The variablesa, b, c, τ, τ1 and τ2 are the fit parameters. The coefficients a and b provide the weight of their respective population in the double-exponential. The coefficient τ describes the average event lifetime for the single-exponential function. The coefficients τ1 and τ2 describe the average event dwell times for shorter- and longer-lived populations of events, respectively. The coefficient c is a constant that gives the y-intercept for the equation. The sum squared error (sse) and R2 values for the fit, which describe the residuals or deviation from the fit and goodness-of-fit, respectively, are used to decide whether a single- or double-exponential fitting is best used. At an sse < 0.05 and R2 > 0.98 for miR-141 detection, a single-exponential was considered a good fit and used. For EGFR exon 19 deletion mutant DNA, an sse < 0.08 and R2 > 0.96 was the cutoff for the single-exponential. When this criterion was not met, a double-exponential fit was used. The average dwell time for double-exponential fits was calculated as τ = (aτ1 + bτ2)/(a + b). This equation calculates a weighted average of both populations that was reported as the average dwell time for the entire dataset.
3. Results and Discussion
3.1. Detection of miR-141 by smFRET-based iSiMREPS
The smFRET-based iSiMREPS sensor was first developed for detecting miR-141 [13], a short non-coding RNA that is part of a larger class of gene regulatory microRNAs (miRNAs) that have emerged as promising cancer biomarkers [26, 27]. The basic architecture of the iSiMREPS sensor design for detecting miR-141 is depicted in Figure 2A. To develop iSiMREPS into a rapid, highly specific, and sensitive technique, we tested several sensors for accurate miR-141 detection that are sketched out in the following to allow the user to pursue optimization of their own target-specific sensor design.
iSiMREPS sensors were named based on several components: the target-binding query sequence (Qa), the query spacer (QSb), the query-binding competitor sequence (Cc), and the competitor spacer (CSd), where a, b, c, and d represent number of nucleotides in each domain (Fig. 2A and B). Four iSiMREPS designs were tested that contained identical query (Q8) and competitor spacer (CS3), but had a different competitor (C6 or C7) or query spacer (QS18 or QS33) (Fig. 2A and B). These sensors were first characterized with P-type TIRF microscopy without invaders or formamide at ~100 pM concentration for all sensor strands and target. While all the sensors generated a kinetic fingerprint in the presence of miR-141, the dwell times of target-bound (high-FRET) and non-target-bound (low-FRET) states were varied based on the length of the competitor and query spacer (Fig. 2B).
The dwell times of all events were extracted with a 2-state HMM model and the averages were calculated by a cumulative frequency fit to an exponential decay function (see Section 2.8.3). Sensor Q8C7QS18CS3 showed the longest average dwell times of both high- and low-FRET states while the sensor Q8C6QS33CS3 showed the shortest values. The dwell time data suggested that as the QS is extended and the C becomes shorter, the query-target interaction is increasingly favored over the query-competitor interaction. This is explained by a longer QS imposing a greater entropic penalty to the query-competitor interaction and a shorter competitor having lower affinity for the query sequence. Overall, the Q8C6QS18CS3 sensor showed the shortest dwell times without a strong bias for one FRET state over the other and was thus chosen as the optimal design for further experiments and optimizations.
Having obtained an effective, optimized design for miR-141 detection, the next step was further improving separation of target signals from background and speeding up detection to a scale of seconds. One possible strategy to shorten the dwell times is reduction of the number of base pairs in the query-target and query-competitor duplexes. However, this would have the undesired consequence of reducing the specificity of the interactions. We therefore decided to use the denaturant formamide, which is known to reduce the melting temperature of nucleic acid duplexes of ~2.4–2.9 °C /mol L−1 depending on the GC content, helix conformation and hydration state [28]. We expected that this would reduce the stability of both duplexes, shorten the dwell times for both states, and that the sensor’s intramolecular arrangement would permit rapid association of any unbound probes and allow for rapid fingerprint generation. We also aimed to use a pair of capture and query invaders that could remove the non-target-bound sensors via TMSD to remove background signal for non-target bound sensors. By adding a pair of invaders before O-type TIRF imaging and using formamide at 10% v/v in the imaging buffer, we obtained much lower background signals, improved signal-to-noise in the single molecule kinetic traces, and much shorter dwell times for smFRET states than without invaders and formamide (Fig. 2C and 2D). At a standard acquisition of 10 s per FOV, the histograms of the number of binding and dissociation events (Nb+d) for the candidate target bound molecules after applying filtering thresholds outlined in Table 1 showed good separation from background at 10% formamide, but poor separation without formamide (Fig. 2C).
Next, we varied the formamide concentration from 0–20% (v/v) to determine the optimal formamide concentration for sensitive detection of miR-141 in a 10 s acquisition window. We observed that the average dwell time gradually decreased, and the change diminished to the point of saturation above 10% formamide (Fig. 2D). The accepted counts also gradually increased and peaked at 10% formamide and decreased after that (Fig. 2E). Based on these results, we considered 10% formamide the optimal condition for quantification of miR-141 in buffer. Using the intramolecular sensor Q8C6QS18CS3 with optimized assay conditions, we obtained an LOD of miR-141 ~1 fM, which is comparable to the conventional (intermolecular) SiMREPS approach [7, 12] (Fig. 2F). However, the standard acquisition time of 10 s per FOV obtained by iSiMREPS is ~60-fold faster than conventional SiMREPS [7, 12].
3.2. Detection of EGFR exon 19 deletion mutant DNA by smFRET-based iSiMREPS
To test the generality of the smFRET-based iSiMREPS approach, we tested a different class of nucleic acid biomarker: cell free tumor derived DNA (ctDNA) [13]. EGFR exon 19 deletion mutation was chosen (COSMIC ID: COSM6225; c. 2235_2249del15) [p.E746_A750delELREA]) as a biomarker with great potential for non-small cell lung cancer (NSCLC) [29]. The initial sensor design (Q8C6QS18CS19) for detecting mutant DNA featured a similar basic architecture as the optimal sensor (Q8C6QS18CS3) for miR-141, but contained a longer CS (CS19) than the miR-141 sensor (CS3). In addition, an auxiliary probe was introduced with the target for efficient capture of target on surface and to increase the specificity[15]. The sensor Q8C6QS18CS19 (Fig. 3A), when imaged under an O-TIRF microscope in 10% v/v imaging buffer in the presence of exon 19 mutant DNA generated a kinetic fingerprint for it that was readily distinguishable from the wild-type DNA and control with no DNA target (Fig. 3B). However, the single-molecule kinetic traces showed slight dominance of high-FRET state over the low-FRET state (Fig. 3B). At a standard acquisition of 10 s per FOV, the Nb+d histograms, after applying filtering thresholds as outlined in Table 1, showed good separation from background.
Next, to generate more even FRET states while detecting mutant DNA, we modified the initial sensor Q8C6QS18CS19 to design four different iSiMREPS sensors that contained either a shorter competitor spacer (CS4 or CS12) to decrease the flexibility of competitor or a longer competitor (C7 or C8) to increase the thermodynamic stability of the query-competitor duplex (Fig. 3C). We expected that variation of these parameters would influence the dwell times of FRET states. But perhaps surprisingly, the results showed that varying CS length has an insignificant effect on both the dynamics of FRET transitions and the accepted counts per FOV during a standard acquisition of 10 s (Fig. 3C). This result may be explained by the fact that iSiMREPS sensors entail a short (4–19 nt), flexible single-stranded spacer (Fig. 3) in the form of a poly-dT stretch that is highly flexible and may supersede changes in the FRET state dwell times. By contrast, increasing the competitor length from 6 to 8 nt increased the dwell times for the low-FRET states significantly and generated more balanced FRET states, but also reduced the accepted counts significantly as Nb+d histograms did not separate well from the background within the 10 s acquisition window. Overall, the Q8C6QS18 design worked well and the choice of CS within the tested range of lengths is somewhat arbitrary. The Q8C6QS18CS19 sensor was chosen for quantification of concentration and for determining sensitivity and specificity. When we tested the performance of the optimized sensor in different concentrations of formamide ranging from 0–20% v/v, the results showed that the average dwell times for the high- and low-FRET states using 10% formamide decreased by a factor of 3.5 and 2.5 respectively compared to when no formamide was used (Fig. 3D). Above 10%, there was an insignificant effect on the average dwell times of the FRET states. The sensor showed the highest accepted counts per FOV at 10% v/v formamide because signals from target-bound sensors separated well from background at ≥ 10% formamide at a standard acquisition time of 10 s. Separation was poor at 0 and 5 % formamide and many true molecules did not pass kinetic filtering at 15 and 20% formamide (Fig. 3E).
Since EGFR exon 19 deletion mutant DNA exists as a double-stranded (ds) DNA form in biofluids, quantifying it necessitated thermally denaturing the target at 90 °C for 3 min and then cooling at room temperature in the presence of an auxiliary probe and the poly-thymidine oligonucleotide dT30, which acted as a carrier strand and was used at a high molar excess (2 μM), as previously described [7]. The iSiMREPS assay for the EGFR exon 19 deletion mutant dsDNA was found to have an LOD of 2.9 fM (Fig. 3F) and a specificity of 99.9999%, permitting detection of mutant DNA at an allelic fraction of 0.0001% (Fig. 3G).
4. Conclusions
We here have presented a practical guide for the detection of two different types of nucleic acid biomarkers using intramolecular smFRET-based single molecule kinetic fingerprinting through iSiMREPS. This technique accelerates kinetic fingerprinting by ~20-fold compared to conventional (intermolecular) SiMREPS without using denaturant (formamide); and 60-fold when aided by 10% v/v formamide, without any change in detection instrumentation. The robust design of iSiMREPS permits removal of non-target-bound fluorescent probes from the TIRF surface using a pair of ssDNA invaders that initiate TMSD. These steps reduce background signal significantly and improve the sensitivity and specificity of iSiMREPS to a level comparable in analytical performance to SiMREPS [7, 12]. iSiMREPS may also allow for spatial encoding in a microarray to facilitate multiplexing. By fine-tuning its different components, multiplex detection of diverse biomarkers such as mutant DNA, miRNA, lncRNA, viral DNA and RNA should be possible to achieve. However, a potential disadvantage of iSiMREPS compared to intermolecular SiMREPS is that the lack of probe exchange renders individual sensors susceptible to photobleaching, which can lower sensitivity. This challenge, in turn, can be addressed using brighter, more photostable fluorophores. We anticipate that iSiMREPS’ combination of rapid analysis; ultrahigh specificity and high sensitivity endow it with potential for early disease detection in clinical diagnostics.
Highlights.
FRET-based intramolecular kinetic fingerprinting (iSiMREPS) is described.
Intramolecular assembly of sensor generates rapid kinetic fingerprints of single molecules.
Kinetic fingerprinting is accelerated using denaturant formamide.
A pair of invader strands aids removal of non-target-bound fluorescent probes and reduces background counts.
Rapid, highly sensitive and ultraspecific detection of diverse nucleic acid biomarkers is enabled.
Acknowledgments
This work was supported by National Institute of Health (NIH) grants R21 CA204560 and R33 CA229023 to N.G.W. The authors acknowledge intellectual contributions and support from Muneesh Tewari in the development of iSiMREPS.
Abbreviations:
- iSiMREPS
intramolecular single-molecule recognition through equilibrium Poisson sampling
- smFRET
single molecule Förster resonance energy transfer
- cfNA
cell-free nucleic acid
- ctDNA
circulating tumor DNA
- A
anchor
- CP
capture probe
- Q
query
- QP
query probe
- C
competitor
- CI
capture invader
- QI
query invader
- QS
query spacer
- CS
competitor spacer
- A647
Alexa Fluor 647
- TMSD
toehold mediated strand displacement
- LNA
locked nucleic acid
- MUT
mutant
- WT
wild-type
- PBS
Phosphate-buffered saline
- O-TIRF
objective-type total internal reflection fluorescence
- P-TIRF
prism-type total internal reflection fluorescence
- EMCCD
electron-multiplying charge coupled device
- ICCD
intensified charge-coupled device
- sCMOS
scientific complementary metal oxide semiconductor
- APTES
(3-aminopropyl) triethoxysilane
- sulfo-DST
sulfo-disuccinimidyltartrate
- OSS
oxygen scavenger system
- FOV
field of view
- HMM
hidden Markov modeling
- Nb+d
number of binding and dissociation events
- sse
sum squared error
- TODP
transition occupancy density plots
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The University of Michigan has filed patent applications related to the SiMREPS technique on which A.J.-B. and N.G.W. are co-inventors. A.J.-B. and N.G.W. are co-founders of a startup company, aLight Sciences Inc., which seeks to commercialize this technology. A.J.-B. is currently an employee of aLight Sciences Inc. The remaining authors declare no competing interests.
References
- [1].Schwarzenbach H, Hoon DS, Pantel K, Cell-free nucleic acids as biomarkers in cancer patients, Nat. Rev. Cancer 11 (2011) 426–37. [DOI] [PubMed] [Google Scholar]
- [2].Anfossi S, Babayan A, Pantel K, Calin GA, Clinical utility of circulating non-coding RNAs - an update, Nat. Rev. Clin. Oncol 15 (2018) 541–563. [DOI] [PubMed] [Google Scholar]
- [3].Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, Higashiyama M, Kodama K, Imamura F, Kato K, Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas, Clin. Cancer. Res 17 (2011) 7808–15. [DOI] [PubMed] [Google Scholar]
- [4].Milbury CA, Zhong Q, Lin J, Williams M, Olson J, Link DR, Hutchison B, Determining lower limits of detection of digital PCR assays for cancer-related gene mutations, Biomol. Detect. Quantif 1 (2014) 8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Postel M, Roosen A, Laurent-Puig P, Taly V, Wang-Renault SF, Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: a cancer diagnostic perspective, Expert. Rev. Mol. Diagn 18 (2018) 7–17. [DOI] [PubMed] [Google Scholar]
- [6].Chen G, Mosier S, Gocke CD, Lin MT, Eshleman JR, Cytosine deamination is a major cause of baseline noise in next-generation sequencing, Mol. Diagn. Ther 18 (2014) 587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hayward SL, Lund PE, Kang Q, Johnson-Buck A, Tewari M, Walter NG, Ultraspecific and Amplification-Free Quantification of Mutant DNA by Single-Molecule Kinetic Fingerprinting, J. Am. Chem. Soc 140 (2018) 11755–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Benes V, Castoldi M, Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available, Methods 50 (2010) 244–9. [DOI] [PubMed] [Google Scholar]
- [9].Wang JS, Zhang DY, Simulation-guided DNA probe design for consistently ultraspecific hybridization, Nat. Chem 7 (2015) 545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cohen L, Hartman MR, Amardey-Wellington A, Walt DR, Digital direct detection of microRNAs using single molecule arrays, Nucleic Acids Res. 45 (2017) e137–e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li Z, Zhou X, Li L, Liu S, Wang C, Yu C, Su X, Probing DNA Hybridization Equilibrium by Cationic Conjugated Polymer for Highly Selective Detection and Imaging of Single-Nucleotide Mutation, Anal. Chem 90 (2018) 6804–6810. [DOI] [PubMed] [Google Scholar]
- [12].Johnson-Buck A, Su X, Giraldez MD, Zhao M, Tewari M, Walter NG, Kinetic fingerprinting to identify and count single nucleic acids, Nat. Biotech 33 (2015) 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Khanna K, Mandal S, Blanchard AT, Tewari M, Johnson-Buck A, Walter NG, Rapid kinetic fingerprinting of single nucleic acid molecules by a FRET-based dynamic nanosensor, Biosens. and Bioelectron (2021) 113433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chatterjee T, Li Z, Khanna K, Montoya K, Tewari M, Walter NG, Johnson-Buck A, Ultraspecific analyte detection by direct kinetic fingerprinting of single molecules, Trends Anal. Chem 123 (2020) 115764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Johnson-Buck A, Li J, Tewari M, Walter NG, A guide to nucleic acid detection by single-molecule kinetic fingerprinting, Methods 153 (2019) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang DY, Seelig G, Dynamic DNA nanotechnology using strand-displacement reactions, Nat. Chem 3 (2011) 103–113. [DOI] [PubMed] [Google Scholar]
- [17].Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, Pierce NA, NUPACK: Analysis and design of nucleic acid systems, J. Comput. Chem 32 (2011) 170–173. [DOI] [PubMed] [Google Scholar]
- [18].Caltech:, NUPACK Analysis (2007). [Google Scholar]
- [19].Zhang DY, Chen SX, Yin P, Optimizing the specificity of nucleic acid hybridization, Nat. Chem 4 (2012) 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha T, Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy, Biophys. J 86 (2004) 2530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li F, Tang Y, Traynor SM, Li X-F, Le XC, Kinetics of Proximity-Induced Intramolecular DNA Strand Displacement, Anal. Chem 88 (2016) 8152–8157. [DOI] [PubMed] [Google Scholar]
- [22].Mandal S, Li Z, Chatterjee T, Khanna K, Montoya K, Dai L, Petersen C, Li L, Tewari M, Johnson-Buck A, Walter NG, Direct Kinetic Fingerprinting for High-Accuracy Single-Molecule Counting of Diverse Disease Biomarkers, Acc. Chem. Res 54 (2021) 388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blanco MR, Johnson-Buck AE, Walter NG, Hidden Markov Modeling in Single-Molecule Biophysics, in: Roberts GCK (Ed.), Encyclopedia of Biophysics, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 971–975. [Google Scholar]
- [24].Nicolai C, Sachs F, Solving ion channel kinetics with the QuB software, Biophys. Rev. Lett 08 (2013) 191–211. [Google Scholar]
- [25].Blanco M, Walter NG, Analysis of complex single-molecule FRET time trajectories, Methods Enzymol. 472 (2010) 153–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M, Circulating microRNAs as stable blood-based markers for cancer detection, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bryant RJ, Pawlowski T, Catto JW, Marsden G, Vessella RL, Rhees B, Kuslich C, Visakorpi T, Hamdy FC, Changes in circulating microRNA levels associated with prostate cancer, Br. J. Cancer 106 (2012) 768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Blake RD, Delcourt SG, Thermodynamic Effects of Formamide on DNA Stability, Nucleic Acids Res. 24 (1996) 2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gazdar AF, Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors, Oncogene 1 (2009) 198. [DOI] [PMC free article] [PubMed] [Google Scholar]