

Abstract
Objective:
Cardioplegic ischemia/reperfusion (CP-I/R) and diabetes mellitus (DM) are correlated with coronary endothelial dysfunction and inactivation of small conductance calcium-activated-potassium (SK) channels. Increased reactive oxidative species (ROS), such as mitochondrial ROS (mROS) may contribute to oxidative injury. Thus, we hypothesized that inhibition of mROS may protect coronary SK channels and endothelial function against CP-I/R-induced injury.
Methods:
Small coronary arteries and endothelial cells from the hearts of mice with and without DM were isolated and examined by using a cardioplegic hypoxia and reoxygenation (CP-H/R) model to determine whether the mitochondria-targeted antioxidant Mito-Tempo (MT) could protect against coronary endothelial and SK channel dysfunction. The microvessels or mouse heart endothelial cells (MHECs) were treated with or without MT (0-10 μM) 5 minutes before and during CP-H/R. Microvascular function was assessed in vitro by vessel myography. K+ currents of MHECs were measured by whole-cell patch clamp. The levels of intracellular cytosolic free calcium (Ca2+) concentration, mROS and SK protein expression of MHECs were measured by Rhod-2 fluorescence staining and MitoSoxTM, Western blotting, respectively.
Results:
CP-H/R significantly attenuated endothelial SK channel activity, caused calcium overload and increased mROS of MHECs in both the ND and DM groups. In addition, treating MHECs with MT (10μM) reduced CP-H/R-induced Ca2+ and mROS overload in both ND and DM groups, respectively (P<0.05). Treatment with MT (10μM) significantly enhanced coronary relaxation responses to ADP and NS309 (P<0.05), and endothelial SK channel currents in both ND and DM groups (P<0.05).
Conclusions:
Administration of MT improves endothelial function and SK channels activity which may contribute to its enhancement of endothelium-dependent vasorelaxation following CP-H/R.
Keywords: Coronary endothelial function, Coronary endothelium-dependent vasodilation, Coronary microcirculation, Endothelial Function, mitochondria, reactive oxygen species, mitochondrial reactive oxygen species, potassium channels, ion channels, small conductance calcium-activated potassium channels, cardioplegia, cardioplegic arrest, Cardioplegic ischemia and reperfusion, ischemia/reperfusion, cardioplegic hypoxia and re-oxygenation, diabetes, Cardiac surgery, Cardiopulmonary bypass
Graphical Abstract
mROS inhibitor protects against coronary endothelial dysfunction following CP-H/R
INTRODUCTION
Cardioplegic arrest and cardiopulmonary bypass (CP/CPB) are used to maintain the circulation during cardiac operations.1, 2 While the majority of heart protection strategies have been shown to be safe and effective, cardioplegic-ischemia/reperfusion (CP-I/R) injury still occurs during CP/CPB. 3, 4 Vascular CP-I/R injury is manifested in part by diminished coronary endothelial function and the impaired coronary relaxation, at times resulting in coronary spasm and myocardial malperfusion early after cardiac surgery.4-6 These disturbances appear to be more pronounced in patients with poorly controlled diabetes (DM). 4, 5, 7, 8 Endothelial cells are involved in various aspects of vessel function, including modulation of inflammation, prevention of coagulation and maintaining the balance between vasodilatation and vasoconstriction. 9 Endothelial dysfunction is characterized by impaired vasodilation, which involves a number of endothelium-derived relaxing factors (EDRF) including nitric oxide (NO), prostacyclin (PGI2), and endothelial-derived hyperpolarizing factors (EDHF).10, 11 The EDHF-induced endothelial hyperpolarization and vasodilation is mediated by small conductance calcium-activated potassium (SK) channels.12 Growing evidence indicates that SK channels also regulate NO and PGI2 in the endothelium. 12-14 Endothelial dysfunction has been associated with impaired SK channel function. 15 We and others have found that inactivation/inhibition of SK channels contributes to coronary endothelial dysfunction in animals and patients after CP-I/R16 or cardioplegic hypoxia and reoxygenation (CP-H/R).17, 18 This effect on endothelial cells and the coronary vasculature is more pronounced in the setting of DM. 18, 19
Both CP/CPB and DM can directly cause an increasing systemic oxidative stress, which results in organ/tissue/cell damage after cardiac surgery.20, 21 Oxidative stress consists of elevated intracellular levels of reactive oxygen species (ROS) that may lead to endothelial dysfunction.4, 22 The excessive production of ROS suppresses the expression and function of EDRF, resulting in impairment of endothelium-dependent vasorelaxation.7, 23 The endothelial cells have an extensive mitochondrial network, 24 suggesting that mitochondrial function may be important in response to the oxidative stress in the endothelium. Increased mitochondrial ROS (mROS) in the endothelium has been reported to play an important role in diabetes-induced endothelial dysfunction.19
Mito-Tempo (MT), which combines the antioxidant piperidine nitroxide (Tempo) with the lipophilic cation triphenylphosphonium (TPP+), was recently reported to be a mitochondria-targeted antioxidant.25 This compound has been shown to protect the mitochondria from oxidative injury in various pathologies, such as endotoxin-induced liver injury, sepsis-induced acute kidney injury, hypertension or colitis. 26-29 However, few have investigated whether application of MT improve coronary endothelial function in the setting of CP-I/R and DM. Therefore, the objective of the present study was to employ a CP-H/R model consisting of coronary endothelial cells and the isolated small coronary arteries from mice with or without DM, and to test whether MT can protect against CP-H/R-induced coronary endothelial dysfunction and SK channel inhibition.
METHODS
Animals and Mouse Heart Tissue Collection
A total of 44 mice were utilized in the study, among which twenty-two C57BL/6J mice were used as non-diabetes (ND) controls (12-16 weeks old, male), and twenty-two BKS. Cg-Dock7m +/+ Leprdb/J mice (12-16 weeks old, male) were studied as a model of diabetic mellitus (DM) (Jackson Laboratory, Bar Harbor, Maine). All experiments were approved by the Institutional Animal Care and Use Committee of the Rhode Island Hospital. Each mouse was anesthetized by inhaled isoflurane after which a thoracotomy was performed. The heart was removed from the chest cavity. The hearts were placed in cold Krebs buffer for the in vitro microvascular experiments or preserved in cell culture medium in preparation for endothelial cell isolation. The surgical procedure of mouse heart tissue collection is detailed in the supplementary files.
Experimetal Groups:
For microvessel study, thirty four mice were divided into following 6 groups for for microvessel study: Group 1: ND mouse coronary microvessels were subjected to CP-H/R without MT treatment [ND(H/R), n = 6]; Group 2: ND mouse coronary microvessels were subjected to CP-H/R with 1μM MT treatment [ND (H/R) +1μMT, n = 5]; Group 3: ND mouse microvessels were subjected to CP-H/R with 10 μM MT treatment [ND (H/R) +1μM MT, n = 6]; Group 4: DM mouse microvessels were subjected to CP-H/R without MT treatment [DM(H/R), n =6]; Group 5: DM mouse microvessels were subjected to CP-H/R with 1μM MT treatment [DM (H/R) + 1μM MT, n = 5]; Group 6: DM mouse microvessels were subjected to CP-H/R with 10μM MT treatment [DM(H/R) + 10μM MT, n =6].
For the endothelial cell experiments, the other ten mice (5 ND and 5 DM) were set for mouse heart endothelial cell (MHECs) isolation. The MHECs were labelled as following 6 experimental groups: Group 1: ND MHECs without H/R or MT treatment (ND); Group 2: ND MHECs were subjected to H/R without MT treatment [ND (H/R)]; Group 3: ND MHECs were subjected to CP-H/R with 10μM MT treatment [ND(H/R) + 10μM MT]; ND MHECs without H/R or MT treatment (DM); Group 5: DM MHECs were subjected to H/R without MT treatment (DM [H/R]); Group 6: DM MHECs were subjected to CP-H/R with 10μM MT treatment [DM(H/R) + 10μM MT]; All the experiments on the cellular level were repeated at least 3 times. All experimental groups were depicted in the Supplementary Figure S1.
Microvessel Dissection and CP-H/R Model of Microvessel
The distal portion of left anterior descending artery (100-150 μm diameter) was dissected from the isolated mouse heart as previously described.18 An in-vitro CP-H/R model of microvessel was employed to simmulate the ischemic cardiac arrest in the operating room. The CP solution, consisting of 110 NaCl, 20 KCl, 16 MgCl2, 1.5 CaCl2, 10 NaHCO3 (in mM, pH 7.4) with or without MT (1μM or 10μM, Enzo Life Science, Farmingdale, NY), replaced Krebs buffer for bathing the microvessel 5 minutes before hypoxia. After the pretreatment which simulates cardioplegia perfusion, the coronary arterioles were cooled in ice and set in the same solution for 60 minutes under hypoxic conditions (5%CO2, 95% N2). Then the vessel was re-oxygenated (95% O2, 5% CO2) for another 60 minutes at 37°C.
Microvascular Vasodilation Assessment
The microvessels were pre-constricted with the thromboxane A2 analog U46619 (4x10−7M-10−6M) to reach a 30%-40% reduction of the baseline diameter. After a stable constriction, microvascular relaxation was measured after using the following vasodilators: the SK channel activator NS309, the endothelium-dependent vasodilator adenosine 5’-diphosphate (ADP)16.18 and the endothelium-independent vasodilator sodium nitroprusside (SNP). The order of drugs was random with 1 or 2 interventions on each vessel. During of each experiment, the responses to NS309 (10−9-10−5 M), ADP (10−9-10−4 M) and SNP (10−9-10−4 M) were recorded.
CP-H/R Model of Endothelial Cells
Mouse heart endothelial cells (MHECs) isolated from the harvested heart were cultured in EGM-2 MV medium (Lonza Biosciences, Alpharetta, GA) and the CP-H/R model of MHECs was used according to previous study, 18 and also summarized in the Supplementary files.
Measurement of Intracellular Calcium
The methods for measurement of intracellular Ca2+ of MHECs 30, 31 aredetailed in the Supplementary files.
Electrophysiological Study of Endothelial Cell K+ Currents
Patch clamp recording techniques were used to measure K+ currents in the whole cell patch-clamp configurations (Supplementary Figure S2). Current-voltage recording conditions and Current-time recording conditions for K+ currents were achieved as described in detail previously15, 18, 19 and in the Supplementary files. For Ca2+ free (low Ca2+ group) pipette solution, no CaCl2 was added. For median Ca2+ (400μM) solution, 1.0 mM CaCl2 was used. For high Ca2+ (2μM) pipette solution, 9.7mM CaCl2 was added. The Free Ca2+ concentration was calculated by using Maxchelator.30 The selective SK activator NS309 on the whole cell K+ currents were examined, and then both SK2/SK3 blocker apamin (10−7M) and the SK4(IK) blocker TRAM34 (10−6M) were applied for testing the specificity of SK channel activation.15, 18, 19
Western Blotting
The methods for whole-cell protein purification, Western blotting, and imaging quantification have been described previously,18, 19 and summarized in the Supplementary files.
Measurement of Mitochondrial ROS
MHECs were stained with 5 μM MitoSox™ Red and 100 nM MitoTracker Green FM (Invitrogen, Waltham, MA) according to the manufacture’s protocol and previously studied.19
Chemicals
ADP, apamin, NS309, and TRAM34 were purchased from Sigma-Aldrich (St. Louis, MO). MitoSox™ Red and MitoTracker Green FM were purchased from Invitrogen (Waltham, MA).
Data Analysis
All data are presented at the mean ± the standard deviation of the mean (SD). Microvessel responses are expressed as the percent relaxation of the pre-constricted diameter. The normality of data were assessed by the Shapiro-Wilk test. The darta of microvascular reactivity, intracellular Ca2+ concentration, patch-clamp, SK protein expression, and mROS were analyzed using one or two-way ANOVA with post hoc tests by GraphPad Prism 7 (GraphPad Software, San Diego, CA). The data of age, bodya weight and blood glucose were analyzed using paired t-test. A values of p<0.05 were considered statistically significant.
RESULTS
Characteristics of DM and ND Mice
The age of ND mice (13.67± 1.53 weeks) have no significant difference with DM mice (13.44± 1.54 weeks). The body weight of the genetically modified DM mice (46.83 ± 3.64 g) was higher than that of ND mice (26.77 ± 4.71g, P<0.01) and the blood glucose levels of DM mice (692.1 ± 17.80 mg/dL) was also higher than that of ND mice (136.4 ± 21.48 mg/dL, P<0.01, Supplementary Figure S3).
Mito-Tempo Improved Endothelium-Dependent Relaxation Response Following CP-H/R
There was no significant difference in the baseline diameter of the microvessels among the 6 groups. The degrees of preconstriction by U46619 were 35.4±2.12% in the ND(H/R) group, 36.4±1.66% in the ND (H/R) +1μM MT group, 32.7±0.98% in the ND(H/R) +10μM MT group, 36.0±2.4% in the DM(H/R) group, 34.1±1.32% in the DM (H/R)+1μM MT group, and 33.4±1.05% in the DM(H/R)−+10μM MT group (P>0.05, respectively). The coronary vascular responses of DM(H/R) mice to the endothelium-dependent vasodilators NS309 (10μM) [P = 0.0002, ND (H/R) vs. DM(H/R), Figure 1A] and ADP (100μM) [P < 0.0001, ND (H/R) vs. DM(H/R), Figure 1B] were significantly decreased compared with ND (H/R) group, respectively. The effect of MT on improvement of vasodilation following CP-H/R is dose-dependent. Treatmemnt with 10μM MT significantly improved NS309 (10μM) [P = 0.002, ND(H/R) +10μM MT vs. ND(H/R); P =0.0329, DM(H/R) +10μM MT vs. DM(H/R), Figure 1A] and ADP (100μM) [P =0.0438, ND(H/R)+10μM MT vs. ND(H/R); P =0.0293, DM(H/R) +10μM MT vs. DM(H/R), Figure 1B] -induced relaxation in both ND (H/R) and DM (H/R) vessels respectively. In contrast, treatmemnt with 1μM MT failed to effect NS309 (Figure 1A) and ADP (Figure 1B)-induced relaxation response (P>0.05), respectively. There were no statistical differences in the vascular responses to SNP among the six groups, respectively (P>0.05, Figure 1C). The data of dose-dependent vasodilatory effects of NS309, ADP and SNP are summarized in the Supplementary Figure S4.
Figure 1. Dose-dependent effects of Mito-Tempo (MT) treatment (0-10μM) on the recovery of relaxation responses of mouse small coronary arteries following CP-H/R.

The diabetic (DM) or non-diabetic (ND) microvessels were pre-constricted with the thromboxane A2 analog U46619 to reach a 30%-40% reduction of the baseline diameter. After a stable constriction, microvascular relaxation in response to the SK activator NS309 (0-10μM); (A), ADP (10μM); (B); and (C) SNP(100μM) in the presence or absence of MT (0-10μM) was measured. ND(H/R) = ND treated by hypoxia/re-oxygenation; ND(H/R)+1μM MT = ND(H/R) + 1μM mito-Tempo; ND(H/R)+10μM MT = ND(H/R) + 10 μM mito-Tempo; DM(H/R) = DM treated by hypoxia/re-oxygenation; DM(H/R) +1μM MT = DM(H/R) + 1μM mito-Tempo; DM(H/R) +10μM MT = DM(H/R) + 10μM mito-Tempo; n= 5-6/group; A, @P =0.0002, ND(H/R) vs. DM(H/R); *P= 0.002 , ND(H/R) + 10μM MT vs. ND (H/R) #P = 0.0329, DM(H/R) + 10μM MT vs. DM(H/R); $P <0.0001, ND(H/R) +10μM MT vs. DM(H/R) +10μM MT; B, @P <0.0001, ND(H/R) vs. DM(H/R); *P = 0.0438, ND(H/R) + 10μM MT vs. ND (H/R); #P =0.0293, DM(H/R) + 10μM MT vs. DM(H/R); $P <0.0001, ND(H/R) +10μM MT vs. DM(H/R) +10μM MT; Mean ± SD, Two-way ANOVA with a post hoc Bonferroni test.
Mito-Tempo Suppressed Intracellular Ca2+ Following CP-H/R
Following CP-H/R, MHECs in ND(H/R) and DM(H/R) showed significant higher intracellular free-Ca2+ concentration than that of the baseline (ND or DM) (P<0.0001, respectively, Figure 2A-B). MT (10μM) treatment significantly suppressed intracellular Ca2+ concentration in ND(H/R)+10μM MT and DM(H/R) + 10μM MT groups compared with ND(H/R) and DM(H/R) groups (P<0.0001, respectively, Figure 2A-B).
Figure 2. Hypoxia/re-oxygenation (H/R) altered Ca2+ homeostasis in ND and DM mouse house endothelial cells (MHECs).

MHECs were isolated from diabetic (DM) or non-diabetic (ND) mice. A, Representative fluorescent of intracellular calcium (Ca2+) of MHECs treated with or without MT (10μM). B, Quantitative analysis of intracellular Ca2+ concentration changes of MHECs in the following experimental groups: ND, n=22; ND(H/R), n=16; ND(H/R) + 10μM MT, n=18; DM, n=20; DM(H/R), n=25; DM(H/R) +10μM MT, n=28; *P <0.0001, ND(H/R) vs. ND; @P <0.0001, ND(H/R) + 10μM MT vs. ND(H/R); &P <0.0001, DM vs. ND; † P <0.0001, DM(H/R) vs. DM; $P <0.0001, DM(H/R) +10μM MT vs. DM(H/R). C, Representative traces of the whole cell currents of MHECs with different free calcium concentration. D, The plots shows Apamin+TRAM34-sensitive component of K+ current at +100 mV (0 calcium, n=4; 400nM Ca2+, n=3; 2μM Ca2+, n=3), *P <0.0001, 400nM Ca2+ vs. 0 Ca2+; @P <0.0001, 400nM Ca2+ vs. 2μM Ca2+; Mean ± SD, ND(H/R) = ND treated by hypoxia/re-oxygenation; DM(H/R) = DM treated by hypoxia/re-oxygenation; ND(H/R) +10μM MT = ND(H/R) + 10μM mito-Tempo; DM(H/R) +10μM MT = DM(H/R) + 10μM mito-Tempo; Mean ±SD, One-way ANOVA with a post hoc Dunnett's multiple comparisons test.
The Biphasic Effects of Intracellular Ca2+ on Endothelial SK Currents
To investigate whether intracellular Ca2+ affects endothelial SK currents, we examined the effects of the low concentration Ca2+-free (0), median concentration Ca2+ (400nM) and high concentration free Ca2+ (2μM), by recording whole-cell apamin + TRAM34-sensitive currents. The SK channel currents were highly activated at median Ca2+ yet inhibited at high Ca2+, and inactived at Ca2+ free (Figure 2C-D), suggesting that intracellular Ca2+ has a biphasic effect on endothelial SK currents.
Mito-Tempo Increased Endothelial SK Currents after CP-H/R
Administration of NS309 significantly increased the total K+ currents of MHECs. Acute MT treatment significantly increased NS309 sensitive currents in CP-H/R model of ND and DM mice [P = 0.0074, ND (H/R)+10μM vs. ND (H/R); P = 0.0162, DM (H/R)+10μM vs. DM(H/R); Figure 3, A, B, and D]. Subsequent application of apamin and TRAM34 abolished NS309-sensitive K+ currents in both groups (Figure 3A, C, E). The MT (10μM) treatment significantly increased SK (apamin +TRAM34 sensitive) channel currents in ND and DM MHECs [P =0.0322, ND (H/R)+10μM MT vs. ND(H/R); P =0.0451, DM (H/R)+10μM MT vs. DM(H/R); Figure 3A, C,E]. Furthermore, this effect of MT was more pronounced in ND (H/R)+10μM MT than DM (H/R)+10μM MT (P<0.05, Figure 3). In the series of time-current relationship experiments, the similar effects of MT (10μM) treatment in ND(H/R)+10μMT and DM(H/R)+10μMT groups were observed by showing noticeably increased in SK currents compared with that non-MT treatment groups, respectively (Figure 4). The data of IV relationship for NS309 and apamin +TRAM34 sensitive currents within each cell/treatment goup are summarized in the supplementary Figure S5.
Figure 3. Mito-Tempo (MT) significantly increases SK channel currents of mouse heart endothelial cells (MHECs) in H/R model from ND and DM mice.

A, Representative traces of the whole cell currents of MHECs treated with or without MT (10μM) at holding potential of −50 mV and test potentials from −100 to +100 mV in 20 mV increments. B, whole-cell I–V relationships sensitive to NS309 in MHECs of ND(H/R) and DM(H/R) with or without MT treatment. C, whole-cell I–V relationships sensitive to TRAM34+Apamin in MHECs of ND(H/R) and DM(H/R) with or without MT treatment. D, Box plots shows NS309-sensitive component of potassium current at +100 mV in ND(H/R) and DM(H/R) MHECs treat with or without MT (n=5/group). *P = 0.0074, ND(H/R) +10μM MT vs. ND(H/R); @P = 0.0162, DM(H/R) +10μM MT vs. DM(H/R). E, Box plots shows TRAM34+Apamin-sensitive component of potassium current at +100 mV in ND(H/R) and DM(H/R) MHECs treat with or without MT (n=5/group). *P =0.0322, ND(H/R) +10μM MT vs. ND(H/R); @P =0.0451, DM(H/R) +10μM MT vs. DM(H/R). ND(H/R) = ND treated by hypoxia/re-oxygenation; DM(H/R) = DM treated by hypoxia/re-oxygenation; ND(H/R) +10μM MT = ND(H/R) + 10μM mito-Tempo; DM(H/R) +10μM MT = DM(H/R) + 10μM mito-Tempo; Mean ± SD, One-way ANOVA with a post hoc Sidak's multiple comparisons test.
Figure 4. Mito-Tempo (MT) significantly increases SK channel currents of MHECs in H/R model from ND and DM mice.

A-D, Time course of the whole-cell current density evoked at +100mV from MHECs using patch clamp. NS309 was added to the bath to activated IK/SK channels, followed by bath application of Apamin and TRAM34 to block them. E, Box plots shows NS309-sensitive component of potassium current at +100 mV in ND(H/R) and DM(H/R) MHECs treat with or without MT (n=3/group); *P =0.0486, ND(H/R)+10μM MT vs. ND(H/R); @P =0.0082, DM(H/R)+10μMT vs. DM(H/R). F, Box plots shows Apamin+TRAM34-sensitive component of potassium current at +100 mV in ND(H/R) and DM(H/R) MHECs treat with or without MT (n=3/group). *P =0.0233, ND(H/R) +10μM MT vs. ND(H/R); @P =0.0203, DM(H/R) +MT vs. DM(H/R); ND(H/R) = ND treated by hypoxia/re-oxygenation; ND(H/R) +10μM MT = ND(H/R) + 10μM mito-Tempo; DM(H/R) = DM treated by hypoxia/re-oxygenation; DM(H/R) +10μM MT = DM(H/R) + 10 μM mito-Tempo; Mean ± SD, One-way ANOVA with Student's t-test.
Effects of H/R and MT treatment on SK Protein Expression
There were no significant differences between ND and DM groups at baseline in the SK3 and SK4 protein expression of the MHECs (Figure 5, P>0.05). Neither H/R nor MT treatment altered endothelial SK3 and SK4 expression in either ND or DM group.
Figure 5. H/R and MT treatment failed to effect SK3 and SK4 protein expression in the mouse heart endothelial cells (MHECs).

A and B, Immunoblot intensity of small conductance calcium activated potassium channels (SK), SK3 (A), SK4 (B) with a GAPDH loading control, C and D, graphs showing densitometric analysis of immunoband intensity of SK3 (C) and SK4(D) protein expression in the experimental groups. ND = non-diabetes, DM- diabetes; ND (H/R) = ND cells treated by hypoxia/re-oxygenation; DM (H/R) = DM cells treated by hypoxia/re-oxygenation; ND (H/R) + 10μM MT = ND(H/R) cells treated with 10μM mito-Tempo; DM (H/R) + 10 μM MT = DM (H/R) cells treated with 10μM mito-Tempo; n =3/group. Mean ± SD, One-way ANOVA and a post hoc Bonferroni test.
Mito-Tempo Reduced mROS Production after CP-H/R
Under the normoxic conditions, DM MHECs exhibited clearly higher levels of mROS than their ND counterparts (P <0.0001, Figure 6). CP-H/R further increased mROS in both ND(H/R) and DM (H/R) (P <0.0001, respectively, Figure 6). In ND(H/R)+10μM MT, the treatment with MT notably reduced mROS (P<0.0001, Figure 6). Similarly, MT treatment was substantially reduced mROS in DM(H/R)+10μM MT (P <0.0001, Figure 6).
Figure 6. The effects of mito-Tempo on the production of mitochondrial ROS (mROS) induced by hypoxia/re-oxygenation in ND and DM MHECs.

Mitochondria localization was visualized using MitoTracker and mROS production was monitored with MitoSOX following exposure to mito-Tempo induced by H/R in ND (A) and DM (B) MHECs. C, Box plots summarizing the data analysis of mROS in the experimental groups, n =5/group. *P <0.0001, ND(H/R) vs. ND; @P <0.0001, ND(H/R) +MT vs. ND(H/R); &P <0.0001, DM vs. ND; †P <0.0001, DM(H/R) vs. DM; $P <0.0001, DM(H/R) +10μM MT vs. DM(H/R). ND = ND cells under normoxic condition; DM = DM cells under normoxic condition; ND(H/R) = ND treated by hypoxia/re-oxygenation; DM(H/R) = DM cells under hypoxia/re-oxygenation; ND(H/R) +10μM MT = ND(H/R) + 10μM mito-Tempo; DM(H/R)+10 μM MT = DM(H/R)+mito-Tempo. Mean ± SD, One-way ANOVA and a post hoc Bonferroni test.
DISCUSSION
There are a number of novel findings in the current study. First, CP-H/R induced significant increase of endothelial mROS and this was more profound in the DM endothelial cells than that of ND. Second, MT (10μM) treatment significantly inhibited endothelial mROS. Third, CP-H/R caused intracellular Ca2+ overload in both ND and DM endothelial cells companied by impairment of SK channel activity. Fourth, adminstration of MT (10 μM) enhanced endothelial SK channel activity of ND and DM in the setting of CP-H/R. Finally, treatment of small coronary artery with MT in the setting of CP-H/R improved the endothelium-dependent relaxation in response to the the SK activator NS309 and the endothelium-/NO-dependent vasodilator ADP in both ND and DM mice; and this effects was MT dose-dependent.
The vascular endothelium has a critical roles in vasomotor regulation, maintenance of tissue perfusion, and prevention of vascular thrombosis. Cardioplegia has been associated with marked endothelial dysfunction 4, 6, 7, 31 and a marked changes in myocardial perfusion in vivo.32 Several mechanisms, such as increased oxidative stress, leukocyte and complement activation contribute to this dysfunction. 4, 31, 33 As mentioned previously, poorly controlled diabetes has been reported to be an important predisposing factor to endothelial dysfunction in previous clinical studies.5, 7 Furthermore, in addition to altered myocardial perfusion, vascular spasm and EKG changes in the perioperative period, endothelial dysfunction can lead to intimal hyperplasia and aggressive atherosclerosis that may arise weeks or year after surgery. Thus, novel strategies to protect the coronary endothelium in the setting of cardiovascular surgery, such as the inhibition of mROS as presented in this study, have considerable short-term and long-term clinical implications.
ROS are formed as a natural byproduct of the normal metabolism of oxygen and have important roles in cell signaling, metabolic adaptation, inflammatory response and homeostasis.34-36 However, the unbalanced ROS production may induce oxidative damage that has been implicated in the pathogenesis of in a variety of disorders, including DM and its complications. Furthermore, accumulated evidence has indicated that the generation of ROS is a major cause of endothelial injury after CP-I/R.37 The mitochondrion is the main sources of ROS generation.24 Importantly, CP-H/R caused a significant increase in endothelial mROS with or without DM. MT is a physicochemical compound as one of the MnSOD mimics, which can pass through lipid bilayers easily and accumulate selectively in mitochondria.27 In the present study, we found that administration of MT significantly inhibited the generation of endothelial mROS induced by CP-H/R.
The in-vitro CP-H/R model was employed to investigate the effects of CP-H/R on coronary endothelium-dependent dilatation in mice with or without DM. Our results demonstrated that the impaired coronary dilation function is associated with a significant increase in endothelial mROS. Multiple studies suggested MT reduced adverse changes in a variety of disorders by inhibiting high levels of mROS.27-29 Therefore, we further investigated the effects of MT on coronary vasodilation of ND and DM vessels in the setting of CP-H/R. Our results demonstrate that MT (10μM) significantly improves the recovery of coronary endothelium-dependent relaxation in both ND and DM vessels. These findings clearly suggest that coronary endothelial dysfunction after CP-H/R is at least in part induced by mROS and MT used during CP-H/R may protect coronary vasculature.
There are four subtypes of SK channels, SK1 (KCa2.1), SK2 (KCa2.2), SK3 (KCa2.3) and SK4 (SK3.1, IK). SK1 and SK2 mainly presents in the neuronal cells, SK2 is also predominately expressed in the cardiomyocytes. SK2 presents in the endothelial nuclei instead of plasma membrane.38 SK3 and SK4 (IK) are predominately in the endothelial cells which are mainly responsible for EDHF-induced endothelial hyperpolarization and microvascular relaxation.12, 16 In the current study, we found that CP-H/R with or without DM did not change SK3/SK4 protein expression in the endothelial cells, and treatment with MT failed to affect the SK3/SK4 protein expression. It suggests that MT may affect SK activity instead of the protein expression amount. Importantly, we found that MT significantly increased endothelial SK channel currents in the setting of CP-H/R in ND and DM cells. Thus, the MT-induced upregulation of endothelial SK activity contributes to its improvement of coronary relaxation by the selective SK activator NS309 in the setting of CP-H/R. However, the improvement of endothelial function by MT can be mediated by cellular/molecular mechanisms other than activation of SK channels. This notion is supported by our finding that MT also improves the vasorelaxation induced by endothelial NO-dependent vasodilator ADP, which suggests that NO signaling pathways may be synergistically involved in MT-improved vascular relaxation in the setting of CP-H/R.
The endothelial hyperpolarization induced by SK activation is directly propagated to the underlying vascular smooth muscle cells via myoendothelial coupling, which leads to the closure of voltage-gated Ca2+ channels and the subsequent vasodilation.39 SK channel is voltage-independent and Ca2+-activated at submicromolar concentrations of intracellular Ca2+ (400-800nM) in the endothelium.40 The current study and others demonstrate that either low intracellular Ca2+ (Ca2+ free) or high intracellular Ca2+ (> 2μM) inactivated or reduced endothelial SK channel activity, particularly in the presence of high concentration of Mg2+.40 In the current study, we observed that H/R caused intracellular Ca2+ overload in both ND and DM cells, coincided with the decreased endothelial SK currents. It is possible that the CP solution containing high K+ (16-25 mM) and Mg2+ (12mM) in the setting of CP-H/R may significantly cause intracellular Ca2+ overload, resulting in the inactivation of endothelial SK channels. In addition, the metabolic alteration in the enhanced activation/expression of Nox, PKC, NADH7, 19 during DM and/or CP-H/R may also reduce SK channel sensitivity to Ca2+ and cause the channel internalization of the damaged endothelial cells.
The inactivation of SK channels may contribute to the CP-I/R and DM-induced endothelial dysfunction in the human coronary arterioles.8, 16 15 Nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox)-dependent ROS overproduction, that could downregulate whole-cell K+ currents, was associated with the damaged SK channels.41 Our recent study found that endothelial SK channel function was regulated by the ratio of NADH and NAD+, and the increase of intracellular NADH in the coronary endothelial cells caused a significant decrease in endothelial SK channel currents.19 These findings suggest that the inactivation of SK channels in CP-H/R may cause by the overproduction of ROS. More evidence was shown in this study, as coronary dilatation and endothelial SK currents induced by the selective SK channels activator were reduced in both ND and DM MHECs after CP-H/R, while MT significantly improved coronary vasorelaxation and SK channel currents., suggesting that MT enhanced the SK channel function by reducing mROS.
There are several limitations in the current study. First, whether MT works specifically through SK3 or SK4 (IK) needs to be further studied. Second, this obese/type-2 diabetic mouse model has been widely used for studying diabetic complications with chronic hyperglycemia. Even though it successfully developed chronic hyperglycemia, the blood glucose levels of the DM mice were much higher than that of patients with poorly controlled DM (250-350 mg/dL). Third, the current study did not show any significant differences in relaxation response to the endothelium-independent vasodilator SNP between ND and DM or the control and MT treatment groups. Further work are needed to address concerning the effect of MT on vascular smooth muscle cells under DM and CP-H/R conditions. Fourth, it is well established that the metabolites like NADH, Nox, PKC, COX-2 and mROS are mutually stimulated during CP-H/R and DM. Therefore, future study should focus on those signaling pathways responsible for MT-enhanced SK channel activity and endothelial protection. Fifth, importantly, the impact of NO, PGI2 and EDHF interactions on the MT-induced endothelial protection also needs to be further elucidated. Finally, the in vitro cell/microvessel findings would be further investigated in an in vivo model of CP/CPB to confirm the cardiovascular protective effects of MT treatment.
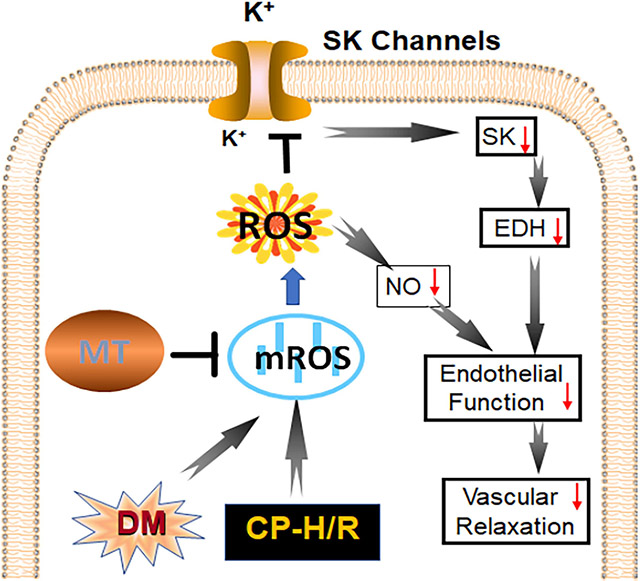
In conclusion, acute administration of MT improves endothelium-dependent vasorelaxation and endothelial SK channel function in the setting of CP-H/R and/or DM by decreasing mROS (Figure 7). This investigation into the mROS impact of CP-H/R in SK channel activity and endothelial function in both non-diabetic and diabetic mice with translational research models may be a fundamental step to connect the bench and the bedside. Our findings suggest that mROS may be a novel therapeutic target to treat vascular complications of CP-I/R, especially in patients with poorly controlled diabetes. These findings may have implications in the recovery of patients early after cardiac surgery.
Figure 7:

Research summary concerning that treatment with the mROS inhibitor, Mito-Tempo (MT) increases endothelial SK-currents and NO signaling pathways, resulting in improving coronary endothelial function, EDH and coronary relaxation in the setting of cardioplegic hypoxia and re-oxygenation (CP-H/R) in mice with or without diabetes (DM); EDH = endothelium-dependent hyperpolarizing; LAD = left anterior descending artery; ND (H/R) = ND treated by hypoxia/re-oxygenation, ND(H/R) + 1μM MT = ND(H/R) + 1μM mito-Tempo; ND(H/R) + 10μM MT = ND(H/R) + 10μM mito-Tempo; DM (H/R) = DM treated by hypoxia/re-oxygenation; DM(H/R) + 1μM MT = DM + 1μM mito-Tempo; DM(H/R) + 10μM MT = DM + 10μM mito-Tempo; CP-H/R = cardioplegic hypoxia/reoxygenation; EDH = endothelial-derived hyperpolarization; LAD = left anterior descending artery; ROS; = reactive oxygen species; mROS = mitochondrial ROS; NO = nitric oxide; SK channels = small conductance calcium-activated potassium channels. @P =0.0002, ND(H/R) vs. DM(H/R); *P= 0.002 , ND(H/R) + 10μM MT vs. ND (H/R); #P = 0.0329, DM(H/R) + 10μM MT vs. DM(H/R); $P <0.0001, ND(H/R) +10μM MT vs. DM(H/R) +10μM MT; n =5-6/group, Mean ± SD, 2-Way-ANOVA repeated measurement.
Supplementary Material
Figure S1: Experimental Groups.
Figure S2: The schematic diagram illustrates patch clamp recording system. A glass pipette seals to the membrane of endothelial cell, once break through the membrane by applying light and short suction pulses with a syringe, then the analog current signal is amplified and digitized so that the signal can be analyzed.
Figure S3 Characteristics of diabetic (DM) and nondiabetic (ND) Mice. A, age, B, Body weight (g), P= 0.0001 DM vs. ND. C, Blood glucose (mg/dL), P = 0.0001 DM vs. ND.
Figure S4: Effects of endothelium-dependent vasodilator on mouse heart microvessel vasodilation. A, Dose-dependent vasodilation of mouse heart microvessels of ND(H/R) and DM(H/R) with or without MT treatment (10μM) in response to the SK channel activator NS309 (10−9–10−5 M), n=6/group. *P <0.0001 (NS309 10−6 M), P <0.0001 (NS309 10−5M), ND(H/R) vs. ND(H/R) +10μM MT; #P =0.0284 (NS309 10−6 M); P =0.0043 (NS309 10−5 M), DM(H/R) vs. DM(H/R) +10μM MT; @P <0.0001 (NS309 10−6 M), P <0.0001 (NS309 10−5M), ND(H/R) vs. DM(H/R). B, Dose-dependent vasodilation of mouse small coronary arteries of ND(H/R) and DM(H/R) with or without MT (10μM) treatment in response to the endothelium-dependent vasodilator ADP (10−9–10−4 M), n=6/group. *P =0.0021(ADP 10−5 M), P =0.0062 (ADP 10−4 M), ND(H/R) vs. ND(H/R) +10μM MT; #P =0.0090 (ADP 10−5 M), P =0.0029 (ADP 10−4 M), DM(H/R) vs. DM(H/R) +10μM MT; @ P <0.0001 (ADP 10−5 M), P <0.0001 (ADP 10 −4 M), ND(H/R) vs. DM(H/R). C, Dose-dependent vasodilation of mouse small coronary arteries of ND(H/R) and DM(H/R) with or without MT treatment (10μM) in response to the endothelium-independent vasodilator SNP (10−9-10−4 M), n=6/group. ND(H/R) = ND treated by hypoxia/re-oxygenation, DM(H/R) = DM treated by hypoxia/re-oxygenation, ND(H/R) +10μM MT = ND(H/R) + 10μM mito-Tempo, DM(H/R) +10μM MT = DM(H/R) + 10μM mito-Tempo.
Figure S5: The data of IV relationship for NS309 and apamin +TRAM34 sensitive currents within each cell/treatment group. MT (10μM) significantly increases SK channel currents of MHECs in H/R model from ND and DM mice. A-D, Effect of the SK channel activator NS309 and SK channel blocker Apamin and TRAM34 on the basal whole-cell K+ currents of MHECs in H/R model from ND and DM mice treatment with or without MT. *NS309 vs. Control, P=0.0071, ND(H/R); P=0.0027, ND(H/R)+10μM MT; P=0.0494, DM(H/R); P<0.001, DM(H/R)+10μM MT, #NS309 vs. NS309+Apamin+TRAM34, P=0.0061, ND(H/R); P=0.0018, ND(H/R) +10μM MT; P=0.038, DM(H/R); P=0.0005, DM(H/R) +10μM MT.
Video: First author Yi Song introduces the aims, hypothesis, methods and results of current study and explains the selective mROS inhibitor mito-Tempo-induced coronary endothelial protection and microvascular relaxation in the setting of cardioplegic hypoxia/reoxygenation and diabetes.
Central Message:
Inhibition of mROS with Mito-Tempo protects coronary endothelial and SK channel function against CP-H/R injury in the mice with or without diabetes.
Perspective Statement:
Inhibition of mROS with Mito-Tempo protects coronary endothelium-dependent relaxation and endothelial SK channel function against CP-H/R injury in the mice with or without diabetes. mROS inhibitors may be a new therapeutic agent for protecting coronary endothelial function during cardioplegic ischemia/reperfusion and cardiac surgery.
Funding:
This research project was mainly supported by the National Institute of Health (NIH) 1R01HL127072-01A1, 1R01 HL136347-01, and National Institute of General Medical Science (NIGMS) of 5P20-GM103652 (Pilot Project) to J.F., and 5P20-GM103652 to E.O.H.and S.R. This work was supported in part by 3R01HL136347-04S1 to J.F., AHA-Grant-in-Aid (#15GRNT25710105) to J.F., R01-HL46716 to F.W.S., and RO1HL128831 to F.W.S.
ABBREVIATIONS:
- ADP
adenosine 5’-diphosphate
- ANOVA
analysis of variance
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- CP
cardioplegia
- CPB
cardiopulmonary bypass
- CP/CPB
cardioplegic arrest and cardiopulmonary bypass
- CP-H/R
cardioplegic hypoxia and re-oxygenation
- CP-I/R
cardioplegic ischemia and reperfusion
- DM
diabetes mellitus
- DM (H/R) + 1μM MT
DM(H/R) + 1μM mito-Tempo
- DM (H/R)
DM vessels or cells treated by hypoxia/re-oxygenation
- DM (H/R) +1μM MT
DM (H/R) + 1μM MT (DM(H/R) + 1μM mito-Tempo
- DM (H/R) +10μM MT
DM (H/R) + 10μM MT (DM(H/R) + 10μM mito-Tempo
- EDHF
endothelium-dependent hyperpolarizing factor
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IK
intermediate conductance calcium-activated potassium channels
- MHECs
mouse heart endothelial cells
- Mito-Tempo
2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl) triphenylphosphonium
- MT
Mito-TEMPO
- mROS
mitochondrial reactive oxygen species
- ND
non-diabetes
- ND (H/R)
ND vessels or cells treated by hypoxia/re-oxygenation
- ND (H/R)
ND (H/R) + 1μM MT (ND(H/R) + 1μM mito-Tempo
- ND (H/R)
ND (H/R) + 10μM MT (ND(H/R) + 10μM mito-Tempo
- NO
nitric oxide
- PGI2
prostacyclin
- ROS
reactive oxygen species
- SK
small conductance calcium-activated potassium channels
- SNP
sodium nitroprusside
- TBST
Tris-buffered saline, 0.1% Tween 20
Biography

Footnotes
Conflict of interests and any other disclosure: Conflict of interests: No any potential conflicts of interest exist for all authors; Any other disclosure: No any potential conflicts of interest exist for all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- 1.Sarkar M and Prabhu V. Basics of cardiopulmonary bypass. Indian journal of anaesthesia. 2017;61:760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tempe DK, Saigal D. Myocardial protection during cardiac surgery: An overview of cardioplegia: In: Sehgal R, Trikha A, eds. Yearbook of Anesthesiology-7. 2018;(Chapter 24):285–305. [Google Scholar]
- 3.Salameh A and Dhein S. Strategies for Pharmacological Organoprotection during Extracorporeal Circulation Targeting Ischemia-Reperfusion Injury. Frontiers in pharmacology. 2015;6:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng J and Sellke F. Microvascular dysfunction in patients with diabetes after cardioplegic arrest and cardiopulmonary bypass. Curr Opin Cardiol. 2016;31:618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sellke N, Kuczmarski A, Lawandy I, Cole VL, Ehsan A, Singh AK, Liu Y, Sellke FW and Feng J. Enhanced coronary arteriolar contraction to vasopressin in patients with diabetes after cardiac surgery. J Thorac Cardiovasc Surg. 2018;156:2098–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robich MP, Araujo EG, Feng J, Osipov RM, Clements RT, Bianchi C and Sellke FW. Altered coronary microvascular serotonin receptor expression after coronary artery bypass grafting with cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2010;139:1033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng J, Liu Y, Chu LM, Singh AK, Dobrilovic N, Fingleton JG, Clements RT, Bianchi C and Sellke FW. Changes in microvascular reactivity after cardiopulmonary bypass in patients with poorly controlled versus controlled diabetes. Circulation. 2012;126:S73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Sellke EW, Feng J, Clements RT, Sodha NR, Khabbaz KR, Senthilnathan V, Alper SL and Sellke FW. Calcium-activated potassium channels contribute to human skeletal muscle microvascular endothelial dysfunction related to cardiopulmonary bypass. Surgery. 2008;144:239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haybar H, Shahrabi S, Rezaeeyan H, Shirzad R and Saki N. Endothelial Cells: From Dysfunction Mechanism to Pharmacological Effect in Cardiovascular Disease. Cardiovascular Toxicology. 2019;19:13–22. [DOI] [PubMed] [Google Scholar]
- 10.Shahreza FD. From oxidative stress to endothelial cell dysfunction. Journal of Preventive Epidemiology. 2016;1:e04–e04. [Google Scholar]
- 11.Vanhoutte PM. Endothelial dysfunction. Circulation Journal. 2009;73:595–601. [DOI] [PubMed] [Google Scholar]
- 12.Félétou M, Vanhoutte PM, Weston AH and Edwards G. EDHF and endothelial potassiun channels: IKCa and SKCa. Br J Pharmacol. 2003;140:225; author reply 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stankevičius E, Dalsgaard T, Kroigaard C, Beck L, Boedtkjer E, Misfeldt MW, Nielsen G, Schjorring O, Hughes A and Simonsen U. Opening of small and intermediate calcium-activated potassium channels induces relaxation mainly mediated by nitric-oxide release in large arteries and endothelium-derived hyperpolarizing factor in small arteries from rat. Journal of Pharmacology and Experimental Therapeutics. 2011;339:842–850. [DOI] [PubMed] [Google Scholar]
- 14.Dalsgaard T, Kroigaard C, Bek T and Simonsen U. Role of calcium-activated potassium channels with small conductance in bradykinin-induced vasodilation of porcine retinal arterioles. Investigative ophthalmology & visual science. 2009;50:3819–3825. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Xie A, Singh AK, Ehsan A, Choudhary G, Dudley S, Sellke FW and Feng J. Inactivation of Endothelial Small/Intermediate Conductance of Calcium-Activated Potassium Channels Contributes to Coronary Arteriolar Dysfunction in Diabetic Patients. Journal of the American Heart Association. 2015;4:e002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng J, Liu Y, Clements RT, Sodha NR, Khabbaz KR, Senthilnathan V, Nishimura KK, Alper SL and Sellke FW. Calcium-Activated Potassium Channels Contribute to Human Coronary Microvascular Dysfunction After Cardioplegic Arrest. Circulation. 2008;118:S46–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Q, Huang JH, Man YB, Yao XQ and He GW. Use of intermediate/small conductance calcium-activated potassium-channel activator for endothelial protection. J Thorac Cardiovasc Surg. 2011;141:501–10, 510 e1. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Shi G, Liu Y, Xing H, Kabakov AY, Zhao AS, Agbortoko V, Kim J, Singh AK, Koren G, Harrington EO, Sellke FW and Feng J. Coronary endothelial dysfunction prevented by small-conductance calcium-activated potassium channel activator in mice and patients with diabetes. J Thorac Cardiovasc Surg. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Kabakov AY, Xie A, Shi G, Singh AK, Sodha NR, Ehsan A, Usheva A, Agbortoko V, Koren G, Dudley SC Jr., Sellke FW and Feng J. Metabolic regulation of endothelial SK channels and human coronary microvascular function. International journal of cardiology. 2020;312:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, Suzuki H, Toyama K, Spin JM and Tsao PS. Diabetic Cardiovascular Disease Induced by Oxidative Stress. International journal of molecular sciences. 2015;16:25234–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura H, Matoba S, Iwai-Kanai E, Kimata M, Hoshino A, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Okigaki M, Adachi S, Tanaka H, Takamatsu T and Matsubara H. p53 promotes cardiac dysfunction in diabetic mellitus caused by excessive mitochondrial respiration-mediated reactive oxygen species generation and lipid accumulation. Circulation Heart failure. 2012;5:106–15. [DOI] [PubMed] [Google Scholar]
- 22.Li T-B, Zhang J-J, Liu B, Luo X-J, Ma Q-L and Peng J. Dysfunction of endothelial progenitor cells in hyperlipidemic rats involves the increase of NADPH oxidase derived reactive oxygen species production. Canadian journal of physiology and pharmacology. 2017;95:474–480. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Z, Shen X, Jiang X, Shan H, Cimini M, Fang P, Ji Y, Park JY, Drosatos K, Yang X, Kevil CG, Kishore R and Wang H. Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: Role of insufficient hydrogen sulfide. Redox Biol. 2018;16:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kluge MA, Fetterman JL and Vita JA. Mitochondria and Endothelial Function. Circulation Research. 2013;112:1171–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trnka J, Blaikie FH, Smith RA and Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radical Biology and Medicine. 2008;44:1406–1419. [DOI] [PubMed] [Google Scholar]
- 26.Choumar A, Tarhuni A, Lettéron P, Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R and Pessayre D. Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxidants & redox signaling. 2011;15:2837–2854. [DOI] [PubMed] [Google Scholar]
- 27.Patil NK, Parajuli N, MacMillan-Crow LA and Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. American journal of physiology Renal physiology. 2014;306:F734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang A, Keita Å V, Phan V, McKay CM, Schoultz I, Lee J, Murphy MP, Fernando M, Ronaghan N, Balce D, Yates R, Dicay M, Beck PL, MacNaughton WK, Söderholm JD and McKay DM. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. The American journal of pathology. 2014;184:2516–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG and Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bers DM, Patton CW and Nuccitelli R. A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol. 2010;99:1–26. [DOI] [PubMed] [Google Scholar]
- 31.Sellke FW, Shafique T, Ely DL and Weintraub RM. Coronary endothelial injury after cardiopulmonary bypass and ischemic cardioplegia is mediated by oxygen-derived free radicals. Circulation. 1993;88:II395–400. [PubMed] [Google Scholar]
- 32.Hiratzka LF, Eastham CL, Carter JG, Moyers JR, Elliott DR, Doty DB, Wright CB and Marcus ML. The effects of cardiopulmonary bypass and cold cardioplegia on coronary flow velocity and the reactive hyperemic response in patients and dogs. Ann Thorac Surg. 1988;45:474–81. [DOI] [PubMed] [Google Scholar]
- 33.Tofukuji M, Stahl GL, Agah A, Metais C, Simons M and Sellke FW. Anti-C5a monoclonal antibody reduces cardiopulmonary bypass and cardioplegia-induced coronary endothelial dysfunction. J Thorac Cardiovasc Surg. 1998;116:1060–8. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y and Dong W. ROS and ROS-Mediated Cellular Signaling. Oxidative Medicine and Cellular Longevity. 2016;2016:4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q and Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res. 2018;122:877–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He L, He T, Farrar S, Ji L, Liu T and Ma X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;44:532–553. [DOI] [PubMed] [Google Scholar]
- 37.Dhalla NS, Elmoselhi AB, Hata T and Makino N. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovascular Research. 2000;47:446–456. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Cole V, Lawandy I, Ehsan A, Sellke FW and Feng J. Decreased coronary arteriolar response to KCa channel opener after cardioplegic arrest in diabetic patients. Mol Cell Biochem. 2018;445:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang JZ, Clark JW, Bryan RMJ and Robertson CS. Mathematical modeling of the nitric oxide/cGMP pathway in the vascular smooth muscle cell. American journal of physiology Heart and circulatory physiology. 2005;289 2:H886–97. [DOI] [PubMed] [Google Scholar]
- 40.Ledoux J, Bonev AD and Nelson MT. Ca2+-activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol. 2008;131:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Gao Q, Jiang L, Feng X, Zhu X, Fan X, Mao C and Xu Z. The NOX2-derived reactive oxygen species damaged endothelial nitric oxide system via suppressed BKCa/SKCa in preeclampsia. Hypertension Research. 2017;40:457–464. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Experimental Groups.
Figure S2: The schematic diagram illustrates patch clamp recording system. A glass pipette seals to the membrane of endothelial cell, once break through the membrane by applying light and short suction pulses with a syringe, then the analog current signal is amplified and digitized so that the signal can be analyzed.
Figure S3 Characteristics of diabetic (DM) and nondiabetic (ND) Mice. A, age, B, Body weight (g), P= 0.0001 DM vs. ND. C, Blood glucose (mg/dL), P = 0.0001 DM vs. ND.
Figure S4: Effects of endothelium-dependent vasodilator on mouse heart microvessel vasodilation. A, Dose-dependent vasodilation of mouse heart microvessels of ND(H/R) and DM(H/R) with or without MT treatment (10μM) in response to the SK channel activator NS309 (10−9–10−5 M), n=6/group. *P <0.0001 (NS309 10−6 M), P <0.0001 (NS309 10−5M), ND(H/R) vs. ND(H/R) +10μM MT; #P =0.0284 (NS309 10−6 M); P =0.0043 (NS309 10−5 M), DM(H/R) vs. DM(H/R) +10μM MT; @P <0.0001 (NS309 10−6 M), P <0.0001 (NS309 10−5M), ND(H/R) vs. DM(H/R). B, Dose-dependent vasodilation of mouse small coronary arteries of ND(H/R) and DM(H/R) with or without MT (10μM) treatment in response to the endothelium-dependent vasodilator ADP (10−9–10−4 M), n=6/group. *P =0.0021(ADP 10−5 M), P =0.0062 (ADP 10−4 M), ND(H/R) vs. ND(H/R) +10μM MT; #P =0.0090 (ADP 10−5 M), P =0.0029 (ADP 10−4 M), DM(H/R) vs. DM(H/R) +10μM MT; @ P <0.0001 (ADP 10−5 M), P <0.0001 (ADP 10 −4 M), ND(H/R) vs. DM(H/R). C, Dose-dependent vasodilation of mouse small coronary arteries of ND(H/R) and DM(H/R) with or without MT treatment (10μM) in response to the endothelium-independent vasodilator SNP (10−9-10−4 M), n=6/group. ND(H/R) = ND treated by hypoxia/re-oxygenation, DM(H/R) = DM treated by hypoxia/re-oxygenation, ND(H/R) +10μM MT = ND(H/R) + 10μM mito-Tempo, DM(H/R) +10μM MT = DM(H/R) + 10μM mito-Tempo.
Figure S5: The data of IV relationship for NS309 and apamin +TRAM34 sensitive currents within each cell/treatment group. MT (10μM) significantly increases SK channel currents of MHECs in H/R model from ND and DM mice. A-D, Effect of the SK channel activator NS309 and SK channel blocker Apamin and TRAM34 on the basal whole-cell K+ currents of MHECs in H/R model from ND and DM mice treatment with or without MT. *NS309 vs. Control, P=0.0071, ND(H/R); P=0.0027, ND(H/R)+10μM MT; P=0.0494, DM(H/R); P<0.001, DM(H/R)+10μM MT, #NS309 vs. NS309+Apamin+TRAM34, P=0.0061, ND(H/R); P=0.0018, ND(H/R) +10μM MT; P=0.038, DM(H/R); P=0.0005, DM(H/R) +10μM MT.
Video: First author Yi Song introduces the aims, hypothesis, methods and results of current study and explains the selective mROS inhibitor mito-Tempo-induced coronary endothelial protection and microvascular relaxation in the setting of cardioplegic hypoxia/reoxygenation and diabetes.