

Abstract
The voltage-gated sodium channel α subunit genes comprise a highly conserved gene family. Mutations of three of these genes, SCN1A, SCN2A and SCN8A, are responsible for a significant burden of neurological disease. Recent progress in identification and functional characterization of patient mutations is generating new insights and novel approaches to therapy for these devastating disorders. In this paper we review the basic elements of sodium channel function that are used to characterize patient mutations. We summarize a large body of work using global and conditional mouse mutants to characterize the in vivo roles of these channels. We provide an overview of the neurological disorders associated with mutations of each of these genes and examples of the effects of patient mutations on channel function. Finally, we highlight therapeutic interventions that are emerging from new insights into mechanisms of sodium channelopathies.
The sodium channel gene family
The voltage-gated sodium channel α subunit gene family is comprised of ten genes in the human genome (Figure 1A). The three sodium channel genes expressed at a high level in neurons of the central nervous system are shown in red. The gene family was generated by whole genome duplication events during early chordate evolution generating four sodium channel loci, followed by tandem gene duplications within the loci on chromosomes 2 and 3 later in vertebrate evolution1,2. The channels are key players in the initiation and propagation of action potentials, the unit of electrophysiological activity in neurons. Sodium channels are among the most highly evolutionarily conserved genes in the human genome, and retain regions of significant sequence identity to invertebrate and prokaryotic sodium channels. Deviations from normal channel function have major clinical consequences that include seizures, intellectual disability, behavioral abnormalities and movement disorders. The genes SCN1A (Nav1.1), SCN2A (Nav1.2) and SCN8A (Nav1.6) together account for >95% of brain sodium channel transcripts and are responsible for most of the known neurological sodium channelopathies. In one survey of 8,565 individuals with epilepsy and neurodevelopmental disorders, 5% carried mutations in one of these three genes 3.
Figure 1. Evolutionary conservation of human sodium channel genes.
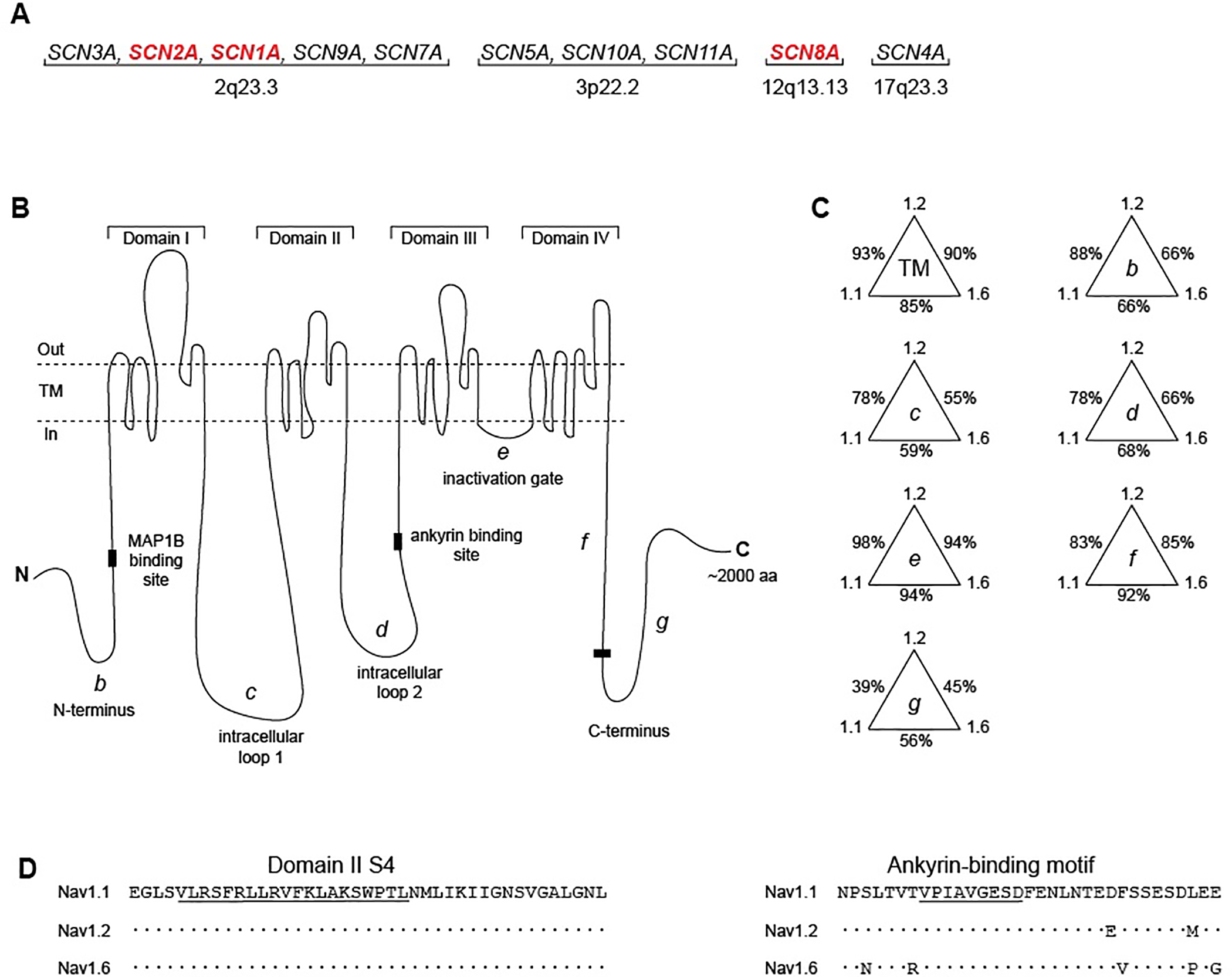
(A) Chromosomal locations of human voltage-gated sodium channel genes. The channels with high expression in the adult CNS (red) are covered in this review. (B) The voltage-gated sodium channel α subunit is composed of four transmembrane domains separated by intracellular loops. TM, transmembrane segments; b, N-terminus; c, cytoplasmic loop 1; d, cytoplasmic loop 2; e, inactivation gate; f, proximal half of C-terminus; g, distal half of C-terminus. (C) Percent conservation of amino acid sequence in the protein domains of SCN1A (Nav1.1), SCN2A (Nav1.2), and SCN8A (Nav1.6). Labels refer to domains in panel B. (D). Examples of regions of high sequence conservation in transmembrane segment DIS4 (left) and around the 9 residue ankyrin binding motif (right) 38. Dots represent amino acid identity.
The structure of the sodium channel protein is represented in Figure 1B. The protein includes four homologous domains each containing six transmembrane segments with high sequence conservation, two large cytoplasmic loops with lower sequence conservation, a highly conserved inactivation gate, and cytoplasmic N-terminal and C-terminal domains. Consistent with their more recent divergence, SCN1A and SCN2A are more closely related to each other than to SCN8A (Figure 1C). The 24 transmembrane (TM) segments exhibit 93% amino acid sequence identity between SCN1A and SCN2A but only 85% and 90% identity to SCN8A. An example of an invariant TM segment is shown in Figure 1D. In the more divergent N-terminus, there is 88% sequence identity between SCN1A and SCN2A but only 66% identity to SCN8A. In spite of extensive sequence conservation, the channels have diverged in function and regulation, and each of these genes is essential in the mammalian genome. In this review, we describe the functions of these closely related channels and examine the clinical consequences of genetic variation. Evolutionary conservation offers a clue to functional impact, and variation at residues that are identical in all three channels (e.g. Figure 1D) tend to be more deleterious than variants at residues that have diverged. Clinical exome sequencing has revealed a major role for rare sodium channel variants in neurodevelopmental disorders (Table 1). Current research is focused on distinguishing between neutral and deleterious variants and understanding the relationship between altered channel function and clinical outcome. Recent progress in physiological, molecular and clinical studies is generating novel therapeutic approaches.
Table 1.
Clinical disorders associated with mutations of SCN1A, SCN2A and SCN8A.
| Gene | Protein | Type of mutation | Common clinical diagnoses | OMIM # |
|---|---|---|---|---|
| SCN1A | Nav1.1 | Gain of function | GEFS+ | 604403 |
| Familial hemiplegic migraine | 609634 | |||
| Loss of function | DEE (Dravet syndrome) | 607208 | ||
| SCN2A | Nav1.2 | Gain of function | DEE | 613721 |
| BFNIS | 607745 | |||
| Episodic ataxia | 618924 | |||
| Loss of function | Autism spectrum disorder | ----- | ||
| Intellectual disability | ----- | |||
| DEE | 613721 | |||
| SCN8A | Nav1.6 | Gain of function | DEE | 614558 |
| Loss of function | Intellectual disability | 614306 | ||
| Movement disorder | 618364 | |||
| Autism spectrum disorder | ----- | |||
DEE, Developmental and Epileptic Encephalopathy; GEFS+, Generalized Epilepsy with Febrile Seizures Plus; BFNIS, Benign Familial Neonatal and Infantile Seizures; OMIM, On-line Mendelian Inheritance in Man (omim.org).
Sodium channel α subunits are associated in the neuronal cell membrane with single-transmembrane β subunits encoded by the genes SCNB1 to SCNB44. The β subunits influence trafficking and electrophysiological properties of the α subunits but do not themselves have channel activity. Their clinical roles have been recently reviewed5 and will not be discussed. SCN3A encodes a sodium channel that is expressed at a high level early in the development of the CNS and at a low level in the adult CNS. A small but growing number of mutations of SCN3A have been identified in patients with epileptic encephalopathy 6 and cortical malformations including polymicrogyria and microcephaly 7,8. These patients were recently reviewed8 and are not included here.
Physiology of Nav1.1, Nav1.2 and Nav1.6.
The voltage-gated sodium channel α subunit is a large protein of 2,000 amino acids with a complex mode of action. It has been selected through evolution to open transiently in response to depolarization of the neuronal membrane and to close within milliseconds, generating a brief inward flow of sodium ions. Single amino acid substitutions frequently change multiple components of channel function, making it difficult to systematically classify mutations. We are still at the stage of identifying rare variants, cataloging functional effects on a few parameters, and looking for correlations with clinical outcomes. We describe some basic features of the human sodium channels for the non-expert reader, as a starting point for the discussion of pathogenic consequences of patient mutations.
Electrophysiology
Sodium channels are located throughout the neuronal cell membrane on axons, dendrites and soma. In response to a depolarizing shift in electrical potential across the membrane, conformational changes in the positively-charged transmembrane segments initiate the transition from closed to open channel state, permitting an influx of sodium ions and initiation of the action potential. Fast inactivation follows within milliseconds: the channel pore is blocked by the inactivation gate and the channel enters the inactive conformation. The influx of sodium ions is thus limited to a brief interval. Recovery from inactivation returns the inactivation gate to its resting position and restores the stable closed conformation. Changes in this progression underlie the pathogenesis of sodium channel mutations.
Electrophysiological measurements used to assess the functional effects of patient mutations are shown in Figure 2. Peak or transient current refers to the maximal inward flow of sodium ions at the beginning of the action potential. The small remaining current at 100 msec after the peak is defined as ‘persistent current’ (Figure 2A). The voltage-dependence of channel activation describes channel opening (Figure 2B), and the voltage dependence of inactivation describes channel closing (Figure 2C); these are altered by many pathogenic mutations. ‘Resurgent current’ is generated when channels open during repolarization after an action potentialm and contributes to repetitive neuronal firing (Figure 2D).
Figure 2. Channel properties frequently used to characterize patient mutations.
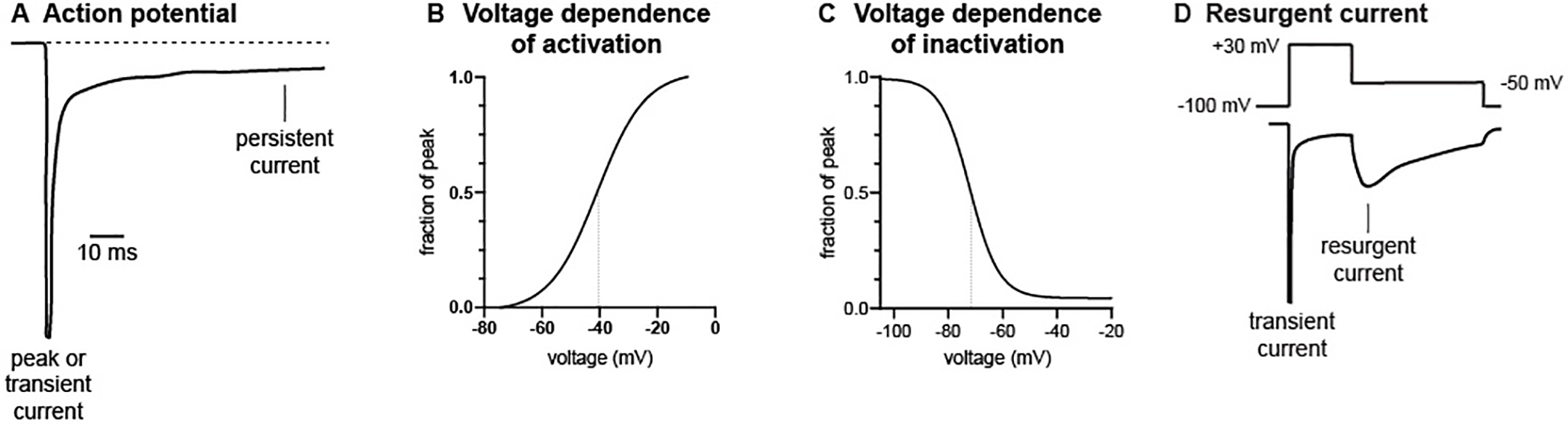
A, peak and persistent current. B, voltage dependence of channel activation. C, voltage dependence of channel inactivation. D, resurgent current. Vertical lines in B and C mark the voltage at which 50% of channels are active.
Partial or complete loss of function mutations are recognized by reduction of peak current. Gain-of-function mutations result in qualitative changes of other parameters. Increased persistent current, due to impaired stability of the closed channel conformation, is frequently associated with seizures. Nav1.6 generates a higher proportion of persistent and resurgent current than Nav1.1 or Nav1.2 in many types of neurons 9,10. A shift the voltage-dependence of activation towards more negative values, a hyperpolarizing shift, leads to premature channel opening and excess neuronal firing. Conversely, depolarizing variants shift the voltage dependence of activation towards more positive potentials and reduce channel activity. The more hyperpolarized voltage dependence of activation of Nav1.6 underlies its role in initiation of action potentials. Delayed channel inactivation can also contribute to excess neuronal activity.
Experimental measurements are influenced by the choice of cells in studies of transfected channels. Cells commonly used include kidney-derived HEK cells, neuroblastoma-derived ND7/23 cells, and cultured primary neurons. In one recent study of Nav1.6 variants, combined analysis in neuroblastoma and primary neuronal cultures was more consistent than either alone in predicting clinical consequences 11. Reprogrammed neurons from patient-derived iPSCs make it possible to assess the function of mutant channels in the context of the patient’s individual genetic background 12. Biophysical properties are influenced by interactions with β-subunits, calmodulin, and other proteins present in different types of neurons13–15 and many pathogenic mutations alter more than one biophysical parameter. These variables make it difficult to classify the functional effects of deleterious mutations assayed in different laboratories. Animal models provide access to the effects of mutations in different types of neurons, but in vivo models are limited to a small number of mutations.
Subcellular localization
The sodium channels are localized in the neuronal cell membrane, with concentration at the axon initial segment (AIS) and nodes of Ranvier of myelinated neurons. Action potentials are initiated at the AIS and propagated in two directions. Forward propagation from the distal AIS (further from the cell body) initiates conduction down the axon to the nerve terminus. Nav1.6 is the major channel in the distal AIS of adult neurons16–23. A recent study employing photoactivation localization microscopy estimated that the concentration of Nav1.6 is 40-fold higher at the AIS than in the soma and proximal dendrites24. The hyperpolarized voltage dependence of Nav1.6 contributes to the initiation of action potentials at the distal AIS; in the absence of Nav1.6 the threshold for initiation of action potentials is increased9,19,25–30.
The proximal AIS (closer to the cell body) is occupied by Nav1.1 or Nav1.2, depending on cell type and stage of development17,20,21. Back propagation from the proximal AIS to the soma and dendrites modulates synaptic strength and mediates learning and memory. Within the hippocampus, Nav1.1 is found at the AIS of interneurons but not excitatory neurons21,22,31. Nav1.2 is localized to the soma and dendrites of pyramidal neurons32.
The concentration of sodium channels at the nodes of Ranvier mediates saltatory conduction in myelinated neurons. Nav1.2 is the major nodal channel during early development, and replaced by Nav1.6 during postnatal development33,34. Nav1.1 is also expressed in some nodes of Ranvier31. In mice lacking Nav1.6, there is reduced transmission at the neuromuscular junction and hind limb paralysis35. In adult unmyelinated neurons, Nav1.2 is localized along the length of the axon16,34,36.
Channel localization is mediated by interaction with protein complexes that include the structural proteins Ankyrin G and MAP1B. Ankyrin G binds a conserved 9-residue motif in intracellular loop 2 of the voltage gated channels (Figure 1B)37–39. Ankyrin G binding is sufficient to localize proteins to the AIS and nodes of Ranvier38,39 and mutation of the ankyrin binding motif prevents localization23,39. The cytoplasmic N-terminus of Nav1.6 contains a binding site for the microtubule-binding protein MAP1B40. Interaction with MAP1B stabilizes Nav1.6 at the AIS by preventing rapid endocytosis41. MAP1B does not interact with Nav1.1 and may play a role in preferential localization of Nav1.6 to the distal AIS40. Neither Ankyrin G nor MAP1B is required for somatodendritic localization of Nav1.624,39,41. The missense mutation p.Ser21Pro in the N-terminus of mouse Scn8a prevents correct localization by trapping the mutant channel in the Golgi42.
Alternative splicing of sodium channel transcripts.
SCN1A, SCN2A and SCN8A undergo two types of alternative splicing. Each gene contains two copies of the 5th exon encoding transmembrane segments 3 and 4 of domain 1. The choice between inclusion of exon 5N (neonatal) versus exon 5A (adult) is developmentally regulated. The peptides encoded by exons 5A and 5N differ by three amino acids in SCN1A43, and by a single amino acid in SCN2A and SCN8A44. In Nav1.2, the voltage dependence of activation of channels expressing exon 5N is more depolarized than channels with exon 5A, resulting in delayed channel opening45. Three pathogenic mutations of SCN2A were reported to have a more severe effect on the neonatal protein than the adult form46. These differences may explain the clinical improvement in some sodium channelopathies after the switch from neonatal to adult splice form.
The gene region encoding domain III contains another set of alternatively spliced exons that are designated “poison exons” because they contain in-frame stop codons that truncate the channel protein. The structure of these exons is similar but not identical in the three genes. In SCN8A, the pair of mutually exclusive exons 18A and 18N encode segments 3 and 4 of domain III, corresponding to exons 5A and 5N in domain I, and indicating a shared evolutionary origin47. Exon 18N contains an in-frame stop codon, and is expressed at a low level in non-neuronal tissues including glia47,48. Transcripts containing 18N are subject to nonsense-mediated decay. Inclusion of exon 18A appears to be restricted to neurons, and is mediated by neuron-specific splice factors including RbFox148-50. Cultured astrocytes and oligodendrocytes express only exon 18N, preventing expression of full-length Nav1.6 protein48. Poison exons may represent a fail-safe mechanism to prevent damage to non-neuronal cells that could result from expression of voltage-gated sodium channels44.
In SCN1A and SCN2A, individual poison exons are also located in regions encoding domain III51,52. These poison exons are potential targets for manipulating gene expression. For example, blocking the inclusion of exon 20N in transcripts of SCN1A increases the expression of Nav1.1 in vivo53; the therapeutic application is discussed later.
Sodium channel function in the mouse.
The evolutionary conservation of the sodium channel gene family in mouse and human has led to a large body of experimental data on the in vivo function of specific sodium channel genes. We discuss below the effects of global knockout of each gene (Table 2A) and the use of conditional knockouts to dissect the pathogenic effects of sodium channel mutations in different classes of neurons (Table 2B and 2C).
Table 2.
Global and regional knock-out of sodium channel genes in the mouse CNS.
| A. Global knock-out | CRE | Specificity | Phenotypes | Ref. | |
|---|---|---|---|---|---|
| Scn1a | +/− | na | Global | Convulsive seizures, strain dependent reduced survival, autistic-behaviors, deficits in spatial learning and memory, hyperactivity | 31,54,57,58,63,166 |
| −/− | na | Global | Convulsive seizures, lethal @ P14 | 31,54 | |
| ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- | |||||
| Scn2a | +/− | na | Global | Absence seizures | 59 |
| Behaviors seen in models of autism and schizophrenia | 118 | ||||
| −/− | na | Global | Lethal @ P2, hypoxia, neuronal cell death | 55,59 | |
| ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- | |||||
| Scn8a | +/− | na | Global | Anxiety-like behaviors; | 62 |
| Protection from induced and genetic seizures |
71,167 | ||||
| −/− | na | Global | Lethal @ P21, hind limb paralysis, muscle atrophy | 56 | |
| B. Regional knock-out | |||||
| Scn1a null | F/+ | Vgat | GABAergic inhibitory neurons | Convulsive seizures, sudden death | 87 |
| F/+ | Dlx1/2-I12b | Forebrain GABAergic interneurons | Convulsive seizures, sudden death | 86 | |
| Autistic-like behaviors, deficits in spatial learning and memory | 57 | ||||
| F/+ | PV | Parvalbumin-expressing neurons | Some convulsive seizures and sudden death, susceptibility to thermally-induced seizures | 87,168 | |
| Autistic-like behaviors, lack of social novelty preference, impaired spatial memory | 60,168 | ||||
| F/+ | SST | Somatostatin -expressing neurons | Hyperactivity without autistic-like behaviors, no spontaneous convulsive seizures, susceptibility to thermally-induced seizures | 60,168 | |
| F/+ | PV+SST | PV and SST-expressing neurons | Deficits in long-term spatial memory, susceptibility to thermally-induced seizures | 168 | |
| F/F | PV | Parvalbumin-expressing neurons | Convulsive seizures, sudden death, ataxia | 87 | |
| F/+ | EmxI | Forebrain excitatory neurons | No seizures | 87 | |
| F/F | AAV-Cre | Hippocampal injection | Spontaneous seizures, nonlethal, susceptibility to thermally-induced seizures, deficits in spatial learning and memory | 88,169 | |
| F/F | AAV-Cre | Cortical injection | Spontaneous seizures, nonlethal, susceptibility to thermally-induced seizures | 169 | |
| ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- | |||||
| Scn2a null | F/+ | EmxI | Forebrain excitatory neurons | Absence seizures | 59 |
| Anxiety-like behaviors, increased vertical activity | 118 | ||||
| F/F | EmxI | Forebrain excitatory neurons | Lethal @ P2 | 59 | |
| F/+ | Vgat | GABAergic inhibitory neurons | No seizures, 30% sudden death | 59 | |
| Anxiety-like behaviors | 118 | ||||
| F/F | Vgat | GABAergic inhibitory neurons | Lethal P2 | 59 | |
| F/F | Trpc4 | Cortical layer 5 pyramidal neurons | Absence seizures | 170 | |
| F/F | Ntsr1 | Cortical layer 6 pyramidal neurons | No seizures | 170 | |
| ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- | |||||
| Scn8a null | F/F | Pcp2 | Purkinje cell | Ataxia, impaired motor functions, reduced firing of Purkinje neurons | 137 |
| Impaired delay conditioning and Morris water maze | 171 | ||||
| F/F | α6 | Cerebellar granule cell | Reduced learning in rotarod test | 137 | |
| F/+ | EmxI | Forebrain excitatory neurons | Protection from induced seizures, flurothyl | 143 | |
| F/+ | Dlx5/6 | Inhibitory neurons | Absence seizures | 143 | |
| F/F | NEX | Forebrain excitatory neurons | No abnormality in vivo, reduced persistent Na+ current in AIS of layer 5 pyramidal neurons | 28 | |
| F/F | Lenti-Cre | Hippocampal injection | Protection from induced seizures | 167 | |
| +/+ | RNAi | Hippocampal injection | Prevents seizure in mouse model of mesial temporal lobe epilepsy (MTLE) | 172 | |
| +/+ | RNAi | Injection of thalamic reticular nucleus cells | Absence seizures | 143 | |
| C. Knock-in of gain of function mutation | |||||
| Scn8a R1872W | R1872W/+ | EIIa | Global | Lethal convulsive seizures @ 2 weeks | 131 |
| EmxI | Forebrain excitatory neurons | Lethal convulsive seizures @ 3 weeks | 131 | ||
| Dlx5/6 | Inhibitory neurons | No abnormality | 131 | ||
| CAG-ER | Inducible, global | Lethal convulsive seizures | 131 | ||
KO, knockout; F/+, floxed heterozygote; F/F, floxed homozygote; lenti, lentivirus injection.
Global knock-out mice.
The distinct in vivo functions of Nav1.1, Nav1.2 and Nav1.6 are evident from comparison of knock-out mice with (null) mutations of each gene. Complete. (homozygous) loss of each gene is lethal, but the phenotypic effects of inactivation differ (Table 2A). Global inactivation of Scn1a results in a seizure disorder with onset at 3 weeks31,54. Global inactivation of Scn2a results in neonatal death due to respiratory failure55. Global inactivation of Scn8a causes failure of the neuromuscular junction and hind limb paralysis 34,56.
Heterozygous null mice with 50% of normal channel gene expression have less severe abnormalities. Haploinsufficiency for Scn1a results in spontaneous convulsive seizures, like the homozygote, with later onset than in homozygotes, 50% lethality, and impaired social interaction and poor spatial learning57,58. Haploinsufficiency for Scn2a causes absence seizures (brief periods of immobility) and behavioral abnormalities with normal survival59,60. Haploinsufficiency for Scn8a results in absence seizures61 and anxiety-like behavior62 with normal life span 35,56.
Genetic modifiers and gene interactions in the mouse.
Genetic divergence between inbred strains of mice has been used to identify modifier genes that influence disease severity. For example haploinsufficiency of Scn1a, a model of Dravet Syndrome, results in spontaneous seizures in strain C57BL/6J but not in strain 12963. This difference was traced to a previously unrecognized splice site variant in the Gabra2 gene in strain C57BL/6J that causes a three-fold reduction in expression of the α2 subunit of the GABAA receptor63,64. This Gabra2 variant also accelerates seizure onset in mice with an epileptogenic mutation of Scn8a65.
Strain C57BL/6J also carries an exonic splice site variant in the gene encoding the splice factor Scnm1, resulting in that exacerbated dystonia in the partial loss-of-function medJ mutant of Scn8a66–68. Variants in the human orthologs of these modifier genes may contribute to observed differences among patients with identical sodium channel mutations69.
Interactions between multiple ion channel variants have also been demonstrated by combining mutants in the mouse. For example, heterozygous loss-of-function of Scn8a is protective against seizures in Scn1a haploinsufficient mice70,71. Variation in the potassium channel Kcnv2 modifies the severity of seizures caused by a gain-of-function variant of Scn2a 72. Conversely, haploinsufficiency of Scn2a mitigates seizures in the Kcn1a−/− mice 73. These observations predict potential gene interactions in patients. In a study of patients with monogenic epilepsy due to cation channel variants, the frequency of secondary deleterious variants in other ion channels was higher than in controls, suggesting exacerbation of the primary pathogenic mutation74.
Sodium channel mutations in human disease.
The past few years has seen a tremendous increase in the association of sodium channelopathies with neurodevelopmental disorders. Not surprisingly in view of their evolutionary and functional similarities, there is considerable overlap among clinical conditions caused by mutations of SCN1A, SCN2A and SCN8A (Table 1). To date, the highest number of sodium channel mutations have been identified in patients with developmental and epileptic encephalopathy (DEE), complex disorders characterized by onset of intractable seizures within the first year of life, intellectual disabilities, movement disorders and elevated risk of sudden unexpected death in epilepsy (SUDEP). Most DEE mutations arise de novo in the patient; a few are inherited from a unaffected mosaic parent75. In addition to DEE, SCN1A, SCN2A and SCN8A are associated with mild seizure disorders and are high confidence genes for autism spectrum disorder (Gene.SFARI.org 2020).
Pathogenic mutations identified by exome or genome sequencing in patients are classified by electrophysiological assay as either ‘loss-of-function’ (LOF), including protein truncation and inactivating missense mutations, or ‘gain-of-function’ (GOF), amino acid substitutions that alter biophysical properties like voltage-dependence, resurgent current, and persistent current (Figure 2). What appear to be minor changes in these parameters in assays often have major impact in vivo. It is medically important to distinguish between GOF and LOF mutations because of their different implication for treatment. Patients with GOF mutations often benefit from sodium channel blockers, while LOF mutations are exacerbated by further reduction in sodium channel activity. Thousands of sodium channel variants have been identified in patients, but only a few hundred have been subjected to functional studies, and most newly described missense variants must still be classified as Variants of Unknown Significance.
In this section we summarize current knowledge regarding the clinical consequences of mutations in the three major sodium channel genes, pointing out overlaps and differences, followed by discussion of new therapies and questions for the future. Databases are available with compiled information about patient variants of SCN1A (www.gzneurosci.com/scn1adatabase/ ), SCN2A and SCN8A (SCN8A.net).
SCN1A.
Developmental and epileptic encephalopathy (DEE).
Dravet Syndrome is the most common of the DEEs, with an incidence of 1/20,900 in the US population 76. Eighty to 90% of patients with Dravet Syndrome have de novo mutations of SCN1A, and more than 1250 unique mutations have been reported77,78. The average age of seizure onset is 6 months. The first seizure is often triggered by fever or other elevated body temperature79,80. Development is often normal during the first year, but most patients develop cognitive, intellectual and motor co-morbidities during the second year of life81. Ataxia is a comorbidity in 60% of patients79.
The major molecular mechanism underlying Dravet Syndrome is haploinsufficiency of SCN1A80. Most mutations are located in coding sequences, and more than half result in protein truncation by frameshift, nonsense or splice site mechanisms82. Missense mutations in Dravet Syndrome also result in loss of channel function, as shown for the patient mutation p.Ser259Arg (Figure 3A)83,84. To explain the 5 to10% of Dravet Syndrome patients lacking mutations in coding exons, attention has been directed to the noncoding sequences of SCN1A. Recently, noncoding variants in intron 20 were demonstrated to reduce SCN1A expression by increasing the inclusion of the ‘poison exon’ 20N, containing an in-frame stop codon, leading to protein truncation85.
Figure 3. Functional effects of patient mutations in SCN1A, SCN2A and SCN3A.
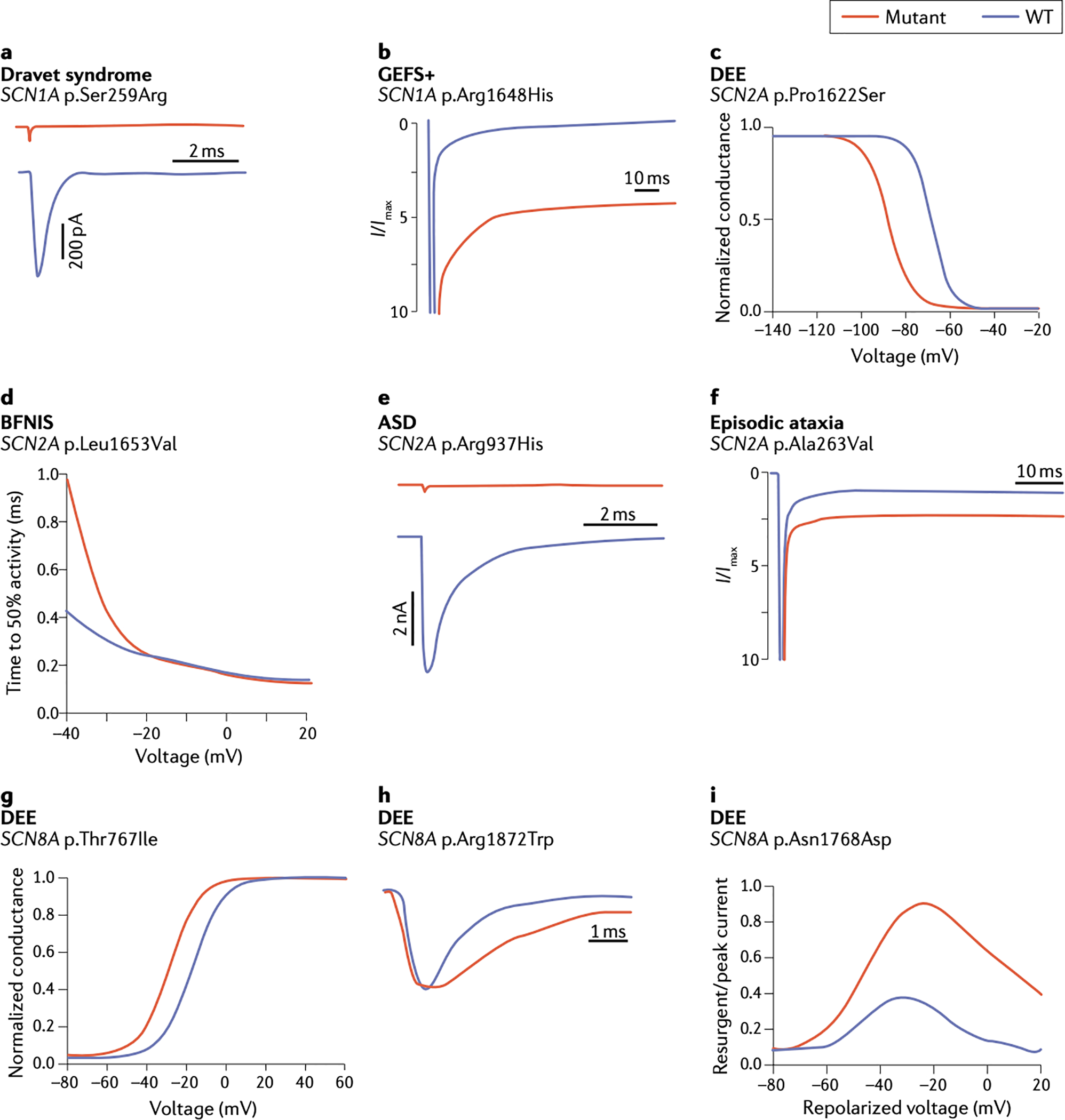
Representative examples adapted from the indicated publications, which contain experimental details. A. The Dravet Syndrome mutation p.S259R in SCN1A causes complete loss of channel function 83. B. The inherited variant p.R1648H in SCN1A in a family with GEFS+ causes increased persistent current 99. C. p.P1622S in SCN2A in a patient with late-onset DEE causes a hyperpolarizing shift in the voltage dependence of inactivation 104. D. p.L1653V in SCN2A in a family with benign familial neonatal-infantile seizures (BFNIS) causes rapid channel activation 114. E. p.R937H in SCN2A in a patient with autism spectrum disorder (ASD) causes loss of channel function 116. F. p.A263V in SCN2A in a patient with episodic ataxia causes increased persistent current 120. G. The mutation p.T767I in SCN8A in a patient with DEE causes premature channel activation 122. H. De novo mutation p.R1872W in SCN8A in a patient with DEE causes delayed channel inactivation 163. I. De novo mutation p.N1768D in SCN8A in a patient with DEE causes elevated resurgent current 164.
Mouse models of Dravet Syndrome with haploinsufficiency of Scn1a reproduce clinical phenotypes including early onset spontaneous tonic-clonic seizures, susceptibility to elevated temperature, and behavioral abnormalities. To identify the neurons contributing to seizures, conditional (floxed) alleles of mouse Scn1a have been combined with neuron-specific CRE recombinase. Seizures can result from loss of Scn1a expression in inhibitory neurons86,87, more specifically in parvalbumin-positive fast-spiking inhibitory neuron 87. Inactivation of Scn1a in the hippocampus by Cre injection causes learning deficits and elevated sensitivity to thermally-induced seizures88. Studies in global haploinsufficient mice suggest that reduced excitability of Purkinje neurons may contribute to ataxia54,89.
The lethal seizure phenotype in the Dravet mouse models exhibits incomplete penetrance, and as many as 50% of Scn1a+/− mice are unaffected. This may be explained by a compensatory up-regulation of other sodium channels around one month of age, with sufficient variability to completely protect some individuals 90,91.
SUDEP is the leading cause of mortality in Dravet Syndrome, accounting for up 20% of deaths92. Scn1a haploinsufficient mice exhibit sudden, early death accompanied by impaired cardiac and respiratory function93–95. Neuronal sodium channels are expressed in cardiomyocytes at approximately 1% of their level in neurons, and expression of Scn1a in cardiomyocytes could mediate a direct effect of pathogenic mutations on cardiac function93. However, in Scn1a haploinsufficient mice, apnea and respiratory failure precede the cessation of cardiac function96.
Other SCN1A seizure disorders.
Mutations of SCN1A were originally identified in patients with GEFS+ (genetic epilepsy with febrile seizures plus)97, which manifests as childhood febrile seizures with afebrile seizures sometimes continuing beyond the age of 6 years80. The majority of SCN1A mutations in GEFS+ are missense mutations, with effects on channel function that range from partial loss of function to gain of function82,98. The p.Arg1648His mutation is a gain-of-function variant identified in GEFS+ that exhibits increased persistent current (Figure 3B)99. Other rare syndromes include epilepsy of infancy with migrating focal seizures, and myoclonic-atonic epilepsy80. The same mutation can generate a range of severity within the same family97,100, suggesting the influence of unidentified genetic modifiers.
Familial hemiplegic migraine.
SCN1A is one of three genes implicated in familial hemiplegic migraine. This rare autosomal dominant disorder is characterized by severe migraine with aura accompanied by transient hemiplegia (unilateral paralysis)101. Functional analysis of ten SCN1A mutations associated with familial hemiplegic migraine demonstrated GOF effects101–103.
SCN2A.
Developmental and epileptic encephalopathy (DEE).
SCN2A-associated DEE is characterized by severe seizures, intellectual disability, and movement disorders including dystonia and chorea104. Many patients exhibit autistic behaviors104. A distinction has been made between early and late onset forms of the disorder. Early seizure onset, prior to 3 months of age, is associated with GOF mutations of SCN2A including increased persistent and peak currents, delayed channel inactivation, and hyperpolarized voltage dependence of activation104. The variants p.Phe1597Leu and p.Ile1473Met both cause a hyperpolarizing shift in voltage dependence of channel activation that results in premature channel opening 104,105. The variant SCN2A- p.Leu1432Pro causes a hyperpolarizing shift in the voltage dependence of activation and inactivation as well as altered channel kinetics, leading to early onset DEE106. Patients with GOF mutations respond to treatment with sodium channel blockers104. In a transgenic model of a GOF mutation in Scn2a, spontaneous seizures are accompanied by elevated activity of hippocampal CA1 and CA3 neurons107,108.
In contrast to the early onset cases, DEE with seizure onset after 3 months of age is associated with partial or complete loss of function of SCN2A, including missense, frameshift, nonsense, and splice-site mutations104,105,109–111. The missense mutation p.Pro1622Ser results in a hyperpolarizing shift of fast inactivation (Figure 3C)104. The protein truncation mutation p.Arg102Stop was identified in a child with onset of intractable seizures at 19 months of age, severe mental decline and autistic behavior109. In patients with partial or complete loss of function of SCN2A, symptoms are exacerbated by sodium channel blockers104.
Benign Familial Neonatal-Infantile Seizures (BFNIS).
Gain of function mutations of SCN2A are also responsible for BFNIS, a transient disorder characterized by seizure onset before 8 months of age, seizure clusters during the first few years of life, and resolution after 2 years of age104,112. Missense mutations in BFNIS are clustered in transmembrane segments S4 and S5113. The p.Leu1653Val variant causes accelerated channel opening (Figure 3D)114. Other observed changes include hyperpolarized voltage dependence of activation and depolarized voltage dependence of inactivation114,115. The GOF variants can be managed with sodium channel blockers and usually resolve with age111. Most BFNIS variants are inherited and less deleterious than the de novo mutations in DEE104,111.
Autism Syndrome Disorder (ASD) and Intellectual Disability (ID).
Mutation of SCN2A is strongly associated with ASD116 and is estimated to account for 7.5 cases of ASD/ID per 100,000 births109,117. These heterozygous mutations result in partial or complete loss of channel function. Protein truncating mutations are common113,116. Missense mutations cluster around the ion selectivity filter of the pore loop; for example, the LOF mutation p.Arg937His causes complete loss of sodium current (Figure 3E)116.
It is not clear why some LOF mutations in SCN2A result in DEE while others lead to autism and intellectual disability. The phenotypes of mice with haploinsufficiency of Scn2a include behavioral abnormalities and absence epilepsy (brief periods of immobility and staring) but no spontaneous convulsive seizures118. Conditional deletion of one copy of Scn2a in excitatory neurons also results in absence seizures and abnormal behavior59,118 (Table 2B). Loss of Scn2a reduces backpropagation of action potentials to the soma and dendrites of excitatory neurons, resulting in synaptic impairment that may contribute to ASD and ID32.
Episodic ataxia.
Another condition caused by gain-of-function mutations of SCN2A is episodic ataxia119,120. Aaxic episodes begin after 10 months of age, last for minutes to hours, and occur on a weekly to monthly basis119. Most affected individuals also experience BFNIS-like seizures by 3 months of age119. The later onset of ataxia compared with seizures may reflect the later initiation of SCN2A expression in cerebellum compared with forebrain120. Many episodic ataxia mutations are located in DIVS4 or the adjacent intracellular linker. The recurrent mutation p.Ala263Val119 causes elevated persistent current and slowed channel inactivation (Figure 3F)120. Homozygous knock-in of p.Ala263Val in the mouse results in seizures and increased mortality121.
SCN8A.
Developmental and epileptic encephalopathy (DEE).
The major class of SCN8A mutations in DEE are de novo GOF mutations causing elevated channel activity with major effects in excitatory neurons. This is more similar to pathogenesis of SCN2A mutations than the haploinsuficiency of SCN1A in inhibitory neurons. DEE mutations in SCN8A are de novo missense mutations122. The average age of onset is 4 months, with multiple seizure types, developmental delay, cognitive impairment, movement disorders and elevated risk of lethality122–124.
SCN8A mutations have been identified in more than 300 patients125. Patient mutations are localized in transmembrane segments, the inactivation gate and the C-terminus of Nav1.6. Electrophysiological consequences of patient mutations include premature channel opening (Figure 3G), impaired channel inactivation (Figure 3H), and elevated resurgent current (Figure 3I), all leading to elevated neuronal activity (Figure 4). The increase of neuronal firing caused by p.Asn1768Asp was demonstrated in transfected hippocampal neurons126 (Figure 4A). In the mouse knock-in model of p.Asn1768Asp, there is spontaneous firing of hippocampal CA1 neurons (Figure 4B)127 and burst firing of neurons of the entorhinal cortex128 (Figure 4C). Neurons in cortical layer 2/3 do not exhibit either of these abnormalities (Figure 4B).
Figure 4. Electrophysiological and cellular mechanisms underlying seizures in an in vivo mouse model of human sodium channelopathy.
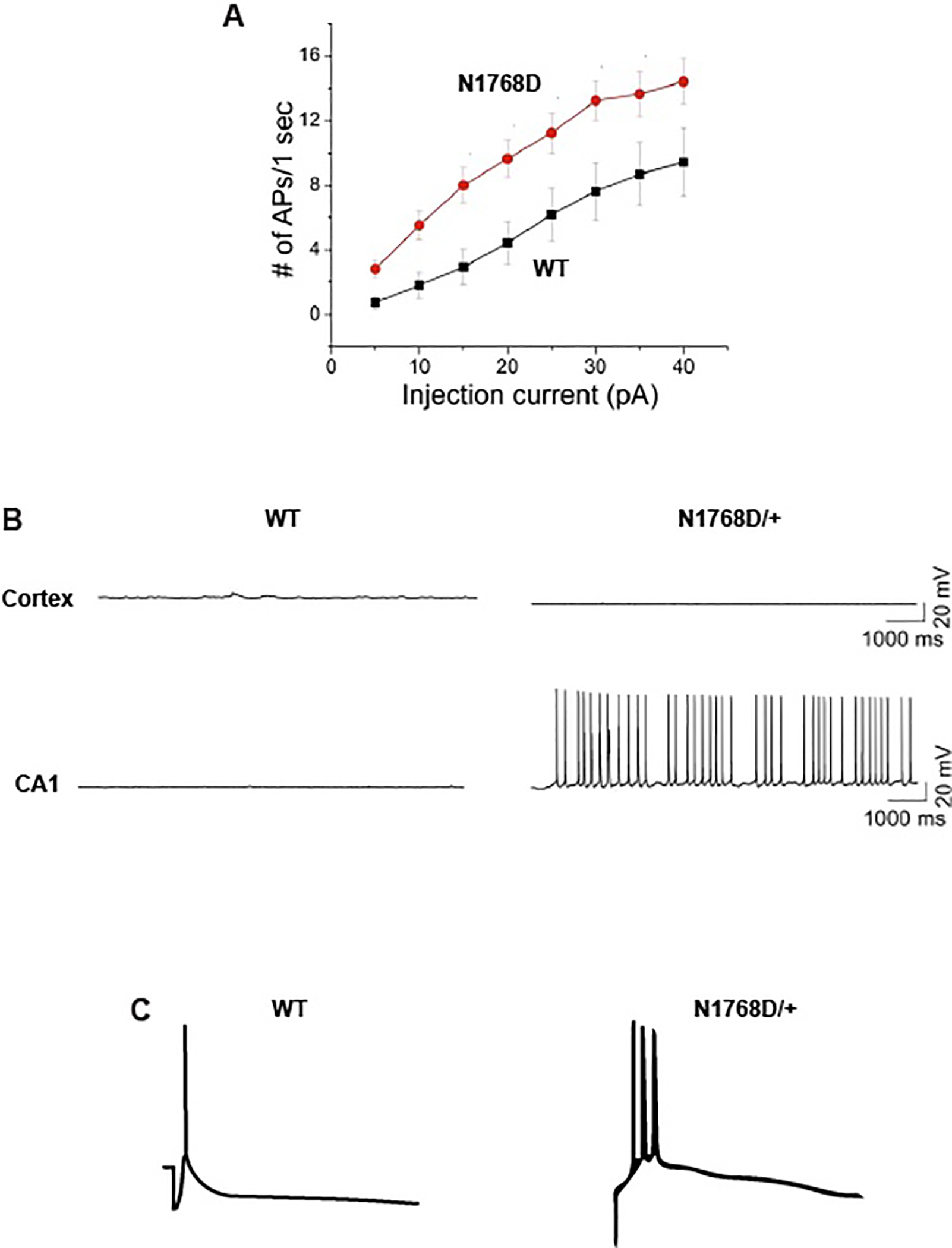
The de novo mutation SCN8A-p.Arg1768Asp was identified in a child with DEE. Functional effects include impaired inactivation, elevated persistent current 165 and elevated resurgent current 164. The altered biophysical properties of SCN8A result in elevated neuronal activity at the cellular level. A. In response to electrical stimulation, cultured hippocampal neurons transfected with the mutant channel generate more action potentials than cells transfected with wildtype channel 126. B. Slice recordings from Scn8aN1768D/+ knock-in mice demonstrate spontaneous firing of hippocampal CA1 neurons 127. Spontaneous firing is not seen in layer 2/3 cortical neurons from the same mice. C. Enterorhinal cortex neurons from the knock-in mouse exhibit burst firing after synaptic stimulation 128.
p.Arg1872Trp is a recurrent de novo mutation of SCN8A that has been observed in 8 unrelated individuals with DEE129. This mutation causes premature channel opening and impaired inactivation (Figure 3H). Substitution of Arg1872 with leucine and glutamine is also recurrent, with more than 20 independent patient mutations reported129. These mutations are predicted to weaken the ionic interaction between the positively charged arginine residue 1872 in the cytoplasmic C-terminus and negatively charged residues in the inactivation gate130 resulting in destabilization of the closed conformation and excess channel activity.
In a conditional mouse model of Scn8a-p.Arg1872Trp, CRE mediated activation of the mutant channel in excitatory neurons of the forebrain is sufficient to initiate spontaneous convulsive seizures and death (Table 2C)131. When the mutant channel was activated in adult mice, lethal seizures began within weeks, demonstrating a likely requirement for life-long treatment.
Movement disorders.
Ataxia may occur alone or in combination with epilepsy in patients with SCN8A mutations132. Movement disorders without seizures have been described in patients with partial or complete loss-of-function mutations133. In the mouse, loss of function of Scn8a may be accompanied by ataxia, dystonia or hind limb paralysis (Table 2B)42,134–137. The ataxia observed with GOF mutations of human SCN8A may result from use-dependent block of firing that mimics LOF in motor pathways.
Autism spectrum disorders and intellectual disability.
Autistic-like behaviors and intellectual disability are common co-morbidities in DEE due to GOF mutations of SCN8A. LOF mutations of SCN8A can cause autism or intellectual disability without seizures124,138,139. Liu et al compared SCN8A variants from patients with seizures and patients with ASD/ID11. When tested in transfected neurons, GOF was associated with seizures and LOF was associated with ASD. Intellectual disability unaccompanied by seizures is seen in patients with LOF mutations of SCN8A140–142.
In a conditional mouse model, inactivation of Scn8a in inhibitory neurons resulted in absence seizures143. RNAi mediated knockdown of Scn8a in the reticular thalamic nucleus also induced absence seizures (Table 2B). Deletion of Scn8a in thalamic reticular neurons was thought to lead to seizures by reducing inhibitory input into the thalamus 143. Inactivation of Scn8a in forebrain excitatory neurons resulted in replacement of Nav1.6 by Nav1.2 at the AIS, and reduced persistent current, but movement was unaffected28.
Protein truncation mutations of SCN8A are under-represented in control and patient populations studied to date. The deficit of protein truncation mutations in the gnomAD database indicates that haploinsufficiency is not tolerated in a neurologically normal population (pLI=1.0, OE=0.07) (gnomad.broadinstitute.org)144. The missing haploinsufficiency may be associate with movement disorders that have not been subjected to large scale sequencing, such as isolated ataxia, dystonia and tremor. It is possible that haploinsufficiency of human SCN8A leads to prenatal or early postnatal lethality, although this is not the case in the mouse.
Overall, there is considerable overlap in clinical consequences of mutations in SCN1A, SCN2A and SCN8A. The interesting differences in molecular mechanisms reflect divergence in aspects of subcellular function and distribution among neuronal circuits that we are just beginning to understand. Mutations in all three genes can result in seizure disorders, autism and intellectual disability. The effects of mutations on individual neurons have been characterized by electrophysiological methods, but the relationship between single-cell function and circuit and network consequences remain to be established. Understanding these processes will have important implications for therapeutic interventions.
New Therapies for Sodium Channelopathies.
Many patients with gain-of-function mutations respond to the classical sodium channel blockers, but most continue to experience some seizures and undesirable side effects. It has been difficult to develop drugs that distinguish among the closely related sodium channels. Genetic therapies can achieve target specificity based on DNA sequence differences among the channels, but their delivery across the blood brain barrier still requires invasive procedures. Advances in diagnosing sodium channel mutations has stimulated increased efforts to develop better treatment for both gain-of-function and loss-of-function disorders, using pharmacology as well as new genetic technologies, briefly reviewed below.
Pharmacology.
Channel-specific activators and inhibitors are predicted to have fewer side effects than the currently-available non-specific sodium channel blockers. The Nav1.6-specific channel blocker NBI-921352 (XEN901), in development by Xenon Pharmaceuticals, is scheduled to begin Phase 2 clinical trials in the United States in 2020. The persistent-current blocker PRAX330 reduces neuronal excitability in vitro and has shown promise in mouse models of SCN1A and SCN8A epilepsy145–148. The discovery that reduced Gabra2 exacerbates SCN1A and SCN8A epilepsies suggests that that positive allosteric modulators of α2 subunit-containing GABA channels could be effective in these disorders63–65. Supporting this prediction, the GABAA activator clobazam reduces susceptibility to febrile seizures in Scn1a+/− mice149. Intraventricular infusion of a spider venom peptide that specifically activates SCN1A was shown to ameliorate seizures in the Dravet mouse 150.
Antisense Oligonucleotides (ASOs).
Allele-specific oligonucleotides (ASOs) targeting specific DNA sequences have been approved for treatment of spinal muscular atrophy151 and Batten’s disease152, and show promise for several types of epilepsy. The SCN1A gene contains an alternatively-spliced “poison exon” with an in-frame stop codon. Approximately 50% of transcripts in young wildtype mice contain the poison exon53. ASOs complementary to the “poison exon” block its inclusion by steric hindrance and increase the abundance of full length transcript53. This ASO rescued seizures in a mouse model of Dravet Syndrome 153 and is currently in clinical trial.
Since SCN8A encephalopathy results from gain-of-function mutations and elevated neuronal activity, appropriate treatment would decrease transcript levels. Intracerebroventricular administration of an antisense ASO reduced Scn8a transcripts by 50%, delayed seizure onset and extended lifespan154. Repeated ASO administration prolonged the effect, suggesting that chronic treatment would be effective154. The Scn8a ASO also rescued the mouse model of Dravet syndrome caused by haploinsufficiency of Scn1a, suggesting that reducing neuronal excitability by reduction of SCN8A could be a general approach to seizures of various etiologies154.
CRISPR-based genetic therapy.
A general approach to treatment of haploinsufficient disorders is to increase the expression of the wildtype gene in the affected heterozygotes. CRISPR-activation technology can be applied for this purpose by fusion of transcriptional activation domains to the dCas9 protein and using an sgRNA to direct the protein to the promoter of the wildtype gene. This approach was tested for treatment of Dravet Syndrome in two mouse models. Intracerebroventricular injection of AAV carrying a transcriptional activator directed to the promoter of the Scn1a gene resulted in elevated activity of inhibitory neurons in vivo and resistance to thermally induced seizures155. Conditional upregulation of the wildtype Scn1a specifically in inhibitory neurons also reduced seizure susceptibility, with a modest effect on prolonging survival156. These examples provide proof of-principle that upregulation of Scn1a could be therapeutic for Dravet Syndrome, when administration of CRISPR to the CNS becomes feasible.
Outstanding issues.
The complexities of sodium channel function and the heterogeneity of channel levels in different types of neurons leave many questions for future investigation. The effects of loss of function in these channels is better understood. For missense mutations, it is difficult to predict the effects on biophysical properties of the channel. When GOF has been shown, it is still difficult to predict clinical clinical prognosis. There is a pressing need for functional analysis of the backlog of patient variants of unknown significance (VUS). The advent of high-throughput electrophysiology for variant analysis will contribute to the solution of this bottleneck157,158. Another large-scale approach is the application of saturation mutagenesis to generate libraries containing every possible missense variant in a gene, followed by pooled functional characterization. The goal of this approach is to generate a complete catalog of functional consequences for each gene that can be consulted after the identification of a novel patient mutation. An effort has been initiated for the cardiac channel SCN5A159. Analysis of reprogrammed neurons from patient-derived iPSC cells is another new development for analysis of patient mutations. In one recent study, seven mutations of SCN8A were studied in reprogrammed neurons; the correction of abnormal currents by riluzole in the cultured cells predicted the therapeutic response subsequently observed in 3 patients 12.
The impact of genetic variants in other genes in the patient genome, along with stochastic events during development, have impacts that are beyond current experimental access. Variation in genetic background may contribute to the surprising observation that, in rare cases, LOF mutations of Nav1.6 may generate seizures 160,161. One approach is to examine variants present in exome sequences of family members with divergent severity, as recently demonstrated for a family with pathogenic mutation of SCN9A 162.
Another challenge for the future is development of better methods for intracellular localization of specific channels in different types of neurons. In addition to filling gaps in basic knowledge, these methods could detect altered localization caused by patient mutations In vivo expression of molecularly tagged channels, such as those developed to study transport in cultured neurons 41, could increase sensitivity and eliminate dependence on immunostaining.
Ultimately, when faced with a newly diagnosed patient with a novel sodium channel mutation, we would like to be able to predict the biophysical effects, clinical course and effective therapy. As we move towards clinical trials for new therapies, family foundations focused on sodium channelopathies are making important contributions towards educating newly diagnosed families, compiling information about natural history for clinical trials, and supporting targeted research. These include the Dravet Syndrome Foundation (www.dravetfoundation.org), FamilieSCN2A Foundation (www.scn2a.org), Wishes for Elliott (www.wishesforelliott.com), and The Cute Syndrome Foundation (TCSF) (thecutesyndrome.com).
In summary, the discovery of monogenic causes underlying complex neurodevelopmantal disorders has clarified etiologies and accelerated efforts to develop targeted treatments. The depth of basic knowledge about the sodium channels have made these disorders early targets for precision medicine efforts. The monogenic epilepsies are particularly amenable to tests of clinical efficacy, because seizure frequency and severity can be acurately quantitated. We are seeing dramatically enhanced prospects for treatment of the sodium channelopathies, approaching the goal of improved quality of life for individuals living with these debilitating disorders.
BOX 1. Definitions.
Ortholog: Evolutionarily corresponding gene in two species, e.g. mouse Scn1a and human SCN1A.
Sodium channel modifier gene: An unrelated gene whose expression can modify the severity of a sodium channel disorder.
Haploinsufficiency: a gene for which 50% of normal expression is insufficient and results in disease.
Gain of Function Variant (GOF): A missense variant with altered amino acid sequence that results in abnormal channel function.
Loss of Function Variant (LOF): A variant that abolishes channel function.
Partial Loss of Function Variant: A variant that retains a reduced level of normal function.
Poison Exon: an alternatively spliced exon that results in protein truncation, for example due to the presence of an in-frame stop codon.
References
- 1.Holland LZ & Ocampo Daza D A new look at an old question: when did the second whole genome duplication occur in vertebrate evolution? Genome Biol 19, 209, doi: 10.1186/s13059-018-1592-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zakon HH Adaptive evolution of voltage-gated sodium channels: the first 800 million years. Proc Natl Acad Sci U S A 109 Suppl 1, 10619–10625, doi: 10.1073/pnas.1201884109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindy AS et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 59, 1062–1071, doi: 10.1111/epi.14074 (2018). [DOI] [PubMed] [Google Scholar]
- 4.O’Malley HA & Isom LL Sodium channel beta subunits: emerging targets in channelopathies. Annu Rev Physiol 77, 481–504, doi: 10.1146/annurev-physiol-021014-071846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouza AA & Isom LL Voltage-Gated Sodium Channel beta Subunits and Their Related Diseases. Handb Exp Pharmacol 246, 423–450, doi: 10.1007/164_2017_48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaman T et al. Mutations in SCN3A cause early infantile epileptic encephalopathy. Ann Neurol 83, 703–717, doi: 10.1002/ana.25188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith RS et al. Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development. Neuron 99, 905–913 e907, doi: 10.1016/j.neuron.2018.07.052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaman T et al. SCN3A-Related Neurodevelopmental Disorder: A Spectrum of Epilepsy and Brain Malformation. Ann Neurol, doi: 10.1002/ana.25809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raman IM, Sprunger LK, Meisler MH & Bean BP Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19, 881–891, doi: 10.1016/s0896-6273(00)80969-1 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Pan Y & Cummins TR Distinct functional alterations in SCN8A epilepsy mutant channels. J Physiol 598, 381–401, doi: 10.1113/JP278952 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 142, 376–390, doi: 10.1093/brain/awy326 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Tidball AM et al. Variant-specific changes in persistent or resurgent sodium current in SCN8A-related epilepsy patient-derived neurons. Brain, doi: 10.1093/brain/awaa247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith MR, Smith RD, Plummer NW, Meisler MH & Goldin AL Functional analysis of the mouse Scn8a sodium channel. J Neurosci 18, 6093–6102 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rush AM, Dib-Hajj SD & Waxman SG Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol 564, 803–815, doi: 10.1113/jphysiol.2005.083089 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calhoun JD & Isom LL The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb Exp Pharmacol 221, 51–89, doi: 10.1007/978-3-642-41588-3_4 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Whitaker WR et al. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res Mol Brain Res 88, 37–53, doi: 10.1016/s0169-328x(00)00289-8 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Boiko T et al. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23, 2306–2313 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Wart A, Trimmer JS & Matthews G Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol 500, 339–352, doi: 10.1002/cne.21173 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Royeck M et al. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol 100, 2361–2380, doi: 10.1152/jn.90332.2008 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Hu W et al. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nature Neuroscience 12, 996–1005 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Lorincz A & Nusser Z Molecular identity of dendritic voltage-gated sodium channels. Science 328, 906–909, doi: 10.1126/science.1187958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian C, Wang K, Ke W, Guo H & Shu Y Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front Cell Neurosci 8, 297, doi: 10.3389/fncel.2014.00297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akin EJ, Sole L, Dib-Hajj SD, Waxman SG & Tamkun MM Preferential targeting of Nav1.6 voltage-gated Na+ Channels to the axon initial segment during development. PLoS One 10, e0124397, doi: 10.1371/journal.pone.0124397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akin EJ et al. Single-Molecule Imaging of Nav1.6 on the Surface of Hippocampal Neurons Reveals Somatic Nanoclusters. Biophys J 111, 1235–1247, doi: 10.1016/j.bpj.2016.08.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Wart A & Matthews G Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J Neurosci 26, 7172–7180, doi: 10.1523/JNEUROSCI.1101-06.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercer JN, Chan CS, Tkatch T, Held J & Surmeier DJ Nav1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. J Neurosci 27, 13552–13566, doi: 10.1523/JNEUROSCI.3430-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu W et al. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci 12, 996–1002, doi: 10.1038/nn.2359 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Katz E et al. Role of sodium channel subtype in action potential generation by neocortical pyramidal neurons. Proc Natl Acad Sci U S A 115, E7184–E7192, doi: 10.1073/pnas.1720493115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maurice N, Tkatch T, Meisler M, Sprunger LK & Surmeier DJ D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21, 2268–2277 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osorio N et al. Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J Physiol 588, 651–670, doi: 10.1113/jphysiol.2010.183798 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogiwara I et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27, 5903–5914, doi: 10.1523/JNEUROSCI.5270-06.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spratt PWE et al. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 103, 673–685 e675, doi: 10.1016/j.neuron.2019.05.037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caldwell JH, Schaller KL, Lasher RS, Peles E & Levinson SR Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A 97, 5616–5620, doi: 10.1073/pnas.090034797 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boiko T et al. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30, 91–104, doi: 10.1016/s0896-6273(01)00265-3 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Meisler MH, Kearney J, Escayg A, MacDonald BT & Sprunger LK Sodium channels and neurological disease: insights from Scn8a mutations in the mouse. Neuroscientist 7, 136–145, doi: 10.1177/107385840100700208 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Westenbroek RE, Merrick DK & Catterall WA Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3, 695–704, doi: 10.1016/0896-6273(89)90238-9 (1989). [DOI] [PubMed] [Google Scholar]
- 37.Jenkins SM & Bennett V Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155, 739–746, doi: 10.1083/jcb.200109026 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemaillet G, Walker B & Lambert S Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem 278, 27333–27339, doi: 10.1074/jbc.M303327200 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Gasser A et al. An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J Neurosci 32, 7232–7243, doi: 10.1523/JNEUROSCI.5434-11.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Brien JE et al. Interaction of voltage-gated sodium channel Nav1.6 (SCN8A) with microtubule-associated protein Map1b. J Biol Chem 287, 18459–18466, doi: 10.1074/jbc.M111.336024 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sole L, Wagnon JL, Akin EJ, Meisler MH & Tamkun MM The MAP1B Binding Domain of Nav1.6 Is Required for Stable Expression at the Axon Initial Segment. J Neurosci 39, 4238–4251, doi: 10.1523/JNEUROSCI.2771-18.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharkey LM, Jones JM, Hedera P & Meisler MH Evaluation of SCN8A as a candidate gene for autosomal dominant essential tremor. Parkinsonism Relat Disord 15, 321–323, doi: 10.1016/j.parkreldis.2008.06.010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tate SK et al. Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc Natl Acad Sci U S A 102, 5507–5512, doi: 10.1073/pnas.0407346102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plummer NW, McBurney MW & Meisler MH Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. J Biol Chem 272, 24008–24015, doi: 10.1074/jbc.272.38.24008 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Sanders SJ et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci 41, 442–456, doi: 10.1016/j.tins.2018.03.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson CH, Ben-Shalom R, Bender KJ & George AL Alternative splicing potentiates dysfunction of early-onset epileptic encephalopathy SCN2A variants. J Gen Physiol 152, doi: 10.1085/jgp.201912442 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plummer NW et al. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics 54, 287–296, doi: 10.1006/geno.1998.5550 (1998). [DOI] [PubMed] [Google Scholar]
- 48.O’Brien JE et al. Rbfox proteins regulate alternative splicing of neuronal sodium channel SCN8A. Mol Cell Neurosci 49, 120–126, doi: 10.1016/j.mcn.2011.10.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zubovic L, Baralle M & Baralle FE Mutually exclusive splicing regulates the Nav 1.6 sodium channel function through a combinatorial mechanism that involves three distinct splicing regulatory elements and their ligands. Nucleic Acids Res 40, 6255–6269, doi: 10.1093/nar/gks249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Gehman LT et al. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26, 445–460, doi: 10.1101/gad.182477.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh Y & Waxman SG Novel splice variants of the voltage-sensitive sodium channel alpha subunit. Neuroreport 9, 1267–1272, doi: 10.1097/00001756-199805110-00002 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Kerr NC, Holmes FE & Wynick D Novel mRNA isoforms of the sodium channels Na(v)1.2, Na(v)1.3 and Na(v)1.7 encode predicted two-domain, truncated proteins. Neuroscience 155, 797–808, doi: 10.1016/j.neuroscience.2008.04.060 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim KH et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat Commun 11, 3501, doi: 10.1038/s41467-020-17093-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu FH et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9, 1142–1149, doi: 10.1038/nn1754 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Planells-Cases R et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J 78, 2878–2891, doi: 10.1016/S0006-3495(00)76829-9 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burgess DL et al. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet 10, 461–465, doi: 10.1038/ng0895-461 (1995). [DOI] [PubMed] [Google Scholar]
- 57.Han S et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390, doi: 10.1038/nature11356 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ito S et al. Mouse with Nav1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis 49, 29–40, doi: 10.1016/j.nbd.2012.08.003 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Ogiwara I et al. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol 1, 96, doi: 10.1038/s42003-018-0099-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tatsukawa T, Ogiwara I, Mazaki E, Shimohata A & Yamakawa K Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin-expressing cells. Neurobiol Dis 112, 24–34, doi: 10.1016/j.nbd.2018.01.009 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Papale LA et al. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Hum Mol Genet 18, 1633–1641, doi: 10.1093/hmg/ddp081 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKinney BC, Chow CY, Meisler MH & Murphy GG Exaggerated emotional behavior in mice heterozygous null for the sodium channel Scn8a (Nav1.6). Genes Brain Behav 7, 629–638, doi: 10.1111/j.1601-183X.2008.00399.x (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller AR, Hawkins NA, McCollom CE & Kearney JA Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 13, 163–172, doi: 10.1111/gbb.12099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulligan MK et al. Identification of a Functional Non-coding Variant in the GABA A Receptor alpha2 Subunit of the C57BL/6J Mouse Reference Genome: Major Implications for Neuroscience Research. Front Genet 10, 188, doi: 10.3389/fgene.2019.00188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu W et al. Gabra2 is a genetic modifier of Scn8a encephalopathy in the mouse. ??? (2020). [DOI] [PMC free article] [PubMed]
- 66.Buchner DA, Trudeau M & Meisler MH SCNM1, a putative RNA splicing factor that modifies disease severity in mice. Science 301, 967–969, doi: 10.1126/science.1086187 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Howell VM et al. Evidence for a direct role of the disease modifier SCNM1 in splicing. Hum Mol Genet 16, 2506–2516, doi: 10.1093/hmg/ddm206 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Howell VM et al. A targeted deleterious allele of the splicing factor SCNM1 in the mouse. Genetics 180, 1419–1427, doi: 10.1534/genetics.108.094227 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagnon JL & Meisler MH Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Front Neurol 6, 104, doi: 10.3389/fneur.2015.00104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hawkins NA, Martin MS, Frankel WN, Kearney JA & Escayg A Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis 41, 655–660, doi: 10.1016/j.nbd.2010.11.016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martin MS et al. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 16, 2892–2899, doi: 10.1093/hmg/ddm248 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Jorge BS et al. Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility. Proc Natl Acad Sci U S A 108, 5443–5448, doi: 10.1073/pnas.1017539108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mishra V et al. Scn2a deletion improves survival and brain-heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP). Hum Mol Genet 26, 2091–2103, doi: 10.1093/hmg/ddx104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Epi25 Collaborative. Electronic address, s. b. u. e. a. & Epi, C. Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals. Am J Hum Genet 105, 267–282, doi: 10.1016/j.ajhg.2019.05.020 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Lange IM et al. Assessment of parental mosaicism in SCN1A-related epilepsy by single-molecule molecular inversion probes and next-generation sequencing. J Med Genet 56, 75–80, doi: 10.1136/jmedgenet-2018-105672 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Wu YW et al. Incidence of Dravet Syndrome in a US Population. Pediatrics 136, e1310–1315, doi: 10.1542/peds.2015-1807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Depienne C et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet 46, 183–191, doi: 10.1136/jmg.2008.062323 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Meng H et al. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat 36, 573–580, doi: 10.1002/humu.22782 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Dravet C The core Dravet syndrome phenotype. Epilepsia 52 Suppl 2, 3–9, doi: 10.1111/j.1528-1167.2011.02994.x (2011). [DOI] [PubMed] [Google Scholar]
- 80.Scheffer IE & Nabbout R SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 60 Suppl 3, S17–S24, doi: 10.1111/epi.16386 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Guerrini R & Falchi M Dravet syndrome and SCN1A gene mutation related-epilepsies: cognitive impairment and its determinants. Dev Med Child Neurol 53 Suppl 2, 11–15, doi: 10.1111/j.1469-8749.2011.03966.x (2011). [DOI] [PubMed] [Google Scholar]
- 82.Escayg A & Goldin AL Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51, 1650–1658, doi: 10.1111/j.1528-1167.2010.02640.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nissenkorn A et al. In vivo, in vitro and in silico correlations of four de novo SCN1A missense mutations. PLoS One 14, e0211901, doi: 10.1371/journal.pone.0211901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kluckova D et al. A Study among the Genotype, Functional Alternations, and Phenotype of 9 SCN1A Mutations in Epilepsy Patients. Sci Rep 10, 10288, doi: 10.1038/s41598-020-67215-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carvill GL et al. Aberrant Inclusion of a Poison Exon Causes Dravet Syndrome and Related SCN1A-Associated Genetic Epilepsies. Am J Hum Genet 103, 1022–1029, doi: 10.1016/j.ajhg.2018.10.023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cheah CS et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 109, 14646–14651, doi: 10.1073/pnas.1211591109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ogiwara I et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 22, 4784–4804, doi: 10.1093/hmg/ddt331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stein RE, Kaplan JS, Li J & Catterall WA Hippocampal deletion of NaV1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet Syndrome. Proc Natl Acad Sci U S A 116, 16571–16576, doi: 10.1073/pnas.1906833116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalume F, Yu FH, Westenbroek RE, Scheuer T & Catterall WA Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci 27, 11065–11074, doi: 10.1523/JNEUROSCI.2162-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mistry AM et al. Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis 65, 1–11, doi: 10.1016/j.nbd.2014.01.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Favero M, Sotuyo NP, Lopez E, Kearney JA & Goldberg EM A Transient Developmental Window of Fast-Spiking Interneuron Dysfunction in a Mouse Model of Dravet Syndrome. J Neurosci 38, 7912–7927, doi: 10.1523/JNEUROSCI.0193-18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cooper MS et al. Mortality in Dravet syndrome. Epilepsy Res 128, 43–47, doi: 10.1016/j.eplepsyres.2016.10.006 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Frasier CR et al. Channelopathy as a SUDEP Biomarker in Dravet Syndrome Patient-Derived Cardiac Myocytes. Stem Cell Reports 11, 626–634, doi: 10.1016/j.stemcr.2018.07.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuo FS, Cleary CM, LoTurco JJ, Chen X & Mulkey DK Disordered breathing in a mouse model of Dravet syndrome. Elife 8, doi: 10.7554/eLife.43387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bagnall RD, Crompton DE & Semsarian C Genetic Basis of Sudden Unexpected Death in Epilepsy. Front Neurol 8, 348, doi: 10.3389/fneur.2017.00348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim Y et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 128, 1141–1153, doi: 10.1172/JCI94999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Escayg A et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24, 343–345, doi: 10.1038/74159 (2000). [DOI] [PubMed] [Google Scholar]
- 98.Spampanato J, Escayg A, Meisler MH & Goldin AL Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci 21, 7481–7490 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lossin C, Wang DW, Rhodes TH, Vanoye CG & George AL Jr. Molecular basis of an inherited epilepsy. Neuron 34, 877–884, doi: 10.1016/s0896-6273(02)00714-6 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Kimura K et al. A missense mutation in SCN1A in brothers with severe myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain Dev 27, 424–430, doi: 10.1016/j.braindev.2004.11.005 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Shao N et al. Familial Hemiplegic Migraine Type 3 (FHM3) With an. Front Neurol 9, 976, doi: 10.3389/fneur.2018.00976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fan C et al. Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia 36, 1238–1247, doi: 10.1177/0333102415608360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dhifallah S et al. Gain of Function for the. Front Mol Neurosci 11, 232, doi: 10.3389/fnmol.2018.00232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolff M et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336, doi: 10.1093/brain/awx054 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Ogiwara I et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology 73, 1046–1053, doi: 10.1212/WNL.0b013e3181b9cebc (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Begemann A et al. Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol Med 25, 6, doi: 10.1186/s10020-019-0073-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kearney JA et al. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 102, 307–317, doi: 10.1016/s0306-4522(00)00479-6 (2001). [DOI] [PubMed] [Google Scholar]
- 108.Kile KB, Tian N & Durand DM Scn2a sodium channel mutation results in hyperexcitability in the hippocampus in vitro. Epilepsia 49, 488–499, doi: 10.1111/j.1528-1167.2007.01413.x (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kamiya K et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci 24, 2690–2698, doi: 10.1523/JNEUROSCI.3089-03.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lossin C, Shi X, Rogawski MA & Hirose S Compromised function in the Na(v)1.2 Dravet syndrome mutation R1312T. Neurobiol Dis 47, 378–384, doi: 10.1016/j.nbd.2012.05.017 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Wolff M, Brunklaus A & Zuberi SM Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 60 Suppl 3, S59–S67, doi: 10.1111/epi.14935 (2019). [DOI] [PubMed] [Google Scholar]
- 112.Heron SE et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 360, 851–852, doi: 10.1016/S0140-6736(02)09968-3 (2002). [DOI] [PubMed] [Google Scholar]
- 113.Reynolds C, King MD & Gorman KM The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol 24, 117–122, doi: 10.1016/j.ejpn.2019.12.016 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Scalmani P et al. Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 26, 10100–10109, doi: 10.1523/JNEUROSCI.2476-06.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liao Y et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133, 1403–1414, doi: 10.1093/brain/awq057 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Ben-Shalom R et al. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol Psychiatry 82, 224–232, doi: 10.1016/j.biopsych.2017.01.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Buxbaum JD et al. The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 76, 1052–1056, doi: 10.1016/j.neuron.2012.12.008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tatsukawa T et al. Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol Autism 10, 15, doi: 10.1186/s13229-019-0265-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schwarz N et al. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur J Paediatr Neurol 23, 438–447, doi: 10.1016/j.ejpn.2019.03.001 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Liao Y et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 75, 1454–1458, doi: 10.1212/WNL.0b013e3181f8812e (2010). [DOI] [PubMed] [Google Scholar]
- 121.Schattling B et al. Activity of NaV1.2 promotes neurodegeneration in an animal model of multiple sclerosis. JCI Insight 1, e89810, doi: 10.1172/jci.insight.89810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meisler MH et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia 57, 1027–1035, doi: 10.1111/epi.13422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Johannesen KM et al. Early mortality in SCN8A-related epilepsies. Epilepsy Res 143, 79–81, doi: 10.1016/j.eplepsyres.2018.04.008 (2018). [DOI] [PubMed] [Google Scholar]
- 124.Larsen J et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 84, 480–489, doi: 10.1212/WNL.0000000000001211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meisler MH SCN8A encephalopathy: Mechanisms and models. Epilepsia 60 Suppl 3, S86–S91, doi: 10.1111/epi.14703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Veeramah KR et al. De Novo Pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. American Journal of Human Genetics 90, 502–510, doi: 10.1016/j.ajhg.2012.01.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lopez-Santiago LF et al. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci U S A 114, 2383–2388, doi: 10.1073/pnas.1616821114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ottolini M, Barker BS, Gaykema RP, Meisler MH & Patel MK Aberrant Sodium Channel Currents and Hyperexcitability of Medial Entorhinal Cortex Neurons in a Mouse Model of SCN8A Encephalopathy. J Neurosci 37, 7643–7655, doi: 10.1523/JNEUROSCI.2709-16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wagnon JL et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 3, 114–123, doi: 10.1002/acn3.276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nguyen HM & Goldin AL Sodium channel carboxyl-terminal residue regulates fast inactivation. J Biol Chem 285, 9077–9089, doi: 10.1074/jbc.M109.054940 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bunton-Stasyshyn RKA et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142, 362–375, doi: 10.1093/brain/awy324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gardella E et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 91, e1112–e1124, doi: 10.1212/WNL.0000000000006199 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Wagnon JL et al. Partial loss-of-function of sodium channel SCN8A in familial isolated myoclonus. Hum Mutat 39, 965–969, doi: 10.1002/humu.23547 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kearney JA et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6). Hum Mol Genet 11, 2765–2775, doi: 10.1093/hmg/11.22.2765 (2002). [DOI] [PubMed] [Google Scholar]
- 135.O’Brien JE & Meisler MH Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet 4, 213, doi: 10.3389/fgene.2013.00213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jones JM et al. Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder. Neurobiol Dis 89, 36–45, doi: 10.1016/j.nbd.2016.01.018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Levin SI et al. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. J Neurophysiol 96, 785–793, doi: 10.1152/jn.01193.2005 (2006). [DOI] [PubMed] [Google Scholar]
- 138.Genton P, Velizarova R & Dravet C Dravet syndrome: the long-term outcome. Epilepsia 52 Suppl 2, 44–49, doi: 10.1111/j.1528-1167.2011.03001.x (2011). [DOI] [PubMed] [Google Scholar]
- 139.Li BM et al. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav 21, 291–295, doi: 10.1016/j.yebeh.2011.04.060 (2011). [DOI] [PubMed] [Google Scholar]
- 140.Trudeau MM, Dalton JC, Day JW, Ranum LP & Meisler MH Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet 43, 527–530, doi: 10.1136/jmg.2005.035667 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wagnon JL et al. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet 3, e170, doi: 10.1212/NXG.0000000000000170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Blanchard MG et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet 52, 330–337, doi: 10.1136/jmedgenet-2014-102813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Makinson CD et al. Regulation of Thalamic and Cortical Network Synchrony by Scn8a. Neuron 93, 1165–1179 e1166, doi: 10.1016/j.neuron.2017.01.031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Karczewski KJ et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-offunction intolerance across human protein-coding genes bioRxiv (2019). [Google Scholar]
- 145.Anderson LL, Hawkins NA, Thompson CH, Kearney JA & George AL Jr. Unexpected Efficacy of a Novel Sodium Channel Modulator in Dravet Syndrome. Sci Rep 7, 1682, doi: 10.1038/s41598-017-01851-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wengert ER, Saga AU, Panchal PS, Barker BS & Patel MK Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology 158, 107699, doi: 10.1016/j.neuropharm.2019.107699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baker EM et al. The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 59, 1166–1176, doi: 10.1111/epi.14196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Anderson LL et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia 55, 1274–1283, doi: 10.1111/epi.12657 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hawkins NA, Zachwieja NJ, Miller AR, Anderson LL & Kearney JA Fine Mapping of a Dravet Syndrome Modifier Locus on Mouse Chromosome 5 and Candidate Gene Analysis by RNA-Seq. PLoS Genet 12, e1006398, doi: 10.1371/journal.pgen.1006398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Richards KL et al. Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc Natl Acad Sci U S A 115, E8077–E8085, doi: 10.1073/pnas.1804764115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Finkel RS et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 377, 1723–1732, doi: 10.1056/NEJMoa1702752 (2017). [DOI] [PubMed] [Google Scholar]
- 152.Kim J et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N Engl J Med 381, 1644–1652, doi: 10.1056/NEJMoa1813279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Han Z et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med 12, doi: 10.1126/scitranslmed.aaz6100 (2020). [DOI] [PubMed] [Google Scholar]
- 154.Lenk GM et al. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A Encephalopathy and Dravet Syndrome. Ann Neurol, doi: 10.1002/ana.25676 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Colasante G et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol Ther 28, 235–253, doi: 10.1016/j.ymthe.2019.08.018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yamagata T et al. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis 141, 104954, doi: 10.1016/j.nbd.2020.104954 (2020). [DOI] [PubMed] [Google Scholar]
- 157.Vanoye CG et al. High-Throughput Functional Evaluation of KCNQ1 Decrypts Variants of Unknown Significance. Circ Genom Precis Med 11, e002345, doi: 10.1161/CIRCGEN.118.002345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Glazer AM et al. High-Throughput Reclassification of SCN5A Variants. Am J Hum Genet 107, 111–123, doi: 10.1016/j.ajhg.2020.05.015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Glazer AM et al. Deep Mutational Scan of an SCN5A Voltage Sensor. Circ Genom Precis Med 13, e002786, doi: 10.1161/CIRCGEN.119.002786 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.de Kovel CG et al. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res 108, 1511–1518, doi: 10.1016/j.eplepsyres.2014.08.020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wengert ER et al. Biallelic inherited SCN8A variants, a rare cause of SCN8A-related developmental and epileptic encephalopathy. Epilepsia 60, 2277–2285, doi: 10.1111/epi.16371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mis MA et al. Resilience to Pain: A Peripheral Component Identified Using Induced Pluripotent Stem Cells and Dynamic Clamp. J Neurosci 39, 382–392, doi: 10.1523/JNEUROSCI.2433-18.2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wagnon JL & Meisler MH Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Frontiers in Neurology 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N & Cummins TR Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 139, 2164–2181, doi: 10.1093/brain/aww129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Veeramah KR et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 90, 502–510, doi: 10.1016/j.ajhg.2012.01.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liautard C et al. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 54, 1251–1261, doi: 10.1111/epi.12213 (2013). [DOI] [PubMed] [Google Scholar]
- 167.Makinson CD, Tanaka BS, Lamar T, Goldin AL & Escayg A Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiol Dis 68, 16–25, doi: 10.1016/j.nbd.2014.03.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rubinstein M et al. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138, 2219–2233, doi: 10.1093/brain/awv142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jansen NA, Dehghani A, Breukel C, Tolner EA & van den Maagdenberg A Focal and generalized seizure activity after local hippocampal or cortical ablation of NaV 1.1 channels in mice. Epilepsia 61, e30–e36, doi: 10.1111/epi.16482 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Miyamoto H et al. Impaired cortico-striatal excitatory transmission triggers epilepsy. Nat Commun 10, 1917, doi: 10.1038/s41467-019-09954-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Woodruff-Pak DS, Green JT, Levin SI & Meisler MH Inactivation of Sodium Channel Scn8a (Nav1.6) in Purkinje Neurons Impairs Learning in Morris Water Maze and Delay but Not Trace Eyeblink Classical Conditioning. Behavioral Neuroscience 120, 229–240, doi: 10.1037/0735-7044.120.2.229 (2006). [DOI] [PubMed] [Google Scholar]
- 172.Wong JC et al. Selective targeting of Scn8a prevents seizure development in a mouse model of mesial temporal lobe epilepsy. Sci Rep 8, 126, doi: 10.1038/s41598-017-17786-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]