

Abstract
PURPOSE
Previous studies have shown an approximately two-fold elevation in the relative risk of urinary bladder cancer (UBC) among people with a family history that could not be entirely explained by shared environmental exposures, thus suggesting a genetic component in its predisposition. Multiple genome-wide association studies and recent gene panel sequencing studies identified several genetic loci that are associated with UBC risk; however, the list of UBC-associated variants and genes is incomplete.
MATERIALS AND METHODS
We exome sequenced eight patients from three multiplex UBC pedigrees and a group of 77 unrelated familial UBC cases matched to 241 cancer-free controls. In addition, we examined pathogenic germline variation in 444 candidate genes in 392 The Cancer Genome Atlas UBC cases.
RESULTS
In the pedigrees, segregating variants were family-specific although the identified genes clustered in common pathways, most notably DNA repair (MLH1 and MSH2) and cellular metabolism (IDH1 and ME1). In the familial UBC group, the proportion of pathogenic and likely pathogenic variants was significantly higher in cases compared with controls (P = .003). Pathogenic and likely pathogenic variant load was also significantly increased in genes involved in cilia biogenesis (P = .001). In addition, a pathogenic variant in CHEK2 (NM_007194.4:c.1100del; p.T367Mfs*15) was over-represented in cases (variant frequency = 2.6%; 95% CI, 0.71 to 6.52) compared with controls (variant frequency = 0.21%; 95% CI, 0.01 to 1.15), but was not statistically significant.
CONCLUSION
These results point to a complex polygenic predisposition to UBC. Despite heterogeneity, the genes cluster in several biologically relevant pathways and processes, for example, DNA repair, cilia biogenesis, and cellular metabolism. Larger studies are required to determine the importance of CHEK2 in UBC etiology.
INTRODUCTION
It is estimated that there will be 83,730 newly diagnosed urinary bladder cancers (UBCs: 64,280 males and 19,450 females) and 17,200 deaths (12,260 males and 4,940 females) in the United States in 2021.1 UBC is typically a slowly developing disease but recurs frequently. It is predominantly observed in older patients (average age at diagnosis = 73 years). A number of environmental risk factors have been identified for this malignancy including smoking, some occupational exposures, and contaminants in drinking water.2 Cigarette smoking is the primary risk factor for bladder cancer, which is estimated to account for approximately 50% of UBC cases in both sexes.2,3 Risk among current smokers is four to five times greater than that in nonsmokers.2,3 Besides environmental factors, a genetic component of predisposition to UBC has been demonstrated as well: the first evidence of genetic susceptibility to UBC was observed in a pedigree of four affected first-degree relatives by Fraumeni and Thomas.4 Subsequent epidemiologic studies have identified an increased relative risk for individuals with family history of UBC,5-10 which could not be fully explained by shared environmental exposure, thus implying a genetic component in the predisposition.11-15 However, familial UBC clustering appears to be rare: a national recruitment effort failed to identify a sufficient number of multiple-case UBC kindreds to warrant a familial cancer study.5
Early important clues for a potential UBC genetic etiology came from studies of hereditary cancer susceptibility disorders such as Lynch (eg, OMIM#120435), Costello (OMIM#218040), Apert (OMIM#101200), and familial adenomatous polyposis 3 (OMIM#616415) syndromes. The presence of UBC in these monogenic disorders5 suggests that rare variants in critical signaling pathways (eg, cell cycle progression and mitogenic signal transduction) play an etiologic role in UBC pathogenesis. A subsequent series of pioneering genome-wide association studies has identified 16 common, low-penetrance polymorphisms associated with elevated UBC risk.2,16-19 A recent genome-wide meta-analysis that investigated the outcomes of non–muscle-invasive UBC identified rs12885353 (near SCFD1), which is significantly associated with UBC recurrence-free survival.20 Most of these variants and genes confer modest increase in UBC risk (odds ratio < 2) and aggregate in xenobiotic metabolism, DNA repair, and cell cycle progression pathways.21
Two recent studies performed cancer gene panel sequencing in predominantly patients with sporadic UBC. Both investigations identified pathogenic and likely pathogenic (P and LP) variants in germlines of 13.7%22 and 24%23 of patients, with BRCA1, BRCA2, MSH2, CHEK2, ERCC3, MLH1, and ATM being most frequently mutated.
Unlike previous sequencing studies, which analyzed candidate gene panels in primarily sporadic UBC cases,22,23 we used an exome-wide approach in patients with familial UBC. We tested the following hypotheses: (1) Familial high-penetrance clusters of bladder cancer are partially driven by shared genetic variants that predispose to higher incidence of the cancer cases in related individuals, (2) Rare deleterious variants segregating among multiple cases in each pedigree could be involved in the etiology of familial bladder cancer, and (3) Rare deleterious variants detected by exome sequencing could have effects that are large enough to be detected in a modestly sized sample set. In this exploratory study, we investigated exomes of eight patients from three multiplex UBC pedigrees and 77 unrelated familial UBC cases that were matched to 241 cancer-free controls. In the pedigrees, we ascertained the segregating pattern of deleterious variants, and in the case-control analysis, we examined a rare-variant association with the UBC risk. In addition, we investigated the germline variation landscape in 392 UBC cases from The Cancer Genome Atlas (TCGA) public database (Data Supplement). The sets were analyzed in parallel, and the results were examined for common variants, genes, and pathways.
MATERIALS AND METHODS
The full version of Materials and Methods can be found in the Data Supplement.
Patients and Sample Collection
All studies were approved by the institutional review board (IRB), the National Cancer Institute (NCI) Special Studies IRB, and participating local IRBs. Clinical information for three pedigrees is summarized in the Data Supplement. Clinical information for 74 familial UBC cases is summarized in the Data Supplement. Cases were matched to controls, and the principal component analysis (PLINK v1.90b4.4)24 was performed on the resulting set to ensure its homogeneity (Data Supplement).
All participants provided written informed consent before enrollment into the NCI DCEG familial cancer protocol “Clinical, Laboratory, and Epidemiologic Characterization of Individuals and Families at High Risk of Cancer” or the parent studies that enrolled the participants. All individual-level data, including clinical data, were deidentified. The authors have modified the pedigree or family tree to avoid potential identification of the family or its members. The authors received and archived written patient consent. This study fully adhered to the principles set out in the Declaration of Helsinki.
Exome Sequencing and Data Processing
Genomic DNA was extracted from blood, whole genome amplified (74 familial UBC cases), exome captured with NimbleGen SeqCap EZ Human Exome Library, and sequenced on the Illumina HiSeq 2000 platform. The human reference genome and the known gene transcript annotation were downloaded from the UCSC database, hg19. Sequencing reads were trimmed (Trimmomatic), and only read pairs with both ends > 36 bp were used. Reads were aligned to the reference genome (NovoAlign). Duplicate reads were removed (MarkDuplicates), and only read pairs mapped in complementary directions at a fragment length of 200-400 bp were used. These alignments were further refined (RealignerTargetCreator and IndelRealigner). Variant discovery and genotype calling were performed on all individuals globally (UnifiedGenotyper, HaplotypeCaller from GATK, and FreeBayes). The three callers were used to call each sample in parallel, and the caller-specific results were generated independently. The ensemble variant calling pipeline was then implemented to integrate the results from the three callers.
Data Filtering and Variant Classification
All noncoding, multiallelic, common variants (> 1% in ExAC or gnomAD) and variants present in this study's controls at frequency above 10% were filtered out. Remaining variants were grouped into three tiers: (1) variants classified in ClinVar as pathogenic or likely pathogenic (tier 1); (2) variants that were unclassified by ClinVar but classified by InterVar as P and LP (tier 2); and (3) all remaining loss-of-function variants and missense variants fulfilling 2 of 3 of the following conditions: CADD_phred_score > 25, REVEL_score > 0.5, MetaSVM_score = D(eleterious) (tier 3). Variants in tier 1 were considered deleterious; remaining variants (tiers 2 and 3) were considered potentially deleterious.
Variant Segregation Pattern in Pedigrees
Tier 1-3 variants found in UBC-affected members of a pedigree were considered as risk variants and were examined further.
Statistical Tests
Differences in frequency between cases and controls were determined by using Fisher's exact test. Rare-variant association tests were performed by using the Cohort Allelic Sums Test, Sequence Kernel Association Test (SKAT), and SKAT optimal test. False discovery rate correction for multiple testing was computed in variant- and gene-based analyses for case-control association tests (q-value < 0.05). Bonferroni correction was applied to pathway-level analyses (0.05/9 = 0.006, P value_corrected < .006).
Ontological Classification of Genes Carrying P and LP Variants
In the familial UBC case-control analysis, genes with tier 1 P and LP variants were stratified by their biologic processes (BPs) as defined in the Gene Ontology database. Related BP terms were further grouped into the following categories: DNA repair, replication, and recombination, gene expression and signal transduction, cellular metabolism, transmembrane transport, protein modifications and metabolism, and cilia biogenesis. Infrequently observed or biologically irrelevant BP categories were placed in the Others group. Genes with unknown BP were placed in the Unknown group.
UBC Gene List Compilation
The list of genes likely involved in the etiology of UBC was compiled by combining genes from published genome-wide association studies, somatic sequencing studies, studies of tumor predisposition syndromes, and all known DNA repair genes (Data Supplement). OncoPrint plots summarize clinical and genomic characteristics for patients carrying tier 1-2 variants in the resulting 444 candidate genes.
TCGA UBC Data Set
Germline sequencing data for UBC-diagnosed participants (N = 392) were downloaded from the Genomic Data Commons. Common variants (> 1%) were filtered out. Tier 1 and 2 variants were used for further analysis.
RESULTS
Variant Segregation Pattern in Three Multiplex UBC Pedigrees
The UBC pedigrees analyzed in this study are shown in Figure 1. Clinical information for these families is summarized in the Data Supplement.
FIG 1.
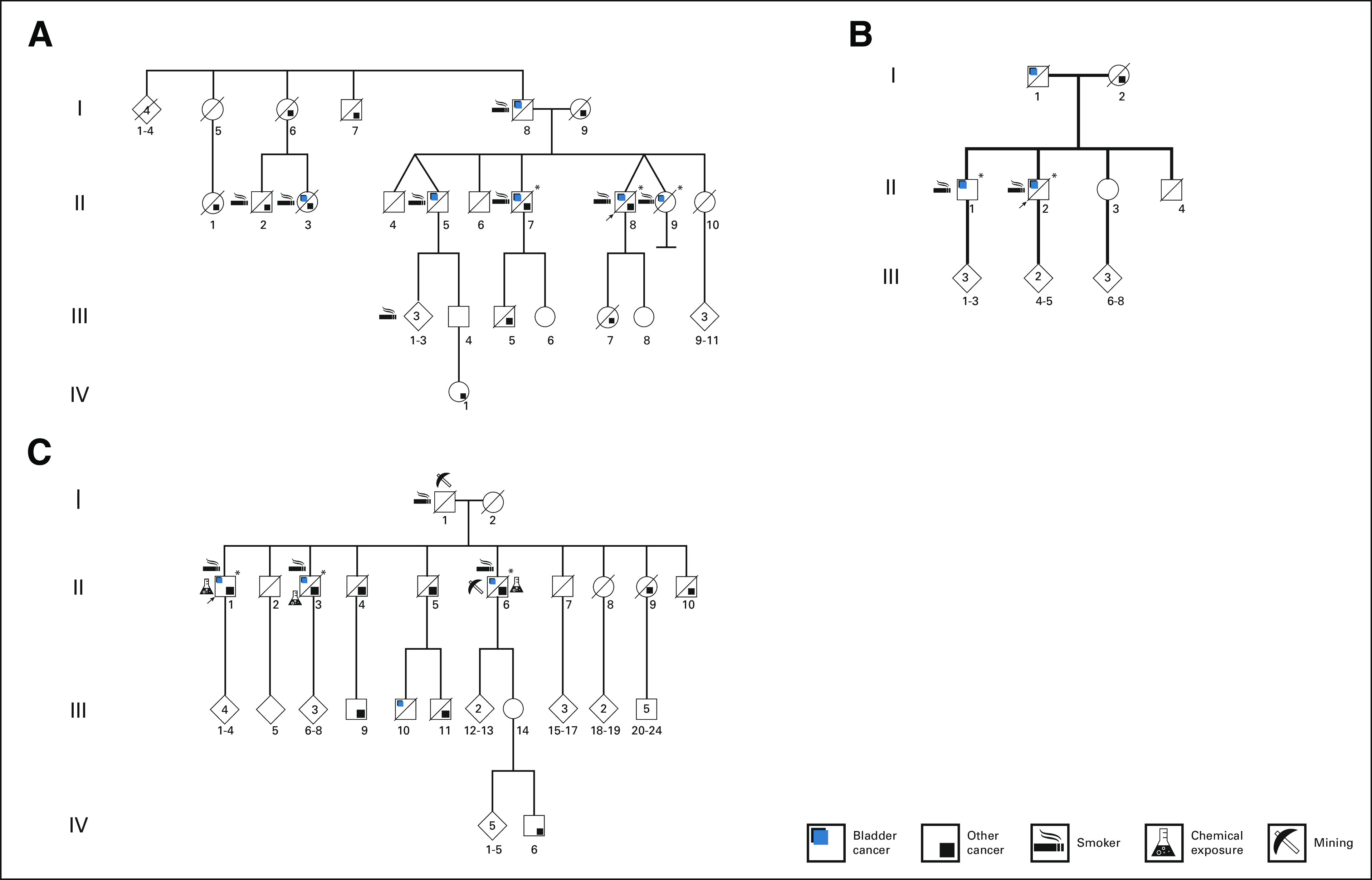
Schematic representation of multiplex urinary bladder cancer pedigrees A, B, and C. Arrows show probands, and asterisks show pedigree members who were exome sequenced.
We exome sequenced germline DNA from three, two, and three UBC-affected members of families A, B, and C, respectively. After ascertainment of variant segregation pattern and assigning the variants to tiers 1, 2, and 3, we identified 4, 15, and 12 tier 1-3 variants in the pedigrees A, B, and C, respectively (Table 1). We detected a single tier 2 variant (P and LP InterVar) in CFTR in family A, two tier 2 variants (IDH1 and ELAC2) in family B, and one tier 1 (ABCA4) and one tier 2 (CHRNE) variants in family C. rs119484086 in ELAC2 has been reported as a prostate cancer susceptibility allele26; however, there were no cases of prostate cancer reported for the members of family B who harbored the variant. ABCA4, CHRNE, and CFTR are expressed at a low level in the bladder, and their known biologic functions (retina-specific membrane transporter, acetylcholine receptor at neuromuscular junctions, and water secretion and absorption in epithelial tissues, respectively) make them candidates unlikely for UBC predisposition. In families B and C, we identified tier 3 variants in mismatch repair genes MLH1 and MSH2, respectively. In addition to a tier 2 variant segregating in family B in IDH1 (one of the key enzymes of carbon metabolism in the cell), we identified a tier 3 variant in ME1 (malic enzyme 1, which connects the glycolytic pathway with the Krebs cycle) that segregated in family A.
TABLE 1.
Segregating Variants in Families A, B, and C

Exome-Wide Analysis of 77 Familial UBC Cases Versus 241 Cancer-Free Controls
Variant-level analysis by Fisher's exact test.
We observed only a single variant in ATP2A1 that reached statistical significance after multiple testing correction (q-value < 0.05; Table 2). ATP2A1 is unexpressed in the urinary bladder and was not investigated further. Notably, the frequency of frameshifting deletion in CHEK2 (NM_007194:c.1100del;p.T367Mfs*15, rs555607708) was elevated among cases (2.6%; 95% CI, 0.71 to 6.52) compared with controls (0.21%; 95% CI, 0.0 to 1.15) by approximately 10-fold; however, it was not significant after multiple testing correction. In several largest public databases, the frequency of c.1100delC among Europeans (excluding Finnish subpopulation) ranged from 0.17% to 0.26%; its frequency varied between different ancestral groups and was highest among Finns (0.87%; 95% CI, 0.76 to 0.99; Table 3).
TABLE 2.
Fisher's Exact Test of Association in the Set of 77 Urinary Bladder Cancer Cases Versus 241 Cancer-Free Controls

TABLE 3.
Frequency of CHEK2 c.1100delC Allele in Sample Sets Used in This Study and in Unaffected Populations

Gene-level analysis by Cohort Allelic Sums Test, SKAT, and SKAT optimal test rare-variant association (burden) tests.
The gene-level analysis identified CC2D2A and GALC at the nominal 0.05 significance level by at least one of the tests, but neither of these genes were significant after multiple testing correction (Data Supplement).
Comparison of P and LP variant loads in 77 cases versus 241 controls.
In addition to variant- and gene-level analyses, we examined the load of tier 1 (ClinVar P and LP) variants in 77 cases and 241 controls. Visual inspection of the distribution of the number of P and LP variants per person in cases and controls revealed a shift toward a higher number of P and LP variants in cases (Fig 2A). We also observed a higher proportion of individuals with at least one P and LP variant among cases as compared with controls (76.6% v 66.4%, P = .003, Table 4). This difference was statistically significant after Bonferroni correction. The total number of unique and overlapping P and LP variants and genes in cases and controls is shown in Figure 2B.
FIG 2.
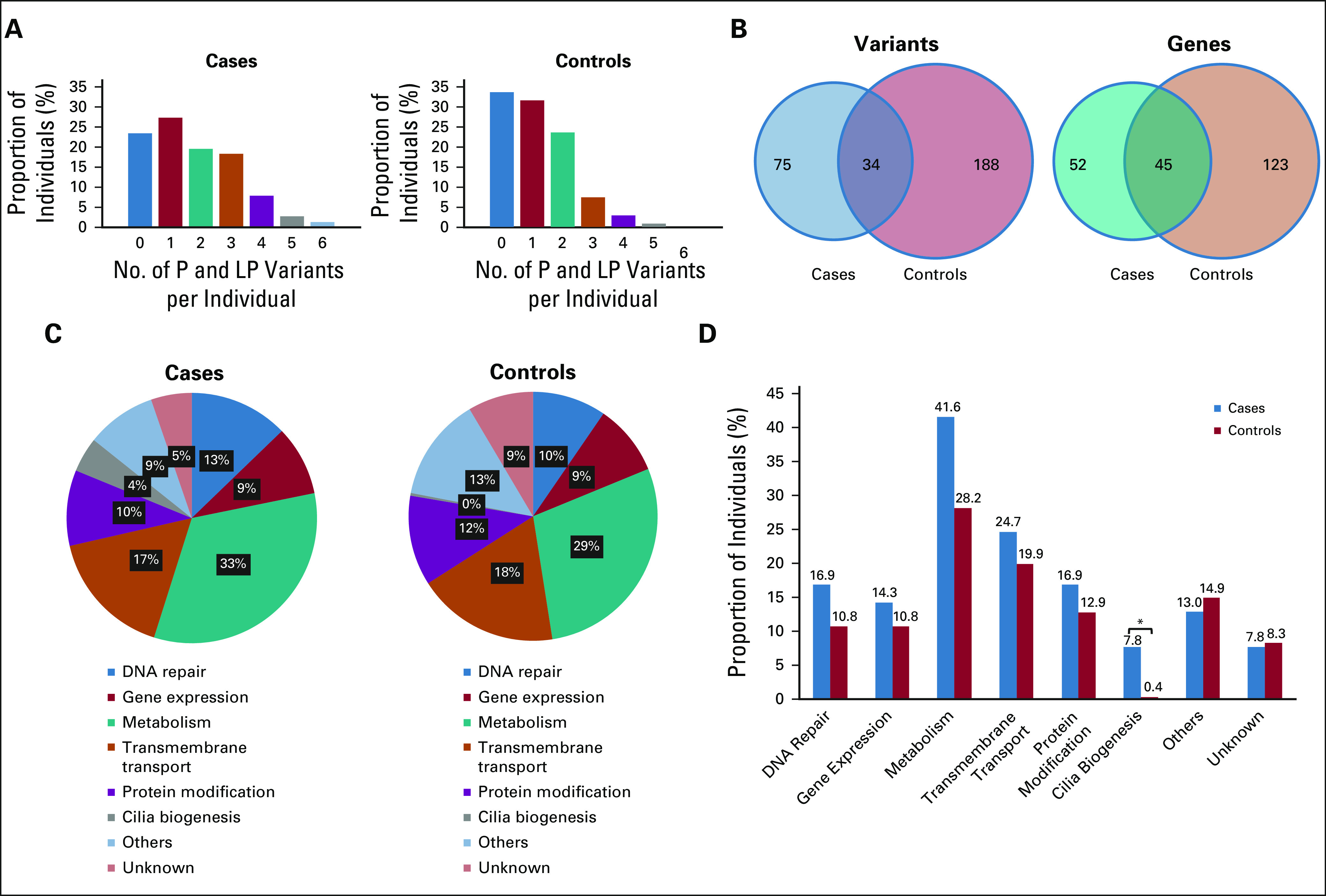
Deleterious variant load and ontological analysis in the familial urinary bladder cancer group (77 cases v 241 controls). (A) Distribution of the number of ClinVar P and LP variants per individual in cases (left) and controls (right). Proportion of individuals (%) carrying 0, 1, 2, 3, 4, 5, or 6 deleterious variants is shown for each group. (B) Venn diagrams showing P and LP variants (left) and genes carrying these variants (right) in cases (smaller circles) and controls (larger circles). (C) Relative abundance of P and LP variants in different ontological categories in cases (left) and controls (right). For each group (cases or controls), the relative abundance of variants in each ontological category was calculated as the sum of P and LP alleles in all individuals and in all genes included in that ontological category and expressed as a percent of total P and LP share in the group. (D) P and LP variant share in cases versus controls in different ontological categories. For each ontological category, the number of cases or controls carrying at least one P and LP variant in any of the genes included in that category was calculated and expressed as percent of total cases or controls. Note that some individuals carried more than one P and LP variant, so the sum of proportions among all ontological categories in cases or controls does not equal 1 (100%). P and LP, pathogenic and likely pathogenic.*, statistically significant after Bonferroni correction, P value < .006.
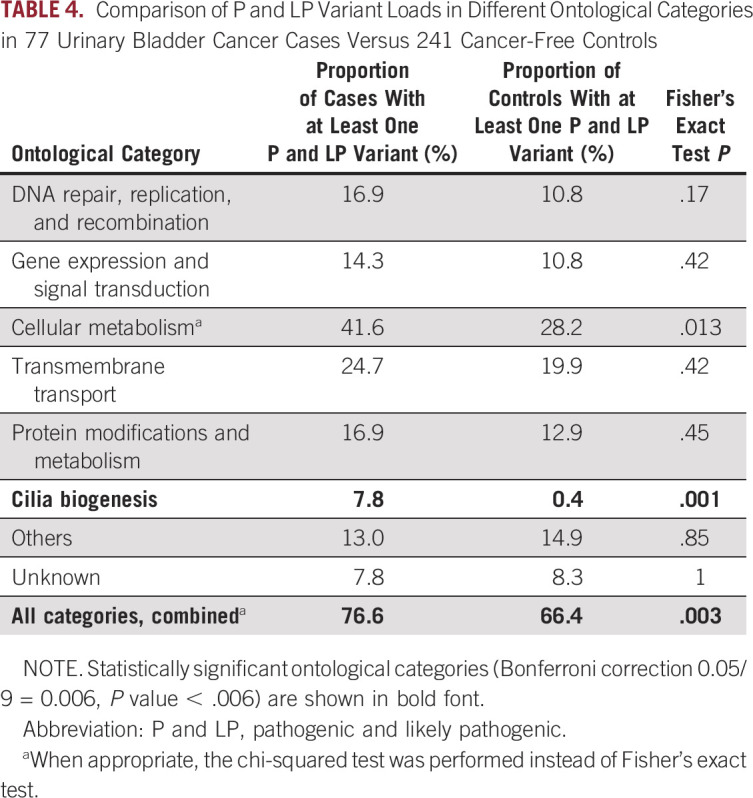
TABLE 4.
Comparison of P and LP Variant Loads in Different Ontological Categories in 77 Urinary Bladder Cancer Cases Versus 241 Cancer-Free Controls
Ontological analysis of tier 1 P and LP variants in 77 cases and 241 controls.
First, we stratified genes harboring P and LP variants into groups with related BP as defined in the Gene Ontology database (Data Supplement). We then determined the proportion of individuals who carried at least one P and LP variant in any of the genes included in an ontological category as referenced above, in both cases and controls. The cases versus controls comparison demonstrated that for most of the ontological categories, the proportion of individuals with at least one P and LP variant was higher among cases; however, after multiple testing correction (Bonferroni), the differences reached statistical significance only in the cilia biogenesis category (P = .001; Table 4 and Figs 2C and 2D).
Analysis of Pathogenic Variant Loads in the Germline of 392 UBC Cases From TCGA Identified an Elevated Frequency of CHEK2 c.1100delC
We also examined ClinVar and InterVar P and LP variants in 444 UBC candidate genes (Data Supplement) found in the germlines of 392 TCGA UBC patients (Data Supplement). In total, we observed 123 tier 1 and 2 variants in 59 genes among TCGA UBC patients. Variants in CHEK2 were observed in 9 of 392 (2.3%) patients, thus making this locus the most frequently altered among 444 candidate genes in the UBC TCGA set (1.15%; 95% CI, 0.53 to 2.17). Notably, 4 of 9 (44.4%) of these CHEK2 pathogenic variants were the deletion c.1100delC identified in the familial UBC group; its frequency among 392 UBC cases was 0.51% (95% CI, 0.14 to 1.30; Table 3).
DISCUSSION
In this exploratory study, we investigated genetic risk factors in familial UBC using exome sequencing data from three multiplex pedigrees and 77 familial UBC cases matched with 241 cancer-free controls from existing epidemiologic studies and examined pathogenic germline variant loads in 444 UBC candidate genes in 392 UBC cases from the TCGA set. In the pedigrees, we identified potentially deleterious variants in mismatch repair DNA repair genes MLH1 and MSH2 that segregated in families B and C, and in the carbon metabolism genes, IDH1 and ME1, in families B and A. In the analysis of the familial UBC cases versus controls, we identified a possible association between the CHEK2 c.1100delC pathogenic variant and UBC, and in the TCGA UBC set, we observed this CHEK2 pathogenic variant at somewhat elevated frequency as well (0.51%; 95% CI, 0.14 to 1.30). Finally, we found that cilia biogenesis genes were significantly enriched with P and LP variants and that the total P and LP variant load was significantly higher in 77 cases with a positive UBC family history compared with controls from the epidemiologic studies. The main limitation of this study was a modest number of samples. This obstacle, which is common in projects involving rare diseases such as familial UBC, precluded us from reaching a sufficient power despite the extensive effort. Future replication studies would benefit from broad collaborations.
The variant segregation pattern in the three pedigrees demonstrated that the variants and the variant-carrying genes were unique to each family. Yet, we identified common ontological categories and biologic pathways affected by these variants in the pedigrees. For instance, we observed potentially deleterious variants in MSH2 and MLH1 segregating in families C and B, respectively. A rare missense MSH2 variant (c.182A>C;p.Q61P) found in family C was also identified in a patient who fulfilled the Bethesda guidelines for Lynch syndrome and who developed an ovarian cancer and colorectal carcinoma at age 44 and 50 years, suggesting a causative role of this variant.27 A deletion-insertion MLH1 variant (rs35502531)28 segregating in family B (c.1852_1853delinsGC;p.K618A), although classified as benign by ClinVar, has been shown to weaken the interaction between MLH1 and PMS2 in functional studies.29
We also observed rare deleterious and potentially deleterious variants in the carbon metabolism genes, IDH1 and ME1, in Families B and A, respectively. The enzymatic activity of IDH1 and ME1 results in increased cellular concentration of nicotinamide adenine dinucleotide phosphate, reduced (NADPH), which could be used to neutralize the excess of reactive oxygen species produced by stress stimuli including xenobiotics.30,31 It should be mentioned that in pedigrees A and B, 11 of 13 patients with UBC were current or former smokers. One possible nexus between mutants ME1 and IDH1 in the etiology of smoking-related UBC could be a consequence of decreased efficiency of these two enzymes in detoxicating xenobiotics produced by tobacco use.
The frameshift deletion (c.1100del;p.T367Mfs*15) in CHEK2 was one of the most frequently observed pathogenic variants in this study. CHEK2 is a serine-threonine kinase that regulates DNA repair through phosphorylation of BRCA2 and arrests progression through the cell cycle via DNA double-strand breaks activation pathway.32 The c.1100delC variant has been shown to eliminate kinase activity of CHEK2 and increase risk of breast cancer 2-fold in women and 10-fold in men.33 In ClinVar, this variant is classified as pathogenic in 37 reports, as a variant of unknown significance in two, and as a risk factor for breast, colorectal, and prostate cancers in another three submissions.34 Despite its apparent pathogenicity, this variant is relatively common in the general population: its global frequency in gnomAD is 0.21% (95% CI, 0.19 to 0.23) and it fluctuates widely in subpopulations and is the highest in Finnish Europeans (0.87%; 95% CI, 0.76 to 0.99).35 Noticeably, in our study, we observed this variant at substantially increased frequency (2.6%; 95% CI, 0.71 to 6.52) among 77 UBC cases of European descent. We also found this variant at somewhat elevated frequency (0.51%; 95% CI, 0.14 to 1.30) among 392 TCGA UBC cases. Contrary to our findings, a recent study by Nassar et al reported the frequency of this variant to be equal to 0.17% (95% CI, 0.04 to 0.51) in their set of UBC samples23; however, their cases included a substantial proportion of non-European samples, which could be a contributing factor to the differences observed in the outcomes. Despite its established role in breast and testicular cancers,33,36-38 no significant association between UBC and c.1100delC has been reported to date. In the Copenhagen general population study, which investigated association of CHEK2 c.1100delC with the risk of breast and other cancers, including UBC, the authors reported a modestly increased hazard ratio of 2.26 (95% CI, 0.94 to 5.43) for UBC, which notwithstanding was nonsignificant (P = .07).39 Another case-control study from Poland compared combined frequency of four pathogenic founder CHEK2 variants, including c.1100delC, and observed a modestly increased but statistically significant odds ratio of 1.9 (95% CI, 1.3 to 2.7; P = .0003).40 Substantially larger studies are needed to estimate penetrance of CHEK2 deleterious variants in various subpopulations and to determine this kinase's role in UBC pathogenesis.
Among other DNA damage repair genes, we observed P and LP variants in BRCA2, ATM, CHEK2, BRIP1, and MUTYH in 16.9% of cases in our familial UBC group. Similar to our findings, two recent papers reported P and LP variants in highly penetrant DNA repair genes in 11.3%22 and 16.7%23 of patients with sporadic high-risk UBC. However, in our familial UBC group, we detected P and LP variants only in moderately penetrant genes (except for BRCA2) such as CHEK2, ATM, BRIP1, and MUTYH, whereas highly penetrant genes were variant-free. This difference may be due to the advanced stage and grade of UBC cases analyzed in the abovementioned reports, whereas most of our cases were predominantly (62%) non–muscle-invasive, stage < T2 tumors.
In the ontological analysis of variants and genes over-represented in cases in our familial UBC group, cilia biogenesis was the only statistically significant category: 7.8% of cases had at least one deleterious variant (CC2D2A, DNAAF4, DNAH5, IQCB1, and RSPH1) versus 0.4% controls (NPHP3; P = .001). There is rapidly accumulating evidence of cilia's involvement in cancer development and progression.41-43 Interestingly, rs8173 in AURKA (involved in regulation of cilia disassembly in mitosis) conferred significantly greater susceptibility to bladder cancer.44
In conclusion, analyses of three distinct data sets revealed multiple biologically plausible genes that may be associated with UBC etiology, pointing to a complex polygenic character of genetic predisposition to this malignancy. Nonetheless, despite the substantial heterogeneity among these genes, they clustered in a limited number of BP, most notably, DNA repair, cilia biogenesis, and cellular metabolism.
ACKNOWLEDGMENT
The authors would like to express their candid gratitude to all members of the Frederick National Laboratory for Cancer Research at the Division of Cancer Epidemiology and Genetics (NCI) for carrying out sample processing and exome sequencing and for providing computational support. The authors express sincere appreciation to all Cancer Prevention Study II Nutrition Cohort and all other participants and to each member of the study and biospecimen management group. The authors would like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention's National Program of Cancer Registries and cancer registries supported by the National Cancer Institute's Surveillance Epidemiology and End Results Program. This work used the computational resources of the NIH High-Performance Computing Biowulf cluster. The results published in this study are in part based upon the data generated by the TCGA Research Network: http://cancergenome.nih.gov/, study ID phs000218.The authors thank Mr Alejandro Lafuente for his help with preparation of the article's figures.
M.S.: Retired.
Talia Wegman-Ostrosky
Travel, Accommodations, Expenses: Pfizer
Bin Zhu
Employment: GeneDx BioReference
Douglas R. Stewart
Employment: Genome Medical
No other potential conflicts of interest were reported.
DISCLAIMER
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
SUPPORT
Supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics of the NCI, Bethesda, MD. The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study II cohort.
A.P. and T.W.-O. contributed equally to this work.
T.W.-O.: Instituto Nacional de Cancerologia, Ciudad de Mexico, Mexico.
DATA SHARING STATEMENT
All relevant data will be deposited to the NLM NCBI database of Genotypes and Phenotypes (dbGaP), phs002326.v1.p1.
AUTHOR CONTRIBUTIONS
Conception and design: Dalsu Baris, Margaret R. Karagas, Neil E. Caporaso, Douglas R. Stewart
Financial support: Douglas R. Stewart
Administrative support: Bin Zhu, Molly Schwenn, Neal D. Freedman, Douglas R. Stewart
Provision of study materials or patients: Stella Koutros, Molly Schwenn, Neal D. Freedman, Demetrius Albanes, Nathaniel Rothman, Neil E. Caporaso
Collection and assembly of data: Alexander Pemov, Stella Koutros, Brenna Douthitt, Kristine Jones, Dalsu Baris, Molly Schwenn, Alison Johnson, Margaret R. Karagas, Brian D. Carter, Marjorie L. McCullough, Maria Teresa Landi, Neal D. Freedman, Demetrius Albanes, Debra T. Silverman, Nathaniel Rothman, Neil E. Caporaso, Mark H. Greene, Douglas R. Stewart
Data analysis and interpretation: Alexander Pemov, Talia Wegman-Ostrosky, Jung Kim, Stella Koutros, Bin Zhu, Molly Schwenn, Neal D. Freedman, Nathaniel Rothman, Neil E. Caporaso, Mark H. Greene, Joseph F. Fraumeni, Douglas R. Stewart
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/go/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Talia Wegman-Ostrosky
Travel, Accommodations, Expenses: Pfizer
Bin Zhu
Employment: GeneDx BioReference
Douglas R. Stewart
Employment: Genome Medical
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, et al. : Cancer statistics, 2021 CA Cancer J Clin 71:7–332021 [DOI] [PubMed] [Google Scholar]
- 2.Silverman DT, Koutros S, Figueroa JD, et al. Thun MJ, Linet MS, Cerhan JR, et al.Schottenfeld and Fraumeni Cancer Epidemiology and Prevention ed 4New York, NY: Oxford University Press; 2018, pp 977–996 [Google Scholar]
- 3.Freedman ND, Silverman DT, Hollenbeck AR, et al. : Association between smoking and risk of bladder cancer among men and women JAMA 306:737–7452011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraumeni JF, Jr, Thomas LB: Malignant bladder tumors in a man and his three sons JAMA 201:507–5091967 [Google Scholar]
- 5.Mueller CM, Caporaso N, Greene MH: Familial and genetic risk of transitional cell carcinoma of the urinary tract Urol Oncol 26:451–4642008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantor AF, Hartge P, Hoover RN, et al. : Familial and environmental interactions in bladder cancer risk Int J Cancer 35:703–7061985 [DOI] [PubMed] [Google Scholar]
- 7.Aben KK, Witjes JA, Schoenberg MP, et al. : Familial aggregation of urothelial cell carcinoma Int J Cancer 98:274–2782002 [DOI] [PubMed] [Google Scholar]
- 8.Murta-Nascimento C, Silverman DT, Kogevinas M, et al. : Risk of bladder cancer associated with family history of cancer: Do low-penetrance polymorphisms account for the increase in risk? Cancer Epidemiol Biomarkers Prev 16:1595–16002007 [DOI] [PubMed] [Google Scholar]
- 9.Plna K, Hemminki K: Familial bladder cancer in the National Swedish Family Cancer Database J Urol 166:2129–21332001 [PubMed] [Google Scholar]
- 10.Randi G, Pelucchi C, Negri E, et al. : Family history of urogenital cancers in patients with bladder, renal cell and prostate cancers Int J Cancer 121:2748–27522007 [DOI] [PubMed] [Google Scholar]
- 11.Czene K, Lichtenstein P, Hemminki K: Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database Int J Cancer 99:260–2662002 [DOI] [PubMed] [Google Scholar]
- 12.Lichtenstein P, Holm NV, Verkasalo PK, et al. : Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland N Engl J Med 343:78–852000 [DOI] [PubMed] [Google Scholar]
- 13.Dong C, Hemminki K: Modification of cancer risks in offspring by sibling and parental cancers from 2,112,616 nuclear families Int J Cancer 92:144–1502001 [PubMed] [Google Scholar]
- 14.Martin C, Leiser CL, O'Neil B, et al. : Familial cancer clustering in urothelial cancer: A population-based case-control study J Natl Cancer Inst 110:527–5332018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer AA, Graham S, Burnett WS, et al. : Familial aggregation of bladder cancer stratified by smoking status Epidemiology 2:145–1481991 [DOI] [PubMed] [Google Scholar]
- 16.Kiemeney LA, Sulem P, Besenbacher S, et al. : A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer Nat Genet 42:415–4192010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiemeney LA, Thorlacius S, Sulem P, et al. : Sequence variant on 8q24 confers susceptibility to urinary bladder cancer Nat Genet 40:1307–13122008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothman N, Garcia-Closas M, Chatterjee N, et al. : A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci Nat Genet 42:978–9842010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X, Ye Y, Kiemeney LA, et al. : Genetic variation in the prostate stem cell antigen gene PSCA confers susceptibility to urinary bladder cancer Nat Genet 41:991–9952009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galesloot TE, Grotenhuis AJ, Kolev D, et al. : Genome-wide meta-analysis identifies novel genes associated with recurrence and progression in non-muscle-invasive bladder cancer Eur Urol Oncol 10.1016/j.euo.2021.07.001[epub ahead of print on August 2, 2021] [DOI] [PubMed] [Google Scholar]
- 21.de Maturana EL, Rava M, Anumudu C, et al. : Bladder cancer genetic susceptibility. A systematic review Bladder Cancer 4:215–2262018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlo MI, Ravichandran V, Srinavasan P, et al. : Cancer susceptibility mutations in patients with urothelial malignancies J Clin Oncol 38:406–4142020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nassar AH, Abou Alaiwi S, AlDubayan SH, et al. : Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma Genet Med 22:709–7182020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.PLINK 1.9 home: https://www.cog-genomics.org/plink/1.9/
- 25.NCBI Gene: https://www.ncbi.nlm.nih.gov/gene
- 26.NCBI ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/variation/5058/evidence/
- 27.Loizidou MA, Neophytou I, Papamichael D, et al. : The mutational spectrum of Lynch syndrome in Cyprus PLoS One 9:e105501.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.NCBI dbSNP: https://www.ncbi.nlm.nih.gov/snp/rs35502531
- 29.Andersen SD, Liberti SE, Lutzen A, et al. : Functional characterization of MLH1 missense variants identified in Lynch syndrome patients Hum Mutat 33:1647–16552012 [DOI] [PubMed] [Google Scholar]
- 30.Gagné F: Chapter 6—Oxidative stress Gagné F.Biochemical Ecotoxicology Oxford, United Kingdom: Academic Press; 2014, pp 103–115 [Google Scholar]
- 31.Vander Heiden MG, DeBerardinis RJ: Understanding the intersections between metabolism and cancer biology Cell 168:657–6692017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.UniProt Knowledgebase: https://www.uniprot.org/uniprot/O96017
- 33.Meijers-Heijboer H, van den Ouweland A, Klijn J, et al. : Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations Nat Genet 31:55–592002 [DOI] [PubMed] [Google Scholar]
- 34.NCBI ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/variation/128042/
- 35.Genome Aggregation Database: https://gnomad.broadinstitute.org/variant/22-29091856-AG-A?dataset=gnomad_r2_1
- 36.Liang M, Zhang Y, Sun C, et al. : Association between CHEK2*1100delC and breast cancer: A systematic review and meta-analysis Mol Diagn Ther 22:397–4072018 [DOI] [PubMed] [Google Scholar]
- 37.Vahteristo P, Bartkova J, Eerola H, et al. : A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer Am J Hum Genet 71:432–4382002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.AlDubayan SH, Pyle LC, Gamulin M, et al. : Association of inherited pathogenic variants in checkpoint kinase 2 (CHEK2) with susceptibility to testicular germ cell tumors JAMA Oncol 5:514–5222019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Näslund-Koch C, Nordestgaard BG, Bojesen SE: Increased risk for other cancers in addition to breast cancer for CHEK2*1100delC heterozygotes estimated from the copenhagen general population study J Clin Oncol 34:1208–12162016 [DOI] [PubMed] [Google Scholar]
- 40.Złowocka E, Cybulski C, Górski B, et al. : Germline mutations in the CHEK2 kinase gene are associated with an increased risk of bladder cancer Int J Cancer 122:583–5862008 [DOI] [PubMed] [Google Scholar]
- 41.Fabbri L, Bost F, Mazure NM: Primary cilium in cancer hallmarks Int J Mol Sci 20:1336.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgins M, Obaidi I, McMorrow T: Primary cilia and their role in cancer Oncol Lett 17:3041–30472019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu H, Kiseleva AA, Golemis EA: Ciliary signalling in cancer Nat Rev Cancer 18:511–5242018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrew AS, Gui J, Sanderson AC, et al. : Bladder cancer SNP panel predicts susceptibility and survival Hum Genet 125:527–5392009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data will be deposited to the NLM NCBI database of Genotypes and Phenotypes (dbGaP), phs002326.v1.p1.