

Abstract
Background
IL-22 promotes epidermal hyperplasia and inhibits skin barrier function.
Objective
Evaluate IL-22 blockade in adults with moderate-to-severe atopic dermatitis (AD).
Methods
Randomized, double-blind, placebo-controlled trial with intravenous fezakinumab monotherapy every 2wks for 10wks, with follow-up assessments until 20wks. SCORAD change from baseline at 12wks served as primary endpoint.
Results
At 12wks, mean SCORAD decline was 13·8±2·7 (fezakinumab) vs. 8·0±3·1 (placebo) for the entire population (p=0·134). In severe patients (baseline SCORAD≥50), SCORAD decline was significantly stronger in drug vs. placebo at 12wks (21·6±3·8 vs. 9·6±4·2, p=0.029) and 20wks (27·4±3·9 vs. 11·5±5·1, p=0·010). At 12wks, improvements were significantly stronger in drug vs. placebo (12·4%±2·4 vs. 6·2%±2·7; p=0·009) for BSA in the entire population, and for IGA in severe patients (0·7±0·2 vs. 0·3±0·1; p=0·034). All scores showed progressive improvements after last dosing (10wks) until end of study (20wks). Common adverse events were upper respiratory tract infections.
Limitations
Limited sample size, lack of EASI and numerical rating scale (NRS) pruritus assessments; significance primarily obtained in severe AD.
Conclusion
Fezakinumab was well-tolerated, with sustained clinical improvements after last drug dosing.
Keywords: Atopic dermatitis, IL-22, fezakinumab, placebo-controlled trial, moderate-to-severe AD
Capsule summary
IL-22 induces epidermal hyperplasia and compromises skin barrier function in model systems.
This clinical trial demonstrates the efficacy of IL-22 blockade in humans, implying its possible therapeutic role in atopic dermatitis/AD.
IL-22/Th22 targeting potentially offers a novel alternative for severe AD patients with limited therapeutic options.
Introduction
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease, with a prevalence of 7-10% in adults.1 It is characterized by pruritus, increased prevalence of allergic manifestations (asthma, allergic rhinitis, food allergies), and a predisposition to cutaneous infections.2 In patients with moderate-to-severe AD (approximately 20% of adult patients),3 the disease often affects large body surface areas, leading to profound effects on patients’ quality of life.4 However, treatment options for moderate-to-severe AD patients are limited, and topical treatments, including emollients, glucocorticosteroids, and calcineurin and phosphodiesterase-4 inhibitors are often unsatisfactory.5 Systemic treatments are largely not FDA-approved for AD (cyclosporine A, azathioprine, mycophenolate-mofetil, methotrexate), with the exception of the recently approved anti-IL-4Rα monoclonal antibody, dupilumab, inhibiting both Th2 cytokines, IL-4 and IL-13.6,7 While dupilumab successfully treats a large portion of AD patients, a considerable subset has insufficient responses,6,7 necessitating further treatment modalities.
Key pathogenic features of AD include a disturbed skin barrier with epidermal hyperplasia and abnormal keratinocyte differentiation, as well as robust activation of the Th2 and Th22 T-cell pathways. In vitro and animal studies suggest that IL-22, the lead Th22 cytokine, promotes hyperplasia and inhibits keratinocyte differentiation and skin barrier formation, two hallmarks of AD.8-13 High levels of IL-22-producing T-cells have also been identified in psoriasis, particularly in children.14 To assess a possible role for IL-22 as a driver cytokine of AD, similar to the established pathogenic role of Th2 cytokines,6,15,16 we investigated the IL-22 antagonist, fezakinumab (ILV-094), in an investigator-initiated clinical trial.
Methods
Study design and oversight
We conducted a phase 2a, randomized, double-blind, placebo-controlled, multicenter clinical trial to evaluate efficacy and safety of fezakinumab in 60 moderate-to-severe AD patients (clinicaltrials.gov #NCT01941537) at the Icahn School of Medicine at Mount Sinai (n=40), and The Rockefeller University (n=20), both in New York City. Patients were randomly assigned to either intravenous fezakinumab or placebo (2:1 drug and placebo randomization, respectively, Figure 1), with a loading dose of 600mg at baseline (day 0), followed by 300mg at weeks 2, 4, 6, 8, and 10 (last dose). Primary outcome measures were assessed at week 12, with follow-up until week 20. Safety was assessed by the incidence of adverse events, vital signs, physical examination, clinical laboratory testing, and electrocardiography. The study protocol (see Supplementary Material) of this investigator-initiated trial was developed by the investigators, and has been approved by the local institutional review boards.
Figure 1. Patient disposition.
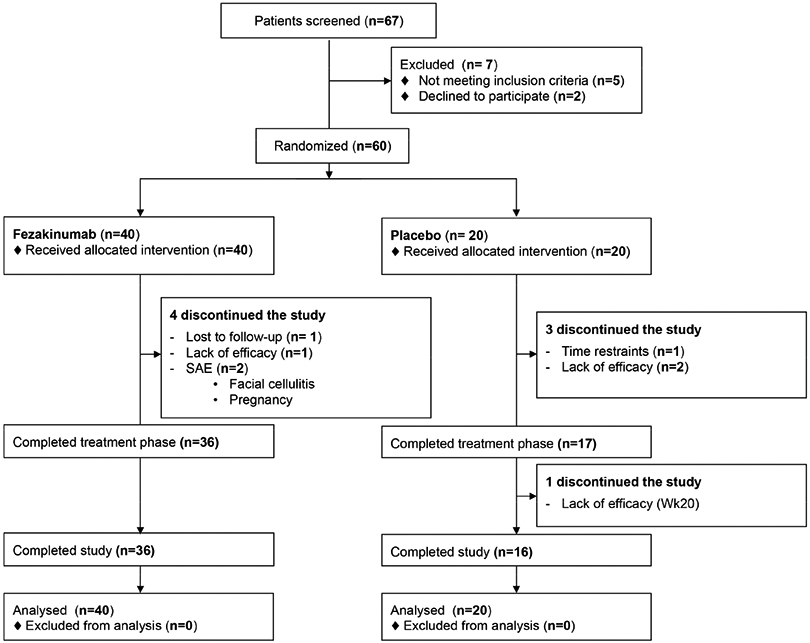
SAE – Serious adverse event; ITT – intention to treat.
Patients
Eligible patients were 18-75 years old, with moderate-to-severe AD for ≥6months, as defined by SCORAD≥30 (SCORing of Atopic Dermatitis), and IGA≥3 (Investigator Global Assessment on a 0 to 5 scale for “clear-0” to “very severe-5”) (Table 1). Patients had to fail or not sustain response with one or more conventional treatments, including topical corticosteroids or calcineurin antagonists, and/or systemic treatments (corticosteroids, phototherapy, cyclosporine, or other immunomodulators). Disease duration was ≥6months. All patients gave written informed consent prior to inclusion.
Table 1.
Demographic and clinical characteristics of the participants at baseline.
| PLACEBO | DRUG | P-VALUE† | |
|---|---|---|---|
| n= 20 | n= 40 | ||
| Age (mean, SD) | 41·3 (16·3) | 40·5 (14·9) | 0·855 |
| BMI (mean, SD) ¶ | 27·4 (6·4) | 27·7 (5·9) | 0·866 |
| Gender (n,%) | 0·360 | ||
| Female | 11 (55·0%) | 17 (42·5%) | |
| Male | 9 (45·0%) | 23 (57·5%) | |
| Race (n, %) | 0·51 | ||
| Asian | 5 (25%) | 10 (25%) | |
| African American | 10 (50%) | 14 (35%) | |
| Caucasian | 5 (25%) | 16 (40%) | |
| IgE Group (n,%) * | 0·620 | ||
| Intrinsic | 4 (20%) | 6 (15%) | |
| Extrinsic | 16 (80%) | 34 (85%) | |
| Total serum IgE, kU/L (mean, SD) | 6,592 (9,720) | 3,646 (4,561) | 0·638 |
| SCORAD (mean, SD) § | 55·5 (13·4) | 53·4 (13·1) | 0·568 |
| SCORAD (range) | 34·5-89 | 36-84·5 | |
| SCORAD <50 (n,%) | 8 (40%) | 20 (50%) | 0.46 |
| SCORAD ≥50 (n,%) | 12 (60%) | 20 (50%) | |
| IGA (n, %) # | 0·66 | ||
| Moderate (3) | 15 | 32 | |
| Severe (4) | 5 | 7 | |
| Very severe (5) | 0 | 1 | |
| BSA (mean, SD) $ | 38·15 (24·26) | 42·68 (27·7) | 0·52 |
| HISTORY OF ASTHMA (n,%) ‡ | 0·89 | ||
| As child only | 4 (20%) | 6 (15%) | |
| No | 10 (50%) | 22 (55%) | |
| Yes | 6 (30%) | 12 (30%) |
Data are given as mean±SD (standard deviation) or as percentage (%).
Body mass index (BMI) is the weight in kilograms divided by the square of the height in meters.
Intrinsic and extrinsic patients were assigned according to baseline total serum IgE levels of <200kU/L or >200kU/L, respectively.
Scores on the SCORAD (SCORing Atopic Dermatitis) range from 0 to 103, with higher scores indicating greater severity; non-severe and severe disease was scored as SCORAD<50 and SCORAD≥50, respectively.
The Investigator’s Global Assessment (IGA) of the severity of atopic dermatitis was scored on a scale of 0 (clear) to 5 (very severe)
Body surface area (BSA) was graded from 0 (no skin involvement) to 100% (total skin involvement)
History of asthma as per patient history.
For numerical variables (Age, BMI, SCORAD, BSA, total serum IgE), differences between the means by treatment were tested using a two-tailed Sudent’s t-test for independent samples. The proportions by treatment for categorical variables (gender, race, IgE group, SCORAD50, IGA, History of Asthma) were compared using a Fisher’s exact test.
Efficacy evaluations
The primary efficacy variable was the change from baseline at week 12 in the AD clinical severity index (SCORAD). The SCORAD combines objective assessments of the extent of body-surface area (BSA) involvement in AD, the severity of erythema, edema/papulation, oozing/crusting, excoriation, lichenification and skin dryness, together with subjective measures such as pruritus and sleep loss, yielding an overall score of 0 (no AD) to 103 (worst possible AD).17 Secondary efficacy endpoints included proportion of patients achieving SCORAD improvements of at least 50% (SCORAD50 responses), percent improvement in SCORAD, decline in BSA and IGA, and IGA-complete-response, defined as “clear” or “almost clear” or a decline ≥2 in IGA. See Supplementary Material for further information.
Statistical analysis
In accordance with study protocol, efficacy variables were analyzed in the modified intention-to-treat (ITT) population, where any patient that started treatment was included in the analysis and patients that stopped treatment earlier than 12 weeks (N=7, 11·6%) were defined as non-responders. Continuous variables measured longitudinally during treatment were analyzed by the mixed-effect model repeated measures (MMRM, using R packages nlme and lme4) approach (with treatment-arm, clinical site, baseline value, visit, treatment-by-visit and treatment-by-severity-by-visit interactions as covariates). For categorical variables, statistical significance of difference between treatment arms was tested by the Fisher’s exact test. To test the robustness of the results we also conducted a per-protocol (PP) analysis, where only patients who completed 12 weeks of treatment were included in the analysis (See Supplementary Material). The thresholds of 30% improvement in SCORAD (SCORAD30) and 15 point decline in BSA were used to define a positive response based on the 95% percentiles of pretreatment variation (absolute difference between screening and baseline visit values) in SCORAD-%-improvement and BSA-decline, and are thus objective indicators of meaningful response (see Supplementary Material). Multivariate analyses of the primary efficacy variable was performed by linear regression for SCORAD decline, with the backward-conditional method, including the co-variates: treatment arm, baseline severity, gender, age, race, BMI, AD duration, baseline IgE, and the combinations of these factors with treatment-arm. Statistical significance was set at a two-tailed p-value<0.05.
Role of the funding source
The study was funded by the National Institutes of Health/NIAMS, project #5UM1AR063917. Fezakinumab was provided by Pfizer Inc. (New York, NY). Data were collected and analyzed by the study investigators only. All authors interpreted the data and collaborated in manuscript preparation, made the decision to submit the manuscript for publication, and vouched for the completeness and accuracy of the data and analyses and the fidelity of the study to the protocol.
Results
Patients
Enrollment and disposition of the patients are shown in Figure 1. The first patient was screened on March 4, 2014, and the entire study was concluded on February 29, 2016. 67 patients were assessed for eligibility, and 60 patients were randomized 2:1 to either fezakinumab (n=40) or placebo (n=20). Demographic and clinical characteristics of the patients at baseline were similar between the study arms (Table 1). Comparable numbers of moderate (30≤SCORAD<50) and severe (SCORAD≥50)18 AD patients at baseline were randomized in both drug and placebo arms. 36 and 17 patients completed treatment and reached the primary endpoint (week 12), and 36 and 16 patients completed the study until week 20 in the fezakinumab and placebo groups, respectively (Figure 1). Two patients discontinued treatment due to serious adverse events, which were deemed non-related to the drug (facial cellulitis after a dental procedure; pregnancy with elective termination, Table 2 and Supplementary Table S1). One patient in the drug-arm was lost to follow-up, and 4 patients discontinued due to lack of efficacy (n=1 in drug-arm, and n=3 in placebo-arm). One patient in the placebo-arm discontinued early (week 2) due to time restraints (Figure 1). All randomized patients were included in the intention-to-treat (ITT) population.
Table 2.
Adverse events.
| Variable | Fezakinumab | Placebo | P- value$ |
|
|---|---|---|---|---|
| N=40 | N=20 | |||
| No. of adverse events | 18 | 10 | ||
| Severity | Mild | 9 | 6 | |
| Moderate | 8 | 3 | ||
| Severe | 1 | 0 | ||
| Mean no. of adverse events per patient | 0·45 | 0·5 | 0·82 | |
| Any adverse event—no. of patients (%) | 14 (35%) | 8 (40%) | 0·78 | |
| Serious adverse event * — no. of patients (%) | 2 (5%)** | 0 | 0·55 | |
| Study discontinuation due to adverse event—no. of patients (%) | 2 (5%) | 0 | 0·55 | |
| Common adverse events: ‡ | ||||
| Upper respiratory infection, viral— no. of patients (%) | 4 (10%) | 0 | 0·29 | |
A serious adverse event was defined as an event that was fatal or life threatening, required or prolonged hospitalization, or caused persistent or substantial disability or incapacity or a congenital anomaly or birth defect or an event that was considered by the investigator to be a medically important event.
Facial cellulitis after a dental procedure; pregnancy with elective termination.
Common adverse events were those that occurred in >5% in any treatment group.
The differences of the proportions were tested using a Fisher’s exact test.
Efficacy
Efficacy endpoints analyzed using a mixed effect model repeated measures (MMRM) approach on the ITT population are listed in Table 3. Similar significance values were also obtained with the per-protocol population (Supplementary Table S2), indicating the robustness of the results. Starting at week 4, the fezakinumab group showed a consistently stronger, and more significant mean SCORAD decline from baseline than the placebo group (Figure 2A), a difference that reached statistical significance between drug and placebo at weeks 6-10 (p<0·05). Differences between drug and placebo extended beyond the last dose (week 10), with non-significant mean±SEM SCORAD reductions of 13·8±2·7 in fezakinumab vs. 8·0±3·1 in placebo (p=0.134) at week 12 (primary endpoint), and progressive reductions seen between weeks 14-20, with significant difference between drug and placebo observed at week 20 (end of study) of 18·8±2·9 vs. 11·7±3·9 (p=0·049), respectively (Figure 2A, Table 3). While higher SCORAD50, and SCORAD30 responses were seen with drug vs. placebo, these were not statistically significant (secondary endpoint; Table 3). While SCORAD comprises both objective and subjective measures (sleep loss and pruritus), BSA is an objective measure. The mean decline in BSA was consistently stronger in the drug group, and was significantly different from placebo from week 8 until the end of study, including at week 12 (p=0·009, Figure 2B, Table 3). Similarly, a BSA decline >15, as defined by less than 15 point pre-treatment variation in BSA between baseline and screening (Supplementary Figure S2B), was present at significantly higher rates in drug vs. placebo arm (Table 3). Also, mean improvements in IGA scores compared to baseline were stronger and earlier with fezakinumab treatment (Figure 2C), significantly different from placebo at week 16 (p<0·001). Higher, but not significant IGA complete response rate (standardly defined as an IGA score of 0 or 1 or IGA decline ≥2 after treatment) were seen in drug vs. placebo-treated patients at week 12 (p=0.119; Table 3). While no significant difference was detected in SCORAD pruritus scores between drug and placebo arms, a sustained treatment effect was observed among patients with baseline pruritus >5 after week 12, and until end of study at week 20, with non-significant exacerbations in placebo treated patients (Supplementary Figure S1).
Table 3.
Efficacy end points by ITT (MMRM) analysis.
| All patients (N=60) | Severe AD (Baseline SCORAD≥50) (N=32) (a) |
Non-Severe AD (Baseline SCORAD<50) (N=28) (a) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug (N=40) |
Placebo (N=20) |
p- value |
Drug (N=20) |
Placebo (N=12) |
p- value |
Drug (N=20) |
Placebo (N=8) |
p- value |
|
| SCORAD Decline (Mean ±SEM) | |||||||||
| Week 12 | −13·8 ±2·7 | −8·0 ±3·1 | 0·134 | −21·6 ±3·8 | −9·6 ±4·2 | 0·029 | −6·0 ±3·2 | −5·7 ±4·6 | 0·764 |
| Week 20 | −18·8 ±2·9 | −11·7 ±3·9 | 0·049 | −27·4 ±3·9 | −11·5 ±5·1 | 0·010 | −10·2 ±3·4 | −11·9 ±6·4 | 0·639 |
| SCORAD Improvement (Mean ±SEM) | |||||||||
| Week 12 | −24·4% ±5·0% | −14·7% ±5·9% | 0·175 | −34·4% ±6·1% | −15·8% ±7·4% | 0·039 | −14·3% ±7·4% | −13·1% ±10·1% | 0·897 |
| Week 20 | −34·1% ±5·4% | −23·0% ±7·4% | 0·072 | −43·9% ±6·6% | −20·6% ±8·8% | 0·028 | −24·2% ±8·0% | −26·6% ±13·8% | 0·794 |
| SCORAD30 (b) | |||||||||
| Week 12 | 42·5% | 20·0% | 0·150 | 55·0% | 16·7% | 0·062 | 30·0% | 25·0% | 1·000 |
| Week 20 | 52·5% | 30·0% | 0·168 | 65·0% | 16·7% | 0·012 | 40·0% | 50·0% | 0·691 |
| SCORAD50 (c) | |||||||||
| Week 12 | 22·5% | 15·0% | 0·734 | 30·0% | 16·7% | 0·676 | 15·0% | 12·5% | 1·000 |
| Week 20 | 37·5% | 25·0% | 0·395 | 45·0% | 16·7% | 0·139 | 30·0% | 37·5% | 1·000 |
| BSA Decline (Mean ±SEM) | |||||||||
| Week 12 | −12·4 ±2·4 | −6·2 ±2·7 | 0·009 | −15·7 ±3·6 | −6·3 ±4·5 | 0·011 | −9·0 ±3·1 | −6·1 ±1·6 | 0·347 |
| Week 20 | −17·7 ±3·2 | −7·6 ±2·9 | 0·001 | −23·3 ±4·7 | −6·8 ±4·7 | 0·009 | −12·2 ±4·2 | −8·8 ±1·9 | 0·357 |
| BSA Decline > 15 (d) | |||||||||
| Week 12 | 37.5% | 10.0% | 0.034 | 45.0% | 16.7% | 0.139 | 30.0% | 0.0% | 0.141 |
| Week 20 | 42.5% | 10.0% | 0.017 | 55.0% | 16.7% | 0.062 | 30.0% | 0.0% | 0.141 |
| IGA Decline (Mean ±SEM) | |||||||||
| Week 12 | −0·6 ±0·1 | −0·3 ±0·1 | 0·119 | −0·7 ±0·2 | −0·3 ±0·1 | 0·034 | −0·5 ±0·2 | −0·4 ±0·3 | 0·684 |
| Week 20 | −0·9 ±0·2 | −0·6 ±0·2 | 0·127 | −1·2 ±0·2 | −0·4 ±0·2 | 0·014 | −0·7 ±0·2 | −0·8 ±0·3 | 0·724 |
| IGA Complete Response (e) | |||||||||
| Week 12 | 15.0% | 5.0% | 0.407 | 20.0% | 0.0% | 0.271 | 10.0% | 12.5% | 1.000 |
| Week 20 | 25.0% | 15.0% | 0.513 | 35.0% | 8.3% | 0.204 | 15.0% | 25.0% | 0.606 |
Summary of responses by treatment arm at primary endpoint (week 12), and at end of study (week 20). Statistical significance of difference in mean decline between treatment arms was assessed by MMRM. Statistical significance of difference in % response between treatment arms was assessed by Fisher’s exact test. Significant results (p≤0·05) are in bold.
Severe patients are defined as patients with a SCORAD≥50 at week 0.
SCORAD30 is defined as SCORAD improvement >30% compared to week 0; a 30% threshold was selected because it is the 95% percentile of pre-treatment variation.
SCORAD50 is defined as SCORAD improvement >50% compared to week 0.
BSA (body surface area) decline threshold of 15 points was selected because it is the 95% percentile of pre-treatment variation.
IGA (Investigator Global Assessment) complete response is defined as IGA≤1 or IGA decline≥2 at the respective end-point.
Figure 2. Time course of efficacy variables in the entire study population.

Clinical responses for SCORAD (A), BSA (B), and IGA (C) are shown for the fezakinumab (red) and placebo (blue) arms. All panels depict the mean ±SEM change from baseline. Data was analyzed by MMRM: red and blue asterisks by each curve indicate significant change from baseline for each arm (*p<0·05, **p<0·01, ***p<0·001); black asterisks at the bottom indicate significant differences between drug and placebo arm; green asterisks indicate the significance of the combined factor: baseline severity and treatment arm.
In a multivariate linear regression analysis considering all relevant baseline factors (treatment arm, severity, race, gender, age, AD duration, BMI, IgE) the combined factor of severity and treatment arm gave the highest (B=15.84) and most significant (p<0.001) association with SCORAD decline at the primary endpoint week 12 (Supplementary Table S3). In fact, the MMRM analysis showed that the severity-by-treatment-arm-by-visit covariate had a significant effect on SCORAD decline at all visits from week 6 to 20 (green stars in Figure 2A), and parallel significant effects on mean BSA change (green stars in Figure 2B), as well as mean IGA change (green stars in Figure 2C) from baseline.
Since severity showed the highest association with treatment response, we further stratified patients according to their baseline disease severity, into moderate (SCORAD<50) and severe (SCORAD≥50) AD. There was a strong drug effect on SCORAD decline in severe patients (Figure 3B, Table 3), which was significantly stronger than in placebo starting at week 6, including at the primary endpoint at week 12 (21·6±3·8 vs. 9·6±4·2; p=0·029), an effect that continued until week 20 (27·4±3·9 vs. 11·5±5·1; p=0·010; Figure 3B, Table 3). SCORAD50 responses were higher in severe drug vs. placebo group but did not show statistical significance (45% vs. 16·7% at week 20; p=0.139; Table 3). SCORAD30 responses at end of study (week 20) were significantly higher in severe drug vs. placebo patients (65·5% vs. 16·7%, p=0·012), but not significant at week 12 (55% vs. 16·7%, p=0·062; Table 3). The threshold of SCORAD30 was derived from less than 30% variation in pre-treatment SCORAD values between screening and baseline (Supplementary Figure S2A).
Figure 3. Time course of efficacy variables stratified for moderate and severe patients.

Patients were stratified as having moderate (SCORAD between 25 and 50) or severe (SCORAD ≥50) AD at baseline (week 0). Panels depict the mean ±SEM change from baseline for SCORAD (A, B), BSA (C, D), and IGA (E, F). Data was analyzed by MMRM: red and blue asterisks by each curve indicate significant change from baseline for each arm (*p<0·05, **p<0·01, ***p<0·001); black asterisks at the bottom indicate significant differences between drug and placebo arm.
BSA decline showed stronger, significant improvements in severe patients treated with drug vs. placebo; p=0.011 at week 12; Figures 3C-3D, Table 3). IGA improvement was stronger in fezakinumab treated severe patients compared to placebo, with significant differences between treatment arms at weeks 8, 10, 12, 14, 16 and 20 (p=0·034 and p=0·014 at weeks 12 and 20, respectively; Figure 3F, Table 3).
None of the efficacy variables showed statistically significant difference between drug and placebo arms in non-severe (moderate) AD patients (Figures 3A,C,E; Table 3). Overall, lower declines in SCORAD, BSA and IGA were observed in drug-treated moderate patients as compared to severe patients, with larger declines in the placebo-treated moderate as compared to placebo severe patients.
Safety
Adverse events occurred with a similar frequency in fezakinumab and placebo groups (Table 2, Supplementary Table S1). All except one adverse event (facial cellulitis) were deemed mild or moderate in severity. 2 serious adverse events occurred in the drug arm (facial cellulitis after dental procedure, and pregnancy), but were deemed as likely unrelated to drug. Most common adverse events were viral upper respiratory tract infections, occurring in 4 patients receiving fezakinumab. A total of 9 and 18 adverse events were reported in the placebo and drug group, respectively, that were not significantly different between arms.
Blood biomarkers
There were neither significant changes over-time per arm nor differences between groups in total serum IgE levels changes during treatment and follow-up (Supplementary Figure S3).
Discussion
This is the first clinical trial investigating IL-22 blockade in patients with AD, and the first to suggest a pathogenic role of IL-22 in any human disease. Fezakinumab treatment in adults with moderate-to-severe AD resulted in consistent improvements in clinical and molecular disease scores as compared to placebo. At week 12, significant clinical improvements in drug-treated compared to placebo-treated patients were best seen in severe patients (baseline SCORAD≥50). Moreover, progressive improvements in all outcome measures were observed until week 20, which was 10 weeks after the last dose, suggesting sustained drug responses beyond end of treatment.
This study is the first evidence in humans that similar to the Th2 cytokines IL-4 and IL-13,6,7,15,16 IL-22 is a key driver of AD. Whereas current monoclonal antibody treatment approaches, either approved or currently tested in trials, target the Th2 pathway in AD,6,19-21 these data provide a completely new mechanism for future therapeutic strategies for AD and other disease where IL-22 may have a role, such as pediatric psoriasis14 or pediatric AD.22
Larger and more significant differences were specifically seen in the severe AD patient group. Possible reasons for reduced statistical significance in the moderate AD cohort might be due to higher variability in this group, and inability to obtain maximal differences due to lower baseline disease, whereas the severe patients started with higher severity, and often show less fluctuations, allowing for greater differences in treatment responses.23-27 The sensitivity of clinical outcome measures generally increases with higher baseline disease activity, and shows less reproducibility with lower scores,23-27 a fact that is now increasingly recognized in trial design. Our study has several limitations: 1) It was designed >6 years ago, and only utilized SCORAD to measure disease severity, similar to many studies at the time,17 while current AD trials often use EASI scores as primary outcomes, limiting the ability to compare to other clinical trials. Nevertheless, recent studies considered both measures, and in these SCORAD appears to be a more stringent disease measure, and changes in EASI scores tend to be much larger than respective SCORAD changes.6,7 2) It was designed prior to emerging data that AD is a heterogeneous disease,22,28-30 which necessitates a study design allowing for analyses of subset populations. To address this, we employed a post-hoc analysis approach separating severe from moderate patients. Larger studies are needed to compare to other treatments, and to confirm efficacy in AD subgroups (moderate vs. severe, different ethnicities, etc.).
This study, supported by progressive clinical improvements with IL-22 antagonism versus placebo, opens a novel therapeutic paradigm for AD, and particularly severe AD, a population that presents the largest unmet need for better therapeutics, due to its debilitating nature and devastating effects on patients’ quality of life.31 Fezakinumab also showed a favorable safety profile with balanced adverse events, and similar study discontinuation rates between treatment arms. While the recently approved dupilumab, that targets Th2 signaling, shows a good safety profile, a large subset of patients show insufficient responses,6 and may benefit from treatment directed at an alternative pathway such as the Th22/IL-22 cytokine pathway.
Supplementary Material
Funding sources:
Supported by NIH/NIAMS grant # 1UM1AR063917. PMB was supported in part by grant # UL1 TR0001866 from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program. Fezakinumab was provided by Pfizer Inc. (New York, NY).
Abbreviation list
- AD
Atopic dermatitis
- BSA
Body surface area
- BMI
Body mass index
- EASI
Eczema Area and Severity Index
- IGA
Investigator Global Assessment
- ITT
Intention to treat
- MMRM
Mixed-effect model repeated measures
- NRS
Numerical Rating Scale
- SCORAD
Scoring Atopic Dermatitis
- Wks
Weeks
Footnotes
IRB approval status: Reviewed and approved by the local IRB of The Rockefeller University (JKR-0766) and the Icahn School of Medicine at Mount Sinai (GCO#12-0424), both in New York City, NY, USA.
The study was registered with the Clinicaltrials.gov listing # NCT01941537.
Declaration of interests: EGY is an employee of Mount Sinai and has received research funds (grants paid to the institution) from: Abbvie, Celgene, Eli Lilly, Janssen, Medimmune/Astra Zeneca, Novartis, Pfizer, Regeneron, Vitae, Glenmark, Galderma, Asana, Innovaderm, Dermira, UCB. EGY is also a consultant for Sanofi Aventis, Regeneron, Stiefel/GlaxoSmithKline, MedImmune, Celgene, Anacor, AnaptysBio, Dermira, Galderma, Glenmark, Novartis, Pfizer, Vitae, Leo Pharma, Abbvie, Eli Lilly, Kyowa, Mitsubishi Tanabe, Asana Biosciences, and Promius. PMB has received personal fees from LEO Pharma and Sanofi. CTH has received research support from Danone Nutricia and personal fees from Novartis and La Roche Posay. JGK is an employee of the Rockefeller University and has received research support (grants paid to his institution) and/or personal fees from Pfizer, Amgen, Janssen, Lilly, Merck, Novartis, Kadmon, Dermira, Boehringer, Innovaderm, Kyowa, BMS, Serono, BiogenIdec, Delenex, AbbVie, Sanofi, Baxter, Paraxel, Xenoport, and Kineta. MGL is an employee of Mount Sinai which receives research funds from: Abbvie, Amgen, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen / Johnson & Johnson, Kadmon, Medimmune/Astra Zeneca, Novartis, Pfizer and ViDac. MGL is also a consultant for Allergan and Promius. The rest of the authors declare that they have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Silverberg JI. Public Health Burden and Epidemiology of Atopic Dermatitis. Dermatol Clin 2017;35:283–9. [DOI] [PubMed] [Google Scholar]
- 2.Weidinger S, Novak N. Atopic dermatitis. Lancet 2016;387:1109–22. [DOI] [PubMed] [Google Scholar]
- 3.DaVeiga SP. Epidemiology of atopic dermatitis: a review. Allergy Asthma Proc 2012;33:227–34. [DOI] [PubMed] [Google Scholar]
- 4.Drucker AM. Atopic dermatitis: Burden of illness, quality of life, and associated complications. Allergy Asthma Proc 2017;38:3–8. [DOI] [PubMed] [Google Scholar]
- 5.Boguniewicz M, Alexis AF, Beck LA, et al. Expert Perspectives on Management of Moderate-to-Severe Atopic Dermatitis: A Multidisciplinary Consensus Addressing Current and Emerging Therapies. J Allergy Clin Immunol Pract 2017;5:1519–1531. [DOI] [PubMed] [Google Scholar]
- 6.Simpson EL, Bieber T, Guttman-Yassky E, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med 2016;375:2335–2348. [DOI] [PubMed] [Google Scholar]
- 7.Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet 2016;387:40–52. [DOI] [PubMed] [Google Scholar]
- 8.Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol 2008;159:1092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werfel T, Allam JP, Biedermann T, et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J Allergy Clin Immunol 2016;138:336–49. [DOI] [PubMed] [Google Scholar]
- 10.Sa SM, Valdez PA, Wu J, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol 2007;178:2229–40. [DOI] [PubMed] [Google Scholar]
- 11.Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol 2005;174:3695–702. [DOI] [PubMed] [Google Scholar]
- 12.Lou H, Lu J, Choi EB, et al. Expression of IL-22 in the Skin Causes Th2-Biased Immunity, Epidermal Barrier Dysfunction, and Pruritus via Stimulating Epithelial Th2 Cytokines and the GRP Pathway. J Immunol 2017;198:2543–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyerich S, Eyerich K, Pennino D, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009;119:3573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cordoro KM, Hitraya-Low M, Taravati K, et al. Skin-infiltrating, interleukin-22-producing T cells differentiate pediatric psoriasis from adult psoriasis. J Am Acad Dermatol 2017;77:417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton JD, Suarez-Farinas M, Dhingra N, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol 2014;134:1293–300. [DOI] [PubMed] [Google Scholar]
- 16.Beck LA, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 2014;371:130–9. [DOI] [PubMed] [Google Scholar]
- 17.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol 2013;132:1337–47. [DOI] [PubMed] [Google Scholar]
- 18.Pucci N, Lombardi E, Novembre E, et al. Urinary eosinophil protein X and serum eosinophil cationic protein in infants and young children with atopic dermatitis: correlation with disease activity. J Allergy Clin Immunol 2000;105:353–7. [DOI] [PubMed] [Google Scholar]
- 19.Ruzicka T, Hanifin JM, Furue M, et al. Anti-Interleukin-31 Receptor A Antibody for Atopic Dermatitis. N Engl J Med 2017;376:826–35. [DOI] [PubMed] [Google Scholar]
- 20.Simpson EL, Flohr C, Eichenfield L. Efficacy and safety of lebrikizumab in patients with atopic dermatitis: a Phase II randomized, controlled trial (TREBLE). 25th European Academy of Dermatology and Venerology Congress 2016. [Google Scholar]
- 21.Wollenberg A, Howell MD, Guttman-Yassky E, et al. A Phase 2b Dose-Ranging Efficacy and Safety Study of Tralokinumab in Adult Patients with Moderate to Severe Atopic Dermatitis (AD). American Academy of Dermatology Annual Meeting 2017. [Google Scholar]
- 22.Esaki H, Brunner PM, Renert-Yuval Y, et al. Early onset pediatric atopic dermatitis is Th2, but also Th17 polarized in skin. J Allergy Clin Immunol 2016;138:1639–1651. [DOI] [PubMed] [Google Scholar]
- 23.Hon KL, Wang SS, Leung TF. What happens to the severity grading by objective SCORAD if we over- or underestimate disease extent or intensity in patients with atopic dermatitis? Int J Dermatol 2012;51:295–9. [DOI] [PubMed] [Google Scholar]
- 24.Bissonnette R, Poulin Y, Zhou Y, et al. Efficacy and safety of topical WBI-1001 in patients with mild to severe atopic dermatitis: results from a 12-week, multicentre, randomized, placebo-controlled double-blind trial. Br J Dermatol 2012;166:853–60. [DOI] [PubMed] [Google Scholar]
- 25.Poole CD, Chambers C, Sidhu MK, Currie CJ. Health-related utility among adults with atopic dermatitis treated with 0.1% tacrolimus ointment as maintenance therapy over the long term: findings from the Protopic CONTROL study. Br J Dermatol 2009;161:1335–40. [DOI] [PubMed] [Google Scholar]
- 26.Nahm DH, Kim ME, Kwon B, Cho SM, Ahn A. Clinical Efficacy of Subcutaneous Allergen Immunotherapy in Patients with Atopic Dermatitis. Yonsei Med J 2016;57:1420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thaci D, Bissonnette R, Maku M, Coughlan D, Climax J. DS107 Clinical Data: A novel oral treatment for moderate to severe atopic dermatitis in adults. 25th Annual Meeting of the European Academy of Dermatology and Venereology, Austria, 2016. [Google Scholar]
- 28.Noda S, Suarez-Farinas M, Ungar B, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol 2015;136:1254–64. [DOI] [PubMed] [Google Scholar]
- 29.Suarez-Farinas M, Dhingra N, Gittler J, et al. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol 2013;132:361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bieber T, D'Erme AM, Akdis CA, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: Where are we, and where should we go? J Allergy Clin Immunol 2017;139:S58–S64. [DOI] [PubMed] [Google Scholar]
- 31.Holm JG, Agner T, Clausen ML, Thomsen SF. Quality of life and disease severity in patients with atopic dermatitis. J Eur Acad Dermatol Venereol 2016;30:1760–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.