

Summary
The highest plateau on Earth, Qinghai-Tibet Plateau, contains thousands of lakes with broad salinity and diverse and unique microbial communities. However, little is known about their co-occurring viruses. Herein, we identify 4,560 viral Operational Taxonomic Units (vOTUs) from six viromes of three saline lakes on Qinghai-Tibet Plateau, with less than 1% that could be classified. Most of the predicted vOTUs were associated with the dominant bacterial and archaeal phyla. Virus-encoded auxiliary metabolic genes suggest that viruses influence microbial metabolisms of carbon, nitrogen, sulfur, and lipid; the antibiotic resistance mediation; and their salinity adaption. The six viromes clustered together with the ice core viromes and bathypelagic ocean viromes and might represent a new viral habitat. This study has revealed the unique characteristics and potential ecological roles of DNA viromes in the lakes of the highest plateau and established a foundation for the recognition of the viral roles in plateau lake ecosystems.
Subject areas: Ecology, Microbial genetics, Microbial metabolism, Viral microbiology
Graphical abstract
Highlights
-
•
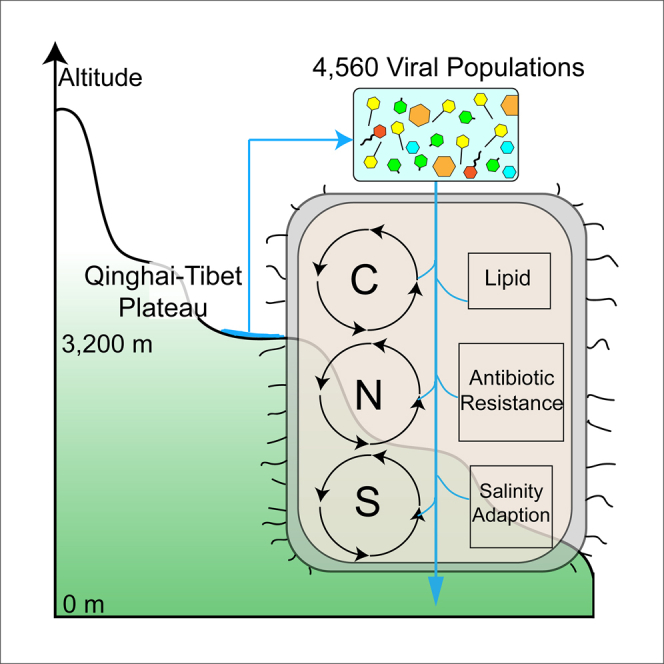
Of all, 4,560 viral populations were identified
-
•
Many virus-encoded auxiliary metabolic genes were predicted
-
•
Qinghai-Tibet plateau and bathypelagic ocean might represent a viral habitat
Ecology; Microbial genetics; Microbial metabolism; Viral microbiology
Introduction
High-altitude plateau areas, which among the least-explored terrestrial biospheres on Earth, are characterized by persistent low temperatures and very sensitive to global warming. The Qinghai-Tibet Plateau is the highest plateau in the world, with an average altitude over 4,000 m. It is the primary water source for south and east Asia (Immerzeel et al., 2010) and contains more than 32,000 lakes, with a total area of 44,993 km2, which comprise the world's largest alpine lake group (Zhang et al., 2011, 2014). In addition, the Qinghai-Tibet Plateau contains 36,793 glaciers, with a total area and volume of 49,873 km2 and 4,561 km3, respectively (Immerzeel et al., 2010; Yao, 2010). In recent decades, under the influence of global climate change, the glaciers of the Qinghai-Tibet Plateau have started to melt and retreat. The melting glacier waters have then increased the total area of lakes in this area (Liu et al., 2019a). Most of these lakes are remote from human disturbance. Together, they have broad environmental gradients, including in salinity (from 0.1 to over 400‰), pH (5.4–10.2), and altitude (Yang et al., 2016; Zheng and Liu, 2009), and provide a unique, natural, and extreme laboratory to study microbial variation and adaption to environmental changes.
Recently, the prokaryotic and eukaryotic communities of soil, sand, lakes, and wetlands on high plateaux, such as the Qinghai-Tibet Plateau, Yunnan Plateau, and Colorado Plateau, have been investigated using high-throughput sequencing techniques (Lee et al., 2016b; Li et al., 2017; Liu et al., 2019a, 2016; Yang et al., 2019b; Zhang et al., 2015, 2020). The composition and function of the microorganisms in these high-altitude environments were found to be significantly different from those of low-altitude environments and were also different between lakes of different environments (Ji et al., 2019; Yang et al., 2016). Microbial diversity in the high-altitude lakes decreased significantly with dilution from melting glaciers (Liu et al., 2019a), whereas the network complexity seemed to increase in the hypersaline lakes (Ji et al., 2019). In addition, abundant microbes in wetland soils of Qinghai-Tibet Plateau have better potential adaptability to environmental change than rarer ones (Wan et al., 2021). However, information on their corresponding viromes in these high-altitude plateau lakes remains rare, especially over 3,000 m.
Viruses are the most abundant and diverse “biological entities” on Earth (Suttle, 2005). They significantly influence the microbial community structure, alter metabolic output through virus-encoded auxiliary metabolic genes (vAMGs), mediate horizontal gene transfer, drive global biogeochemical cycles, and contribute to carbon sequestration through viral lysing and aggregation (Suttle, 2005, 2007). During the past decade, the importance of viruses in global biogeochemical cycles and the composition of the viral community at a genome level has been explored using metagenomics and high-throughput sequencing, covering ocean, soil, and low-altitude lake environments (Brum and Sullivan, 2015; Chow and Suttle, 2015; Coutinho et al., 2020; Gregory et al., 2019b; Jin et al., 2019). Recently, 2,314,329 uncultivated viral genomes were identified with their relationship to habitat type from 21,075 public datasets in IMG/VR v3 (Roux et al., 2021). Most of the understanding of these viruses has been reported from marine environments. Our knowledge of high-altitude plateaux viromes remains limited (Eissler et al., 2020; Zhong et al., 2020). In particular, until now, there has been no information on viromes at a genome level from lakes on the Qinghai-Tibet Plateau.
Here we use the metagenomic method to examine the genetic composition of viruses in three Qinghai-Tibet Plateau high-altitude lakes, Qinghai Lake (QhL), Da Qaidam Lake (DQL), and Xiao Qaidam Lake (XQL), which span a salinity gradient from 13 to 246. This study aims to characterize the viral assemblages by assessing viral diversity, virus-host interaction, and virus-encoded auxiliary metabolic genes (vAMGs) and explore their adaption to different salinities.
Results
Overview of six viromes of three lakes on Qinghai-Tibet Plateau
Six viral samples were collected from the surface waters of three lakes, with distinct environmental parameters, on the Qinghai-Tibet Plateau; this includes four samples around QhL, one from XQL and one from DQL (Figure 1). All samples were alkaline with pH values from 7.8 to 9.1. Salinities ranged from 246 in DQL, 79 in XQL, and from 13 to 17‰ in QhL (Table S1). In DQL, sodium and chloride ions accounted for about three-quarters of the total dissolved solids.
Figure 1.

Sampling stations and community structure of classified viral operational taxonomic units (vOTUs) of the six viromes of three lakes on Qinghai-Tibet Plateau
The pie charts show relative abundance on virus taxonomic composition, assessed for all vOTUs at the family level (except haloviruses) based on the results of CAT. The proportion of annotated vOTUs in all predicted vOTUs of each sample is shown below the names of stations.
Six viral metagenomic libraries of three lakes on Qinghai-Tibet Plateau were obtained after Illumina sequencing and contained about 72G bases raw data (a total of 239,539,878 raw paired-end reads [150bp each-end]). After quality control, these six viromes contained about 210 million paired clean reads. A total of 120,515 contigs longer than 1.5 kilobases were assembled. Combined with the prediction outputs of VirSorter, VirFinder, and CAT, a total of 4,835 viral contigs were detected (Gregory et al., 2019b; Ren et al., 2017; Roux et al., 2015; von Meijenfeldt et al., 2019). After redundant contigs were removed through alignment, 4,560 viral Operational Taxonomic Units (vOTUs) were classified (Table S2). Based on the results of VIBRANT, a fraction of the vOTUs were predicted to be temperate phages (Kieft et al., 2020). The proportion of lytic vOTUs in the four QhL samples (92.4%–98.2%) were lower than in XQL (98.7%) and DQL (99.2%, Figure S1), which was consistent with the salinity pattern.
Viral community structure
Shannon index values of the six viromes were calculated based on the relative abundance of vOTUs (Table S1); these varied from the lowest value of 4.03 at QhL1 to the highest of 6.88 at QhL10. However, the alpha diversities of the prokaryotic communities were different from those of the viruses. The sample from the hypersaline DQL had the lowest Shannon index value (0.80) and XQL sample had the highest (3.37) (Table S1, X. Li, Q. Liu, C. Zhang, X. Zhou, C. Gu, X. Yu, M. Wang, H. Shao, J. Li, Y. Jiang, unpublished data). The most abundant vOTUs differed between the three lakes (Figure S2) but the four viromes of QhL were more similar to each other than the other lakes.
At a family level, vOTUs were annotated using CAT, based on the lowest common ancestor (LCA) algorithm, and represented an average relative abundance of 10.9% (von Meijenfeldt et al., 2019). Siphoviridae (33.8%–74.0%) and Podoviridae (17.9%–55.9%) were the abundant families, although DQL mainly consisted of haloviruses (71%, Figure 1). This observation is in accordance with the dominance of Halobacteria in the prokaryotic community of DQL (Li Xianrong et al., unpublished data). In addition, Microviridae, a nonenveloped ssDNA virus, accounted for a moderate proportion of viruses (21.2% in QhL3, 1.0% in QhL10, and 11.8% in DQL; Figure 1) (Fard, 2016). Nucleocytoplasmic large DNA viruses (NCLDVs, mainly Mimiviridae and Phycodnaviridae) were detected in QhL3, QhL10, and XQL by CAT (Figure 1) and also detected in QhL10 and XQL by NCLDVs marker gene (major capsid protein, MCP) using hmmsearch (Figure S4B) (Mistry et al., 2013; Schulz et al., 2020; von Meijenfeldt et al., 2019). In addition, Lavidaviridae, virophages related to NCLDVs, was detected either by CAT (QhL3, QhL10, QhL11, and XQL) or by hmmsearch against virophage marker gene MCP (QhL1, QhL3, and QhL10) (Mistry et al., 2013; Paez-Espino et al., 2019; von Meijenfeldt et al., 2019). The relative abundances of NCLDVs and virophages were very low and less than 1% (Figure 1).
The representative sequences of vOTUs of six viromes were searched against NCBI RefSeq genomes of viruses using BLASTN to classify the vOTUs at species level. However, no hit met the threshold (≥95% average nucleotide identity [ANI], ≥ 85% alignment fraction, ≤ 1 × 10−3 e-value). In addition, the vOTUs were searched against the IMG/VR v3 dataset, which is the largest available public viral dataset consisting of over 2,314,329 uncultivated viral genomes viral sequences, and against the Global Ocean Viromes (GOV 2.0) dataset with over 190,000 viral populations, with the same threshold to test the distinctiveness of the six viromes (Gregory et al., 2019b; Roux et al., 2021). However, again there was no hit against GOV 2.0, and only 0.7% of the vOTUs were aligned with the IMG/VR v3 dataset, most of which were linked to vOTUs from nonmarine saline and alkaline, marine, and freshwater environments (Figure 2C).
Figure 2.

Taxonomic diversity of the six viromes of three lakes on Qinghai-Tibet Plateau
(A) Gene-sharing network of viral sequence based on sequences (≥10k) of the six viromes and IMG/VR v3 (seawater, freshwater, nonmarine saline, and alpine and terrestrial habitats). Nodes represent viral genomes, and edges indicate similarity based on shared protein clusters.
(B) Venn diagram of shared viral clusters among the five different environmental habitats.
(C) Assignment of vOTUs of the six viromes of three lakes on Qinghai-Tibet Plateau against IMG/VR v3.
To investigate the relationship between vOTUs of six viromes and viral sequences from other habitats, 261 viral contigs longer than 10k were selected and compared with over 6,000 reference and predicted complete viral genomes of seawater, freshwater, nonmarine saline and alkaline, and terrestrial environment in IMG/VR v3 dataset using vContact 2 (Figure 2A) (Bin Jang et al., 2019). Seventy-four viral clusters (VCs, with a very high probability of the same genus in a VC) were detected in the six viromes, whereas 14 VCs (18.9%) were not found in the other four habitats of the IMG/VR v3 dataset (Figure 2B) (Roux et al., 2021).
Phylogenetic analysis of viral contigs and complete and near-complete viral genomes
A total of 24 complete and four near-complete viral metagenome-assembled genomes (MAGs) were assembled from the six viromes, according to the thresholds of the GOV dataset (Tara Oceans Coordinators et al., 2016). Eighteen complete viral genomes were shorter than 10 kbp. The marker gene of the ssDNA Microviridae, the MCP gene, was recognized in 13 complete viral genomes using DIAMOND BLASTP against all Microviridae MCP and major coat protein downloaded from NCBI (Buchfink et al., 2015). The phylogenetic tree was constructed based on 1,418 MCP gene sequences of Microviridae (and major coat protein for Bullavirinae) (Figure 3A). Most MCP sequences of Microviridae were divided into four superclades, and the 13 sequences from the six viromes were all classified into superclade one. This superclade included all reported viruses in the Bullavirinae subfamily and did not include any virus in the Gokushovirinae subfamily, suggesting the close taxonomic relationship between the new-found microviruses of the six viromes and Bullavirinae. These results extended our knowledge of the diversity of Microviridae.
Figure 3.

Phylogenetic and comparative genomics-based synteny analysis of microviruses
(A) The phylogenetic tree of maximum likelihood was generated from the major capsid protein. Only the nodes with the bootstrap test value higher than 95 were shown. The star indicated the new-found microviruses detected in the six viromes of three lakes on Qinghai-Tibet Plateau.
(B) Comparative genomics-based synteny analysis of 13 microviruses from the viromes and one reference genome sequence and relative abundance of microviruses of the viromes.
To further understand the genomic similarity of superclade one of Microviridae, 24 microvirus genomes were chosen to compare the ANI using OrthoANI (Lee et al., 2016a). A heatmap of ANI showed that most genomes in the same clade were homologous (Figure S9). DQL_55, QhL10_5914, and QhL3_4981 were very similar with an ANI higher than 99%, which is the same for DQL_44, QhL10_5533, and QhL3_4669, suggesting that these genomes might represent two species of Microviridae. Therefore, three microviruses (DQL_55 group, DQL_44 group, and QhL3_4806) were classified into clade 1–2. In addition, because of their high ANI values, three sequences (DQL_40, QhL10_5371, and QhL3_4528) might represent the same species, and two other sequences (DQL_151 and QhL3_7004) could be an additional species. Furthermore, the virome from the XQL was different from those of the other two lakes, and no complete microvirus genome was detected. Alignment between the 13 complete genomes and the reference (Antarctic microvirus TYR_006_V_SP_13) is shown in Figure 3B (Sommers et al., 2019). All 14 genomes had three conserved genes, including MCP, DNA pilot protein, and replication initiation protein. The results of alignment were consistent with the ANI results (Figure S9).
To further investigate the diversity of the two main groups of dsDNA viruses, Caudovirales and NCLDVs, phylogenetic trees, based on the gene sequences of Caudovirales terminase large subunit and NCLDVs MCP, are shown in Figure S4. A total of 281 phage terminase sequences was detected from the six viromes of salt lakes. The phylogenetic tree shows that most of putative phage terminase in the high diversity were closely related to Siphoviridae and Myoviridae (Figure S4A). Also, many viral clades consisting of terminase have only been detected in these six viromes, which suggests that these might represent an endemic Qinghai-Tibet Plateau Caudovirales taxa.
By comparison, only 14 NCLDVs MCP sequences were detected in QhL and XQL and none in hypersaline DQL. The phylogenetic tree shows that most NCLDVs of the saline lake viromes were close to several algal viruses belonging to Phycodnaviridae, such as Micromonas sp. Rcc1109 virus mpv1, Phaeocystis globosa virus 12T, Bathycoccos sp. RCC1105 virus BpV1, and Yellowstone lake phycodnavirus 3 (Figure S4B).
Host prediction of viruses
After comparing sequence similarity, tRNA sequences, CRISPR spacers, and oligonucleotide frequencies between prokaryotic metagenome-assembled genomes and vOTUs, putative hosts were predicted for 358 vOTUs of the six viromes (Figure 4), accounting for a relative abundance of 13.5% (Li et al., 2021). Most of the predicted vOTUs had narrow host ranges, with only 11 potentially exhibiting a broader host range across several classes. Interestingly, 25 vOTUs in the DQL were linked to the dominant archaea, Halobacteria, but no vOTUs were predicted to link to archaeal host in the other two lakes. Predicted prokaryotic hosts spanned one archaeal and 17 bacterial class-level taxa, with Alphaproteobacteria (34 vOTUs, accounting for 35.5% relative abundance of predicted vOTUs) being the most frequently predicted (Figure 4). The most abundant vOTU of the six viromes (vOTU_1374) was also linked to Alphaproteobacteria, which was the dominant prokaryotic class in QhL. Most of the vOTUs were linked to Proteobacteria and Bacteroidetes, which were the dominant prokaryotes in QhL and XQL at phylum level.
Figure 4.

Host prediction of viral operational taxonomic units (vOTUs) of the six viromes of three lakes on Qinghai-Tibet Plateau against prokaryotic metagenome-assembled genomes
Undirected virus-host prediction network with nodes that represent vOTUs (gray small nodes) and their putative hosts (big nodes), colored on the basis of taxonomy at the class level. Edges represent in silico virus-host linkages and are colored on the basis of host. If a node of vOTU is connected to different host nodes, it means this vOTU is predicted to link to different host classes, indicating potential broad host range.
Focusing on the 1,244 most abundant vOTUs with a cumulative relative abundance of 80% (Figure S2A), putative hosts were predicted for 123 with a relative abundance of 15.4%. Fifteen of 123 vOTUs were classified into Podoviridae and haloviruses. Most of the 123 vOTUs were predicted to link to Bacteroidetes (41), Gammaproteobacteria (23), and Halobacteria (23). In addition, vOTU_13 with a broad putative host range and linking to Alphaproteobacteria and Betaproteobacteria, coexisted in DQL and QhL. vOTU_285 with a putative host (Bacteroidetes) was detected in XQL and QhL.
Functional analysis of protein clusters
A total of 27,321 putative open reading frames (ORFs) in the six viromes of saline lakes were detected using metaProdigal (Hyatt et al., 2010). According to the NCBI COG functional categories of genes annotated by eggNOG-Mapper (Figure S5), genes with an unknown function were the most abundant annotated ORFs (Huerta-Cepas et al., 2017). In addition, genes dealing with information storage and processing and genes related to replication, recombination, and repair were also abundant categories.
To analyze the functional diversity of genes of viruses in the six viromes, all-verses-all BLAST was run by DIAMOND against all predicted genes (Buchfink et al., 2015; Zhao et al., 2013). This method detected reciprocal best-BLAST hits and received putative orthologs information, which was used to classify protein clusters (PCs). A total of 22,367 PCs were classified (Figure 5A). However, only 3.67% of PCs were shared among different lakes and no PC was detected in all three lakes, which suggests that there were considerable differences in virus functional genes between the different lakes. The diversity of viral PCs in the six viromes was the highest in the XQL virome, which is about 3.6 times higher than in the DQL virome (Figure 5A).
Figure 5.

Functional protein clusters of viral contigs of the six viromes of three lakes on Qinghai-Tibet Plateau
(A) Venn graph of viral protein clusters of three different lakes.
(B) KEGG functional categories of different viromes of three saline lakes.
(C) Number of protein clusters of different categories of virus-encoded auxiliary metabolic genes.
To better understand the functional genes in the six viromes, the relative abundance of each functional gene in each sample detected by GhostKOALA, except the group “not included in pathway or brite”, is shown in Figure 5B (Kanehisa et al., 2016). In brief, only a small proportion of the PCs (less than 15%) were annotated as genes with classified functions. The three categories of “BRITE hierarchies,” “metabolism,” and “genetic information processing” were all abundant (Figure 5B). The category “BRITE hierarchies”, which contains noncoding RNAs and protein families of metabolism, genetic information processes, and signaling and cellular processes, was the most abundant category, except in QhL3.
Virus-encoded auxiliary metabolic genes
Several kinds of vAMGs were annotated after comparing with different databases; these mainly included osmoregulation, carbohydrate-active, nitrogen metabolism, sulfur metabolism, lipid metabolism, and antibiotic resistance genes (Figure 5C). Carbohydrate active and lipid metabolism genes were the most diverse vAMGs. For carbohydrate-active genes, no polysaccharide lyases or carbohydrate esterases were detected in the six viromes. Glycoside hydrolases, which hydrolyze or rearrange glycosidic bonds and glycosyltransferases to form glycosidic bonds, were more diverse in QhL than the other two lakes. Interestingly, three adjacent glycoside hydrolase genes were found in the viral contig DQL-84, suggesting the potential viral mediation of carbohydrate metabolism of the host cells. For nitrogen metabolism, three tandem glutamine synthetase genes were detected in the viral contig QhL10-122, suggesting the potential for viral mediation of glutamine metabolism in the host cells. Moreover, no nitrogen metabolism genes were found in the DQL virome and no sulfur metabolism genes were detected in the viromes from QhL. Furthermore, three associated sox genes, L-cysteine S-thiosulfotransferase (soxA), S-sulfosulfanyl-L-cysteine sulfohydrolase (soxB), and sulfur-oxidizing protein (soxZ), were detected in the viral contig XQL-17618, which shows its potential mediation ability involving sulfur oxidation in the host cells. Many kinds of lipid metabolism genes were detected, especially for fatty acid biosynthesis and glycerophospholipid metabolism (Figure 5C). These genes might contribute to the formation of the components of the host's cell membrane or the virus envelope.
Interestingly, some antibiotic resistance genes, relating to different mechanisms of resistance to antibiotics, were detected in XQL and QhL, such as antibiotic efflux, antibiotic inactivation, and antibiotic target alteration (Figure 5C). In addition, many viral osmoregulation genes were detected in the six viromes, which might be beneficial for osmotic pressure regulation in the host. However, the lower the salinity of the lake, the more kinds of glucan exporter associated genes were detected. None were detected in the hypersaline DQL virome, whereas one was found in the XQL viromes and three in QhL.
Ribosomal genes and potential CRISPR-like sequence
Twenty different ribosomal genes were encoded by viruses in QhL and XQL but none were detected in DQL (Figure S6). These genes, such as L1 and S1, are different parts of different ribosomal components, but they did not consist of an integrated component.
A putative CRISPR-like sequence was detected from one near-complete viral genome (XQL_135) (Figure S7A). In the predicted ORF20 of XQL_135, a CRISPR-like sequence consisting of nine similar repeats and eight spacers was detected. However, not only the repeats but also the spacers had no hits with the IMG/M dataset. Using BLASTP hits against NCBI nr database, XQL_135 was found to be most similar to Lake Baikal phage Baikal-20-5-C28. Both viral genomes had a low GC content and a similar gene order (Cabello-Yeves et al., 2017). There was no ortholog fragment between the ORF20 of XQL_135 and Baikal-20-5-C28, whereas some sequences before and after this ORF contained reciprocal orthologs with Baikal-20-5-C28. According to the tetranucleotide analysis, the region of CRISPR-like sequence in ORF20 of XQL_135 had lower tetranucleotide adaptability compared with the whole sequence (Figure S7B), whereas the corresponding region in Baikal-20-5-C28 had a higher self-adaptability (Figure S7C). XQL_135 contained four T4-like genes (terminase large subunit T4 headful, portal protein Gp20, RNA polymerase sigma factor Gp55, and DNA polymerase clamp loader subunit Gp62), suggesting its close relationship with genus “T4-like viruses” of the Myoviridae family. No tRNA sequences were detected in XQL_135, although six were found in Baikal-20-5-C28. In addition, five carbohydrate-associated vAMGs were detected in XQL_135, whereas vAMGs associated with the iron-sulfur cluster were detected in Baikal-20-5-C28 (Figure S7).
Relationship with environmental factors
Canonical correspondence analysis (CCA) was conducted to examine the relationship between the viral communities and environmental factors (Figure 6B). The results show that the four QhL viromes were clustered together, whereas viromes of the other two lakes were separate; this is similar to the result of PCoA analysis (Figure 6A). After forward selection, four environmental factors, elevation, latitude, and concentrations of magnesium and calcium, were found to be related to variability in the viral community structure (P < 0.05). The QhL viromes were mainly correlated with elevation, whereas the DQL virome correlated with magnesium concentration, and the XQL virome was mainly correlated with calcium concentration (Figure 6B).
Figure 6.

Comparisons among the six viromes of three lakes on Qinghai-Tibet Plateau and other environmental viromes
(A) Principal co-ordinate analysis (PCoA) of the six viromes of six viromes.
(B) Canonical correspondence analysis (CCA) of the relationship between viral populations and environmental factors.
(C) Nonmetric multidimensional scaling (NMDS) analysis of the six viromes of three lakes on Qinghai-Tibet Plateau and other environmental viromes. According to habitats, viromes are divided into four groups, bathypelagic (BATHY), other marine (Epi and Meso), Qinghai-Tibetan Plateau (QTP), and other terrestrial (LAND). Group BATHY is inside Group QTP.
Pearson's correlation analysis was used to examine the relationships between viral community structure, environmental factors, and prokaryotic alpha diversity (Shannon Index) (Figure S3). Viral alpha diversity indices were not significantly correlated with any factor (P > 0.05), although the correlation coefficient between prokaryotic and viral alpha diversity was relatively high (R = 0.775).
Comparison between the six viromes and other environmental viromes
To compare the six viromes of three lakes on Qinghai-Tibet Plateau with other environmental viromes, a nonmetric multidimensional scaling (NMDS) analysis was conducted, which showed that the 40 virome samples could be divided into three groups (Figure 6C). The first group consisted of viromes from the Arctic, Antarctic, temperate, and tropical epi- (0–200 meters below surface, mbs) and mesopelagic (200–1000 mbs) zones in GOV 2.0 (Gregory et al., 2019b), whereas the second group contained nonmarine viromes and XQL viromes. The third group included the high-altitude aquatic viromes from the Qinghai-Tibet Plateau (four viromes from QhL and the viromes from DQL), two viromes from northwestern Qinghai-Tibet Plateau glaciers, and ten viromes from the bathypelagic zone (1000–4000 mbs) of the ocean (Gregory et al., 2019b; Zhong et al., 2020). In addition, the DQL virome is similar to a virome from the alpine, hypersaline Lake Salar de Uyuni in Bolivia, South America (Figure 6C) (Eissler et al., 2020; Ramos-Barbero et al., 2019).
Discussion
Although metagenomic analyses greatly expanded our understanding of viromes, our knowledge of high-altitude saline lake viral communities is still sparse. In this study, 4,560 vOTUs with 24 complete and four near-complete viral MAGs from six viromes of three saline lakes on the Qinghai-Tibet Plateau were detected. Almost all vOTUs of the six viromes were not cultured, based on a comparison of results with the NCBI viral RefSeq genomes dataset, GOV 2.0, and IMG/VR v3 dataset (Gregory et al., 2019b; Roux et al., 2021). These vOTUs contained many vAMGs associated with biogeochemical cycles, ARGs, and the osmotic pressure regulation of host cells. Multivariate analysis showed that the viromes from the Qinghai-Tibet Plateau were significantly different from viromes of land and epi- and mesopelagic oceans. Overall, the Qinghai-Tibet Plateau probably harbors many unique viruses that had not been found in other natural environments because of the unique alpine and saline-alkaline aquatic environments.
Qinghai-Tibet Plateau harbor unique alpine and saline-alkaline viral assemblages
No identified hit was found for the six viromes of three lakes on Qinghai-Tibet Plateau against the NCBI viral RefSeq genomes dataset and GOV 2.0. On average, only 0.7% vOTU sequences could be found in the IMG/VR v3 dataset, which included diverse habitats, such as freshwater, different marine zones, and nonmarine saline and alpine waters (Figure 2B) (Roux et al., 2021). These results suggest that these viromes were distinct from currently reported viromes from other environments and reflect the inadequate research and absence of datasets of viromes from high-altitude lakes, especially from the Qinghai-Tibet Plateau. The proportion of lysogenic vOTUs in the saline lakes was less than 7% in the six viromes (Figure S1) and decreased with increasing salinity, suggesting that the viruses inhabiting these alpine, saline-alkaline lakes prefer a lytic life strategy.
Only a few viral sequences of the six viromes could be assigned to a taxonomic rank, which is similar to previous viral metagenomic studies (Liang et al., 2019; Roux et al., 2016; Yang et al., 2019a). Caudovirales were abundant in QhL and XQL, which had low to moderate salinities (Ackermann, 1998). This distribution is similar to other environments, including seawater, freshwater lakes, soil, and marine sediments (Fancello et al., 2013; Jasna et al., 2018; Kim et al., 2017; Parmar et al., 2018; Segobola et al., 2018; Skvortsov et al., 2016; Yang et al., 2019a; Zhang et al., 2021a; Zheng et al., 2021). However, viruses of DQL, a hypersaline lake, were unique and dominated by haloviruses (Figure 1). Hypersaline environments are a reservoir of viruses with diverse shapes and have the highest reported abundance in water (up to 1010 mL−1) (Boujelben et al., 2012; Santos et al., 2012). However, only a limited number of tailed haloviruses have been isolated from hypersaline environments, which is not in accordance with the reports based on transmission electron microscope observations (Atanasova et al., 2015; Santos et al., 2012). This paradox suggests that previous reports may be incomplete and biased because only isolated haloviruses were analyzed in hypersaline viromes. Viruses within Siphoviridae, with long and noncontractile tails, were the most abundant viral family in the saline lakes (Figure 1) and many areas of seawater (Jasna et al., 2018; Suttle, 2005; Zhang et al., 2021a). Siphoviruses tend to infect host cells with a lysogenic life strategy and are not often the most dominant viral family in natural aquatic environments (Parmar et al., 2018; Suttle, 2005). In contrast, myoviruses, with contractile, long tails and a relatively broader host range, are more commonly observed as the dominant family in aquatic environment (Parmar et al., 2018; Suttle, 2005; Zhang et al., 2021a). However, Myoviridae was relatively rare in the six viromes (Figure 1); this suggests the unique environmental characteristics of lakes on the Qinghai-Tibet Plateau, such as the presence of a broad salinity gradient and high altitude, might shape the host communities. So the presence of viruses with a broad host range would not be conducive to precise control of the host community, which would be supported by the results of host's prediction of viruses (Figure 4) (Ji et al., 2019; Liu et al., 2016, 2019b; Wang et al., 2011; Yang et al., 2016).
Some NCLDVs were detected in XQL and QhL10, which had moderate salinities (Figure 1); most of these were similar to phycodnaviruses that infect microalgae (Figure S4B) (Schulz et al., 2020). This occurrence pattern is similar to marine viral communities (Yang et al., 2019a). The NCLDV supergroup is commonly abundant and diverse in oceans but can also be found in freshwater and associated with most major eukaryotic lineages; it might be the most important contributor to the control of primary production in hypersaline environments despite the low biomass (Schulz et al., 2020; Uritskiy et al., 2019). However, only a limited number of NCLDVs have been isolated from hypersaline environments (Atanasova et al., 2015). This study has expanded the number of NCLDVs found in hypersaline environments.
In addition, without multiple displacement amplification (MDA) some short putative ssDNA complete genomes were detected and 13 complete microvirus genomes were selected for phylogenetic and comparative genomic analysis. The ssDNA viruses are abundant in environments, such as sediments and the gut (Creasy et al., 2018; Gregory et al., 2019a; Yoshida et al., 2018; Yu et al., 2018). ssDNA viruses that have a small genome, only approximately 2–15 kb, may encode only a single structural protein and a single protein involved in their DNA replication (O'Carroll and Rein, 2016). The Microviridae contains two subfamilies, Gokushovirinae and Bullavirinae (Cherwa and Fane, 2011). Unlike dsDNA Caudovirales, no visible tails could be detected on microviruses (Cherwa and Fane, 2011). By comparison with MCP genes of microviruses in GenBank, four major superclades were clustered on the phylogenetic tree (Figure 3A). New microviruses in the six viromes and the isolated bullaviruses were classified into superclade one, without any gokushoviruses, suggesting a close relationship between these new-found microviruses and the Bullavirinae subfamily. Three annotated coding sequences, MCP, DNA pilot protein, and replication initiation protein, were detected in all selected microviruses (Figure 3B); this suggests that these microviruses probably had a common ancestor. Our study defined and expanded the diversity of Microviridae to a global scale.
From the host prediction of viruses, abundant vOTUs were linked to the dominant bacterial and archaeal populations. Three hundred fifty-eight of 4,560 vOTUs could be linked to putative hosts, which represents 13.5% of the viral abundance in the six viromes of three lakes. This abundance is lower than in other environments, such as cold, deep-sea seep sediments, suggesting the uniqueness of the viral and microbial communities in the Qinghai-Tibet Plateau lakes (Li et al., 2021). Although most predicted vOTUs were linked to Bacteroidetes (90), Gammaproteobacteria (73), and other bacteria (51), the most abundant vOTUs (35.5% in relative abundance) were linked to Alphaproteobacteria, including the most abundant vOTU (vOTU_1374). Alphaproteobacteria, which contains many important and abundant microbes, can utilize terrestrial-derived dissolved organic matter, especially that related to CHO and CHOS mixtures (Kang et al., 2013, p. 116; Yang et al., 2020; Zhao et al., 2013, p. 11). These results suggest that these abundant viruses linked to Alphaproteobacteria might have an important role in Qinghai-Tibetan Plateau lakes’ biogeochemical cycles.
In addition, 25 vOTUs linked to the dominant archaea, Halobacteria, were detected in DQL, the hypersaline lake (Figure 4). This observation is consistent with the community structure of viruses and prokaryotes in DQL (Figure 1, Li Xianrong et al., unpublished data), suggesting that DQL harbors special archaeal viruses.
Potential virus-mediated biogeochemical cycling, antibiotic resistance, and osmotic pressure regulation of host cells
In this study, 21 different categories of vAMGs related to carbon, nitrogen, sulfur, and lipid metabolism were detected. As viruses can carry and express some vAMGs to directly mediate the metabolism of the host cells, such as genes involved in photosynthesis, phosphate removal, and nitrogen metabolism, and indirectly affect biogeochemical cycles (Brum and Sullivan, 2015; Zimmerman et al., 2020), the presence of vAMGs suggests that viruses might mediate the metabolism of host cells and influence biogeochemical cycles in this area. Glycoside hydrolases, which hydrolyze or rearrange glycosidic bonds and glycosyltransferases to form glycosidic bonds, were more diverse in QhL than the other two lakes. The presence of these amino acids suggests that the viruses in the Qinghai-Tibet Plateau lakes might participate in carbon cycling through the mediation of carbohydrate synthesis and decomposition metabolism of the host cells by gene expression of vAMGs. In these lakes, viruses might mediate the carbon cycle, through lysis of the host cells, i.e. the “viral shunt,” increasing aggregation and vertical carbon flux to deeper waters by viral lysates, named the “viral shuttle,” and manipulation of the hosts' metabolic pathways by the expression of virus-encoded carbohydrate-associated genes (Sullivan et al., 2017; Suttle, 2007; Zimmerman et al., 2020).
Interestingly, viral antibiotic resistance genes (ARGs) were detected in the six viromes, indicating that viruses might help their host survive environmental antibiotics introduced by human activities (Moon et al., 2020; Yang et al., 2019b). Previous studies have shown that viruses can be key vehicles of ARG transfer in bacteria. This might be a significant threat to global health, as viral ARGs are common in urban water, oceans, and animal feces (Debroas and Siguret, 2019). ARGs have been observed in the wetlands across the Qinghai-Tibet Plateau but with lower abundance than in coastal estuaries and sites associated with human activity (Yang et al., 2019b). It is suggested that viruses with these genes might help the host cell survive the relatively high concentrations of antibiotics. The antibiotic concentrations in QhL (1.14–17.3 ng/L) were higher than in input rivers (Li et al., 2020; Yang et al., 2018). These results suggest that the viruses on the pristine Qinghai-Tibet Plateau are an important gene pool for ARGs, and so special attention should be paid to the usage of antibiotics in this area.
Viruses might also provide assistance to host cells for the osmotic pressure regulation, which is important for bacterial adaption to saline environments (Singh et al., 2013). Different virus-encoded osmoregulation genes were detected in saline lakes (Figure 6). These genes could help their host adapt to changing osmotic pressure in two different ways. The first approach, “salt-in-cytoplasm mechanism,” is to accumulate salt to match the environmental osmolarity (Kunte, 2006; Ma et al., 2010). The second approach, “organic-osmolyte mechanism,” requires highly water-soluble and uncharged organic compounds to maintain an osmotic equilibrium with the surrounding environment (Kunte, 2006; Ma et al., 2010). Interestingly, three and one vAMG encoding glucan exporter ATP-binding protein was detected in QhL, with low osmotic pressure, and in XQL, with moderate osmotic pressure, respectively, suggesting that viruses might help host cells to export glucan from the cytoplasm to periplasm to adapt to the low osmotic pressure (Lu et al., 2020).
Qinghai-Tibet Plateau harbors viral-encoded ribosomal protein genes and CRISPR-like sequence
Twenty virus-encoded elements of ribosomal proteins, which might control host transcription, were found (Figure S6). These genes, which have been detected in viral genomes, might replace some components of host ribosomes to moderate host translation and might give a functional fitness advantage during phage evolution (Mizuno et al., 2019).
One putative CRISPR-like sequence, consisting of nine similar repeats and eight spacers, was detected from the near-complete viral genome XQL_135 (ORF20), which might belong to the “T4-like virus” genus of the Myoviridae family; it was most similar to the Lake Baikal phage Baikal-20-5-C28 (Figure S7) (Cabello-Yeves et al., 2017; Chen et al., 2019). However, neither repeats nor spacers had hits with the IMG/M dataset and Baikal-20-5-C28, whereas some sequences before and after the ORF20 contained reciprocal orthologs with Baikal-20-5-C28. This indicates that the putative CRISPR-like sequence might be one of the inserted or removed parts of their common ancestor. According to the tetranucleotide analysis, the region of the CRISPR-like sequence in ORF20 of XQL_135 had lower tetranucleotide adaptability compared with the whole sequence, whereas the corresponding region in Baikal-20-5-C28 had a higher self-adaptability, suggesting that this region in XQL_135 might have been inserted later (Duhaime et al., 2011).
Qinghai-Tibet Plateau might represent a viral ecological zone
Marine viral communities are separated into five distinct viral ecological zones (VEZs), according to GOV 2.0 (Gregory et al., 2019b). However, few comparisons have been made between the five marine VEZs and terrestrial viromes, especially from alpine and saline-alkaline lakes. Here we compared the similarity of the six viromes, two viromes from glacier ice of the Qinghai-Tibet Plateau, eight other nonmarine viromes, and 24 viromes of GOV 2.0 (Adriaenssens et al., 2017; Coutinho et al., 2020; Eissler et al., 2020; Gregory et al., 2019b; Skvortsov et al., 2016; Zhong et al., 2020). According to the NMDS analysis and hierarchical clustering, the viromes could be divided into three groups. The first group consisted of viromes from the Arctic, Antarctic, temperate and tropical epipelagic zones, and mesopelagic zones in GOV 2.0, whereas the second group contained other nonmarine viromes and XQL viromes. However, the other seven Qinghai-Tibet Plateau viromes and the bathypelagic viromes clustered together forming the third group. As these represent the highest and lowest parts of the Earth, it is very interesting that viruses from these two areas are clustered together. Given the great environmental differences between the Qinghai-Tibet Plateau and the bathypelagic zone, it is speculated that viruses of these two environments might either have similar origins or possess adaption strategies to the cold and high-/low-pressure environments, and these two environments together might constitute a VEZ for viruses. However, the virome from XQL, which was assigned to another group, was closed to other QTP viromes (Figure 6C) and might represent a transitional state from the VEZ to other aquatic environments.
A study of marine viral communities has found that temperature is one of the most important environmental factors influencing viruses (Gregory et al., 2019b). The main factor influencing the planktonic viral communities in estuaries, however, was salinity (Zhang et al., 2021a). Four environmental factors, elevation, latitude, and concentrations of magnesium and calcium, were related to the variations of viral community structures. Elevation and latitude contributed to the variation of temperature, which might be an important factor affecting viral diversity. Alternatively, the concentrations of calcium and magnesium ions varied between lakes. Adsorption, as the first step of viral infection cycles, is influenced by divalent cations, and so calcium ions might inhibit the combination of phage QHHSV-1 with its host Halomonas ventosae QH52-2 (Fu et al., 2016). Thus, magnesium and calcium ions might further influence viruses through adsorption and therefore the beginning of the infection cycle (Shao and Wang, 2008).
Conclusions
Lakes on the Qinghai-Tibet Plateau harbor diverse and unique microbes, yet their associated viruses and influences on lake microbiomes have not yet been identified. In this study, for the first time, six viromes from three lakes with altitude above 3,000 m on the Qinghai-Tibet Plateau are revealed. The unique high-altitude plateau, which contains lakes spanning a broad salinity gradient, results in a unique and distinctive viral community and distribution. Almost all vOTUs of alpine saline lakes were different from those of other environments and most had predicted hosts that were linked to bacteria, whereas 25 linked to the dominant archaea, Halobacteria, which was only detected in the DQL, the hypersaline lake. Phylogenetic analysis expanded our knowledge of the Caudovirales, NCLDVs, and redefined the ssDNA Microviridae on a global scale. The life strategies of the viruses inhabiting the high-altitude plateau seem to indicate that they have adopted a lytic life cycle and potentially help their host adapt to osmotic pressure through the expression of virus-encoded osmoregulation genes. It also suggests that the six viromes of three lakes on Qinghai-Tibet Plateau might be delineated separately from land and epi- and mesopelagic ocean viromes and form a VEZ with bathypelagic ocean viromes. These data begin to fill the information gap on viruses from lakes on the highest Qinghai-Tibet Plateau and shed light on their potential influences on the associated microbiomes. Future studies expanding viral spatial-temporal dynamics of more lakes with deeper sequencing, covering a larger area and longer time span on the Qinghai-Tibet Plateau, will provide a better understanding of viral diversity and co-evolution and interactions with host cells.
Limitations of the study
In this study, only six viromes of three lakes were analyzed. Overall, little is known about viral communities in lakes on the whole plateau. In addition, viral communities in these lakes at different depths of different seasons are not known. Future studies about viral spatial-temporal dynamics of more lakes would help us understand viral diversity on Qinghai-Tibet Plateau comprehensively.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Min Wang (mingwang@ouc.edu.cn)
Materials availability
This study did not generate new unique reagents.
Method details
Experimental model and subject details
Six samples were collected form QhL (4 surface stations), XQL (1 surface) and DQL (1 surface), three saline lakes in the Qinghai Province of China, during May 2019 (Figure 1). The methods used for measuring environmental factors and prokaryotic alpha diversity based on 16s amplicons were as before (Xiong et al., 2012). Descriptions of all six samples is shown in Table S1.
In brief, six lake water samples, 50L each, were collected for the following study.
Virus concentration, DNA extraction and metagenomic sequencing
Lake water samples were collected and filtered through 3.0 μm and 0.2 μm polycarbonate membrane filters (Millipore, MA, USA). 50 L of the filtrate was added to 2.5 mL of 10 g/L Fe Stock (FeCl3) Solution. After shaking and settling, the mixed water was filtered through 0.8 μm polycarbonate membrane filters (Millipore, MA, USA). The 0.8 μm polycarbonate membrane filters were stored at – 80°C (John et al., 2011).
The viral particles on the filters were resuspended in fresh 0.1 M EDTA-0.2 M MgCl2 buffer (pH 6.0) at 4°C. Viruses were concentrated by centrifugal ultrafiltration into 400 μL. The QIAamp DNA Mini Kit (QIAGEN) was used to extract DNA from the concentrated solution following manufacturer handling instructions. Library construction and next generation sequencing of the viral DNA was carried out by Novogene (Nanjing, China) using Illumina NovaSeq 6000 (pair-end sequencing, 2 × 150 bp). In this study, the RNA component of the viromes was not considered as only DNA was extracted.
Quality control of reads, assembly and identification
The clean reads were picked from the raw reads by Novogene. Then, the high-quality paired-end reads were filtered using cutadapt and Perl scripts using the following conditions: (1) without N; (2) no more than 20% bases with a quality score less than 20; (3) no more than 30% bases with a quality score less than 30 (Martin, 2017; Yang et al., 2019a).
High-quality reads in each sample were assembled using metaSPAdes (v 3.12.0). Contigs which are less than 500 bp were removed (Nurk et al., 2017). MetaQUAST (v 5.0.2) was used to evaluate these metagenomic assemblies (Mikheenko et al., 2016). Then, contigs ≥ 1.5 kb were piped through VirFinder and VirSorter (v 1.0.6) and contigs that sorted with a VirFinder score ≥ 0.7 and p < 0.05 or VirSorter categories 1-6 were selected for further viral identification (Ren et al., 2017; Roux et al., 2015). The contigs with the following conditions were considered as viral contigs: (1) contigs sorted by VirFinder with a score ≥ 0.9 and p < 0.05; (2) contigs sorted by VirSorter in categories 1 and 2; (3) contigs sorted by VirFinder with a score ≥ 0.7 and p < 0.05 and VirSorter categories 1-6; (4) the remaining contigs were run through CAT (v 5.0.3) and contigs with CAT results of superkingdom not classified and the viruses were selected (Gregory et al., 2019b; von Meijenfeldt et al., 2019).
Dereplication, calculating relative abundances and taxonomic profiling
Viral contigs of each sample were piped through CD-hit-est (v 4.8.1) separately (Fu et al., 2012). Viral contigs were considered to be the same viral population if they shared ≥ 95% nucleotide identity across ≥ 80% of short viral contigs and the longest contig in each population was considered as a representative sequence. In order to calculate the relative abundances of viral populations in each sample, clean reads were mapped to representatives of the viral population using bowtie2 and counted using SAMtools (v 1.9) (Langmead and Salzberg, 2012; Li et al., 2009). Then, relative abundances of viral populations were calculated as transcripts per million (TPM) reads mapped (Li et al., 2010).
The drafts of taxonomic information of vOTUs were generated by CAT (von Meijenfeldt et al., 2019). As the CAT software found some NCLDVs and Lavidaviridae, all predicted genes of vOTUs were searched against NCLDV and virophage major capsid with Hidden Markov Models using hmmsearch (v 3.1b2, -E 1e-10) separately to test the results (Paez-Espino et al., 2019; Schulz et al., 2020).
VIBRANT (v 1.2.1) was used to detect the lifestyle of viruses with the default parameters (except -virome) to filter some contigs which were hard to detect (Kieft et al., 2020).
Identification of shared vOTUs
The viral populations in all six samples were piped through CD-hit-est in the same way as dereplication. Viral populations in different samples were considered to have shared vOTUs if they shared ≥ 95% nucleotide identity across ≥ 80%, while other viral populations were considered as individual vOTUs.
Virus–host prediction
Microbiome sequencing was performed by Shanghai Biozeron Biotechnology Co., Ltd. (Shanghai, China.) using 0.2 μm polycarbonate membrane filters (Li Xianrong et al., unpublished data). Reads from the quality control (by Trimmomatic v 0.36) were assembled using metaSPAdes (Bolger et al., 2014; Nurk et al., 2017). Contigs were binned (parameters: --metabat2 --maxbin2 --concoct) and refined (parameters: -c 70 -x 5) using metaWRAP (v 1.3.2) (Uritskiy et al., 2018). Taxonomy of each prokaryotic MAG was assigned by CAT (von Meijenfeldt et al., 2019).
Four different in silico methods were used to predict virus-host linkages, nucleotide sequence homology, oligonucleotide frequency, tRNA match and CRISPR spacer match (Li et al., 2021). (1) Sequences of vOTUs were compared with prokaryotic MAGs using BLASTN (≥70% minimum nucleotide identity, ≥75% coverage of viral sequences, ≥50 bit score and ≤0.001 e-value). (2) Sequences of vOTUs and prokaryotic MAGs were piped into VirHostMatcher (v1.0) to detect linkages (d2∗ values ≤0.2) (Ahlgren et al., 2017). (3) ARAGORN (v 1.2.38) was used to identify tRNAs from sequences of vOTUs and prokaryotic MAGs (parameters: -t) (Laslett, 2004). Each Match required ≥90% length with identity ≥90% of the sequences by reciprocal BLASTN (Coutinho et al., 2017; Zhao et al., 2013, p. 11). (4) CRISPR arrays were assembled from prokaryotic quality-controlled reads by crass (v 1.0.1) and CRISPR spacers were compared with viral contigs using BLASTN with ≤1 mismatch over the whole spacer (Li et al., 2021; Skennerton et al., 2013). For each match, BLASTN was used to compare the repeat and prokaryotic MAGs with the same parameters.
Comparison between the six viromes and other environmental viromes
To examine the relationship between six viromes of three lakes on Qinghai-Tibet Plateau and other environmental viral communities, 34 viral metagenomes from different environmental habitats were selected and download from NCBI Sequence Read Archive (SRA) data (https://www.ncbi.nlm.nih.gov/sra/) and CyVerse Data Commons (https://datacommons.cyverse.org/) (Table S3, Figure S8). Frequencies of di-, tri- and tetranucleotides, calculated by Python script.
The vOTUs were BLASTN (BLAST+ v 2.9.0) searched against the NCBI virus reference genomes with 95% identity, 85% of alignment length and an evalue of 0.001 were used to modify the drafts (Roux et al., 2019). In addition, vOTUs were blasted to the Integrated Microbial Genome/Virus (IMG/VR) system v.3 dataset with the same threshold in order to obtain further information (Figure 2C) (Roux et al., 2021). The vOTUs of the viromes were also BLASTN against GOV 2.0 contigs, over 10 k and circular, with the same threshold.
All vOTUs more than 10 k were piped into vConTACT 2 to with 113 reference genomes of freshwater, 3679 of seawater, 69 terrestrial viral reference genomes and 2187 complete, high quality viral sequences with IMG/VR v3 (Roux et al., 2021). All open reading frames (ORFs) were predicted by metaProdigal (v 2.6.3) (Hyatt et al., 2010). All proteins were compared using all-verses-all DIAMOND (v 0.9.29.130) BLASTP (e-value ≤1 × 10−5, query coverage ≥50%, identity ≥25%) (Buchfink et al., 2015; Zhang et al., 2021b). vConTACT 2 was then used to calculate the network (--db 'None' --pcs-mode MCL --vcs-mode ClusterONE) (Bin Jang et al., 2019). The network is illustrated using Gephi (v 0.9.2) (Figure 2A) (Bastian et al., 2009). The number of VCs were plotted using the VennDiagram in R (Figure 2B) (Chen and Boutros, 2011).
Open reading frames prediction and functional analyses
ORFs were recognized from vOTUs in each sample using metaProdigal with default parameters (Hyatt et al., 2010). To remove the contamination of microbial fragments, the non-viral ORFs in the contigs with provirus predicted by VIBRANT were removed (Kieft et al., 2020). Relative abundance of ORFs was calculated with the relative abundance of vOTUs.
ORFs were compared to the NCBI nonredundant protein database to evaluate the percentage of known genes using DIAMOND (v 0.9.29.130) BLASTP (Buchfink et al., 2015). In order to obtain the proportion of various functional genes, ORFs were piped through eggNOG V5.0 (http://eggnog5.embl.de/#/app/home) to identify COG functional classifications with default parameters, through PfamScan (e value < 1 × 10−5; bit score > 40) to identify domains and hidden Markov models (HMMs) and through KEGG GhostKOALA (https://www.kegg.jp/ghostkoala/) and KEGG KAAS (https://www.genome.jp/tools/kaas/) to predict KEGG Orthology pathways (Huerta-Cepas et al., 2017, 2019; Kanehisa et al., 2016; Mistry et al., 2013; Moriya et al., 2007; Tara Oceans Coordinators et al., 2016). ORFs were BLASTP against Carbohydrate-active enzymes (CAZy) database (http://www.cazy.org/) to find glycol-metabolism related genes using DIAMOND (e value < 1 × 10−5; > 60% identity with > 60% length of the query amino acid sequence) (Cao et al., 2020; Lombard et al., 2014). RGI (v 5.1.1) was run locally to detect antibiotic resistance genes excluding loose hits but nudging more than 95% identity loose hits to strict based on CARD reference data (Alcock et al., 2019). For further analysis of function, all ORFs were RPSBLAST against Conserved Domain Database (CDD) to annotate (Lu et al., 2020). All ORFs were all-verses-all BLAST to detect reciprocal BLAST hits (e value < 1 × 10−5; > 60% identity with > 60% length of the query amino acid sequence) (Cao et al., 2020; Zhao et al., 2013, p. 11). Query sequence and subject sequence of each reciprocal BLAST hit were considered to belong to the same protein cluster with the same function. Heatmaps of predicted ORFs function were plotted by TBtools (Chen et al., 2020).
Analysis of complete and near-complete viral genomes
All circular contigs detected by VirSorter and VIBRANT were viewed as complete viral genomes and all contigs more than 50 k with terminase detected by PfamScan and eggNOG-mapper were considered as near-complete viral genomes (Huerta-Cepas et al., 2017; Kieft et al., 2020; Roux et al., 2015; Tara Oceans Coordinators et al., 2016). These sequences were piped into Rapid Annotation using Subsystem Technology (RAST) (https://rast.nmpdr.org/) to annotate genome features (Overbeek et al., 2014).
MetaCRT (modified from CRT1.2) was used to detect CRISPR-like components of these high quality draft genomes (Rho et al., 2012). Only XQL-135 had a CRISPR-like sequence and not only repeaters but also spacers had no identical hit when BLASTN against RefSeq, CRISPR databases using CRISPRs web server and IMG/M (Chen et al., 2019; Grissa et al., 2007). All genes of XQL-135 predicted by prodigal were aligned against NCBI-nr database using DIAMOND to obtain the best hit of each gene (Buchfink et al., 2015). TBLASTX was run to detect homology parts plotted by Easyfig with Lake Baikal phage Baikal-20-5m-C28 (Cabello-Yeves et al., 2017; Sullivan et al., 2011). Tetranucleotide frequencies of two sequences were calculated by Python script with 10 kbp window size and 1 kbp step size and were normalized by z-scoring (Duhaime et al., 2011). The Pearson’s correlation coefficient was calculated by script from either the array of each fragment and the whole genome.
In order to obtain more credible predicted ORFs of shorter complete genomes, all complete genomes shorter than 10k were upload to GeneMarkS (http://topaz.gatech.edu/GeneMark/genemarks.cgi, sequence type: virus) (Besemer, 2001). These predicted protein sequences were aligned against all Microviridae MCPs and major coat proteins downloaded from NCBI using DIAMOND (e value < 1e-10) and two protein sequences at the beginning and end of the same genomes were overlapped if both of them had hits. 13 genomes of predicted microviruses were overlapped and adjusted to positive sequences starting at predicted MCP genes using the Sequence Manipulation Suite Version 2 (Stothard, 2000). All downloaded sequences dereplicated on the amino acid sequence combined with 13 new-found MCP genes were aligned with MAFFT (v 7.471) (auto mode) and trimmed using trimAL (v 1.4.rev15) to remove position with gaps over 90% (Capella-Gutierrez et al., 2009; Katoh and Standley, 2016; Schulz et al., 2020). The maximum-likelihood phylogenetic tree was computed by IQ-TREE (v 2.0.3) after choosing model (LG+F+R10) (Minh et al., 2020). The phylogenetic tree was visualized with iTOL v5 (https://itol.embl.de/) (Letunic and Bork, 2021).
24 genomes of microviruses were chosen to compare ANI using OrthoANI, which was plotted by TBtools (Chen et al., 2020; Lee et al., 2016a).
Phylogenetic analysis of Caudovirales and NCLDVs
All terminase sequences over 300 amino acids of Caudovirales were downloaded from NCBI virus and dereplicated. Terminase genes of the six viromes of three lakes on Qinghai-Tibet Plateau were detected by BLASTP using DIAMOND (-e 1 × 10−5) against downloaded sequences (Buchfink et al., 2015). Sequences with less than 300 amino acids were removed.
The MCP sequences of the six viromes were detected by hmmsearch (-E 1e-10) against MCP genes of NCLDVs dataset (Schulz et al., 2020). Sequences less than 100 amino acids were removed.
Dereplicated sequences combined with reference sequences were aligned with MAFFT (v 7.471) (auto mode) and trimmed using trimAL (v 1.4.rev15) to remove position with gaps over 90% (Capella-Gutierrez et al., 2009; Katoh and Standley, 2016; Schulz et al., 2020). The maximum-likelihood phylogenetic tree was computed by IQ-TREE (v 2.0.3) after choosing model (terminase: LG+F+R10; MCP: LG+F+R8) (Minh et al., 2020). The phylogenetic tree was visualized with iTOL v5 (Letunic and Bork, 2021).
Quantification and statistical analysis
The α- diversity (Shannon’s H) and β- diversity (Bray-Curtis dissimilarity) statistics were performed using vegan (v 2.5-6) in Rstudio (v 1.2.5001; R Stats v 3.6.1), based on the relative abundances of vOTUs (Dixon, 2003). PCoA based on Bray-Curtis dissimilarity matrices was conducted using cmdscale (k = 3, eig=T) in Rstudio. Figures were generated by ggplot2 (v 3.2.1) (Figure 6A). CCA was conducted by Canoco 5 with iterative selection of environmental factors (Figure 6B) (Jiangshan, 2014). The Pearson’s correlation between environmental factors and viral alpha diversity was calculated using IBM SPSS Statistics v 25.
Oligonucleotide frequencies of 40 viromes were piped into Rstudio and analyzed to compute euclidian distance and hierarchical cluster using vegan and pvclust (v 2.2-0) (method.hclust = “average”, method.dist = “cor”, nboot = 1000) libraries respectively (Gregory et al., 2019b; Roux et al., 2014; Suzuki and Shimodaira, 2006). The boundaries of groups were based on approximately unbiased (AU) p-value threshold 0.70 using function pvrect. The non-metric multidimensional scaling was based on frequencies using the metaMDS function (distance = “euclidean”, k = 2, trymax = 1000) and plotted using ggplot2 (Figure 6C).
Acknowledgments
The research was funded by the Marine Scientific and Technological Innovation Project financially supported by Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018SDKJ0406-6), National Key Research and Development Program of China (2018YFC1406704), Research Funds for the Central Universities (202072002 and 201812002), Talent project of Youth Innovation Promotion Association, Chinese Academy of Sciences (2020428), and Natural Science Foundation of China (42120104006, 42176111, 41976117, 41972258). The work was carried out at Marine Big Data Center of Institute for Advanced Ocean Study of Ocean University of China. We appreciate the computing resources provided by IEMB-1, a high-performance computing cluster operated by the Institute of Evolution and Marine Biodiversity. We also thank for the support of the high-performance servers of Frontiers Science Center for Deep Ocean Multispheres and Earth System. We thank for the support of the high-performance servers of Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao).
Author contributions
Min Wang, Yantao Liang, and Jiwei Tian: Conceptualization and Project administration. Chengxiang Gu, Yantao Liang, and Min Wang: Methodology, Software, Formal analysis, Visualization, and Writing—Original Draft. Jiansen Li, Hongbing Shao, Yong Jiang, Xinhao Zhou, Xianrong Li, and Wenjing Zhang: Resources and Investigation. Cui Guo, Hui He, and Hualong Wang: Data Curation. Yeong Yik Sung, Wen Jye Mok, Li Lian Wong, Andrew McMinn, Curtis A Suttle, and Jiwei Tian: Writing—Review and Editing.
Declaration of interests
The authors declare that they have no competing interests.
Published: December 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103439.
Contributor Information
Yantao Liang, Email: liangyantao@ouc.edu.cn.
Jiwei Tian, Email: tianjw@ouc.edu.cn.
Min Wang, Email: mingwang@ouc.edu.cn.
Supplemental information
Data and code availability
All original code describing the data analysis process are available on GitHub at https://github.com/amitaleth/lake-virome. The clean reads data reported in this paper have been deposited in the Genome Sequence Archive (Wang et al., 2017) in National Genomics Data Center (CNCB-NGDC Members and Partners et al., 2021), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences, under Bioproject number PRJCA005626 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ackermann H.-W. Tailed bacteriophages: the order Caudovirales. Adv. Virus Res. 1998;51:135–201. doi: 10.1016/S0065-3527(08)60785-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriaenssens E.M., Kramer R., Van Goethem M.W., Makhalanyane T.P., Hogg I., Cowan D.A. Environmental drivers of viral community composition in Antarctic soils identified by viromics. Microbiome. 2017;5:83. doi: 10.1186/s40168-017-0301-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlgren N.A., Ren J., Lu Y.Y., Fuhrman J.A., Sun F. Alignment-free $d_2ˆ∗$ oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res. 2017;45:39–53. doi: 10.1093/nar/gkw1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcock B.P., Raphenya A.R., Lau T.T.Y., Tsang K.K., Bouchard M., Edalatmand A., Huynh W., Nguyen A.-L.V., Cheng A.A., Liu S., et al. Card 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019:gkz935. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasova N.S., Oksanen H.M., Bamford D.H. Haloviruses of archaea, bacteria, and eukaryotes. Curr. Opin. Microbiol. 2015;25:40–48. doi: 10.1016/j.mib.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Bastian M., Heymann S., Jacomy M. Gephi: an open source software for exploring and manipulating networks. Proc. Int. AAAI Conf. Web Soc. Media. 2009;3:361–362. [Google Scholar]
- Besemer J. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin Jang H., Bolduc B., Zablocki O., Kuhn J.H., Roux S., Adriaenssens E.M., Brister J.R., Kropinski A.M., Krupovic M., Lavigne R., et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019;37:632–639. doi: 10.1038/s41587-019-0100-8. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boujelben I., Yarza P., Almansa C., Villamor J., Maalej S., Antón J., Santos F. Virioplankton community structure in Tunisian solar Salterns. Appl. Environ. Microbiol. 2012;78:7429–7437. doi: 10.1128/AEM.01793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum J.R., Sullivan M.B. Rising to the challenge: accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015;13:147–159. doi: 10.1038/nrmicro3404. [DOI] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Cabello-Yeves P.J., Zemskaya T.I., Rosselli R., Coutinho F.H., Zakharenko A.S., Blinov V.V., Rodriguez-Valera F. Genomes of novel microbial lineages assembled from the Sub-ice waters of lake Baikal. Appl. Environ. Microbiol. 2017;84 doi: 10.1128/AEM.02132-17. e02132–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S., Zhang W., Ding W., Wang M., Fan S., Yang B., Mcminn A., Wang M., Xie B., Qin Q.-L., et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome. 2020;8:47. doi: 10.1186/s40168-020-00826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Boutros P.C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.-M.A., Chu K., Palaniappan K., Pillay M., Ratner A., Huang J., Huntemann M., Varghese N., White J.R., Seshadri R., et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2019;47:D666–D677. doi: 10.1093/nar/gky901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Chen H., Zhang Y., Thomas H.R., Frank M.H., He Y., Xia R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020;13:1194–1202. doi: 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- Cherwa J.E., Fane B.A. In: Microviridae : Microviruses and Gokushoviruses. John Wiley & Sons, Ltd, editor. John Wiley & Sons, Ltd; 2011. p. a0000781. pub2. [DOI] [Google Scholar]
- Chow C.-E.T., Suttle C.A. Biogeography of viruses in the sea. Annu. Rev. Virol. 2015;2:41–66. doi: 10.1146/annurev-virology-031413-085540. [DOI] [PubMed] [Google Scholar]
- CNCB-NGDC Members and Partners, Xue Y., Bao Y., Zhang Z., Zhao W., Xiao J., He S., Zhang G., Li Y., Zhao G., et al. Database resources of the National genomics data center, China National center for Bioinformation in 2021. Nucleic Acids Res. 2021;49:D18–D28. doi: 10.1093/nar/gkaa1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho F.H., Silveira C.B., Gregoracci G.B., Thompson C.C., Edwards R.A., Brussaard C.P.D., Dutilh B.E., Thompson F.L. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017;8:15955. doi: 10.1038/ncomms15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho F.H., Cabello-Yeves P.J., Gonzalez-Serrano R., Rosselli R., López-Pérez M., Zemskaya T.I., Zakharenko A.S., Ivanov V.G., Rodriguez-Valera F. New viral biogeochemical roles revealed through metagenomic analysis of Lake Baikal. Microbiome. 2020;8:163. doi: 10.1186/s40168-020-00936-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasy A., Rosario K., Leigh B., Dishaw L., Breitbart M. Unprecedented diversity of ssDNA phages from the family Microviridae detected within the gut of a Protochordate model organism (Ciona robusta) Viruses. 2018;10:404. doi: 10.3390/v10080404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debroas D., Siguret C. Viruses as key reservoirs of antibiotic resistance genes in the environment. ISME J. 2019;13:2856–2867. doi: 10.1038/s41396-019-0478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003;14:927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- Duhaime M.B., Wichels A., Waldmann J., Teeling H., Glöckner F.O. Ecogenomics and genome landscapes of marine Pseudoalteromonas phage H105/1. ISME J. 2011;5:107–121. doi: 10.1038/ismej.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissler Y., Dorador C., Kieft B., Molina V., Hengst M. Virus and potential host microbes from viral-Enriched metagenomic characterization in the high-altitude wetland, salar de Huasco, Chile. Microorganisms. 2020;8:1077. doi: 10.3390/microorganisms8071077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancello L., Trape S., Robert C., Boyer M., Popgeorgiev N., Raoult D., Desnues C. Viruses in the desert: a metagenomic survey of viral communities in four perennial ponds of the Mauritanian Sahara. ISME J. 2013;7:359–369. doi: 10.1038/ismej.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fard R.M.N. A short introduction to bacteriophages. Trends Pept. Protein Sci. 2016;1:7. [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C.-Q., Zhao Q., Li Z.-Y., Wang Y.-X., Zhang S.-Y., Lai Y.-H., Xiao W., Cui X.-L. A novel Halomonas ventosae-specific virulent halovirus isolated from the Qiaohou salt mine in Yunnan, Southwest China. Extremophiles. 2016;20:101–110. doi: 10.1007/s00792-015-0802-x. [DOI] [PubMed] [Google Scholar]
- Gregory A.C., Zablocki O., Howell A., Bolduc B., Sullivan M.B. The human gut virome database (preprint) Bioinformatics. 2019 doi: 10.1101/655910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A.C., Zayed A.A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C., et al. Marine DNA viral Macro- and Microdiversity from Pole to Pole. Cell. 2019;177:1109–1123.e14. doi: 10.1016/j.cell.2019.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I., Vergnaud G., Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J., Forslund K., Coelho L.P., Szklarczyk D., Jensen L.J., von Mering C., Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017;34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund S.K., Cook H., Mende D.R., Letunic I., Rattei T., Jensen L.J., et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D., Chen G.-L., LoCascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immerzeel W.W., van Beek L.P.H., Bierkens M.F.P. Climate change will affect the Asian water Towers. Science. 2010;328:1382–1385. doi: 10.1126/science.1183188. [DOI] [PubMed] [Google Scholar]
- Jasna V., Parvathi A., Dash A. Genetic and functional diversity of double-stranded DNA viruses in a tropical monsoonal estuary, India. Sci. Rep. 2018;8:16036. doi: 10.1038/s41598-018-34332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M., Kong W., Yue L., Wang J., Deng Y., Zhu L. Salinity reduces bacterial diversity, but increases network complexity in Tibetan Plateau lakes. FEMS Microbiol. Ecol. 2019;95:fiz190. doi: 10.1093/femsec/fiz190. [DOI] [PubMed] [Google Scholar]
- Jiangshan L. Canoco 5: a new version of an ecological multivariate data ordination program. Biodivers. Sci. 2014;21:765–768. doi: 10.3724/SP.J.1003.2013.04133. [DOI] [Google Scholar]
- Jin M., Guo X., Zhang R., Qu W., Gao B., Zeng R. Diversities and potential biogeochemical impacts of mangrove soil viruses. Microbiome. 2019;7:58. doi: 10.1186/s40168-019-0675-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S.G., Mendez C.B., Deng L., Poulos B., Kauffman A.K.M., Kern S., Brum J., Polz M.F., Boyle E.A., Sullivan M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation: virus concentration by flocculation with iron. Environ. Microbiol. Rep. 2011;3:195–202. doi: 10.1111/j.1758-2229.2010.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Sato Y., Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016;428:726–731. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Kang I., Oh H.-M., Kang D., Cho J.-C. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc. Natl. Acad. Sci. U S A. 2013;110:12343–12348. doi: 10.1073/pnas.1219930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics. 2016;32:1933–1942. doi: 10.1093/bioinformatics/btw108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft K., Zhou Z., Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020;8:90. doi: 10.1186/s40168-020-00867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Van Bonn W., Aw T.G., Rose J.B. Aquarium viromes: viromes of human-managed aquatic systems. Front. Microbiol. 2017;8:1231. doi: 10.3389/fmicb.2017.01231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte H.J. Osmoregulation in bacteria: compatible solute accumulation and osmosensing. Environ. Chem. 2006;3:94. doi: 10.1071/EN06016. [DOI] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I., Ouk Kim Y., Park S.-C., Chun J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016;66:1100–1103. doi: 10.1099/ijsem.0.000760. [DOI] [PubMed] [Google Scholar]
- Lee K.C., Archer S.D.J., Boyle R.H., Lacap-Bugler D.C., Belnap J., Pointing S.B. Niche filtering of bacteria in soil and rock habitats of the Colorado Plateau Desert, Utah, USA. Front. Microbiol. 2016;7:1489. doi: 10.3389/fmicb.2016.01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021:gkab301. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 genome project data processing Subgroup, 2009. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Ruotti V., Stewart R.M., Thomson J.A., Dewey C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Adams J., Shi Y., Wang H., He J.-S., Chu H. Distinct Soil Microbial Communities in habitats of differing soil water balance on the Tibetan Plateau. Sci. Rep. 2017;7:46407. doi: 10.1038/srep46407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Kuang Y., Hu J., You M., Guo X., Gao Q., Yang X., Chen Q., Sun W., Ni J. Enrichment of antibiotics in an inland lake water. Environ. Res. 2020;190:110029. doi: 10.1016/j.envres.2020.110029. [DOI] [PubMed] [Google Scholar]
- Li Z., Pan D., Wei G., Pi W., Zhang C., Wang J.-H., Peng Y., Zhang L., Wang Y., Hubert C.R.J., et al. Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. ISME J. 2021:1–13. doi: 10.1038/s41396-021-00932-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Wang L., Wang Z., Zhao J., Yang Q., Wang M., Yang K., Zhang L., Jiao N., Zhang Y. Metagenomic analysis of the diversity of DNA viruses in the surface and deep sea of the south China sea. Front. Microbiol. 2019;10:1951. doi: 10.3389/fmicb.2019.01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Hou W., Dong H., Wang S., Jiang H., Wu G., Yang J., Li G. Distribution and diversity of Cyanobacteria and eukaryotic Algae in Qinghai–Tibetan lakes. Geomicrobiol. J. 2016;33:860–869. doi: 10.1080/01490451.2015.1120368. [DOI] [Google Scholar]
- Liu K., Liu Y., Han B.-P., Xu B., Zhu L., Ju J., Jiao N., Xiong J. Bacterial community changes in a glacial-fed Tibetan lake are correlated with glacial melting. Sci. Total Environ. 2019;651:2059–2067. doi: 10.1016/j.scitotenv.2018.10.104. [DOI] [PubMed] [Google Scholar]
- Liu K., Yao T., Liu Y., Xu B., Hu A., Chen Y. Elevational patterns of abundant and rare bacterial diversity and composition in mountain streams in the southeast of the Tibetan Plateau. Sci. China Earth Sci. 2019;62:853–862. doi: 10.1007/s11430-018-9316-6. [DOI] [Google Scholar]
- Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucl. Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Wang J., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R., Gwadz M., Hurwitz D.I., Marchler G.H., Song J.S., et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–D268. doi: 10.1093/nar/gkz991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Galinski E.A., Grant W.D., Oren A., Ventosa A. Halophiles 2010: life in saline environments. AEM. 2010;76:6971–6981. doi: 10.1128/AEM.01868-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2017;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Mikheenko A., Saveliev V., Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2016;32:1088–1090. doi: 10.1093/bioinformatics/btv697. [DOI] [PubMed] [Google Scholar]
- Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J., Finn R.D., Eddy S.R., Bateman A., Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41:e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno C.M., Guyomar C., Roux S., Lavigne R., Rodriguez-Valera F., Sullivan M.B., Gillet R., Forterre P., Krupovic M. Numerous cultivated and uncultivated viruses encode ribosomal proteins. Nat. Commun. 2019;10:752. doi: 10.1038/s41467-019-08672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon K., Jeon J.H., Kang I., Park K.S., Lee K., Cha C.-J., Lee S.H., Cho J.-C. Freshwater viral metagenome reveals novel and functional phage-borne antibiotic resistance genes. Microbiome. 2020;8:75. doi: 10.1186/s40168-020-00863-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll I.P., Rein A. Encyclopedia of Cell Biology. Elsevier; 2016. Viral nucleic acids; pp. 517–524. [DOI] [Google Scholar]
- Overbeek R., Olson R., Pusch G.D., Olsen G.J., Davis J.J., Disz T., Edwards R.A., Gerdes S., Parrello B., Shukla M., et al. The SEED and the Rapid annotation of microbial genomes using Subsystems Technology (RAST) Nucl. Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D., Zhou J., Roux S., Nayfach S., Pavlopoulos G.A., Schulz F., McMahon K.D., Walsh D., Woyke T., Ivanova N.N., et al. Diversity, evolution, and classification of virophages uncovered through global metagenomics. Microbiome. 2019;7:157. doi: 10.1186/s40168-019-0768-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar K., Dafale N., Pal R., Tikariha H., Purohit H. An insight into phage diversity at environmental habitats using comparative metagenomics approach. Curr. Microbiol. 2018;75:132–141. doi: 10.1007/s00284-017-1357-0. [DOI] [PubMed] [Google Scholar]
- Ramos-Barbero M.D., Martínez J.M., Almansa C., Rodríguez N., Villamor J., Gomariz M., Escudero C., Rubin S.D.C., Antón J., Martínez-García M., et al. Prokaryotic and viral community structure in the singular chaotropic salt lake Salar de Uyuni. Environ. Microbiol. 2019;21:2029–2042. doi: 10.1111/1462-2920.14549. [DOI] [PubMed] [Google Scholar]
- Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017;5:69. doi: 10.1186/s40168-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M., Wu Y.-W., Tang H., Doak T.G., Ye Y. Diverse CRISPRs Evolving in human microbiomes. PLoS Genet. 2012;8:e1002441. doi: 10.1371/journal.pgen.1002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Tournayre J., Mahul A., Debroas D., Enault F. Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics. 2014;15:76. doi: 10.1186/1471-2105-15-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Enault F., Hurwitz B.L., Sullivan M.B. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015;3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Enault F., Ravet V., Colombet J., Bettarel Y., Auguet J.-C., Bouvier T., Lucas-Staat S., Vellet A., Prangishvili D., et al. Analysis of metagenomic data reveals common features of halophilic viral communities across continents: halovirus genomes are homogeneous across continents. Environ. Microbiol. 2016;18:889–903. doi: 10.1111/1462-2920.13084. [DOI] [PubMed] [Google Scholar]
- Roux S., Adriaenssens E.M., Dutilh B.E., Koonin E.V., Kropinski A.M., Krupovic M., Kuhn J.H., Lavigne R., Brister J.R., Varsani A., et al. Minimum information about an uncultivated virus genome (MIUViG) Nat. Biotechnol. 2019;37:9. doi: 10.1038/nbt.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Páez-Espino D., Chen I.-M.A., Palaniappan K., Ratner A., Chu K., Reddy T.B.K., Nayfach S., Schulz F., Call L., et al. IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021;49:D764–D775. doi: 10.1093/nar/gkaa946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F., Yarza P., Parro V., Meseguer I., Rosselló-Móra R., Antón J. Culture-independent approaches for studying viruses from hypersaline environments. Appl. Environ. Microbiol. 2012;78:1635–1643. doi: 10.1128/AEM.07175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz F., Roux S., Paez-Espino D., Jungbluth S., Walsh D.A., Denef V.J., McMahon K.D., Konstantinidis K.T., Eloe-Fadrosh E.A., Kyrpides N.C., et al. Giant virus diversity and host interactions through global metagenomics. Nature. 2020;578:432–436. doi: 10.1038/s41586-020-1957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segobola J., Adriaenssens E., Tsekoa T., Rashamuse K., Cowan D. Exploring viral diversity in a unique South African soil habitat. Sci. Rep. 2018;8:111. doi: 10.1038/s41598-017-18461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y., Wang I.-N. Bacteriophage adsorption rate and optimal lysis time. Genetics. 2008;180:471–482. doi: 10.1534/genetics.108.090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.P., Raval V., Purohit M.K. In: Plant Acclimation to Environmental Stress. Tuteja N., Singh Gill S., editors. Springer New York; 2013. Strategies for the salt tolerance in bacteria and archeae and its implications in developing crops for adverse conditions; pp. 85–99. [DOI] [Google Scholar]
- Skennerton C.T., Imelfort M., Tyson G.W. Crass: identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 2013;41:e105. doi: 10.1093/nar/gkt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsov T., de Leeuwe C., Quinn J.P., McGrath J.W., Allen C.C.R., McElarney Y., Watson C., Arkhipova K., Lavigne R., Kulakov L.A. Metagenomic characterisation of the viral community of Lough Neagh, the largest freshwater lake in Ireland. PLoS One. 2016;11:e0150361. doi: 10.1371/journal.pone.0150361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommers P., Fontenele R.S., Kringen T., Kraberger S., Porazinska D.L., Darcy J.L., Schmidt S.K., Varsani A. Single-stranded DNA viruses in antarctic cryoconite holes. Viruses. 2019;11:1022. doi: 10.3390/v11111022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P. The sequence manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques. 2000;28:1102–1104. doi: 10.2144/00286ir01. [DOI] [PubMed] [Google Scholar]
- Sullivan M.J., Petty N.K., Beatson S.A. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M.B., Weitz J.S., Wilhelm S. Viral ecology comes of age: crystal ball. Environ. Microbiol. Rep. 2017;9:33–35. doi: 10.1111/1758-2229.12504. [DOI] [PubMed] [Google Scholar]
- Suttle C.A. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- Suttle C.A. Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- Suzuki R., Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- Tara Oceans Coordinators, Roux S., Brum J.R., Dutilh B.E., Sunagawa S., Duhaime M.B., Loy A., Poulos B.T., Solonenko N., Lara E., et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature. 2016;537:689–693. doi: 10.1038/nature19366. [DOI] [PubMed] [Google Scholar]
- Uritskiy G.V., DiRuggiero J., Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uritskiy G., Tisza M.J., Gelsinger D.R., Munn A., Taylor J., DiRuggiero J. Cellular life from the three domains and viruses are transcriptionally active in a hypersaline desert community (preprint) Microbiology. 2019;7:3401–3417. doi: 10.1101/839134. [DOI] [PubMed] [Google Scholar]
- von Meijenfeldt F.A.B., Arkhipova K., Cambuy D.D., Coutinho F.H., Dutilh B.E. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 2019;20:217. doi: 10.1186/s13059-019-1817-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W., Gadd G.M., Yang Y., Yuan W., Gu J., Ye L., Liu W. Environmental adaptation is stronger for abundant rather than rare microorganisms in wetland soils from the Qinghai-Tibet Plateau. Mol. Ecol. 2021;30:2390–2403. doi: 10.1111/mec.15882. [DOI] [PubMed] [Google Scholar]
- Wang J., Soininen J., Zhang Y., Wang B., Yang X., Shen J. Contrasting patterns in elevational diversity between microorganisms and macroorganisms: patterns in elevational diversity. J. Biogeogr. 2011;38:595–603. doi: 10.1111/j.1365-2699.2010.02423.x. [DOI] [Google Scholar]
- Wang Y., Song F., Zhu J., Zhang S., Yang Y., Chen T., Tang B., Dong L., Ding N., Zhang Q., et al. GSA: genome sequence archive. Genomics Proteomics Bioinformatics. 2017;15:14–18. doi: 10.1016/j.gpb.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J., Liu Y., Lin X., Zhang H., Zeng J., Hou J., Yang Y., Yao T., Knight R., Chu H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012;14:2457–2466. doi: 10.1111/j.1462-2920.2012.02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Ma L., Jiang H., Wu G., Dong H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 2016;6:25078. doi: 10.1038/srep25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Song W., Lin H., Wang W., Du L., Xing W. Antibiotics and antibiotic resistance genes in global lakes: a review and meta-analysis. Environ. Int. 2018;116:60–73. doi: 10.1016/j.envint.2018.04.011. [DOI] [PubMed] [Google Scholar]
- Yang Q., Gao C., Jiang Y., Wang M., Zhou X., Shao H., Gong Z., McMinn A. Metagenomic characterization of the viral community of the south Scotia ridge. Viruses. 2019;11:95. doi: 10.3390/v11020095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liu G., Ye C., Liu W. Bacterial community and climate change implication affected the diversity and abundance of antibiotic resistance genes in wetlands on the Qinghai-Tibetan Plateau. J. Hazard Mater. 2019;361:283–293. doi: 10.1016/j.jhazmat.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Yang J., Jiang H., Liu W., Huang L., Huang J., Wang B., Dong H., Chu R.K., Tolic N. Potential utilization of terrestrially derived dissolved organic matter by aquatic microbial communities in saline lakes. ISME J. 2020;14:2313–2324. doi: 10.1038/s41396-020-0689-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T. Glacial fluctuations and its impacts on lakes in the southern Tibetan Plateau. Chin. Sci. Bull. 2010;55:2071. doi: 10.1007/s11434-010-4327-5. [DOI] [Google Scholar]
- Yoshida M., Mochizuki T., Urayama S.-I., Yoshida-Takashima Y., Nishi S., Hirai M., Nomaki H., Takaki Y., Nunoura T., Takai K. Quantitative viral community DNA analysis reveals the dominance of single-stranded DNA viruses in offshore upper bathyal sediment from Tohoku, Japan. Front. Microbiol. 2018;9:75. doi: 10.3389/fmicb.2018.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.-T., Han L.-L., Zhang L.-M., He J.-Z. Diversity and distribution characteristics of viruses in soils of a marine-terrestrial Ecotone in east China. Microb. Ecol. 2018;75:375–386. doi: 10.1007/s00248-017-1049-0. [DOI] [PubMed] [Google Scholar]
- Zhang G., Xie H., Kang S., Yi D., Ackley S.F. Monitoring lake level changes on the Tibetan Plateau using ICESat altimetry data (2003–2009) Remote Sens. Environ. 2011;115:1733–1742. doi: 10.1016/j.rse.2011.03.005. [DOI] [Google Scholar]
- Zhang G., Yao T., Xie H., Zhang K., Zhu F. Lakes’ state and abundance across the Tibetan Plateau. Chin. Sci. Bull. 2014;59:3010–3021. doi: 10.1007/s11434-014-0258-x. [DOI] [Google Scholar]
- Zhang J., Yang Y., Zhao L., Li Y., Xie S., Liu Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015;99:3291–3302. doi: 10.1007/s00253-014-6262-x. [DOI] [PubMed] [Google Scholar]
- Zhang S., Qin W., Xia X., Xia L., Li S., Zhang L., Bai Y., Wang G. Ammonia oxidizers in river sediments of the Qinghai-Tibet Plateau and their adaptations to high-elevation conditions. Water Res. 2020;173:115589. doi: 10.1016/j.watres.2020.115589. [DOI] [PubMed] [Google Scholar]
- Zhang C., Du X.-P., Zeng Y.-H., Zhu J.-M., Zhang S.-J., Cai Z.-H., Zhou J. The communities and functional profiles of virioplankton along a salinity gradient in a subtropical estuary. Sci. Total Environ. 2021;759:143499. doi: 10.1016/j.scitotenv.2020.143499. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Qin F., Chen F., Chu X., Luo H., Zhang R., Du S., Tian Z., Zhao Y. Culturing novel and abundant pelagiphages in the ocean. Environ. Microbiol. 2021;23:1145–1161. doi: 10.1111/1462-2920.15272. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Temperton B., Thrash J.C., Schwalbach M.S., Vergin K.L., Landry Z.C., Ellisman M., Deerinck T., Sullivan M.B., Giovannoni S.J. Abundant SAR11 viruses in the ocean. Nature. 2013;494:357–360. doi: 10.1038/nature11921. [DOI] [PubMed] [Google Scholar]
- Zheng M., Liu X. Hydrochemistry of salt lakes of the Qinghai-Tibet Plateau, China. Aquat. Geochem. 2009;15:293–320. doi: 10.1007/s10498-008-9055-y. [DOI] [Google Scholar]
- Zheng X., Liu W., Dai X., Zhu Y., Wang J., Zhu Y., Zheng H., Huang Y., Dong Z., Du W., et al. Extraordinary diversity of viruses in deep-sea sediments as revealed by metagenomics without prior virion separation. Environ. Microbiol. 2021;23:728–743. doi: 10.1111/1462-2920.15154. [DOI] [PubMed] [Google Scholar]
- Zhong Z.-P., Solonenko N.E., Li Y.-F., Gazitúa M.C., Roux S., Davis M.E., Van Etten J.L., Mosley-Thompson E., Rich V.I., Sullivan M.B., et al. Glacier ice archives fifteen-thousand-year-old viruses (preprint) Ecology. 2020 doi: 10.1101/2020.01.03.894675. [DOI] [Google Scholar]
- Zimmerman A.E., Howard-Varona C., Needham D.M., John S.G., Worden A.Z., Sullivan M.B., Waldbauer J.R., Coleman M.L. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbiol. 2020;18:21–34. doi: 10.1038/s41579-019-0270-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original code describing the data analysis process are available on GitHub at https://github.com/amitaleth/lake-virome. The clean reads data reported in this paper have been deposited in the Genome Sequence Archive (Wang et al., 2017) in National Genomics Data Center (CNCB-NGDC Members and Partners et al., 2021), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences, under Bioproject number PRJCA005626 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.