

Abstract
No approved therapies are available for nonalcoholic steatohepatitis (NASH). Adenosine monophosphate–activated protein kinase (AMPK) is a central regulator of cell metabolism; its activation has been suggested as a therapeutic approach to NASH. Here we aimed to fully characterize the potential for direct AMPK activation in preclinical models and to determine mechanisms that could contribute to efficacy for this disease. A novel small‐molecule direct AMPK activator, PXL770, was used. Enzyme activity was measured with recombinant complexes. De novo lipogenesis (DNL) was quantitated in vivo and in mouse and human primary hepatocytes. Metabolic efficacy was assessed in ob/ob and high‐fat diet–fed mice. Liver histology, biochemical measures, and immune cell profiling were assessed in diet‐induced NASH mice. Direct effects on inflammation and fibrogenesis were assessed using primary mouse and human hepatic stellate cells, mouse adipose tissue explants, and human immune cells. PXL770 directly activated AMPK in vitro and reduced DNL in primary hepatocytes. In rodent models with metabolic syndrome, PXL770 improved glycemia, dyslipidemia, and insulin resistance. In mice with NASH, PXL770 reduced hepatic steatosis, ballooning, inflammation, and fibrogenesis. PXL770 exhibited direct inhibitory effects on pro‐inflammatory cytokine production and activation of primary hepatic stellate cells. Conclusion: In rodent models, direct activation of AMPK is sufficient to produce improvements in all core components of NASH and to ameliorate related hyperglycemia, dyslipidemia, and systemic inflammation. Novel properties of direct AMPK activation were also unveiled: improved insulin resistance and direct suppression of inflammation and fibrogenesis. Given effects also documented in human cells (reduced DNL, suppression of inflammation and stellate cell activation), these studies support the potential for direct AMPK activation to effectively treat patients with NASH.
AMPK is a key cellular sensor that is activated in response to a variety of conditions that deplete energy levels. AMPK activity is reduced in NASH. Direct AMPK activation using PXL770, a clinical‐stage investigational drug, improves the hallmarks of NASH and type 2 diabetes in rodent models.

Abbreviations
- ADaM
allosteric drug and metabolite
- AMPK
AMP‐activated protein kinase
- ANOVA
analysis of variance
- CBM
carbohydrate‐binding module
- DC
dendritic cell
- DIO‐NASH
diet‐induced obesity–NASH
- DNL
de novo lipogenesis
- FFA
free fatty acid
- GPR
glucose production rate
- HbA1C
hemoglobin A1c
- HFD
high‐fat diet
- HSC
hepatic stellate cell
- HSL
hormone‐sensitive lipase
- IHC
immunohistochemistry
- IL
interleukin
- LPS
lipopolysaccharide
- MAFLD
metabolic dysfunction–associated fatty liver disease
- MCP1
monocyte chemoattractant protein 1
- moDC
monocyte‐differentiated dendritic cell
- moMac
monocyte‐differentiated macrophage
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NF‐κB
nuclear factor kappa B
- SSGIR
steady‐state glucose infusion rate
- T2DM
type 2 diabetes mellitus
- TG
triglycerides
- TGF‐β
transforming growth factor β
- TNF‐α
tumor necrosis factor α
- α‐SMA
smooth muscle actin
Nonalcoholic fatty liver disease (NAFLD) is the most common hepatic disorder worldwide. Overnutrition and a sedentary lifestyle are principal causes; however, onset and progression are influenced by other factors such as genetics and comorbid conditions.( 1 )
Approximately 10%‐30% of patients with NAFLD progress to nonalcoholic steatohepatitis (NASH), manifested by the presence of lobular inflammation, hepatocellular damage, and fibrosis; NASH can lead to liver failure and hepatocellular carcinoma.( 1 )
Importantly, NAFLD/NASH are associated with multiple comorbidities. Type 2 diabetes mellitus (T2DM) has the strongest overlap; 20%‐30% of patients with T2DM have NASH, and up to 47% of patients with NASH also have T2DM.( 2 , 3 ) Furthermore, pathways contributing to their pathophysiology are highly convergent.( 4 ) The importance of metabolic dysfunction in NAFLD pathophysiology was highlighted by experts who suggested new nomenclature: redefining NAFLD as metabolic dysfunction–associated fatty liver disease or MAFLD.( 1 , 5 )
Increased free fatty acid (FFA) flux from adipose tissue lipolysis along with an increase in de novo lipogenesis (DNL) in hepatocytes are major contributors to steatosis.( 6 ) Insulin resistance is a driver of this pathophysiology through impaired inhibition of lipolysis( 7 ) and stimulation of DNL from hyperinsulinemia.( 8 ) Insulin resistance, dyslipidemia, and peripheral adipose tissue dysfunction also contribute to a systemic proinflammatory state, a major cause of the progression to NASH.( 6 )
AMP‐activated protein kinase (AMPK) is a heterotrimeric enzyme complex consisting of catalytic α (α1 and α2), regulatory β (β1 and β2), and γ (γ1, γ2, and γ3) subunits that controls key metabolic pathways involved in the development of NASH.( 9 ) AMPK activity is tightly regulated by posttranslational modifications and allosteric effects mediated through multiple domains and interactions, including the carbohydrate‐binding module (CBM) and the allosteric drug and metabolite (ADaM) site. Recently, a class of direct pharmacological AMPK activators has been shown to bind through the ADaM site.( 10 , 11 ) In the liver, AMPK inhibits DNL and increases fatty acid oxidation, and its activation has been shown to reduce steatosis.( 12 ) Interestingly, AMPK activation has been implicated as a means of inhibiting lipolysis in adipocytes.( 13 ) However, this potential effect is controversial.( 14 ) In adipose tissue macrophages, AMPK inhibits multiple pro‐inflammatory signaling pathways, including interleukin (IL)‐1β and tumor necrosis factor α (TNF‐α), and its activation has been proposed to limit liver inflammation.( 15 , 16 ) AMPK activation has also been suggested as an approach to attenuate hepatic fibrogenesis by inhibiting primary inflammatory injury, extracellular matrix secretion, and the induction of hepatic stellate cells (HSCs).( 17 , 18 , 19 , 20 )
Taken together, these elements point toward AMPK activation as an attractive approach to therapeutics targeting MAFLD/NAFLD and NASH. Consistent with this concept, constitutively active AMPK( 21 , 22 ) or small‐molecule direct AMPK activators A769662,( 12 , 23 ) C13,( 24 ) PF‐249/739,( 25 ) PF‐06409577,( 26 ) and Compound 1( 27 ) have been shown to decrease liver fat content in rodents.
Despite extensive efforts, there are still no approved therapies for NASH. PXL770 is an orally bioavailable thienopyridone small molecule that is currently in clinical development for NASH treatment (ClinTrials.gov; NCT03763877); it is also the first direct AMPK activator that is advanced into human efficacy studies. PXL770 was discovered through chemical optimization of one initial lead compound identified during a classical discovery high‐throughput screening process performed using a recombinant AMPK isoform (α2β1γ1). Physical chemistry, pharmacokinetics, and efficacy properties were part of the criteria for the chemical optimization. Here, we show that PXL770 directly increases AMPK activity by both allosteric activation and protection from dephosphorylation. We confirmed the benefit of direct AMPK activation to improve glucose metabolism and dyslipidemia in rodent models of metabolic syndrome, and to reduce hepatic DNL in vitro and in vivo. Using euglycemic hyperinsulinemic clamp methods, we showed for the first time that direct AMPK activation enhances insulin sensitivity in vivo. In a diet‐induced (DIO) mouse NASH model, we showed that direct AMPK activation ameliorated each of the classical NASH hallmarks, including steatosis, ballooning, inflammation, and fibrogenesis. Importantly, we unveiled that direct AMPK activation can produce independent direct effects in human cells to reduce DNL (hepatocytes), suppress inflammation (macrophages and dendritic cells [DCs]), and inhibit stellate cell activation. Thus, results obtained with PXL770 provide evidence for new attributes linked to direct AMPK activation and suggest the potential for translation to humans, supporting prospects in the treatment of NASH and NASH‐related comorbidities.
Materials and Methods
All procedures were performed in accordance with the principles and guidelines established by the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (Council of Europe, ETS 123; 1991).
In Vitro Assays
Direct AMPK Assays
For direct AMPK activation, recombinant AMPK isoforms were obtained from Sino Biological (Beijing, China), 2B Scientific (Oxfordshire, United Kingdom), and SignalChem Lifesciences Corp. (Richmond, BC). AMPK activity was measured using Delfia Fluorescent–based technology (PerkinElmer, Waltham, MA) as described in the Supporting Information. To assess the interaction with CBM domain, wild‐type AMPK (α1β1γ1) or AMPK harboring a deletion or mutation was incubated in the presence or absence of recombinant PP2C, and AMPK activity was measured as described previously.
Lipogenesis Measurement in Primary Human and Mouse Hepatocytes
Primary mouse hepatocytes were prepared from C57BL/6J mice (Harlan, Houston, TX), and primary human hepatocytes were purchased from Biopredic International (Ref. #HEP200B0; Saint Grégoire, France), both cultured as described in the Supporting Information. Effects of PXL770 on basal and insulin‐stimulated lipogenesis were determined by measuring incorporation of [1‐14C]‐acetate into total lipids as described in the Supporting Information.
Intracellular Triglyceride Content Determination in Control or AMPKα1α2‐Null Hepatocytes
AMPKα1α2‐null mice were obtained as described in the Supporting Information. Hepatocytes were incubated in a medium containing 25 mM glucose with 100 nM insulin (lipogenic condition), with indicated concentrations of PXL770, and triglyceride (TG) content was assayed using a commercial kit (Diasys, Waterbury, CT).
Assessment of Inflammation in ob/ob Mouse Adipose Tissue Explants
Epidydimal fat pads from ob/ob mice were excised and treated with IL‐1β or lipopolysaccharide (LPS) in the absence or the presence of 50 µM PXL770. Nuclear factor kappa B (NF‐κB) activity was assessed using a commercial kit from Abcam (Cambridge, United Kingdom). IL‐6 and IL‐1β concentrations in cell culture supernatants were determined using commercial kits (R&D Systems, Minneapolis, MN). Details are described in the Supporting Information.
Human Macrophage and DC Assays
Human CD14+ monocytes were isolated from healthy volunteers and differentiated toward either macrophages (monocyte‐differentiated macrophages, moMacs) or dendritic cells, DCs (monocyte‐differentiated DCs, moDCs), as described in the Supporting Information. Polarized moMacs were exposed to PXL770 as also described in the Supporting Information. moDCs were stimulated and exposed to PXL770 followed by phenotypic profiling, as described in the Supporting Information. All of the results were expressed as a percentage of vehicle‐control treated cells.
In Vitro HSC Assays
Primary HSCs were obtained from human donors and C57Bl6 mice (Supporting Information). HSCs were activated with transforming growth factor β (TGF‐β) alone and exposed to PXL770 or LY‐364947, a selective inhibitor of TGF‐β type 1 receptor. Phenotyping of HSCs to quantitate pathways relating to fibrogenesis and AMPK target engagement was performed using gene‐expression measurements, western blotting, and an assessment of DNL. All methods are described in the Supporting Information.
In Vivo Studies
Hepatic Lipogenesis Measurement in C57BL/6J Mice
Mice were fasted (24 hours) and refed (12 hours) and treated with PXL770 35 mg/kg or 75 mg/kg. The rate of hepatic lipid synthesis was quantified by determining the rate of incorporation of 3H2O into lipids by liquid scintillation counting. Details are described in the Supporting Information. Twice daily (bid) dosing was used in all in vivo studies to ensure 24‐hour coverage, given an estimated terminal t1/2 of 8.9 hours (data not shown).
ob/ob Mouse Model
ob/ob mice were treated by oral gavage for 5 days or 5 weeks with 75 mg/kg or 25 and 50 mg/kg of PXL770 twice daily, respectively. After 5 days of treatment, an oral glucose tolerance test was performed and plasma glucose, and insulin levels were measured at indicated timepoints. After 5 weeks of treatment, hemoglobin A1c (HbA1C) and plasma TG, FFAs, and glycerol were measured (details in Supporting Information).
Euglycemic Hyperinsulinemic Clamp
After 10 weeks of exposure to a high‐fat diet (HFD; ssniff Spezialdiaten, Soest, Germany; 60% of energy from fat), mice were treated with vehicle (control HFD), PXL770 75 mg/kg twice daily, or pioglitazone 25 mg/kg once daily for 6 weeks. Standard diet (SD) mice were treated with vehicle. A euglycemic hyperinsulinemic clamp with glucose production/turnover measurements was then performed in fasted mice as described in the Supporting Information.
DIO‐NASH Mouse Model
The diet‐induced obesity (DIO)–NASH mouse model( 28 , 29 ) was established as described in the Supporting Information.
Mice were fed with a diet high in fat for 41 weeks before starting the treatment with 35 mg/kg or 75 mg/kg PXL770 for 8 weeks. At the end of treatment, blood glucose levels (EKF Diagnostics, Cardiff, United Kingdom), plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), TG, and total cholesterol were measured using commercial kits (Roche Diagnostics, Basel, Switzerland). Plasma FFAs were determined using a commercial kit (WACO Chemicals, Richmond, VA).
Terminal liver samples were analyzed for TG content (Roche Diagnostics). Liver histology (hematoxylin and eosin and sirius red staining) and measurements of specific protein levels (immunohistochemistry [IHC]) were performed as described in the Supporting Information. The expression level of specific genes was assessed through RNA sequencing using RNA extracts from terminal liver samples.( 28 , 29 ) Flow cytometry was used for measurements of selected immune cells present in liver tissue, as described in the Supporting Information.
Statistical Analysis
Results are expressed as means ± SEM. Groups were compared in unpaired two‐tailed Student t tests or one‐way analysis of variance ANOVA (or Kruskal Wallis test) with Dunnett’s post hoc test for multiple comparisons, where appropriate. Differences between groups were considered statistically significant if P < 0.05. IC50s were calculated by nonlinear regression analysis and correlations by simple linear regression. GraphPad Prism 8.0 software was used.
Results
PXL770 Is a Direct AMPK Activator
The small molecule PXL770 (Fig. 1A) directly activated recombinant AMPK heterotrimeric proteins, allosterically and/or by protecting AMPK from dephosphorylation induced by PP2C (Table 1). In general, higher potency for AMPK complexes that contain the β1 subunit was clearly evident (Table 1). Similar potency was observed whether the catalytic subunit was α1 versus α2. Potential off‐target activity was excluded by screening against a panel of 56 receptors, ion channels, and transporters (Eurofins Cerep, Celle‐Lévescault, France); only one target showed a possible effect. In an assay of CCK1 receptor binding, modest inhibition was noted (data not shown).
FIG. 1.
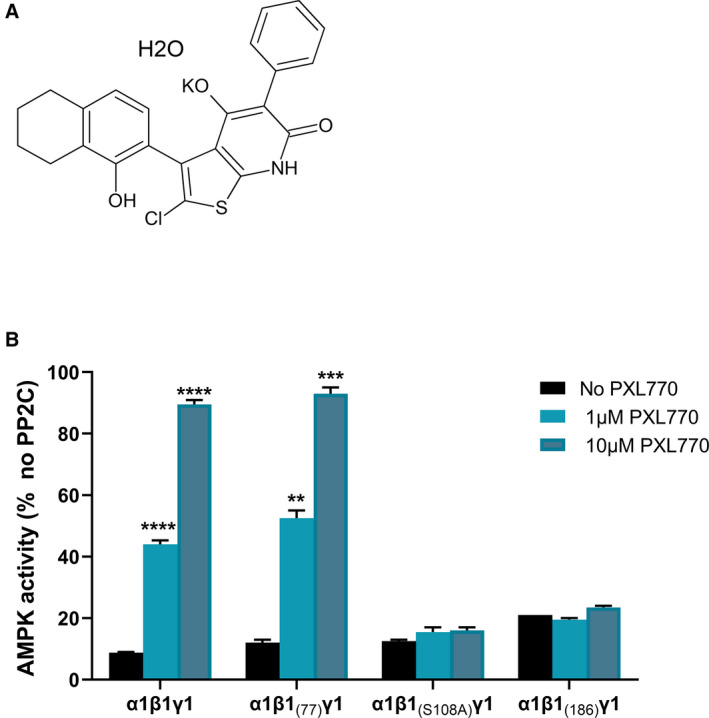
PXL770 activates AMPK. (A) Chemical structure of PXL770 monohydrate potassium salt. (B) Effect of mutations within β1 on protection against dephosphorylation: Wild‐type AMPK (α1β1γ1) or AMPK harboring deletion of the N‐terminal 76 (α1β 1(77) γ1) or N‐terminal 185 (α1β1 (186) γ1) residues of β1, or a point mutation of S108A in β1 (α1β1 [S108A] γ1). PXL770 had no effect with mutations in β1 N‐terminal 185 or S108A. **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus no PXL770 (one‐way ANOVA); n = 2‐4 replicates/condition.
TABLE 1.
In vitro Activity of PXL770 on Recombinant AMPK Heterotrimeric Proteins
| Heterotrimerics AMPK | AMPK Activity | |
|---|---|---|
| EC50 | Emax % | |
| α1β1γ1 | 16.2 nM | 155 |
| α1β1γ2 | 42.1 nM | 200 |
| α1β1γ3 | 64 nM | 450 |
| α2β1γ1 | 1,338 nM | 240 |
| α2β1γ2 | 68.7 nM | 700 |
| α2β1γ3 | 41.5 nM | 550 |
| α1β2γ1 | 1.3µM | 115 |
| α1β2γ3 | >µM | 130 |
| α2β2γ1 | >µM | >500 |
| α2β2γ2 | >µM | 820 |
| α2β2γ3 | >µM | 210 |
Emax, the maximal stimulation achieved from basal activity, is expressed as a percentage of basal activity.
Activation by PXL770 Requires an Intact ADaM Binding Site Within AMPK
PXL770’s effect to protect AMPK from dephosphorylation was abolished by mutation of S108 within the CBM of β1 to an alanine (Fig. 1B). This result is consistent with that reported with another AMPK activator (A769662).( 30 ) We also found that deletion of the N‐terminal 185 residues of β1, which includes the CBM, abolished protection by PXL770. In contrast, deletion of the N‐terminal 76 residues of β1, which leaves the CBM intact, does not alter this effect of PXL770. We further observed that PXL770 does not bind competitively to nucleotide binding sites 1 or 3 of the γ‐subunit (data not shown). Collectively, these data are consistent with an allosteric mechanism for activation mediated by binding to the AdaM site.
PXL770 Improves Glycemia and Insulin Sensitivity in Rodents With Metabolic Syndrome
In ob/ob mice, short‐term treatment with PXL770 75 mg/kg (5‐day) improved glucose tolerance (Fig. 2A); no effect on body weight was observed (data not shown). Given the lack of an increase in insulinemia (Fig. 2A), an improvement of insulin sensitivity was suggested. Five weeks of treatment with PXL770 50 mg/kg decreased HbA1c (Fig. 2B) and plasma TG, plasma FFA, and plasma glycerol levels (Fig. 2B).
FIG. 2.
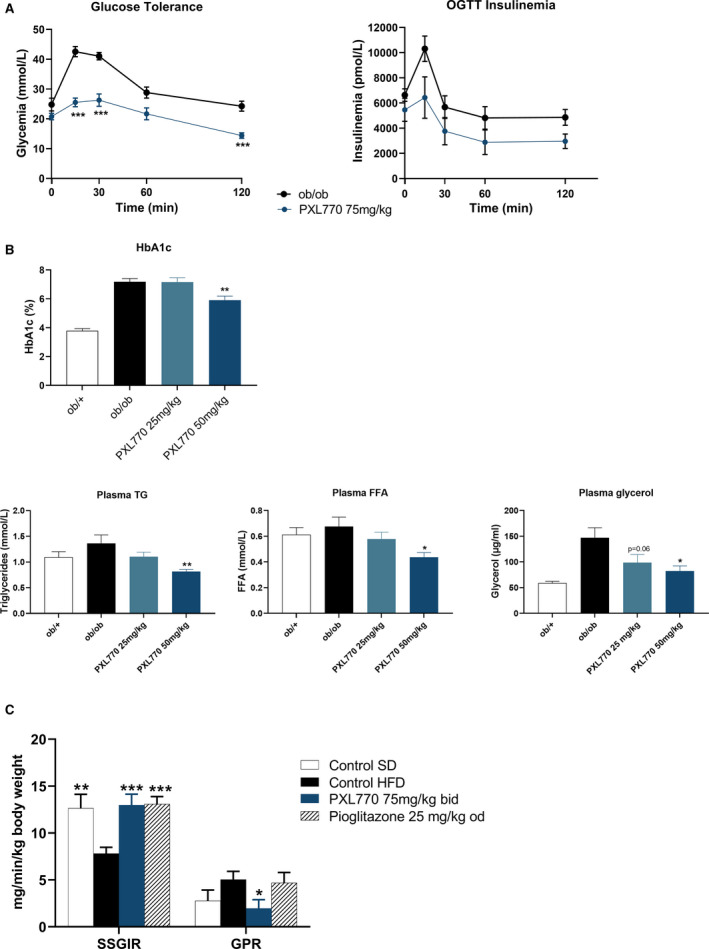
Metabolic profile of PXL770 ob/ob mice. (A) Oral glucose tolerance test with glucose and insulin levels, after 5 days of treatment. (B) HbA1C and plasma TGs, FFAs, and glycerol after 5 weeks of treatment. *P < 0.05, **P < 0.01, and ***P < 0.001 versus vehicle‐treated ob/ob mice (Kruskal‐Wallis); n = 9‐12 /group. (C) Measure of insulin sensitivity in HFD mice: Both SSGIR and GPR rates are shown; *P < 0.05, **P < 0.01, and ***P < 0.001 versus control HFD (one‐way ANOVA); n = 8‐21/group. Abbreviation: OGTT, oral glucose tolerance test.
To definitively assess whether PXL770 could enhance insulin sensitivity, a euglycemic hyperinsulinemic clamp was performed in HFD mice. After 6 weeks of treatment, insulin sensitivity was increased in PXL770‐treated mice compared with vehicle‐treated mice (Fig. 2C, Supporting Table S1). The steady‐state glucose infusion rate (SSGIR) increased, and a strong decrease in (hepatic) glucose production rate (GPR) in response to insulin infusion was observed (Fig. 2C). As expected, pioglitazone (used as an insulin sensitizer reference) increased SSGIR without any effect on GPR (Fig. 2C, Supporting Table S1). Interestingly, the SSGIR in both PXL770 and pioglitazone groups was normalized, reaching the level measured in SD mice.
PXL770 Reduces DNL in Primary Mouse and Human Hepatocytes and In Vivo
DNL is a key pathway contributing to steatosis,( 31 ) and AMPK is known to inhibit DNL.( 32 ) In isolated primary mouse and human hepatocytes, treatment with PXL770 induced dose‐dependent inhibition of DNL (Fig. 3A). Importantly, similar potency of PXL770 was evident in both mouse and human primary hepatocytes with IC50 values of 2.8 µM and 2.6 µM, respectively (Fig. 3A).
FIG. 3.
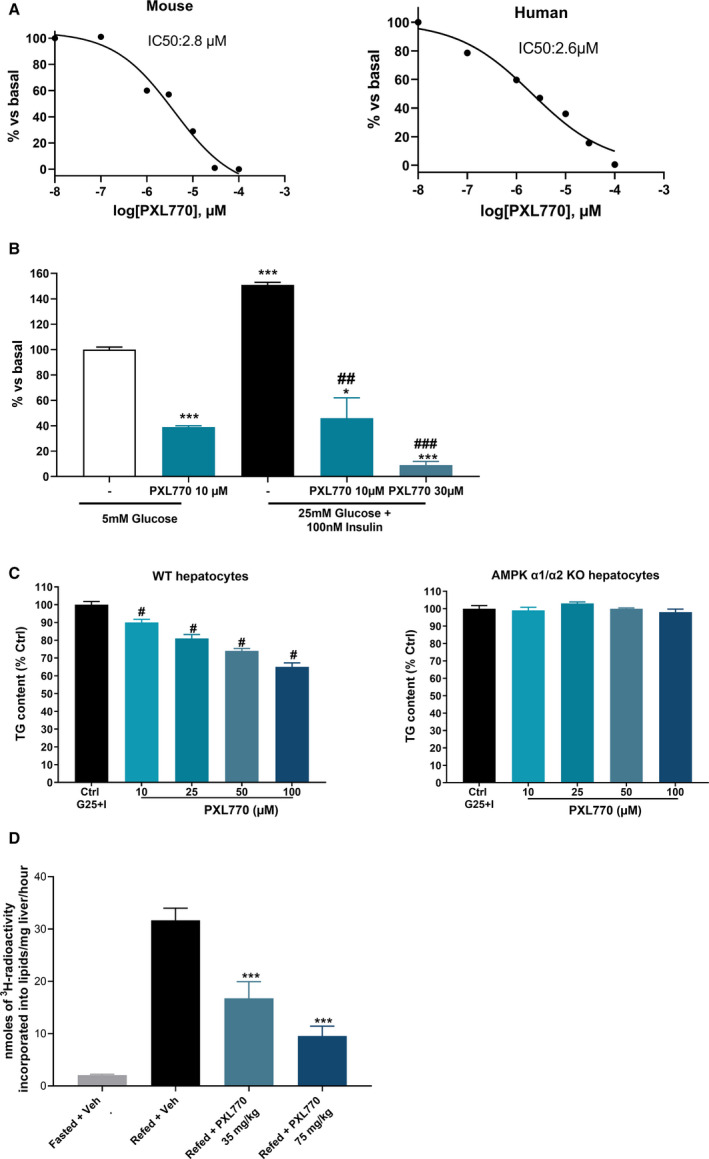
Effects of PXL770 on DNL. (A) Basal DNL in mouse and human hepatocytes; n = 3 replicates. (B) Insulin‐stimulated DNL in human hepatocytes; *P < 0.05 and ***P < 0.001 versus glucose alone, and ## P < 0.01 and ### P < 0.001 versus glucose plus insulin (one‐way ANOVA); n = 3 replicates. (C) TG content in wild‐type and AMPK knockout mouse hepatocytes; # P < 0.05 versus control (one‐way ANOVA); n = 3 replicates. (D) In vivo DNL in fasted/refeeding C57/BL6J mice; ### P < 0.001 versus fasted *** P < 0.001 versus refed (Student t test, one‐way ANOVA); n = 5 fasted mice, n = 10 refed mice/group.
The effect of PXL770 was also assessed in insulin‐stimulated human hepatocytes. In this context, PXL770 decreased lipid synthesis by 70%‐90% (Fig. 3B), in line with dose‐dependent increases in ACC S79/221 phosphorylation and AMPKα Thr172 phosphorylation (Supporting Fig. S1). In these experiments, we also confirmed that PXL770 did not impede the activation of insulin signaling as measured by Akt phosphorylation (Supporting Fig. S1).
To examine the requirement of AMPK for PXL770’s inhibitory effects, DNL studies were conducted using AMPKα1α2‐null hepatocytes. Treatment with PXL770 decreased intracellular TG accumulation in control hepatocytes, whereas this effect was severely blunted in AMPKα1α2‐null hepatocytes (Fig. 3C).
To determine whether oral dosing with PXL770 could suppress liver [3H]‐H2O incorporation into lipids, in vivo DNL was assessed during refeeding stimulation in mice. As expected, a strong (15‐fold) induction of DNL was observed in refed vehicle‐treated animals, and PXL770 treatment inhibited DNL at both doses studied (Fig. 3D).
PXL770 Improves Hallmarks of Liver Pathology in a Rodent NASH Model
To test the hypothesis that metabolic effects of PXL770 might affect NASH, several experiments were conducted using a relevant and validated model: the amylin liver NASH (AMLN) DIO‐NASH mouse.( 28 , 29 ) After 8 weeks of treatment, PXL770 dose‐dependently increased AMPK activation in liver, adipose tissue, and whole blood (Fig. 4A). Interestingly, compared with lean chow‐fed mice, vehicle‐treated DIO‐NASH mice exhibited lower basal AMPK activity (pAMPK) in both epididymal adipose tissue (22%) and in blood (17%); however, no change in liver was evident (Fig. 4A). These findings are consistent with the concept of reduced endogenous AMPK tone that occurs in the context of overnutrition, systemic metabolic dysfunction, and inflammation.( 33 , 34 )
FIG. 4.
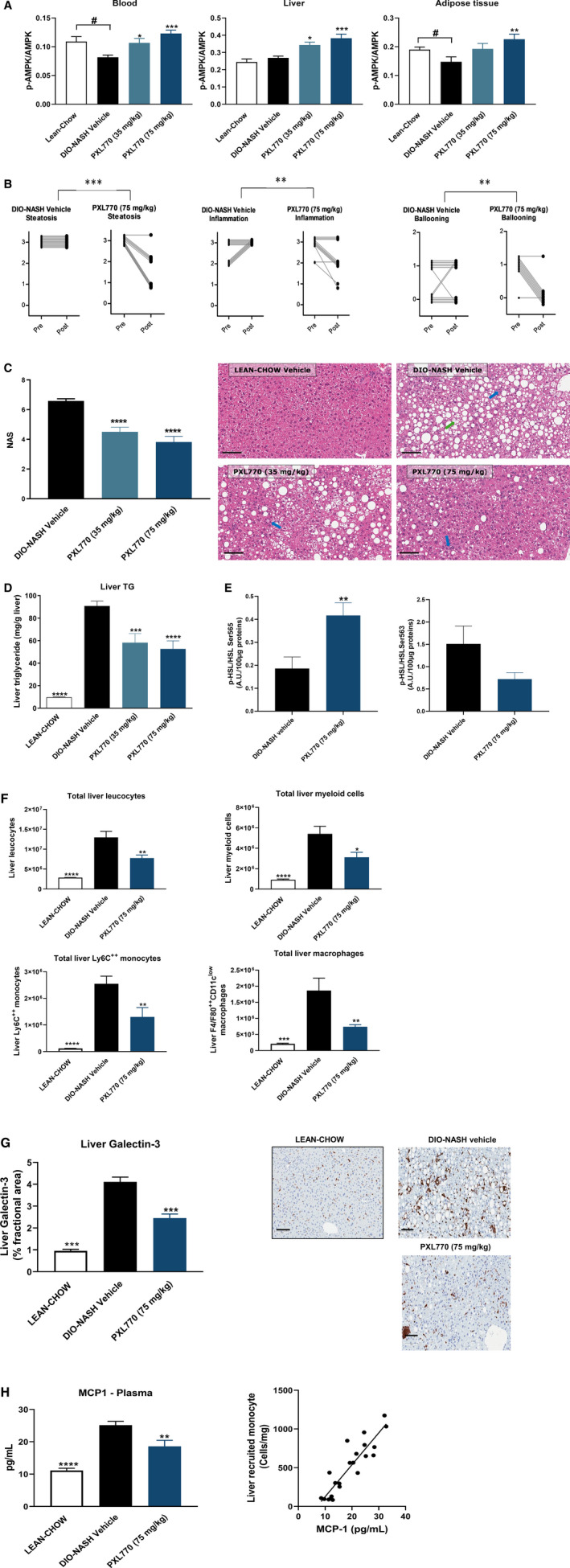
PXL770 improves steatohepatitis in DIO‐NASH mice. (A) AMPK phosphorylation in blood, liver, and adipose tissue measured by enzyme‐linked immunosorbent assay; # P < 0.05 versus vehicle (Student t test) (B) Liver steatosis, inflammation, and hepatocellular ballooning scores before (pre) and after (post) PXL770 treatment for each mouse. **P < 0.01, differences in progression of prescores to postscores between vehicle‐treated and PXL770‐treated groups (Fisher’s exact test). (C) NASH activity score; representative images of liver hematoxylin and eosin staining (magnification, ×20; scale bar, 100 µm); blue arrows, inflammatory foci; green arrow, ballooning degeneration. (D) Liver triglyceride content. (E) p‐HSL/HSLSer565/563 levels in adipose tissue. (F) Liver inflammatory cell counts assessed by flow cytometry. (G) Quantitation of relative galectin‐3 levels; representative images of liver galectin‐3 IHC (magnification, ×20; scale bar, 100 µm). (H) Plasma MCP1 and correlation with total liver LyC++ monocytes. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus vehicle (one‐way ANOVA, Student t test); n = 11‐12/group.
Vehicle‐treated DIO‐NASH mice demonstrated substantial liver steatosis and inflammation scores (Fig. 4B). PXL770, at both doses, reduced liver steatosis, liver inflammation, and hepatocellular ballooning scores (Fig. 4B, Supporting Fig. S2), leading to a reduction of the NASH score (NAS, Fig. 4C). The decrease in liver steatosis was also confirmed by a decrease in liver TG content (Fig. 4D).
These effects of PXL770 were also associated with substantial improvements in liver injury markers: plasma ALT and AST levels (Supporting Table S2). PXL770 also reduced liver weight (Supporting Table S2). Consistent with results in ob/ob mice, plasma FFA levels were also decreased by PXL770 (Supporting Table S2). To explore whether lipolysis could be inhibited, ex vivo adipose hormone‐sensitive lipase (HSL) phosphorylation was monitored and shown to be modulated by PXL770 in this NASH model: A clear reciprocal increase in Ser565 phosphorylation versus decreased Ser563 phosphorylation was evident (Fig. 4E). Interestingly, this effect was also observed in vitro in isolated human and mouse adipocytes. PXL770 significantly increased HSL phosphorylation at Ser565 in both species (Supporting Fig. S3A,B). HSL phosphorylation at Ser563 tended to be modestly suppressed in human adipocytes (Supporting Fig. S3A).
PXL770 Reduces Liver and Adipose Tissue Inflammation in DIO‐NASH Mice
Resident liver macrophages and freshly recruited monocyte‐derived macrophages have a key role in driving inflammation and fibrogenesis in NASH.( 35 ) We therefore used flow cytometry to assess specific effects of PXL770 on immune cells and mediators of inflammation in the liver. Compared with healthy mice, DIO‐NASH mice exhibited an increase in total liver leucocytes (Fig. 4F). In response to PXL770 treatment, total liver leucocytes were reduced, including reductions in total liver monocytes and resident macrophages (Fig. 4F). Consistent with these findings, PXL770 decreased levels of galectin‐3 (Fig. 4G) and CD68, a pan‐macrophage marker (Supporting Fig. S4), as well as reduced monocyte chemoattractant protein 1 (MCP1) gene expression (Supporting Fig. S4). Importantly, plasma MCP1 was also suppressed (Fig. 4H), and a strong correlation between plasma MCP1 levels and total liver monocytes was established (Fig. 4H). In adipose tissue of DIO‐NASH mice, PXL770 decreased MCP1 gene expression without modifying markers of macrophages (CD68) or (CD163) (Supporting Fig. S4).
PXL770 Directly Suppresses Markers of Inflammation in ob/ob Mouse Adipose Tissue Explants
Having seen in vivo benefits of PXL770 on adipose inflammation in DIO‐NASH mice, we assessed the potential for direct anti‐inflammatory effects. In explants of ob/ob epididymal adipose tissue, incubation with IL1‐β increased NF‐κB nuclear activity. PXL770 prevented this induction (Fig. 5A). Incubation of adipose explants with PXL770 also inhibited the response to LPS; the induction of secreted IL‐1β and IL‐6 were significantly attenuated (Fig. 5A). Basal IL‐6 secretion was also decreased by PXL770 (Fig. 5A).
FIG. 5.
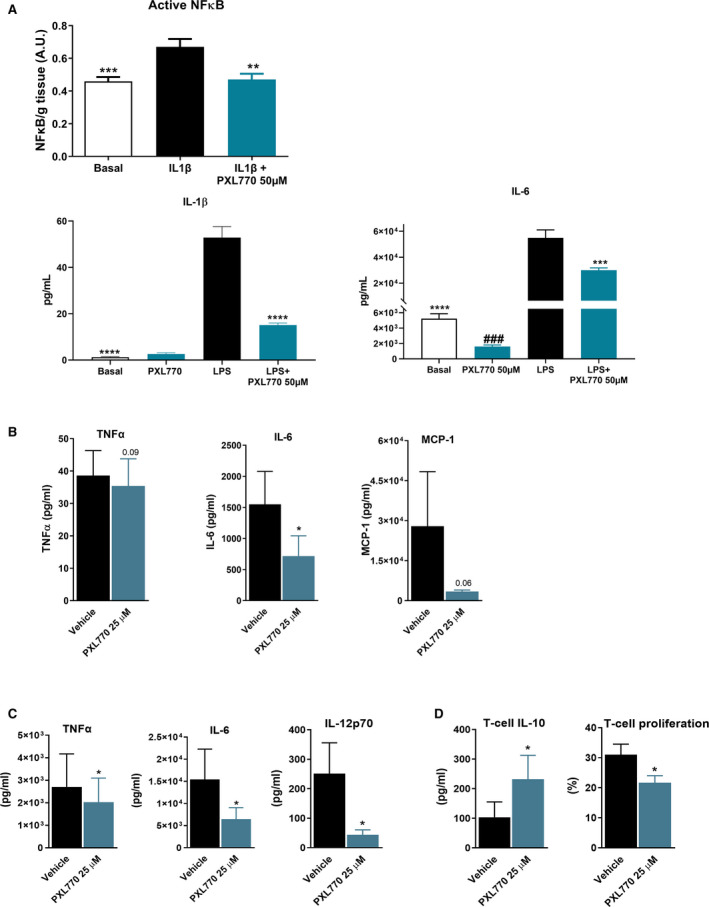
PXL770 exerts direct anti‐inflammatory effects. (A) ob/ob mice epididymal adipose tissue explants: active nuclear NF‐kB activity measured (enzyme‐linked immunosorbent assay) in tissue and cytokine levels in supernatants; ###P < 0.001 versus basal, *P < 0.05, **P < 0.01, and ***P < 0.001 versus IL‐1β or LPS alone (Student t test, one‐way ANOVA/Mann‐Whitney U test); n = 10‐11 observations, n = 7 mice. (B) Cytokine production from human M1 macrophages; n = 5 donors; nonsignificant P value noted for TNF‐α and MCP1. (C) Cytokine production from LPS‐primed human DCs (moDCs; n = 5 donors). (D) Induction of IL‐10‐producing T cells by LPS‐primed moDCs and T‐cell proliferation assay (n = 3 donors); *P < 0.05 versus vehicle (Student t test) for (B), (C), and (D).
PXL770 Reduces Pro‐inflammatory Cytokine Production in Human Macrophages and Promotes Tolerogenic dendritic cells
To address whether AMPK activation may exert direct anti‐inflammatory effects, human macrophages (moMacs) and dendritic cells (moDCs) were used. PXL770 increased AMPK activity in a dose‐dependent manner in M1‐polarized inflammatory moMacs (Supporting Fig S5A) and simultaneously reduced the production of pro‐inflammatory cytokines (TNF‐α and IL‐6) and chemokine (MCP1) (Fig. 5B). In LPS‐primed moDCs, PXL770 also increased AMPK activity in a dose‐dependent manner (Supporting Fig S5B) and reduced pro‐inflammatory cytokines (TNFα, IL‐6, and IL‐12p70) (Fig. 5C). Furthermore, PXL770 promoted DC‐mediated priming of IL‐10‐producing CD4 T cells with enhanced regulatory capacity (Fig. 5D). This was associated with decreased expression of the co‐stimulatory molecules CD40, CD80 and CD86, and up‐regulation of the CD103 tolerogenic marker (Supporting Fig. S5C), whereas neither major histocompatibility complex class II and IL‐T3 expression nor DC‐mediated Th1, Th2, and Th17 priming were affected (Supporting Fig. S5C,D). Thus, PXL770 has the capacity to directly suppress inflammation while also augmenting anti‐inflammatory pathways and cell types.
PXL770 Reduces Liver Fibrogenesis in DIO‐NASH Mice
Vehicle‐treated DIO‐NASH mice exhibited only mild fibrosis with a score between 1 and 2 (Supporting Fig. S6). Nevertheless, the expression of profibrogenic genes, including those encoding TGF‐β and platelet‐derived growth factor (PDGF), were increased along with increases in smooth muscle actin (α‐SMA), a marker of HSC activation, and an increase in collagen types I and II gene expression (Fig. 6A,B). In parallel, expression levels for tissue inhibitors of matrix metalloproteinases such as tissue inhibitor of metalloproteinase 1 (TIMP1) and TIMP2 were substantially increased (Fig. 6A).
FIG. 6.
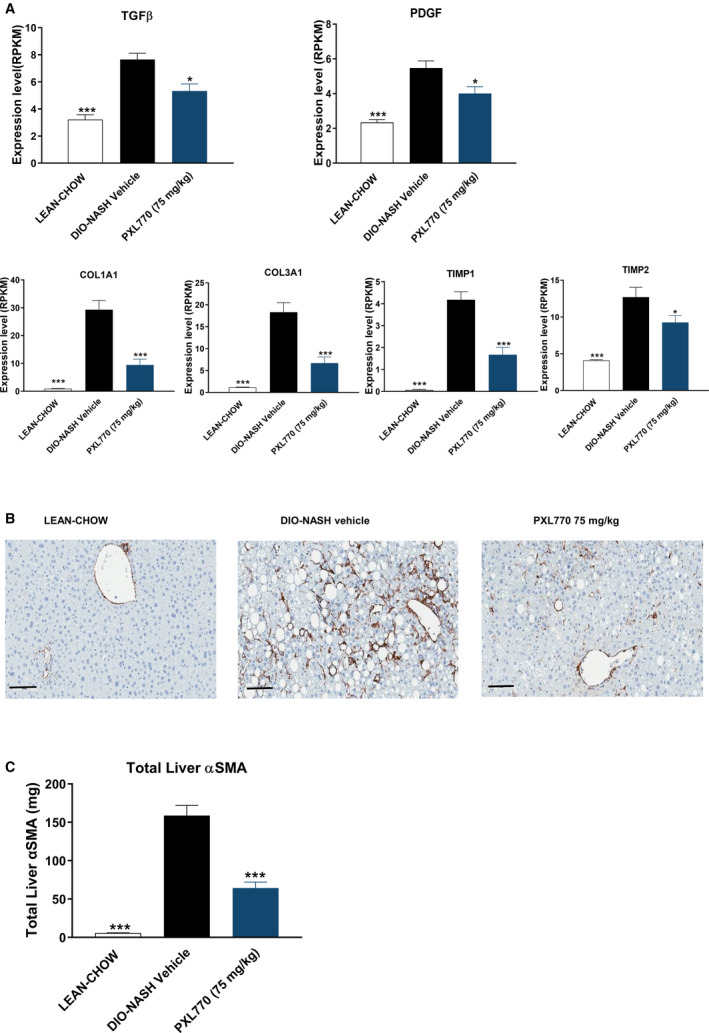
PXL770 exerts antifibrogenic effects in DIO‐NASH mice. (A) Relative messenger RNA expression of indicated genes in liver. (B) α‐SMA IHC in liver‐tissue sections (magnification, ×20; scale bar, 100 µm). (C) Quantitation of liver α‐SMA levels. *P < 0.05 and ***P < 0.001 versus vehicle‐treated DIO‐NASH (one‐way ANOVA); n = 8‐12/group.
In response to PXL770 treatment, changes in several of the aforementioned parameters were partially reversed. This included reductions of TGF‐β1 and PDGF gene expression (Fig. 6A) and decreases in α‐SMA protein levels, as assessed by histomorphometry (Fig. 6B). Furthermore, elevated levels of collagen 1A1 and 3A1 gene expression were substantially lowered along with reductions in mean TIMP1 and TIMP2 messenger RNA levels (Fig. 6A).
PXL770 Reduces Activation and Proliferation of HSCs
Having documented in vivo effects on fibrogenesis, we assessed potential direct effects to inhibit HSC activation. In human HSCs, PXL770 suppressed the expression of key activation markers, ACTA2 and COL1A1, along with a strong reduction in procollagen alpha 1 protein secretion with an IC50 ≈ 3 µM (Fig. 7A,B). In addition, PXL770 inhibited TGF‐β‐induced cell proliferation and the expression of the proliferation marker MIKI67 (Supporting Fig. S7A). NF‐κB activation can promote stellate cell activation/proliferation through induction of TNF‐α,( 36 ) while SMAD7 inhibits TGF‐β‐induced stellate cell activation.( 37 ) PXL770 reduced TNF‐α and increased SMAD7 gene expression (Fig. 7C), suggesting that this may be important for reducing stellate cell activation/proliferation and would need further investigation. Similar effects were observed in mouse HSCs (Supporting Fig. S7D).
FIG. 7.
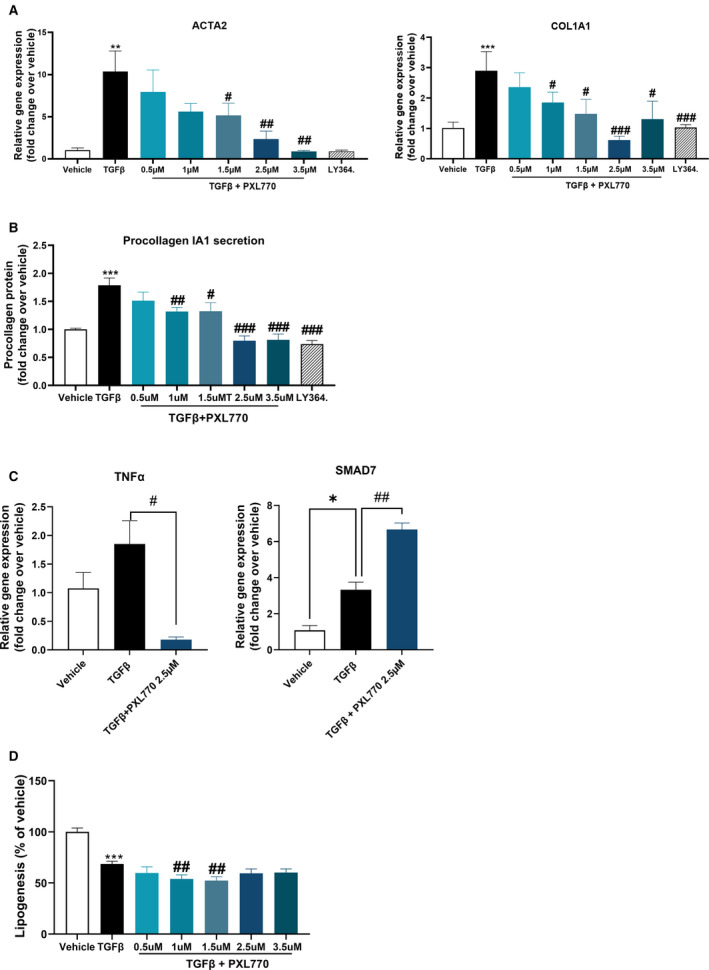
PXL770 effects on human HSCs. (A) Relative gene expression of ACTA2 and COL1A1 gene. (B) Procollagen 1A1 protein secretion in supernatant. (C) Relative gene expression of TNF‐α and SMAD7 gene. (D) Relative lipogenesis. *P < 0.05, **P < 0.01, and ***P < 0.001 versus vehicle. # P < 0.05, ## P < 0.01, and ### P < 0.001 versus TGF‐β alone (one‐way ANOVA); n = 3 donors, three replicates/donor. (LY364 = LY364947, selective TGF‐β receptor 1 inhibitor).
Importantly, the dose‐response for markers of fibrogenesis and for measures of target engagement (pAMPK and pACC) were well aligned and were similar with respect to potency in mouse versus human cells (Supporting Fig. S7B,E). As expected from reduced pACC, PXL770 also reduced lipogenesis in HSCs (Fig. 7D, Supporting Fig. S7D). PXL770 also reduced the phosphorylation of p70S6‐kinase, a downstream substrate of mammalian target of rapamycin (mTOR), a critical pathway controlling cell growth and proliferation (Supporting Fig. S7B,E).
Discussion
We considered and pursued AMPK as a potential therapeutic target for NASH, given its well‐described role as a key metabolic sensor where activation has been implicated in counteracting selected features of NASH pathophysiology.( 9 , 38 ) Importantly, endogenous AMPK activity is reduced in the context of metabolic dysfunction or inflammation, including in liver and adipose tissue.( 39 , 40 )
PXL770 is a small molecule and, to our knowledge, is the only direct AMPK activator that has advanced into human trials in NASH. Our results showed that PXL770 is a direct allosteric AMPK activator, and experiments with site‐directed mutants implicate binding to the AMPK β1 CBM domain. These results are consistent with data reported for A769662, a direct AMPK activator that appears to also bind to the ADaM site.( 10 ) AMPK activity was increased by 1.5‐fold to 8‐fold with PXL770 for most isoforms; this degree of activation is similar to the maximal 2‐5‐fold effect achieved with AMP—the endogenous activator—in our experiments and in the literature.( 41 ) In contrast, other direct activators have been reported to induce supra‐physiologic AMPK activation.( 42 , 43 ) Potential safety liabilities have been reported with MK‐8722,( 42 ) which might relate to markedly greater degrees of activation by this molecule.
Having established PXL770 as a direct AMPK activator, we then tested the hypothesis that PXL770 could favorably affect metabolic parameters that underlie the pathophysiology of NASH. In ob/ob mice, a classical obese T2DM model, we observed substantial improvements in HbA1c and elevated plasma TG levels as well as reduced hepatic steatosis. These effects are generally consistent with another report using murine (or non‐human primate) models.( 42 ) We also provided definitive evidence of reduced insulin resistance through effects seen during the euglycemic hyperinsulinemic clamp. This represents the first clear evidence of a bona fide increase in insulin sensitivity with direct pharmacologic activation of AMPK in vivo. This is of particular importance, as insulin resistance is a critical component of the pathogenesis of metabolic diseases including T2DM and NAFLD and a major driver of NASH.( 4 )
Data from genetic mouse models leading to AMPK deficiency or activation and pharmacological activators clearly indicate that AMPK inhibits hepatic DNL.( 12 , 22 , 40 , 41 ) Here, we confirmed that PXL770 could suppress DNL in vitro and in vivo in mice during carbohydrate diet refeeding. Our results also suggest that the pro‐lipogenic insulin signaling pathway does not override PXL770’s effect. The potential for this effect of direct AMPK activation to translate to humans was evident using human hepatocytes, where suppression of DNL occurred with similar potency as in mouse hepatocytes. The significance of this effect is derived from the knowledge that hepatic DNL is increased in patients with NAFLD and accounts for a significant portion of liver lipid accumulation.( 31 )
Flux of adipose‐derived FFAs to liver represents an additional important contributor to steatosis.( 4 ) In vivo, we observed that PXL770 decreases plasma FFA and glycerol levels, suggesting an effect of PXL770 to inhibit lipolysis. In addition, changes in HSL phosphorylation, shown with PXL770, support this hypothesis. Although our data are consistent with a reduction in lipolysis, more direct measurements would be required to confirm that this mechanism is involved in reducing steatosis.
Using the well‐characterized AMLN diet NASH model,( 28 , 29 ) we showed that PXL770 improved all hallmarks of NASH pathology, reducing each component of the NASH activity score and several markers of fibrogenesis (including activation of HSCs). Other direct AMPK activators have been shown to ameliorate steatosis in rodents. One such molecule (A‐769662) was also shown to improve liver fibrosis in a HFD mouse model,( 18 ) and markers associated with fibrosis were reportedly improved with PF‐06409577( 26 ) and Compound 1( 27 ); however, no effects on inflammation and ballooning were observed. Thus, PXL770 appears to more broadly affect NASH‐related parameters in rodents when compared to historical observations with other direct AMPK activators.
Inflammation is a major driver of NASH. A potential role for AMPK was shown in mice lacking the AMPK β1 subunit, where systemic inflammation including increased macrophage infiltration in liver was reported.( 15 ) In addition to reducing hepatic inflammation scores, we documented robust effects of PXL770 to decrease several immune cell types in diseased liver, including monocytes and macrophages (Kupffer cells). Monocytes and macrophages have a major role in NASH, and monocyte‐derived cells can develop into macrophages or into DCs, responsible for antigen presentation to adaptative immune cells.( 35 ) Monocyte flux to the liver is driven by the chemokine CCL2 (MCP1).( 44 , 45 ) Thus, reductions in MCP1 may have contributed to the reduction in infiltration of monocytes into liver. To test the hypothesis that PXL770’s anti‐inflammatory effects were (at least in part) direct (vs. secondary to chronic improvements in insulin resistance and steatosis), we performed several in vitro experiments. Using ob/ob mouse adipose tissue explants, PXL770 was shown to abrogate activation of NF‐κB and concomitantly reduce secretion of several inflammatory cytokines/chemokines: IL‐1β, IL‐6, and MCP1. Importantly, we also demonstrated clear direct effects to reduce pro‐inflammatory cytokines in both human macrophages and DCs. A potential link between AMPK activation and anti‐inflammatory effects in macrophages has been previously reported, although in vitro effects of direct AMPK activators were not described.( 15 , 46 , 47 ) In the current study we also provide the first evidence that direct AMPK activators could exert immunomodulatory effects on human DCs, leading to an “anti‐inflammatory” tolerogenic profile characterized by reduced maturation and production of pro‐inflammatory cytokines, and an enhanced capacity to skew T‐cell responses toward a regulatory phenotype (Tregs). This may be beneficial in the context of NASH, where increased immunogenic “pro‐inflammatory” DCs and reduced Tregs have been suggested to contribute to hepatic inflammation.( 48 , 49 )
It is well established that inflammation leads to hepatic fibrosis. To ascertain whether antifibrogenic effects observed in liver of DIO‐NASH mice could occur through an independent action on HSCs, direct effects of PXL770 were assessed in vitro. In both primary mouse and human HSCs, PXL770 strongly reduced activation. Direct AMPK activators have not been previously shown to independently affect HSCs; however, other molecules that promote AMPK activity (e.g., AICAR) have been shown to exert similar effects in mouse HSCs.( 50 ) In addition, PXL770 reduced DNL in this cell type; this effect may contribute to reduced HSC activation, as shown with an ACC inhibitor that was noted to reduce HSC activity.( 51 )
In summary, we report that activation of AMPK in relevant animal models using PXL770, a direct allosteric activator, targets root causes of NASH including insulin resistance, T2DM, adipose inflammation, and dyslipidemia. Moreover, core features of disease in liver were ameliorated (steatosis, inflammation, and hepatic ballooning). In addition to known effects of direct AMPK activators to modulate DNL, as well as a likely decrease in FFA flux to the liver from the adipose tissue, we demonstrated direct effects to attenuate inflammation and fibrogenesis. These findings, which also included equipotent in vitro effects in human versus mouse cells, provide novel insights into the mechanisms underlying potential benefits of AMPK activation in NASH and metabolic syndrome, and support the concept that AMPK activators could have therapeutic utility for the treatment of NASH and related comorbid conditions.
Supporting information
Supporting Materials
Acknowledgment
The authors thank Sanne Skovgård Veidal and Michael Feigh (Gubra, Hørsholm, Denmark) for the in vivo DIO‐NASH studies, and Micheline Kergoat, Sophie Raynal, and Armel Nijman (Metabrain Research, Maisons‐Alfort, France) for the HSL assessment in human/mouse adipocytes and DIO‐NASH, ob/ob adipose tissue explants (cytokines and NF‐kB), in vitro AMPK activation, pAMPK ex vivo in DIO‐NASH mice, in vivo treatment, and euglycemic clamp in ob/ob mice.
Supported by Poxel SA.
Potential conflict of interest: Dr. Bolze owns stock in and is employed by Poxel SA. Dr. Gluais‐Dagorn owns stock in and is employed by Poxel SA. Dr. Hallakou‐Bozec owns stock in and is employed by Poxel SA. Dr. Moller owns stock in and is employed by Poxel SA. Dr. Monternier owns stock in and is employed by Poxel SA. Dr. Steinberg consults, advises, and received grants from Poxel SA and Esperion Therapeutics. He owns stock in, is employed by, consults for, advises, and received grants from Espervita Therapeutics.
References
Author names in bold designate shared co‐first authorship.
- 1. Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology 2020;158:1851‐1864. [DOI] [PubMed] [Google Scholar]
- 2. Cusi K, Sanyal AJ, Zhang S, Hartman ML, Bue‐Valleskey JM, Hoogwerf BJ, et al. Non‐alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes Metab 2017;19:1630‐1634. [DOI] [PubMed] [Google Scholar]
- 3. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 4. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 2012;142:711‐725.e6. [DOI] [PubMed] [Google Scholar]
- 5. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero‐Gomez M, et al. A new definition for metabolic dysfunction‐associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202‐209. [DOI] [PubMed] [Google Scholar]
- 6. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349‐364. [DOI] [PubMed] [Google Scholar]
- 7. Bril F, Cusi K. Liver fat accumulation as a barometer of insulin responsiveness again points to adipose tissue as the culprit. Hepatology 2017;66:296‐297. [DOI] [PubMed] [Google Scholar]
- 8. Kim KH, Lee MS. Pathogenesis of nonalcoholic steatohepatitis and hormone‐based therapeutic approaches. Front Endocrinol (Lausanne) 2018;9:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steinberg GR, Carling D. AMP‐activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 2019;18:527‐551. [DOI] [PubMed] [Google Scholar]
- 10. Gu X, Bridges MD, Yan Y, de Waal PW, Zhou XE, Suino‐Powell KM, et al. Conformational heterogeneity of the allosteric drug and metabolite (ADaM) site in AMP‐activated protein kinase (AMPK). J Biol Chem 2018;293:16994‐17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun 2013;4:3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M. AMPK re‐activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 2018;28:194‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim SJ, Tang T, Abbott M, Viscarra JA, Wang Y, Sul HS. AMPK phosphorylates desnutrin/ATGL and hormone‐sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol Cell Biol 2016;36:1961‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kopietz F, Berggreen C, Larsson S, Säll J, Ekelund M, Sakamoto K, et al. AMPK activation by A‐769662 and 991 does not affect catecholamine‐induced lipolysis in human adipocytes. Am J Physiol Endocrinol Metab 2018;315:E1075‐E1085. [DOI] [PubMed] [Google Scholar]
- 15. Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest 2011;121:4903‐4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mancini SJ, White AD, Bijland S, Rutherford C, Graham D, Richter EA, et al. Activation of AMP‐activated protein kinase rapidly suppresses multiple pro‐inflammatory pathways in adipocytes including IL‐1 receptor‐associated kinase‐4 phosphorylation. Mol Cell Endocrinol 2017;440:44‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang Z, Li T, Jiang S, Xu J, Di W, Yang Z, et al. AMPK: a novel target for treating hepatic fibrosis. Oncotarget 2017;8:62780‐62792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao P, Sun X, Chaggan C, Liao Z, in Wong K, He F, et al. An AMPK‐caspase‐6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020;367:652‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, et al. Adenosine monophosphate‐activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology 2008;47:668‐676. [DOI] [PubMed] [Google Scholar]
- 20. Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate‐activated protein kinase. Hepatology 2008;47:677‐685. [DOI] [PubMed] [Google Scholar]
- 21. Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly‐Y M, et al. Liver‐specific activation of AMPK prevents steatosis on a high‐fructose diet. Cell Rep 2017;18:3043‐3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia D, Hellberg K, Chaix A, Wallace M, Herzig S, Badur MG, et al. Genetic liver‐specific AMPK activation protects against diet‐induced obesity and NAFLD. Cell Rep 2019;26:192‐208.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006;3:403‐416. [DOI] [PubMed] [Google Scholar]
- 24. Gómez‐Galeno JE, Dang Q, Nguyen TH, Boyer SH, Grote MP, Sun Z, et al. A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med Chem Lett 2010;1:478‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjøbsted R, Jørgensen NO, et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non‐human primates and mice. Cell Metab 2017;25:1147‐1159.e10. [DOI] [PubMed] [Google Scholar]
- 26. Esquejo RM, Salatto CT, Delmore J, Albuquerque B, Reyes A, Shi Y, et al. Activation of liver AMPK with PF‐06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine 2018;31:122‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmoll D, Ziegler N, Viollet B, Foretz M, Even PC, Azzout‐Marniche D, et al. Activation of adenosine monophosphate‐activated protein kinase reduces the onset of diet‐induced hepatocellular carcinoma in mice. Hepatol Commun 2020;4:1056‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kristiansen MNB, Veidal SS, Rigbolt KTG, Tølbøl KS, Roth JD, Jelsing J, et al. Obese diet‐induced mouse models of nonalcoholic steatohepatitis‐tracking disease by liver biopsy. World J Hepatol 2016;8:673‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tølbøl KS, Kristiansen MNB, Hansen HH, Veidal SS, Rigbolt KTG, Gillum MP, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet‐induced obese mouse models of biopsy‐confirmed nonalcoholic steatohepatitis. World J Gastroenterol 2018;24:179‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP‐activated protein kinase by the small molecule A‐769662, a member of the thienopyridone family. J Biol Chem 2007;282:32539‐32548. [DOI] [PubMed] [Google Scholar]
- 31. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen Z‐P, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin‐sensitizing effects of metformin. Nat Med 2013;19:1649‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, et al. Tumor necrosis factor alpha‐induced skeletal muscle insulin resistance involves suppression of AMP‐kinase signaling. Cell Metab 2006;4:465‐474. [DOI] [PubMed] [Google Scholar]
- 34. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 2013;123:2764‐2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vonghia L, Van Herck MA, Weyler J, Francque S. Targeting myeloid‐derived cells: new frontiers in the treatment of non‐alcoholic and alcoholic liver disease. Front Immunol 2019;10:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luedde T, Schwabe RF. NF‐κB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011;8:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta‐inducible antagonist of TGF‐beta signalling. Nature 1997;389:631‐635. [DOI] [PubMed] [Google Scholar]
- 38. Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab 2016;311:E730‐E740. [DOI] [PubMed] [Google Scholar]
- 39. Gauthier M‐S, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, et al. Decreased AMP‐activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole‐body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun 2011;404:382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu XJ, Gauthier M‐S, Hess DT, Apovian CM, Cacicedo JM, Gokce N, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot‐specific changes in gene expression in adipose tissue. J Lipid Res 2012;53:792‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP‐activated protein kinase cascade. Biochem J 2007;403:139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Myers RW, Guan H‐P, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan‐AMPK activator MK‐8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017;357:507‐511. [DOI] [PubMed] [Google Scholar]
- 43. Scott J, Ling N, Issa S, Dite T, O’Brien M, Chen Z‐P, et al. Small molecule drug A‐769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem Biol 2014;21:619‐627. [DOI] [PubMed] [Google Scholar]
- 44. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 2019;16:145‐159. [DOI] [PubMed] [Google Scholar]
- 45. Zhao P, Saltiel AR. From overnutrition to liver injury: AMP‐activated protein kinase in nonalcoholic fatty liver diseases. J Biol Chem 2020;295:12279‐12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5'‐monophosphate‐activated protein kinase promotes macrophage polarization to an anti‐inflammatory functional phenotype. J Immunol 2008;181:8633‐8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5'‐monophosphate‐activated protein kinase regulates IL‐10‐mediated anti‐inflammatory signaling pathways in macrophages. J Immunol 2015;194:584‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013;58:589‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The differential roles of T cells in non‐alcoholic fatty liver disease and obesity. Front Immunol 2019;10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. da Silva Morais A, Abarca‐Quinones J, Guigas B, Viollet B, Starkel P, Horsmans Y, et al. Development of hepatic fibrosis occurs normally in AMPK‐deficient mice. Clin Sci (Lond) 2009;118:411‐420. [DOI] [PubMed] [Google Scholar]
- 51. Bates J, Vijayakumar A, Ghoshal S, Marchand B, Yi S, Kornyeyev D, et al. Acetyl‐CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J Hepatol 2020;73:896‐905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Materials