

Summary
Achnatherum splendens Trin. (Gramineae) is a constructive species of the arid grassland ecosystem in Northwest China and is a major forage grass. It has good tolerance of salt and drought stress in alkaline habitats. Here, we report its chromosome‐level genome, determined through a combination of Illumina HiSeq sequencing, PacBio sequencing and Hi‐C technology. The final assembly of the ~1.17 Gb genome sequence had a super‐scaffold N50 of 40.3 Mb. A total of 57 374 protein‐coding genes were annotated, of which 54 426 (94.5%) genes have functional protein annotations. Approximately 735 Mb (62.37%) of the assembly were identified as repetitive elements, and among these, LTRs (40.53%) constitute the highest proportion, having made a major contribution to the expansion of genome size in A. splendens. Phylogenetic analysis revealed that A. splendens diverged from the Brachypodium distachyon–Hordeum vulgare–Aegilops tauschii subclade around 37 million years ago (Ma) and that a clade comprising these four species diverged from the Phyllostachys edulis clade ~47 Ma. Genomic synteny indicates that A. splendens underwent an additional species‐specific whole‐genome duplication (WGD) 18–20 Ma, which further promoted an increase in copies of numerous saline–alkali‐related gene families in the A. splendens genome. By transcriptomic analysis, we further found that many of these duplicated genes from this extra WGD exhibited distinct functional divergence in response to salt stress. This WGD, therefore, contributed to the strong resistance to salt stress and widespread arid adaptation of A. splendens.
Keywords: Achnatherum splendens, genome assembly, whole‐genome duplication, transcriptome, stress tolerance
Introduction
Soil salinity, which is one of the major environmental stresses, limits plant growth (Mahajan and Tuteja, 2005; Zhu, 2001). It has been reported that ∼20% of global land plants are seriously affected by salinity (Morton et al., 2019; Qadir et al., 2014). High concentrations of salt trigger ion imbalance and hyperosmotic stress in plants, and possibly cause secondary stresses such as oxidative damage (Møller and Tester, 2007; Shabala and Cuin, 2008; Zhu, 2001). These stresses eventually lead to poor growth or plant death. Based on their responses to soil salinity, plants can be divided into glycophytes and halophytes. While glycophytes are highly sensitive to salinity, halophytes can survive in saline–alkali habitats (Flowers and Colmer, 2008; Flowers et al., 2010; Gong et al., 2020). It is, therefore, necessary to understand the genomic make‐up of halophytes, which may be helpful in the management and usage of these special wild resources (Wu et al., 2012).
Achnatherum splendens Trin. (Gramineae) (2n = 48; Guo et al., 2003) is a perennial halophyte, which is a constructive species of the alkaline grassland in northwest China (e.g. Figure 1a; Huai et al., 2008; Jiang et al., 2017). Through long‐term adaptation to the extremely alkaline environment, this species has developed a considerable level of resistance to salt and drought stress (e.g. Figure 1b; Haixia et al., 2008; Irfan et al., 2016). In addition, the highly developed underground roots of this species help to improve sand fixation and water conservation in saline areas (Zhang et al., 2010). This constructive species also provides a major forage resource with good palatability and high nutritive value for local livestock in the arid alkaline regions, and its stalks have also been used as superior raw material for paper making for a long time (You et al., 2020).
Figure 1.
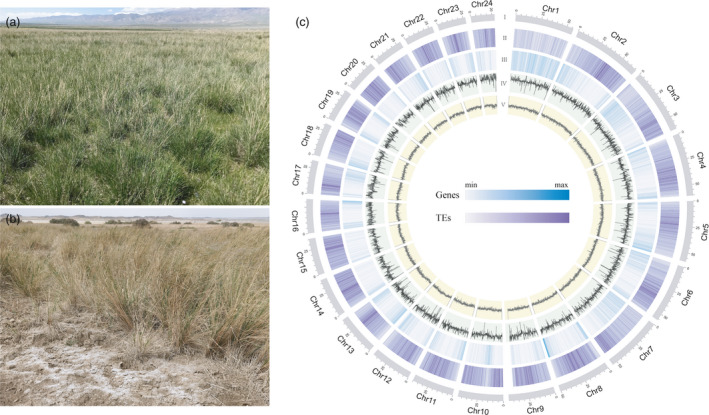
(a) and (b) Photographs of A. splendens as a constructive species of the alkaline grassland. (c) The landscape of assembly and annotation of the A. splendens genome. Tracks from outside to the inside correspond to I, pseudochromosomes; II, repeat density; III, gene density; IV, depth density and V, GC density.
In this study, we assembled de novo a chromosome‐scale genome for A. splendens through a combination of various sequencing strategies, involving Illumina HiSeq sequencing, single‐molecule real‐time (SMRT) sequencing and chromosome conformation capture (Hi‐C) technology. RNA‐seq‐based transcriptomics were used to identify differential expressions of the paralogous genes under salt treatments. Using gene annotation, phylogenetic analysis and transcriptomic analysis, we aimed to clarify the genomic basis of how this species adapts to alkaline habitats.
Results
Genome sequencing, size estimation and assembly
We estimated the genome size based on K‐mer distribution. In total, 57 879 333 379 k‐mers (size 17) were identified. The major peak was around a k‐mer depth of 52, and another clear peak was detected as being located at half of the expected depth, and inferred to be a heterozygous peak. The genome size of A. splendens was estimated to be 1113 Mb (Figure S1 and Table S1).
A total of 83.81 Gb of subreads (75× genome coverage) from the PacBio Sequel platform and 142.82 Gb of short reads (128× genome coverage) from the Illumina HiSeq platform were generated, representing almost 200‐fold coverage of the A. splendens genome (Table S2). Contigs were first self‐corrected and initially assembled with high‐quality PacBio subreads, and then polished using the Illumina reads. The improved contigs were further assembled into scaffolds with a scaffold N50 of 1.08 Mb. To assign scaffolds to the correct chromosomal positions, a total of 123.42 Gb Hi‐C reads were produced based on genomic proximity. 94.85% of the whole‐genome assembly was successfully anchored into 24 pseudochromosomes and 92.28% was ordered (Figure S2 and Table S3). The final assembled genome size was 1178 Mb with a super‐scaffold N50 length of 40.3 Mb (Table 1). This is little larger than the predicated genome size, probably as a result of the comparatively high heterozygosity of the genome.
Table 1.
Features of the A. splendens genome assembly
| Genome assembly | Value |
|---|---|
| Estimated genome size (Gb) | 1.13 |
| Total length of scaffolds (Gb) | 1.17 |
| Number of scaffolds | 2297 |
| N50 of scaffolds (Mb) | 1.08 |
| N90 of scaffolds (kb) | 271.02 |
| Longest scaffolds (Mb) | 6.08 |
| Average scaffold length (kb) | 513.07 |
| Anchored to chromosome (Gb) | 1.12 |
| N50 of super‐scaffold (Mb) | 40.3 |
| GC content (%) | 45.5 |
| Undetermined bases (%) | 0.66 |
| Genome annotation | |
| Repetitive sequences (Mb) | 731.95 |
| Number of protein‐coding genes | 57 374 |
| Genes in pseudochromosomes | 50 069 (87.30%) |
| Number of non‐coding RNAs | 289 938 |
The completeness and accuracy of the assembled genome were assessed by BUSCO and RNA sequencing. We found that 1401 of 1440 (97.2%) conserved protein genes were completely captured in the genome (Table S4). Moreover, 74 546 isoforms were generated by 1.37 Gb of PacBio full‐length cDNA sequencing data (Table S5), approximately 98.9% (73 770) of which could be well aligned to the genome (Table S6). These assessments indicate that the genome of A. splendens was well assembled, with high quality, completeness and accuracy.
Genome annotation
A total of ~732 Mb (62.56%) of the A. splendens genome assembly was identified as repetitive elements based on de novo and homology‐based methods (Figure 1c and Table S7). As in most plant genomes, long terminal repeat retrotransposons (LTR‐RTs), accounting for 40.53% of the genome, are the most abundant elements. Among the LTR‐RTs, Gypsy and Copia are the most common super‐families, representing 27.4% and 12.4% of the total assembly respectively (Table S7). The ratio of Gypsy elements to Copia elements is 2.23, similar to that in other species in the Gramineae, except for O. sativa which has a ratio of 5.06 (Table S8).
For gene annotation, a high‐confidence set of 57 374 protein‐coding genes was predicted by integrating results from de novo, homology‐based and transcript‐based approaches (Table S9), and of these, 87.3% (50 069) genes were anchored into 24 pseudochromosomes. On average, protein‐coding genes in the A. splendens genome are 3250 bp long and cover 4.85 exons, and these features are similar to those in other species in the Gramineae (Figure S3; Table S9). The predicted genes were then annotated for functional proteins they encode by a consensus method, applying InterPro (Hunter et al., 2009), Gene Ontology (GO; Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto, 2000) and Swiss‐Prot (Boeckmann et al., 2003). In the aggregate, 54 426 genes (94.8% of the predicted genes) were identified as having known homologs in protein databases (Table S10). In addition, we detected 644 microRNA (miRNA), 898 transfer RNA (tRNA), 885 ribosomal RNA (rRNA) and 447 small nuclear (snRNA) genes in the genome sequence (Table S11).
Comparative genomic analysis
Based on sequence homology, a total of 310 214 genes were clustered into 177 606 families, of which 7129 families were shared by all 10 species and 1116 were specific to the A. splendens genome (Figure 2c,d and Table S12). These unique gene families were predicted to have functions involved in ‘response to oxidative stress’ (GO:0006979) and ‘oxidoreductase activity, acting on peroxide as acceptor’ (GO:0016684) (Table S13).
Figure 2.
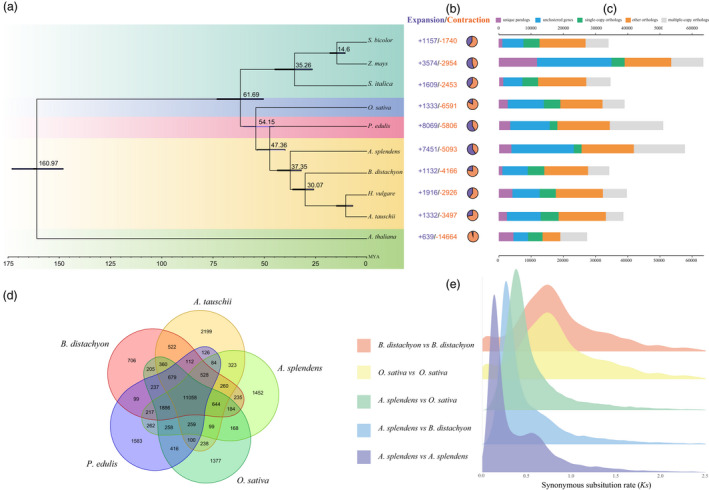
(a) Phylogenetic relationships and times of divergence between A. splendens and other Gramineae species. (b) Expansions and contractions of gene families. Purple and yellow indicate expanded and contracted gene families respectively. (c) Clusters of orthologous and paralogous gene families in A. splendens and other Gramineae species. Only the longest isoform for each gene is shown. (d) Venn diagram of orthologous genes shared among the five species A. splendens, O. sativa, B. distachyon, P. edulis and A. tauschii. © Distribution of average synonymous substitution levels (Ks) between syntenic blocks.
To infer the phylogeny of A. splendens, 963 single‐copy orthologous genes from 10 species were identified and used to construct a phylogenetic tree. The resulted phylogeny indicated that within the monocot lineage comprising six species (O. sativa, P. edulis, A. splendens, B. distachyon, H. vulgare and A. tauschii), the subclade containing only A. splendens diverged from the other one comprising the other three species (B. distachyon, H. vulgare and A. tauschii), which together diverged from P. edulis and further from O. sativa. Based on the MCMCtree and fossil calibration, the estimated divergence times were around 54.16 million years ago (Ma) between O. sativa and the other five monocots, 47.36 Ma between P. edulis and the remaining four species and 37.35 Ma between A. splendens and the other three species (Figure 2a). These divergences were in agreement with estimates from previous reports (Varshney et al., 2017).
We identified 7451 gene families that had expanded while 5093 had contracted in the A. splendens genome compared to the other plant species (Figure 2b). GO analysis revealed that the expanded orthogroups were significantly enriched in the functional terms ‘transmembrane transport’, ‘transmembrane transporter activity’ and ‘adenyl ribonucleotide binding’ (Figure S4 and Table S14). Several markedly expanded gene families were functionally associated with maintenance of intracellular osmolality and pH homeostasis.
Whole‐genome duplication and chromosome composition
Whole‐genome duplication (WGD) events, which have happened frequently in most plants, are commonly regarded as a major evolutionary force (De Peer et al., 2009; Lynch and Conery, 2000; Salmanminkov et al., 2016). The additional genetic material provided by WGD enhances the adaptive plasticity of plants and contribute to species diversification and functional innovations (Paterson et al., 2004; Tang et al., 2008). By revealing preserved ancestral gene order, gene collinearity analysis is crucial in uncovering changes in and evolution of a plant genome (Wang et al., 2006). We examined gene collinearity among the A. splendens, B. distachyon and O. sativa genomes. B. distachyon and O. sativa were chosen as references because both have relatively simple genome structures, with only one WGD, common to all grass species (cWGD), found in them (Paterson et al., 2009; Wang et al., 2015).
In order to infer intra‐genomic gene collinearity, we identified a total of 15 371 gene pairs in 677 homologous blocks of the A. splendens genome. Using the same criteria, we detected 324 and 657 homologous blocks from the O. sativa and B. distachyon genomes, which contained 3770 and 5602 collinear gene pairs respectively. The largest homologous block in A. splendens, with 1211 gene pairs, was located in chromosomes 2 and 3 (Figure S5), while the largest in O. sativa and B. distachyon had, respectively, 280 and 265 gene pairs (Table S15). Thus, considerably more homologous gene pairs and more complete homologous regions were found in the A. splendens genome than in O. sativa or B. distachyon, suggesting that one or more additional WGD events had occurred in the A. splendens genome. For inter‐genomic gene collinearity, there were 1553 homologous blocks involving 35 753 collinear gene pairs between the A. splendens and O. sativa genomes and 1664 homologous blocks involving 35 923 collinear gene pairs between the A. splendens and B. distachyon genomes, revealing well‐preserved syntenic features between the A. splendens genome and the two reference genomes (Table S15).
Through mapping the O. sativa genome sequence onto the A. splendens genome, we generated a homologous gene dotplot (Figure 3), which was helpful in locating homologous correspondence and determining orthologs (established from the O. sativa––A. splendens split) and outparalogs (established after the cWGD). Without taking into account any gene loss, if there was no additional WGD in A. splendens after the cWGD, we would expect to find each O. sativa gene (or chromosomal region) having one best matched orthologous A. splendens gene (or chromosomal region), and one outparalogous A. splendens gene (or chromosomal region). If there had been an A. splendens‐specific WGD, we would expect to find each O. sativa gene (or chromosomal region) having two best matched orthologous A. splendens genes (or chromosomal regions), and two outparalogous A. splendens genes (or chromosomal regions). In the dotplots and genomic synteny between O. sativa and A. splendens (Figure 3), we found that almost every rice chromosome had two best matched chromosomal regions in A. splendens. For instance, O. sativa chromosomes 1 and 5 are homologous (paralogous) after the cWGD event (Figure 4) and we could identify two more orthologs and two more outparalogs in A. splendens (Figure 3b). O. sativa chromosome 1 is best matched with (or orthologous to) regions in the A. splendens chromosomes 2 and 3, and each orthologous region is outparalogous to O. sativa chromosome 5. Using the same strategy, we drew homologous dotplots between A. splendens and B. distachyon, which exhibited a pattern analogous to that for O. sativa (ortholog ratio 1 : 2), further supporting an additional WGD in A. splendens (Figure S6).
Figure 3.
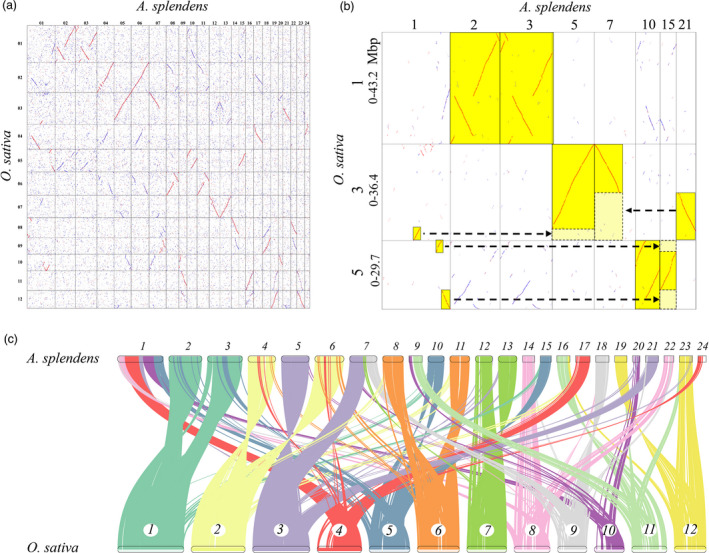
(a) Dotplots showing genes homologous between the A. splendens and O. sativa genomes. (b) Example of homologous gene dotplots for A. splendens and O. sativa. Chromosome numbers and regions (in Mbp) are shown. Best hit (orthologous) genes are red dots, secondary hits (outparalogous) are blue dots and the others are shown in grey. Highlights show the best matched chromosomal regions. Arrows show correspondence produced by chromosome breakages during evolution. (c) Gene synteny between the A. splendens and O. sativa genomes.
Figure 4.
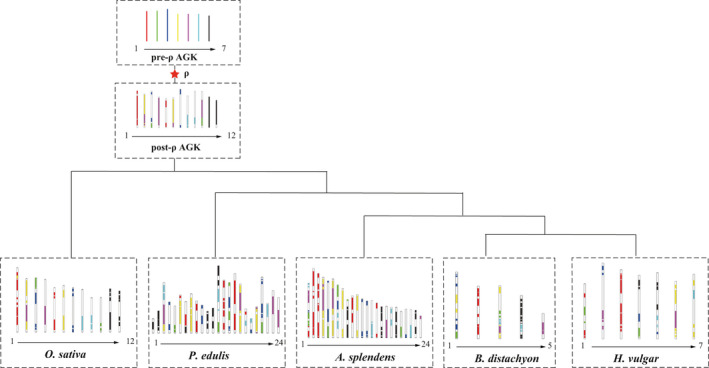
Construction of ancestral chromosomes for A. splendens, O. sativa, B. distachyon, H. vulgare and P. edulis. AGK indicates the ancestral grass karyotype. The red star indicates a whole‐genome duplication event.
Furthermore, the synonymous substitution divergence (Ks) of each collinear gene pair within a genome or between genomes was characterized. The Ks distribution of paralogs within O. sativa and B. distachyon suggested only one peak at Ks ~0.6. In contrast, the paralogous peaks in A. splendens showed a clear bimodal feature, having one peak at Ks ~0.15 and another at ~0.5 (Figure 2e), implying at least two WGD events. The paralogous peak at ~0.6 corresponds to the cWGD event, which has been dated to ~96 Ma (Wang et al., 2015). Based on this calibrated time, the A. splendens‐specific WGD event was inferred to have occurred ~18–20 Ma. The orthologous peaks for A. splendens–O. sativa and –B. distachyon, corresponding to Ks values of ~0.45 and ~0.3, were dated to 50 and 35 Ma, respectively, consistent with phylogenetic results.
Genomes often underwent massive gene loss and chromosomal reorganization after WGD events (Wang et al., 2005, 2009). Previous studies have suggested that Gramineae genomes evolved from a pre‐ρ ancestral grass karyotype (AGK) with 7 protochromosomes to a post‐ρ AGK with 12 protochromosomes (Murat et al., 2010, 2017). To unmask the details of chromosomal reorganizations in A. splendens, the AGK genes were downloaded and mapped onto the chromosomes of five Gramineae species (A. splendens, O. sativa, B. distachyon, P. edulis and H. vulgare) (Figure 4). It is obvious that O. sativa is most similar to the ancestral chromosomal composition; ancestral chromosomes 1, 6 and 7 were well conserved and duplicated to generate chromosomes 1 and 5, 8 and 9, and 11 and 12 respectively. In A. splendens, apart from six chromosomes (1, 4, 5, 6, 7 and 9) that had experienced chromosome fusions, each chromosome originated from only one ancestral chromosome, suggesting that there was little chromosomal rearrangement after the additional WGD event in A. splendens. In addition, each species examined here inherited one relatively conserved chromosome (AGK 1); for example, it was present as chromosomes 1 and 5 in O. sativa, chromosomes 7, 9, 14 and 16 in H. vulgare, chromosomes 2, 3, 5 and 15 in A. splendens, chromosome 2 in B. distachyon and chromosome 3 in H. vulgare. It is likely that genes on this conserved chromosome may have played a significant role in major aspects of development and regulation and have therefore avoided recursive chromosomal fusion and fission.
WGD contributed to adaptation to the saline–alkali environment
In addition to a common WGD event shared by all monocots and another common WGD event shared by Gramineae species (Paterson et al., 2004), A. splendens experienced an additional species‐specific WGD event after its divergence from other Gramineae species (see above results). Although massive gene loss may have occurred after these WGD events, we still found that more than 55% of the annotated 57 374 genes were duplicate genes in the WGD regions (Table S16). Especially, most of these duplicate genes resulted mainly from the species‐specific WGD event (Figure 3 and Figure S6). To investigate whether this extra WGD event had contributed to the adaptation of A. splendens to a saline–alkali environment, we first, based on the functional annotations in the A. thaliana genome, identified gene families that are closely related to resistance to salt stress and transcription factor (TF) families in the A. splendens, O. sativa and B. distachyon genomes. We found that copy numbers of the salt‐tolerant–related gene families and TF families in A. splendens were, respectively, 1.77 and 1.73 times more than those in O. sativa, and 1.77 and 1.67 times more than those in B. distachyon (Table S17 and Table S18). The increase in copy number of these gene families may have played a role in the evolution of salt tolerance in A. splendens.
Hierarchical cluster analysis of transcriptomic data showed clustering of the four repeats for the shoot or root at each time point (Figure S7), except for one shoot sample at 24 h, which was discarded for downstream analysis. Differentially expressed genes (DEGs) were identified under salt treatment by comparing each time point (6 and 24 h) with 0 h. A total of 2866, 2517, 571 and 2587 DEGs were identified in the 6 h root, 24 h root, 6 h shoot and 24 h shoot respectively (Figure 5a). Only 571 DEGs were detected under salt treatment of 6 h shoot, while more than four times DEGs were identified in 24 h shoot compared to 6 h shoot, indicating that the early stage of salt stress does not considerably affect the shoot of A. splendens. Of the total 5917 DEGs, majority of them were specific to each of the four samples and only 170 DEGs were shared by all samples (Figure 5a,b). Co‐expression analysis showed that the DEGs formed eight major clusters in root and shoot respectively (Figure 6). The patterns of some clusters may be correlated with the adaptation to a saline–alkali environment of this species.
Figure 5.
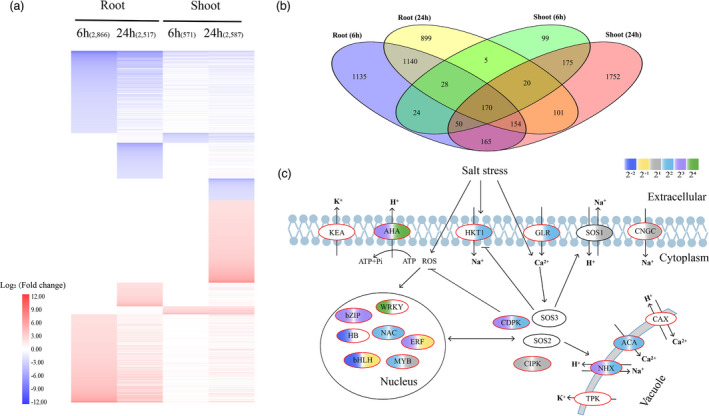
Transcriptomics of A. splendens under salt stress. (a) Expression of DEGs identified in root and shoot at each time point. The heatmap was generated from hierarchical cluster analysis of genes. (b) Venn diagram of the number of DEGs in root and shoot at each time point. (c) Expression of key salt tolerance‐related genes in A. splendens. Ellipses with black borders indicate single gene, while red borders indicate gene or TF families. For each family, we only selected one copy to show its expression under salt treatment. Detailed expression information of these family members is shown in Table S21. The filled colours correspond to their degree of regulation (TPMtreatment/TPMcontrol) at 6 h in root (left) and at 24 h in shoot (right) in response to salt stress.
Figure 6.
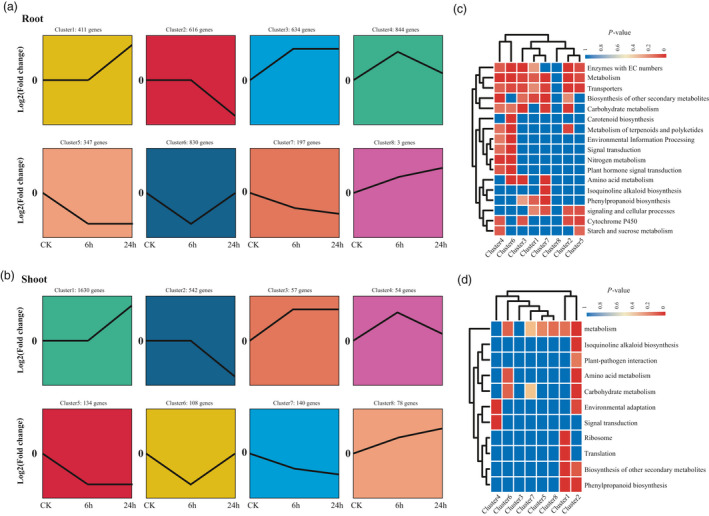
Expression patterns and enrichment analysis of DEGs under salt stress. (a–b) Cluster analysis of DEGs displaying a log2 fold change (with absolute value >2) of transcripts during salt stress at 6 and 24 h in root and shoot respectively. The comparisons include control versus control (CK), 6 h versus control (6 h) and 24 h versus control (24 h). (c–d) Heat maps of significantly enriched pathways during salt stress. The blue and red colours indicate the P‐values for significantly enriched pathways for each cluster.
In root, cluster 4 (844 DEGs) and cluster 6 (830 DEGs; Figure 6a) are the two biggest modules containing up‐regulated and down‐regulated genes respectively. DEGs of both clusters were induced only at 6 h after salt treatment and are mainly enriched in metabolism of carbohydrate, terpenoids, polyketides and nitrogen, environmental information processing, signal transduction and plant hormone signal transduction. Therefore, phytohormone signals and metabolism processes were turned on or off at the early phase of salt stress. Pathways that play an important role in response to abiotic stresses, such as cytochrome P450, metabolism of amino acid, starch and sucrose and phenylpropanoid biosynthesis, were also enriched by other clusters (Figure 6c). In shoot, the biggest cluster 1, containing 1630 DEGs, was up‐regulated in the late phase (24 h). It contains genes for ribosome, translation and biosynthesis of other secondary metabolites and phenylpropanoid biosynthesis. The second biggest cluster (cluster 2: 542 DEGs) was also induced in the late phase, comprising genes enriched in metabolism of carbohydrate and amino acid, environmental adaptation and plant–pathogen interaction (Figure 6b,d). Therefore, the major clusters and DEGs revealed in root and shoot were different (Figures 5a and 6b,d), probably in relation to diverse adaptive mechanisms in different tissues.
We then investigated expression patterns of paralogous genes in A. splendens under salt treatment. We focused on the paralogous genes that have two copies in A. splendens with one synteny copy in O. sativa. A total of 6796 paralogous gene pairs were identified. Among them, 5347 (78.68%) pairs of genes had contrasted differential expressions and 1047 (15.41%) pairs had one DEG between different tissues in each pair. A total of 402 pairs of genes with differential expression (Table S19) are enriched in many functional categories that are related to salt stress, such as ‘oxidoreductase activity’, ‘antioxidant activity’, ‘transcription regulator activity’ and ‘response to oxidative stress’ (Figure S8). Within these 402 gene pairs, we further detected the differential expression pattern of two paralogous copies in each pair. We did not consider the expression changes in different time points in order to reduce the number of patterns. We recognized four patterns: pattern I, gene functional redundancy; pattern II, one copy was DEG in a tissue, while the other copy was DEG in a different tissue; pattern III, the two copies were differentially expressed in the same tissue, but in different way (i.e. up‐ vs. down‐regulation) and pattern IV: a combination of patterns II and III. We found that 258 (64.18%) gene pairs showed pattern I, 108 gene pairs (26.86%) for pattern II, 10 pairs for (2.49%) pattern III and 26 pairs (6.47%) for pattern IV (Table S20). These results indicated that many duplicated paralogous genes from the extra WGD in A. splendens had evolved the new functions during its saline–alkali adaptation. Finally, we examined the expression of the duplicated genes, especially in the TF families related to the salt stress response pathway of this species. We found that many duplicate genes of these families, for examples, bZIP, NAC and WRKY, important TFs in the regulation of gene expression in response to abiotic stress and the key genes controlling ion homeostasis (e.g. NHX and ACA) were extensively up‐regulated in the salt‐stressed samples (Figure 5c and Table S21). However, the SOS (salt overly sensitive) genes with relative high expression level (Table S21) that are critical for salt tolerance (Gong et al., 2020) were not differentially expressed in A. splendens, which is consistent with previous result (Liu et al., 2016).
Discussion
Abiotic stress, especially salinity and alkali, severely limits plant growth and biomass productivity worldwide, and this is becoming an enormous threat because of increases in climatic changes and human activities (Yamaguchi and Blumwald, 2005; Zhu, 2001). Previously researchers have conducted genomic analyses of salt‐tolerant plants, but our current knowledge remains limited in the case of constructive forage species of alkaline grassland (Huang et al., 2020; Ma et al., 2013; Zhu et al., 2007). Such key species are likely to have developed more adaptive features than other halophytes. In this study, we assembled a high‐quality genome for A. splendens, a constructive species of the alkaline grassland in Northwest China.
We combined Illumina HiSeq sequencing, PacBio sequencing and Hi‐C technology to generate a high‐quality chromosome‐scale genome assembly for A. splendens. The final assembled genome size was around 1.17 Gb with a super‐scaffold N50 of 40.3 Mb, and more than 94% of the genome sequences could be located on 24 pseudochromosomes. Frequent activation of repetitive elements is one of the main driving forces that act to increase genome sizes (Flavell et al., 1974; Levin and Moran, 2011). A total of 731 Mb was identified as repetitive elements, representing 62.11% of the total genome. LTR‐RTs dominated primarily, as in other plants (Vicient et al., 1999). Most LTR‐RTs had expanded and accumulated recently in the A. splendens genome. It should be noted that TE activity may also contribute to plant stress responses and, thus, strengthen the adaptation of A. splendens to harsh environments (Wendel, 2000). We identified a total of 57 374 protein‐coding genes from the assembled genome of A. splendens. This number is greater than those in O. sativa (39 049) and B. distachyon (34 310), which have not undergone an additional WGD, and other Gramineae species.
whole‐genome duplications or polyploidizations play critical roles in plant adaptations to stressful habitats (Adams and Wendel, 2005; Chen et al., 2020; Li et al., 2021; Soltis et al., 2014; Wang et al., 2021). They provide functional innovations through gene duplication, chromosome rearrangements and genome repatterning events (Murat et al., 2010). WGDs are, therefore, closely linked to species evolution and diversification. Recent research has revealed that all monocots experienced a common WGD event and that Gramineae species shared another common WGD event (Paterson et al., 2004). Genomic analyses indicated that A. splendens experienced an additional WGD event after its divergence from other monocots, which was estimated to happen at 18–22 Ma. We also established lists of orthologous and paralogous genes resulting from this recent WGD event, which may be useful for comparative genomics analyses in future research. This WGD may have resulted in the evolution of salt and drought tolerance in A. splendens through increasing the copy numbers of gene families followed by functional redundancy or innovations related to the regulation of ionic and osmotic equilibrium and the ABA pathway. For example, we identified numerous pairs of paralogous genes (i.e. two copies in A. splendens vs. one copy in O. sativa) in A. splendens, which arose from this species‐specific WGD. We focused on the duplicate genes that both are identified as DEGs in each pair. We identified a total of 402 pairs of such duplicate genes, some of which are enriched in response to oxidative stress and cell wall organization or biogenesis, suggesting that these duplicated genes should have played an important role in response to salt stress of this species. Of these 402 paralogous gene pairs, more than 35% gene pairs exhibit functional divergence by differential expression in different tissues or in different ways. Such an increase in gene family members and their functional redundancy or innovations may have enhanced the adaptation of this constructive species to large‐scale alkaline environments. The WGD‐driven salt resistance mechanisms revealed for this species differ from those in other genome‐sequenced halophytes, in which tandem copy increases in gene family members and positive mutations in genes involved in salt stress have enhanced their salt tolerance (Huang et al., 2020; Ma et al., 2013; Zhu et al., 2007). In summary, the availability of a high‐quality genome and transcriptomic data under salt treatment for A. splendens provides genomic insights into salt adaptation in this species and new genetic resources for evolutionary and comparative genomics analyses with other monocots in the future.
Methods
Plant material and growth conditions
One live A. splendens plant was collected from Xining, Qinghai Province, China, and then transferred to a greenhouse at the State Key Laboratory of Grassland and Agro‐ecosystems, School of Life Sciences, Lanzhou University. This plant was grown under controlled temperature (24 ± 1 °C) and humidity (45%–55%) conditions with a 14 h−10 h day−night cycle and used for genomic sequencing. Seeds of A. splendens from the same population were collected and used for karyotype analysis and RNA‐seq analysis under salt treatment. Seeds were first treated with a 4% sodium hypochlorite solution for 15 min, then rinsed thoroughly with sterile water. After chilled at 4 °C for 2 days, seeds were put in moist Petri dishes to germinate at 25 °C until the radicals were approximately 2 cm. Then, the seedlings were transferred into plant cultivation boxes (96 holes, aperture 5 mm) with 1 litre of MS (Murashige and Skoog Basal Medium with Vitamins, Phyto Technology Laboratories, Usage: 4.43 grams per litre) fluid nutrient medium, and grown in greenhouse under the same controlled conditions as the live A. splendens plant. The 4‐week‐old seedlings were treated with 300 mm NaCl (Liu et al., 2016). Roots and shoots with salt treatment for 0, 6 and 24 h were collected, immediately frozen in liquid nitrogen and stored at −80 °C. For each time point, ten seedlings from one cultivation box were pooled and used as a single sample. There were four biological replicates for each sample.
DNA extraction, genome and Hi‐C sequencing
To overcome the difficulty caused by the high heterozygosity and repetitive sequence content of the A. splendens genome (see Results), a combination of three different sequencing strategies (short read from Illumina, subread from PacBio and interaction read from Hi‐C) was employed to obtain a high‐quality reference genome. Genomic DNA was extracted by a standard CTAB method (Porebski et al., 1997) from fresh leaves which were harvested and frozen immediately in liquid nitrogen. Selected high‐quality DNA samples were sent to BGI for both Illumina and PacBio sequencing. For the Illumina sequencing, a library with an insert size of 270 bp was constructed and sequenced using an Illumina HiSeq 4000 platform (Illumina, San Diego, CA) to produce a short‐read sequencing library. The 20‐kb PacBio SMRT libraries were prepared according to the manufacturer’s standard protocol and sequenced on a PacBio‐sequel platform (PacBio, Menlo Park, CA) with 18 cells. Through mapping interactions between chromatin regions, the Hi‐C approach has been proved to be more effective than traditional genetic mapping for genome assembly, especially in terms of specificity, coverage and cost. A Hi‐C experiment was conducted according to a published procedure that briefly included cell cross‐linking, endonuclease digestion, terminal repair, cyclization, DNA purification and capture. We generated a total of 120 Gb of clean Hi‐C data (Table S2) from the Illumina HiSeq instrument, accounting for 100× coverage of the estimated A. splendens genome size. All sequencing reads were trimmed to remove adaptors and enhance quality.
Transcriptome sequencing
Fresh tissues of the same plant of A. splendens (leaf, stem, flower, seed and root) were mixed and sampled for PacBio full‐length cDNA sequencing. Two ISO‐Seq libraries were constructed for the mixed sample from these tissues and sequenced in two cells with a Pacific Bioscience RS II platform, generating 1 371 202 253 raw data (475 753 reads) and details are summarized in Table S3. For salt‐treated samples, total RNA extraction, library construction and sequencing were performed by BGI‐Shenzhen Company (Wuhan, China) on the MGI2000 platform by 2 × 150 bp pair‐end mode.
Genome size estimation
Genome size was estimated by K‐mer frequency distribution analysis (Genome Size = K‐mer_num/Peak_depth) (Li et al., 2010). In order to calculate and plot the K‐mer frequency distribution, a total of 67.28 Gb Illumina short reads were used to determine the total number of k‐mers of length 17 by means of jellyfish (Marçais and Kingsford, 2011). The selection of 17 mers to plot the frequency distribution was based on genome characteristics and the pattern of the Poisson distribution (Figure S1).
Genome assembly
The PacBio reads, corrected by CANU v1.8 (Koren et al., 2017), were first applied to generate the primary contigs via FALCON v3.1 (Chin et al., 2016) with the parameters length_cutoff = 15kb, length_cutoff_pr = 15kb, pa_HPCdaligner_option = ‘‐v ‐B128 ‐t16 ‐w8 ‐M32 ‐e0.75 ‐k16 ‐T32 ‐h320 ‐l3200 ‐s1000’ and ovlp_HPCdaligner_option = ‘‐v ‐B128 ‐k16 ‐T32 ‐h480 ‐e0.96 ‐l1600 ‐s1000’. Subsequently, contigs were enhanced by mapping all filtered PacBio reads through QUIVER (Chin et al., 2013) and were further corrected by Pilon v1.22 (Sun et al., 2021; Walker et al., 2014) with default parameters to obtain the consensus sequences. These improved contigs were then assembled into scaffolds by SSPACE‐LongRead v1.1 (Boetzer and Pirovano, 2014). Finally, potential duplicate haplotypes were removed based on PURGE HAPLOTIGS v1.1.0 with the option ‘‐a 80’ (Roach et al., 2018).
Chromosome assembly using Hi‐C data
For chromosome‐level assembly, the Hi‐C data were first aligned to the scaffolds by BWA v0.7.17 (Li and Durbin, 2009). They were classified as valid interaction pairs or invalid interaction pairs using HiC‐Pro v2.11.1 (Servant et al., 2015), and only valid interaction pairs were retained for subsequent assembly (Sun et al., 2021). Any two segments that presented connections inconsistent with the information from the raw contig were corrected manually. Furthermore, scaffolds were divided into subgroups and sorted and oriented into 24 pseudochromosomes using LACHESIS (Burton et al., 2013) with parameters CLUSTER_MIN_RE_SITES = 22, CLUSTER_MAX_LINK_DENSITY = 2, CLUSTER_NONINFORMATIVE_RATIO = 2, ORDER_MIN_N_RES_IN_TRUN = 10 and ORDER_MIN_N_RES_IN_SHREDS = 10.
Assessment of genome assembly
The completeness of the genome assembly was evaluated by both transcriptome data and Benchmarking Universal Single‐Copy Orthologs (BUSCO). Isoforms generated by PacBio full‐length cDNA sequencing were remapped to the final genome assembly by GMAP with default parameters (Wu and Watanabe, 2005). In addition, 1440 conserved single‐copy orthologs in the BUSCO embryophyta odb9 dataset were searched against the genome sequences through BUSCO with the default parameters.
Repeat annotation
Repetitive elements, including tandem repeats and transposable elements (TEs), were predicted in the A. splendens genome. For tandem repeats, Tandem Repeats Finder v4.09 (Benson, 1999) was implemented with the settings Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 50 and MaxPeriod = 2000. For identification of TEs, a combination of homology‐based and de novo approaches was mainly used. We first used RepeatMasker v4.0.5 (Tarailo‐Graovac and Chen, 2009) with the Repbase TE library (Jurka et al., 2005) and RepeatProteinMasker (Tarailo‐Graovac and Chen, 2009) with the TE protein database to search for homologous repeat sequences in the genome. Then, de novo‐based identification was performed by RepeatModeler v5.8.8 (Tarailo‐Graovac and Chen, 2009) and LTR_FINDER (Xu and Wang, 2007) to predict the repeat element boundaries and family relationships from genome data. Finally, all repeat identification results from the different software packages were integrated and redundancy was eliminated to produce the final repeat annotation.
Gene annotation
Three complementary methods were employed to predict protein‐coding genes: homology‐based, de novo and transcriptome‐based predictions. For homology‐based predictions, protein sequences from seven different species (A. thaliana, O. sativa, S. bicolor, S. italica, Z. mays, B. distachyon and H. vulgare) were downloaded and aligned with the repeat‐masked A. splendens genome by TBALSTN with an E‐value cut‐off of 1e‐5. The aligned sequences and candidate genomic regions were corrected and optimized by GeneWise v2.4.1 (Birney et al., 2004) for further prediction of exact protein coding gene structures. For de novo prediction, 3000 full‐length genes were selected randomly from the homology‐based predictions to train gene models of A. splendens. Four de novo prediction programs (Augustus v2.5.5, Stanke et al., 2008; Geneid v1.4, Blanco et al., 2007; GeneMark v3.54, Lukashin and Borodovsky, 1998; SNAP v2006‐07‐28, Korf, 2004) were utilized with A. splendens gene models for de novo prediction. Genes with coding sequences of <150 bp were discarded. For transcriptome‐based predictions, all RNA‐seq data from mixed samples (flower, leaf, stem, root and seed) were mapped to the A. splendens genome by PASA v2.3.3 (Haas et al., 2003), then TransDecoder (http://transdecoder.sourceforge.net/) was used to produce the annotation file. Finally, all predictions of gene models yielded by the above approaches were integrated using EVidenceModeler (EVM) v1.1.1 (Haas et al., 2008) to generate a consensus gene set.
Functional annotations of the predicted protein‐coding genes were applied by carrying out BLASTP (E‐value 1e‐5) against publicly available protein databases including KEGG (Kanehisa and Goto, 2000), SwissProt (Boeckmann et al., 2003) and InterProScan v5.36 (Zdobnov and Apweiler, 2001). GO annotations were accomplished using the Blast2GO pipeline v3.1.3 (Conesa et al., 2005).
Annotation of non‐coding RNAs (ncRNAs)
Transfer RNAs (tRNAs) were predicted by tRNAscan‐SE v1.3.1 (Lowe and Eddy, 1997) with eukaryote parameters. Four types of ncRNA (microRNA, ribosomal RNA, small nuclear RNA and small nucleolar RNA) were identified by Infernal cmscan v1.1.1 (Nawrocki and Eddy, 2013) against the Rfam database v12.0 (Kozomara and Griffiths‐Jones, 2013).
Gene family clusters and phylogenetic tree reconstruction
To examine evolution and divergence of the A. splendens genome, protein‐coding gene sequences from eight species of Gramineae (Oryza sativa, Sorghum bicolor, Setaria italica, Zea mays, Brachypodium distachyon, Hordeum vulgare, Aegilops tauschii and Phyllostachys edulis) and one eudicot (Arabidopsis thaliana) were downloaded from Phytozome v12 (https://phytozome.jgi.doe.gov/pz/portal.html) and the NCBI website (https://www.ncbi.nlm.nih.gov/) for comparative analyses. When one gene had multiple transcripts, only the longest transcript in the coding region was kept for further analysis. An all‐against‐all BLASTP search (Camacho et al., 2009) was applied to calculate pairwise sequence similarities with the default settings (E‐value ≤1e‐5 and a minimum match length of 50%). BLASTP results were then clustered into paralogs and orthologs using the OrthoMCL v2.0.9 method (Li et al., 2003). A Venn diagram was drawn to show the numbers of shared and species‐specific gene families among five species of Gramineae (A. splendens, O. sativa, B. distachyon, P. edulis and A. tauschii).
Single‐copy orthologous genes were extracted from the OrthoMCL clustering results and MUSCLE v3.8.31 (Edgar, 2004) was used to align the protein sequences. Guided by these alignments, protein sequences were transformed into coding DNA sequences (CDS). Fourfold degenerate sites were extracted from CDS alignments and concatenated to give a super‐gene matrix. RAxML v8.1.17 (Stamatakis, 2014) was used to reconstruct a phylogenetic tree with the GTR+G+I model and a bootstrap value of 100. The resulting phylogeny was visualized by iTOL (https://itol.embl.de/).
Estimation of divergence time
Species divergence time was inferred by MCMCtree in the PAML program v4.5 (Yang, 2007) with the ‘relaxed‐clock (clock = 2)’ model and ‘F84’ model. The divergence times for A. thaliana–O. sativa (mean: 150.0 Mya, std dev: 4.0) and for O. sativa–Z. mays (mean: 50.0 Mya, std dev: 4.0) obtained from the TimeTree database (http://www.timetree.org) were applied to calibrate the divergence times.
Expansion and contraction of gene families
To analyse the expansion and contraction of gene families, the Computational Analysis of gene Family Evolution (CAFÉ) program v3.1 (De Bie et al., 2006) was run to compute changes in gene families along each lineage of the phylogenetic tree under a random birth‐and‐death model. The clustering results and the information from the estimated divergence times were used. Using conditional likelihood as the test statistic, the corresponding P‐values of each lineage were calculated and a P‐value of 0.01 was regarded significant. Additionally, the expanded and contracted gene families were subjected to GO enrichment to determine their functions.
Genome synteny and whole‐genome duplication
Protein sequences within and between genomes were searched against one another to detect putative homologous genes (E‐value < 1e‐5) by BLASTP. With information about homologous genes as input, CollinearScan (Wang et al., 2006) and MCscanX (Wang et al., 2012) were implemented to infer homologous blocks involving collinear genes within and between genomes. The maximal gap length between collinear genes along a chromosome region was set to 50 genes (Wang et al., 2017, 2018). Then, homology dotplots were constructed by a perl script to reveal genomic correspondence. Non‐synonymous (Ka) and synonymous (Ks) substitution rates for gene pairs were calculated with KaKs Calculator v2.0 (Wang et al., 2010) under a YN model after converting protein alignments into codon alignments by PAL2NAL v14 (Suyama et al., 2006).
Identification of genes related to saline–alkali tolerance
Gene families related to the regulation of stress tolerance were collected manually from published A. thaliana papers. Transcription factor families from A. thaliana were further downloaded from PlantTFDB v5.0 (Jin et al., 2016). All genes of the genomes of A. splendens and related species were first searched against A. thaliana by BLASTP with an E‐value cut‐off of 1e‐5, then domains of candidate genes were identified by HMMER v3.2.1 (Finn et al., 2011) by searching the Pfam database (Punta et al., 2011). Only genes having protein domains identical to those of A. thaliana genes were selected as orthologs.
Differential expression analysis
To examine genome‐wide responses to salt stress of A. splendens, we performed transcriptomic analysis under 300 mm NaCl treatment for 6 and 24 h (h) for both shoot and root tissues. For raw sequencing reads, adapters, reads containing poly‐N and lower‐quality reads (<Q30) were removed to obtain clean reads, which were then mapped to the A. splendens genome using HISAT2 (Daehwan et al., 2015). StringTie v1.3.1 (Mihaela et al., 2015) was used to calculate transcripts per million (TPM) of mRNAs in each sample. TPM of genes was computed by summing the TPMs of transcripts in each gene group. We used DESeq R package v1.10.1 (negative binomial distribution) to perform differential expression analysis. DEGs were defined as those having at least twofold change in expression compared with those in 0 h (false discovery rate, FDR < 0.05). K‐cluster analysis of DEGs was performed using the OmicShare tools (http://www.omicshare.com/tools). Genes in each cluster were subjected to KEGG enrichment analysis.
Conflicts of interest
The authors declare no competing interests.
Author contributions
J.Q.L. conceived and designed the project. Y.Y.J. collected the genomic materials. G.P.R. and M.Y. conducted salt treatment experiment and collected the transcriptomic materials. G.P.R. and Y.Y.J. assembled the genome and performed gene annotation, gene family and evolutionary analyses, and genomic analyses with help from W.J.M. and Y.W. A.L. and M.J.L. conducted the transcriptomic analyses. G.P.R., Y.Y.J. and J.Q.L. wrote the manuscript. All authors read and approved the final version of the manuscript.
Supporting information
Figure S1 K‐mer analysis of the A. splendens genome by using K‐mer = 17.
Figure S2 Hi‐C‐assisted assembly of A. splendens pseudochromosomes.
Figure S3 Comparison of (a) CDS length, (b) mRNA length, (c) exon length and (d) intron length between A. splendens and other six related species.
Figure S4 Gene ontology (GO) enrichment of the expanded gene families in A. splendens.
Figure S5 Overview of dotplots within the A. splendens genome paralogous genes.
Figure S6 (a) Overview of dotplots between A. splendens and B. distachyon genome homologous genes. (b) Example of homologous gene dotplots between A. splendens and B. distachyon. Chromosome numbers and regions (in Mbp) were shown. Best hit (orthologous) genes are red dots, secondary hits (outparalogous) are blue dots and the others are shown in grey. Highlights show the best matched chromosomal regions. Arrows show complement correspondence produced by chromosome breakages during evolution.
Figure S7 Hierarchical cluster analysis of gene expression in root and shoot tissues.
Figure S8 Gene ontology (GO) enrichment of the 402 pairs of paralogous genes in A. splendens.
Table S1 Estimation of the A. splendens genome size based on 17‐mer statistics.
Table S2 Summary of DNA sequencing data.
Table S3 Assembly statistics based on Hi‐C data.
Table S4 Genome assembly completeness evaluation by BUSCO.
Table S5 Summary of PacBio full‐length cDNA sequencing.
Table S6 The length distribution of full‐length (FL) transcripts.
Table S7 Classification of interspersed repeats in the assembled A. splendens genome.
Table S8 LTR subclass ratio in A. splendens and other five Gramineae genomes.
Table S9 Summary of predicted protein‐coding gene annotations and their supporting evidence types.
Table S10 Functional annotation of predicted genes in the A. splendens genome.
Table S11 Summary statistics of non‐coding RNAs in the A. splendens genome.
Table S12 Summary of gene family clustering.
Table S13 Gene ontology (GO) enrichment analysis of the unique families in A. splendens.
Table S14 Gene ontology (GO) enrichment analysis of the expanded families in A. splendens.
Table S15 Number of gene pairs and homologous blocks within six Gramineae genomes or between the A. splendens and other five genomes.
Table S16 Statistics of duplication types of annotated genes in A. splendens.
Table S17 Copy numbers of gene families that are involved in salt–saline tolerance in the A. thaliana, O. sativa, B. distachyon and A. splendens genomes.
Table S18 Copy numbers of transcription factor families identified in the A. thaliana, O. sativa, B. distachyon and A. splendens genomes.
Table S19 The present pattern of DEGs in each of the total 6,796 paralogous gene (i.e., two copies in A. splendens with one synteny copy in O. sativa) pairs in A. splendens.
Table S20 The differential expression pattern of 402 paralogous gene pairs in both root and shoot.
Table S21 The expression (TPM) of the salt tolerance‐related DEGs in root and shoot of A. splendens.
Acknowledgements
This work was supported equally by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB31010300), as well as by the National Natural Science Foundation of China (31971391, 41901056). We received support for computational work from the Big Data Computing Platform for Western Ecological Environment and Regional Development of Lanzhou University.
Ren, G. , Jiang, Y. , Li, A. , Yin, M. , Li, M. , Mu, W. , Wu, Y. and Liu, J. (2022) The genome sequence provides insights into salt tolerance of Achnatherum splendens (Gramineae), a constructive species of alkaline grassland. Plant Biotechnol. J., 10.1111/pbi.13699
Data availability statement
The whole‐genome sequence data (including Illumina short‐gun reads, PacBio SMRT reads and Hi‐C interaction reads), the PacBio full‐length transcriptomes and salt treatment of RNA‐seq data have been deposited at NCBI under the Bioproject ID: PRJNA755179. The final assembled genome and genome annotations have been deposited in the National Genomics Data Center (https://bigd.big.ac.cn/?lang=en), under accession PRJCA006214.
References
- Adams, K.L. and Wendel, J.F. (2005) Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8, 135–141. [DOI] [PubMed] [Google Scholar]
- Ashburner, M. , Ball, C.A. , Blake, J.A. , Botstein, D. , Butler, H. , Cherry, J.M. , Davis, A.P. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25(1), 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to pomixe DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. and Durbin, R. (2004) GeneWise and genomewise. Genome Res. 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, E. , Parra, G. and Guigó, R. (2007) Using geneid to identify genes. Curr. Protoc. Bioinformatics, 18, 4.3.1–4.3.28. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B. , Bairoch, A. , Apweiler, R. , Blatter, M.‐C. , Estreicher, A. , Gasteiger, E. , Martin, M.J. et al. (2003) The SWISS‐PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer, M. and Pirovano, W. (2014) SSPACE‐LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics, 15, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, J.N. , Adey, A. , Patwardhan, R.P. , Qiu, R. , Kitzman, J.O. and Shendure, J. .(2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol., 31(12), 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. and Madden, T.L. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Ma, T. , Zhang, L. , Kang, M. , Zhang, Z. , Zheng, Z. , Sun, P. et al. (2020) Genomic analyses of a “living fossil”: the endangered dove‐tree. Mol. Ecol. Resour. 20, 756–769. [DOI] [PubMed] [Google Scholar]
- Chin, C.‐S. , Alexander, D.H. , Marks, P. , Klammer, A.A. , Drake, J. , Heiner, C. , Clum, A. et al. (2013) Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Chin, C.‐S. , Peluso, P. , Sedlazeck, F.J. , Nattestad, M. , Concepcion, G.T. , Clum, A. , Dunn, C. et al. (2016) Phased diploid genome assembly with single‐molecule real‐time sequencing. Nat. Methods, 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. , Götz, S. , García‐Gómez, J.M. , Terol, J. , Talón, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Daehwan, K. , Ben, L. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- De Peer, Y.V. , Maere, S. and Meyer, A. (2009) The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Clements, J. and Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell, R. , Bennett, M. , Smith, J. and Smith, D. (1974) Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 12, 257–269. [DOI] [PubMed] [Google Scholar]
- Flowers, T.J. and Colmer, T.D. (2008) Salinity tolerance in halophytes. New Phytol. 179, 945–963. [DOI] [PubMed] [Google Scholar]
- Flowers, T.J. , Galal, H.K. and Bromham, L. (2010) Evolution of halophytes: multiple origins of salt tolerance in land plants. Funct. Plant Biol. 37, 604–612. [Google Scholar]
- Gong, Z. , Xiong, L. , Shi, H. , Yang, S. , Herrera‐Estrella, L.R. , Xu, G. , Chao, D.‐Y. et al. (2020) Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 63, 635–674. [DOI] [PubMed] [Google Scholar]
- Guo, Y.T. , Wei, L.J. and Yan, P. (2003) Analysis on karyotype of Achnatherum splendens . J. Shiehzi Univ. 7, 3. (in Chinese). [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. , Wortman, J.R. , Smith, R.K. Jr , Hannick, L.I. , Maiti, R. et al. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al. (2008) Automated eukaryotic gene structure annotation using EvidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haixia, X. , Xia, Z. , Shaoming, W. , Ping, Y. and Jinzhou, D. (2008) Genetic diversity of Achnatherum splendens . Agric. Sci. Technol. 36, 6223–6224. [Google Scholar]
- Huai, H. , Wei, W. and Zhang, Y. (2008) Characteristics of the Achnatherum splendens community along the Qinghai‐Tibet Railway, China. Front. Biol. China, 3, 477–483. [Google Scholar]
- Huang, L. , Feng, G. , Yan, H. , Zhang, Z. , Bushman, B.S. , Wang, J. , Bombarely, A. et al. (2020) Genome assembly provides insights into the genome evolution and flowering regulation of orchardgrass. Plant Biotechnol. J. 18, 373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, S. , Apweiler, R. , Attwood, T.K. , Bairoch, A. , Bateman, A. , Binns, D. , Bork, P. et al. (2009) InterPro: the integrative protein signature database. Nucleic Acids Res., 37, D211–D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irfan, M. , Chen, Q. , Yue, Y. , Pang, R. , Lin, Q. , Zhao, X. and Chen, H. (2016) Co‐production of biochar, bio‐oil and syngas from halophyte grass (Achnatherum splendens L.) under three different pyrolysis temperatures. Bioresour. Technol. 211, 457–463. [DOI] [PubMed] [Google Scholar]
- Jiang, Z.‐Y. , Li, X.‐Y. , Wu, H.‐W. , Zhang, S.‐Y. , Zhao, G.‐Q. and Wei, J.‐Q. (2017) Linking spatial distributions of the patchy grass Achnatherum splendens with dynamics of soil water and salt using electromagnetic induction. Catena, 149, 261–272. [Google Scholar]
- Jin, J. , Tian, F. , Yang, D.‐C. , Meng, Y.‐Q. , Kong, L. , Luo, J. and Gao, G. (2016) PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M. and Goto, S. (2000) KEGG: pomi encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara, A. and Griffiths‐Jones, S. (2013) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, H.L. and Moran, J.V. (2011) Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.J. , Yang, Y.Z. , Xu, R.P. , Mu, W.J. , Li, Y. , Mao, X.X. , Zheng, Z.Y. et al. (2021) A chromosome‐level genome assembly for the Tertiary relict plant Tetracentron sinense Oliv. (Trochodendraceae). Mol. Ecol. Resour. 21, 1186–1199. [DOI] [PubMed] [Google Scholar]
- Li, R. , Fan, W. , Tian, G. , Zhu, H. , He, L. , Cai, J. , Huang, Q. et al. (2010) The sequence and de novo assembly of the giant panda genome. Nature, 463, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Zhou, Y. , Luo, C. , Xiang, Y. and An, L. (2016) De novo transcriptome sequencing of desert herbaceous Achnatherum splendens (Achnatherum) seedlings and identification of salt tolerance genes. Genes, 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. (1997) tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin, A.V. and Borodovsky, M. (1998) GeneMark. Hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. and Conery, J.S. (2000) The evolutionary fate and consequences of duplicate genes. Science, 290, 1151–1155. [DOI] [PubMed] [Google Scholar]
- Ma, T. , Wang, J. , Zhou, G. , Yue, Z. , Hu, Q. , Chen, Y. , Liu, B. et al. (2013) Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 4, 1–9. [DOI] [PubMed] [Google Scholar]
- Mahajan, S. and Tuteja, N. (2005) Cold, salinity and drought stresses: an overview. Archi. Biochem. Biophys. 444, 139–158. [DOI] [PubMed] [Google Scholar]
- Marçais, G. and Kingsford, C. (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics, 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaela, P. , Pertea, G.M. , Antonescu, C.M. , Tsung‐Cheng, C. , Mendell, J.T. and Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller, I.S. and Tester, M. (2007) Salinity tolerance of Arabidopsis: a good model for cereals? Trends Plant Sci. 12, 534–540. [DOI] [PubMed] [Google Scholar]
- Morton, M.J. , Awlia, M. , Al‐Tamimi, N. , Saade, S. , Pailles, Y. , Negrão, S. and Tester, M. (2019) Salt stress under the scalpel–dissecting the genetics of salt tolerance. Plant J. 97, 148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat, F. , Armero, A. , Pont, C. , Klopp, C. and Salse, J. (2017) Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 49, 490–496. [DOI] [PubMed] [Google Scholar]
- Murat, F. , Xu, J. , Tannier, E. , Abrouk, M. , Guilhot, N. , Pont, C. , Messing, J. et al. (2010) Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Res. 20, 1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki, E.P. and Eddy, S.R. (2013) Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A.H. , Bowers, J.E. and Chapman, B. (2004) Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA, 101, 9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A.H. , Bowers, J.E. , Bruggmann, R. , Dubchak, I. , Grimwood, J. , Gundlach, H. , Haberer, G. et al. (2009) The Sorghum bicolor genome and the diversification of grasses. Nature, 457, 551–556. [DOI] [PubMed] [Google Scholar]
- Porebski, S. , Bailey, L.G. and Baum, B.R. (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. [Google Scholar]
- Punta, M. , Coggill, P.C. , Eberhardt, R.Y. , Mistry, J. , Tate, J. , Boursnell, C. , Pang, N. et al. (2011) The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadir, M. , Quillerou, E. , Nangia, V. , Murtaza, G. , Singh, M. , Thomas, R.J. , Drechsel, P. et al. (2014) Economics of salt‐induced land degradation and restoration. Nat. Resour. Forum, 38, 282–295. [Google Scholar]
- Roach, M.J. , Schmidt, S.A. and Borneman, A.R. (2018) Purge Haplotigs: allelic contig reassignment for third‐gen diploid genome assemblies. BMC Bioinformatics, 19, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmanminkov, A. , Sabath, N. and Mayrose, I. (2016) Whole‐genome duplication as a key factor in crop domestication. Nat. Plants, 2, 16115. [DOI] [PubMed] [Google Scholar]
- Servant, N. , Varoquaux, N. , Lajoie, B.R. , Viara, E. , Chen, C.J. , Vert, J.P. , Heard, E. et al. (2015) HiC‐Pro: an optimized and flexible pipeline for Hi‐C data processing. Genome Biol., 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabala, S. and Cuin, T.A. (2008) Potassium transport and plant salt tolerance. Physiol. Plant, 133, 651–669. [DOI] [PubMed] [Google Scholar]
- Soltis, D.E. , Visger, C.J. and Soltis, P.S. (2014) The polyploidy revolution then… and now: Stebbins revisited. Am. J. Bot. 101, 1057–1078. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RaxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Diekhans, M. , Baertsch, R. and Haussler, D. (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Sun, P.C. , Jiao, B.B. , Yang, Y.Z. , Shan, L.X. , Li, T. , Li, X.N. , Xi, Z.X. et al. (2021) WGDI: a user‐friendly toolkit for evolutionary analyses of whole‐genome duplications and ancestral karyotypes. BioRxiv, 10.1101/2021.04.29.441969 [DOI] [PubMed] [Google Scholar]
- Suyama, M. , Torrents, D. and Bork, P. (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Bowers, J.E. , Wang, X. , Ming, R. , Alam, M. and Paterson, A.H. (2008) Synteny and collinearity in plant genomes. Science, 320, 486–488. [DOI] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, 25, 4.10.11–14.10.14. [DOI] [PubMed] [Google Scholar]
- Varshney, R.K. , Shi, C. , Thudi, M. , Mariac, C. , Wallace, J. , Qi, P. , Zhang, H. et al. (2017) Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. 35, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicient, C.M. , Suoniemi, A. , Anamthawat‐Jónsson, K. , Tanskanen, J. , Beharav, A. , Nevo, E. and Schulman, A.H. (1999) Retrotransposon BARE‐1 and its role in genome evolution in the genus Hordeum. Plant Cell, 11, 1769–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Zhang, Y. , Zhang, Z. , Zhu, J. and Yu, J. (2010) KaKs_Calculator 2.0: a toolkit incorporating gamma‐series methods and sliding window strategies. Genomics Proteomics Bioinformatics, 8, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Sun, P. , Li, Y. , Liu, Y. , Yu, J. , Ma, X. , Sun, S. et al. (2017) Hierarchically aligning 10 legume genomes establishes a family‐level genomics platform. Plant Physiol. 174, 284–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.‐P. , Yu, J.‐G. , Li, J. , Sun, P.‐C. , Wang, L.I. , Yuan, J.‐Q. , Meng, F.‐B. et al. (2018) Two likely auto‐tetraploidization events shaped kiwifruit genome and contributed to establishment of the Actinidiaceae family. iScience, 7, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Tong, S. , Ma, T. , Xi, Z. and Liu, J.Q. (2021) Chromosome‐level genome assembly of Sichuan pepper provides insights into pomixes, drought tolerance, and alkaloid biosynthesis. Mol. Ecol. Resour. 21, 2533–2545. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Shi, X. , Hao, B. , Ge, S. and Luo, J. (2005) Duplication and DNA segmental loss in the rice genome: implications for diploidization. New Phytol. 165, 937–946. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Shi, X. , Li, Z. , Zhu, Q. , Kong, L. , Tang, W.Y. , Ge, S. et al. (2006) Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinformatics, 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Tang, H. , Bowers, J.E. and Paterson, A.H. (2009) Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 19, 1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Wang, J. , Jin, D. , Guo, H. , Lee, T. , Liu, T. and Paterson, A.H. (2015) Genome alignment spanning major poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Mol. Plant, 8, 885–898. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.‐H. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J.F. (2000) Genome evolution in polyploids. Plant Mol. Biol. 42, 225–249. [PubMed] [Google Scholar]
- Wu, H.‐J. , Zhang, Z. , Wang, J.‐Y. , Oh, D.‐H. , Dassanayake, M. , Liu, B. , Huang, Q. et al. (2012) Insights into salt tolerance from the genome of Thellungiella salsuginea . Proc. Natl. Acad. Sci. USA, 109, 12219–12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T.D. and Watanabe, C.K. (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics, 21, 1859–1875. [DOI] [PubMed] [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, T. and Blumwald, E. (2005) Developing salt‐tolerant crop plants: challenges and opportunities. Trends Plant Sci. 10, 615–620. [DOI] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol, 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- You, L. , Liu, B. and Bussmann, R.W. (2020) Achnatherum splendens (Trin.) Nevski P oaceae. Ethnobotany of the Mountain Regions of Central Asia and Altai, 1–3. [Google Scholar]
- Zdobnov, E.M. and Apweiler, R. (2001) InterProScan–an integration platform for the signature‐recognition methods in InterPro. Bioinformatics, 17, 847–848. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Liang, C. , Wei, W. , Wang, L. , Peng, J. , Yan, J. and Jia, C. (2010) Soil salinity and Achnatherum splendens distribution. Chin. J. Ecol. 29, 2438–2443. [Google Scholar]
- Zhu, J. (2001) Plant salt tolerance. Trends Plant Sci. 6, 66–71. [DOI] [PubMed] [Google Scholar]
- Zhu, J. , Fu, X. , Koo, Y.D. , Zhu, J.‐K. , Jenney, F.E. , Adams, M.W. , Zhu, Y. et al. (2007) An enhancer mutant of Arabidopsis salt overly sensitive 3 mediates both ion homeostasis and the oxidative stress response. Mol. Cell. Biol. 27, 5214–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 K‐mer analysis of the A. splendens genome by using K‐mer = 17.
Figure S2 Hi‐C‐assisted assembly of A. splendens pseudochromosomes.
Figure S3 Comparison of (a) CDS length, (b) mRNA length, (c) exon length and (d) intron length between A. splendens and other six related species.
Figure S4 Gene ontology (GO) enrichment of the expanded gene families in A. splendens.
Figure S5 Overview of dotplots within the A. splendens genome paralogous genes.
Figure S6 (a) Overview of dotplots between A. splendens and B. distachyon genome homologous genes. (b) Example of homologous gene dotplots between A. splendens and B. distachyon. Chromosome numbers and regions (in Mbp) were shown. Best hit (orthologous) genes are red dots, secondary hits (outparalogous) are blue dots and the others are shown in grey. Highlights show the best matched chromosomal regions. Arrows show complement correspondence produced by chromosome breakages during evolution.
Figure S7 Hierarchical cluster analysis of gene expression in root and shoot tissues.
Figure S8 Gene ontology (GO) enrichment of the 402 pairs of paralogous genes in A. splendens.
Table S1 Estimation of the A. splendens genome size based on 17‐mer statistics.
Table S2 Summary of DNA sequencing data.
Table S3 Assembly statistics based on Hi‐C data.
Table S4 Genome assembly completeness evaluation by BUSCO.
Table S5 Summary of PacBio full‐length cDNA sequencing.
Table S6 The length distribution of full‐length (FL) transcripts.
Table S7 Classification of interspersed repeats in the assembled A. splendens genome.
Table S8 LTR subclass ratio in A. splendens and other five Gramineae genomes.
Table S9 Summary of predicted protein‐coding gene annotations and their supporting evidence types.
Table S10 Functional annotation of predicted genes in the A. splendens genome.
Table S11 Summary statistics of non‐coding RNAs in the A. splendens genome.
Table S12 Summary of gene family clustering.
Table S13 Gene ontology (GO) enrichment analysis of the unique families in A. splendens.
Table S14 Gene ontology (GO) enrichment analysis of the expanded families in A. splendens.
Table S15 Number of gene pairs and homologous blocks within six Gramineae genomes or between the A. splendens and other five genomes.
Table S16 Statistics of duplication types of annotated genes in A. splendens.
Table S17 Copy numbers of gene families that are involved in salt–saline tolerance in the A. thaliana, O. sativa, B. distachyon and A. splendens genomes.
Table S18 Copy numbers of transcription factor families identified in the A. thaliana, O. sativa, B. distachyon and A. splendens genomes.
Table S19 The present pattern of DEGs in each of the total 6,796 paralogous gene (i.e., two copies in A. splendens with one synteny copy in O. sativa) pairs in A. splendens.
Table S20 The differential expression pattern of 402 paralogous gene pairs in both root and shoot.
Table S21 The expression (TPM) of the salt tolerance‐related DEGs in root and shoot of A. splendens.
Data Availability Statement
The whole‐genome sequence data (including Illumina short‐gun reads, PacBio SMRT reads and Hi‐C interaction reads), the PacBio full‐length transcriptomes and salt treatment of RNA‐seq data have been deposited at NCBI under the Bioproject ID: PRJNA755179. The final assembled genome and genome annotations have been deposited in the National Genomics Data Center (https://bigd.big.ac.cn/?lang=en), under accession PRJCA006214.