

Abstract
Purpose:
Human papillomavirus (HPV) plays a major role in oncogenesis and circular extrachromosomal DNA (ecDNA) is found in many cancers. However, the relationship between HPV and circular ecDNA in human cancer is not understood.
Experimental design:
Forty-four primary tumor tissue samples were obtained from a cohort of HPV-positive OPSCC patients. Twenty-eight additional HPVOPC tumors from the Cancer Genome Atlas (TCGA) project were analyzed as a separate validation cohort Genomic, transcriptomic, proteomic, computational, and functional analyses of HPV oropharyngeal cancer (HPVOPC) were applied to these datasets.
Results:
Our analysis revealed circular, oncogenic DNA in nearly all HPVOPC, with circular human and human-viral hybrid ecDNA present in over a third of HPVOPC and viral circular DNA in remaining tumors. Hybrid ecDNA highly express fusion transcripts from HPV promoters and HPV oncogenes linked to downstream human transcripts that drive oncogenic transformation and immune evasion, and splice multiple, diverse human acceptors to a canonical SA880 viral donor site. HPVOPC have high E6*I expression with specific viral oncogene expression pattern related to viral or hybrid ecDNA composition.
Conclusions:
Non-chromosomal circular oncogenic DNA is a dominant feature of HPVOPC, revealing an unanticipated link between HPV and ecDNA that leverages the power of extra-chromosomal inheritance to drive HPV and somatic oncogene expression.
Keywords: HPV, human papillomavirus, head and neck, transcription, integration, ecDNA
Introduction
Oropharynx cancer has become the second-fastest growing cause of cancer death and the third-fastest growing in frequency among solid organ cancers in the U.S (1,2). The main histology is oropharynx squamous cell carcinoma (OPSCC) which is driven by high risk human papillomavirus (HPV) type 16 (3,4). The annual number of HPV-related oropharynx carcinoma (HPVOPC) cases has already surpassed the number of cervical cancer cases in the US in 2009, and by 2030 approximately half of all head and neck cancers in the US are predicted to be HPV-related (3). Although HPVOPC exhibits an improved clinical prognosis compared to HPV-negative OPSCC, 20–35% of tumors exhibit an aggressive course despite multimodality therapy (5). A major hurdle to understanding HPV-mediated oncogenesis is an incomplete understanding of the role of viral and viral-human hybrid transcripts and viral-human DNA integration.
Extrachromosomal DNA (ecDNA) has recently been shown to play a critical role in human cancer (6–9). Because of its non-chromosomal mechanism of inheritance, ecDNA can drive high copy number while promoting intratumoral heterogeneity, promoting accelerated tumor evolution and drug resistance (6,10). Moreover, chromatin rewiring on ecDNA allows for higher accessibility and increased expression of oncogenes (6,7,11). More recent reports have conjectured that hybrid human-virus ecDNA formation could be a possible mechanism for increased copy number of the HPV oncogenes E6, E7 (12–16). Our previous studies have demonstrated that the HPVOPC cell line UPCI:SCC090 features hybrid human-viral circular ecDNA containing FOXE1 and HPV-16 through conventional and long-read whole genome sequencing, and which we verified in vitro using fluorescent in situ hybridization (FISH) (11).
Given these data, we hypothesized that the genetic structure and viral gene expression in primary HPVOPC as well as the expression of human viral hybrid transcripts may be related to ecDNA. We combined whole genome sequencing, conventional RNA-seq and long-read RNA-seq to analyze HPV and human viral hybrid genomic and transcriptomic structure in the context of HPVOPC (17). Analysis of ecDNA and associated transcript structure clarified HPV transcript structure and the role of viral, human, and hybrid ecDNA in enhancing expression of diverse and oncogenic viral, human, and hybrid transcripts with functional validation.
Methods
Patient samples
Forty-four primary tumor tissue samples were obtained from a cohort of HPV-positive OPSCC patients from the Johns Hopkins Tissue Core (institutional review board protocol #NA_00–36235) and Moores Cancer Center Biorepository and Tissue technology shared at University of California, San Diego Human Research Protections Program (institutional review board approved protocol HRPP# 181755). Pathology of the primary tumors confirmed by two independent pathologists and tumor tissue was microdissected to yield at least 80% tumor purity. HPV tumor status was determined by in situ hybridization or p16 immunohistochemistry. In equivocal cases, HPV-16 E6 and E7 viral oncoproteins were detected via PCR for confirmation. Whole genome sequencing using paired-end Illumina sequencing along with conventional RNA-seq was acquired for 37 samples. Full clinical characteristics of the cohort are presented in Table S1.
Whole genome sequencing
DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen) for high-quality extraction per the manufacturer instructions. DNA samples from tumor were quantified using a Qubit (ThermoFisher Scientific). Greater than 1ug of each sample was prepared using a sonication based library construction and enrichment method per the Beijing Genomics Institute (BGI) as previously described (18).
DNA was isolated from 0.35 mm thick frozen tissue cuts digested in 1% SDS (Sigma-Aldrich, St. Louis, MO) and 50 μg/ml proteinase K (Invitrogen, Carlsbad, CA) solution at 48°C for 48 hours. The DNA was purified by phenol-chloroform extraction and ethanol precipitation. DNA was resuspended in LoTE buffer, and the DNA concentration was quantified using the NanoDrop spectrophotometer. Sequencing was performed with the Illumina Hiseq Xten 151PE strategy with 350bp insert library. The pipeline steps included preparation of HPV reference genome file, performance of quality control on BAM files, extraction of unmapped read pairs, conversion of unmapped read pairs to FASTQ format, alignment of unmapped read pairs to the HPV reference genomes (accession number: AY686584.1).
RNA preparation
Frozen tissue specimens were cut into 0.35-mm thick sections and RNA was extracted according to the Qiagen RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). RNA concentration was verified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). The absorbance ratio of 260 nm to 280 nm was used to verify adequate quality, defined as > 1.8. An RNA Integrity Number (RIN) of 7.0 or greater was required for quality assessment. RNA from all eleven tumors passed quality assessment.
cDNA library preparation, long read RNAseq, and alignment to HPV16 genome
Briefly, the RNA was extracted from 0.35 mm thick frozen tissue sections and a stranded RNA library was prepared using the Illumina TruSeq stranded total RNA seq poly A+ Gold kit (San Diego, CA) following the manufacturer’s recommendations. Long-read RNAseq of full-length transcripts was performed on 2 non-integrated and 3 integrated tumors according to the PacBio Iso-Seq pipeline (Menlo Park, CA). Briefly, 500 ng of purified RNA was used to prepare cDNA was using the Clontech SMARTer PCR cDNA synthesis kit (Mountain View, CA) and cDNA was then repaired. Large-scale PCR was performed using the Blue-Pippin size selection system for three sized cDNA libraries (<1.5kb, 1.5 – 2.5 kb, >2.5 kb). SMRTbell templates were then purified and sequenced on the PacBio SMRT Sequencing platform. The general SMARTer IIA oligonucleotide was used to anneal to the polyA tail of transcripts during cDNA sample preparation. Junction-spanning reads covered by fewer than 5 reads were dropped from analysis.
The Spliced Transcripts Alignment to a Reference (STAR) software was used to align long-read RNA seq reads to the HPV16 reference genome (GenBank: AY686584.1) (19). Full length transcripts were visualized with IGV for confirmation, and erroneously mapping transcripts were removed from analysis.
Short read RNA-seq alignment and analysis
Standard (short read) RNA-seq was performed as previously described.(20) A ribosomal RNA reduction was performed and the purified RNA was fragmented, then converted to double stranded cDNA, and the cDNA was 3′ adenylated and ligated with barcode adapters. The library was then enriched using PCR and AMPure XP bead purification. Sequencing was then performed using the HiSeq 2500 platform sequencer (Illumina), and the TruSeq Cluster Kit for 2×100 bp sequencing. The reads were trimmed to remove adapter sequences and low-quality reads using Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/).
The RNA sequences were aligned to the HPV16 genome (GenBank: AY686584.1) and hg19 assembly using MapSplice2 version 2.0.1.9. Integration and expression of HPV genes were identified by taking reads in RNA-seq data aligned to a combined database of human reference genome and high-risk HPV16, HPV33, HPV35 reference genomes using MapSplice (https://github.com/favorov/viruses-in-sequencing) (21). MapSplice was run with the default command line arguments. RNA-seq reads were aligned to the HPV16 genome and reads spanning canonical HPV16 splice junctions were extracted. For splicing analysis, RNA-seq reads were normalized by dividing by the total number of junction-spanning reads in the sample. Junction-spanning reads were discarded if the junction constituted < 1% of all junctions in the sample.
Quantification of RNA-seq expression was performed following the HISAT, StringTie, and Ballgown pipeline (22). Briefly, HISAT2 was used to align the RNA-seq reads to the hg19+HPV reference genomes. StringTie was run on each individual alignment to identify the assembled transcripts within the sample. All identified transcripts across all samples were merged using StringTie to create a consistent set of reference transcripts across the entire dataset. Abundances for each transcript for each sample was re-estimated using StringTie, and Ballgown was run on the resulting output to obtain read counts, coverage, and expression data across all samples.
Detection of focal amplifications with AmpliconArchitect (AA)
WGS reads were aligned to hg19 and 337 viral genomes using BWA-MEM (23). AA seed detection was performed with CNVKit (24). Copy number amplification regions matching low complexity, repetitive or poorly mappable genomic regions were filtered using the AA database. Some unfiltered regions corresponding to repetitive genomic regions still existed after this step and were shared across multiple samples. We removed any such regions existing in 10% or more of samples. Remaining copy number seed intervals larger than 10 kbp and with estimated CN > 4.3 were used as input to AA. Resulting amplicons generated by AA were examined for the presence of breakpoint graph cycles containing solely amplified human DNA (human ecDNA) and viral breakpoint edges linking HPV to an amplified cyclic human DNA structure (human-viral, ‘hybrid’, ecDNA).
Integrative Genome Viewer confirmation
Putative full-length transcripts and splice junctions were visualized using Integrative Genome Viewer (IGV/ Broad Institute, version 2.4.5) (25). From long-read and short-read RNA sequencing data, BAM files were loaded into IGV and visualized at the start and end of each junction. Long-read isoforms from IGV were individually verified and isoforms with mapping error were removed from analysis.
Integration, fusion, and splicing analysis
WGS and RNA-seq reads were aligned to hg19 and viral genomes using BWA-MEM. ViFi was run on each aligned BAM file to detect viral integration and transcription fusion location. Several transcription fusion events did not have a proximal viral integration event. Closer inspection of these fusion transcripts revealed that they had much lower support (mean 74 RNA-seq reads supporting the fusion event) compared to fusion transcripts that were proximal to a viral integration event (mean 955 RNA-seq reads supporting the fusion event). As such, we removed all fusion transcripts without supporting genomic integration.
Samples were classified according to the presence of viral integration and transcript fusion events. Samples containing a viral integration were classified as Hybrid-DNA. Samples that also contained a fusion transcript were classified as Hybrid-RNA.
In-depth splicing analysis was performed by taking the reads aligned to the HPV16 reference genome and counting the number of splice events detected by the alignment denoted by HPV16 donor and acceptor site pair (i.e., SDx-SAy). For the cases in which the splice junction started with an HPV16 donor site and ended in the human genome, it was denoted by the HPV16 donor site to human splice event (i.e., SDx-Human). From this data, we generated a splicing matrix where each row is a sample and each column is a splicing event, and the entries in the matrix are the total number of times that splicing event was observed within the sample. We performed principal component analysis on the splicing matrix in order to examine how the samples clustered.
Unsupervised hierarchical clustering and splicing analysis
Conventional RNA-seq reads spanning canonical HPV16 splice donor and acceptor sites (SD226, SA409, SA526, SA742, SD880, SD1302, SA2582, SA2709, SA3358, SD3632, SA5639) from the Institutional and TCGA cohorts were extracted and normalized to the number of reads specific to that tumor. Tumors with fewer than 500 mapped RNA-seq reads were excluded from inclusion in the heatmap (T14 – 241, T12 – 4, T30 – 2). RNAseq reads spanning HPV16 splice donor to human acceptor sites were also extracted and normalized to the number of reads specific to that tumor. To calculate the proportion of E6 protein reads with truncation to E6*I in each tumor, the following formula was applied to each tumor:
We also quantified the relative frequency of HPV16 SD880 to human splicing events with the following:
Insertion of HPV16 into Human Genome Analysis
Integration breakpoints and intragenomic viral breakpoints were identified with ViFi and AA. For DNA-based breakpoints both ViFi and AA were used to identify integration sites. To detect intragenomic viral breakpoints, AA alone was used. For RNA-seq data ViFi and MapSplice2 were used to identify splicing and human-viral chimeric sequences.
Analysis of non-canonical HPV16 structures
Canonical and non-canonical circular viral genome structure status was determined by AmpliconArchitect analysis. Tumor samples which did not have hybrid ecDNA were classified as non-canonical if they contained a cyclic AA graph decomposition and > 100 bp of rearranged genomic content (including indels), while canonical circular status was assigned if no such large rearrangements were present and cyclic AA graph decomposition of virus was present.
Quantification of splice acceptor cluster ranges for hybrid splicing events
For measurement of splice cluster ranges, splice clusters were defined via k-means clustering. Any group of donor, sample, and chromosome with fewer than 10 samples was excluded from consideration. Clustering was repeated 30 times at a given threshold to account for random seeding, with the optimal clustering at a given k threshold determined via silhouette score. The number of clusters was increased until a loss in performance was observed, and the number of clusters was confirmed by visual inspection. Clusters containing fewer than five observations were then filtered out, and the range of splicing sites was then computed on each remaining cluster.
Hybrid RNAseq reads from both Institutional and TCGA cohorts were extracted and mapped to specific viral splice donor and human acceptor sites for each tumor. The location of the human acceptor (chromosome and nucleotide) for each read was then determined using split reads, which were identified as having a primary alignment to HPV and a secondary alignment to the human genome. Histograms were created to map the distribution of splice acceptor sites for a given HPV16 donor (i.e. SD226, SD880, etc.) across the human genome for each tumor. Samples with viral read counts < 10 were removed from analysis.
Functional Studies
Proliferation of HCT116 and NOKSI cells was investigated in the presence of empty vector (negative control), as well as E6E7 (positive controls), and parent/daughter constructs. Cells were seeded in 96 well plates at a density of 3,000 cells/well for NOKSI and 4,000 cells/well for HCT116. Individual vectors composed of daughter constructs were then transfected by X-tremeGENE9 (Roche). Proliferation was measured as a ratio of relative absorbance two days after transfection vs. day of transfection.
The effect of FOXE1 siRNA was investigated on cell line SCC090. Cells were seeded at a density of 2,000 cells/well. SiFOXE1 (Santa Cruz Biotechnology) was added at a concentration of 10nM. Percent viability was measured using % viability = (absorbance of siRNA)/(absorbance of vehicle) x 100. For proliferation experiments each datapoint is the average of five replicates with standard error represented by error bars, and all experiments were repeated at least three times demonstrating consistent results.
HCT116 and SCC090 cells were obtained from ATCC, NOKSI was provided as a gift by the Silvio Gutkind Lab (University of California, San Diego, Department of Pharmacology. Cell lines were used for between 4 to 20 passages after thawing from frozen stock. Mycoplasma testing was conducted monthly using the MycoAlert-Plus Mycoplasma Detection Kit (Lonza).
Results
Forty-four primary tumors were acquired from a cohort of HPV-positive OPSCC patients. HPV tumor status was determined by in situ hybridization or p16 immunohistochemistry (see Table S1 for clinical information). In equivocal cases, HPV-16 E6 and E7 viral DNA were detected via PCR for confirmation. Forty of 44 samples were HPV16-positive, three were HPV33-positive, and one was HPV35-positive. Whole genome sequencing (WGS) using paired-end Illumina reads at mean coverage of 30x along with RNA-seq was acquired for 38 HPV16 samples (20), and long-read RNA-seq of full-length transcripts was generated for 5 samples using PacBio Iso-Seq technology (26). Twenty-eight additional HPVOPC tumors from the Cancer Genome Atlas (TCGA) project were analyzed as a separate validation cohort. WGS and RNA-seq data were mapped to the hg19 reference and analyzed using ViFi (27).
EcDNA that carry oncogenes are common in HPVOPC
As we had previously demonstrated the presence of ecDNA in an HPVOPC cell line (11), we hypothesized that ecDNA may be present in primary HPVOPC. A recently developed method, Amplicon Architect (AA) analyzes whole genome sequences to predict ecDNA with 85% precision and 83% sensitivity, as well as reconstruct the fine structure of the amplicons (8). We applied AA to the 28 samples from the HPVOPC cohort (Figure S1) (11). Remarkably, we found six hybrid viral-human ecDNA, and another six with human-only ecDNA, (one tumor exhibited both hybrid and human ecDNA; T14 - Figure 1A) in our institutional cohort. Additionally, eighteen tumors contained only HPV viral circular DNA (vcDNA). One tumor was not classified due to low viral copy number (T26). HPV vcDNA was present in an intact form including a complete, non-rearranged (canonical) form in 16 tumors. Interestingly, another 15 tumors contained a non-canonical truncated vcDNA with deletions mostly in the L1 and L2 region (Figure S2) suggesting that a significant fraction of HPVOPC tumors contain HPV genomes which have undergone substantial genomic rearrangement prior to enrichment in copy number.
Figure 1. HPV oropharynx cancers display distinct patterns of hybrid status including extrachromosomal DNA structures.
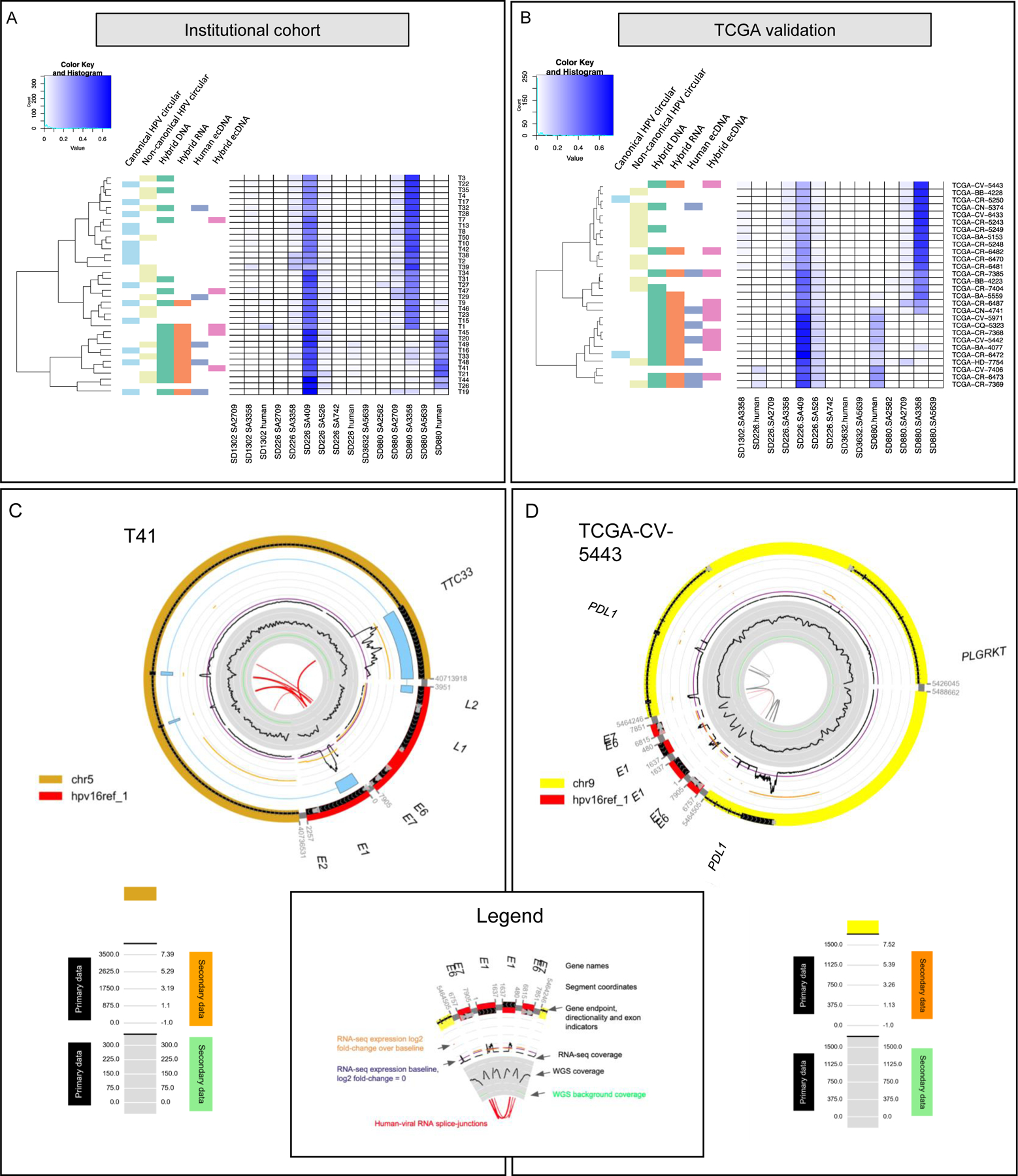
A) Unsupervised hierarchical clustering of splicing patterns for HPV16+ tumors (n=37). Hybrid genome (DNA) and transcriptome (RNA) status were assigned based on ViFi. Human ecDNA (extrachromosomal DNA) and hybrid ecDNA status were assigned based on AmpliconArchitect analysis. The accompanying heatmap displays splice junction coverage normalized as a percent of RNA-seq reads per tumor. Clustering of hybrid genome and hybrid transcriptome depended on splice junction coverage. Tumor T14 had evidence of hybrid ecDNA but had too few reads (241 RNA-seq reads mapping to HPV16) to integrate into the heatmap. Canonical and non-canonical circular viral genome structure status was determined from AmpliconArchitect analysis. Tumor samples which did not have hybrid ecDNA were classified as non-canonical if they contained a cyclic decomposition and > 100 bp of rearranged genomic content (including indels), while canonical circular status was assigned if no such large rearrangements were present.
B) Unsupervised hierarchical clustering of splicing patterns of HPV16+ tumors in the TCGA cohort (n=28) validates the presence of hybrid, human, or viral ecDNA in the HPV16 cohort tumors using aforementioned ViFi and AmpliconArchitect. Tumor CV-7406 had HPV16 genomic copy number < 1.
C) Circular genome structure of a 28 kbp human-viral hybrid ecDNA in T41. The circular genome CycleViz plot shows the following properties from outside to inside. Outer: putative genomic structure of the ecDNA, with genes and gene directions. In light blue a long-read transcript mapping to the circular ecDNA is shown, connecting viral regions to TTC33, then back to viral regions again. Middle track: black primary data indicates the RNA-seq positional coverage. Orange secondary data indicates the log2 ratio of T41’s exon-level FPKM values to the median FPKM values for those locations in study samples without ecDNA or viral integration. The purple line indicates a log2 positional FPKM ratio of 0. Inner track: black primary data indicates the WGS positional coverage. Light green secondary data indicates the mean WGS coverage for the chromosome from which the ecDNA was derived. Numeric values for the light grey tick lines are shown in the legend. The orange log2 positional FPKM ratio values in the middle track demonstrate increased expression of the regions compared to the median FPKM of study samples without ecDNA at those locations.
D) Circular genome structure of a 67 kbp human-viral hybrid ecDNA in TCGA-CV-5443. The circular genome CycleViz plot shows the following properties from outside to inside. Outer: putative genomic structure of the ecDNA, with genes and gene directions. Middle track: black primary data indicates the RNA-seq positional coverage. Orange secondary data indicates the log2 value of the ratio of TCGA-CV-5443’s exon-level FPKM values to the median FPKM values for those locations in study samples without ecDNA or viral integration. The purple line indicates a log2 positional FPKM ratio of 0. Inner track: black primary data indicates the WGS positional coverage. Light green secondary data indicates the mean WGS coverage for the chromosome from which the ecDNA was derived. Numeric values for the light grey tick lines are shown in the legend. The orange log2 positional FPKM ratio values in the middle track demonstrate increased expression of the regions compared to the median FPKM of study samples without ecDNA at those locations.
To test that the prediction of ecDNA in HNSC samples was not specific to the institutional cohort, we analyzed WGS samples from the HNSC data in the Cancer Genome Atlas (TCGA). Ten samples contained viral-human hybrid ecDNA and 8 contained human-only ecDNA (four tumors exhibited both; Figure 1B). VcDNA was also prevalent, with two tumors containing canonical vcDNA while 13 other tumors contained non-canonical truncated HPV vcDNA. One tumor was not classified due to low viral copy number (CV-7406).
EcDNA do not carry centromeres and therefore segregate independently, allowing tumors to rapidly modulate copy numbers of genes on ecDNA, specifically when the genes provide a growth or proliferative advantage (28). Consistent with this hypothesis, seven of the ecDNA+ tumors (both hybrid and human-only) in the institutional cohort carried oncogenic protein-coding genes or ncRNA (Supplementary Table S1), including many known oncogenes: EGFR, SEC61G, VOPP1, VSTM2A on chr7 in T1; DUSP4 and KIF138 on chr8 as also CD93 in T29; and, CST1 and THBD on chr20 in T14. The TCGA cohort revealed similar findings, with protein-coding genes found in five hybridecDNA-carrying samples, and three human only ecDNA samples (Supplementary Table S2). The structures were largely sample specific. However, we did observe two samples with ecDNA segments on chromosome 11 carrying six oncogenes (ANO1, CTTN, FADD, MIR548K, PPFIA1, SHANK2). Tumor TCGA-CQ-5323 contained an ecDNA with 13 cancer-associated genes, including ANO1, CCND1, CPT1A, CTTN, TRPC4AP, FADD, IGHMP2, MIR548K, MRPL21, ORAOV1, PPFIA1, and SHANK2 (Table S2). In TCGA-CV-5443, a hybrid ecDNA amplicon containing and amplifying the immune regulating ligand PDL1 was identified (Figure 1D). PDL1 amplification occurs in a subset of lung, kidney, bladder, and head and neck cancers. The PD1 checkpoint is the most common immunotherapeutic target in solid tumors currently and PD1/PDL1 directed immunotherapy has gained FDA approval for first-line treatment of unresectable or metastatic head and neck cancer (29).
Overlaying RNA -seq data on to the hybrid ecDNA structure in the institutional cohort showed hybrid RNA combining HPV-16 E6, E7, E1, E4, L1, and L2 with a multitude of human sequences containing genes EGFL7, TBCD (chr17; T1), SOX2-OT (chr3; T19), TTC33 (chr5; T41 see Figure 1C, PVT1 chr8; T14), LINC01363 (chr1; T47), and TBC1D16 (chr17; T49), As one interesting example, we identified an ecDNA in T41 amplicon that connected viral promoter to multiple exons in TTC33 (Figure 1C). Hybrid splicing was additionally confirmed using long-read Iso-Seq data (Figure 1C) consistent with the circular hybrid ecDNA structure. The TTC33 gene (tetraticopeptide repeat domain 33) has been implicated as an mRNA chimera in breast, ovarian, stomach, colon, kidney, and uterine cancer (30). These interesting patterns suggested a possible rewiring of the regulatory circuitry in hybrid ecDNA.
Hybrid ecDNA is associated with increased human gene expression.
Prior data have shown that ecDNA mediate increased expression of oncogenic human transcripts contained within ecDNA structure. To examine the effect of viral DNA genomic integration and viral integration in ecDNA, we examined expression of both viral and human transcripts in these contexts. For each gene on an ecDNA, we computed the ratio of its expression (in FPKM units) in the target sample to its mean expression value in all samples where the gene was not on an ecDNA (Methods) and called it the ‘FPKM-ratio’. Genes associated with the 38 ecDNA amplicons in the institutional cohort and the TCGA cohort were upregulated nearly 150X, with a mean FPKM-ratio of 149.8 (SD 1,015); median 4.26 (IQR 1.67– 8.92) (Figure 2A, B, Table S2). Thirty-three of 38 (86.8%) of these genes were oncogenes or associated with oncogenic phenotypes. For example, oncogene TNFSF4 on chromosome 1 in TCGA-CR-6473 was upregulated to FPKM-ratio of 116.58. TNFSF4 has been reported to be upregulated in brain metastases (31). EGFR on chromosome 7 in T48 was also upregulated with FPKM-ratio 30.7. PVT1 transcripts are found at a high level in lung cancer and contribute to VEGFC expression (32). CD274/PDL1, associated with immune checkpoint activation, was one of the most upregulated genes by tumor TCGA-CV-5443 in the TCGA cohort with FPKM-ratio 24.4.
Figure 2. Oncogene expression is upregulated in extrachromosomal DNA structures.
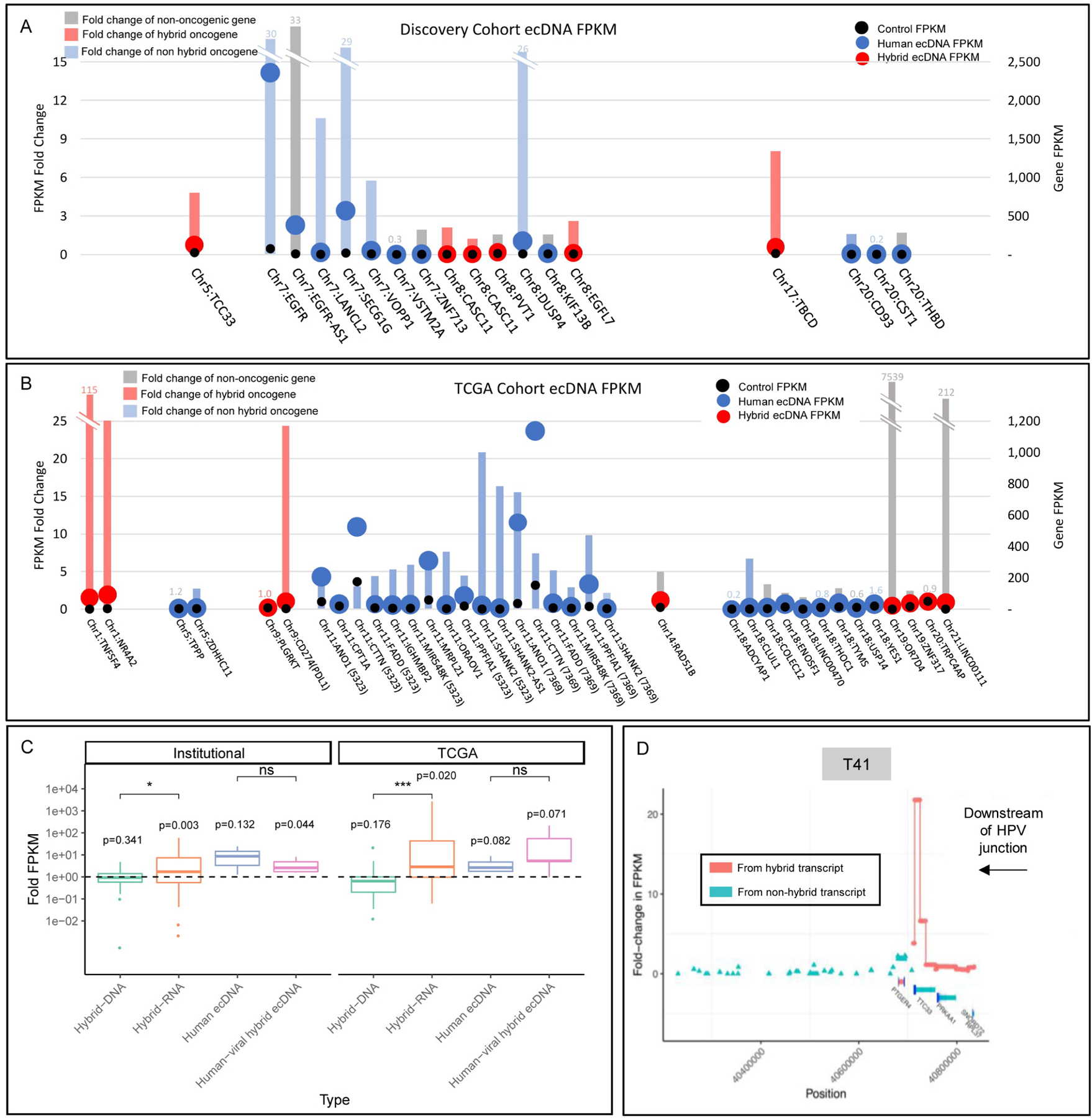
For A) and B), x-axis groups ecDNA structures for individual tumor samples and orders by chromosomal location of the gene. The left axis corresponds to the bars, which are colored by the gene’s status as hybrid ecDNA oncogene (red), human ecDNA oncogene (blue), or not an established oncogene (grey) and indicates the fold-change in expression (FPKM) of the ecDNA+ tumor’s gene (g) against the expression of g in other tumors where the gene is not found on ecDNA (FPKM of g in the ecDNA+ tumor )/(mean FPKM of g in tumors where g is not on ecDNA). The right axis uses overlaid bubbles defining FPKM of each gene of interest in ecDNA in individual tumors colored by ecDNA type (blue for human or red for hybrid) and also for mean FPKM in tumors that do not contain the gene of interest on ecDNA (black dots). FPKM is corrected for depth and fragment length to provide a representative coverage of the gene, normalized against the read depth of the sequenced sample and corrected for overrepresentation of shorter reads.
A) EcDNA associated with human genes from the Institutional cohort exhibited upregulation of associated genes, with a mean FPKM-ratio of 9.59 (SD 12.06); median 2.59 (IQR 1.58– 10.60).
B) EcDNA associated with human genes from TCGA exhibited upregulation of associated genes, with a mean FPKM sample:control ratio of 212.54 (SD 1,221.3); median 4.32 (IQR 2.14– 7.57). Genes (parentheses) refers to TCGA tumor affiliated with ecDNA gene.
C) Human genes proximal to the viral human junction showed increased expression in association with both hybrid genomes and hybrid transcriptomes for both the institutional and TCGA cohorts. Human-viral hybrid ecDNA was associated with increased expression of associated genes in the Institutional cohort. Hybrid RNA tumors showed higher upregulation of associated genes than hybrid DNA tumors in both Institutional and TCGA cohorts.
D) T41 shows dramatic increase in TTC33 expression downstream of the HPV-human hybrid sequence junction.
Importantly, human genes associated with hybrid transcriptomes showed increased expression for both the institutional and TCGA cohorts (Figure 2C). The increased expression was most pronounced in tumors with hybrid transcripts, but also increased expression was noted for all genes located on hybrid ecDNA. To further explore this relationship, we assembled and annotated human reads overlapping with human viral splice junctions, and spatially defined expression along genomic fusions. We noted that strand-specific expression of human genes downstream of viral sequences in fusion DNA structures could exceed 30-fold compared to surrounding genes. For example, T41 shows dramatic increase in TTC33 expression downstream of HPV-human hybrid sequence (Figure 2D). A similar phenomenon was noted in the context of other hybrid ecDNA structures (Figure S3).
Viral transcripts show diverse isoforms in hybrid ecDNA and hybrid transcript expression
Unsupervised hierarchical clustering based on frequency of HPV16 splicing junctions in mapped RNA reads showed a number of distinct patterns. First, we observed that SD226-SA409 represent a significant portion of junction-spanning reads in the HPV16 cohort, occurring in every sample with high frequency 36.3%, range 18.3% to 63.5%, SD 9.7% of junction-spanning reads. The use of this junction creates a shortened form of E6, called E6*I, which results in a premature stop codon (33). The mean fraction of reads demonstrating truncation of E6 to E6*I was 81.1% (SD 12.0%; min 44%.5 max 94.7%) across the cohort of HPV16-positive tumors (see Methods), demonstrating that E6*I is more commonly expressed than full-length E6 across all HPVOPC. Our results are consistent with previous results identifying E6*I in cervical dysplasia, cervical cancer, and HPVOPC (33–37). We calculated the proportion of E6 transcripts that were E6*I in the institutional cohort and found that tumors with either form of ecDNA had reduced E6*I production compared to the non-ecDNA tumors [0.72 (0.15) vs. 0.82 (0.09); p=0.0197 by T-test; mean (SD)]. Human ecDNA tumors had reduced E6*I production compared to the non-human ecDNA tumors [0.69 (0.13) vs. 0.81 (0.11); p=0.0288 by T-test; mean (SD)]. Hybrid ecDNA tumors did not have reduced E6*I production compared to the non-human ecDNA tumors [0.75 (0.18) vs. 0.80 (0.11); p=0.42 by T-test; mean (SD)]. To validate these findings, we examined the TCGA cohort of 28 HPVOPSCC tumors. Similar to the institutional cohort, the majority of E6 was truncated to E6*I (mean 0.89, SD 0.05, median 0.89 IQR 0.87–0.92), although we did not detect a difference in proportion of E6*I based on ecDNA status. This does confirm that, contrary to the classic model of HPV carcinogenesis, E6*I, rather than E6, is the most common viral transcript in HPVOPC.
Second, although splicing of the 5’ SD880 splice donor site to the 3’ SA3358 site had been described as the most frequent splicing event in HPV-16 cervical cancers and in cell lines (38–40), we found that 10 of 37 tumors (27%) preferentially spliced from SD880 to a human splice acceptor site instead of the canonical SA3358 (Figure 1A). In these 10 tumors, 47% (mean; SD=11%) of reads spanning the SD880 splice site spliced to a human locus, and only 3% (mean; SD=3%) of SD880 reads spliced to the canonical SA3358 receptor (p<0.001 by T-test). This questions previous findings that efficient usage of SA3358 is necessary for production of E6, E7, E4, E5, L1, and possibly L2 proteins (38). However, we did not detect preferential splicing of SD880 to a human acceptor to be associated with ecDNA status (40% of any ecDNA tumors preferentially spliced SD880 to human vs. 22% non-ecDNA; p=0.28; 40% of hybrid ecDNA tumors preferentially spliced SD880 to human vs. 25% non-hybrid ecDNA; p=0.482; 40% of human ecDNA tumors preferentially spliced SD880 to human vs. 25% non-ecDNA; p=0.482). We also analyzed splicing patterns of SD880 in TCGA and found that 10/28 (35%) preferentially spliced to a human splice acceptor rather than canonical SA3358. Tumors with either form of ecDNA were significantly more likely to splice to a human acceptor from SD880 (8/14; 57% vs. 2/14; 14%, p=0.018).
We also noted a strong association of splicing patterns depending on hybrid DNA or RNA status in the institutional cohort, irrespective of ecDNA (Figure 1A, B). Hybrid-DNA tumors (n=18) exhibited a greater fraction of RNA reads covering SD880-human junctions than non-hybrid-DNA tumors [n=19; 0.26 (0.25) vs. 0.02 (0.01); p<0.001; and fewer RNA reads covering the SD880-SA3358 junction [0.21 (0.21) vs. 0.42 (0.12); p<0.001 by T-test]. Similarly, hybrid-RNA tumors (n=12) exhibited a greater fraction of RNA reads covering SD880-human junctions than non-hybrid-RNA tumors [n=25; 0.38 (0.21) vs. 0.01 (0.05); p<0.001 by T-test] and fewer RNA reads covering the SD880-SA3358 junction [0.09 (0.12) vs. 0.43 (0.12); p<0.001 by T-test]. Principal component analysis of splicing patterns in hybrid RNA tumors also demonstrated a distinct subset based on splicing signature, in which HPV splicing pattern most closely relates to the presence of viral-human hybrid transcripts (Figure 3A and Figure S4). We observed selective enrichment of E6/E7 regions in both WGS and RNA data, which is pronounced in hybrid RNA tumors compared to non-hybrid RNA tumors (Figure 3B), as well as depletion of L2 in hybrid samples.
Figure 3. Hybrid genome and transcriptome status results in distinct splicing patterns and HPV oncogene expression levels.
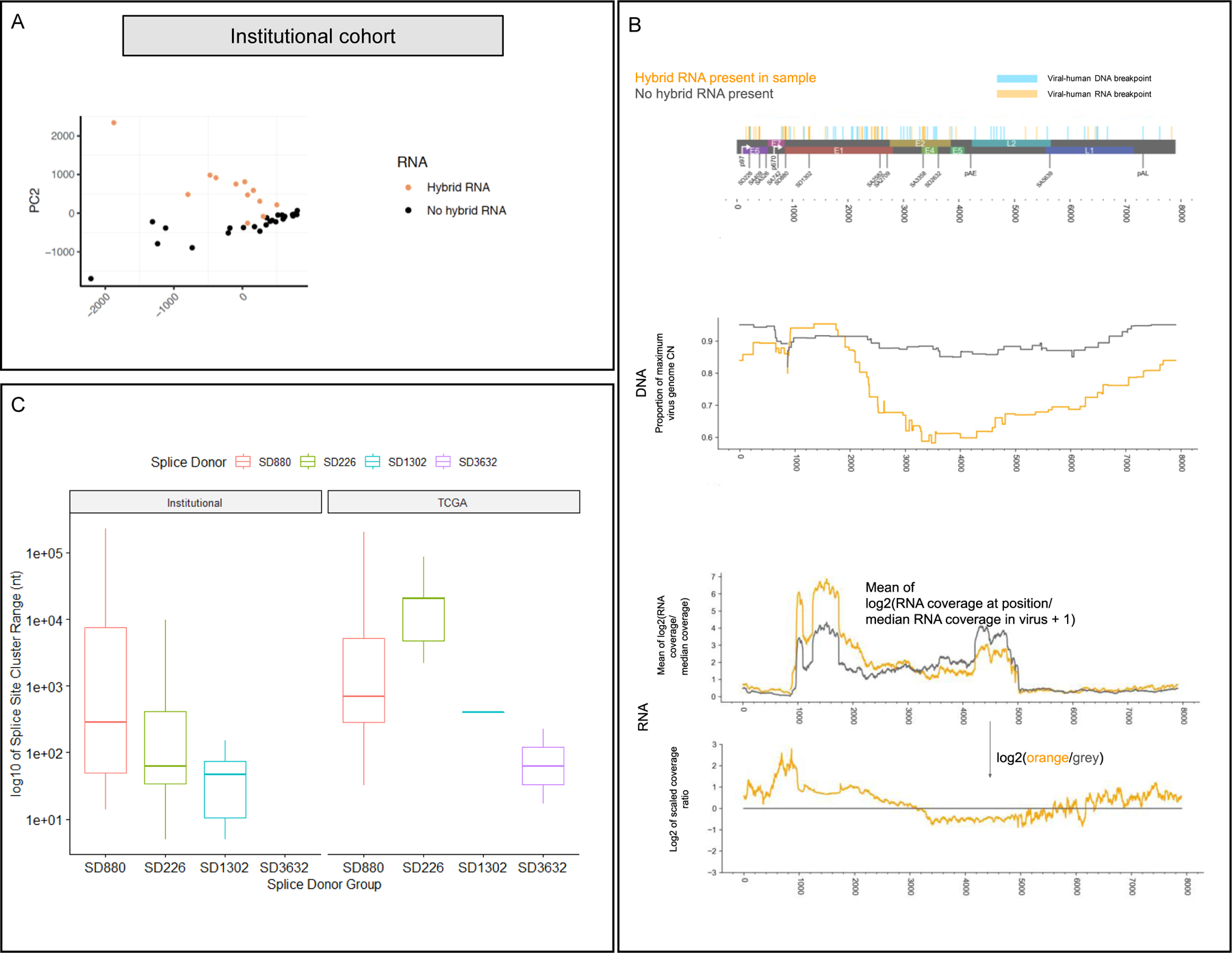
A) Principal component analysis of splice junction expression in individual tumors with annotation of tumors by presence or absence of human-viral hybrid transcripts. PCA of the tumor cohort supports the finding that hybrid transcriptome tumors (orange dots) exhibit distinct splicing patterns compared to non-hybrid transcriptome tumors (black dots).
B) Top: genome map of HPV16, indicating human-viral DNA breakpoints and RNA breakpoints. Middle (DNA): Mean proportion of the CN at each position over the maximum HPV16 CN for each sample with hybrid RNA transcripts (orange) and those without (grey), based on AA copy number estimates. We observed selective enrichment of viral genomic copy number in the E6/E7 region, and 5’ end of E1 in hybrid RNA tumors, while those without hybrid RNA showed much more uniform enrichment of viral copy number throughout the genome. Bottom (RNA): RNA-seq coverage for hybrid RNA tumors (orange) and those without hybrid RNA (grey). Coverage is rescaled by the median coverage across the virus, and the log2 value is shown. The lower plot shows the log2 ratio of scaled coverage between orange and grey from the upper plot, representing a log2 fold-change in mean scaled coverage. The highest peak overlaps SD880, indicating selectively increased transcription of that location in tumors with hybrid RNA. We also observed that the E4/E5 region, including the 5’ end of L2 were far less likely to have selective enrichment for genomic copy number in hybrid RNA tumors, and that there was decreased expression of those regions as compared to tumors without hybrid RNA.
C) Splice acceptor cluster quantification of hybrid transcripts for Institutional and TCGA cohorts stratified based on splice donor location. We found that the median splice acceptor range varied from 29 to 20,399 nucleotides wide across the TCGA and institutional cohort, and were narrowest for SD1302 (median 28.8 in TCGA; 406 in institutional) and widest for SD226 (20,399 in institutional cohort).
We defined aggregate splice donors in HPV and hybrid transcripts, and noted that SD226, SD880, and other known splice sites in the early region of HPV16 genome were strongly preserved and limited to a single donor canonical nucleotide. However, splice acceptors in hybrid transcripts showed broad variation. In the institutional cohort, the mean variation of the SD880 splice acceptor was 11,060 nucleotides (SD 37,217), but tighter for SD226 (mean 885, SD 2,290) and SD1302 (mean 48, SD 58). In TCGA, the degree of variation was similar (Figure 3C). We also noted that splicing patterns varied depending on hybrid DNA or RNA status in TCGA, irrespective of ecDNA. Sixty-four percent (18/28) exhibited a hybrid genome and 53% (15/28) exhibited hybrid transcriptomes, and hybrid DNA was a prerequisite for hybrid RNA (p<0.001). Similarly, SD226-SA409 represented a significant portion 40.0% (range 19.1 – 73.2%, SD 17.1%) of junction-spanning reads, TCGA tumors also preferentially expressed E6*I compared to full length E6 (89.2% E6*I reads (SD 5.3%)), and hybrid genome tumors (n=18) exhibited significantly higher percentage of splicing events from SD880 to a human locus [11.0 (10.0%) vs. 2.9 (9.0%); mean (SD); p=0.0508] and fewer reads covering the SD880-SA3358 junction [22.2 (23.9%) vs. 50.0 (19.2%); p=0.005]. Hybrid transcriptome tumors (n=15) also exhibited significantly higher fraction of splicing events from SD880 to a human locus [13.2 (10.0%) vs. 2.2 (8.0%); mean (SD); p=0.0041] and fewer reads covering the SD880-SA3358 junction [16.5 (21%) vs. 50.0 (17.4%); p=0.001] (Table S3, Table S4).
Novel HPV transcript structures related to ecDNA are found in HPVOPC
Long read polyA RNA sequencing provides direct sequencing of full-length transcripts and can avoid artifacts introduced by short read transcript assembly. To provide a more precise understanding of HPV transcripts in HPVOPC, we performed long-read RNA whole genome polyA transcript sequencing on a subset of two tumors without hybrid transcripts (T2 and T38), one human ecDNA tumor with hybrid transcript expression (T19), and one hybrid ecDNA tumor with hybrid transcripts (T45). (Figure 4A and Table S5).
Figure 4. HPV16 full length transcript structure in primary HPVOPC tumors.

A) Integrated Genome Viewer graphics are displayed of long-read RNA whole genome poly A transcript sequencing on a subset of two tumors without hybrid transcripts (T2 and T38), one human ecDNA tumor with hybrid transcript expression (T19), and one hybrid ecDNA tumor with hybrid transcripts (T45).
B) The most common long read Iso-Seq transcripts mapping to HPV exclusively are depicted with their coding potential. The single most common full-length transcript in non-hybrid tumors was 1,476 nt long, beginning at the p97 promoter with splicing at SD226-SA409 and SD880-SA3358 extending to the early polyA tail, with coding potential for the E6 oncoprotein variant E6*I defined by SD226-SA409, full-length E7, full-length E4, and full-length E5.
C) Poly-A tail-based long-read RNA sequencing of non-integrated and integrated tumors with mapping of full-length transcripts to HPV-16 genome demonstrates the transcriptome patterns in primary tumors. Identical isoforms have been color-coded. Read counts fewer than 5 were discarded. Full length coverage counts per Iso-Seq protocol are not proportionate to transcript quantity. Tumors without hybrid transcripts (T2 and T38) have distinct transcriptomes from those with hybrid transcripts and with ecDNA (T19, T45).
Long read sequencing confirmed a divergent transcript structure of hybrid ecDNA and vcDNA HPV transcripts. For example, T2 and T38 vcDNA (non-hybrid DNA/transcriptome tumors) exhibited 0% of conventional RNA-seq reads covering SD880 spliced to a human junction. By contrast, T19 (human ecDNA / hybrid transcripts) exhibited 19.5% and T45 (hybrid ecDNA/hybrid-RNA tumors) 100% of conventional RNA-seq reads of SD880 to be spliced to a human splice acceptor. Of interest, even though long-read RNA-seq is not quantitative in nature, we observed that both T19, a non-hybrid ecDNA tumor that expressed hybrid transcripts, and T45, a hybrid ecDNA/hybrid transcript tumor, essentially displayed no full-length transcripts that mapped to HPV alone; rather the transcripts were all hybrid. In T19, 36/40,474 (0.1%) long-reads mapped to HPV16 and in hybrid ecDNA tumor T45 17/55,814 (0.03%) long-reads mapped to HPV16. Conversely, the two non-hybrid vcDNA tumors T2 and T38 carried more full-length HPV-only transcripts. In T2, 515/19,220 (2.6%) long reads mapped to HPV and in T38 405/38,026 (1.1%) long reads mapped to HPV; (p<0.001 between non-hybrid and hybrid tumors).
Long-read data confirmed the presence of canonical splicing events seen in conventional RNA sequencing. The most common full-length transcript in non-hybrid tumors was 1,476 nt long, beginning at the p97 promoter with splicing at SD226-SA409 and SD880-SA3358 extending to the early polyA tail, with coding potential for the E6 oncoprotein variant E6*I defined by SD226-SA409, full-length E7, full-length E4, and full-length E5 (Fig 4B, C). This transcript was observed in 55% of full length reads mapped to HPV16 in T2 and 65% of full length reads mapped to HPV16 in T38, and included splice-junctions commonly observed in RNA-seq data, including SD226-SA409.
The long-read results and the RNA-seq data from the institutional and TCGA cohorts suggested that the predominant form of E6 in full-length transcripts found in HPVOPC was not the full-length E6 isoform, but truncated version E6*I defined by SD226-SA409. Additional isoforms E6*II, and E6*III(41,42), were also noted in long read transcripts. Full-length E7 was also common and present in the majority of HPV coding transcripts (91%). Finally, E1^E4, which is the result of SD880-SA3358 splicing was observed in 11/23 (48%) of distinct full-length isoforms.
ecDNA Hybrid HPV transcripts are functionally active
We selected a tumor (T41) for functional characterization of transcripts due to presence of fusion RNA reads, in addition to the fusion WGS reads and observation of up to 512x increased expression of segments associated on this ecDNA structure. After confirming the presence of AmpliconArchitect-predicted hybrid junctions using RT-PCR and sequencing (Figure S5), we cloned the entire transcript (E6-E7-E1-TTC33*-E5*) as well as daughter constructs into a pcDNA 3.1(+)-myc-His A vector (Genscript, Inc) (Figure 5A–B), as well as the most common full-length HPV16 in the form of component HPV gene transcripts, into the same backbone. To explore the functional effects of these transcripts, we transfected these constructs into HPV null diploid HCT116 (p53 and Rb wt MMR deficient colorectal carcinoma) cells as well as an HPV null normal oral keratinocyte NOKSI (spontaneously immortalized oral keratinocyte) cell lines that respond to HPV E6/E7 gene expression with enhanced proliferation, to provide an assessment of the effects of transcripts from this primary tumor (43,44). In HCT116 cells, E6*I, E6E7, E6*I/E7, E6*I/E7/E4/E5, E7, TTC33, E6E7E1 and the entire hybrid transcript from T41 induced significant growth compared to the empty vector (p=0.02, 0.02, 6x10−3, 0.03, 3x10−4, 2x10−4, 7x10−3, 0.01, respectively, Student’s t-test) (Figure 5C). Similarly, in NOKSI cells, E6*I, E6E7, E6E7E1 and the entire intact hybrid transcript from T41 increased proliferation (p=0.02, 0.01, 0.03, and 0.04, respectively, Student’s t-test) (Figure 5D).
Figure 5. Functional activity of hybrid ecDNA structures and full-length HPV transcripts.
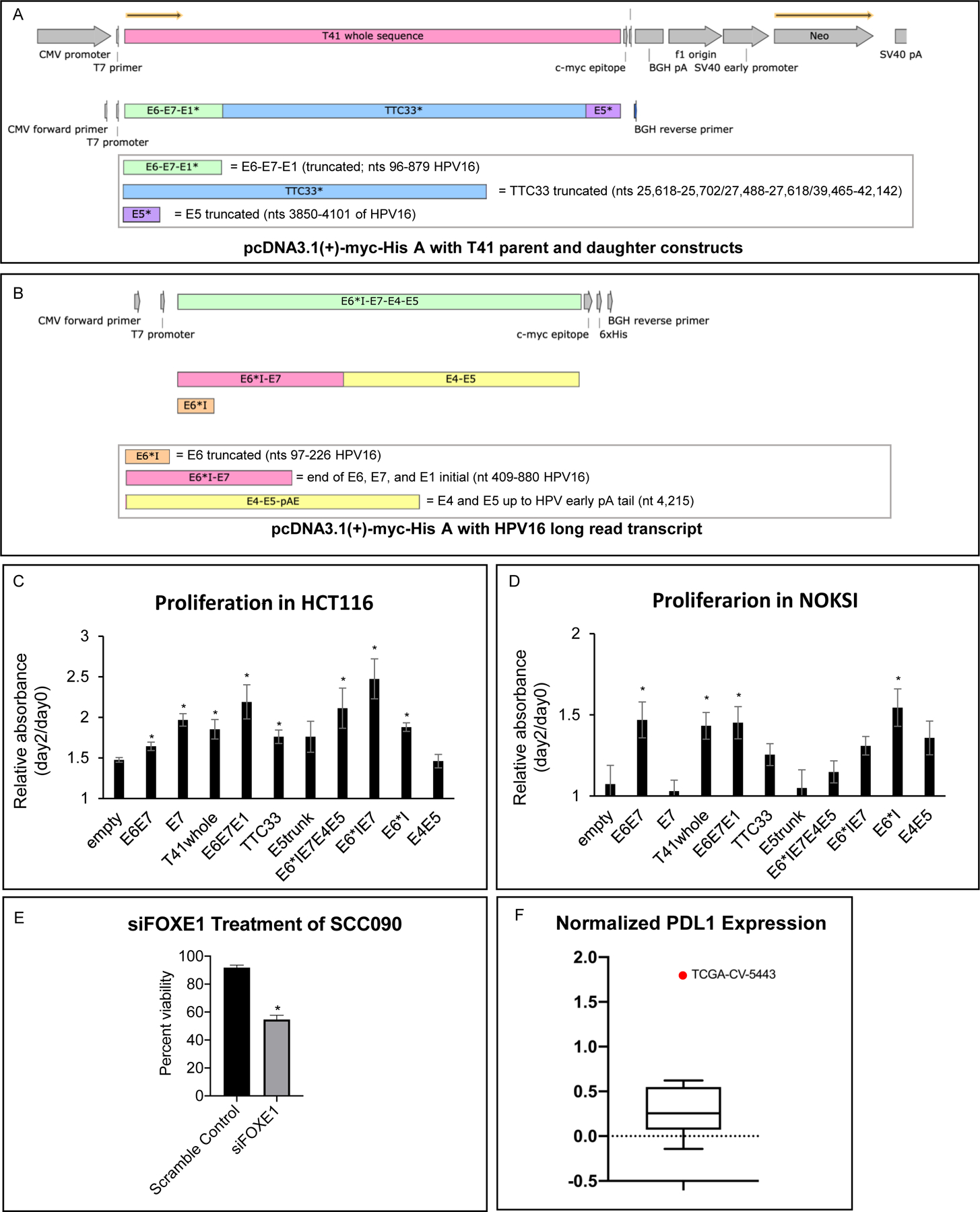
A) T41 entire transcript (E6-E7-E1-TTC33*-E5*) as well as daughter constructs were cloned into a pcDNA 3.1(+)-myc-His A vector (Genscript, Inc)
B) Most common full length HPV16 RNA (E6*I-E7-E4-E5) as well as daughter constructs were cloned into a pcDNA 3.1(+)-myc-His A vector (Genscript, Inc)
C) Transfection of pcDNA cloned with T41 fusion transcript and HPV transcript promoted the proliferation in HCT116. The proliferation assays after transient transfection of pcDNA3.1 cloned with empty, E6*I, E7, E6*IE7, E6E7, E6*IE7E4E5, TTC33, E6E7E1, E5truncated, E4E5 and T41 vector (E6-E7-E1-TTC33*-E5*) were performed on HCT116. Proliferation was normalized to day0, and relative absorbance of day2/day0 data was shown. E6*I, E7, E6*IE7, E6E7, E6*IE7E4E5, TTC33, E6E7E1 and T41 vector promoted the proliferation significantly compared to the empty vector (p =0.02, 3x10−4, 6x10−3, 0.02, 0.03, 2x10−4, 7x10−3, and 0.01, respectively, *p<0.05, Student’s t-test. Error bars represent standard error.
D) Transfection of pcDNA cloned with T41 fusion transcript and HPV transcript promoted the proliferation in NOKSI cells. The proliferation assays after transient transfection of pcDNA3.1 cloned with empty, E6*I, E7, E6*IE7, E6E7, E6*IE7E4E5, TTC33, E6E7E1, E5truncated, E4E5 and T41 vector (E6-E7-E1-TTC33*-E5*) were performed on NOKSI cells. Proliferation was normalized to day0, and relative absorbance of day2/day0 data was shown. E6*I, E6E7, E6E7E1 and the entire intact hybrid transcript from T41 increased proliferation (p=0.02, 0.01, 0.03, and 0.04, respectively, *p<0.05, Student’s t-test. Error bars represent standard error.
E) Proliferation assay after knockdown of FOXE1 was performed on SCC090. Knockdown of FOXE1 using siRNA of FOXE1 (Santa Cruz biotech) significantly suppressed the proliferation compared to the scramble control (Santa Cruz biotech). Percent viability was normalized to day0. *p<0.05, Student’s t-test. Error bars represent standard error.
F) Data derived via reverse phase protein arrays (RPPA) corresponding to head and neck squamous cell carcinoma samples included in TCGA were extracted from The Cancer Proteome Atlas (TCPA). Nine tumor samples identified as being HPV+ in the TCGA cohort had RPPA-derived expression data available in TCPA. Mean PDL1 protein expression in TCGA-CV-5443 (red data point outlier) was increased 7.6 X relative to the mean of the other eight TCGA tumors represented by standard box and whisker plot including median horizontal line inside box, box spanning interquartile range, and whiskers extending to highest and lowest value of these eight tumors (one sample T-test p<0.001).
We have previously reported on the presence of ecDNA in an HPOPC cell line UPCI;SCC090, demonstrating reconstruction of a complex hybrid structure (>100 kbp) containing the oncogene FOXE1 as well as highly expressing HPV16 sequence, and shown the presence of FOXE1 in both chromosomal HSRs as well as in ecDNA using FISH probes in metaphase imaging (11). To examine the functional contribution of FOXE1 in an ecDNA context, we treated SCC090 cells with siRNA for FOXE1 demonstrating significant inhibition of growth, indicating that overexpression of FOXE1 via ecDNA mediated mechanisms is a major driver of growth in SCC090 cells (Figure 5E).
To define the potential for hybrid ecDNA in HPVOPC to drive protein expression related to immune evasion, data derived via reverse phase protein arrays (RPPA) corresponding to head and neck squamous cell carcinoma samples included in TCGA were extracted from The Cancer Proteome Atlas (TCPA) (45). Nine tumor samples identified as being HPV+ in the TCGA cohort had RPPA-derived expression data available in TCPA. Mean PDL1 protein expression in the tumor with PDL1 present in a hybrid ecDNA structure (TCGA-CV-5443) was increased 7.6x relative to the mean of the other eight TCGA tumors (one sample T-test p<0.001; Figure 5F).
We analyzed the presence of hybrid DNA, hybrid RNA, hybrid ecDNA, and human circular ecDNA as compared to clinical features in the institutional cohort and in TCGA. In the institutional cohort there was no significant correlation with aggressive pathologic features (perineural invasion, lymphovascular invasion, or extranodal extension), poorly differentiated tumors, smoking status. Hybrid RNA status was more likely in patients with a drinking history (11/11 vs. 18/26; p = 0.038), and in patients who had a drinking history and had more than 10 pack years of smoking (6/11 vs. 3/23, p = 0.010). There was no association with the above groups and AJCC v7 staging. We then compared overall and recurrence-free survival based on presence of hybrid DNA, hybrid RNA, hybrid circular ecDNA, and human circular ecDNA. There were ten deaths, with the median time to death being 121 months. There were eleven recurrences and/or deaths, with the median time to event being 104 months. We found no significant difference in overall or recurrence free survival. In a multivariable proportional hazards model adjusting for age, stage, and treatment modality (surgery, radiation, and/or chemotherapy), we found no significant relationship between recurrence-free survival and hybrid DNA (p = 0.105), hybrid RNA (p = 0.540), hybrid ecDNA (p = 0.486), or human circular ecDNA (p = 0.282). Nor did we find a significant relationship between overall survival and hybrid DNA (p = 0.150), hybrid RNA (p = 0.525), hybrid ecDNA (p = 0.667), or human circular ecDNA (p = 0.195).
In TCGA, there were 7 deaths in the 28 patients for whom survival data was available. Multivariable survival analysis was not possible due to the size of the study population, however univariable analysis showed that neither patients with hybrid DNA (p=0.172) or human circular ecDNA (p = 0.649) were more likely to die. Patients with hybrid ecDNA had decreased likelihood of death (0/7 deaths vs. 7/21 deaths in non-hybrid ecDNA; p = 0.078), as did patients with hybrid RNA (1/15 deaths vs. 6/13 in non-hybrid RNA; p = 0.016).
It is critical to note that neither our cohort nor TCGA was not powered to detect an expected difference in survival. We were unable to perform multivariable analyses to adjust for additional clinical factors due to the small cohort and infrequency of failure events.
Discussion
Intrachromosomal oncogene transcription and amplification have been the dominant paradigm for oncogene mediated transformation for decades. Similarly, HPV viral integration into human chromosomes and expression of oncoproteins E6 and E7 have been the classic mechanism for HPV-mediated oncogenesis (4). The recent definition of ecDNA as a driver of oncogene amplification and overexpression that is found in nearly of half of all human cancers has altered the paradigm of the role of extrachromosomal circular DNA in carcinogenesis (6,11). Recently non-integrated HPV has been reported in HPVOPC, indicating that HPV may exert oncogenic effects while maintaining status as vcDNA (46). The data we present describes an unexpected interaction of the HPV genome with cancer associated ecDNA. Specifically, our results suggest that HPVOPC frequently employ ecDNA in human as well as human-viral hybrid forms that leverage ecDNA mediated viral gene transcription to drive high expression of hybrid viral-human oncogene transcripts. Furthermore, HPVOPC ecDNA structures express a broad variety of human and hybrid cancer related transcripts including functional oncogenic noncoding RNA and immune evasion checkpoint proteins, in addition to traditional oncogenes. HPVOPC hybrid transcripts drive high expression of E6*I as well as other noncanonical HPV transcripts. This confirms and expands the spectrum of oncogenic HPV gene expression beyond the traditional expression of E6 and E7. In addition, HPV vcDNA is found in HPVOPC in deletion structures that lack coding for viral capsid proteins without evidence of integration into host DNA, as well as in the traditional full length episomal state. Most strikingly, nearly all HPVOPC contain transcriptionally active, circular, oncogenic DNA outside of host chromosomes, including vcDNA, human ecDNA, or viral-human hybrid ecDNA.
As noted, HPVOPC tumors express hybrid human viral sequences with increased human and viral gene expression driven by HPV promoters from integrated and ecDNA structures. These hybrid transcripts often include human genes implicated in carcinogenesis, and ecDNA hybrid transcripts are often expressed at a higher level than hybrid transcripts expressed from genomic HPV integration. Our data show that E6 and E7 expression is slightly enhanced in tumors in the context of HPV integration into chromosomal loci, and E1, E2, E4, and E5 expression is only slightly decreased. However, hybrid genomes incorporated into circular ecDNA show with significant upregulation of E6 and E7 as well as dramatic upregulation of human genes within 10kb up and down-stream of the recombined region, with loss of E2, E4, and E5 expression. Taken together, these data indicate that the mechanism for the classic model of HPV carcinogenesis characterized by E6 and E7 overexpression is often driven by the formation of viral-human hybrid ecDNA structures, facilitating amplification of E6 and E7 and associated human genes.
The near universal presence of oncogenic, circular DNA that expresses oncogenic transcripts, whether viral, human, or hybrid, indicates that HPVOPC create a genomic context that is generally permissive for the stable maintenance of non-chromosomal, transcriptionally active, circular DNA. Traditionally, the E2 protein has been shown to be the regulator of episomal maintenance for high-risk HPV (47,48). However, we describe hybrid ecDNA structures that exclude E2 in tumors that have dominant or exclusive expression of E6 and E7. It is possible that high risk HPV gene products other than E2 may have effects on genomic maintenance and chromosomal structure, allowing for perturbations in DNA homeostasis that facilitate stability, replication, and heritability of circular, non-chromosomal DNA structures. The evolution and progression of benign HPV infection to HPVOPC, therefore, requires ongoing maintenance of stable ecDNA or vcDNA as part of carcinogenesis. In this context, these data show three main viral genomic/transcriptomic HPVOPC pathways: 1) nonintegrated HPV vcDNA in a canonical or non-canonical form with broad expression of HPV gene products, 2) chromosomal integration of HPV with retained expression of HPV gene products and overexpression of human hybrid transcripts, and 3) formation of viral-human hybrid ecDNA as well as integrated viral DNA with dramatic amplification and overexpression of E6 and E7 as part of human viral fusion transcripts, with dramatic reduction in early E2, E4, and E5 HPV gene product expression. These pathways are independently supported by recent data showing HPV integration does not necessarily result in high levels of E6 and E7 transcripts, and that tumors with non-integrated HPV16 actually overexpress E6 compared to tumors with integrated or mixed genomes (49,50).
We identified the most common viral HPVOPC mRNAs as polycistronic transcripts typically > 1,000 nt in length, and that the most common full-length transcript in viral ecDNA HPVOPC is 1,476 nt long, beginning at the p97 promoter with splicing at SD226-SA409 and SD880-SA3358 extending to the early polyA tail, with coding potential for E6*I, full-length E7, E1^E4, and full-length E5. The function of the E6*I protein (a truncated protein with 43 residues) remains incomplete, although E6*I is found predominantly in high risk HPV strains and not in low risk strains (51). Studies suggest it may have opposing functions to full-length E6 with respect to p53 and, through procaspase 8, the cellular response to the TNF-family of cytokines (52,53). Meanwhile, the E6*I RNA (which contains the E7 ORF) functions as a source of E7 mRNA and increases the efficiency of E7 protein translation.(36,54) E6*I has been observed in E6-expressing keratinocytes to upregulate IL6, a key cytokine in tumorigenesis and inflammation which functions via the JAK-STAT pathway (55). In addition, E6*I expressing cells demonstrated increased p53 levels as well as increased reactive oxygen species levels which has the effect of increasing DNA damage in cells (56). E6*I RNA levels were significantly higher in cervical samples from patients who had higher grades of dysplasia (57). In our cell line systems, we were able to demonstrate that the most common transcript expressing E6*I, full-length E7, E1^E4, and full-length E5 was able to induce proliferation, indicating that the net biologic effect of coordinated expression of these genes can support a malignant phenotype. Conventional and long-read RNA-seq of HPVOPC also suggest that mechanisms of oncogenesis independent of E6 exist, as we found the truncated form E6*I to be more common than full length E6 in mRNAs. This finding is of interest because the crystal structure of the ternary complex that inactivates p53 is comprised of full length E6, not E6*I (58). We found E5 protein was commonly expressed, and E5 is increasingly being recognized as a driver of HPV-mediated tumorigenesis (59) and mediates resistance to checkpoint inhibitor blockade in head and neck SCC and can be targeted (60). Finally, we have recently published data demonstrating a similar proliferative cellular phenotype in systems where E2/E4/E5 are expressed in comparison to E6/E7 expression, indicating that E6/E7 gene products may not be as critical for HPV mediated carcinogenesis (61).
We have noted a striking propensity for canonical splicing junctions to be preserved in HPV viral transcripts, and that these canonical junctions serve as donors for acceptors to diverse human hybrid transcripts, resulting in consistent expression of specific HPV transcripts. Previous studies have identified hybrid viral-human transcripts in HPV anogenital lesions with the HPV splice donor being in the E1 ORF, but these findings have not yet been described in HPVOPC (62). We found that a unifying feature of tumors with hybrid genomes was preferential use of a human splice acceptor site for SD880, rather than the SA3358 HPV site. This is of interest because SA3358 has been documented as the primary splice acceptor involved in transcripts coding for E4, E5, E6, and E7 (38). Together, these data show that alternatively spliced forms of HPV genes, including E6*I, are in fact more highly expressed than conventional transcripts. These transcripts may be key to HPV carcinogenesis with potential value as therapeutic targets. In addition, the proliferation data we provide as well as prior reports of E2, E4, and E5 cooperative ability to support carcinogenesis indicate that coordinated expression of multiple transcripts may be necessary to reproduce phenotypic characteristics of malignancy (61). Similarly, we have found that the overexpressed human transcripts that are associated with human and hybrid ecDNA are quite diverse, comprising traditional oncogenes, e.g. CCND1, EGFR, oncogenic long non-coding RNAs like PVT1, as well as immune modulatory genes, including PDL1. In addition, circular HPV DNA molecules have often been assumed to represent traditional intact, full-length forms found in intact virus. However, we found that non-canonical HPV genome, including deletion in the L1/L2 region, were noted in the majority of tumors that contained HPV vcDNA, and that full-length intact HPV genome was not found in these tumors with non-canonical HPV vcDNA.
These data do have limitations and present opportunities for further investigation. The effect of viral promoters and genes that facilitate high expression of hybrid transcripts and human oncogene expression in hybrid ecDNA structures is presumably facilitated by chromatin modification that is found in human ecDNA (7). However, the additional effect of viral promoters in an ecDNA context may facilitate chromatin structural effects. Although we have previously noted that human ecDNA is associated with poorer prognosis for human cancers in general, we have not seen worse prognosis associated with human ecDNA in HPVOPC, perhaps due to the small sample sizes available for ecDNA characterization, as well as the general favorable prognosis of HPVOPC.
The understanding of the role of ecDNA in HPVOPC has direct implications for development of HPVOPC therapy as ecDNA-mediated amplification has been recognized as a potential mechanism of therapeutic resistance and specific, overexpressed oncogenes on ecDNA may be targeted with precision medicine approaches (6). EcDNA may also have interactions with chromosomes, affecting stability and integration that may also provide therapeutic opportunities for HPVOPC (63), including the presence of ecDNA as a marker for susceptibility to DNA repair inhibition. Indeed, HPVOPC has been shown to be sensitive to Wee-1 and CHK1/2 inhibition, and SCC090 cells that we have identified as ecDNA driven in this manuscript have shown dramatic apoptotic response to ionizing radiation in combination with the Chk1/2 inhibitor prexasertib (64–66). Non-chromosomal circular oncogenic DNA is present and transcriptionally active in nearly all HPVOPC in the form of ecDNA and vcDNA, and the mechanisms of circular DNA formation and maintenance themselves may be potential therapeutic targets. It is possible that the HPV gene products that facilitate general mechanisms of non-chromosomal circular DNA formation may be targeted in all HPVOPC, to target vcDNA as well as human and hybrid ecDNA that transcribe a variety of oncogenic products. Finally, human genes overexpressed via ecDNA in individual tumors have key driver oncogenic roles as well as roles in immune evasion that may serve as targets for personalized therapy.
Supplementary Material
Translational relevance.
The incidence of HPV-driven oropharynx cancer has increased in recent years, and in the U.S., its frequency is the second-fastest growing of solid tumors. We reveal that human-viral extrachromosomal circular DNA is a strong driver of oncogenic HPV transcription, and that circular HPV genomes may exist in rearranged, non-canonical states in the cancer genome. These circular oncogenic DNA structures enable expression of oncogenic elements in nearly all HPVOPC. The formation and maintenance of oncogenic circular DNA are tasks which are both unique to cancer, and thus represent potential therapeutic targets. Furthermore, such elements are not constricted by the rules of Mendelian inheritance, and they enable more rapid tumor evolution, even in the face of targeted therapies. As a result, detecting and profiling circular DNA in cancers presents an important potential prognostic indicator for patient outcome.
Acknowledgments:
The following funding sources are noted:
John Pang - NIH/NIDCD T32 DC000028
Joseph Califano - NIH/NIDCR R01DE023347—04, NIH 1R01 CA204264–01
Kathleen Fisch – NIH/CTSA UL1TR001442
Vineet Bafna - R01GM114362
In addition, we acknowledge Amanda Birmingham for assistance with data processing within the UCSD Center for Computational Biology.
Footnotes
Conflict of interest statement:
V.B. is a co-founder, serves on the scientific advisory board and has equity interest in Boundless Bio, Inc. (BB) and Digital Proteomics, LLC (DP), and receives income from DP. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. P.S.M. is a co-founder of Boundless Bio, Inc. He has equity and chairs the scientific advisory board, for which he is compensated. N.N is currently employed by Boundless Bio, Inc.
Data and Materials Availability
AmpliconArchitect amplicon visualizations, breakpoint graphs, and amplicon decompositions have been uploaded to FigShare (doi: 10.6084/m9.figshare.13520087; URL: dx.doi.org/10.6084/m9.figshare.13520087).
Plasmids generated in this study are available upon request.
The codebases utilized in this study are available at:
https://github.com/namphuon/ViFi
References
- 1.Annual Report to the Nation 2019: Overall Cancer Statistics. National Cancer Institute: Surveillance, Epidemiology, and End Results Program. https://seer.cancer.gov/report_to_nation/statistics.html. Accessed March 1, 2020.
- 2.04/22/2020. American Cancer Society. Cancer Facts & Figures 2020. Atlanta: American Cancer Society; 2020. <https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf>. 04/22/2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011;29(32):4294–301 doi 10.1200/jco.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham SV. Human papillomavirus: gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol 2010;5(10):1493–506 doi 10.2217/fmb.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine 2010;363(1):24–35 doi 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017;543(7643):122–5 doi 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019;575(7784):699–703 doi 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 2020;52(9):891–7 doi 10.1038/s41588-020-0678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey C, Shoura MJ, Mischel PS, Swanton C. Extrachromosomal DNA-relieving heredity constraints, accelerating tumour evolution. Ann Oncol 2020;31(7):884–93 doi 10.1016/j.annonc.2020.03.303. [DOI] [PubMed] [Google Scholar]
- 10.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 2014;343(6166):72–6 doi 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshpande V, Luebeck J, Nguyen ND, Bakhtiari M, Turner KM, Schwab R, et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 2019;10(1):392 doi 10.1038/s41467-018-08200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groves IJ, Coleman N. Human papillomavirus genome integration in squamous carcinogenesis: what have next-generation sequencing studies taught us? J Pathol 2018;245(1):9–18 doi 10.1002/path.5058. [DOI] [PubMed] [Google Scholar]
- 13.Morgan IM, DiNardo LJ, Windle B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017;9(8) doi 10.3390/v9080208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nulton TJ, Olex AL, Dozmorov M, Morgan IM, Windle B. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017;8(11):17684–99 doi 10.18632/oncotarget.15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res 2014;24(2):185–99 doi 10.1101/gr.164806.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmes A, Lameiras S, Jeannot E, Marie Y, Castera L, Sastre-Garau X, et al. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom Med 2016;1:16004 doi 10.1038/npjgenmed.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, Davis J, Li X, Smith KS, Battle A, Montgomery SB. High-resolution transcriptome analysis with long-read RNA sequencing. PLoS One 2014;9(9):e108095 doi 10.1371/journal.pone.0108095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patch AM, Nones K, Kazakoff SH, Newell F, Wood S, Leonard C, et al. Germline and somatic variant identification using BGISEQ-500 and HiSeq X Ten whole genome sequencing. PLoS One 2018;13(1):e0190264 doi 10.1371/journal.pone.0190264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo T, Sakai A, Afsari B, Considine M, Danilova L, Favorov AV, et al. A Novel Functional Splice Variant of AKT3 Defined by Analysis of Alternative Splice Expression in HPV-Positive Oropharyngeal Cancers. Cancer Res 2017;77(19):5248–58 doi 10.1158/0008-5472.can-16-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010;38(18):e178 doi 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 2016;11(9):1650–67 doi 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Quantitative Biology 2013(https://arxiv.org/abs/1303.3997).
- 24.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 2016;12(4):e1004873 doi 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29(1):24–6 doi 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ardui S, Ameur A, Vermeesch JR, Hestand MS. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res 2018;46(5):2159–68 doi 10.1093/nar/gky066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen ND, Deshpande V, Luebeck J, Mischel PS, Bafna V. ViFi: accurate detection of viral integration and mRNA fusion reveals indiscriminate and unregulated transcription in proximal genomic regions in cervical cancer. Nucleic Acids Res 2018;46(7):3309–25 doi 10.1093/nar/gky180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu S, Bafna V, Mischel PS. Extrachromosomal DNA (ecDNA) in cancer pathogenesis. Curr Opin Genet Dev 2021;66:78–82 doi 10.1016/j.gde.2021.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther 2015;14(4):847–56 doi 10.1158/1535-7163.Mct-14-0983. [DOI] [PubMed] [Google Scholar]
- 30.Plebani R, Oliver GR, Trerotola M, Guerra E, Cantanelli P, Apicella L, et al. Long-range transcriptome sequencing reveals cancer cell growth regulatory chimeric mRNA. Neoplasia 2012;14(11):1087–96 doi 10.1593/neo.121342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosonaga M, Saya H, Arima Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer and Metastasis Reviews 2020;39(3):711–20 doi 10.1007/s10555-020-09881-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan Y, Liu L, Cheng Y, Yu J, Feng Y. Amplified LncRNA PVT1 promotes lung cancer proliferation and metastasis by facilitating VEGFC expression. Biochem Cell Biol 2020:1–7 doi 10.1139/bcb-2019-0435. [DOI] [PubMed] [Google Scholar]
- 33.Smotkin D, Prokoph H, Wettstein FO. Oncogenic and nononcogenic human genital papillomaviruses generate the E7 mRNA by different mechanisms. J Virol 1989;63(3):1441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillison ML, Akagi K, Xiao W, Jiang B, Pickard RKL, Li J, et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res 2019;29(1):1–17 doi 10.1101/gr.241141.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cornelissen MT, Smits HL, Briet MA, van den Tweel JG, Struyk AP, van der Noordaa J, et al. Uniformity of the splicing pattern of the E6/E7 transcripts in human papillomavirus type 16-transformed human fibroblasts, human cervical premalignant lesions and carcinomas. J Gen Virol 1990;71 (Pt 5):1243–6 doi 10.1099/0022-1317-71-5-1243. [DOI] [PubMed] [Google Scholar]
- 36.Tang S, Tao M, McCoy JP Jr., Zheng ZM. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol 2006;80(9):4249–63 doi 10.1128/jvi.80.9.4249-4263.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olmedo-Nieva L, Munoz-Bello JO, Contreras-Paredes A, Lizano M. The Role of E6 Spliced Isoforms (E6*) in Human Papillomavirus-Induced Carcinogenesis. Viruses 2018;10(1) doi 10.3390/v10010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Johansson C, Cardoso Palacios C, Mossberg A, Dhanjal S, Bergvall M, et al. Eight nucleotide substitutions inhibit splicing to HPV-16 3’-splice site SA3358 and reduce the efficiency by which HPV-16 increases the life span of primary human keratinocytes. PLoS One 2013;8(9):e72776 doi 10.1371/journal.pone.0072776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somberg M, Schwartz S. Multiple ASF/SF2 sites in the human papillomavirus type 16 (HPV-16) E4-coding region promote splicing to the most commonly used 3’-splice site on the HPV-16 genome. J Virol 2010;84(16):8219–30 doi 10.1128/jvi.00462-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson C, Schwartz S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat Rev Microbiol 2013;11(4):239–51 doi 10.1038/nrmicro2984. [DOI] [PubMed] [Google Scholar]
- 41.Vaisman CE, Del Moral-Hernandez O, Moreno-Campuzano S, Arechaga-Ocampo E, Bonilla-Moreno R, Garcia-Aguiar I, et al. C33-A cells transfected with E6*I or E6*II the short forms of HPV-16 E6, displayed opposite effects on cisplatin-induced apoptosis. Virus Res 2018;247:94–101 doi 10.1016/j.virusres.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Nindl I, Rindfleisch K, Lotz B, Schneider A, Dürst M. Uniform distribution of HPV 16 E6 and E7 variants in patients with normal histology, cervical intra-epithelial neoplasia and cervical cancer. Int J Cancer 1999;82(2):203–7 doi 10.1002/(sici)1097-0215(19990719)82:2<203::aid-ijc9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 43.Brattain MG, Fine WD, Khaled FM, Thompson J, Brattain DE. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res 1981;41(5):1751–6. [PubMed] [Google Scholar]
- 44.Martin D, Abba MC, Molinolo AA, Vitale-Cross L, Wang Z, Zaida M, et al. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget 2014;5(19):8906–23 doi 10.18632/oncotarget.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, et al. TCPA: a resource for cancer functional proteomics data. Nat Methods 2013;10(11):1046–7 doi 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A 2014;111(43):15544–9 doi 10.1073/pnas.1416074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Leo A, Calderon A, Lieberman PM. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol 2020;28(2):150–62 doi 10.1016/j.tim.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jose L, Androphy EJ, DeSmet M. Phosphorylation of the Human Papillomavirus E2 Protein at Tyrosine 138 Regulates Episomal Replication. J Virol 2020;94(14) doi 10.1128/JVI.00488-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hafner N, Driesch C, Gajda M, Jansen L, Kirchmayr R, Runnebaum IB, et al. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene 2008;27(11):1610–7 doi 10.1038/sj.onc.1210791. [DOI] [PubMed] [Google Scholar]
- 50.Hong D, Liu J, Hu Y, Lu X, Li B, Li Y, et al. Viral E6 is overexpressed via high viral load in invasive cervical cancer with episomal HPV16. BMC Cancer 2017;17(1):136 doi 10.1186/s12885-017-3124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paget-Bailly P, Meznad K, Bruyere D, Perrard J, Herfs M, Jung AC, et al. Comparative RNA sequencing reveals that HPV16 E6 abrogates the effect of E6*I on ROS metabolism. Sci Rep 2019;9(1):5938 doi 10.1038/s41598-019-42393-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pim D, Massimi P, Banks L. Alternatively spliced HPV-18 E6* protein inhibits E6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene 1997;15(3):257–64 doi 10.1038/sj.onc.1201202. [DOI] [PubMed] [Google Scholar]
- 53.Filippova M, Johnson MM, Bautista M, Filippov V, Fodor N, Tungteakkhun SS, et al. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J Virol 2007;81(8):4116–29 doi 10.1128/jvi.01924-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roggenbuck B, Larsen PM, Fey SJ, Bartsch D, Gissmann L, Schwarz E. Human papillomavirus type 18 E6*, E6, and E7 protein synthesis in cell-free translation systems and comparison of E6 and E7 in vitro translation products to proteins immunoprecipitated from human epithelial cells. J Virol 1991;65(9):5068–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Artaza-Irigaray C, Molina-Pineda A, Aguilar-Lemarroy A, Ortiz-Lazareno P, Limon-Toledo LP, Pereira-Suarez AL, et al. E6/E7 and E6(*) From HPV16 and HPV18 Upregulate IL-6 Expression Independently of p53 in Keratinocytes. Front Immunol 2019;10:1676 doi 10.3389/fimmu.2019.01676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams VM, Filippova M, Filippov V, Payne KJ, Duerksen-Hughes P. Human papillomavirus type 16 E6* induces oxidative stress and DNA damage. J Virol 2014;88(12):6751–61 doi 10.1128/jvi.03355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kosel S, Burggraf S, Engelhardt W, Olgemoller B. Increased levels of HPV16 E6*I transcripts in high-grade cervical cytology and histology (CIN II+) detected by rapid real-time RT-PCR amplification. Cytopathology 2007;18(5):290–9 doi 10.1111/j.1365-2303.2007.00481.x. [DOI] [PubMed] [Google Scholar]
- 58.Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016;529(7587):541–5 doi 10.1038/nature16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hemmat N, Baghi HB. Human papillomavirus E5 protein, the undercover culprit of tumorigenesis. Infect Agent Cancer 2018;13:31 doi 10.1186/s13027-018-0208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyauchi S, Sanders PD, Guram K, Kim SS, Paolini F, Venuti A, et al. HPV16 E5 Mediates Resistance to PD-L1 Blockade and Can Be Targeted with Rimantadine in Head and Neck Cancer. Cancer Res 2020;80(4):732–46 doi 10.1158/0008-5472.Can-19-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren S, Gaykalova DA, Guo T, Favorov AV, Fertig EJ, Tamayo P, et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 2020;39(40):6327–39 doi 10.1038/s41388-020-01431-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wentzensen N, Ridder R, Klaes R, Vinokurova S, Schaefer U, Doeberitz M. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002;21(3):419–26 doi 10.1038/sj.onc.1205104. [DOI] [PubMed] [Google Scholar]
- 63.Zhu Y, Gujar AD, Wong CH, Tjong H, Ngan CY, Gong L, et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 2021;39(5):694–707.e7 doi 10.1016/j.ccell.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Molkentine JM, Molkentine DP, Bridges KA, Xie T, Yang L, Sheth A, et al. Targeting DNA damage response in head and neck cancers through abrogation of cell cycle checkpoints. Int J Radiat Biol 2020:1–8 doi 10.1080/09553002.2020.1730014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeng L, Nikolaev A, Xing C, Della Manna DL, Yang ES. CHK1/2 Inhibitor Prexasertib Suppresses NOTCH Signaling and Enhances Cytotoxicity of Cisplatin and Radiation in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther 2020;19(6):1279–88 doi 10.1158/1535-7163.Mct-19-0946. [DOI] [PubMed] [Google Scholar]
- 66.Tanaka N, Patel AA, Wang J, Frederick MJ, Kalu NN, Zhao M, et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clin Cancer Res 2015;21(21):4831–44 doi 10.1158/1078-0432.Ccr-15-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.