

Abstract
Chromosome rearrangement is one of the hallmarks of human malignancies. Gene fusion is one of the consequences of chromosome rearrangements. In this report, we show that gene fusion between solute carrier family 45 member 2 (SLC45A2) and alpha‐methylacyl‐coenzyme A racemase (AMACR) occurs in eight different types of human malignancies, with frequencies ranging from 45% to 97%. The chimeric protein is translocated to the lysosomal membrane and activates the extracellular signal‐regulated kinase signaling cascade. The fusion protein promotes cell growth, accelerates migration, resists serum starvation‐induced cell death, and is essential for cancer growth in mouse xenograft cancer models. Introduction of SLC45A2‐AMACR into the mouse liver using a sleeping beauty transposon system and somatic knockout of phosphatase and TENsin homolog (Pten) generated spontaneous liver cancers within a short period. Conclusion: The gene fusion between SLC45A2 and AMACR may be a driving event for human liver cancer development.

Abbreviations
- aa
amino acid
- AMACR
alpha‐methylacyl‐coenzyme A racemase
- BD
binding domain
- BP
breakpoint
- cDNA
complementary DNA
- CMV
cytomegalovirus
- CRISPR
clustered regularly interspaced short palindromic repeats
- del
deleted
- ERK
extracellular signal‐regulated kinase
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GST
glutathione‐S‐transferase
- HCC
hepatocellular carcinoma
- his
histidine
- IC50
median inhibitory concentration
- IgG
immunoglobulin G
- KO
knockout
- LAMP1
lysosomal‐associated membrane protein 1
- LoxP
locus of x‐over, P1
- Lys
lysosome
- MEK
mitogen‐activated protein kinase/extracellular signal‐regulated kinase
- mTOR
mammalian target of rapamycin
- p
plasmid
- PCa
prostate cancer
- phospho
phosphorylated
- Pten
phosphatase and TENsin homolog
- SB
sleeping beauty
- SCID
severe combined immunodeficient
- SLAM
SLC45A2‐AMACR
- SLC45A2
solute carrier family 45 member 2
- Tet
ten‐eleven translocation
Human cancer is one of the leading causes of death in the world. Approximately 8.2 million people worldwide die from cancers annually.( 1 , 2 ) The development of human cancer is dependent on critical alterations of the human genome. Chromosome alterations, including mutations, copy number changes, and rearrangement, are some of the fundamental changes underlying cancer development. Identifying the driver chromosome alterations for cancer is the key to developing therapeutic interventions to treat cancer and to reduce the mortality of the disease.
Previously, through ultradeep transcriptome and whole‐genome sequencings of prostate cancer (PCa) samples, we identified a panel of cancer‐specific fusion genes.( 3 ) One of these fusion genes, solute carrier family 45 member 2 (SLC45A2)–alpha‐methylacyl‐coenzyme A racemase (AMACR), was present in a significant number of PCa samples. Subsequent analyses showed that SLC45A2‐AMACR gene fusion was present in urothelial carcinoma.( 4 ) High expression of SLC45A2‐AMACR was found in the non‐small cell lung cancer cell line H2198.( 5 ) Separately, the SLC45A2‐AMACR transcript was discovered in up to 7% of samples from patients with PCa in Asia.( 6 , 7 ) Furthermore, the SLC45A2‐AMACR fusion transcript was readily detectable in the serum samples of 33% of patients with liver cancer.( 8 ) The analysis of the matched liver tumor samples suggests that SLC45A2‐AMACR may be common in liver cancers. However, studies on SLC45A2‐AMACR are fragmented and lack insight into the function of gene fusion. The biological role of SLC45A2‐AMACR remains uncharacterized. In this study, we showed that SLC45A2‐AMACR gene fusion is present in eight different types of human malignancies and plays crucial roles in cancer transformation in both humans and mice. Its oncogenic activity is mediated by its interaction with and activation of extracellular signal‐regulated kinase (ERK).
Materials and Methods
Tissue Samples
We obtained 815 tissue specimens from the University of Pittsburgh Tissue Bank in compliance with institutional regulatory guidelines and approved by the Institutional Review Board of the University of Pittsburgh. Tissues comprised 219 PCa samples and 56 lymph nodes; 102 non‐small cell lung cancer samples; 61 ovarian cancer samples and 30 lymph nodes; 60 colon cancer samples and 30 lymph nodes; 70 liver cancer samples; 150 glioblastoma samples; 60 breast cancer samples and 30 lymph nodes; and 34 esophageal adenocarcinomas (Supporting Tables S1‐S8; Supporting Fig. S1). Cancer tissues that were obtained from other institutions included 16 non‐small cell lung cancer samples from the University of Kansas and 28 non‐small cell lung cancer samples from the University of Iowa. These samples were obtained in accordance with guidelines approved by the institutional review boards of the respective institutions. The cell lines used in the study were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia) and were cultured and maintained following the recommendations of the manufacturer.
For detailed descriptions of SLC45A2‐AMACR detection, fusion gene breakpoint discovery, yeast two‐hybrid screening,( 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 ) SLC45A2‐AMACR disruption in HUH7 and H1299 cells, colony formation and bromodeoxyuridine cell‐cycle assays,( 10 , 12 , 14 , 17 , 18 , 19 , 20 ) serum starvation cell death assay, wound healing assay, and 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay, please see the Supporting Methods.
Results
Chromosome Rearrangement Underlies SLC45A2‐AMACR Gene Fusion
SLC45A2 is a transporter protein known to be overexpressed in melanoma,( 21 , 22 ) while AMACR is an enzyme involved in the metabolism of branched fatty acids( 23 ) and is known for its overexpression in several human malignancies.( 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 ) In normal cells, AMACR is located on chromosome 5p13, while SLC45A2 is located on chromosome 5p13.2; both genes are located on the minor strand of the chromosome. However, in the fusion, the relative positions of these two genes are reversed (Fig. 1A). Chromosome breakpoints were identified between intron 2 of SLC45A2 and intron 1 of AMACR in primary cancer samples as well as cancer cell lines. A breakpoint between intron 2 of SLC45A2 and intron 1 of AMACR was identified. Interestingly, the same breakpoint was found in all cancer cell lines and primary liver cancer and PCa samples that were positive for the SLC45A2‐AMACR fusion. The gene fusion generates a chimeric protein with 187 amino acids from the N‐terminus of SLC45A2 and 311 amino acids from the C‐terminus of AMACR. As a result, eight transmembrane helical segments from SLC45A2 are replaced with the C‐terminus of AMACR, which contains an intact racemase domain (Fig. 1B).
FIG. 1.
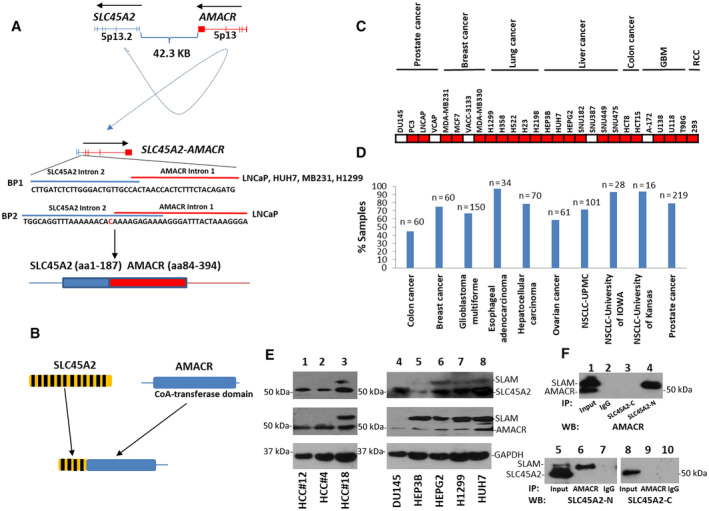
SLC45A2‐AMACR gene fusion in human malignancies. (A) Schematic diagram of the SLC45A2‐AMACR gene fusion. Top: SLC45A2 and AMACR on chromosome 5. The transcription directions are indicated. Mid: Result of chromosome rearrangement of SLC45A2 intron 2 and AMACR intron 1. The breakpoint sequences between SLC45A2 and AMACR are shown. Representative chromatogram of Sanger’s sequencing from HUH7 cells are shown. Bottom: Translation product of SLC45A2‐AMACR gene fusion; blue is the SLC45A2 domain, red is the AMACR domain. (B) Structure of the SLC45A2, AMACR, and SLC45A2‐AMACR proteins; yellow is SLC45A2, blue is the AMACR racemase domain, black stripes show the transmembrane helical segments. (C) Distribution of SLC45A2‐AMACR in human cancer cell lines; red is SLC45A2‐AMACR positive, white is SLC45A2‐AMACR negative. The names and origins of the malignancies of the cell lines are indicated. (D) Frequencies of SLC45A2‐AMACR in primary human malignancies. Each type of sample cohort is indicated. The number of samples is indicated at the top of the bar. (E) Expression of SLC45A2‐AMACR proteins in human cancers. Lanes 1‐3 are the primary HCC samples. HCC#12 and HCC#4 are negative for SLC45A2‐AMACR, while HCC#18 is positive. Lanes 4‐8: the human cancer cell lines DU145 (PCa), HEP3B (liver cancer), HEPG2 (liver cancer), H1299 (lung cancer), and HUH7 (liver cancer). (F) Immunoprecipitation to identify the SLC45A2‐AMACR chimeric protein. Top: Immunoblotting using an antibody specific for the C‐terminus of AMACR in the immunoprecipitates of antibodies specific for the N‐terminus of SLC45A2, the C‐terminus of SLC45A2, or IgG control of protein lysates from HUH7 cells. Bottom: Immunoblotting using an antibody specific for the N‐terminus (left) or C‐terminus (right) of SLC45A2 on the immunoprecipitates of antibodies specific for the C‐terminus of AMACR or IgG control of protein lysates from HUH7 cells. Abbreviations: CoA, coenzyme A; IP, immunoprecipitation; NSCLC, non‐small cell lung cancer; RCC, renal cell carcinoma; SLC45A2‐C, C‐terminus of SLC45A2; SCL45A2‐N, N‐terminus of SLC45A2; WB, western blot.
SLC45A2‐AMACR Gene Fusion is Common in Human Cancers
To examine the frequency of SLC45A2‐AMACR gene fusion in human cancers, we analyzed 27 human cancer cell lines, including PCa (DU145, PC3, LNCaP, and VCaP), breast cancer (MDA‐MB231, MCF7, VACC‐3133, and MDA‐MB330), lung cancer (H1299, H358, H522, H23, and H2198), liver cancer (HEP3B, HuH7, HEPG2, SNU182, SNU387, SNU449, and SNU475), colon cancer (HCT8 and HCT15), glioblastoma multiforme (GBM) (A‐172, U138, U118, and T98G), and renal cell carcinoma (293) (Fig. 1C; Supporting Fig. S2A). All lung cancer cell lines were positive for the SLC45A2‐AMACR fusion (5/5), while six of seven liver cancer cell lines were positive for the fusion gene. Cell lines originating from ductal carcinoma of the breast (MCF7, MDA‐MB231, and MDA‐MB330) were positive for the fusion, while the lobular type (VACC‐3133) was negative. Fifty percent of PCa cell lines were positive for the SLC45A2‐AMACR fusion (2/4) as were 75% of the GBM cell lines. Two colon cancer cell lines and one renal cell carcinoma cell line were also positive for the fusion gene.
The high positive rate of SLC45A2‐AMACR in a variety of human cancer cell lines suggests that many primary human cancer samples might be positive for the fusion gene. Indeed, the examination of 799 human cancer samples revealed various rates of SLC45A2‐AMACR in these cancers, ranging from 45% of colon cancer samples to 97% of esophageal adenocarcinoma samples (Fig. 1D; Supporting Tables S1‐S8; Supporting Fig. S1). Interestingly, colon cancer positivity for SLC45A2‐AMACR was associated with poor differentiation of the colon cancer (86% [6/7] versus 38% [20/52], P = 0.037). In contrast, patients with non‐small cell lung cancer who were negative for SLC45A2‐AMACR had a lower 90‐month survival rate (0%, 0/15) than patients with positive cancer samples (29.4%, 15/51; P = 0.02). In a cohort from the University of Iowa, patients with non‐small cell lung cancer who were negative for SLC45A2‐AMACR showed associations with metastasis (P = 0.0048) and more advanced clinical stages (P = 0.021). For lung cancer, samples from patients who were white had a higher frequency of SLC45A2‐AMACR (74%, 62/84) than from those who were black (36%, 4/11; P = 0.03).
SLC45A2‐AMACR is Expressed as a Chimeric Protein
To identify the isoform of SLC45A2‐AMACR, extended long reverse‐transcription polymerase chain reaction (RT‐PCR) was performed on three liver cancer samples and several human cancer cell lines, including HUH7, using primers encompassing the entire coding sequence of the fusion gene. A 1.6‐kDa complementary DNA (cDNA) was found in the samples positive for SLC45A2‐AMACR (Supporting Fig. S2B). Sanger sequencing revealed that the AMACR domain in the fusion was identical to the truncated variant 3 of AMACR, while the SLC45A2 domain was identical to the 5' end of variants 1‐3 of SLC45A2 transcripts. To examine whether SLC45A2‐AMACR is expressed as a chimeric protein in cancer cells, immunoblotting using an antibody specific for the N‐terminus of SLC45A2 was performed. Hepatocellular carcinoma (HCC) samples positive for SLC45A2‐AMACR (HCC#18) showed a protein of 60 kDa detected by the antibody in addition to the SLC45A2 protein (55 kDa) (Fig. 1E). In contrast, the 60‐kDa protein was absent in HCC samples (HCC#4 and HCC#12) that were negative for the fusion gene. The 60‐kDa protein was also present in cell lines that were positive for the SLC45A2‐AMACR fusion (HEP3B, HEPG2, H1299, and HUH7) but absent in the cell line negative for the fusion gene (DU145). When an antibody specific for the C‐terminus of AMACR was applied, the 60‐kDa protein was detected in the HCC sample that was positive for SLC45A2‐AMACR in addition to the AMACR protein (47 kDa) but was not detected in the HCC samples that were negative for the fusion gene. Similar results were obtained for the cancer cell lines. Cells positive for SLC45A2‐AMACR contained a 60‐kDa protein and a 47‐kDa AMACR detected by the AMACR antibody, while cells negative for the fusion contained only the 47‐kDa AMACR band in the immunoblotting analysis (Fig. 1E). To examine whether the 60‐kDa protein is the translation product of the SLC45A2‐AMACR fusion gene, the protein extracts from HUH7 cells were immunoprecipitated with an antibody specific for the SLC45A2 N‐terminus. The immunoprecipitates were blotted with an antibody specific for AMACR. The same 60‐kDa protein band was visualized in the immunoprecipitate specific for the N‐terminus of SLC45A2 but not for the C‐terminus or immunoglobulin G (IgG) control (Fig. 1F). The 60‐kDa protein was similarly identified on AMACR immunoprecipitation using the antibody specific for the N‐terminus of SLC45A2 but not using the antibody specific for the C‐terminus of SLC45A2. Based on these analyses, we concluded that the 60‐kDa band represents the protein product of the SLC45A2‐AMACR gene fusion.
SLC45A2‐AMACR Gene Product Exhibits Lysosomal Membrane Translocation
SLC45A2 is a membrane‐associated protein mostly located in the lysosome/melanosome membrane.( 39 ) This protein is involved in the biosynthesis of melanin and the transport of sucrose.( 40 ) In contrast, AMACR is a racemase located in the mitochondria that are involved in the metabolism of branched fatty acids( 41 , 42 ) and is essential for bile acid biosynthesis and metabolism.( 43 ) The fusion between SLC45A2 and AMACR maintains four transmembrane helical segments of SLC45A2 in the N‐terminus while replacing the C‐terminus with the racemase domain of AMACR. As a result, the chimeric protein may be translocated to the lysosomal membrane. To test this hypothesis, NIH3T3 and HUH7 cells were transformed with plasmid (p)CDNA4‐SLC45A2‐AMACR‐FLAG/pCDNA6TO to induce the expression of SLC45A2‐AMACR‐FLAG. Coimmunostaining was performed with the lysosomal residential enzyme lysosomal‐associated membrane protein 1 (LAMP1). Colocalization between LAMP1 and SLC45A2‐AMACR‐FLAG was apparent in both cell lines (Fig. 2A,B). To validate the imaging analyses, HUH7 cells were fractionated into mitochondrial, lysosomal, and nuclear fractions (Fig. 2C). AMACR was readily detected in the mitochondria, while endogenous SLC45A2‐AMACR was located in the lysosome in HUH7 cells. When NIH3T3 cells were transformed with the SLC45A2‐AMACR‐FLAG construct, the transfected SLC45A2‐AMACR‐FLAG was exclusively located in the lysosome. Interestingly, when the SLC45A2 N‐terminus was removed from the chimeric protein, the truncated AMACR protein was retained in the mitochondria (Fig. 2C). Thus, the translocation of the SLC45A2‐AMACR chimeric protein to the lysosomal membrane is dependent on the N‐terminus of SLC45A2.
FIG. 2.
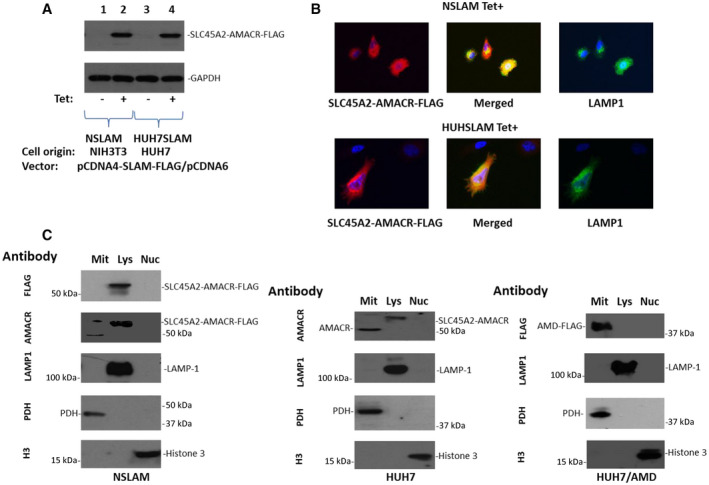
SLC45A2‐AMACR is localized in the lysosome membrane. (A) Immunoblotting of SLC45A2‐AMACR‐FLAG using an antibody specific for FLAG in NIH3T3 (NSLAM) and HUH7 (HUH7SLAM) cells transformed with pCDNA4‐SLC45A2‐AMACR‐FLAG/pCDNA6TO, treated with or without tetracycline. Antibodies specific for GAPDH are for the loading control. (B) Colocalization of SLC45A2‐AMACR‐FLAG and the lysosome resident protein LAMP1 in NSLAM and HUH7SLAM cells using an antibody specific for FLAG and LAMP1 proteins (20x images). (C) Lysosomes, mitochondria, and nucleus fractionation of SLC45A2, AMACR, and SLC45A2‐AMACR proteins from HUH7, NSLAM, and HUH7‐AMD (HUH7 transformed with pCDNA4‐ΔAMACRaa84‐394‐FLAG) cells. The antibody for each immunoblot is indicated. Abbreviations: H3, histone H3; Mit, mitochondria; Nuc, nucleus; PDH, pyruvate dehydrogenase.
SLC45A2‐AMACR Shows Oncogenic Activity
Most cancer cell lines are positive for the SLC45A2‐AMACR gene fusion. To examine the impact of SLC45A2‐AMACR fusion on the carcinogenesis of the cancer cell lines, we chose to disrupt the gene fusion between SLC45A2 and AMACR in H1299 (lung cancer) and HuH7 (liver cancer) cells by inserting an mCherry‐zeo cassette into the chromosomal breakpoint SLC45A2‐AMACR, using clustered regularly interspaced short palindromic repeats (CRISPR)‐Cas9 gene‐editing technology (Fig. 3A). Cell lines displaying mCherry fluorescence and zeocin resistance were selected. These cell lines were verified to have disrupted SLC45A2‐AMACR fusion transcripts (Supporting Fig. S3). The disruption of SLC45A2‐AMACR in H1299 and HuH7 cells reduced colony formation by 5.1‐fold and 4.6‐fold, respectively (Fig. 3B). The migration of these cancer cells was retarded by 2.1‐fold for H1299 and 2.9‐fold for HuH7 cells when SLC45A2‐AMACR was knocked out in the cell lines (Fig. 3C). Interestingly, disruption of SLC45A2‐AMACR in H1299 cells sensitized the cancer cells to serum starvation; only 29% of cells survived after serum starvation for 7 days when SLC45A2‐AMACR was disrupted versus approximately 85% of wild‐type H1299 cells that survived the same period (P < 0.01) (Fig. 3D). Similar results of increased sensitivity to serum starvation were found in HuH7 cells when the SLC45A2‐AMACR fusion was knocked out (62.3% versus 41.2% on day 10, P < 0.01). When cancer cells were xenografted into the subcutaneous regions of severe combined immunodeficient (SCID) mice, H1299 cells with the knockout of SLC45A2‐AMACR failed to form tumors in the animals in 8 weeks while the wild‐type H1299 cells formed tumors with an average size of 4,206 mm3 (Fig. 3E). HuH7 cells with SLC45A2‐AMACR had a significant delay in forming tumors and had an average tumor size 51‐fold smaller than the wild‐type control (3,935 mm3 versus 76 mm3, P < 0.01; Fig. 3E). Extensive metastases were found in mice xenografted with wild‐type HuH7 (100%; Fig. 3F) and H1299 (62.5%) tumors. In contrast, no apparent metastasis was found in any of the mice xenografted with HuH7‐knockout (KO) or H1299KO2 cells. All animals xenografted with HuH7 cells succumbed to tumors in 3 weeks (Fig. 3G; Supporting Table S9), while none of the SLC45A2‐AMACR‐knockout counterparts died in the same time frame after the xenografting.
FIG. 3.
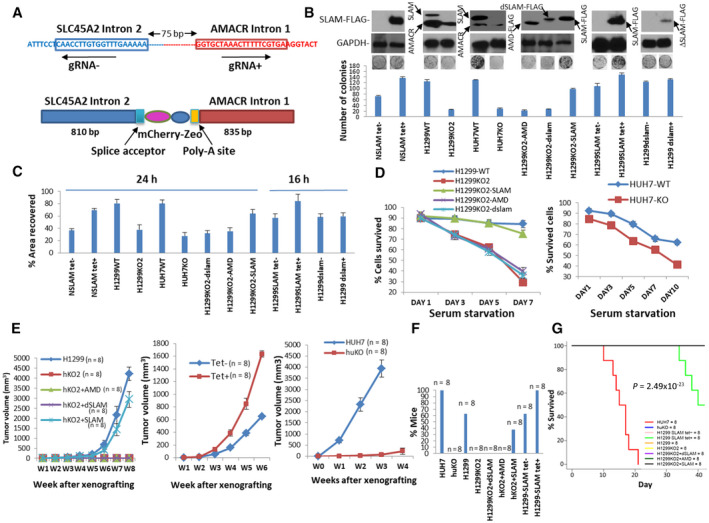
Oncogenic activity of SLC45A2‐AMACR in vitro. (A) Schematic diagram of CRISPR‐Cas9 knockout for SLC45A2‐AMACR. Top: SLC45A2‐AMACR breakpoint region (BP1). The gRNA target sequences are boxed. The direction of gRNA is indicated. Bottom: SLC45A2‐AMACR‐knockout cassette. (B) Colony formation assays of cells with forced expression of SLC45A2‐AMACR or knockout of endogenous SLC45A2‐AMACR. NSLAM‐NIH3T3 cells transformed with pCDNA4‐ SLC45A2‐AMACR‐FLAG/pDNA6TO; H1299WT‐H1299 cells with control vector; H1299KO1‐H1299 cells with genome disruption of SLC45A2‐AMACR fusion; HuH7WT‐HuH7 with control vector; HuH7KO‐HuH7 cells with genome disruption of SLC45A2‐AMACR fusion; H1299KO2‐dSLAM (H1299KO2 cells rescued with FLAG‐SLC45A2‐AMACRdelaa368‐397); H1299KO2‐AMD (H1299KO1 cells rescued with FLAG‐AMACRaa84‐394); H1299KO2‐SLAM (H1299KO1 cells rescued with pCMV‐SLC45A2‐AMACR‐FLAG); H1299SLAM‐H1299 cells transformed with pCDNA4‐SLC45A2‐AMACR‐FLAG/pCDNA6TO; H1299dSLAM‐H1299 cells transformed with pCDNA4‐ΔSLC45A2‐AMACRdelaa368‐397‐FLAG/pCDNA6TO. Data show mean ± SD. (C) Wound healing assays of cells from (B). Data show mean ± SD. (D) Serum starvation assays of cancer cells with or without SLC45A2‐AMACR. Left: Serum starvation of H1299 cells and their mutant counterparts. Right: Serum starvation of HUH7 cells with or without SLC45A2‐AMACR. Data show mean ± SD.(E) SCID mouse xenografted tumor models of SLC45A2‐AMACR. Left panel: H1299 and its SLC45A2 knockout and knockout‐rescued models. Middle panel: H1299 cells with overexpression of SLC45A2‐AMACR models. Right panel: HUH7 cells with or without SLC45A2‐AMACR. Data show mean ± SD.(F) Frequency of metastasis of (E). (G) Kaplan‐Meier analysis of mouse xenografts with HuH7 and H1299 cells and their mutant counterparts. Abbreviations: bp, base pair; gRNA, guide RNA; h, hours; W, weeks; WT, wild type.
In contrast to the disruption of SLC45A2‐AMACR, overexpression of SLC45A2‐AMACR increased colony formation and migration rates of NIH3T3 and H1299 cells (Fig. 3B,C). Overexpression of SLC45A2‐AMACR in H1299 cells also increased tumor sizes by an average of 2.5‐fold (P < 0.05) (Fig. 3E) and the mortality of the animals (50% versus 0%, P < 0.05). Interestingly, when H1299ko cells were reintroduced with SLC45A2‐AMACR by transforming the cells with the plasmid cytomegalovirus (pCMV)‐SLC45A2‐AMACR‐FLAG construct, the oncogenic activity of the cell line reappeared; a 4.6‐fold increase in colony formation and a 2‐fold increase in migration over the SLC45A2‐AMACR‐knockout control were observed. In addition, over 75% of SLC45A2‐AMACR‐FLAG‐rescued cells survived 10 days under serum starvation versus 29% of the SLC45A2‐AMACR‐knockout counterparts. Unlike the SLC45A2‐AMACR‐knockout controls that generated no identifiable tumors, the rescued H1299KO2 cells produced large tumors (2,935 mm3 average at 8 weeks after xenografting) in SCID mice and had a 37.5% metastasis rate. In contrast, H1299KO2 cells rescued with the AMACR domain only (amino acids [aa] 84‐394) showed no obvious impact on colony formation, migration, resistance to serum starvation, and tumor generation in the xenograft model compared with the controls. These results suggest that the subcellular localization of SLC45A2‐AMACR may be crucial for its oncogenic activities.
SLC45A2‐AMACR Interacts With and Activates ERK In Vitro and In Vivo
To investigate the oncogenic mechanism of SLC45A2‐AMACR, a yeast two‐hybrid screening of a prostate cDNA library using plasmid binding domain (pBD)‐SLC45A2‐AMACR was performed. After three rounds of nutrient selection, 15 colonies containing unique cDNA sequences were identified. One of these cDNAs was identified as ERK2. To identify the domain in SLC45A2‐AMACR that interacts with ERK2, the SLC45A2 domain and AMACR domain of the chimeric protein were separately ligated into pBD. The cotransfection results showed that the AMACR domain of the fusion chimera protein was responsible for the interaction with ERK2 (Fig. 4A). To validate the interaction results from the yeast two‐hybrid assay, coimmunoprecipitation was performed on NIH3T3 cells transformed with SLC45A2‐AMACR‐FLAG. Coimmunoprecipitation of SLC45A2‐AMACR‐FLAG and ERK1/2 was readily detectable in both the ERK and FLAG immunoprecipitates (Fig. 4B). These coimmunoprecipitation results were reproduced in HUH7 cells using an antibody specific for ERK or the SLC45A2 N‐terminus. Colocalization between ERK1/2 and SLC45A2‐AMACR‐FLAG was identified in NIH3T3 cells transformed with the fusion gene (Fig. 4C). To rule out the possibility that a bridge protein is required for the interaction between ERK2 and SLC45A2‐AMACR, glutathione‐S‐transferase (GST)‐SLC45A2‐AMACR recombinant protein was produced from Escherichia coli. The purified recombinant GST‐SLC45A2‐AMACR bound directly to recombinant histidine (His)TAG‐ERK2 (Fig. 4D,E). To identify the binding motif in the AMACR domain that binds with the recombinant ERK2, serial deletions of the AMACR domain were made. The binding analyses indicated that aa 368‐397 of SLC45A2‐AMACR were responsible for binding with ERK2 (Fig. 4E).
FIG. 4.
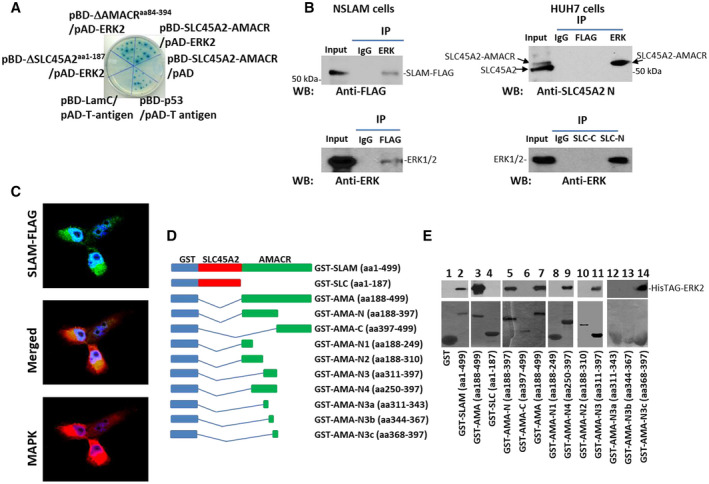
SLC45A2‐AMACR interaction with ERK. (A) Yeast two‐hybrid validation of the interaction between SLC45A2‐AMACR and ERK. Cotransfection of pBD‐SLC45A2‐AMACR and pAD‐ERK2 resulted in colonies grown in SD‐Ade/‐His/‐Leu/Trp agar plates and positive for α‐galactosidase. The interaction between BD‐p53 and activation domain (AD)‐T‐antigen was used as the positive control, while the interaction between BD‐Lamin C (LamC) and AD‐T‐antigen was used as the negative control. (B) Coimmunoprecipitation of SLC45A2‐AMACR and ERK in cell cultures. Left panel: Coimmunoprecipitation in NSLAM cells induced to express SLC45A2‐AMACR‐FLAG (SLAM‐FLAG). Top left: Immunoblotting using an antibody specific for FLAG on the immunoprecipitate of ERK antibody or control mouse IgG. Bottom left: Immunoblotting using antibody specific for anti‐ERK on the immunoprecipitate of FLAG antibody or control mouse IgG. Right panel: Coimmunoprecipitation in HUH7 cells. Top right: Immunoblotting using an antibody specific for the N‐terminus of SLC45A2 on the immunoprecipitate of ERK antibody or control mouse IgG or FLAG antibody. Bottom right: Immunoblotting using antibody specific for ERK on the immunoprecipitate by the antibody specific for the N‐terminus of SLC45A2 or control mouse IgG or SLC45A2 C‐terminus antibody. (C) Colocalization between SLC45A2‐AMACR‐FLAG and ERK using antibodies specific for ERK and FLAG (20x images). (D) Schematic diagrams of deletion constructs of GST‐SLC45A2‐AMACR. The GST, SLC45A2, and AMACR domains are indicated. (E) Binding assays between GST‐SLC45A2‐AMACR mutants and recombinant HisTAG‐ERK2. Top: Immunoblotting of HisTAG‐ERK2 to detect the binding between HisTAG‐ERK2 and SLC45A2‐AMACR mutants using an antibody specific for ERK. Bottom: Coomassie blue staining of GST‐SLC45A2‐AMACR mutants. Abbreviations: AD‐T, AMA, AMACR; IP, immunoprecipitation; LamC, SD‐Ade/‐His/‐Leu/Trp, synthetic‐defined adenine, histidine, leucine and tryptophan; SLC, SLC45A2; SLC‐C, SLC45A2 C‐terminus; SLC‐N, N‐terminus of SLC45A2; WB, western blot.
SLC45A2‐AMACR Activates ERK
We performed in vitro kinase assays using recombinant ERK2 to investigate the impact of SLC45A2‐AMACR binding on the kinase activity of ERK. The presence of GST‐SLC45A2‐AMACR increased the kinase activity of ERK2 by 2.6‐fold (Fig. 5A). The AMACR domain also increased the kinase activity of ERK by 2.7‐fold. However, when the assay was applied with the ERK2‐binding motif only from SLC45A2‐AMACR, the kinase activity of ERK2 was not changed. These results indicated that SLC45A2‐AMACR is a partner of ERK2 and that the binding between them enhances the kinase activity of ERK. To examine whether SLC45A2‐AMACR also enhances the kinase activity of ERK2 in vivo, HUH7 and its SLC45A2‐AMACR‐knockout counterpart were analyzed for their ERK activation. The disruption of SLC45A2‐AMACR dramatically reduced the phosphorylation of threonine 202 and tyrosine 204 of ERK (Fig. 5B). This change was accompanied by the elimination of mitogen‐activated protein kinase (MAPK)/ERK (MEK; pS217/221) and mammalian target of rapamycin (mTOR; pS2448) phosphorylation. Similar results were found for H1299KO2 cells where SLC45A2‐AMACR was knocked out in H1299 cells; knockout of SLC45A2‐AMACR largely eliminated the activation of ERK, MEK, and mTOR in the H1299 cell line. When HUH7 cells were forced to overexpress SLC45A2‐AMACR, the phosphorylation of ERK, MEK, and mTOR increased. However, no change in the activation status of ERK, MEK, and mTOR was found when HUH7 cells were forced to express the mutant ΔSLC45A2‐AMACRdeleted (del)aa368‐397, where the binding motif for ERK2 was deleted. When SLC45A2‐AMACR was reintroduced into H1299ko cells, ERK was reactivated along with MEK activation (Fig. 5C). The reactivation of ERK and MEK, however, was not found when mutant ΔSLC45A2‐AMACRdelaa368‐397 or ΔAMACRaa84‐394 was introduced. These results suggested that the activation of the ERK signaling pathway by SLC45A2‐AMACR (Fig. 5D) in vivo requires both the lysosomal membrane localization and the binding of ERK.
FIG. 5.
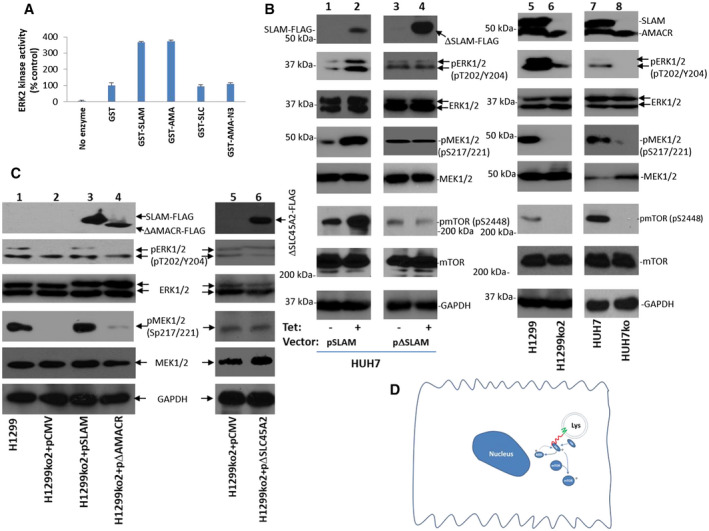
SLC45A2‐AMACR activates ERK kinase in vitro and in vivo. (A) GST‐SLC45A2‐AMACR (GST‐SLAM) enhanced the kinase activity of recombinant ERK2 on basic myelin protein. The kinase activity is calculated as percentage recombinant ERK2 kinase activity without any added GST recombinant protein component in a kinase assay. Standard deviations are indicated. (B) SLC45A2‐AMACR activates the kinase cascade of ERK in vivo. Overexpression of SLC45A2‐AMACR but not its mutant with a deletion of the ERK binding motif enhanced the activation of ERK1/2 kinase activity and its cascade signaling molecules MEK and mTOR in HUH7 cells (lanes 1‐4). Knockout of SLC45A2‐AMACR deactivates the ERK kinase cascade in H1299 and HuH7 cells (lanes 5‐8). (C) Rescue of H1299KO2 cells with SLC45A2‐AMACR‐FLAG reactivates ERK kinase cascades but not with the SLC45A2‐AMACR mutant that lacks the ERK binding motif or the mutant that lacks the SLC45A2 domain. (D) Diagram of SLC45A2‐AMACR signaling cascade; green line shows the SLC45A2 domain, red line shows the AMACR domain.
To examine whether ERK‐MEK activation by SLC45A2‐AMACR is also reflected in human cancer samples, immunostainings of phosphorylated (phospho)‐ERK1/2 and phospho‐MEK were performed on liver cancer samples positive for SLC45A2‐AMACR. The results showed that the presence of SLC45A2‐AMACR increased the levels of phospho‐ERK1/2 and phospho‐MEK in liver cancer samples (Supporting Figs. S4 and S5).
Next, we analyzed whether SLC45A2‐AMACR fusion plays a role in sensitizing cancer cells to ERK inhibitor. HUH7 and H1299 cells and their corresponding SLC45A2‐AMACR‐knockout counterparts were treated with SCH772984, a potent ERK inhibitor. Knockout of SLC45A2‐AMACR abrogated the sensitivity of HUH7 (median inhibitory concentration [IC50], 41.6 nM versus >1,000 nM) and H1299 cells (IC50, 572.3 nM versus >1,000 nM) to SCH772984, while overexpression of SLC45A2‐AMACR in H1299 cells decreased the IC50 of SCH772984 by 9.7‐fold (59 nM versus 572.3 nM) (Supporting Fig. S6). These results lend further support that ERK plays a key role in SLC45A2‐AMACR‐mediated transformation.
SLC45A2‐AMACR Fusion and Somatic Phosphatase and TENsin Homolog Knockout Induced Spontaneous Liver Cancer in Mice
Phosphatase and TENsin homolog (Pten) deletion and loss of expression in HCC are frequent (up to 50%).( 44 , 45 , 46 ) The analysis of our previous publication showed that Pten loss occurred in 31% (17/55) of HCC samples that were positive for SLC45A2‐AMACR.( 47 ) Thus, the combination of Pten loss and gene fusion between SLC45A2 and AMACR may play a critical role in HCC generation. To investigate this hypothesis directly, we cloned SLC45A2‐AMACR‐FLAG into pT3‐E5α to create pT3‐ SLC45A2‐AMACR‐FLAG. We somatically knocked out Pten in the liver by treating C57Bllocus of x‐over, P1 (LoxP)‐Pten‐LoxP mice with adeno‐associated virus 8 (AAV8)‐cre. We then delivered pT3‐ SLC45A2‐AMACR‐FLAG/plasmid sleeping beauty (pSB) into 3%‐5% hepatocytes by hydrodynamic tail vein injection. Cancer nodules in the livers were identified from 9 to 16 weeks in all animals (6/6) treated with pT3‐ SLC45A2‐AMACR‐FLAG/pSB (Fig. 6A‐C). In contrast, none of the animals with somatic Pten loss and treated with pT3/pSB developed cancer in the same period. The liver to body ratios were significantly higher due to the presence of the cancer (Fig. 6D). Derangement of liver architecture, deposition of fat vacuoles, and activation of MEK were found in the cancer samples induced by SLC45A2‐AMACR (Supporting Fig. S7). Cancer nodules had a 15.6‐fold higher frequency of Ki67‐positive cells on average (Fig. 6E). The tumor nodules displayed similar higher levels of ERK, MEK1/2, and mTOR activation compared with the controls (Fig. 6F). These results indicate that SLC45A2‐AMACR fusion is a driver of mouse liver cancer development.
FIG. 6.
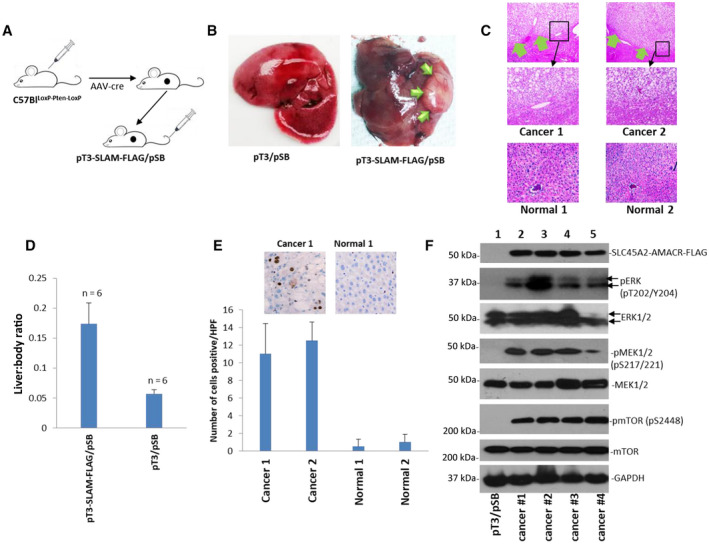
SLC45A2‐AMACR induced spontaneous liver cancer. (A) Schematic diagram of the hydrodynamic injection of pT3‐SLC45A2‐AMACR‐FLAG/pSB to induce liver cancer in C57Blloxp‐Pten‐Loxp mice treated with AAV8‐cre. (B) Representative picture of mouse liver treated with pT3/pSB or pT3‐SLC45A2‐AMACR‐FLAG/pSB (0.5 images). Liver cancer nodules are indicated by green arrows. (C) Hematoxylin and eosin staining of liver samples from pT3‐SLC45A2‐AMACR‐FLAG/pSB‐treated (9 weeks) mice (top, 4x images) or pT3/pSB‐treated (9 weeks) mice (bottom, 20x images). Liver cancer is indicated by green arrows. Middle panels represent the high magnification (20x) images of the indicated areas of the top panel. (D) SLC45A2‐AMACR increased the liver to body ratio. Data show mean ± SD.(E) SLC45A2‐AMACR‐FLAG increased Ki67‐positive cells in liver cancer (20x images). (F) SLC45A2‐AMACR‐FLAG enhanced the activation of ERK kinase and its signaling molecules MEK and mTOR kinases. Abbreviations: AAV8, adeno‐associated virus 8; HPF, high‐power field.
Discussion
Chromosome rearrangement is one of the main features of human cancer. It includes translocation, deletion, duplication, and inversion of the DNA sequences in the chromosome(s). SLC45A2‐AMACR gene fusion was discovered in the transcriptome sequencing studies in several research group data sets in a significant number of PCa samples( 3 , 6 , 7 ) and in liver cancers,( 8 ) bladder cancer( 4 ) and lung cancer cell lines.( 5 ) Using sensitive TaqMan qRT‐PCR and Sanger sequencing, we showed that SLC45A2‐AMACR is present in eight different types of human cancers with high frequencies. Interestingly, most lymph node metastases retain or gain SLC45A2‐AMACR expression compared with the primary cancers (Supporting Fig. S2). Characterizations of SLC45A2‐AMACR in our studies suggest SLC45A2‐AMACR is crucial in the development of human cancer. First, the disruption of SLC45A2‐AMACR in cancer cell lines dramatically reduced the carcinogenesis of the cell lines. In H1299 cells, the knockout of this fusion gene eliminated the ability of the cancer cells to form tumors in SCID mice. In contrast, forced overexpression of SLC45A2‐AMACR in the same cell line increased tumor growth and the rate of metastasis in the xenograft animal model. The results suggest that the transformation of the cancer cell line is dependent on this gene fusion. Second, most lymph node metastases of colon cancer, PCa, ovarian cancer, and breast cancer retained SLC45A2‐AMACR gene fusion in comparison with the matched primary cancer samples. Significant numbers of samples had a gain of SLC45A2‐AMACR gene fusion in the metastatic sites over the matched primary cancer samples, implying a critical role of this gene fusion in metastasis. Third, the introduction of SLC45A2‐AMACR into the livers of mice along with somatic Pten knockout induced a high rate of spontaneous liver cancer (6/6, 100%) in 16 weeks while none of the control animals displayed any neoplasm. Interestingly, introduction of pT3‐SLC45A2‐AMACR‐FLAG/pSB alone did not induce liver cancer in a similar period (Supporting Fig. S8), suggesting a requirement of “two‐hits” for liver cancer generation, similar to most carcinogenesis in humans.( 48 ) In light of the high rate of SLC45A2‐AMACR in various human cancers, the presence of this fusion gene may be essential to human cancer development.
Topology analysis showed that SLC45A2 is a multipass membrane protein.( 39 ) Truncation at aa 187 abrogates the transmembrane pass that traverses toward the lumen of lysosome and leaves the AMACR domain exposed in the cytoplasm compartment (Fig. 5D). Thus, the AMACR domain of the SLC45A2‐AMACR chimera protein is accessible to cytosolic signaling molecules. Our analyses revealed that SLC45A2‐AMACR binds ERK2 through aa 368‐397. This motif is critical to the cancer transforming activity of SLC45A2‐AMACR because the fusion protein mutant with deletion of this sequence did not result in transformation. Interestingly, this sequence is outside the boundary of the racemase motifs in the AMACR domain. The lysosomal membrane location is also critical to the transformation activity of SLC45A2‐AMACR. The cancer cell line with SLC45A2‐AMACR knockout lost most of the cancer phenotype but regained the phenotype after the cells were transfected with SLC45A2‐AMACR. On the other hand, these SLC45A2‐AMACR‐knockout cells failed to recover most of the cancer transformation with the expression of mitochondrial‐localized AMACR domain. Although the truncated AMACR domain protein retains the ERK2 binding motif, mitochondria localization may prevent its interaction with ERK. ERK, an effector, converges several tyrosine kinase signaling activities, like epidermal growth factor receptor, platelet‐derived growth factor receptor, and hepatocyte growth factor receptor.( 49 ) Our study suggests that ERK kinase activation is short circuited by SLC45A2‐AMACR/ERK interaction, resulting in activation of the ERK pathway without the upstream signaling. Interestingly, MEK kinase was activated along with ERK by SLC45A2‐AMACR, raising the possibility of overflow activation of kinase pathways. Because MEK is widely recognized as the upstream kinase for ERK activation, the activation of MEK by ERK may create a self‐reinforced kinase activation circuit to amply the progrowth signals. Thus, SLC45A2‐AMACR may play a critical role in the transformation of cancer cells by promoting cell growth, motility, and survival without the excessive activation of upstream growth factor receptors. Future transcriptome and proteomic analyses on SLC45A2‐AMACR‐transformed cells may gain additional insight into the mechanisms of this oncogene.
To our knowledge, SLC45A2‐AMACR is one of the most widespread fusion genes in human malignancies. Overexpression of AMACR was found in a variety of human cancers. Some of the incidences of AMACR overexpression in cancers may be overlapped with SLC45A2‐AMACR fusion because the fusion retains most RNA sequence and protein domains of AMACR, including an intact racemase domain. Because the transforming activity of SLC45A2‐AMACR is ERK activation dependent, approaches targeting ERK and its downstream signaling molecules should be considered. The uniqueness of the SLC45A2‐AMACR gene in cancer cells would be a potential target in our fusion gene targeting approach using CRISPR‐Cas9 genome editing to insert a suicide gene into the breakpoint region of a fusion gene. Such an approach achieved partial remissions of xenografted cancers that contain fusion gene breakpoints.( 50 ) The high frequencies of SLC45A2‐AMACR and its near uniform breakpoint region make it feasible to develop a highly selective and specific genome intervention in the treatment of human cancers.
Supporting information
Supplementary Material
Acknowledgment
We thank Songyang Zheng and Meijuan Zou for technical support.
Supported in part by the National Cancer Institute (grants RO1 CA098249 and 1R56CA229262‐01 to J.H.L.), Department of Defense (W81XWH‐16‐1‐0541 to J.H.L.), and University of Pittsburgh Medical Center Enterprises, Prediction of Prostate Cancer Outcomes by Fusion Genes (grants to J.H.L., J.N., and G.M.).
Potential conflict of interest: Dr. Zhang consults for AstraZeneca, Biodesix, Bayer, Daiichi Sankyo, and Novocure; he is on the speakers’ bureau for AstraZeneca and MJH Life Sciences; he received grants from AbbVie, AstraZeneca, Genentech, Hengrui, Mirati, and Novartis. Dr. Luketich owns stock in Cigna Corporation and Intuitive Surgical Inc.; he consults for Intuitive Surgical Inc. and Medtronics; he is on the speakers’ bureau for Covidien. The other authors have nothing to report.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69‐90.Erratum in: CA Cancer J Clin 2011;61:134. [DOI] [PubMed] [Google Scholar]
- 2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 3. Yu YP, Ding Y, Chen Z, Liu S, Michalopoulos A, Chen R, et al. Novel fusion transcripts associate with progressive prostate cancer. Am J Pathol 2014;184:2840‐2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshihara K, Wang Q, Torres‐Garcia W, Zheng S, Vegesna R, Kim H, et al. The landscape and therapeutic relevance of cancer‐associated transcript fusions. Oncogene 2015;34:4845‐4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 2015;33:306‐312. [DOI] [PubMed] [Google Scholar]
- 6. Yang J, Chen Y, Lu J, Wang X, Wang L, Liang J, et al. Identification and characterization of novel fusion genes in prostate cancer by targeted RNA capture and next‐generation sequencing. Acta Biochim Biophys Sin (Shanghai) 2018;50:1166‐1172. [DOI] [PubMed] [Google Scholar]
- 7. Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature 2020;580:93‐99. [DOI] [PubMed] [Google Scholar]
- 8. Yu YP, Tsung A, Liu S, Nalesnick M, Geller D, Michalopoulos G, et al. Detection of fusion transcripts in the serum samples of patients with hepatocellular carcinoma. Oncotarget 2019;10:3352‐3360. [PMC free article] [PubMed] [Google Scholar]
- 9. He D‐M, Ren B‐G, Liu S, Tan L‐Z, Cieply K, Tseng G, et al. Oncogenic activity of amplified miniature chromosome maintenance 8 in human malignancies. Oncogene 2017;36:3629‐3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen ZH, Yu YP, Michalopoulos G, Nelson J, Luo JH. The DNA replication licensing factor miniature chromosome maintenance 7 is essential for RNA splicing of epidermal growth factor receptor, c‐Met, and platelet‐derived growth factor receptor. J Biol Chem 2015;290:1404‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang XY, Tang LZ, Ren BG, Yu YP, Nelson J, Michalopoulos G, et al. Interaction of MCM7 and RACK1 for activation of MCM7 and cell growth. Am J Pathol 2013;182:796‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han YC, Zheng ZL, Zuo ZH, Yu YP, Chen R, Tseng GC, et al. Metallothionein 1 h tumour suppressor activity in prostate cancer is mediated by euchromatin methyltransferase 1. J Pathol 2013;230:184‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han YC, Yu YP, Nelson J, Wu C, Wang H, Michalopoulos GK, et al. Interaction of integrin‐linked kinase and miniature chromosome maintenance 7‐mediating integrin {alpha}7 induced cell growth suppression. Cancer Res 2010;70:4375‐4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi YK, Yu YP, Zhu ZH, Han YC, Ren B, Nelson JB, et al. MCM7 interacts with androgen receptor. Am J Pathol 2008;173:1758‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu YP, Luo JH. Myopodin‐mediated suppression of prostate cancer cell migration involves interaction with zyxin. Can Res 2006;66:7414‐7419. [DOI] [PubMed] [Google Scholar]
- 16. Zuo ZH, Yu YP, Martin A, Luo JH. Cellular stress response 1 down‐regulates the expression of epidermal growth factor receptor and platelet‐derived growth factor receptor through inactivation of splicing factor 3A3. Mol Carcinog 2017;56:315‐324. [DOI] [PubMed] [Google Scholar]
- 17. Lin F, Yu YP, Woods J, Cieply K, Gooding B, Finkelstein P, et al. Myopodin, a synaptopodin homologue, is frequently deleted in invasive prostate cancers. Am J Pathol 2001;159:1603‐1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, et al. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 2006;25:1090‐1098. [DOI] [PubMed] [Google Scholar]
- 19. Zhu ZH, Yu YP, Shi YK, Nelson JB, Luo JH. CSR1 induces cell death through inactivation of CPSF3. Oncogene 2009;28:41‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo J‐H, Liu S, Tao J, Ren B‐G, Luo K, Chen Z‐H, et al. Pten‐NOLC1 fusion promotes cancers involving MET and EGFR signalings. Oncogene 2021;40:1064‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Graf J, Voisey J, Hughes I, van Daal A. Promoter polymorphisms in the MATP (SLC45A2) gene are associated with normal human skin color variation. Hum Mutat 2007;28:710‐717. [DOI] [PubMed] [Google Scholar]
- 22. Fernandez LP, Milne RL, Pita G, Aviles JA, Lazaro P, Benitez J, et al. SLC45A2: a novel malignant melanoma‐associated gene. Hum Mutat 2008;29:1161‐1167. [DOI] [PubMed] [Google Scholar]
- 23. Schmitz W, Albers C, Fingerhut R, Conzelmann E. Purification and characterization of an alpha‐methylacyl‐CoA racemase from human liver. Eur J Biochem 1995;231:815‐822. [DOI] [PubMed] [Google Scholar]
- 24. Kuefer R, Varambally S, Zhou M, Lucas PC, Loeffler M, Wolter H, et al. alpha‐Methylacyl‐CoA racemase: expression levels of this novel cancer biomarker depend on tumor differentiation. Am J Pathol 2002;161:841‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC, Rubin MA. Alpha‐Methylacyl‐CoA racemase: a novel tumor marker over‐expressed in several human cancers and their precursor lesions. Am J Surg Pathol 2002;26:926‐931. [DOI] [PubMed] [Google Scholar]
- 26. Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ, et al. Alpha‐methylacyl‐CoA racemase: a new molecular marker for prostate cancer. Cancer Res 2002;62:2220‐2226. [PubMed] [Google Scholar]
- 27. Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, et al. alpha‐Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 2002;287:1662‐1670. [DOI] [PubMed] [Google Scholar]
- 28. Tretiakova MS, Sahoo S, Takahashi M, Turkyilmaz M, Vogelzang NJ, Lin F, et al. Expression of alpha‐methylacyl‐CoA racemase in papillary renal cell carcinoma. Am J Surg Pathol 2004;28:69‐76. [DOI] [PubMed] [Google Scholar]
- 29. Fellegara G, Gabba S, Dorji T, De Luca G, Colecchia M. Observations on Aron et al's "Utility of a triple antibody cocktail intraurothelial neoplasm‐3 (IUN‐3 CK20/CD44s/p53) and alpha‐methylacyl‐CoA racemase (AMACR) in the distinction of urothelial carcinoma in situ (CIS) and reactive urothelial atypia". Am J Surg Pathol 2014;38:1013‐1015. [DOI] [PubMed] [Google Scholar]
- 30. Aron M, Luthringer DJ, McKenney JK, Hansel DE, Westfall DE, Parakh R, et al. Utility of a triple antibody cocktail intraurothelial neoplasm‐3 (IUN‐3‐CK20/CD44s/p53) and alpha‐methylacyl‐CoA racemase (AMACR) in the distinction of urothelial carcinoma in situ (CIS) and reactive urothelial atypia. Am J Surg Pathol 2013;37:1815‐1823. [DOI] [PubMed] [Google Scholar]
- 31. Shilo K, Dracheva T, Mani H, Fukuoka J, Sesterhenn IA, Chu WS, et al. Alpha‐methylacyl CoA racemase in pulmonary adenocarcinoma, squamous cell carcinoma, and neuroendocrine tumors: expression and survival analysis. Arch Pathol Lab Med 2007;131:1555‐1560. [DOI] [PubMed] [Google Scholar]
- 32. Kastelein F, Biermann K, Steyerberg EW, Verheij J, Kalisvaart M, Looijenga LH, et al.; ProBar study group . Value of alpha‐methylacyl‐CoA racemase immunochemistry for predicting neoplastic progression in Barrett's oesophagus. Histopathology 2013;63:630‐639. [DOI] [PubMed] [Google Scholar]
- 33. Scheil‐Bertram S, Lorenz D, Ell C, Sheremet E, Fisseler‐Eckhoff A. Expression of alpha‐methylacyl coenzyme A racemase in the dysplasia carcinoma sequence associated with Barrett's esophagus. Mod Pathol 2008;21:961‐967. [DOI] [PubMed] [Google Scholar]
- 34. Huang W, Zhao J, Li L, Huang Y, Yang X, Wang J, et al. a‐Methylacyl coenzyme A racemase is highly expressed in the intestinal‐type adenocarcinoma and high‐grade dysplasia lesions of the stomach. Histol Histopathol 2008;23:1315‐1320. [DOI] [PubMed] [Google Scholar]
- 35. Lee WA. Alpha‐methylacyl‐CoA‐racemase expression in adenocarcinoma, dysplasia and non‐neoplastic epithelium of the stomach. Oncology 2006;71:246‐250. [DOI] [PubMed] [Google Scholar]
- 36. Li W, Cagle PT, Botero RC, Liang JJ, Zhang Z, Tan D. Significance of overexpression of alpha methylacyl‐coenzyme A racemase in hepatocellular carcinoma. J Exp Clin Cancer Res 2008;27:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ha YS, Kim YW, Min BD, Lee OJ, Kim YJ, Yun SJ, et al. Alpha‐methylacyl‐coenzyme a racemase‐expressing urachal adenocarcinoma of the abdominal wall. Korean J Urol 2010:51:498‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang Z, Fanger GR, Banner BF, Woda BA, Algate P, Dresser K, et al. A dietary enzyme: alpha‐methylacyl‐CoA racemase/P504S is overexpressed in colon carcinoma. Cancer Detect Prev 2003;27:422‐426. [DOI] [PubMed] [Google Scholar]
- 39. Chi AN, Valencia JC, Hu Z‐Z, Watabe H, Yamaguchi H, Mangini NJ, et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res 2006;5:3135‐3144. [DOI] [PubMed] [Google Scholar]
- 40. Bartolke R, Heinisch JJ, Wieczorek H, Vitavska O. Proton‐associated sucrose transport of mammalian solute carrier family 45: an analysis in Saccharomyces cerevisiae. Biochem J 2014;464:193‐201. [DOI] [PubMed] [Google Scholar]
- 41. Mobley JA, Leav I, Zielie P, Wotkowitz C, Evans J, Lam YW, et al. Branched fatty acids in dairy and beef products markedly enhance alpha‐methylacyl‐CoA racemase expression in prostate cancer cells in vitro. Cancer Epidemiol Biomarkers Prev 2003;12:775‐783. [PubMed] [Google Scholar]
- 42. Wilson BA, Wang H, Nacev BA, Mease RC, Liu JO, Pomper MG, et al. High‐throughput screen identifies novel inhibitors of cancer biomarker alpha‐methylacyl coenzyme A racemase (AMACR/P504S). Mol Cancer Ther 2011;10:825‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Savolainen K, Kotti TJ, Schmitz W, Savolainen TI, Sormunen RT, Ilves M, et al. A mouse model for alpha‐methylacyl‐CoA racemase deficiency: adjustment of bile acid synthesis and intolerance to dietary methyl‐branched lipids. Hum Mol Genet 2004;13:955‐965. [DOI] [PubMed] [Google Scholar]
- 44. Kawamura N, Nagai H, Bando K, Koyama M, Matsumoto S, Tajiri T, et al. PTEN/MMAC1 mutations in hepatocellular carcinomas: somatic inactivation of both alleles in tumors. Jpn J Cancer Res 1999;90:413‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yao YJ, Ping XL, Zhang H, Chen FF, Lee PK, Ahsan H, et al. PTEN/MMAC1 mutations in hepatocellular carcinomas. Oncogene 1999;18:3181‐3185. [DOI] [PubMed] [Google Scholar]
- 46. Hu T‐H, Huang C‐C, Lin P‐R, Chang H‐W, Ger L‐P, Lin Y‐W, et al. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer 2003;97:1929‐1940. [DOI] [PubMed] [Google Scholar]
- 47. Nalesnik MA, Tseng G, Ding Y, Xiang G‐S, Zheng Z‐L, Yu YanPing, et al. Gene deletions and amplifications in human hepatocellular carcinomas: correlation with hepatocyte growth regulation. Am J Pathol 2012;180:1495‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 2007;1773:1213‐1226. [DOI] [PubMed] [Google Scholar]
- 50. Chen Z‐H, Yu YP, Zuo Z‐H, Nelson JB, Michalopoulos GK, Monga S, et al. Targeting genomic rearrangements in tumor cells through Cas9‐mediated insertion of a suicide gene. Nat Biotechnol 2017;35:543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material