

Abstract
Liver cancers consist primarily of hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC). Immune checkpoint inhibitors have emerged as promising therapeutic agents against liver cancers. Programmed cell death protein 1 (PD‐1) is an immunoinhibitory receptor present on T cells that interacts with its ligand programmed death‐ligand 1 (PD‐L1) found on cancer cells. Blocking PD‐1/PD‐L1 binding improves T‐cell survival, proliferation and cytotoxicity, which enhances their antitumor activity. Better understanding of the molecular mechanisms governing PD‐1/PD‐L1 response is essential to the development of predictive markers and therapeutic combinations that could improve the efficiency of anti‐PD‐1/PD‐L1 treatment. Chemokine‐like factor (CKLF)–like MARVEL transmembrane domain–containing 6 (CMTM6) has been recently identified as a major regulator of PD‐L1. Another member in the CMTM family, CKLF‐like MARVEL transmembrane domain–containing 4 (CMTM4), has been shown to compensate for the effects of CMTM6 when CMTM6 is lost. Interestingly, we found that CMTM4 is the major regulator of PD‐L1 in the context of liver cancer. Up‐regulated CMTM4 in patients with HCC and ICC is associated with poor patient survival, potentially due to its function in stabilizing PD‐L1 expression, hence facilitating escape from T cell–mediated cytotoxicity. We confirmed the role of CMTM4 as a positive regulator of PD‐L1 in multiple HCC and ICC cell lines and demonstrated that CMTM4 stabilizes PD‐L1 through posttranslational mechanisms. In vivo, suppression of Cmtm4 inhibited HCC growth and increased CD8+ T‐cell infiltration in immunocompetent mice. Furthermore, we found that depletion of CMTM4 sensitized HCC tumor to anti‐PD‐L1 treatment compared with control. This suggests that CMTM4 expression level could be a predictive marker for patient response to anti‐PD‐L1 treatment, and CMTM4 depletion can potentially be used to enhance the clinical benefits of anti‐PD‐L1 immunotherapy in patients with liver cancer.
CMTM4 stabilizes PD‐L1 on liver cancer cells. CMTM4‐low ICC/HCC, which expresses lower PD‐L1, could be completely blocked by anti‐PD‐L1 mAb.

Abbreviations
- CMTM4
CKLF‐like MARVEL transmembrane domain–containing 4
- CMTM6
CKLF‐like MARVEL transmembrane domain–containing 6
- CTLA4
cytotoxic T lymphocyte antigen 4
- EV
empty vector
- HCC
hepatocellular carcinoma
- HDTV
hydrodynamic tail‐vein
- HKU‐QMH
The University of Hong Kong, Queen Mary Hospital
- ICC
intrahepatic cholangiocarcinoma
- ICI
immune checkpoint inhibitor
- IFN‐γ
interferon‐γ
- IHC
immunohistochemical
- KEAP1
kelch‐like ECH‐associated protein 1
- mAb
monoclonal antibody
- MFI
median fluorescence intensity
- mRNA
messenger RNA
- NT
nontumor
- NTC
nontarget control
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death‐ligand 1
- PTM
posttranslational modification
- RT‐PCR
real‐time polymerase chain reaction
- sg
single guide
- sh
short hairpin
- TCGA
The Cancer Genome Atlas
Hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC) are the two major types of primary liver cancer. Primary liver cancer is highly prevalent in Asian countries and is currently ranked the fourth most common cause of cancer deaths globally.( 1 , 2 ) HCC originates from hepatocytes, whereas ICC originates from cholangiocytes. HCC and ICC are often diagnosed at late stages; hence, the disease prognosis is poor with a 5‐year relative survival rate of 18%.( 3 ) The survival benefits for currently available treatments for HCC and ICC are modest. Moreover, global liver cancer incidence and death rates are projected to rise for the next decade, partly attributed to the rising incidence in nonalcoholic fatty liver disease–associated HCC.( 4 )
Multiple kinase inhibitors, including sorafenib and lenvatinib, can repress HCC growth, although often some cancer cells acquire drug resistance by relying on alternative signaling pathways and tumor growth will progress.( 5 ) On the contrary, immunotherapy can unleash the intrinsic potential of the immune system to recognize and destroy cancer cells, achieving a durable response in some patients with advanced‐stage cancer.( 6 ) Currently, immune checkpoint blockade is the most established immunotherapy against cancer. Immune checkpoints refer to T‐cell inhibitory receptors, which relay signals to inhibit T‐cell activation after binding to their ligands to prevent overactivation T cells, which would otherwise induce autoimmunity. Programmed cell death protein 1 (PD‐1) is an immune checkpoint receptor expressed on antigen‐stimulated T cells; it binds to programmed death‐ligand 1 (PD‐L1), which is commonly expressed on antigen‐presenting cells and cancer cells. PD‐1/PD‐L1 signaling inhibits the proliferation, cytokine production, cytotoxicity, and survival of T cells. Immune checkpoint inhibitors (ICIs) such as anti‐PD‐1 and anti‐PD‐L1 monoclonal antibodies (mAb) are used widely in cancer treatment to reactivate T cell–mediated antitumor immunity.( 7 ) Nivolumab or pembrolizumab (anti‐PD‐1) as monotherapy and nivolumab in combination with ipilimumab (anti–cytotoxic T lymphocyte antigen 4 [CTLA4]) have been approved for second‐line treatment of advanced HCC. Furthermore, atezolizumab (anti‐PD‐L1) in combination with bevacizumab (anti–vascular endothelial growth factor) demonstrated superior efficacy over sorafenib and has become the first immunotherapy to be approved for first‐line treatment of HCC.( 8 ) The unprecedented clinical success of anti‐PD1/PD‐L1 in HCC has paved the way for more ICIs and combination therapies involving ICIs to be used as standard treatments for patients with liver cancer in the next decade.
Although it is encouraging to see PD‐1/PD‐L1 inhibitors being effective against advanced‐stage HCC in some patients, it is important to understand why most patients with HCC did not benefit from PD‐1/PD‐L1 inhibitors,( 9 ) a phenomenon observed across many types of cancers.( 10 ) PD‐L1 expression level is a factor that determines anti‐PD‐1/PD‐L1 efficiency; hence, modulation of PD‐L1 expression on cancer cells can potentially enhance the efficacy of ICIs.( 11 , 12 , 13 ) PD‐L1 up‐regulation in cancer cells is driven primarily by interferon‐γ (IFN‐γ), whereas other mechanisms such as microRNAs,( 14 ) ubiquitination,( 11 , 15 ) glycosylation,( 12 , 16 ) and oncogenic signaling pathways( 13 , 17 ) act in concert to regulate the expression of PD‐L1.
Recently, two chemokine‐like factor (CKLF)–like MARVEL transmembrane domain‐containing (CMTM) family members (ie, CMTM4 and CMTM6) were identified as regulators of PD‐L1.( 18 , 19 ) MARVEL domain‐containing proteins, characterized by their four transmembrane helices, are often involved in vesicular transport and regulation of tight junction.( 20 ) The molecular functions of the eight CMTM family members are still largely unknown. CMTM1, 3, 4, and 7 were reported to be associated with oncogenesis,( 21 , 22 , 23 ) while CMTM3 and 4 regulate angiogenesis in endothelial cells.( 24 , 25 ) CMTM6 was brought to light when Burr et al. and Mezzadra et al. independently identified it as a PD‐L1 stabilizing protein using CRISPR library screening and haploid genetic screening, respectively.( 18 , 19 ) It was found that CMTM6 stabilized PD‐L1 membrane expression by favoring PD‐L1 membrane recycling and/or preventing ubiquitin‐mediated proteasomal degradation. Moreover, among all CMTM members, only CMTM4 plays a compensatory role in PD‐L1 stabilization under a CMTM6‐deficient condition.( 18 )
These two seminal studies focused on demonstrating CMTM6 as a positive PD‐L1 regulator by co‐localizing with PD‐L1 in the plasma membrane and recycling endosomes. Whether CMTM6 or CMTM4 regulates the immune microenvironment in liver cancers and affects PD‐1/PD‐L1 blockade response in patients with liver cancer are two important questions to be addressed. Furthermore, the regulatory mechanism that drives CMTM4/6 up‐regulation in cancer remains unclear. In this study, we focused on the role of CMTM4 in HCC and ICC, and explored how CMTM4 inhibition alters the tumor microenvironment and affects the efficacy of anti‐PD‐L1 in HCC.
Materials and Methods
Patient Samples
Human HCC tumor and their paired nontumor (NT) liver samples were collected from patients with HCC undergoing surgical resection at the Queen Mary Hospital, Pamela Youde Nethersole Eastern Hospital, and Queen Elizabeth Hospital. The tissue samples were snap‐frozen in liquid nitrogen and stored at −80°C. The use of human samples was approved by the institutional review board of the University of Hong Kong and Hospital Authority Hong Kong (Ref. No. UW 09‐158). Consent for the use of resected tissues for research purposes was obtained from patients before collection.
Cell Culture
Human HCC cell lines CLC5 and HepG2, human ICC cell lines KKU‐213, HuCCT1, SSP‐25 and RBE, and a mouse HCC cell line Hepa1‐6 were used in this study. CLC5 was a gift from Prof. Lijian Hui (Chinese Academy of Sciences, Shanghai, China). HepG2 and Hepa1‐6 were purchased from the American Type Culture Collection (Manassas, VA). KKU‐213 and HuCCT1 were purchased from the Japanese Collection of Research Bioresources cell bank. SSP‐25 and RBE were purchased from RIKEN BioResource Research Center (Kyoto, Japan). CLC5 was cultured in Roswell Park Memorial Institute 1640 medium (RPMI‐1640 medium) supplemented with 40 ng/mL endothelial growth factor and 1% insulin‐transferrin‐selenium. HuCCT1 and RBE were cultured in RPMI‐1640 medium with 25 mM HEPES (4‐[2‐hydroxyethyl]‐1‐piperazine ethanesulfonic acid). HepG2, KKU213, and SSP‐25 were cultured in Dulbecco’s modified Eagle medium. All growth media were supplemented with 1% penicillin‐streptomycin and 10% fetal bovine serum (all reagents from Gibco by Life Technologies, Grand Island, NY). All cell lines were cultured in a 37°C humidified incubator supplied with 5% CO2. Authentication for all cell lines used in this study was performed using the AuthentiFiler PCR Amplification Kit (Applied Biosystems, Inc., Foster City, CA). All cell lines were thawed from the authenticated cell stock and used within five passages. Mycoplasma detection was performed regularly.
Animal Studies
Animal experiments throughout the study were performed on male C57BL/6N mice. For subcutaneous implantation, 3 × 106 Hepa1‐6 cells and 50 μL Matrigel (Corning, New York, NY) were injected into the dorsal flanks of 5‐7‐week‐old mice with a 27‐gauge syringe. A dose of 10 mg/kg anti‐mouse PD‐L1 (InVivoMAb clone 10F.9G2; Bio X Cell, Lebanon, NH) was administered intraperitoneally every 3‐4 days, 1 week following tumor inoculation, for a total of four doses. Tumor volumes were measured with a caliper twice weekly and calculated as length × width × height × 0.52 mm3. For orthotopic implantation, 3.5 × 106 Hepa1‐6 cells and 15 μL Matrigel (Corning) were injected into the left lobe of the livers of 5‐7‐week‐old mice with a microliter syringe, and the peritoneum and abdominal wall were closed with sutures. Tumors were harvested for histological and flow‐cytometric analysis 12 days after tumor inoculation. For hydrodynamic tail‐vein (HDTV) injection model, DNA plasmids were mixed with sterile saline to a total volume corresponding to 10% body weight of 8‐10‐week‐old mice. The plasmid–saline mixture was injected through the lateral tail vein within 5‐7 seconds. DNA plasmids entered primarily into hepatocytes due to hydrodynamic pressure from the heart, causing retrograde flow of solution into the liver through the hepatic vein. c‐Myc was overexpressed using the Sleeping Beauty transposon system, and kelch‐like ECH‐associated protein 1 (Keap1) and Cmtm4 were knocked out using the CRISPR‐Cas9 system. Tumors were harvested for histological and flow‐cytometric analysis 7 weeks following injection.
All animal procedures were approved by the Committee on the Use of Live Animals in Teaching and Research of the University of Hong Kong and performed under the Animals (Control of Experiments) Ordinance of Hong Kong. All animal experiments were performed under the UK Coordinating Committee on Cancer Research PMID: 9459138 Guidelines for the Welfare of Animals in Experimental Neoplasia, to ensure minimal suffering of the animals throughout the procedures.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software, Inc., La Jolla, CA). All functional assays are representative of at least three independent experiments and are expressed as mean ± SD unless otherwise specified. A P value < 0.05 was considered statistically significant.
Data Availability Statement
Messenger RNA (mRNA) expression levels of CMTM4 and CMTM6 of human HCC and ICC samples with their corresponding NT liver tissues were retrieved from transcriptome sequencing data from The Cancer Genome Atlas (TCGA) through cBioPortal (http://www.cbioportal.org). All data supporting the findings of this study are available within the article, in the Supporting Information, or from the corresponding author upon reasonable request.
Descriptions of the establishment of knockdown and knockout cells, western blotting, preparation of tumor samples into single‐cell suspension, flow cytometry, immunohistochemistry, RNA extraction, reverse‐transcription and quantitative real‐time polymerase chain reaction (RT‐PCR), copy number analysis, clinicopathological correlation and survival analysis, antibodies and recombinant proteins, short hairpin RNA (shRNA), single‐guide RNA (sgRNA), and primer sequences are provided in the Supporting Information. Sources of antibodies and recombinant proteins are listed in Supporting Table S1. Sequences of shRNAs and sgRNAs are listed in Supporting Table S2. Sequences of quantitative RT‐PCR primers are listed in Supporting Table S3.
Results
CMTM4 Is Overexpressed in Human HCC and ICC
Transcriptome sequencing data retrieved from TCGA showed significant down‐regulation of CMTM6 mRNA expression in human HCC or ICC tumor samples compared with paired NT liver tissues (Fig. 1A). On the other hand, CMTM4 mRNA expression was up‐regulated in both HCC and ICC (Fig. 1B). Fifty‐five percent of HCC samples from TCGA showed CMTM4 overexpression by at least two‐fold compared with NT samples (Fig. 1C). CMTM4 mRNA overexpression in HCC was further confirmed in 75 pairs of patient samples from our institute by quantitative RT‐PCR (Fig. 1E, F). Up‐regulation of CMTM4 protein in human HCC tissue was confirmed by immunohistochemical (IHC) staining (Fig. 1F). Data from TCGA indicated a high mRNA expression of CMTM4 was significantly associated with poor disease‐free survival (P = 0.021) in patients with HCC (Fig. 1G). Clinicopathological analysis of our in‐house data on patients with HCC showed that overexpression of CMTM4 was significantly correlated with higher Edmondson and Steiner grading, corresponding to poorly differentiated cellular phenotype, which is often associated with more aggressive disease (Fig. 1H; Supporting Fig. S1 and Supporting Table S4). Next, we investigated the mechanism contributing to CMTM4 overexpression in HCC. TCGA data showed that CMTM4 copy‐number gain correlated with increased CMTM4 mRNA expression (Fig. 1I). Indeed, CMTM4 copy‐number analysis in 18 paired HCC and NT samples showed that CMTM4 copy‐number gain (three copies or above) was present in over half of the HCC tumor tissues; among these, many have more than or equal to five copies (Fig. 1J). In NT tissues, copy‐number gain was detected in 20% of samples, none having more than or equal to five copies.
FIG. 1.
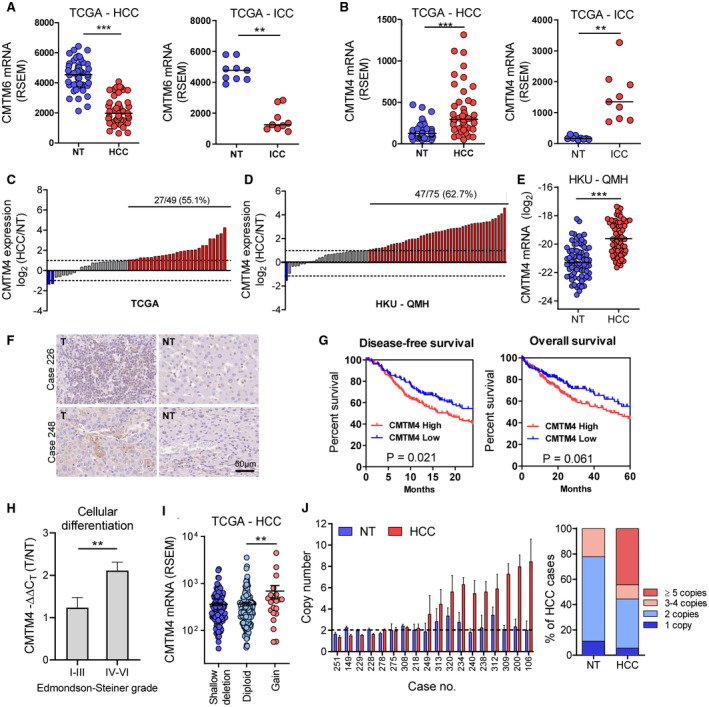
CMTM4 is up‐regulated in HCC and ICC and is associated with poor prognosis. CMTM6 (A) and CMTM4 (B) mRNA expression in 49 pairs of HCC (left) or 9 pairs of ICC (right) are shown with their corresponding NT liver tissues from TCGA database. (C,D) Waterfall plot showing CMTM4 overexpression by at least two‐fold in 55% and 63% of patients with HCC from TCGA database (C) and the University of Hong Kong, Queen Mary Hospital (HKU‐QMH) (D), respectively. (E) CMTM4 mRNA expression in 75 cases of paired HCC and NT tissues from HKU‐QMH patients. (F) Representative images of IHC staining of CMTM4 in two pairs of human HCC and NT tissues. (G) Kaplan‐Meier curves showed that high CMTM4 mRNA expression is associated with poor disease‐free (left) and overall survival (right) in patients with HCC from TCGA. (H) Correlation of CMTM4 expression with degrees of cellular differentiation in HKU‐QMH HCC patients. (I) Correlation of CMTM4 mRNA expression with CMTM4 copy‐number alterations in HCC tissues from TCGA. (J) CMTM4 copy‐number analysis in genomic DNA extracted from paired HCC and NT tissues from 18 HKU‐QMH patients with HCC. (A,B,E) Lines indicate median. (H‐J) Error bars indicate mean ± SEM. **P < 0.01, ***P < 0.001. (A,B,E) Wilcoxon signed‐rank test. (H,I) Student t test. (G) Log rank test. Abbreviation: T, tumor.
CMTM4 Stabilized PD‐L1 Surface Expression in HCC and ICC Cell Lines
To study the functional role of CMTM4 in HCC and ICC, we established multiple stable CMTM4 or Cmtm4 knockdown and knockout cell lines, including human HCC, human ICC, as well as mouse HCC cell line, using two independent shRNA or sgRNA sequences. CMTM4 depletion in knockdown and knockout cells was validated by quantitative RT‐PCR and western blot (Fig. 2A,C,E; Supporting Fig. S2). CMTM4 knockdown or knockout in multiple HCC and ICC cells resulted in significant down‐regulation of surface PD‐L1 expression compared to nontarget control (NTC), with or without IFN‐γ stimulation, before flow cytometry analysis (Fig. 2B,D,F; Supporting Figs. S3 and S4), confirming CMTM4 as a universal regulator of PD‐L1 protein expression. Similar to a previous report, we confirmed that inhibition of CMTM4 suppressed PD‐L1 expression in an IFN‐γ‐independent manner.( 18 )
FIG. 2.
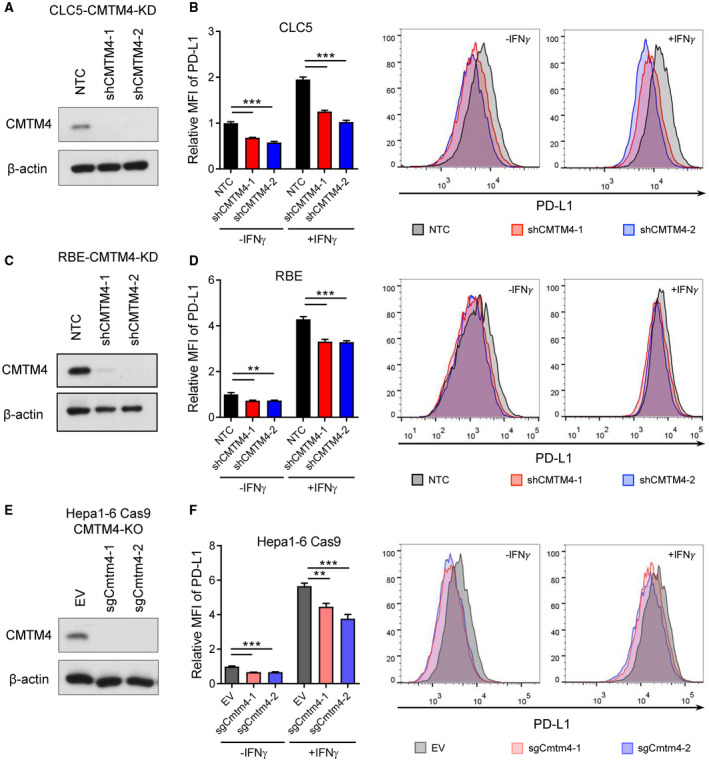
CMTM4 stabilized PD‐L1 surface expression in HCC and ICC cell lines. (A,C) CMTM4 knockdown efficiency in CLC5 and RBE cells confirmed by western blot. (B,D) Relative median fluorescence intensity (MFI) of surface PD‐L1 (left) and representative histograms (right) from flow cytometry analysis showing PD‐L1 expression in −shCMTM4 cells compared to −NTC cells, with or without IFN‐γ stimulation. (E) Cmtm4 knockout efficiency in mouse syngeneic HCC cell line Hepa1‐6 transduced with Cas9. (F) Relative MFI (left) and representative histograms (right) of PD‐L1 surface expression in Hepa1‐6‐Cas9 −sgCmtm4 cells compared with −EV cells. (See Supporting Figs. S1 and S2 for the full data set.) Cells were pretreated with 25 ng/mL human or mouse IFN‐γ for 24 hours before analysis. Error bars indicate mean ± SD. **P < 0.01, ***P < 0.001. (B,D,F) Student t test.
CMTM4 Stabilized PD‐L1 Through Posttranslational Mechanisms
Next, we examined how CMTM4 stabilizes PD‐L1. CMTM4 knockdown did not suppress PD‐L1 mRNA expression, suggesting that CMTM4 does not regulate PD‐L1 at a transcriptional level (Fig. 3A‐C). It has been reported previously that CMTM6 participates in regulating the endocytic trafficking of PD‐L1,( 19 ) whereas another report suggested that CMTM6 prevents ubiquitin‐dependent proteasomal degradation of PD‐L1.( 18 ) We investigated whether CMTM4 has similar molecular functions as described for CMTM6. Treatment of clathrin‐dependent endocytosis inhibitor Dyngo4a or proteasome inhibitor MG132 in CLC5 and RBE‐CMTM4 knockdown cells partially rescued PD‐L1 surface expression, whereas combination treatment of Dyngo4a and MG132 completely rescued PD‐L1 surface expression to the level comparable with NTC cells (Fig. 3D, E). This suggests that the stabilization of PD‐L1 by CMTM4 can be attributed to the prevention of PD‐L1 degradation by the endosome‐lysosomal pathway and proteasomal pathway.
FIG. 3.
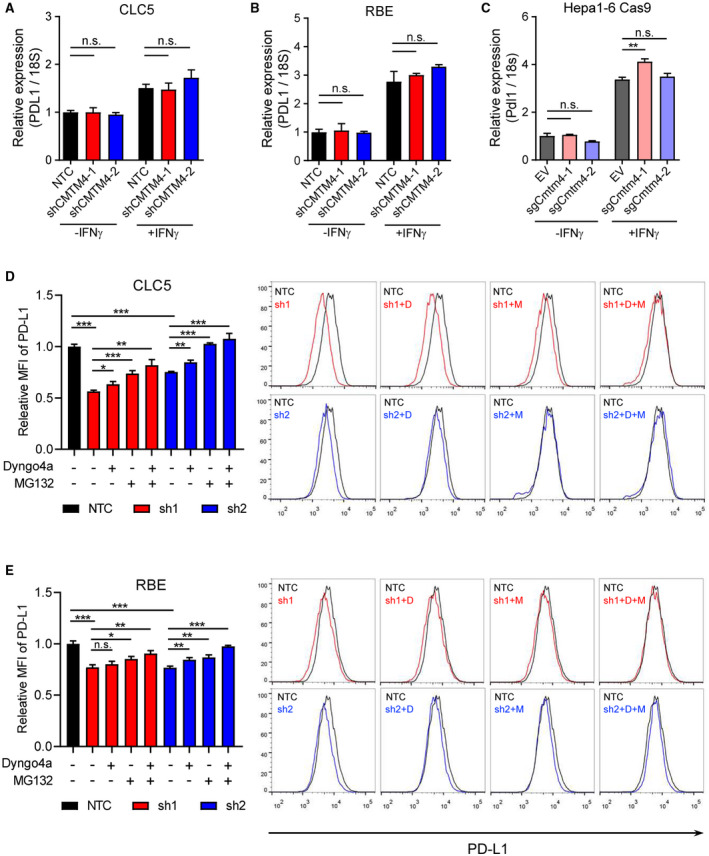
CMTM4 stabilized PD‐L1 through posttranslational mechanisms. (A‐C) PD‐L1 mRNA expressions measured by quantitative RT‐PCR in control and CMTM4‐knockdown or CMTM4‐knockout CLC5, RBE, and Hepa1‐6‐Cas9 cells with or without IFN‐γ stimulation (24 hours). CLC5 (D) and RBE (E) control and CMTM4‐knockdown cells were stimulated with IFN‐γ (24 hours). A total of 30 μM Dyngo4a (clathrin‐dependent endocytosis inhibitor) and/or 8 μM MG132 (proteasome inhibitor) were added into culture medium 24 hours and 6 hours before flow cytometry analysis, respectively. Relative MFI (left) and representative histograms (right) of surface PD‐L1 level. Error bars indicate mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (A‐C) Student t test. Abbreviations: D, Dyngo4a; M, MG132; n.s., not significant.
Inhibition of CMTM4 Improved Antitumor Immune Response and Suppressed HCC Growth In Vivo
As CMTM4 regulates PD‐L1 expression in HCC, and PD‐L1 has known inhibitory function on T cells, we asked whether CMTM4 modulates antitumor activity of T cells in vivo. To investigate the tumor immune microenvironment, we used a syngeneic mouse HCC model by orthotopically implanting empty vector control (EV) or Cmtm4‐knockdown Hepa1‐6 cells into the liver of immunocompetent mice (Fig. 4A). Knockdown efficiency of Cmtm4‐knockdown Hepa1‐6 cells was confirmed (Supporting Fig. S5A). HCC tumors derived from Hepa1‐6‐shCmtm4‐2 cells were significantly smaller in size compared with control, although shCmtm4‐1 did not reach statistical significance (Fig. 4B). Flow cytometry analysis also revealed that more CD8+ T cells were found in shCmtm4‐1 tumors (Fig. 4C), suggesting that Cmtm4 plays an immunosuppressive role in HCC. Flow cytometry analysis also confirmed that HCC cells, as indicated by the CD45‐ population from Cmtm4 knockdown tumors, expressed a lower level of PD‐L1 (Supporting Fig. S5B). However, expressions of T‐cell exhaustion markers including PD‐1, LAG‐3, and TIM‐3 showed no significant difference (Supporting Fig. S5C), suggesting that the exhausted T cells might have undergone apoptosis, rendering them undetectable in the tumor bulk. Interestingly, in this mouse HCC model, we could not observe a significant difference in CD4+ T cells (Supporting Fig. S5D,E). To confirm that our model is not limited to Hepa1‐6 cells, we also used the HDTV injection model, in which HCC was induced in vivo through the injection of genome‐editing plasmids, comprised of the Sleeping Beauty transposon system and the CRISPR‐Cas9 system, to generate c‐Myc‐overexpressing, Keap1‐knockout, Cmtm4‐wild‐type/Cmtm4‐knockout (c‐MycOE Keap1KO Cmtm4WT or c‐MycOE Keap1KO Cmtm4KO) HCCs (Fig. 4D). Knockout of Cmtm4 with sgCmtm4‐1 and sgCmtm4‐2 significantly reduced tumor size and suppressed HCC formation compared with control (Fig. 4E). Moreover, infiltration of CD4+ and CD8+ cells in tumor were significantly up‐regulated in Cmtm4 knockout tumors confirmed by IHC staining (Fig. 4F), further validating that reactivation of T cell–mediated antitumor immunity following CMTM4 inhibition is effective in suppressing HCC growth. CD8+ T cells are the cytotoxic T cells that directly kill target cells following their recognition of antigens. Therefore, major effector cells mediate cancer cell killing at tumor sites in multiple ways, such as perforin and granzyme B secretion.( 26 ) Indeed, a study on patients with melanoma who were treated with anti‐PD‐1 blocking antibody showed increased CD8+ T‐cell infiltration in the response group, but not progression group.( 27 ) It is also reported that high CD8+ T‐cell infiltration was correlated with better prognosis in patients with HCC.( 28 , 29 , 30 ) Therefore, it is reasonable that increased CD8+ T cells could be found consistently in the two mouse HCC models.
FIG. 4.
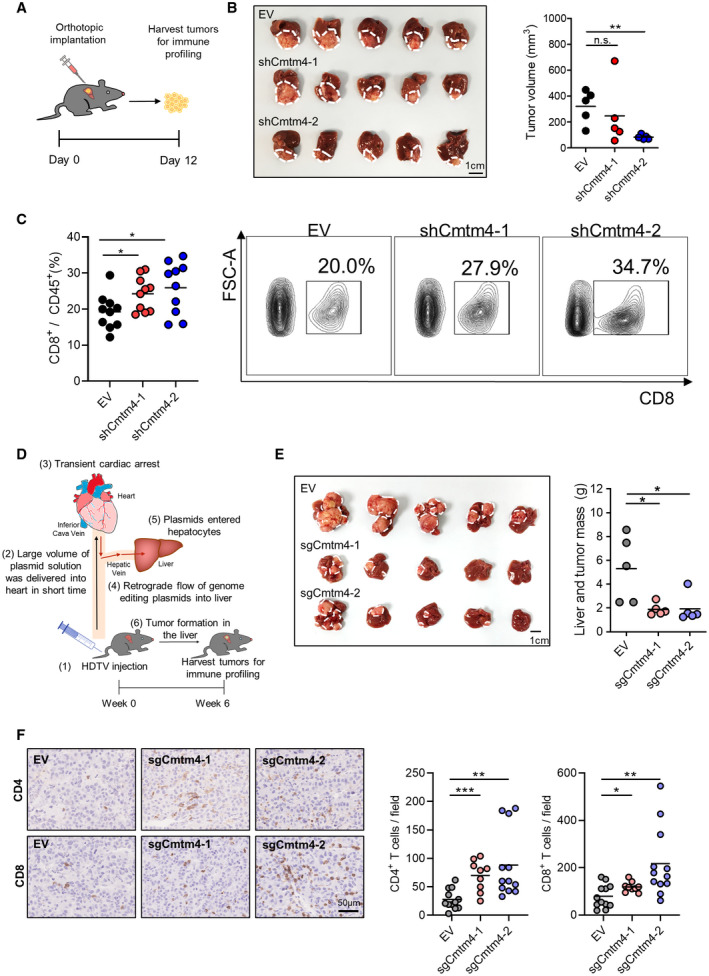
Inhibition of Cmtm4‐suppressed tumor growth in mouse by increasing T‐cell recruitment. (A) 3.5 × 106 of control (EV) or Cmtm4‐knockdown (shCmtm4‐1, shCmtm4‐2) Hepa1‐6 cells were orthotopically implanted into the liver of wild‐type mice. Tumors were harvested 12 days following implantation and dissociated for immune profiling. (B) Picture (left) and volume (right) of orthotopically implanted tumors. (C) Flow cytometry analysis of CD8+ T cells in the tumors. Quantification of the percentage of CD8+ T cells over total tumor‐infiltrating leukocytes (left) and representative contour plots showing the gating of CD8+ T cells (right). (D) HDTV injection model for the generation of HCC tumor. Genome editing plasmids were injected into mice through lateral veins. Large volume of plasmid solution was delivered into heart within 6‐8 seconds, causing transient cardiac arrest and retrograde flow of plasmids into the mouse liver. Plasmids entered hepatocytes by high pressure created by HDTV injection. Tumors developed in the liver spontaneously, and mice were killed 6 weeks after HDTV injection. (E) Picture (left) and mass (right) of control (EV) or Cmtm4‐knockout (sgCmtm4‐1, sgCmtm4‐2) liver tumors. (F) CD4 and CD8 were stained by IHC in formalin‐fixed paraffin‐embedded HDTV tumor specimens. Representative pictures of CD4 and CD8 staining in EV and sgCmtm4 tumors (left) and quantification of CD4+ and CD8+ T cells in the tumors (right). Cells with positive staining at three random regions per sample were counted. (B,C,E) Lines indicate mean. (F) Lines indicate mean, and error bars indicate SD. *P < 0.05, **P < 0.01. (B,C,E,F) Student t test. Abbreviations: FSC‐A, forward scatter; n.s., not significant.
Inhibition of CMTM4 Sensitized HCC Toward PD‐L1 Blockade
We further investigated whether CMTM4 affects response toward anti‐PD‐L1 immune checkpoint blockade. We hypothesized that HCC tumors expressing a lower level of CMTM4 would also express a lower level of surface PD‐L1 (i.e., shCmtm4 tumors can be more effectively targeted by anti‐PD‐L1 mAb). To test this hypothesis, we subcutaneously implanted Hepa1‐6 Cmtm4‐knockdown or EV control cells into the left and right flanks of the same wild‐type mouse, respectively. The mice were divided into control (Ctrl) or anti‐PD‐L1 mAb treatment group (Fig. 5A). PD‐L1 blockade significantly reduced tumor growth in both EV and Cmtm4‐knockdown tumors (Fig. 5B). More importantly, shCmtm4 tumors regressed more substantially than EV tumors, as seen from the growth curves of individual tumors, with more shCmtm4 achieving complete regression after PD‐L1 blockade (Fig. 5B). These results indicate that suppressing PD‐L1 level by inhibiting CMTM4 could sensitize response toward anti‐PD‐L1 in vivo (Fig. 5C), thereby establishing CMTM4 inhibition as a promising mechanism to enhance the efficacy of PD‐L1 blockade.
FIG. 5.
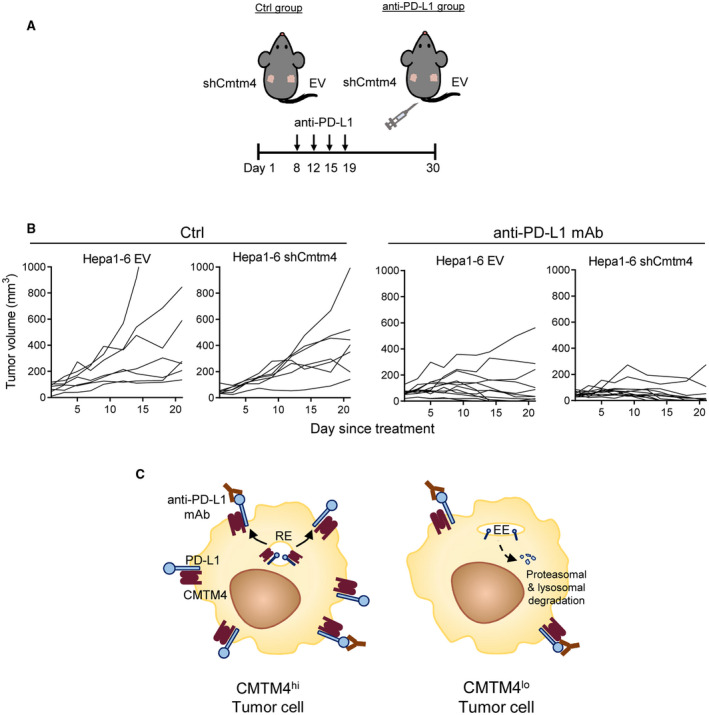
Inhibition of CMTM4 sensitized HCC toward PD‐L1 blockade in mouse. (A) Scheme of experimental design. A total of 3 × 106 of Hepa1‐6 EV and Cmtm4‐knockdown (shCmtm4‐1) cells were subcutaneously implanted into the right and left sides of the lateral flank of wild‐type mice, respectively. Four doses of anti‐PD‐L1 mAb or saline control were administered by intraperitoneal injection during weeks 2‐3 (10 mg/kg/dose). Control group, n = 7; anti‐PD‐L1, group n = 12. (B) Growth curves of individual tumors, measured from the commencement of anti‐PD‐L1 treatment onward. (C) Schematic diagram explaining the therapeutic effect of CMTM4 inhibition in anti‐PD‐L1 immunotherapy. In CMTM4hi tumor cells, anti‐PD‐L1 monoclonal antibodies fail to occupy all PD‐L1 binding sites (left), whereas with lowered PD‐L1 surface expression in CMTM4lo tumor cells, anti‐PD‐L1 mAb is able to completely abrogate PD‐1/PD‐L1 signaling between T cells and tumor cells and hence fully unleash T cell–mediated antitumor immunity (right). Abbreviations: Ctrl, saline control; EE, early endosome; RE, recycling endosome.
Discussion
In recent years, an increasing number of anti‐PD‐1/anti‐PD‐L1 ICIs have been approved by the Food and Drug Administration as monotherapy or combination therapy for the treatment of HCC,( 31 ) marking the beginning of a change in the treatment paradigm for HCC. Although immunotherapy has not been approved for treatment in patients with ICC yet, it has been reported that PD‐1 and PD‐L1 were up‐regulated in ICC tissues compared with adjacent nontumor tissues, and PD‐L1 high expression was associated with low CD8+ T‐cell infiltration.( 32 ) This suggests that PD‐1/PD‐L1 blockade could also be effective in ICC, and the field is actively exploring the use of ICIs for treating advanced ICC.( 33 )
PD‐L1 expression is dependent on transcriptional regulation, posttranslational modifications (PTMs), and protein degradation.( 34 ) In cancer, PD‐L1 is often transcriptionally up‐regulated by IFN‐γ. Alternatively, activation of oncogenic signaling pathways such as epidermal growth factor receptor (EGFR),( 35 ) MYC,( 36 ) and RAS( 17 ) also induce PD‐L1 expression. Furthermore, multiple PTMs and protein‐degradation pathways are responsible for the tight regulation and rapid turnover of PD‐L1.( 37 ) PD‐L1 protein is posttranslationally modified by N‐linked glycosylation, serine/threonine phosphorylation, and polyubiquitination. PD‐L1 is glycosylated at asparagine residues before being transported to the plasma membrane. Glycosylation stabilizes PD‐L1 by preventing its internalization and degradation by the proteasome.( 12 ) Phosphorylation of PD‐L1 by GSK3β facilitates PD‐L1 binding to an E3 ubiquitin ligase. Poly‐ubiquitinated PD‐L1 is marked for subsequent proteasomal degradation,( 11 ) but this process can be reversed by de‐ubiquitinates such as COP9 signalosome 5, which was reported to be responsible for stabilizing PD‐L1 in breast cancer.( 15 ) Although the ubiquitin–proteasome pathway is the major protein degradation pathway for removal of immature, misfolded, and unwanted cytosolic proteins, the endosome–lysosome pathway is considered the major means by which membrane proteins are degraded.( 38 ) Stabilization of PD‐L1 after lysosomal inhibition with primaquine or chloroquine provided key evidence that lysosomes are involved in the regulation of PD‐L1 turnover.( 19 , 39 ) Palmitoylation of PD‐L1 was recently identified as a PTM mechanism that stabilizes PD‐L1 by preventing PD‐L1 mono‐ubiquitination and its subsequent lysosomal targeting.( 39 )
Among these PD‐L1 posttranslational regulators, the CMTM family of proteins is the most poorly studied. Particularly in liver cancer, the molecular functions and clinical implications of CMTM family proteins still require further investigation. Burr et al. discovered CMTM6 to be a PD‐L1 posttranslational regulator that physically associates with PD‐L1 at the plasma membrane and in endosomes, promotes PD‐L1 membrane recycling, and prevents its lysosomal degradation.( 19 ) Mezzadra et al. also found that CMTM6 stabilizes PD‐L1 protein by preventing the poly‐ubiquitination by STUB1 E3 ubiquitin ligase, and hence the subsequent proteasomal degradation.( 18 ) Mezzadra et al. also discovered that under a CMTM6‐deficient context, CMTM4 acts as a second PD‐L1 regulator.( 18 ) In this study, we showed that CMTM4 contributed to immune evasion by stabilizing PD‐L1 expression, and its inhibition improved the efficacy of anti‐PD‐L1 checkpoint inhibitors in liver cancer.
In HCC and ICC, we found that CMTM4 but not CMTM6 was up‐regulated, suggesting that—different from other types of cancers in which CMTM6 is dominant—CMTM4 may play a more important role in liver cancer. More importantly, a high expression of CMTM4 is associated with poor prognosis. We further showed that CMTM4 up‐regulation in patients with HCC is due to CMTM4 copy‐number gain. CMTM4 depletion suppressed PD‐L1 protein expression in the eight HCC and ICC cell lines we tested, confirming CMTM4 as a universal PD‐L1 regulator. The CMTM family of proteins share a common approximate 130‐residue membrane‐associating domain called the MARVEL domain, which is important for membrane fusion, vesicle trafficking, and cell–cell junction regulation.( 20 ) It has been reported that CMTM4 co‐localizes with endosome compartments and the Golgi apparatus in endothelial cells and HeLa cells, respectively.( 22 , 25 ) We showed that by inhibiting clathrin‐dependent endocytosis and proteasomal activity, CMTM4 knockdown–mediated PD‐L1 suppression could be completely restored, confirming the role of CMTM4 in stabilizing PD‐L1 by protecting it from endosome‐lysosome‐mediated and proteasome‐mediated proteolysis. Inhibition of CMTM4 suppressed tumor growth in different mouse HCC models and facilitated tumor infiltration of CD4+ and CD8+ T cells. This suggests that suppressing PD‐L1 through CMTM4 can reverse the immunosuppressive tumor microenvironment and enhance the antitumor activity of T cells in HCC.
Various predictive markers have been identified to better guide the stratification of patients for PD‐1/PD‐L1 blockade. These include tumor mutational burden,( 40 ) expression of neoantigens,( 41 ) microsatellite instability,( 42 ) and the extent of tumor infiltration by T cells and macrophages.( 43 ) It has been reported that pretreatment PD‐L1 expression is associated with better objective response toward PD‐1 blockade in multiple types of cancer.( 44 ) However, conflicting studies have also shown that the expression level of PD‐L1 may not be associated with clinical response of anti‐PD‐1/PD‐L1.( 45 ) A possible explanation for this is false‐negative IHC staining of PD‐L1, due to the obstruction of binding of anti‐PD‐L1 antibodies to glycosylated PD‐L1. A study showed that enzyme‐mediated deglycosylation before IHC staining greatly improves PD‐L1 detection.( 46 ) As CMTM4 directly regulates the level of PD‐L1, CMTM4 expression provides information on PD‐L1 turnover rate. In patients who are PD‐L1‐positive, indicative of immune activation, low CMTM4 could predict better response to anti‐PD‐L1 treatment in liver cancer. This is because PD‐L1 is more prone to intrinsic protein degradation when in the absence of CMTM4; hence, there is a lower overall PD‐L1 abundance, raising the likelihood of complete inhibition of PD‐1/PD‐L1 signaling at the cell surface with anti‐PD‐L1 treatment. Thus, CMTM4 expression may also be a predictive marker for anti‐PD‐L1 response.
To date, the major caveat of PD‐1/PD‐L1 blockade is that most patients with cancer do not benefit from anti‐PD‐1/PD‐L1 due to innate and acquired resistance. A pitfall of using mAb is that its large size might hinder penetration into the tumor microenvironment, and hence fail to block PD‐1/PD‐L1 binding sites efficiently. Moreover, PD‐L1 may be highly expressed in certain regions of a solid tumor where T cells are activated. In such regions, anti‐PD‐1/PD‐L1 mAb may not be able to fully occupy PD‐1/PD‐L1 binding sites.( 34 ) In this study, we validated that the depletion of PD‐L1 protein through CMTM4 to lower PD‐L1 membrane expression could be one mechanism to circumvent such resistance (Fig. 6). A previous report showed that reducing PD‐L1 membrane expression could render cancer more susceptible to ICIs.( 15 ) CMTM4 stabilized PD‐L1 independent of IFN‐γ stimulation, suggesting that inhibiting CMTM4 can suppress PD‐L1 expression driven by IFN‐γ or other oncogenic signaling pathways activated by drugs such as EGFR,( 12 ) cyclin‐dependent 4/6,( 11 ) or PARP (poly[adenosine diphosphate ribose] polymerase) inhibitors.( 47 ) We demonstrated that suppressing PD‐L1 expression by inhibition of CMTM4 enhanced the efficacy of anti‐PD‐L1 immunotherapy by achieving a more complete inhibition of PD‐1/PD‐L1 signaling between tumor cells and T cells, suggesting that targeting CMTM4 can have a synergistic effect with PD‐L1 blockade in liver cancer. High doses of ICIs often lead to immune‐related adverse effects, causing the termination of the treatments. In a phase 2 clinical trial in patients with melanoma, drug‐related adverse effects were more frequently observed in patients treated with higher dose of anti‐CTLA4 blocking antibody (NCT00289640).( 48 ) In a phase 1 clinical trial investigating antitumor activity and safety of anti‐PD‐1 blocking antibody in multiple cancer types (NCT00730639), patients were treated with escalating dose of anti‐PD‐1 blocking antibody, ranging from 0.1 mg/kg to 10 mg/kg. There was a dose‐dependent increase in occurrence of adverse events. Due to immune‐related adverse effects, treatments were terminated in 19% and 13% of patients receiving 10 mg/kg anti‐PD‐1 and 1 mg/kg anti‐PD‐1, respectively.( 49 ) Notably, PD‐1 inhibitors were associated with higher risk of hepatotoxicity compared with the control regimen in a meta‐analysis study.( 50 ) This information is particularly relevant for HCC treatments, as most patients with HCC have impaired liver functions, suggesting that low doses of PD‐1/PD‐L1 inhibitors should be considered in them. When low doses of PD‐1/PD‐L1 inhibitors are administered, the availability of the antibodies becomes limited. This might explain our result that CMTM4‐low tumors that express a lower level of PD‐L1 are more responsive to anti‐PD‐L1 mAb. Therefore, the general hypothesis that tumors with high PD‐L1 expression might be more responsive to anti‐PD‐L1 immunotherapy might also be dependent on the doses of antibodies used.
FIG. 6.
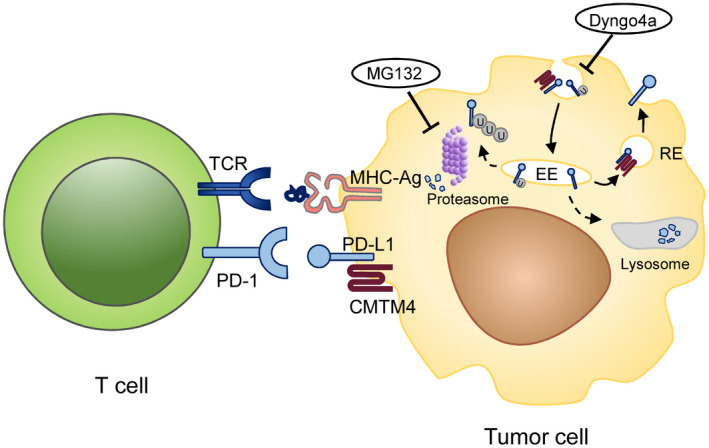
Schematic diagram of the regulation of PD‐1/PD‐L1 signaling by CMTM4 in liver cancer. CMTM4 physically interacts with PD‐L1 in tumor cells to promote and sustain PD‐L1 expression at the plasma membrane by shuttling PD‐L1 to recycling endosomes, preventing it from being degraded by the lysosome or proteasome. As a result, CMTM4 potentiates the binding of PD‐L1 on tumor cells to PD‐1 on T cells, inhibiting T‐cell activation and facilitating immune escape. Inhibition of proteasomal activity by MG132 and inhibition of clathrin‐dependent endocytosis by Dyngo4a restored PD‐L1 surface expression after CMTM4 inhibition. Abbreviations: EE, early endosome; RE, recycling endosome; MHC‐Ag, MHC class I conjugated with tumor antigen; TCR, T‐cell receptor; U, ubiquitin.
Our study revealed that CMTM4 up‐regulation in liver cancer contributes to an immunosuppressive tumor microenvironment that facilitates escape from antitumor T‐cell immunity and tumor growth. Depleting PD‐L1 by promoting its degradation through CMTM4 inhibition suppressed HCC growth and enhanced the efficacy of PD‐L1 checkpoint inhibitors. From the example of CMTM4, one could infer that other posttranslational regulators that promote protein internalization and degradation are also likely to be relevant to other immune checkpoints, such as CTLA4, LAG3, and TIGIT. By reducing the overall abundance of other immune checkpoint receptors or their respective ligands, the inhibitory effect of ICIs can be further enhanced, bringing hope that more patients with cancer can benefit from immune checkpoint blockade therapy.
Supporting information
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Acknowledgment
We thank the Core Facility and Center for Genomic Sciences of the Li Ka Shing Faculty of Medicine, The University of Hong Kong, for the technical support. We also thank the Laboratory Animal Unit for animal housing.
Supported by the Research Grant Council Theme‐Based Research Scheme (T12‐704/16‐R), Croucher Foundation (2017), and the University of Hong Kong (OYRA2017).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 4. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411‐428. [DOI] [PubMed] [Google Scholar]
- 5. Ramos P, Bentires‐Alj M. Mechanism‐based cancer therapy: resistance to therapy, therapy for resistance. Oncogene 2015;34:3617‐3626. [DOI] [PubMed] [Google Scholar]
- 6. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T‐Y, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 9. El‐Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD‐1 inhibition. New Engl J Med 2017;377:2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D‐CDK4 kinase destabilizes PD‐L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018;553:91‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand‐1 suppresses T‐cell activity. Nat Commun 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cerezo M, Guemiri R, Druillennec S, Girault I, Malka‐Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F–STAT1–PD‐L1 axis in melanoma. Nat Med 2018;24:1877‐1886. [DOI] [PubMed] [Google Scholar]
- 14. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y‐H, Byers LA, et al. Metastasis is regulated via microRNA‐200/ZEB1 axis control of tumour cell PD‐L1 expression and intratumoral immunosuppression. Nat Commun 2014;5:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lim S‐O, Li C‐W, Xia W, Cha J‐H, Chan L‐C, Wu Y, et al. Deubiquitination and stabilization of PD‐L1 by CSN5. Cancer Cell 2016;30:925‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu J‐M, Xia W, Hsu Y‐H, Chan L‐C, Yu W‐H, Cha J‐H, et al. STT3‐dependent PD‐L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun 2018;9:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina‐Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD‐L1 mRNA. Immunity 2017;47:1083‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mezzadra R, Sun C, Jae LT, Gomez‐Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD‐L1 protein regulators. Nature 2017;549:106‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burr ML, Sparbier CE, Chan Y‐C, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD‐L1 and regulates anti‐tumour immunity. Nature 2017;549:101‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sánchez‐Pulido L, Martín‐Belmonte F, Valencia A, Alonso MA. A conserved domain involved in membrane apposition events. Trends Biochem Sci 2002;27:599‐601. [DOI] [PubMed] [Google Scholar]
- 21. Delic S, Thuy A, Schulze M, Proescholdt MA, Dietrich P, Bosserhoff A‐K, et al. Systematic investigation of CMTM family genes suggests relevance to glioblastoma pathogenesis and CMTM 1 and CMTM 3 as priority targets. Genes Chromosom Cancer 2015;54:433‐443. [DOI] [PubMed] [Google Scholar]
- 22. Plate M, Li T, Wang YU, Mo X, Zhang Y, Ma D, et al. Identification and characterization of CMTM4, a novel gene with inhibitory effects on HeLa cell growth through Inducing G2/M phase accumulation. Mol Cells 2010;29:355‐361. [DOI] [PubMed] [Google Scholar]
- 23. Li H, Li J, Su Y, Fan Y, Guo X, Li L, et al. A novel 3p22.3 gene CMTM7 represses oncogenic EGFR signaling and inhibits cancer cell growth. Oncogene 2014;33:3109‐3118. [DOI] [PubMed] [Google Scholar]
- 24. Chrifi I, Louzao‐Martinez L, Brandt M, van Dijk CGM, Burgisser P, Zhu C, et al. CMTM3 (CKLF‐like marvel transmembrane domain 3) mediates angiogenesis by regulating cell surface availability of ve‐cadherin in endothelial adherens junctions. Arterioscler Thromb Vasc Biol 2017;37:1098‐1114. [DOI] [PubMed] [Google Scholar]
- 25. Chrifi I, Louzao‐Martinez L, Brandt MM, van Dijk CGM, Bürgisser PE, Zhu C, et al. CMTM4 regulates angiogenesis by promoting cell surface recycling of VE‐cadherin to endothelial adherens junctions. Angiogenesis 2019;22:75‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer 2021;124:359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sideras K, Biermann K, Verheij J, Takkenberg BR, Mancham S, Hansen BE, et al. PD‐L1, Galectin‐9 and CD8+ tumor‐infiltrating lymphocytes are associated with survival in hepatocellular carcinoma. Oncoimmunology 2017;6:e1273309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao Q, Qiu S‐J, Fan J, Zhou J, Wang X‐Y, Xiao Y‐S, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007;25:2586‐2593. [DOI] [PubMed] [Google Scholar]
- 30. Sun C, Xu J, Song J, Liu CQ, Wang J, Weng C, et al. The predictive value of centre tumour CD8+ T cells in patients with hepatocellular carcinoma: comparison with Immunoscore. Oncotarget 2015;6:35602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu AX, Kang Y‐K, Yen C‐J, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α‐fetoprotein concentrations (REACH‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2019;20:282‐296. [DOI] [PubMed] [Google Scholar]
- 32. Ye Y, Zhou L, Xie X, Jiang G, Xie H, Zheng S. Interaction of B7–H1 on intrahepatic cholangiocarcinoma cells with PD‐1 on tumor‐infiltrating T cells as a mechanism of immune evasion. J Surg Oncol 2009;100:500‐504. [DOI] [PubMed] [Google Scholar]
- 33. Liu X, Yao J, Song L, Zhang S, Huang T, Li Y. Local and abscopal responses in advanced intrahepatic cholangiocarcinoma with low TMB, MSS, pMMR and negative PD‐L1 expression following combined therapy of SBRT with PD‐1 blockade. JITC 2019;7:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD‐L1 checkpoint. Immunity 2018;48:434‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD‐L1 expression via the IL‐6/JAK/STAT3 signaling pathway in EGFR‐mutated non‐small cell lung cancer. Int J Oncol 2016;49:1360‐1368. [DOI] [PubMed] [Google Scholar]
- 36. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD‐L1. Science 2016;352:227‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD‐L1 and their applications in cancer therapy. Can Res 2018;78:6349‐6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell 2010;143:682‐685. [DOI] [PubMed] [Google Scholar]
- 39. Yao H, Lan J, Li C, Shi H, Brosseau J‐P, Wang H, et al. Inhibiting PD‐L1 palmitoylation enhances T‐cell immune responses against tumours. Nature Biomed Eng 2019;3:306‐317. [DOI] [PubMed] [Google Scholar]
- 40. Roszik J, Haydu LE, Hess KR, Oba J, Joon AY, Siroy AE, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med 2016;14:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017;357:409‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, et al. Dynamic changes in PD‐L1 expression and immune infiltrates early during treatment predict response to PD‐1 blockade in melanoma. Clin Cancer Res 2017;23:5024‐5033. [DOI] [PubMed] [Google Scholar]
- 44. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti–PD‐1 therapy. Clin Cancer Res 2014;20:5064‐5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davis AA, Patel VG. The role of PD‐L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer 2019;7:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee H‐H, Wang Y‐N, Xia W, Chen C‐H, Rau K‐M, Ye L, et al. Removal of N‐linked glycosylation enhances PD‐L1 detection and predicts anti‐PD‐1/PD‐L1 therapeutic efficacy. Cancer Cell 2019;36:168‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M‐K, Hsu J‐M, et al. PARP inhibitor upregulates PD‐L1 expression and enhances cancer‐associated immunosuppression. Clin Cancer Res 2017;23:3711‐3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double‐blind, multicentre, phase 2, dose‐ranging study. Lancet Oncol 2010;11:155‐164. [DOI] [PubMed] [Google Scholar]
- 49. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD‐1 antibody in cancer. N Engl J Med 2012;366:2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang W, Lie P, Guo M, He J. Risk of hepatotoxicity in cancer patients treated with immune checkpoint inhibitors: a systematic review and meta‐analysis of published data. Int J Cancer 2017;141:1018‐1028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Data Availability Statement
Messenger RNA (mRNA) expression levels of CMTM4 and CMTM6 of human HCC and ICC samples with their corresponding NT liver tissues were retrieved from transcriptome sequencing data from The Cancer Genome Atlas (TCGA) through cBioPortal (http://www.cbioportal.org). All data supporting the findings of this study are available within the article, in the Supporting Information, or from the corresponding author upon reasonable request.
Descriptions of the establishment of knockdown and knockout cells, western blotting, preparation of tumor samples into single‐cell suspension, flow cytometry, immunohistochemistry, RNA extraction, reverse‐transcription and quantitative real‐time polymerase chain reaction (RT‐PCR), copy number analysis, clinicopathological correlation and survival analysis, antibodies and recombinant proteins, short hairpin RNA (shRNA), single‐guide RNA (sgRNA), and primer sequences are provided in the Supporting Information. Sources of antibodies and recombinant proteins are listed in Supporting Table S1. Sequences of shRNAs and sgRNAs are listed in Supporting Table S2. Sequences of quantitative RT‐PCR primers are listed in Supporting Table S3.