

Abstract
Background
Trials have assessed bile acids for patients with viral hepatitis, but no consensus has been reached regarding their usefulness.
Objectives
To assess the beneficial and harmful effects of bile acids for viral hepatitis.
Search methods
Searches were performed in The Cochrane Hepato‐Biliary Group Controlled Trials Register (July 2007), The Cochrane Library (Issue 1, 2007), MEDLINE (July 2007), EMBASE (July 2007), Science Citation Index Expanded (July 2007), and Chinese Biomedical Database (July 2007).
Selection criteria
Randomised clinical trials comparing any dose or duration of bile acids versus placebo or no intervention for viral hepatitis were included, irrespective of language, publication status, or blinding. Co‐interventions were allowed in the included randomised clinical trials.
Data collection and analysis
Two authors extracted the data independently. The methodological quality of the trials was evaluated with respect to generation of the allocation sequence, allocation concealment, double blinding, and follow‐up. The outcomes were presented as relative risks (RR) or weighted mean differences (WMD) with 95% confidence intervals (CI).
Main results
We identified 29 randomised trials of bile acids for hepatitis B or C; none were of high methodological quality. We were unable to extract data from two trials. In one trial, ursodeoxycholic acid (UDCA) versus placebo for acute hepatitis B significantly reduced the risk of hepatitis B surface antigen positivity at the end of treatment and serum HBV DNA level at the end of follow‐up. In another trial, UDCA versus no intervention for chronic hepatitis B significantly reduced the risk of having abnormal serum transaminase activities at the end of treatment. Twenty‐five trials compared bile acids (21 trials UDCA; four trials tauro‐UDCA) versus placebo or no intervention with or without co‐interventions for chronic hepatitis C. Bile acids did not significantly reduce the risk of having detectable serum HCV RNA (RR 0.99, 95% CI 0.91 to 1.07), cirrhosis, or portal and periportal inflammation score at the end of treatment. Bile acids significantly decreased the risk of having abnormal serum alanine aminotransferase activity at the end of treatment (RR 0.82, 95% CI 0.76 to 0.90) and follow‐up (RR 0.91, 95% CI 0.85 to 0.98). Bile acids significantly increased the Knodell score (WMD 0.20, 95% CI 0.08 to 0.31) at the end of treatment. No severe adverse events were reported. We did not identify trials including patients with hepatitis A, acute hepatitis C, hepatitis D, or hepatitis E.
Authors' conclusions
Bile acids lead to a significant improvement in serum transaminase activities in hepatitis B and C but have no effects on the clearance of virus. There is insufficient evidence either to support or to refute effects on long‐term outcomes including hepatocellular carcinoma, hepatic decompensation, and liver related mortality. Randomised trials with high methodological quality are required before clinical use is considered.
Plain language summary
Bile acids may improve liver biochemistry of patients with hepatitis B or C, but there is insufficient evidence about long‐term beneficial effects
Viral hepatitis causes significant morbidity and mortality. Based on this Cochrane systematic review, bile acids may decrease serum transaminase activities in patients with acute hepatitis B, chronic hepatitis B, or chronic hepatitis C. However, bile acids have no effects in eradicating viral markers. There is insufficient evidence either to support or to refute effects on the long‐term outcomes that include hepatocellular carcinoma, decompensated cirrhosis, and/or liver related mortality.
No clinical trials have evaluated bile acids for patients with hepatitis A, acute hepatitis C, hepatitis D, or hepatitis E. Accordingly, bile acids should first be evaluated in adequately sized and adequately conducted randomised trials before clinical use is considered.
Background
Viral hepatitis is liver inflammation that can be caused by hepatitis A, B, C, D, or E virus (Wolfram 1999). All of these viruses can cause an acute disease lasting several weeks including jaundice, dark urine, extreme fatigue, nausea, vomiting, and abdominal pain (Zuckermann 1993). After acute hepatitis, Hepatitis B virus (HBV), hepatitis C virus (HCV), and hepatitis D virus (which can only infect patients already infected by hepatitis B virus) also cause chronic infection, which may develop into hepatic cirrhosis and/or liver cancer (Rizzetto 1986; Fattovich 1995; Brechot 1996). Viral hepatitis is considered prevalent disease associated with significant morbidity, mortality, and socio‐economic loss (Stapleton 1994). It has been estimated that more than 350 million have chronic HBV infection (WHO 2000a) and 170 million persons are chronically infected with HCV (WHO 2000b).
Current treatment of acute viral hepatitis is based on supportive measures, relief of symptoms, and avoidance of further liver injury (Maddrey 1980). At present, antiviral agents are not advocated for patients with typical acute viral hepatitis (Alam 1995) apart from the use of interferon for acute hepatitis C (Myers 2001a; Seeff 2002). For chronic viral hepatitis, treatment recommendations vary with the viruses. Interferon or lamivudine are used for chronic hepatitis B (Liaw 1999; Lok 2001). Combination therapy with interferon and ribavirin is recommended for patients with chronic hepatitis C (Farrell 1999; Seeff 2002; Brok 2005). Interferon can induce good biochemical responses in patients with chronic hepatitis D, but in the great majority of responders there is relapse after discontinuation of the drug (Wolfram 1999). Interferon exerts a direct antiviral effect and enhances the immunomodulatory response of the host (Hoffnagle 1997; Poynard 1997; Farrell 1999; Liaw 1999). Unfortunately, only a limited number of patients with chronic viral hepatitis are suitable for interferon therapy (Myers 2002b). Even the most effective therapy available, pegylated interferon alpha and ribavirin, leads to a sustained virologic response in about 50% of patients with genotype 1 virus, the major genotype for chronic HCV in western countries (Manns 2001; Fried 2002). New options to treat viral hepatitis have been explored and bile acids have been investigated for the possible effects on viral hepatitis. Moreover, treatment is costly and entails a high rate of adverse events (Renault 1989; Brok 2005). Effective, well tolerated, and economic drugs for the treatment of viral hepatitis are needed. Bile acids include chenodeoxycholic acid, deoxycholic acid, lithocholic acid, and ursodeoxycholic acid (UDCA) (Fuchs 1999). In the course of cholestasis, intrahepatic accumulation of hydrophobic bile acids such as chenodeoxycholic acid and deoxycholic acid is thought to induce liver damage (Palmer 1972) due to their detergent activity, which appears to dissolve phospholipid and cholesterol in cell membranes (Sholmerich 1984). The hepatocyte toxicity of hydrophobic bile acids has been observed both in animals and human beings (Jaeschke 2002). These effects can be prevented by the addition of UDCA, which modifies the bile acid pool, resulting in an increase of the hydrophilic fraction, and stabilises cell membranes (Armstrong 1982). The mechanisms of UDCA action include reduction of toxic endogenous bile acids, membrane stabilising activity, and an immunomodulatory effect (Calmus 1990). Another bile acid, tauro‐ursodeoxycholic acid (TUDCA), has also been used for 'liver protection' (Podda 1996).
UDCA and TUDCA have been used for the treatment of cholestatic liver diseases including primary biliary cirrhosis (Gluud 2001b), primary sclerosis cholangitis (Chen 2002), and other chronic cholestatic diseases (Larghi 1997). Several randomised clinical trials have shown that UDCA improves the serum biochemical indexes of cholestasis and cytolysis in patients with chronic liver diseases (Nakagawa 1990; Gluud 2001b). Moreover, there is some evidence that UDCA, used alone or in combination with interferon, is able to improve liver enzyme levels in patients with chronic hepatitis C (Boucher 1995). TUDCA has physiochemical and metabolic properties, which favour its use as an alternative to UDCA for chronic cholestatic liver disease (Gallo 1993). TUDCA may prove of benefit also for necroinflammatory liver diseases, especially for HCV‐related chronic hepatitis in which bile duct damage is frequently seen (Fallon 1997).
This Cochrane systematic review represents an update of our previous review, in which we found moderate beneficial effects of bile acids for viral hepatitis in trials with high risk of bias (Chen 2003).
Objectives
The objectives were to evaluate the beneficial and harmful effects of bile acids for viral hepatitis caused by one or more of the hepatotrophic viruses (A to E) based on the results of randomised clinical trials.
Methods
Criteria for considering studies for this review
Types of studies
Randomised clinical trials were included irrespective of blinding, publication status, or language. Quasi‐randomised trials (eg, using date of birth) and observational studies were excluded. However, for the evaluation of rare, serious adverse events, we also considered large cohort studies and previous meta‐analyses/systematic reviews, as such events are rarely captured in randomised clinical trials due to their sample size.
Types of participants
Patients with viral hepatitis diagnosed by any method according to the following definitions were included: (1) Acute hepatitis A defined as: serum transaminase activities (alanine aminotransferase (ALT); aspartate aminotransferase (AST); gamma glutamyltranspeptidase (GGT)) above the upper normal limits; seropositivity for immunoglobulin M (IgM) anti‐hepatitis A virus. (2) Acute hepatitis B defined as: serum transaminase activities above twice the upper normal limits; seropositivity for hepatitis B surface antigen (HBsAg), hepatitis B 'e' antigen (HBeAg), and IgM antibody to hepatitis B core antigen. (3) Chronic hepatitis B defined as: serum transaminase activities above the upper normal limits; serum HBsAg positivity and HBeAg positivity or HBV DNA positivity for more than six months. (4) Acute hepatitis C defined as: serum transaminase activities above twice the upper normal limits and serum antibody to HCV (anti‐HCV) and/or serum HCV RNA positivity. (5) Chronic hepatitis C defined as: serum transaminase activities above the upper normal limits for more than six months and/or liver biopsy findings compatible with chronic hepatitis C plus HCV‐RNA seropositivity for more than six months. (6) Acute hepatitis D defined as: serum transaminase activities above the upper normal limits and seropositivity for hepatitis D virus RNA and IgM anti‐hepatitis D virus. (7) Chronic hepatitis D defined as: serum transaminase activities above the upper normal limits and hepatitis D virus RNA positivity for more than six months. (8) Acute hepatitis E defined as: serum transaminase activities above the upper normal limits and seropositivity for hepatitis E virus RNA and IgM anti‐hepatitis E virus.
Types of interventions
This review includes randomised comparisons of any dose or duration of bile acid (UDCA or TUDCA) versus placebo or no intervention. Co‐interventions were allowed if received by both intervention arms.
Types of outcome measures
For each individual type of viral hepatitis, the primary outcome measures were: (1) Virological response:
acute hepatitis A: detectable hepatitis A virus at the end of treatment and at the maximal follow‐up;
acute hepatitis B: detectable HBsAg, HBV DNA measured by polymerase chain reaction (PCR) technique, HBeAg and seroconversion from HBeAg to anti‐HBe at the end of treatment and at the maximal follow‐up;
chronic hepatitis B: detectable HBsAg, HBV DNA measured by PCR technique, HBeAg and seroconversion from HBeAg to anti‐HBe at the end of treatment and at the maximal follow‐up;
acute hepatitis C: detectable HCV RNA measured by a validated PCR technique at the end of treatment and at the maximal follow‐up;
chronic hepatitis C: detectable HCV RNA measured by a validated PCR technique at the end of treatment and at the maximal follow‐up;
acute hepatitis D: detectable hepatitis D virus RNA measured by a validated PCR technique at the end of treatment and at the maximal follow‐up;
chronic hepatitis D: detectable hepatitis D virus RNA measured by a validated PCR technique at the end of treatment and at the maximal follow‐up;
acute hepatitis E: detectable hepatitis E virus RNA measured by a validated PCR technique at the end of treatment and at the maximal follow‐up.
(2) All‐cause mortality. (3) Liver‐related morbidity: decompensated cirrhosis, hepatocellular carcinoma, or liver transplantation.
The secondary outcome measures were: (1) Biochemical non‐responders: number of patients without normalisation of serum transaminase activities at the end of treatment and/or maximal follow‐up. (2) Histological response: histological activity index or fibrosis score at the end of treatment and/or the maximal follow‐up. (3) Quality of life. (4) Adverse events. Number and type of adverse events, defined as any untoward medical occurrence in a patient. This occurrence should not necessarily have a causal relationship with the treatment, but should have resulted in the discontinuation of treatment. We have defined serious adverse events according to the International Conference on Harmonisation (ICH) Guidelines (ICH‐GCP 1997) as any event that leads to death, is life‐threatening, requires in‐patient hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability, and any important medical event which may have jeopardised the patient or requires intervention to prevent it. All other adverse events were considered non‐serious. (5) Cost‐effectiveness.
Search methods for identification of studies
The Cochrane Hepato‐Biliary Group Controlled Trials Register (July 2007) and The Cochrane Controlled Trials Register (Cochrane Library, Issue 1, 2007) were searched using the terms 'hepatitis' and the names of individual bile acids (lithocholic acid, or chenodeoxycholic acid, or ursodeoxycholic acid, or deoxycholic acid, or dehydrocholic acid, or tauro‐ursodeoxycholic acid). MEDLINE (1950 to July 2007) was searched using the search strategy of The Cochrane Hepato‐Biliary Group (see Collaborative Review Group for details) combined with the terms 'hepatitis' and the bile acids mentioned above. EMBASE (January 1980 ‐ July 2007) and Science Citation Index Expanded (1945 ‐ July 2007) were searched using the same search terms. In addition, Chinese Biomedical Database (1980 ‐ 2007) was searched by using similar search strategy (Appendix 1).
The principal authors of the included trials and pharmaceutical companies involved in the production of bile acids were contacted about additional published or unpublished randomised clinical trials on the topic.
Data collection and analysis
The meta‐analysis was conducted according to our previously published protocol (Chen 2001) following the recommendations given by The Cochrane Reviewers' Handbook (Higgins 2005). Identified trials were listed and two contributors independently evaluated whether the trials fulfilled the inclusion criteria. Excluded trials were listed with the reasons for exclusion.
Methodological quality Methodological quality was defined as the confidence that the design and report restrict bias in the intervention comparison (Moher 1998). The methodological quality was assessed by quality components, ie, adequacy of generation of the allocation sequence, allocation concealment, double blinding, and follow‐up (Schulz 1995; Moher 1998; Kjaergard 2001). Trials were assessed as having high methodological quality, ie, low risk of bias, if all quality components were adequate.
The quality components were assessed according to Kjaergard et al (Kjaergard 2001):
Generation of the allocation sequence
Adequate, if the allocation sequence was generated by a computer or random number table. Drawing of lots, tossing of a coin, shuffling of cards, or throwing dice was considered as adequate if a person who was not otherwise involved in the recruitment of participants performed the procedure.
Unclear, if the trial was described as randomised, but the method used for the allocation sequence generation was not described.
Inadequate, if a system involving dates, names, or admittance numbers were used for the allocation of patients. Such quasi‐randomised studies were excluded.
Allocation concealment
Adequate, if the allocation of patients involved a central independent unit, on‐site locked computer, identically appearing numbered drug bottles or containers prepared by an independent pharmacist or investigator, or serially numbered, sealed, and opaque envelopes.
Unclear, if the trial was described as randomised, but the method used to conceal the allocation was not described.
Inadequate, if the allocation sequence was known to the investigators who assigned participants or if the study was quasi‐randomised. The latter studies were excluded.
Blinding
Adequate, if the trial was described as double blind and the method of blinding involved identical placebo or active drugs.
Unclear, if the trial was described as double blind, but the method of blinding was not described.
Not performed, if the trial was not double blind.
Follow‐up
Adequate, if the numbers and reasons for dropouts and withdrawals in all intervention groups were described or if it was specified that there were no dropouts or withdrawals.
Unclear, if the report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated.
Inadequate, if the number or reasons for dropouts and withdrawals were not described.
Further, we registered whether or not the trials reported to have used an intention‐to‐treat analysis and performed a sample size calculation (Gluud 2001a).
Data extraction The data were extracted by WC and JL independently. The data were validated by CG. Disagreements were resolved by discussion. The following characteristics were extracted from each trial: primary author, number of patients randomised, patient inclusion and exclusion criteria, methodological quality, sample size calculation, intention‐to‐treat analysis, intervention regimens, mean age, proportion of males, proportion of patients with cholestasis, proportion of patients with cirrhosis, proportion of virus genotypes, number of outcomes, and number and type of adverse events in the intervention and the control group. Further information was sought by correspondence with the principal investigators of the included trials in case the relevant data were not published.
Statistical methods The analyses were performed in Review Manager 4.1. The analyses for binary outcomes included all patients irrespective of compliance or follow‐up ('intention‐to‐treat') using the last reported observed response ('carry forward'). The analyses for continuous outcomes included only the patients with available data. We used both random‐effects (DerSimonian 1986) and fixed‐effect models (DeMets 1987) in the meta‐analyses. When we did not find a difference between the results of the two methods, we only reported the results of the fixed‐effect model. Evidence of publication bias was analysed by funnel plot analyses (Egger 1997). Binary outcome measures were analysed by relative risks (RR) with 95% confidence intervals (CI) and continuous outcome measures by weighted mean differences (WMD) and 95% CI.
In our protocol (Chen 2001), we planned to perform sensitivity analyses according to the (1) methodological quality of the included trials (adequate compared to inadequate); (2) dose and duration of treatment with bile acid; (3) treatment with or without co‐intervention; and (4) publication status. The trials included in our review were of low methodological quality; the dosages of UDCA applied in the trials were in the range of 10 to 15 mg/kg/day; and we were unable to identify any unpublished trials. Accordingly, sensitivity analyses were only performed according to the duration of treatment with bile acids. The outcome measures used for sensitivity analyses were the risk of having serum HCV RNA at the end of follow‐up and the risk of abnormal serum ALT at the end of treatment and follow‐up.
We planned to do meta‐regression if a sufficient number of trials were identified to explore the effect of patient characteristics, intervention regimens, and methodological quality of the trials on the virological response at the end of treatment. However, we decided not to perform such analyses due to the general low quality of the included trials in this review.
Results
Description of studies
Our initial searches identified 174 studies; 154 from the electronic searches and 20 from the handsearches. After reading titles and abstracts, 124 of these articles were excluded because they were duplicates, non‐clinical studies, or had study objectives different from our review. A total of 50 articles published in three languages (English, Chinese, and Italian) were retrieved for further assessment. Of these, 17 studies were excluded because they did not meet the inclusion criteria. The reasons for exclusion are listed in the table 'Characteristics of excluded studies'.
The remaining 33 publications were randomised clinical trials that reported the random allocation of patients with viral hepatitis to bile acids versus control, including four double publications (Angelico 1995a; Boucher 1995; Puoti 1995; Fabbri 2000). Accordingly, the searches identified 29 trials that met all pre‐specified inclusion criteria. These trials are listed in the table 'Characteristics of included studies'. Twenty‐four trials were published in English and five in Chinese. Ten trials were from Italy, five from China, five from Japan, four from France, two from Argentina, and one each from the Czech Republic, USA, and Turkey.
We were unable to supplement our electronic searches and handsearches with additional data from personal communications. Only one author replied and this author was not able to supply the information because the original data were lost. No responses were received from pharmaceutical companies (Axcan Pharma Inc and Dr. Falk Pharma).
Patients' origin and sample size One trial (Galský 1999) included in‐patients, three trials (Puoti 1995; Abdelmalek 1998; Fabbri 2000) included out‐patients, and the other 25 trials did not specify the origin of the patients. The average size of the trial was 69 patients (range 31 to 167 patients).
Diagnoses Inclusion criteria were explicit viral hepatitis diagnosed by virological markers, clinical manifestations, and biochemical variables. Thirteen trials confirmed the diagnosis by liver biopsy. Galský1999 included patients with acute hepatitis including A, B, and/or C. Zhou 1995 included patients with chronic hepatitis B. Twenty‐five trials included patients with chronic hepatitis C. Two trials (Qing 1999; Cadranel 2003) included patients with chronic hepatitis B or C. We were unable to extract data on the respective diagnostic groups, nor did we receive a reply from the authors when requested for separate data on the patients with hepatitis B and C. Accordingly, outcome data from these two trials could not be included in this review. However, they are listed in the table 'Characteristics of included studies' and the section regarding the methodological quality of the included studies. No trials were found evaluating bile acids for hepatitis A, acute hepatitis C, acute or chronic hepatitis D, or hepatitis E.
Bile acids and collateral interventions Twenty‐five trials evaluated UDCA, and four trials evaluated TUDCA. The median duration of treatment was nine months (range, three to 18 months). The median duration of follow‐up was nine months (range six to 18 months).
Acute or chronic hepatitis B One trial (Galský1999) compared UDCA versus placebo for acute hepatitis, which included hepatitis A, B, and/or C. Most of patients included in this trial had acute hepatitis B. The dose of UDCA was 750 mg/day and the duration of treatment was three months. The follow‐up after treatment was nine months.
One trial (Zhou 1995) compared UDCA versus no intervention for chronic hepatitis B. Patients in the UDCA group received 400 mg/day for three months. The follow‐up time was three months.
Chronic hepatitis C Five trials (Leri 1994; Takano 1994; Zhu 1994; Puoti 1995; Scotto 1997) compared UDCA versus placebo or no intervention for chronic hepatitis C. The dose of UDCA ranged from 400 to 800 mg/day. The duration of treatment ranged from three to 12 months. The duration of follow‐up after treatment ranged from three to six months.
Two trials compared TUDCA versus placebo (Crosignani 1998) or no treatment (Belloni 1999) for chronic hepatitis C. The dose of TUDCA ranged from 500 to 750 mg/day; the duration of treatment ranged from four to six months.
Fifteen trials compared UDCA combined with interferon versus interferon monotherapy in patients with chronic hepatitis C. Eleven trials (Clerici 1994; Kawata 1994; Angelico 1995a; Boucher 1995; Tanaka 1996; Ge 1997; Huang 1997; Kiso 1997; Senturk 1997; Abdelmalek 1998; Viola 1998) included patients who were naive to interferon, and four trials (Bonnand 1996; Fabbri 2000; Poupon 2000; Viola 1996) included patients who were non‐responders to interferon. The dose of UDCA ranged from 10 to 15 mg/kg body weight/day, and the dose of interferon ranged from three to six million units three times a week. The duration of treatment ranged from six to 18 months. The duration of follow‐up after treatment ranged from six to 18 months.
Two trials (Picciotto 1994; Pigozzi 1997) compared TUDCA plus interferon versus interferon monotherapy for chronic hepatitis C. The dose of TUDCA ranged from 500 mg/day to 10 mg/kg body weight/day. The dose of interferon was three million units three times a week. The duration of treatment was six months and the follow‐up after treatment ranged from three to six months.
One trial (Tsubota 1999) compared UDCA combined with glycyrrhizin versus glycyrrhizin monotherapy in patients with chronic hepatitis C. The dose of UDCA was 600 mg/day and the dose of glycyrrhizin was 300 ml/week. The duration of treatment was six months.
Chronic hepatitis B and C Two trials compared UDCA versus placebo (Cadranel 2003) or no intervention (Qing 1999) for chronic hepatitis (including hepatitis B and C). Patients in the Cadranel 2003 trial received 800 mg/day UDCA for 12 months, and patients in the Qing 1999 trial received 450 mg/day UDCA for three months.
Outcome measures The outcome measures reported by most trials were virological markers and/or biochemical variables. None of the 29 trials reported mortality, hepatocellular carcinoma, liver transplantation, quality of life, or cost‐effectiveness.
Risk of bias in included studies
Of the 29 included trials, one trials (3.5%) (Angelico 1995a) reported both adequate generation of the allocation sequence and adequate allocation concealment. One trial (Boucher 1995) reported only adequate generation of the allocation sequence, and two trials (Picciotto 1994; Tsubota 1999) reported only adequate allocation concealment. The remaining trials had unclear generation of the allocation sequence and allocation concealment. Eight trials (27.6%) (Bonnand 1996; Cadranel 1996; Viola 1996; Abdelmalek 1998; Crosignani 1998; Viola 1998; Galský 1999; Poupon 2000) used double blinding, which was assessed as adequate. One trial (Scotto 1997) reported single blinding, but it was unclear who was blinded. The remaining trials were conducted without blinding. Accordingly, none of the trials were of high methodological quality, ie, having adequate generation of the allocation sequence, allocation concealment, double blinding, and follow‐up. Therefore, all trials were considered as having risk of bias.
Only one trial reported a sample size calculation (3.4%) (Angelico 1995a), and only eight trials (27.6%) (Picciotto 1994; Angelico 1995a; Boucher 1995; Viola 1996; Kiso 1997; Viola 1998; Poupon 2000; Cadranel 2003) performed intention‐to‐treat analyses. Nine trials (31.0%) (Picciotto 1994; Boucher 1995; Kiso 1997; Senturk 1997; Abdelmalek 1998; Crosignani 1998; Galský 1999; Tsubota 1999; Poupon 2000) reported the numbers of patients withdrawn and reasons for withdrawal.
Of the 29 trials, 17 trials (58.6%) (Clerici 1994; Kawata 1994; Picciotto 1994; Angelico 1995a; Boucher 1995; Zhou 1995; Bonnand 1996; Ge 1997; Huang 1997; Kiso 1997; Gracielle 2002; Scotto 1997; Abdelmalek 1998; Galský 1999; Fabbri 2000; Poupon 2000) reported follow‐up after the end of intervention with bile acids. The duration of follow‐up varied from three to 18 months.
Effects of interventions
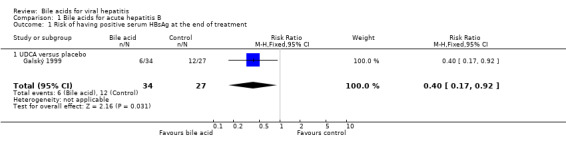
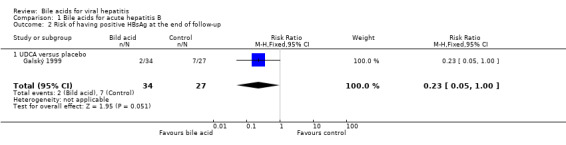
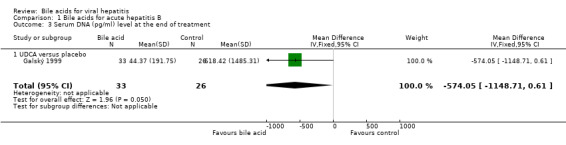
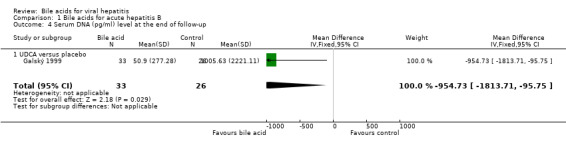
Bile acids for acute hepatitis B One trial (Galský 1999) evaluated UDCA versus placebo in patients with acute hepatitis (12 HAV, 61 HBV, 4 HCV, and 1 HBV + HCV). However, only the outcome measures of patients with acute hepatitis B were reported in this trial. UDCA significantly decreased the risk of having positive serum HBsAg at the end of treatment (6/34 (17.6%) versus 12/27 (44.4%); RR 0.40, 95% CI 0.17 to 0.92) (Comparison 01‐01). A non‐significant tendency favouring UDCA was shown at the end of follow‐up (2/34 (5.9%) versus 7/27 (25.9%); RR 0.23, 95% CI 0.05 to 1.00) (Comparison 01‐02). UDCA did not significantly decrease the serum HBV DNA level at the end of treatment (WMD ‐574 pg/ml, 95% CI ‐1149 to 1) (Comparison 01‐03). At the end of follow‐up, UDCA significantly decreased the serum HBV DNA level (WMD ‐955 pg/ml, 95% CI ‐1814 to ‐96) (Comparison 01‐04).
No data were available regarding clinical outcomes including mortality or histological outcomes.
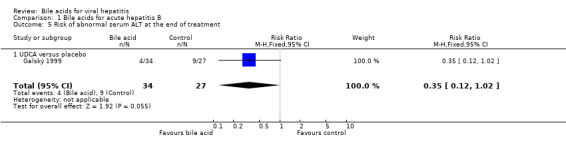
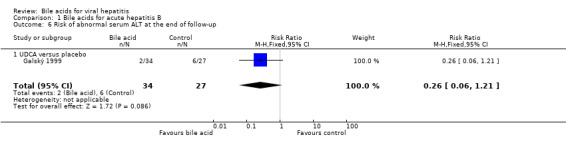
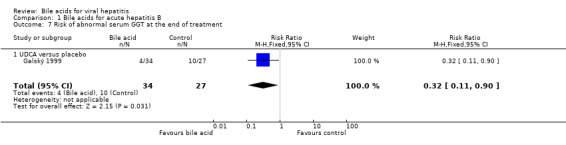
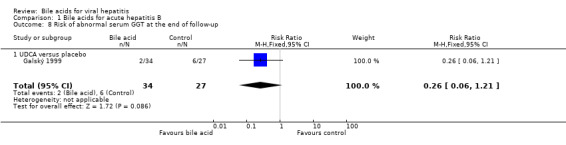
UDCA significantly decreased the risk of abnormal serum GGT activity (4/34 (11.8%) versus 10/27 (37%); RR 0.32, 95% CI 0.11 to 0.90) (Comparison 01‐07), but not ALT activity (4/34 (11.8%) versus 9/27 (33.3%); RR 0.35, 95% CI 0.12 to 1.02) at the end of treatment (Comparison 01‐05). UDCA did not significantly decrease the risk of abnormal serum ALT (2/34 (5.9%) versus 6/27 (22.2%); RR 0.26, 95% CI 0.06 to 1.21) (Comparison 01‐06) or GGT (2/34 (5.9%) versus 6/27 (22.2%); RR 0.26, 95% CI 0.06 to 1.21) activities at the end of follow‐up (Comparison 01‐08).
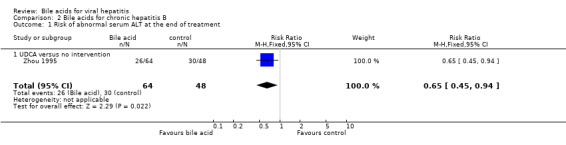
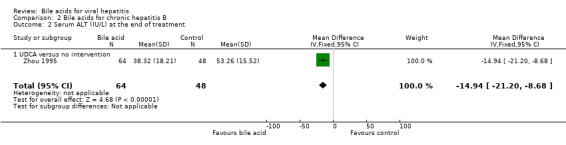
Bile acids for chronic hepatitis B One trial (Zhou 1995) evaluated UDCA versus no intervention in 112 patients with chronic hepatitis B. There were no data on virological, clinical, or histological outcomes. UDCA significantly reduced the risk of having abnormal serum ALT activity (26/64 (40.6%) versus 30/48 (62.5%); RR 0.65, 95% CI 0.45 to 0.94) (Comparison 02‐01) and serum ALT activity (WMD ‐15 IU/L, 95% CI ‐21 to ‐9) at the end of treatment (Comparison 02‐02).
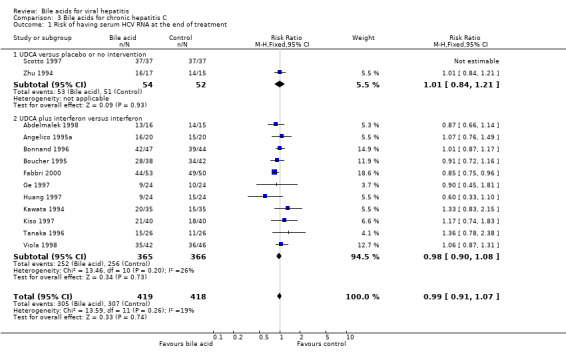
Bile acids for chronic hepatitis C Risk of being serum HCV RNA positive at the end of treatment Thirteen trials were included in this comparison. Two trials (106 patients) compared UDCA versus placebo (Scotto 1997) or no intervention (Zhu 1994), and 11 trials (831 patients) compared UDCA plus interferon versus interferon (Kawata 1994; Angelico 1995a; Boucher 1995; Bonnand 1996; Tanaka 1996; Ge 1997; Huang 1997; Abdelmalek 1998; Viola 1998; Fabbri 2000). UDCA did not significantly reduce the risk of having serum HCV RNA at the end of treatment (305/419 (72.8%) versus 307/418 (73.4%); RR 0.99, 95% CI 0.91 to 1.07). The trials comparing UDCA versus placebo or no intervention (53/54 (98.2%) versus 51/52 (98.1%); RR 1.01, 95% CI 0.84 to 1.21) and UDCA plus interferon versus interferon (252/365 (69%) versus 256/366 (70%); RR 0.98, 95% CI 0.90 to 1.08) did not individually show significant effects of UDCA (Comparison 03‐01).
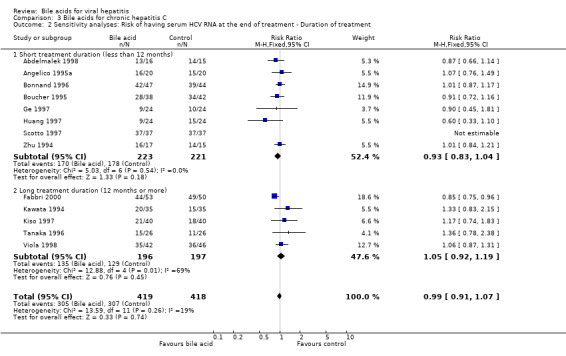
We performed a sensitivity analysis according to the duration of treatment. Eight trials (Zhu 1994; Angelico 1995a; Boucher 1995; Bonnand 1996; Ge 1997; Huang 1997; Scotto 1997; Abdelmalek 1998) evaluated short treatment duration (less than 12 months) and five trials (Kawata 1994; Tanaka 1996; Kiso 1997; Viola 1998; Fabbri 2000) evaluated long treatment duration (12 months or more). In none of the subgroups did UDCA significantly decrease the risk of having serum HCV RNA at the end of treatment (short treatment duration: 170/223 (76.2%) versus 178/221 (80.5%); RR 0.93, 95% CI 0.83 to 1.04 and long treatment duration: 135/196 (68.9%) versus 129/197 (65.5%); RR 1.05, 95% CI 0.92 to 1.19) (Comparison 03‐02).
Risk of being serum HCV RNA positive at the end of follow‐up Ten trials (Kawata 1994; Angelico 1995a; Boucher 1995; Tanaka 1996; Ge 1997; Kiso 1997; Senturk 1997; Abdelmalek 1998; Viola 1998; Fabbri 2000) compared UDCA plus interferon versus interferon (676 patients). UDCA plus interferon did not significantly decrease the risk of having serum HCV RNA at the end of follow‐up (268/333 (80.5%) versus 294/343 (85.7%); RR 0.93, 95% CI 0.87 to 1.00) (Comparison 03‐03).
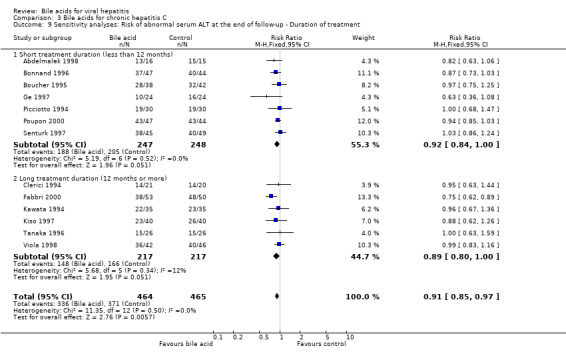
We performed a sensitivity analysis regarding treatment duration. Five trials (Angelico 1995a; Boucher 1995; Ge 1997; Senturk 1997; Abdelmalek 1998) evaluated short treatment duration (less than 12 months) and showed that UDCA did not significantly decrease the risk of having serum HCV RNA at the end of follow‐up (116/137 (84.7%) versus 125/146 (85.6%); RR 0.99, 95% CI 0.87 to 1.12). Five trials (Kawata 1994; Tanaka 1996; Kiso 1997; Viola 1998; Fabbri 2000) evaluated long treatment duration (12 months or more) and showed that UDCA significantly decreased the risk of serum HCV RNA at the end of follow‐up (152/196 (77.6%) versus 169/197 (85.8%); RR 0.90, 95% CI 0.83 to 0.99) (Comparison 03‐04).
Mortality We were not able to find any data related to mortality in the trials included in our review.
Risk of cirrhosis at the end of treatment Fabbri 2000 compared UDCA plus interferon versus interferon alone. UDCA plus interferon did not significantly reduce the risk of cirrhosis at the end of treatment (19/53 (35.9%) versus 10/50 (20%); RR 1.79, 95% CI 0.93 to 3.47) (Comparison 03‐05).
Risk of abnormal serum ALT activity at the end of treatment Twenty one trials (1582 patients) were included in this comparison. Bile acids significantly decreased the risk of having abnormal serum ALT activity (425/812 (52.3%) versus 467/770 (60.6%); RR 0.83, 95% CI 0.77 to 0.90) (Comparison 03‐06). Three trials (Takano 1994; Zhu 1994; Puoti 1995) compared UDCA versus placebo or no intervention, 15 trials (Clerici 1994; Kawata 1994; Angelico 1995a; Boucher 1995; Bonnand 1996; Tanaka 1996; Viola 1996; Ge 1997; Huang 1997; Kiso 1997; Senturk 1997; Abdelmalek 1998; Viola 1998; Fabbri 2000; Poupon 2000) compared UDCA plus interferon versus interferon, two trials (Picciotto 1994; Gracielle 2002) compared TUDCA plus interferon versus interferon, and one trial (Tsubota 1999) compared UDCA plus glycyrrhizin versus glycyrrhizin. UDCA significantly decreased the risk of having abnormal serum ALT activity at the end of treatment in the comparisons of UDCA versus placebo or no intervention (97/141 (68.8%) versus 81/98 (82.7%); RR 0.77, 95% CI 0.68 to 0.87) and UDCA plus interferon versus interferon (236/501 (47.1%) versus 291/506 (57.5%); RR 0.81, 95% CI 0.73 to 0.91). Bile acids (UDCA and TUDCA) did not significantly decrease the risk of having abnormal serum ALT activity in the comparison of TUDCA plus interferon versus interferon (43/85 (50.6%) versus 44/81(54.3%); RR 0.93, 95% CI 0.75 to 1.24) and UDCA plus glycyrrhizin versus glycyrrhizin (49/85 (57.7%) versus 51/85 (60%); RR 0.96, 95% CI 0.75 to 1.24) (Comparison 03‐06).
A sensitivity analysis was performed regarding treatment duration. Fifteen trials (Clerici 1994; Picciotto 1994; Takano 1994; Zhu 1994; Angelico 1995a; Boucher 1995; Puoti 1995; Bonnand 1996; Viola 1996; Ge 1997; Huang 1997; Senturk 1997; Abdelmalek 1998; Tsubota 1999; Poupon 2000) showed that short‐term treatment (less than 12 months) with bile acids (UDCA or TUDCA) significantly decreased the risk of having abnormal serum ALT activity at the end of treatment (301/561 (53.7%) versus 324/522 (62.1%); RR 0.82, 95% CI 0.75 to 0.91). Five trials (Kawata 1994; Tanaka 1996; Kiso 1997; Viola 1998; Fabbri 2000) showed that long‐term treatment (12 months or more) with UDCA significantly decreased the risk of having abnormal serum ALT activity at the end of treatment (93/196 (47.4%) versus 113/197 (57.4%); RR 0.83, 95% CI 0.69 to 0.99) (Comparison 03‐07). No significant difference was observed between short‐term treatment and long‐term treatment (P = 0.89).
Risk of abnormal serum ALT at the end of follow‐up Fourteen trials (997 patients) were included in this comparison. Bile acids (UDCA and TUDCA) significantly decreased the risk of abnormal serum ALT activity at the end of follow‐up (351/500 (70.2%) versus 384/497 (77.3%); RR 0.91, 95% CI 0.85 to 0.98) (Comparison 03‐08). Twelve trials (Clerici 1994; Kawata 1994; Boucher 1995; Bonnand 1996; Tanaka 1996; Ge 1997; Kiso 1997; Senturk 1997; Abdelmalek 1998; Viola 1998; Fabbri 2000; Poupon 2000) compared UDCA plus interferon versus interferon and two trials (Picciotto 1994; Pigozzi 1997) compared TUDCA plus interferon versus interferon. UDCA plus interferon significantly decreased the risk of abnormal serum ALT at the end of follow‐up (317/434 (73%) versus 352/435 (80.9%); RR 0.90, 95% CI 0.84 to 0.97), while TUDCA plus interferon did not show this effect (34/66 (51.5%) versus 32/62 (51.6%); RR 1.01, 95% CI 0.73 to 1.40).
Sensitivity analysis was performed regarding treatment duration. Eight trials (Picciotto 1994; Boucher 1995; Bonnand 1996; Ge 1997; Pigozzi 1997; Senturk 1997; Abdelmalek 1998; Poupon 2000) showed that short‐term treatment (less than 12 months) with bile acids (UDCA or TUDCA) did not significantly decrease the risk of abnormal serum ALT activity at the end of follow‐up (203/283 (71.7%) versus 218/280 (77.9%); RR 0.92, 95% CI 0.85 to 1.01). Six trials (Clerici 1994; Kawata 1994; Tanaka 1996; Kiso 1997; Viola 1998; Fabbri 2000) showed that long‐term treatment (12 months or more) with UDCA tended to significantly decrease the risk of abnormal serum ALT activity at the end of follow‐up (148/217 (68.2%) versus 166/217 (76.5%); RR 0.89, 95% CI 0.80 to 1.00) (Comparison 03‐09). No significant difference was observed between short‐term treatment and long‐term treatment (P = 0.67).
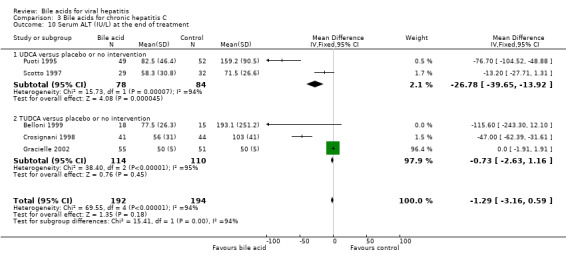
Serum ALT activity at the end of treatment Two trials (Puoti 1995; Scotto 1997) compared UDCA versus placebo or no intervention and three trials including 224 patients (Crosignani 1998; Belloni 1999) compared TUDCA versus placebo or no intervention. Bile acids did not significantly decrease serum ALT activity at the end of treatment (WMD ‐1.29 IU/L, 95% CI ‐3.16 to ‐0.59) (Comparison 03‐10). However, there was significant heterogeneity in this comparison (Chi‐square = 69.55, df = 4, P < 0.0001). In subgroup analyses, UDCA (WMD ‐27 IU/L, 95% CI ‐40 to ‐14) but not TUDCA (WMD ‐0,73 IU/L, 95% CI ‐2.63 to ‐1.16) significantly decreased serum ALT activity at the end of treatment. The subgroup analysis regarding UDCA had significant heterogeneity (Chi‐square = 15.73, df = 1, P = 0.0001).
Serum AST activity at the end of treatment One trial (Scotto 1997) compared UDCA versus placebo and one trial (Crosignani 1998) compared TUDCA versus placebo. Bile acids significantly decreased serum AST activity at the end of treatment (WMD ‐25 IU/L, 95% CI ‐34 to ‐16) (Comparison 03‐11). Both UDCA (WMD ‐21 IU/L, 95% CI ‐32 to ‐9) and TUDCA (WMD ‐32 IU/L, 95% CI ‐46 to ‐18) significantly decreased serum AST activity at the end of treatment.
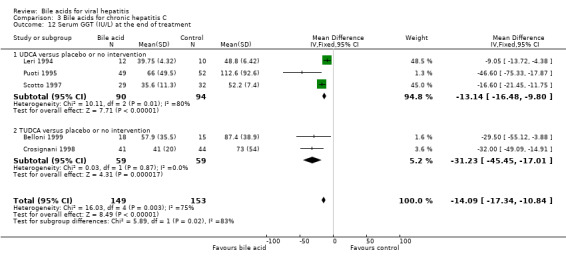
Serum GGT activity at the end of treatment Three trials (Leri 1994; Puoti 1995; Scotto 1997) compared UDCA versus placebo or no intervention and two trials (Crosignani 1998; Belloni 1999) compared TUDCA versus placebo or no intervention. Bile acids significantly decreased serum GGT activity at the end of treatment (WMD ‐14 IU/L, 95% CI ‐17 to ‐11) (Comparison 03‐12). However, there was significant heterogeneity in this comparison (Chi‐square = 16.03, df = 4, P = 0.003). In subgroup analyses, both UDCA (WMD ‐13 IU/L, 95% CI ‐16 to ‐10) and TUDCA (WMD ‐31 IU/L, 95% CI ‐46 to ‐17) significantly decreased serum GGT activity at the end of treatment. The subgroup analysis regarding UDCA had significant heterogeneity (Chi‐square = 10.11, df = 2, P = 0.0064).
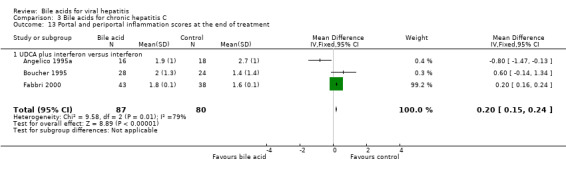
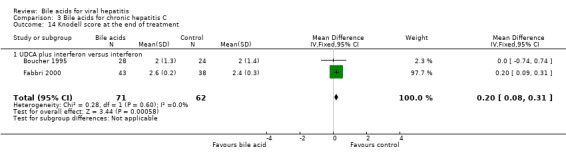
Liver histology Three trials (Angelico 1995a; Boucher 1995; Fabbri 2000) compared the effects of UDCA plus interferon versus interferon on the portal and periportal inflammation score. At the end of treatment, UDCA significantly increased portal and periportal inflammation score (WMD 0.20, 95% CI 0.15 to 0.24) based on a fixed‐effect model, but not a random‐effects model (WMD 0.02, 95% CI ‐0.62 to 0.66) (Comparison 03‐13). There was significant heterogeneity in this comparison (Chi‐square = 9.58, df = 2, P = 0.0083). Two trials (Boucher 1995; Fabbri 2000) compared the effects of UDCA plus interferon versus interferon on the Knodell score (Knodell 1981). At the end of treatment, UDCA plus interferon significantly increased the Knodell score (WMD 0.20, 95% CI 0.08 to 0.31) (Comparison 03‐14).
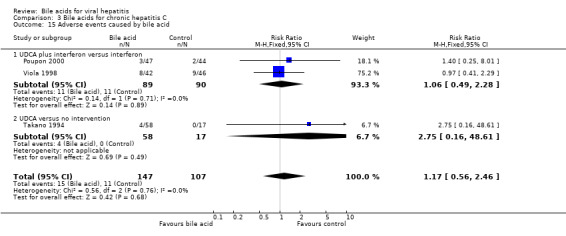
Adverse events No serious adverse events were reported in the included trials. UDCA did not significantly increase the proportion of adverse events (15/147 (10.2%) versus 11/107 (10.3%); RR 1.17, 95% CI 0.56 to 2.46) (Comparison 03‐15). Two trials (Viola 1998; Poupon 2000), which compared UDCA plus interferon versus interferon, reported adverse events (11/89 (12.4%) versus 11/90 (12.2%); RR 1.06, 95% CI 0.49 to 2.28). In Poupon 2000, five adverse events (diarrhea in all cases) were reported (three in the UCLA plus interferon group and two in the placebo plus interferon group). In Viola 1998, the adverse events were reported without providing the types. One trial (Takano 1994), which compared UDCA versus no intervention, reported adverse events (4/58 (6.9%) versus 0/17 (0%); RR 2.75, 95% CI 0.16 to 48.61), which included three patients with abdominal discomfort and one patient with icterus in the UDCA group.
Funnel plot asymmetry A funnel plot assessing the trials effect estimates on the outcome of having serum HCV RNA at the end of treatment for patients with chronic hepatitis C revealed no significant funnel plot asymmetry (Intercept ‐0.276, 95% CI ‐1.702 to 1.150, P = 0.676).
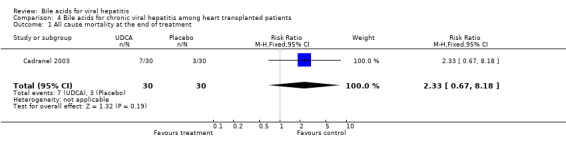
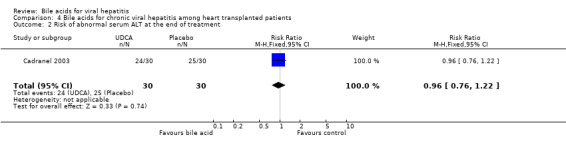
Bile acids for chronic viral hepatitis among heart‐transplanted patients There was one trial assessing the effects of UDCA on chronic viral hepatitis including B and C among 60 heart‐transplanted patients after one year treatment. No significant differences between UDCA and placebo were found in terms of all cause mortality (7/30 (23.2%) versus 3/30 (10%); RR 2.74, 95% CI 0.63 to 11.82), raised serum ALT (24/30 (80.0%) versus 25/30 (83.3%); RR 0.96, 95% CI 0.76 to 1.22), Knodell score improvement (at least one point) (6/30 (20.0%) versus 13/30 (43.3%); RR 0.46, 95% CI 0.20 to 1.05), and graft rejection (8/30 (26.7%) versus 3/30 (10.0%); RR 2.67, 95% CI 0.78 to 9.09) at the end of treatment.
Discussion
We identified only two trials evaluating UDCA for acute or chronic hepatitis B. However, we identified a large number of trials evaluating UDCA or TUDCA for chronic hepatitis C. We were unable to identify any randomised clinical trials evaluating bile acids for hepatitis A, acute hepatitis C, acute or chronic hepatitis D, or hepatitis E. None of the included trials could be considered of high methodological quality with low risk of bias, ie, having adequate generation of the allocation sequence, allocation concealment, double blinding, and follow‐up (Schulz 1995; Moher 1998; Kjaergard 2001). Accordingly, the findings of our review must be interpreted with caution. Furthermore, the observed effects of bile acids were small and affected only surrogate outcomes (Gluud 2007). We have no evidence that these effects can be translated into meaningful clinical outcomes for patients. However, our findings suggest that UDCA may normalise or decrease serum transaminase activities in patients with acute or chronic hepatitis B and chronic hepatitis C. One single trial observed beneficial effects of UDCA on HBV markers in patients with acute hepatitis B. Further, a subgroup analysis of the trials evaluating long‐term UDCA treatment of chronic hepatitis C suggested a beneficial effect on clearance of HCV RNA. However, we need large randomised clinical trials conducted with adequate methodology to confirm these observations. We were mostly unable to identify data on mortality, liver related morbidity (ie, hepatocellular carcinoma and end‐stage liver disease), quality of life, or cost‐effectiveness. Bile acid treatment appeared safe; no serious adverse events were reported. Most of the adverse events which caused withdrawal of patients, such as depression, weight loss, and severe flu‐like syndrome, probably arose from co‐intervention with interferon (Brok 2005).
Bile acids for hepatitis B One trial in patients with acute hepatitis B reported that UDCA significantly reduced the risk of having positive HBsAg at the end of treatment (but not the end of follow‐up) and significantly decreased HBV DNA levels at the end of follow‐up (but not the end of treatment). This observation requires replication for several reasons. First, UDCA had no consistent effects on serum enzyme activities at the end of treatment or follow‐up. Second, generation of the allocation sequence and allocation concealment were considered inadequate in this trial. Third, although the trial used placebo, we cannot be sure that this resulted in adequate blinding. Since trials involving bile acids may be difficult to blind sufficiently (Gluud 2001b), it cannot be excluded that the trial may have been biased. Therefore, the observations of Galský1999 need confirmation before any therapeutic recommendation can be made.
One trial (Zhou 1995) compared UDCA versus no intervention for patients with chronic hepatitis B. In this trial, UDCA significantly decreased the risk of abnormal serum ALT and serum ALT activities at the end of treatment. However, the trial did not provide evidence for an effect of UDCA on serum viral markers or clinical or histological outcomes. Therefore, we also need more trials to investigate the effects of UDCA for chronic hepatitis B.
Bile acids for chronic hepatitis C Overall, bile acids did not significantly affect the risk of being HCV RNA positive at the end of treatment or at the end of follow‐up. In a sensitivity analysis considering treatment duration, bile acid treatment for 12 months or more was associated with a significant decrease in the risk of having HCV RNA at the end of follow‐up, but not at the end of treatment. Before considering therapeutic actions based on this finding, one should consider the following. First, why should bile acids, which are quickly metabolised (Fuchs 1999), affect HCV RNA levels at the end of follow‐up, but not at the end of treatment? Second, subgroup results, particularly when post hoc in nature, should be evaluated cautiously (Hahn 2000). Third, due to the low methodological quality of the trials, significant overestimation of intervention effects cannot be excluded (Schulz 1995; Moher 1998; Kjaergard 2001). Our observation should lead to proper assessment of bile acids in large scale randomised trials using adequate methods to control bias. The quest for such trials is further supported by the significant effects of bile acids on transaminase activities, whether analysed at the end of treatment or at the end of follow‐up.
Although we observed significant effects of UDCA on biochemical variables such as ALT, AST, and GGT, we could not find evidence to prove that the improvement of these variables could be translated into meaningful improvement of clinical outcomes such as mortality, incidence of cirrhosis, and hepatocellular carcinoma. In clinical practice, some physicians continue to base therapeutic decisions on surrogate measures such as the biochemical variables mentioned above (Gluud 2007). The results of our present review are comparable to two Cochrane systematic reviews on the effect of bile acids for primary biliary cirrhosis and primary sclerosing cholangitis, which reported significant improvement of biochemical variables, but not on 'hard' outcomes, such as mortality and liver transplantation (Gluud 2001b; Chen 2003). A new trial to investigate the effects of UDCA on meaningful clinical outcomes seems to be needed. Quality of life is another very important aspect that needs to be studied in patients with viral hepatitis. None of the included trials in our review assessed quality of life specifically.
Chronic hepatitis C infection can be characterised by histological changes, which run from relatively mild hepatic inflammatory activity and a low degree of fibrosis to full blown cirrhosis with liver failure. Hepatic lesions may be accompanied by bile duct damage, intraportal lymphoid aggregates, steatosis, or a combination of these manifestations (Mihm 1997). The pathological changes of the bile duct system caused by viral hepatitis contribute the formation of cholangitis and accumulation of hydrophobic bile acids in the liver tissue (Rodrigues 2000). Therefore, one possible explanation for the liver biochemistry improvement obtained by administering UDCA would be that it replaces the more hydrophobic human bile acids, which cause liver cell damage (Batta 1989; Chretien 1989). The protection of UDCA may also result from stimulation of hepato‐biliary secretion (Paumgartner 2002). Furthermore, recent data from basic research demonstrate that bile acids, including UDCA, appear to affect both 'death receptors' (Caspase 8/10) and 'cell survival cascades' (UDCA‐epidermal growth factor‐mitogen activated protein kinase) (Guicciardi 2002; Qiao 2002). The beneficial effects of UDCA on liver biochemistry variables suggest that in patients with viral hepatitis (the present review), primary biliary cirrhosis (Gluud 2001b), and primary sclerosing cholangitis (Chen 2003), UDCA's stimulus of the 'cell survival cascades' seems to overpower UDCA's stimulus of the cell 'death receptors'. However, beneficially affecting cell death of, for example hepatocytes, may not be important enough to arrest or slow down the progression of these liver diseases. From the meta‐analyses in our review, we observed that UDCA did have significant effects on improving the biochemical variables such as ALT, AST and GGT, which may result from the mechanisms of UDCA mentioned above. However, in this present review and another review on UDCA for primary biliary cirrhosis (Gluud 2001b), we found that bile acids may worsen liver histology. More evidence regarding this finding is needed.
As indicated, the reported methodological quality of the reviewed trials was low and in some trials we had difficulties extracting the relevant data. In order to get detailed data on the trials, 17 letters were sent to the first authors. Only one author replied and this author could not supply the information because the original data were lost. In the future, trials ought to report more fully on clinically relevant outcomes and follow recommended guidelines for the reporting of trials (CONSORT ‐ Consolidated Standards of Reporting Trials: www.consort‐statement.org).
We did not observe any therapeutic advantage of using TUDCA instead of UDCA. We therefore advocate that UDCA should be the bile acid being evaluated for patients with viral hepatitis, as this bile acid has been most widely used. Accordingly, we know the adverse event profile of this bile acid best. If it turns out to be possible to demonstrate beneficial clinical effects with UDCA, it may then become worthwhile considering 'head‐to‐head' comparisons of UDCA versus TUDCA in randomised clinical trials.
UDCA is still being used for primary biliary cirrhosis, although the evidence for its clinical effects have been questioned in a meta‐analysis (Goulis 1999) and in a Cochrane review (Gluud 2001b). In primary biliary cirrhosis, UDCA compared to placebo/no intervention leads to a decrease in serum bilirubin of about 10 µmol/L and a decrease of serum ALT of about 48 IU/L (Gluud 2001b). These decreases are similar to those observed in the present review in patients with chronic hepatitis B or C considering the dosage administered and the duration of treatment. Bile acids do not seem to differ regarding the lack of significant clinical effects in both primary biliary cirrhosis and viral hepatitis. It is, therefore, a question why we as clinicians are not more consistent as UDCA is often used for primary biliary cirrhosis and it is very seldom that UDCA (or other bile acids) are used for viral hepatitis.
This review demonstrates the necessity to search for trials in databases that are not normally included in Cochrane Reviews and other reviews. We identified five Chinese trials by searching the Chinese Biomedical Database. Although the methodological quality of Chinese clinical trials is generally low (Liu 2002), the rising activity in performing randomised clinical trials in China must be considered in the West.
In the above meta‐analyses we have disregarded the risks of random errors in meta‐analysis. Evidence shows that much errors may be substantial (Wetterslev 2007). If these risks were taken properly into consideration, several of our significant findings are likely to come out insignificant (Wetterslev 2007).
Authors' conclusions
Implications for practice.
There is not significant evidence to support or refute beneficial effects of bile acids for viral hepatitis. UDCA or TUDCA may improve liver transaminase activities, but there is no compelling evidence showing that these bile acids beneficially affect viral markers, mortality, cirrhosis development, need for liver transplantation, or liver histology in patients with acute or chronic hepatitis B and chronic hepatitis C. We have no knowledge about the potential beneficial and harmful effects of bile acids for hepatitis A, acute hepatitis C, hepatitis D, or hepatitis E.
Implications for research.
Acute hepatitis B Due to the potential beneficial effect of UDCA on viral markers in acute hepatitis B, it seems worthwhile to test this in randomised placebo controlled clinical trials with sufficient power and long‐term follow‐up after the end of treatment. If such trials should show positive results, further trials addressing clinical effects seem warranted.
Chronic hepatitis B Due to the potential beneficial effect of UDCA on viral markers in acute hepatitis B and on liver biochemistry in chronic hepatitis B, it seems worthwhile to test if similar effects could be observed in chronic hepatitis B patients included in randomised placebo controlled clinical trials with sufficient power and long‐term follow‐up after the end of treatment. Co‐intervention in both arms of the trial could be used. If such trials should show positive results on viral markers, further trials addressing clinical effects seem warranted.
Chronic hepatitis C Due to the potential beneficial effect of bile acids on viral markers in chronic hepatitis C with co‐intervention of interferon, it seems worthwhile to test this potential effect in randomised placebo controlled clinical trials with sufficient power and long‐term follow‐up after the end of treatment. Co‐intervention in both arms of the trial could be pegylated interferon plus ribavirin. If such trials should show positive results, further trials addressing clinical effects seem warranted.
Other types of viral hepatitis We suggest that trials on bile acids for other forms of viral hepatitis should await the results of trials demonstrating clear virological and/or clinical effects of bile acids for patients with hepatitis B and/or C.
General aspects We do not have any data examining the potential advantage of using TUDCA instead of UDCA. However, before such 'head‐to‐head' comparative trials are considered, the beneficial clinical effects of UDCA ought to be established.
Researchers wishing to examine the effects of bile acids for viral hepatitis ought to use adequate trial methodology and follow the guidelines for reporting of trials (CONSORT ‐ Consolidated Standards of Reporting Trials: www.consort‐statement.org).
Detrimental effect of bile acids on liver histology cannot be excluded. Therefore, an independent Data Monitoring and Safety Committee should effectively monitor such trials, including the results of liver histology.
What's new
| Date | Event | Description |
|---|---|---|
| 17 October 2008 | Amended | Converted to new review format. |
Notes
According to the advice from the latest workshop on how to edit systematic reviews, we performed the following change in this review.
We used both a random‐effects (DerSimonian 1986) and fixed effect‐model (DeMets 1987) in the meta‐analysis. We reported the results of the fixed‐effect model if there was no difference in the results of the two models. Otherwise, we would have reported the results produced by both models.
Acknowledgements
The main acknowledgement is to the patients with hepatitis who took part in the clinical trials reviewed and to the researchers who conducted the trials. Special thanks to Li Lin who helped us retrieve original Chinese publications as well as searching The Chinese Biomedical Database. We also want to express our thanks to Ronald L Koretz, Rob Myers (Contact Editor for this review), and the other peer reviewers for their valuable comments.
Appendices
Appendix 1. Search Strategies
| Data base | Search strategy | Date of search | |
| The Cochrane Hepato‐Biliary Group Controlled Trials Register | #1: 'hepatitis' and 'chenodeoxycholic' #2: 'hepatitis' and 'cholic' #3: 'hepatitis' and 'deoxycholic' #4: 'hepatitis' and 'glycochenodeoxycholic' #5: 'hepatitis' and 'glycocholic' #6: 'hepatitis' and 'glycodeoxycholic' #7: 'hepatitis' and 'glycolithocholic' #8: 'hepatitis' and 'hyodeoxycholic' #9: 'hepatitis' and 'lithocholic' #10: 'hepatitis' and 'taurochenodeoxycholic' #11: 'hepatitis' and 'taurocholic' #12: 'hepatitis' and 'taurodehydrocholic' #13: 'hepatitis' and 'taurodeoxycholic' #14: 'hepatitis' and 'tauroglycocholic' #15: 'hepatitis' and 'taurolithocholic' #16: 'hepatitis' and 'tauroselcholic' #17: 'hepatitis' and 'tauroursocholic' #18: 'hepatitis' and 'tauroursodeoxycholic' #19: 'hepatitis' and 'ursocholic' #20: 'hepatitis' and 'ursodeoxycholic' | July 2007. | |
| The Cochrane Controlled Trials Register | #1: HEPATITIS*:ME #2: HEPATITIS #3: BILE ACID *:ME #4: CHENODEOXYCHOLIC or CHOLIC or DEOXYCHOLIC or GLYCOCHENODEOXYCHOLIC or GLYCOCHOLIC or GLYCODEOXYCHOLIC or GLYCOLITHOCHOLIC or HYODEOXYCHOLIC or LITHOCHOLIC or TAUROCHENODEOXYCHOLIC or TAUROCHOLIC or TAURODEHYDROCHOLIC or TAURODEOXYCHOLIC or TAUROGLYCOCHOLIC or TAUROLITHOCHOLIC or TAUROSELCHOLIC or TAUROURSOCHOLIC or TAUROURSODEOXYCHOLIC or URSOCHOLIC or URSODEOXYCHOLIC #5: #1 or #2 #6: #3 or #4 #7: #5 and #6 | July 2007. | |
| MEDLINE | #1: HEPATITIS *:ME #2: HEPATITIS #3: BILE ACID *:ME #4: CHENODEOXYCHOLIC or CHOLIC or DEOXYCHOLIC or GLYCOCHENODEOXYCHOLIC or GLYCOCHOLIC or GLYCODEOXYCHOLIC or GLYCOLITHOCHOLIC or HYODEOXYCHOLIC or LITHOCHOLIC or TAUROCHENODEOXYCHOLIC or TAUROCHOLIC or TAURODEHYDROCHOLIC or TAURODEOXYCHOLIC or TAUROGLYCOCHOLIC or TAUROLITHOCHOLIC or TAUROSELCHOLIC or TAUROURSOCHOLIC or TAUROURSODEOXYCHOLIC or URSOCHOLIC or URSODEOXYCHOLIC #5: #1 or #2 #6: #3 or #4 #7: #5 and #6 #8: RANDOMIZED‐CONTROLLED‐TRIAL *:ME #9: RANDOM* #10: #8 or #9 #11: #7 and #10 | July 2007. | |
| EMBASE | #1: HEPATITIS *:ME #2: HEPATITIS #3: BILE ACID *:ME #4: CHENODEOXYCHOLIC or CHOLIC or DEOXYCHOLIC or GLYCOCHENODEOXYCHOLIC or GLYCOCHOLIC or GLYCODEOXYCHOLIC or GLYCOLITHOCHOLIC or HYODEOXYCHOLIC or LITHOCHOLIC or TAUROCHENODEOXYCHOLIC or TAUROCHOLIC or TAURODEHYDROCHOLIC or TAURODEOXYCHOLIC or TAUROGLYCOCHOLIC or TAUROLITHOCHOLIC or TAUROSELCHOLIC or TAUROURSOCHOLIC or TAUROURSODEOXYCHOLIC or URSOCHOLIC or URSODEOXYCHOLIC #5: #1 or #2 #6: #3 or #4 #7: #5 and #6 #8: RANDOMIZED‐CONTROLLED‐TRIAL *:ME #9: RANDOM* #10: #8 or #9 #11: #7 and #10 | July 2007. | |
| The Chinese Biomedical Database | #1: HEPATITIS *:ME #2: HEPATITIS #3: BILE ACID *:ME #4: CHENODEOXYCHOLIC or CHOLIC or DEOXYCHOLIC or GLYCOCHENODEOXYCHOLIC or GLYCOCHOLIC or GLYCODEOXYCHOLIC or GLYCOLITHOCHOLIC or HYODEOXYCHOLIC or LITHOCHOLIC or TAUROCHENODEOXYCHOLIC or TAUROCHOLIC or TAURODEHYDROCHOLIC or TAURODEOXYCHOLIC or TAUROGLYCOCHOLIC or TAUROLITHOCHOLIC or TAUROSELCHOLIC or TAUROURSOCHOLIC or TAUROURSODEOXYCHOLIC or URSOCHOLIC or URSODEOXYCHOLIC #5: #1 or #2 #6: #3 or #4 #7: #5 and #6 #8: RANDOMIZED‐CONTROLLED‐TRIAL *:ME #9: RANDOM* #10: #8 or #9 #11: #7 and #10 | July 2007. | |
| Science Citation Index Expanded | #1: HEPATITIS *:ME #2: HEPATITIS #3: BILE ACID *:ME #4: CHENODEOXYCHOLIC or CHOLIC or DEOXYCHOLIC or GLYCOCHENODEOXYCHOLIC or GLYCOCHOLIC or GLYCODEOXYCHOLIC or GLYCOLITHOCHOLIC or HYODEOXYCHOLIC or LITHOCHOLIC or TAUROCHENODEOXYCHOLIC or TAUROCHOLIC or TAURODEHYDROCHOLIC or TAURODEOXYCHOLIC or TAUROGLYCOCHOLIC or TAUROLITHOCHOLIC or TAUROSELCHOLIC or TAUROURSOCHOLIC or TAUROURSODEOXYCHOLIC or URSOCHOLIC or URSODEOXYCHOLIC #5: #1 or #2 #6: #3 or #4 #7: #5 and #6 #8: RANDOMIZED‐CONTROLLED‐TRIAL *:ME #9: RANDOM* #10: #8 or #9 #11: #7 and #10 | July 2007. |
Data and analyses
Comparison 1. Bile acids for acute hepatitis B.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Risk of having positive serum HBsAg at the end of treatment | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.17, 0.92] |
| 1.1 UDCA versus placebo | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.17, 0.92] |
| 2 Risk of having positive HBsAg at the end of follow‐up | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.05, 1.00] |
| 2.1 UDCA versus placebo | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.05, 1.00] |
| 3 Serum DNA (pg/ml) level at the end of treatment | 1 | 59 | Mean Difference (IV, Fixed, 95% CI) | ‐574.05 [‐1148.71, 0.61] |
| 3.1 UDCA versus placebo | 1 | 59 | Mean Difference (IV, Fixed, 95% CI) | ‐574.05 [‐1148.71, 0.61] |
| 4 Serum DNA (pg/ml) level at the end of follow‐up | 1 | 59 | Mean Difference (IV, Fixed, 95% CI) | ‐954.73 [‐1813.71, ‐95.75] |
| 4.1 UDCA versus placebo | 1 | 59 | Mean Difference (IV, Fixed, 95% CI) | ‐954.73 [‐1813.71, ‐95.75] |
| 5 Risk of abnormal serum ALT at the end of treatment | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.12, 1.02] |
| 5.1 UDCA versus placebo | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.12, 1.02] |
| 6 Risk of abnormal serum ALT at the end of follow‐up | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.21] |
| 6.1 UDCA versus placebo | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.21] |
| 7 Risk of abnormal serum GGT at the end of treatment | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.11, 0.90] |
| 7.1 UDCA versus placebo | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.11, 0.90] |
| 8 Risk of abnormal serum GGT at the end of follow‐up | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.21] |
| 8.1 UDCA versus placebo | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.21] |
1.1. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 1 Risk of having positive serum HBsAg at the end of treatment.
1.2. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 2 Risk of having positive HBsAg at the end of follow‐up.
1.3. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 3 Serum DNA (pg/ml) level at the end of treatment.
1.4. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 4 Serum DNA (pg/ml) level at the end of follow‐up.
1.5. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 5 Risk of abnormal serum ALT at the end of treatment.
1.6. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 6 Risk of abnormal serum ALT at the end of follow‐up.
1.7. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 7 Risk of abnormal serum GGT at the end of treatment.
1.8. Analysis.
Comparison 1 Bile acids for acute hepatitis B, Outcome 8 Risk of abnormal serum GGT at the end of follow‐up.
Comparison 2. Bile acids for chronic hepatitis B.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Risk of abnormal serum ALT at the end of treatment | 1 | 112 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.45, 0.94] |
| 1.1 UDCA versus no intervention | 1 | 112 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.45, 0.94] |
| 2 Serum ALT (IU/L) at the end of treatment | 1 | 112 | Mean Difference (IV, Fixed, 95% CI) | ‐14.94 [‐21.20, ‐8.68] |
| 2.1 UDCA versus no intervention | 1 | 112 | Mean Difference (IV, Fixed, 95% CI) | ‐14.94 [‐21.20, ‐8.68] |
2.1. Analysis.
Comparison 2 Bile acids for chronic hepatitis B, Outcome 1 Risk of abnormal serum ALT at the end of treatment.
2.2. Analysis.
Comparison 2 Bile acids for chronic hepatitis B, Outcome 2 Serum ALT (IU/L) at the end of treatment.
Comparison 3. Bile acids for chronic hepatitis C.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Risk of having serum HCV RNA at the end of treatment | 13 | 837 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.91, 1.07] |
| 1.1 UDCA versus placebo or no intervention | 2 | 106 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.84, 1.21] |
| 1.2 UDCA plus interferon versus interferon | 11 | 731 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.90, 1.08] |
| 2 Sensitivity analyses: Risk of having serum HCV RNA at the end of treatment ‐ Duration of treatment | 13 | 837 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.91, 1.07] |
| 2.1 Short treatment duration (less than 12 months) | 8 | 444 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.83, 1.04] |
| 2.2 Long treatment duration (12 months or more) | 5 | 393 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.92, 1.19] |
| 3 Risk of having serum HCV RNA at the end of follow‐up | 10 | 676 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.87, 1.00] |
| 3.1 UDCA plus interferon versus interferon | 10 | 676 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.87, 1.00] |
| 4 Sensitivity analyses: Risk of having serum HCV RNA at the end of follow‐up ‐ Duration of treatment | 10 | 676 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.87, 1.00] |
| 4.1 Short treatment duration (less than 12 months) | 5 | 283 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.87, 1.12] |
| 4.2 Long treatment duration (12 months or more) | 5 | 393 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.83, 0.99] |
| 5 Risk of cirrhosis at the end of treatment | 1 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [0.93, 3.47] |
| 5.1 UDCA plus interferon versus interferon | 1 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [0.93, 3.47] |
| 6 Risk of abnormal serum ALT at the end of treatment | 21 | 1582 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.77, 0.90] |
| 6.1 UDCA versus placebo or no intervention | 3 | 239 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.69, 0.88] |
| 6.2 UDCA plus interferon versus interferon | 15 | 1007 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.73, 0.91] |
| 6.3 TUDCA plus interferon versus interferon | 2 | 166 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.70, 1.24] |
| 6.4 UDCA plus glycyrrhizin versus glycyrrhizin | 1 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.75, 1.24] |
| 7 Sensitivity analyses: Risk of abnormal serum ALT at the end of treatment ‐ Duration of treatment | 21 | 1582 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.77, 0.90] |
| 7.1 Short treatment duration (less than 12 months) | 16 | 1189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.76, 0.92] |
| 7.2 Long treatment duration (12 months or more) | 5 | 393 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.69, 0.99] |
| 8 Risk of abnormal serum ALT at the end of follow‐up | 13 | 929 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.85, 0.97] |
| 8.1 UDCA plus interferon versus interferon | 12 | 869 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.84, 0.97] |
| 8.2 TUDCA plus interferon versus interferon | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.68, 1.47] |
| 9 Sensitivity analyses: Risk of abnormal serum ALT at the end of follow‐up ‐ Duration of treatment | 13 | 929 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.85, 0.97] |
| 9.1 Short treatment duration (less than 12 months) | 7 | 495 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.84, 1.00] |
| 9.2 Long treatment duration (12 months or more) | 6 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.80, 1.00] |
| 10 Serum ALT (IU/L) at the end of treatment | 5 | 386 | Mean Difference (IV, Fixed, 95% CI) | ‐1.29 [‐3.16, 0.59] |
| 10.1 UDCA versus placebo or no intervention | 2 | 162 | Mean Difference (IV, Fixed, 95% CI) | ‐26.78 [‐39.65, ‐13.92] |
| 10.2 TUDCA versus placebo or no intervention | 3 | 224 | Mean Difference (IV, Fixed, 95% CI) | ‐0.73 [‐2.63, 1.16] |
| 11 Serum AST (IU/L) at the end of treatment | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐25.16 [‐34.02, ‐16.30] |
| 11.1 UDCA versus placebo or no intervention | 1 | 61 | Mean Difference (IV, Fixed, 95% CI) | ‐20.70 [‐32.08, ‐9.32] |
| 11.2 TUDCA versus placebo or no intervention | 1 | 85 | Mean Difference (IV, Fixed, 95% CI) | ‐32.0 [‐46.10, ‐17.90] |
| 12 Serum GGT (IU/L) at the end of treatment | 5 | 302 | Mean Difference (IV, Fixed, 95% CI) | ‐14.09 [‐17.34, ‐10.84] |
| 12.1 UDCA versus placebo or no intervention | 3 | 184 | Mean Difference (IV, Fixed, 95% CI) | ‐13.14 [‐16.48, ‐9.80] |
| 12.2 TUDCA versus placebo or no intervention | 2 | 118 | Mean Difference (IV, Fixed, 95% CI) | ‐31.23 [‐45.45, ‐17.01] |
| 13 Portal and periportal inflammation scores at the end of treatment | 3 | 167 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.15, 0.24] |
| 13.1 UDCA plus interferon versus interferon | 3 | 167 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.15, 0.24] |
| 14 Knodell score at the end of treatment | 2 | 133 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.08, 0.31] |
| 14.1 UDCA plus interferon versus interferon | 2 | 133 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.08, 0.31] |
| 15 Adverse events caused by bile acid | 3 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.56, 2.46] |
| 15.1 UDCA plus interferon versus interferon | 2 | 179 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.49, 2.28] |
| 15.2 UDCA versus no intervention | 1 | 75 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [0.16, 48.61] |
3.1. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 1 Risk of having serum HCV RNA at the end of treatment.
3.2. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 2 Sensitivity analyses: Risk of having serum HCV RNA at the end of treatment ‐ Duration of treatment.
3.3. Analysis.
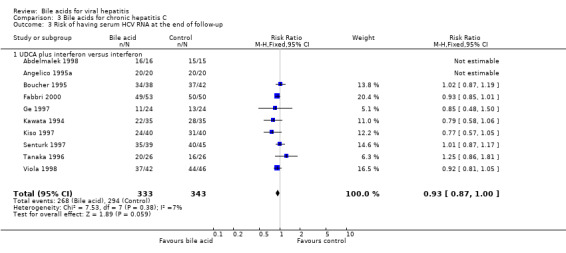
Comparison 3 Bile acids for chronic hepatitis C, Outcome 3 Risk of having serum HCV RNA at the end of follow‐up.
3.4. Analysis.

Comparison 3 Bile acids for chronic hepatitis C, Outcome 4 Sensitivity analyses: Risk of having serum HCV RNA at the end of follow‐up ‐ Duration of treatment.
3.5. Analysis.
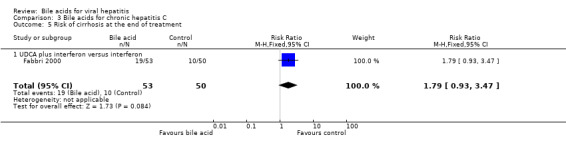
Comparison 3 Bile acids for chronic hepatitis C, Outcome 5 Risk of cirrhosis at the end of treatment.
3.6. Analysis.
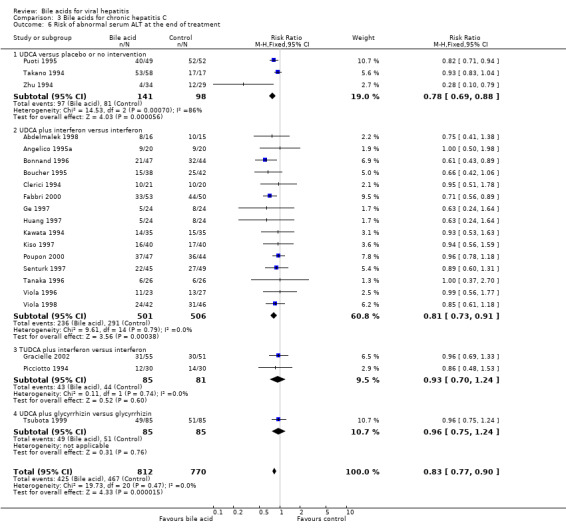
Comparison 3 Bile acids for chronic hepatitis C, Outcome 6 Risk of abnormal serum ALT at the end of treatment.
3.7. Analysis.
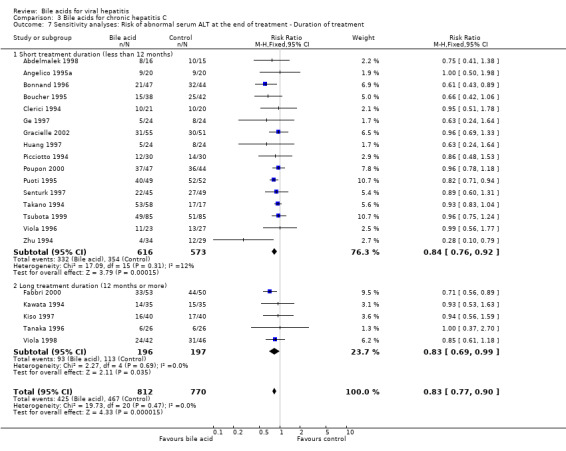
Comparison 3 Bile acids for chronic hepatitis C, Outcome 7 Sensitivity analyses: Risk of abnormal serum ALT at the end of treatment ‐ Duration of treatment.
3.8. Analysis.

Comparison 3 Bile acids for chronic hepatitis C, Outcome 8 Risk of abnormal serum ALT at the end of follow‐up.
3.9. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 9 Sensitivity analyses: Risk of abnormal serum ALT at the end of follow‐up ‐ Duration of treatment.
3.10. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 10 Serum ALT (IU/L) at the end of treatment.
3.11. Analysis.

Comparison 3 Bile acids for chronic hepatitis C, Outcome 11 Serum AST (IU/L) at the end of treatment.
3.12. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 12 Serum GGT (IU/L) at the end of treatment.
3.13. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 13 Portal and periportal inflammation scores at the end of treatment.
3.14. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 14 Knodell score at the end of treatment.
3.15. Analysis.
Comparison 3 Bile acids for chronic hepatitis C, Outcome 15 Adverse events caused by bile acid.
Comparison 4. Bile acids for chronic viral hepatitis among heart transplanted patients.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All cause mortality at the end of treatment | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.33 [0.67, 8.18] |
| 2 Risk of abnormal serum ALT at the end of treatment | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.76, 1.22] |
| 3 Improvement of total Knodell score at the end of treatment | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.20, 1.05] |
| 4 Number of graft rejection at the end of treatment | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.78, 9.09] |
4.1. Analysis.
Comparison 4 Bile acids for chronic viral hepatitis among heart transplanted patients, Outcome 1 All cause mortality at the end of treatment.
4.2. Analysis.
Comparison 4 Bile acids for chronic viral hepatitis among heart transplanted patients, Outcome 2 Risk of abnormal serum ALT at the end of treatment.
4.3. Analysis.
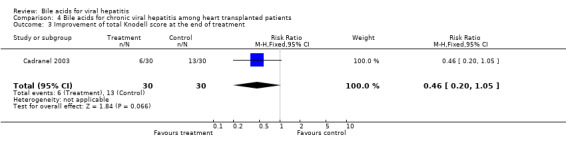
Comparison 4 Bile acids for chronic viral hepatitis among heart transplanted patients, Outcome 3 Improvement of total Knodell score at the end of treatment.
4.4. Analysis.

Comparison 4 Bile acids for chronic viral hepatitis among heart transplanted patients, Outcome 4 Number of graft rejection at the end of treatment.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Abdelmalek 1998.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Follow up: adequate ( UDCA plus IFN group (one patient); placebo plus IFN group (one patient). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: United States. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ outpatients; ‐ persistent serum ALT elevation (at least three times the upper normal limit) on two or more occasions during six months before enrolment; ‐ serum HCV RNA positive; ‐ negative for HBsAg and HIV; ‐ no history of exposure to hepatotoxins, alcohol intake greater than 20 g/day, or illicit drug use. EXCLUSION CRITERIA ‐ other causes of chronic liver disease; ‐ decompensated liver disease, severe concomitant medical or psychiatric illness, malignancy, untreated thyroid disease, or renal insufficiency; ‐ previous treatment with any type of IFN, UDCA, or immunosuppressive or antiviral agents within six months of entry into the study. PARTICIPANTS ‐ UDCA plus IFN group (n = 16): Mean age (years +/‐ SD) 42.5 +/‐ 1.8. Ratio of sex (M/F) 10/6. ‐ Placebo plus IFN group (n = 15): Mean age (years +/‐ SD) 46.2 +/‐ 3.1. Ratio of sex (M/F) 11/4. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 13‐15 mg/kg/day;
‐ Route: orally;
‐ Duration: six months.
Alpha IFN 2b
‐ Dose: three million units three times a week;
‐ Route: subcutaneously;
‐ Duration: six months. Placebo plus IFN group: Identical placebo. Alpha IFN‐2b ‐ Dose: three million units three times a week; ‐ Route: subcutaneously; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Decrease of at least three points in the Knodell score at the end of treatment. ‐ Absence of HCV RNA in serum at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: six months after the end of treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Angelico 1995a.
| Methods | Generation of allocation sequence: adequate (computer‐generated randomisation list). Allocation concealment: adequate (sealed envelopes). Double blinding: inadequate (not blinding). Follow up: adequate (one patient in each group). Sample size calculation: yes. Intention‐to‐treat analysis: yes. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ age between 18 and 65 years; ‐ persistent serum ALT elevation during the six months before the study, with value three times above the upper normal limit for at least three months; ‐ serum anti‐HCV positive; ‐ histological evidence of chronic active or persistent hepatitis within three months of entry, with or without cirrhosis. EXCLUSION CRITERIA ‐ grade B and C cirrhosis, according to Child‐Pugh classification; ‐ presence of HBsAg in serum; ‐ recent use of corticosteroids or previous IFN medications; ‐ recent history of alcohol or drug abuse; ‐ presence of autoimmune, genetic, or other types of liver diseases; ‐ platelet count below 100,000/µl and or leukocyte count below 3,000/µl; ‐ malignancies or renal, hematological, cardiopulmonary, neurological, and gastrointestinal diseases; ‐ marked abnormalities in liver function tests (serum bilirubin > 70 µmol/l and/or serum albumin < 30 g/l and/or prothrombin time < 50%); ‐ serum positivity for anti‐HIV antibody. PARTICIPANTS ‐ UDCA plus IFN group (n = 20): Mean age (years +/‐ SD) 49 +/‐ 14. Ratio of sex (M/F) 18/2. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN group (n = 20): Mean age (years +/‐ SD) 44 +/‐ 15. Ratio of sex (M/F) 16/4. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 10 mg/kg/day;
‐ Route: orally;
‐ Timing: two times a day;
‐ Duration: nine months and 15 days.
Alpha IFN 2a
‐ Dose: 3 million units three times a week;
‐ Route: subcutaneously;
‐ Duration: six months. IFN group: Alpha IFN‐2a ‐ Dose: three million units three times a week; ‐ Route: subcutaneously; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Absence of HCV RNA in serum at the end of treatment and follow‐up. ‐ Histological changes six months after cessation of IFN therapy. | |
| Notes | Follow‐up time: 18 months after stopping alpha IFN treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Belloni 1999.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate. Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA: ‐ HCV‐related chronic hepatitis; ‐ negative HBsAg; ‐no history for alcohol abuse, IVDA, use of hepatotoxic drugs and previous treatment with IFN; ‐ HCV antibodies. EXCLUSION CRITERIA: ‐ not described. PARTICIPANTS ‐ TUDCA group (n = 18): Mean age not stated. Ratio of sex (M/F) 13/5. Genotype 12 with genotype 1b, six with genotype 2a/2c. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Control group (n = 15): Mean age not stated. Ratio of sex (M/F) 8/7. Genotype six with genotype 1b, nine with genotype 2a/2c. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | TUDCA group:
TUDCA
‐ Dose:
500 mg/day;
‐ Route: orally;
‐ Timing: two times a day;
‐ Duration: four months. Control group: ‐ No treatment. |
|
| Outcomes | ‐ Serum ALT and GGT activities at the end of treatment. ‐ Serum HCV RNA level at the end of treatment. | |
| Notes | We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Bonnand 1996.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Sample size calculation: no information. Follow up: adequate (five patients from UDCA group, four patients from control group). Intention‐to‐treat analysis: no information. | |
| Participants | Country: France. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ anti‐HCV positive; ‐ chronic hepatitis; ‐ ALT activities persistently greater than two times the upper normal limit. EXCLUSION CRITERIA: not described. PARTICIPANTS ‐ UDCA plus IFN group (n = 47); ‐ IFN group (n = 44). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 13‐15 mg/kg/day;
‐ Route: orally;
‐ Duration: six months.
IFN
‐ Dose: three million units three times a week;
‐ Route: subcutaneously;
‐ Duration: six months. IFN group: ‐ Dose: three million units three times a week; ‐ Route: subcutaneously; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of ALT at the end of treatment and follow‐up. ‐ Absence of serum HCV at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: six months after the end of IFN treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Boucher 1995.
| Methods | Generation of allocation sequence: adequate (table of random permutations). Allocation concealment: unclear (no description). Double blinding: unclear (not blinding). Follow up: adequate (one patient from UDCA plus IFN group; four patients from IFN group). Sample size calculation: no information. Intention‐to‐treat analysis: yes. | |
| Participants | Country: France. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ age between 18 and 70 years old; ‐ persistent serum ALT elevation of 1.5 times or more of the upper normal limit for at least six months preceding treatment; ‐ serum anti‐HCV positive; ‐ absence of other causes of chronic active hepatitis; ‐ liver biopsy findings of chronic hepatitis; ‐ white blood cell count > 1500/µl and platelets count > 50,000/µl; ‐ no previous corticosteroid, immunosuppressive, or UDCA treatment. EXCLUSION CRITERIA ‐ history of decompensated chronic liver disease; ‐ marked abnormalities in liver function tests (serum bilirubin > 70 µmol/l and/or serum albumin < 30 g/l and/or prothrombin time < 50%) ‐ serum positivity for anti‐HIV antibody. PARTICIPANTS ‐ UDCA plus IFN group (n = 38): Mean age (years +/‐ SD) 49 +/‐ 14. Ratio of sex (M/F) 20/18. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 10.5%. ‐ IFN group (n = 42): Mean age (years +/‐ SD) 44 +/‐ 15. Ratio of sex (M/F) 26/16. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 13.5%. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 10 mg/kg/day;
‐ Route: orally;
‐ Timing: not described;
‐ Duration: nine months.
IFN
‐ Alpha IFN‐2b five million units three times a week for one month; three million units three times a week for the second month, and three or five million units during the last four months according to serum ALT. IFN group: ‐Alpha IFN‐2b five million units three times a week for one month; three million units three times a week for the second month, and three or five million units during the last four months according to serum ALT. |
|
| Outcomes | ‐ Normalisation of ALT at the end of treatment and follow‐up. ‐ HCV RNA negative at the end of treatment and follow‐up. ‐ Histological changes six months after cessation of IFN therapy. | |
| Notes | Follow‐up time: nine months after the end of the treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Cadranel 2003.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Sample size calculation: no information. Follow up: adequate. Intention‐to‐treat analysis: yes. | |
| Participants | Country: France. Type of hepatitis: chronic viral hepatitis. INCLUSION CRITERIA ‐ heart transplanted patients; ‐ biopsy proven chronic viral hepatitis; PARTICIPANTS ‐ UDCA group (n = 30). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Placebo group (n = 30). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA group: UDCA ‐ Dose: 800 mg/day; ‐ Route: orally; ‐ Duration: 12 months. Placebo group: ‐ Identical placebo for 12 months. | |
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | D ‐ Not used |
Clerici 1994.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: unclear (no information). Sample size calculation: no information. Follow up: unclear (no information). Intention‐to‐treat analysis: no information. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ anti‐HCV positive; ‐ chronic hepatitis; ‐ ALT activities persistently greater than two times the upper normal limit. PARTICIPANTS ‐ UDCA plus IFN group (n = 21). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN group (n = 20). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Duration: 18 months.
IFN
‐ Dose: 3 million units three times a week;
‐ Route: subcutaneously;
‐ Duration: six months. IFN group: ‐ Dose: three million units three times a week; ‐ Route: subcutaneously; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: 12 months after the end of IFN treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Crosignani 1998.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Follow up: adequate (nine patients from TUDCA group; six patients from placebo group). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA: ‐ histological and clinical diagnosis of HCV‐related chronic hepatitis; ‐ persistent abnormal serum ALT in two consecutive determinations during the four months before the study; ‐ contraindication or had no response to IFN. EXCLUSION CRITERIA: ‐ histological and/or clinical evidence of liver cirrhosis; ‐ presence of serological markers of HIV or HBV infection; ‐ recent history of drug abuse or alcohol intake higher than 40 g/day; ‐ use of corticosteroids or IFN within the previous six months; ‐ malignancies or renal, haematological, cardiopulmonary, neurological diseases; ‐ presence of liver tumours or extrahepatic biliary obstruction. PARTICIPANTS ‐ TUDCA group (n = 41): Mean age (years+/‐ SD) 51 +/‐ 7. Ratio of sex (M/F) 26/15. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 0. ‐ Placebo group (n = 44): Mean age (years +/‐ SD) 48 +/‐12. Ratio of sex (M/F) 35/9. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 0. | |
| Interventions | TUDCA group: TUDCA ‐ Dose: 750 mg/day; ‐ Route: orally; ‐ Timing: three times per day; ‐ Duration: six months. Placebo group: ‐ Placebo. | |
| Outcomes | ‐ Serum ALT, AST, and GGT activities at the end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Fabbri 2000.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: no information. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ serum HCV RNA positive; ‐ liver biopsy findings of chronic hepatitis; ‐ no response to alpha IFN treatment for four consecutive months; ‐ persistent elevations of serum ALT levels (at least 1.5‐fold the upper normal limit). EXCLUSION CRITERIA: ‐ HBV or HIV co‐infection; ‐ genetic liver disease; ‐ evidence of auto‐immune disorder (auto‐antibody titre greater than 1:40); ‐ malignancy, or renal, haematological, cardiac or pulmonary diseases, or was consuming alcohol. PARTICIPANTS ‐ UDCA plus IFN group (n = 53): Mean age (years +/‐ SD) 52.6 +/‐ 1.8. Ratio of sex (M/F) 34/19. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN group (n = 50): Mean age (years +/‐ SD) 45.8 +/‐ 1.8. Ratio of sex (M/F) 26/24. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | Alpha IFN 2b was administered to all patients at a dose of three million units three times weekly for four months firstly. UDCA plus IFN group: UDCA ‐ Dose: 600 mg/day; ‐ Route: orally; ‐ Timing: two times a day; ‐ Duration: 14 months. IFN Alpha IFN 2b ‐ Dose: three million units three times a week; ‐ Route: subcutaneously; ‐ Duration: eight months. IFN group: Alpha IFN 2b ‐ Dose: three million units three times a week; ‐ Route: subcutaneously; ‐ Duration: eight months. |
|
| Outcomes | ‐ Normalization of serum ALT at the end of treatment. ‐ Absence of serum HCV RNA at the end of treatment. ‐ Liver histology changes. | |
| Notes | Follow‐up time is six months after the end of treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Galský 1999.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Follow up: adequate (one in each group). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Czech Republic.
Type of hepatitis: acute hepatitis.
INCLUSION CRITERIA:
‐ elevation of ALT above three times the upper normal limit twice within the same week;
‐ serological confirmation of acute viral hepatitis A, B or C. EXCLUSION CRITERIA: ‐ other viral infections causing hepatitis, chronic hepatitis, alcoholic liver disease, drug‐induced liver injury; ‐ treatment with immunosuppressive or hepatoprotective agents; ‐ pregnancy. PARTICIPANTS ‐ UDCA group (n = 40): Mean age (years +/‐ SD) 41.8+/‐ 16.8. Ratio of sex (M/F) 20/20. Type of hepatitis HAV(4), HBV(34), HCV(1), HBV+HCV(1). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Control group (n = 38): Mean age (years +/‐ SD) 41.2 +/‐ 17.4; Ratio of sex (M/F) 19/19; Types of hepatitis. HAV(8), HBV (27), HCV (3). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. |
|
| Interventions | UDCA group:
UDCA
‐ Dose: 250 mg;
‐ Route: orally;
‐ Timing: three times per day;
‐ Duration: three months. Placebo group: ‐ Placebo. |
|
| Outcomes | ‐ Normalisation of serum ALT and GGT at the end of treatment and follow‐up. ‐ Absence of serum HBsAg at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: nine months after the end of treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, the principle author replied without applying the information we wanted. The results in this trial were based on the patients with acute hepatitis B. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Ge 1997.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: unclear (no information). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: China. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ abnormal serum ALT over six months; ‐ serum anti HCV and HCV RNA positivity; ‐ no other causes for chronic hepatitis. EXCLUSION CRITERIA: ‐ not described. PARTICIPANTS Mean age (years) 34.3 (15‐57). Ratio of sex (M/F) 32/16. 24 patients in each group. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 10 mg/kg/day;
‐ Route: orally;
‐ Duration: six months.
Alpha IFN
‐ Dose: three million units;
‐ Route: subcutaneously;
‐ Timing: three times per week;
‐ Duration: six months. IFN group: Alpha IFN ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. | |
| Notes | Follow‐up time:
six months after the end of treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Gracielle 2002.
| Methods | Generation of allocation sequence: adequate (block). Allocation concealment: adequate (sealed envelopes). Double blinding: inadequate (not blind). Follow up: adequate. Sample size calculation: calculated. Intention‐to‐treat analysis: stated and performed. | |
| Participants | Country: France.
Type of hepatitis: chronic hepatitis C.
INCLUSION CRITERIA
‐ Histological evidence of chronic hepatitis as judged on a liver biopsy performed no longer than 6 months prior to enrolment, and confirmation of HCV infection by R.I.B.A. second and third generation.
Exclusion criteria: age lower than 18 years or older than 65 years, pregnancy or lack of appropriate contraceptive measures in women of child bearing age, previous treatment with antiviral or immunosuppressive drugs, current or previous drug addiction, alcoholism, positive HBsAg or HIV testing, histological evidence of cirrhosis, concomitant metabolic, autoimmune or neoplastic liver diseases, severe concomitant diseases other than liver diseases, leukocyte count lower than 3000/dl, platelet count lower than 75000/dl and serum albumin lower than 3g/dl. PARTICIPANTS ‐ TUDCA plus IFN group (n = 55): Male/female 35/20; age 47+/‐20; body mass index 24.0 +/‐ 0.6; Post‐transfusional 16%; History surgery 18%; HCV genotype 1 68%; histological staging 1.6+/‐0.1. ‐ IFN group (n = 51). Male/female 29/22; age 47+/‐20; body mass index 24.7 +/‐ 0.6; Post‐transfusional 14%; History surgery 17%; HCV genotype 1 47%; histological staging 1.2+/‐0.1. |
|
| Interventions | TUDCA plus IFN group: TUDCA ‐ Dose: 10mg/kg/day; ‐ Route: orally; ‐ Duration: six months. IFN IFN alpha ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: six months. IFN group: IFN alpha ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: six months. | |
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Changes in liver histology. | |
| Notes | Follow‐up time: six months after the end of IFN treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | D ‐ Not used |
Huang 1997.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: unclear (no information). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: not used. | |
| Participants | Country: China. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ abnormal serum ALT over six months; ‐ both serum HCV RNA and HCV antibodies positivity; ‐ no other causes for liver diseases; ‐ no previous treatment with antiviral agents and/or corticosteroids and/or UDCA. EXCLUSION CRITERIA ‐ no described. PARTICIPANTS Mean age (years) 39.6 (18‐65). Ratio of sex (M/F) 48/24. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 10 mg/kg/day;
‐ Route: orally;
‐ Timing: not described;
‐ Duration: six months.
Alpha IFN
‐ Dose: three million units;
‐ Route: subcutaneously;
‐ Timing: three times per week;
‐ Duration: six months. IFN group: Alpha IFN ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ HCV RNA and HCV antibodies negativity at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: six months after the end of treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Kawata 1994.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: unclear (no information). Withdrawal: unclear (no information). Sample size calculation: no information. Intention‐to‐treat analysis: no information. | |
| Participants | Country: Japan. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ anti‐HCV positive; ‐ chronic hepatitis; ‐ ALT levels persistently greater than two times the upper normal limit. PARTICIPANTS UDCA plus IFN group (n = 35). IFN group (n = 35). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Duration: 48 weeks.
IFN
‐ IFN alpha 2a six million units a day for two weeks then thrice weekly for 22 weeks. IFN group: ‐ IFN alpha 2a six million units a day for two weeks then thrice weekly for 22 weeks. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Absence of serum HCV RNA at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: six months after the end of IFN treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Kiso 1997.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: unclear (no information). Follow up: adequate (one patient in each group). Sample size calculation: no information. Intention‐to‐treat analysis: yes. | |
| Participants | Country: Japan. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA: ‐ abnormal serum ALT activities (at least two times the upper normal limit) on more than one occasion during the three months prior to the enrolment; ‐ absence of HBs Ag; ‐ serum HCV RNA positive; ‐ absence of HBs Ag; ‐ liver biopsy findings of chronic hepatitis. EXCLUSION CRITERIA: ‐ other causes of chronic hepatitis. PARTICIPANTS ‐ IFN group (n = 40): Mean age (years +/‐ SD) 54.0 +/‐ 7.6. Ratio of sex (M/F) 24/16. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ UDCA plus IFN group (n = 40): Mean age (years +/‐ SD) 52.4 +/‐ 9.3; Ratio of sex (M/F) 27/13. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Duration: 48 weeks.
Alpha IFN
‐ six million units intramuscular injection (i.m) daily for two weeks, and then three times a week for 22 weeks. IFN group: Alpha IFN ‐ six million units. i.m. three times a week for six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Absence of serum HCV RNA at the end of treatment and follow‐up. ‐ Rate of sustained complete response in relation to liver histology. | |
| Notes | Follow‐up time:
six months after the end of treatment. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Leri 1994.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: unclear (no description). Sample size calculation: no. Follow up: adequate (no drop out). Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ anti‐HCV positive; ‐ liver biopsy scoring were assessed before the treatment and showed periportal necrosis, lobular and portal inflammation, according to chronic hepatitis. PARTICIPANTS ‐ UDCA group (n = 12): Mean age (years) 59.17. Ratio of sex (M/F) 5/7. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Placebo group (n = 10): Mean age (years) 56.40; Ratio of sex (M/F) 5/5. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: Orally;
‐ Timing: no information;
‐ Duration: six months. Placebo group: ‐ Placebo. |
|
| Outcomes | ‐ Serum AST, ALT, and GGT activities at the end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Picciotto 1994.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: adequate (sealed envelopes). Double blinding: inadequate (not blinding). Sample size calculation: no information. Follow up: adequate (two patients in each group). Intention‐to‐treat analysis: yes. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA: ‐ anti‐HCV positive; ‐ biopsy‐proven chronic hepatitis; ‐ ALT activities persistently greater than two times the upper normal limit. EXCLUSION CRITERIA: ‐ autoimmune hepatitis, alcoholic‐ or drug‐related liver diseases, hemochromatosis, Wilson's disease, or alpha‐1‐antitrypsin deficiency; ‐ bilirubin levels > three mg/dl, albumin levels< three g/dL, prothrombin time > three seconds longer than that of normal value, serum creatinine levels > 1.7 mg%, platelet count < 100,000/µl, granulocyte count < 1500/µl. PARTICIPANTS ‐ TUDCA plus IFN group (n = 30): Mean age (years+/‐ SD) 49.4+/‐11.3. Ratio of sex (M/F) 23/7. ‐ IFN group (n = 30): Mean age (years+/‐ SD) 49.5+/‐16; Ratio of sex (M/F) 21/9. | |
| Interventions | TUDCA plus IFN group:
TUDCA
‐ Dose: 500 mg/day;
‐ Route: orally;
‐ Timing: two times a day;
‐ Duration: nine months.
IFN
IFN alpha‐2b
‐ Dose: three million units;
‐ Route: subcutaneously;
‐ Timing: three times per week;
‐ Duration: six months. IFN group: IFN alpha‐2b ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: three months after the end of IFN treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Poupon 2000.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Follow up: adequate (five and four patients withdrew from the combination and the monotherapy groups, respectively). Sample size calculation: no. Intention‐to‐treat analysis: yes. | |
| Participants | Country: France. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ age between 18 and 65 years; ‐ persistent serum ALT elevation higher than 1.5 times the upper normal limit for more than six months; ‐ serum anti‐HCV positive; ‐ histological confirmation in the year preceding inclusion; ‐ resistant to IFN on the basis of having received IFN at a dosage of at least three million units three times weekly for at least six months. EXCLUSION CRITERIA ‐ autoimmune hepatitis type I or type II, other autoimmune diseases; ‐ associated liver diseases, other serious illnesses, signs of intolerance to IFN during previous treatment, haemophilia and other constitutional coagulation disorders, serum creatinine higher than 150 µmol/l, polymorphonuclear neutrophils lower than 1000/µl, platelets lower than 80,000/µl, signs of decompensated cirrhosis; alcohol consumption more than 40 g/day; history of psychiatric illness or treatment with antidepressant drugs; human immunodeficiency seropositivity; drug addiction in previous year; pregnancy, and diabetes requiring treatment with oral antidiabetic drugs or insulin. PARTICIPANTS ‐ UDCA plus IFN group (n = 47): Mean age (years +/‐ SD) 45 +/‐ 2. Ratio of sex (M/F) 37/10. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 14 patients. ‐ Placebo plus IFN group (n = 44): Mean age (years +/‐ SD) 52 +/‐ 1. Ratio of sex (M/F) 32/12. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 14 patients. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 13‐15 mg/kg/day;
‐ Route: orally;
‐ Timing: two times per day;
‐ Duration: six months.
Alpha IFN 2a
‐ Dose: three million units;
‐ Route: subcutaneously;
‐ Timing: three times per week;
‐ Duration: six months. Placebo plus IFN group: Alpha IFN‐2a ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Serum ALT, AST, GGT, and alkaline phosphatases activities and serum total bilirubin concentrations at the end of treatment and follow‐up. ‐ Absence of HCV RNA in serum at the end of treatment and follow‐up. ‐ Histological changes six months after cessation of IFN. | |
| Notes | Follow‐up time: six months after stopping alpha IFN treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Puoti 1995.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA: ‐ positive for anti‐HCV antibodies; ‐ biopsy‐proven or biochemical and clinical evidence of chronic liver disease; ‐ serum ALT levels at least twice the upper limit of the normal range on three different occasions in the last 12 months. EXCLUSION CRITERIA: ‐ other forms of chronic liver disease; ‐ history of alcohol abuse; ‐ presence of severe liver disease and concomitant treatment with drugs which could interfere with hepatic metabolism or with bile acid absorption; ‐ previous interferon treatment; ‐ positive to HIV antibodies or HBs Ag; ‐ presence of hepatic tumours. PARTICIPANTS 101 outpatients (52 men and 49 women); mean age 45.1 years old. ‐ UDCA group (n = 49): Ratio of sex (M/F) 25/24. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 13 patients. ‐ Control group (n = 52): Ratio of sex (M/F) 27/25. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: 14 patients. | |
| Interventions | UDCA group:
UDCA
‐ Dose: 450 mg/day;
‐ Route: orally;
‐ Timing: single bedtime dose;
‐ Duration: six months. Control group ‐ No treatment. |
|
| Outcomes | ‐ Serum ALT and GGT activities at the end of treatment. ‐ Normalisation of serum ALT and GGT at the end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Qing 1999.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: China. Type of hepatitis: chronic hepatitis B or C. INCLUSION CRITERIA ‐ the 5th China Clinical Diagnosis Guideline for Infectious Diseases in 1995. EXCLUSION CRITERIA ‐ not described. PARTICIPANTS ‐ UDCA group (n = 30): Mean age (years +/‐ SD) 42.7+ / ‐ 12.3. Ratio of sex (M/F) 25/5. Mean time of infection 5.3 years. Genotypes of virus: HCV(1) and HBV(29). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Control group (n = 30): Mean age (years) 44.1. Ratio of sex (M/F) 24/6. Mean time of infection 6.3 years. Genotypes of virus: HCV (2) and HBV (28). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA group:
UDCA
‐ Dose: 450 mg/day;
‐ Route: orally;
‐ Timing: three times per day;
‐ Duration: three months. Control group: ‐No special treatment. |
|
| Outcomes | ‐ Serum ALT, AST, GGT, and alkaline phosphatases activities at the end of treatment. ‐ Absence of serum HBeAg, HBe antibodies, and HCV antibodies at the end of treatment. | |
| Notes | We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Scotto 1997.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (eight patients from UDCA group, five patients from control group). Sample size calculation: no information. Intention‐to‐treat analysis: no. | |
| Participants | Country: Italy. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA: ‐ increase in serum ALT activities three‐fold higher than normal values over the previous six months; ‐ no other cause of chronic liver diseases (viral hepatitis B, Wilson's disease, autoimmune hepatitis, etc.); ‐ positivity for AB‐HCV; III generation confirmatory test; HCV‐RNA; ‐ histologic pattern of either chronic hepatitis or compensated cirrhosis, according to international standard criteria; ‐ no previous antiviral treatment; ‐ exclusion of subjects with anti‐HIV positivity and drug addicts. PARTICIPANTS ‐ UDCA group (n = 37): Means age (years) 47.7. Ratio of sex (M/F) 26/11. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: five patients. Placebo group (n = 37): Means age (years) 48.1. Ratio of sex (M/F) 25/12. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: four patients. | |
| Interventions | UDCA group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Timing: twice a day;
‐ Duration: 12 months. Control group: ‐ placebo for 12 months. |
|
| Outcomes | ‐ Serum ALT, AST, and GGT activities at the end of treatment and follow‐up. ‐ Absense of HCV RNA at the end of treatment. | |
| Notes | Follow‐up time: six months after the end of treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | D ‐ Not used |
Senturk 1997.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (six patients from UDCA plus IFN group; four patients from IFN group). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Turkey. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ persistent serum ALT elevation of two times or more of the upper normal limit for at least six months preceding treatment; ‐ serum anti‐HCV and HCV RNA positive; ‐ absence of HBsAg; ‐ liver biopsy findings of chronic hepatitis. EXCLUSION CRITERIA ‐ patients with signs of decompensated cirrhosis such as ascites, oesophageal varices, and hepatic encephalopathy. PARTICIPANTS ‐ UDCA plus IFN group (n =45): Mean age (years) 50 (19‐69). Ratio of sex (M/F) 22/23. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN group: Mean age (years) 46 (23‐73). Ratio of sex (M/F) 23/26. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 500 mg/day;
‐ Route: orally;
‐ Timing: two times per day;
‐ Duration: six months.
Alpha IFN
‐ Dose: three million units;
‐ Route: no described;
‐ Timing: three times per week;
‐ Duration: six months. IFN group: Alpha IFN ‐ Dose: three million units; ‐ Route: no described; ‐ Timing: three times per week; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of six months treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Takano 1994.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Sample size calculation: no information. Follow up: unclear (no information). Intention‐to‐treat analysis: no information. | |
| Participants | Country: Japan. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ serum anti‐HCV antibody positive at least three times. EXCLUSION CRITERIA ‐ liver dysfunction was a result of other reasons (e.g., the use of hepatotoxic drugs or severe right‐sided heart failure); ‐ serum ALT was normal within three months before the initiation of treatment; ‐ a history of hepatic encephalopathy, bleeding oesophageal varices or ascites; ‐ receiving immunomodulatory or antiviral agents within six months before the start of treatment; ‐ with lesions in the liver, except benign hemangioma. PARTICIPANTS ‐ Low dose UDCA group (n = 21). Mean age (years) 54.3. Ratio of sex (M/F) 13/8. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Intermediate dose UDCA group (n = 18). Mean age (years) 57.7. Ratio of sex (M/F) 9/9. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ High dose UDCA group (n = 19). Mean age (years) 51.8. Ratio of sex (M/F) 14/5. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Control group (n = 17). Mean age (years) 53.7. Ratio of sex (M/F) 15/2. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | Low dose UDCA group:
UDCA
‐ Dose: 150 mg/day;
‐ Route: orally;
‐ Duration: 16 weeks. Intermediate dose UDCA group: UDCA ‐ Dose: 600 mg/day; ‐ Route: orally; ‐ Duration: 16 weeks. High dose UDCA group: UDCA ‐ Dose: 900 mg/day; ‐ Route: orally; ‐ Duration: 16 weeks. Control group: ‐ No treatment. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment. ‐ Adverse events at the end of treatment. | |
| Notes | We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Tanaka 1996.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (no information). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: Japan. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ serum HCV RNA positive at least three times. ‐ liver biopsy findings of chronic hepatitis. EXCLUSION CRITERIA ‐ previously received corticosteroid, immunosuppressive, UDCA or antiviral treatment; ‐ history of alcohol or drug abuse or evidence of metabolic or autoimmune disorders. PARTICIPANTS ‐ UDCA plus IFN group (n = 26): Mean age (years +/‐ SD) 53 +/‐ 10; Ratio of sex (M/F) 14/12. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN group (n =26): Mean age (years +/‐ SD) 49 +/‐ 10. Ratio of sex (M/F) 17/9. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Timing: not described;
‐ Duration: 18 months.
Alpha IFN
‐ six million units intramuscular injection daily for two weeks, and then three times a week for 22 weeks. IFN group: Alpha IFN ‐ six million units intramuscular injection daily for two weeks, and then three times a week for 22 weeks. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Absence of serum HCV RNA negativity at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: 12 months. We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Tsubota 1999.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: adequate (sealed, opaque, numbered envelopes). Double blinding: inadequate (not blinding). Follow up: adequate (one patient from UDCA plus glycyrrhizin group; two patients from glycyrrhizin group). Sample size calculation: no information. Intention‐to‐treat analysis: not used. | |
| Participants | Country: Japan. Type of hepatitis: chronic hepatitis C INCLUSION CRITERIA: ‐ persistent serum ALT elevation (at least 1.5 times the upper normal limit) on two or more occasions for at least six months before enrolment; ‐ liver biopsy evidence for chronic hepatitis; ‐ serum HCV RNA and anti‐HCV antibodies positive; ‐ age between 20 and 70 years old. EXCLUSION CRITERIA: ‐ liver cancer or severe liver failure; ‐ any other form of liver disease, coexistence of any other serious medical illness; ‐ previous course of IFN therapy or any other antiviral or immunomodulant therapy administered within the previous year; ‐ HBsAg positive; ‐ pregnancy or lactation. PARTICIPANTS ‐ UDCA plus glycyrrhizin group (n = 83): Mean age (years +/‐ SD) 56.3 +/‐ 10.8. Ratio of sex (M/F) 57/26. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Glycyrrhizin group (n = 84): Mean age (years +/‐ SD) 58.7 +/‐ 8.1. Ratio of sex (M/F) 47/37. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus glycyrrhizin group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Timing: three times per day;
‐ Duration: six months.
Glycyrrhizin
‐ 100 ml three times weekly intravenous injection for six months. Glycyrrhizin group: Glycyrrhizin ‐ 100 ml three times weekly intravenous injection for six months. |
|
| Outcomes | ‐ Serum ALT, AST and GGT activities at the end of treatment. ‐ Changes in serum HCV RNA level. ‐ Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Viola 1996.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Withdrawal: no. Sample size calculation: no information. Intention‐to‐treat analysis: yes. | |
| Participants | Country: Argentina. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ chronic hepatitis C; ‐ persistent ALT elevation. EXCLUSION CRITERIA ‐ not described. PARTICIPANTS ‐ UDCA plus IFN group (n = 23). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN group (n = 27). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Duration: six months.
Alpha IFN 2b
‐ Dose: three million units;
‐ Route: subcutaneously;
‐ Timing: three times per week;
‐ Duration: six months. IFN group: Alpha IFN 2b ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: six months. |
|
| Outcomes | ‐ Normalisation of serum ALT and AST activities at the end of treatment. ‐ Absence of serum HCV RNA at the end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Viola 1998.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: adequate (identical placebo). Sample size calculation: no information. Follow up: unclear (no information). Intention‐to‐treat analysis: yes. | |
| Participants | Country: Argentina. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ anti‐HCV and HCV RNA positive; ‐ chronic hepatitis. PARTICIPANTS ‐ UDCA plus IFN group (n = 42). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ IFN plus placebo group (n = 46). Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA plus IFN group:
UDCA
‐ Dose: 600 mg/day;
‐ Route: orally;
‐ Duration: 12 months.
IFN
IFN alpha 2b
‐ Dose: three million units;
‐ Route: subcutaneously;
‐ Timing: three times per week;
‐ Duration: 12 months. IFN plus placebo group: IFN alpha 2b ‐ Dose: three million units; ‐ Route: subcutaneously; ‐ Timing: three times per week; ‐ Duration: 12 months. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment. ‐ Absence of serum HCV RNA at the end of treatment. | |
| Notes | We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Zhou 1995.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: China. Type of hepatitis: chronic hepatitis B. INCLUSION CRITERIA ‐ China Clinical Diagnosis Guideline for Viral Hepatitis in 1990; ‐ Serum ALT un‐normalisation over 12 months; ‐ Serum HBsAg positivity. EXCLUSION CRITERIA: ‐ not described. PARTICIPANTS ‐ UDCA group (n = 64): Age (years) 23‐45. Duration of infection 1‐2 years. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Control group (n = 48): Age (years) 22‐45. Duration of infection: 1‐2 years. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA group:
UDCA
‐ Dose: 400 mg/day;
‐ Route: orally;
‐ Timing: four times per day;
‐ Duration: three months. Control group ‐ No specific treatment. |
|
| Outcomes | ‐ Normalisation of serum ALT at the end of treatment and follow‐up. ‐ Serum ALT activities at the end of treatment and follow‐up. ‐ HBV serum antibodies (Ig G, Ig M) and antigens (HBs Ag and HBe Ag) titers changes at the end of treatment and follow‐up. | |
| Notes | Follow‐up time: three months after the end of treatment.
We sent letter for more information on trial methodological quality and missing data on April 17, 2001, but no response was received. The trial was described as randomised, but it is not explained why they randomised 64 patients to the UDCA arm and 48 patients to the control arm. We have inquired about this, but no response has been obtained from the authors. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Zhu 1994.
| Methods | Generation of allocation sequence: unclear (no description). Allocation concealment: unclear (no description). Double blinding: inadequate (not blinding). Follow up: adequate (no drop out). Sample size calculation: no information. Intention‐to‐treat analysis: not performed. | |
| Participants | Country: China. Type of hepatitis: chronic hepatitis C. INCLUSION CRITERIA ‐ abnormal serum ALT over six months; ‐ serum HCV IgM or HCV RNA positivity. EXCLUSION CRITERIA ‐ chronic hepatitis caused by other viruses. PARTICIPANTS ‐ UDCA group (n = 34): Mean age (years) 35. Ratio of sex (M/F) 34/0. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. ‐ Control group (n = 29): Mean age (years) 37. Ratio of sex (M/F) 29/0. Proportion of patients with cholestasis: no information. Proportion of patients with cirrhosis: no information. | |
| Interventions | UDCA group:
UDCA
‐ Dose: 450 mg/day;
‐ Route: orally;
‐ Timing: three times per day;
‐ Duration: three months. Control group: ‐ No specific treatment. |
|
| Outcomes | ‐ Normalisation of serum ALT activities and total bilirubin concentrations at the end of treatment. ‐ Serum IgM anti HCV and HCV RNA negativity at the end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
ALT: alanine aminotransferase. AST: aspartate aminotransferase. GGT: gamma glutamyltranspeptidase. HIV: human immunodefeciency virus. IFN: interferon. IVDA: intravenous drug abuse. TUDCA: tauro‐ursodeoxycholic acid. UDCA: ursodeoxycholic acid.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Angelico 1995b | This study is a randomised cross‐over clinical trial. Forty anti‐HCV positive patients were randomised to receive IFN alpha, from day 0 to 180, plus TUDCA, from day 61 to 240; or TUDCA, from day 0 to 180, plus IFN alpha from day 61 to 240. The two interventions did not differ significantly. The trial does not fulfill our inclusion criteria. |
| Attilli 1994 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. Thirty‐six patients with chronic active hepatitis were included. Patients were randomly allocated to receive 600 mg UDCA per day or placebo. Clinical and biochemical follow‐up was performed at three‐month intervals. A percutaneous liver biopsy was performed before and after one year of treatment. Multifactorial covariance analysis showed that the reductions in ALT, AST, and GGT were significantly higher in UDCA group than in the placebo group. Biochemical remission was not observed in either group. No differences were found in the two groups before or after treatment in histological activity index. |
| Buzzelli 1991 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. A total of 40 patients with chronic active hepatitis were randomised to oral UDCA 300 mg two times a day or S‐adenosyl‐methionine (200 mg two times a day) for six months. Treatment with UDCA produced a statistically significant reduction in ALT, AST, and GGT compared with SAMe. |
| Buzzelli 1992 | This study is a randomised clinical trial, but the type of viral hepatitis can not be identified. A total of 36 patients with chronic hepatitis B and/or C were included. Eighteen were randomised to 300 mg of UDCA‐hemisuccinate orally twice a day for six months and 18 randomised to 200 mg of S‐adenosyl‐methionine twice a day for six months. Treatment with UDCA produced a statistically significant reduction in ALT, AST and GGT compared with SAMe. |
| Crosignani 1991 | This study is not a randomised clinical trial, but a case series. The effects of UDCA on liver function tests and on bile acid metabolism were investigated in 18 patients with chronic active hepatitis. Three different doses of UDCA ‐ 250 mg, 500 mg, and 750 mg ‐ were administered daily to each patient for two months. A significant decrease in serum aminotransferase occurred with the lowest dose of UDCA, which corresponded to four mg/kg body weight/day, and no further significant decrease in serum aminotransferases with the higher doses was seen. |
| Crosignani 1998(a) | This study is a randomised clinical trial, but the type of viral hepatitis can not be identified. One hundred and fifty‐five patients with chronic active hepatitis were randomly assigned to receive TUDCA at the daily doses of 250, 500, and 1000 mg, or no treatment for six months. Serum aminotransferase and GGT activities decreased with each dose of TUDCA compared with controls (P < 0.001). The 1000 mg dose was followed by more marked improvement compared with the 250 mg dose (P < 0.05). An improvement of ALT with time (P < 0.05) was found only with the two higher doses. |
| Del Vecchio 1982 | This study is a randomised clinical trial, but the type of viral hepatitis can not be identified. Forty‐four in‐patients with low activity chronic hepatitis were included in this study. Patients were randomly allocated to receive in a double‐blind way UDCA (10 mg/kg/day) or placebo in divided doses for three months. In both groups there was a significant improvement in all biochemical variables but without a statistically significant difference between treatments. |
| Italian 1990 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. Sixty‐three chronic active hepatitis patients were enrolled in three participating centres: 34 received UDCA and 29 placebo. Serum AST and GGT activities were significantly reduced (P < 0.05 and P < 0.01, respectively) during the treatment with UDCA but not with placebo. Liver histology remained substantially unchanged in terms of periportal necrosis, intralobular degeneration, portal inflammation, and fibrosis in patients treated with UDCA or placebo. |
| Kadayifci 1997 | This study is a case‐report. A 38‐year‐old man had pruritus and jaundice of eight weeks, initially compatible with acute hepatitis B. The patient took 10 mg/kg bodyweight/day UDCA, and the symptoms began to improve after two weeks. Bilirubin levels also gradually reduced. After 14 weeks of UDCA therapy, bilirubin levels and all other laboratory parameters returned to normal, hepatitis B surface antigen disappeared, and hepatitis B antibody became positive. |
| Lu 1995 | This study is not a randomised clinical trial, but a case‐control study. It was conducted to evaluate the efficacy of UDCA in the treatment of Chinese patients with chronic hepatitis C. Patients who failed to have sustained responses to IFN therapy, refused to take IFN, or were unsuitable for IFN treatment, were enrolled. Fifteen patients received UDCA 600 mg orally per day for six months. Another fifteen patients were chosen as the control group. After the treatment period, the mean serum ALT activities in both groups were not significantly different and mean serum ALT activities in the UDCA‐treated group did not decrease after the treatment. |
| Pinto 1992 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. Forty patients (15 M, 25 F) with biopsy proven chronic liver disease were randomly allocated to two treatment groups. Twenty patients received UDCA 600 mg/day and 20 patients received placebo for six months. Serum ALT activities were significantly reduced in the treated group when compared with placebo group at the end of treatment. |
| Podda 1989 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. Forty‐eight patients (30 with PBC, six with PSC and 12 with chronic hepatitis) were included in this trial. UDCA was administered at dosages of 250, 500, and 750 mg/day for two months. Highly significant decreases in serum aminotransferase activities were observed in all groups, but there is no significant difference for the decrease in serum aminotransferase activities between the 500 and the 750 mg/day doses. |
| Podda 1990 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. The trial compared the effects of UDCA, taurine, or a combination of the two on indices of liver injury in 24 patients with chronic active hepatitis. They were assigned at random to two of the four following treatments: UDCA (600 mg/day), taurine (1.5 g/day), UDCA (600 mg/day) plus taurine (1.5 g/day) or placebo, given in two successive cycles of two months each. UDCA and the combination of UDCA and taurine significantly decreased serum AST, ALT, and GGT at the end of treatment when compared with taurine alone. |
| Podda 1995 | This study is a randomised clinical trial, but the type of viral hepatitis can not be identified. In seven Italian centres, 155 patients with histological diagnosis of chronic active hepatitis were enrolled. They were randomly assigned to receive a six‐months course of TUDCA at the daily doses of 250 mg, 500 mg, 1000 mg, or no treatment. After six months of treatment, AST, ALT, and GGT decreased in patients administered TUDCA compared to patients receiving no treatment (P < 0.001). |
| Portincasa 1993 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. A total of 53 patients with histologic evidence of chronic active hepatitis were enrolled in the study. TUDCA 500 mg/day divided into two doses with meals was given to 27 patients; 26 patients served as controls. Follow‐ups were performed for one month, two months, and the end of the study period. TUDCA significantly lowered AST (‐44%), ALT (‐49%), and GGT(‐38%). Throughout the study, serum levels of alkaline phosphatases and serum bilirubin concentration remained within normal range in all patients. |
| Rolandi 1991 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. Twenty‐six patients with serum ALT values at least twice the upper normal limit in two of three pre‐treatment tests received UDCA 450 mg/day or a placebo for twelve weeks. In all UDCA‐treated patients, serum AST, ALT, GGT and alkaline phosphatases fell significantly after four weeks of treatment when compared with patients in placebo group. Four weeks after suspension of therapy, there was no significant difference between UDCA group and placebo group in serum aminotransferase. |
| Song 1998 | This study is a randomised clinical trial, but the type of viral hepatitis cannot be identified. Seventy‐nine patients with chronic active hepatitis were divided into two groups. Forty‐two patients in the treatment group received UDCA 300‐450 mg a day and thirty‐seven patients in control group received no treatment. The treatment group improved the serum GGT activities significantly when compared to the control group. |
| Zhu 1997 | This study is not a randomised clinical trial, but a control series. Fifty‐three patients with chronic hepatitis C received UDCA 600 mg a day with the combination of ribaviram 900 mg a day and polyinosine polycytidylic acid four mg once in two days. Forty‐seven patients with chronic hepatitis C received ribaviram 900 mg a day and polyinosine polycytidylic acid four mg once in two days as control group. A significant improvement on biochemical response at the end of treatment in UDCA group was observed when compared with the control group. |
ALT: alanine aminotransferase. AST: aspartate aminotransferase. GGT: gamma glutamyltranspeptidase. IFN: interferon. TUDCA: tauro‐ursodeoxycholic acid. UDCA: ursodeoxycholic acid.
Contributions of authors
Wendong Chen drafted and revised the protocol and the review, co‐developed the search strategy, and performed handsearches and data extraction. Jianping Liu developed the search stategy, extracted data, and revised the protocol and the review. Christian Gluud formulated the idea for the review, evaluated the data extraction, and revised the protocol and the reveiw.
Sources of support
Internal sources
Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Denmark.
External sources
The Danish Medical Research Council's Grant on Getting Research into Practice (GRIP), Denmark.
Declarations of interest
None known.
Edited (no change to conclusions)
References
References to studies included in this review
Abdelmalek 1998 {published data only}
- Abdelmalek MF, Harrison ME, Gross JB, Poterucha JJ, Gossard AA, Spivey JR, et al. Treatment of chronic hepatitis C with interferon with or without ursodeoxycholic acid. Journal of Clinical Gastroenterology 1998;26(2):130‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Angelico 1995a {published data only}
- Angelico M, Gandin C, Pescarmona E, Rapicetta M, Vecchio C, Goffredo F. Recombinant interferon‐alpha and ursodeoxycholic acid versus interferon‐alpha alone in the treatment of chronic hepatitis C: a randomized clinical trial. Italian Journal of Gastroenterology 1993; Vol. 25:446. [PubMed]
- Angelico M, Gandin C, Pescarmona E, Rapicetta M, Vecchio CD, Bini A, et al. Recombinant interferon alpha and ursodeoxycholic acid versus interferon alpha alone in the treatment of chronic hepatitis C: a randomized clinical trial with long‐term follow‐up. The American Journal of Gastroenterology 1995;90(2):263‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Belloni 1999 {published data only}
- Belloni G, Ferrari L, Balduzzi C, Barbieri F, Insolia R, Klersy C, et al. Effect of tauroursodeoxycholic acid on ALT, GGT and viral load in patients with hepatitis C virus‐related chronic hepatitis with reference to the genotype. The Italian Journal of Gastroenterology and Hepatology 1999;31(5):427. [MEDLINE: ] [PubMed] [Google Scholar]
Bonnand 1996 {published data only}
- Bonnand AM, Poupon R, Queneau PE, Trepo C, Cohard M, Vetter D, et al. Interferon alfa and ursodeoxycholic acid combination versus interferon alone in the treatment of chronic hepatitis C: Results of a randomized trial [AASLD abstract]. Hepatology 1996;24(4 Pt.2):403A. [Google Scholar]
Boucher 1995 {published data only}
- Boucher E, Andre P, Jouanolle H, Moirand R, Guyader D, Ruffault A, et al. Treatment of chronic viral C hepatitis by interferon plus ursodeoxycholic acid: results from a controlled randomized trial in 80 patients. Hepatology 1994; Vol. 20, issue 4:169A. [PubMed]
- Boucher E, Jouanolle H, Andre P, Ruffault A, Guyader D, Moirand R, et al. Interferon and ursodeoxycholic acid combined therapy in the treatment of chronic viral C hepatitis: results from a controlled randomized trial in 80 patients. Hepatology 1995;21(2):322‐7. [MEDLINE: ] [PubMed] [Google Scholar]
Cadranel 2003 {published data only}
- Cadranel JF, Martino V, Dorent R, Bernard B, Hoang C, Myara A, et al. Effects of ursodeoxycholic acid (ursodiol) treatment on chronic viral hepatitis in heart transplant patients: results of a prospective, double‐blind, placebo‐randomized study. Transplantation 2003;75(7):977‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Clerici 1994 {published data only}
- Clerici C, Distrutti E, Gentili G, Solinas A, Miglietti M, Balo S. Interferon plus ursodeoxycholic acid versus interferon in the treatment of chronic C viral hepatitis. Journal of Hepatology 1994;21(Suppl 1):S102. [PubMed] [Google Scholar]
Crosignani 1998 {published data only}
- Crosignani A, Budillon G, Cimino L, Vecchio Blanco C, Loguercio C, Ideo G, et al. Tauroursodeoxycholic acid for the treatment of HCV‐related chronic hepatitis: a multicenter placebo‐controlled study. Hepato‐Gastroenterology 1998;45(23):1624‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Fabbri 2000 {published data only}
- Fabbri C, Marchetto S, Pezzoli A, Accogli E, Fusaroli E, Azzaroli F, et al. Efficacy of ursodeoxycholic acid in association with interferon alpha for chronic hepatitis C in interferon alpha non‐responder patients. European Journal of Gastroenterology & Hepatology 2000;12(5):511‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Fabbri C, Mazzella G, Marchetto A, Pezzoli E, Accogli P, Fusaroli F, et al. Efficacy of ursodeoxycholic acid administration as adjuvant therapy together with alfa‐interferon for chronic hepatitis C. Journal of Hepatology 1997; Vol. 26:S179. [DOI] [PubMed]
Galský 1999 {published data only}
- Galský J, Bansky G, Holubova T, Konig J. Effect of ursodeoxycholic acid in acute viral hepatitis. Journal of Clinical Gastroenterology 1999;28(3):249‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ge 1997 {published data only}
- Ge Q, Huang Y. Combined interferon and ursodeoxycholic acid therapy in 48 chronic hepatitis C patients [(original title in Chinese)]. Journal of New Gastroenterology 1997;5(5):338‐9. [Google Scholar]
Gracielle 2002 {published data only}
- Gracielle P, Roberta S, Ornella B, Luciana D, Alessandro R, Daniela Q, et al. A controlled randomised trial of t‐UDCA as adjuvant to interferon for treatment of chronic hepatitis C: an interferon sparing effect of t‐UDCA. Hepatology Research 2002;23(4):227‐36. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Huang 1997 {published data only}
- Huang Y, Huang T, Ge Q. Ursodeoxycholic acid therapy in chronic hepatitis C patients [(original title in Chinese)]. Acta Academic Medicine CPAPF (Chinese People Armed Police Force) 1997;5(4):32‐3. [Google Scholar]
Kawata 1994 {published data only}
- Kawata S, Imai Y, Tamura S, Inada M, Ito N, Inui Y, et al. Efficacy of combination therapy of interferon with ursodeoxycholic acid in chronic hepatitis C: sustained clearance of viremia after interferon discontinuation. Hepatology 1994;20(4):363A. [Google Scholar]
Kiso 1997 {published data only}
- Kiso S, Kawata S, Tamura S, Imai Y, Inui Y, Nagase T, et al. Efficacy of combination therapy of interferon alpha with ursodeoxycholic acid in chronic hepatitis C: a randomized controlled clinical trial. Journal of Gastroenterology 1997;32(1):56‐62. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Leri 1994 {published data only}
- Leri O, Luca D, Sinopoli MT, Parparo BS, Tubili S, Calaf HN, et al. Ursodeoxycholic acid, interferon‐alfa, versus interferon‐alfa + ursodeoxycholic acid in chronic C hepatitis. Mediterranean Journal of Infectious and Parasitic Diseases 1994;9:15‐8. [Google Scholar]
Picciotto 1994 {published data only}
- Picciotto A, Savarino V, Bardellini E, Borzone S, Pireddu M, Sinelli N, et al. Effect of therapy with combined interferon and tauroursodeoxycholic acid in chronic hepatitis C: biochemical and virologic evaluation. Current Therapeutic Research 1994;55(6):675‐81. [Google Scholar]
Poupon 2000 {published data only}
- Poupon RE, Bonnand AM, Queneau PE, Trepo C, Zarski JP, Raabe JJ. Randomized trial of interferon‐alpha plus ursodeoxycholic acid versus interferon plus placebo in patients with chronic hepatitis C resistant to interferon. Scandinavian Journal of Gastroenterology 2000;35(6):642‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Puoti 1995 {published data only}
- Puoti C, Magrini A, Filippi T, Annovazzi G, Pannullo A. Effects of ursodeoxycholic acid on serum liver enzymes in patients with hepatitis C virus‐related chronic liver disease. European Journal of Gastroenterology & Hepatology 1995;7(2):151‐4. [MEDLINE: ] [PubMed] [Google Scholar]
- Puoti C, Pannullo A, Annovazzi G, Filippi T, Magrini A. Ursodeoxycholic acid and chronic hepatitis C infection. The Lancet 1993; Vol. 341:1413‐4. [DOI] [PubMed]
Qing 1999 {published data only}
- Qing Y, Chen Y, Ma L, Liu L, Xie W, Tu X, et al. The effeicacy of ursodeoxycholic acid on chronic hepatitis [(original title in Chinese)]. Journal of North Sichuan Medical College 1999;14(1):48‐50. [Google Scholar]
Scotto 1997 {published data only}
- Scotto G. The ursodeoxycholic acid for the treatment of HCV infections [L'acido ursodeossicolico per il trattamento delle infezioni da HCV]. Infezioni in Medicine 1997;3:168‐73. [PubMed] [Google Scholar]
Senturk 1997 {published data only}
- Senturk H, Uzunalimoglu O, Batur Y, Simsek I, Mert A, Ozbay G, et al. Long‐term efficacy of interferon alpha and ursodeoxycholic acid in treatment of chronic type C hepatitis. Digestive Diseases and Sciences 1997;42(7):1438‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Takano 1994 {published data only}
- Takano S, Nakamura K, Yokosuka O, Hirota K, Omata M. A multi‐center randomized controlled dose study of ursodeoxycholic acid for chronic hepatitis C. Hepatology 1994;19(4):1291. [PubMed] [Google Scholar]
Tanaka 1996 {published data only}
- Tanaka K, Kondo M, Sakaguchi T, Saito S, Arata S, Ikeda M, et al. Efficacy of ursodeoxycholic acid in combination with interferon alpha in treating chronic hepatitis C: results of a long‐term follow‐up trial. Journal of Gastroenterology & Hepatology 1996;11(12):1155‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tsubota 1999 {published data only}
- Tsubota A, Kumada H, Arase Y, Chayama K, Saitoh S, Ikeda K, et al. Combined ursodeoxycholic acid and glycyrrhizin therapy for chronic hepatitis C virus infection: a randomized controlled trial in 170 patients. European Journal of Gastroenterology & Hepatology 1999;11(10):1077‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Viola 1996 {published data only}
- Viola L, Perez V, Tanno H, Silva M, Welz G, Vilar J, et al. Predictive factors of non‐response to recombinant interferon alfa 2b in patients with chronic hepatitis C (AASLD abstract). Hepatology 1996;24(4 Pt.2):594A. [Google Scholar]
Viola 1998 {published data only}
- Viola L, Fernandez JL, Tanno H, Silva M, Welz G, Ferraz M, Rendo P. A randomized controlled trial of 12‐month treatment with interferon alfa 2b and ursodeoxycholic acid for chronic hepatitis C. Hepatology 1998;28(4):290A. [Google Scholar]
Zhou 1995 {published data only}
- Zhou J, Wang C, Wu S. Efficacy of ursodeoxycholic acid therapy on chronic hepatitis [(original title in Chinese)]. Journal of Clinical Hepato‐Biliary Diseases 1995;11(3):148‐50. [Google Scholar]
Zhu 1994 {published data only}
- Zhu Y. Efficacy of ursodeoxycholic acid therapy on chronic hepatitis C [(original title in Chinese)]. Bulletin of Nantong Medical College 1994;14(2):237‐8. [Google Scholar]
References to studies excluded from this review
Angelico 1995b {published data only}
- Angelico M, Gandin C, Vecchio CD, Plat C, Bini A, Rosa TL, et al. Interferon‐alpha and tauroursodeoxycholic acid in chronic hepatitis C: a novel therapeutic combination (AASLD abstract). Hepatology 1995;22(4 Pt 2):120A. [Google Scholar]
Attilli 1994 {published data only}
- Attilli AF, Rusticali A, Varriale M, Carli L, Repice AM, Callea F, et al. The effect of ursodeoxycholic acid on serum enzymes and liver histology in patients with chronic active hepatitis. Journal of Hepatology 1994;20(3):315‐20. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Buzzelli 1991 {published data only}
- Buzzelli G, Moscarella S, Focardi G, Dattolo P, Giusti A, Relli P, et al. Long‐term treatment with ursodeoxycholic acid in patients with chronic active hepatitis. Current Therapeutic Research 1991;50(5):635‐42. [Google Scholar]
Buzzelli 1992 {published data only}
- Buzzelli G, Moscarella S, Focardi G, Dattolo P, Giusti A, Calviani L, et al. Ursodeoxycholic acid‐hemisuccinate in the treatment of chronic active hepatitis. A controlled clinical study. Minerva medica 1992;83:537‐40. [PubMed] [Google Scholar]
Crosignani 1991 {published data only}
- Crosignani A, Battezzati PM, Setchell KDR, Camisasca M, Bertolini E, Roda A, et al. Effects of ursodeoxycholic acid on serum liver enzymes and bile acid metabolism in chronic active hepatitis: a dose‐response study. Hepatology 1991;13(2):339‐44. [MEDLINE: ] [PubMed] [Google Scholar]
Crosignani 1998(a) {published data only}
- Crosignani A, Battezzati PM, Cestari C, Stabilini R, Conte D, Zappala F, et al. Tauroursodeoxycholic acid for the treatment of chronic hepatitis: a multicentre dose‐response study. Hepatology Research 1998;13:10‐9. [Google Scholar]
Del Vecchio 1982 {published data only}
- Vecchio Blanco C, Caporaso N, Gentile S, Rinaldi M, Pucci R. Safe use of ursodeoxycholic acid in the treatment of dyspeptic symptoms in patients with chronic active hepatitis: A double‐blind controlled trial. The Journal of International Medical Research 1982;10(4):278‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Italian 1990 {published data only}
- Italian Multicenter Group for Ursodeoxycholic Therapy in Chronic Hepatitis. UDCA improves serum liver enzymes but not liver histology in chronic hepatitis. Journal of Hepatology 1990;11(Suppl 2):S32. [Google Scholar]
Kadayifci 1997 {published data only}
- Kadayifci A, Savas MC, Arsian S, Gullu IH. Ursodeoxycholic acid in the management of prolonged cholestasis of acute hepatitis B. Journal of Clinical Gastroenterology 1997;24(2):125‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lu 1995 {published data only}
- Lu CL, Chan CY, Hwang SJ, Lu RH, Lee SD. Efficacy of ursodeoxycholic acid in the treatment of patients with chronic hepatitis C. Journal of Gastroenterology and Hepatology 1995;10(4):432‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pinto 1992 {published data only}
- Pinto A, Parini P, Novelli V, Zagari M, Sangermano A, Orsini M, et al. Effect of therapy with bis‐hemisuccinate of ursodeoxycholic acid bisodic salt in patients with chronic liver diseases. Minerva Medica 1992;83(6):359‐61. [MEDLINE: ] [PubMed] [Google Scholar]
Podda 1989 {published data only}
- Podda M, Ghezzi C, Battezzati PM, Bertolini E, Crosignani A, Petroni ML, et al. Effect of different doses of ursodeoxycholic acid in chronic liver disease. Digestive Diseases and Sciences 1989;34(12):59‐65. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Podda 1990 {published data only}
- Podda M, Ghezzi C, Battezzati PM, Crosignani A, Zuin M, Roda A. Effects of ursodeoxycholic acid and taurine on serum liver enzymes and bile acids in chronic hepatitis. Gastroenterology 1990;98(4):1044‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Podda 1995 {published data only}
- Podda M, Crosignani A, Battezzati PM, Buscarini L, Conte D, Notarbartolo A, et al. Tauroursodeoxycholic acid for the treatment of chronic hepatitis: A dose‐response study (AASLD abstract). Hepatology 1995;22(4 Pt 2):120A. [Google Scholar]
Portincasa 1993 {published data only}
- Portincasa P, Palmieri V, Doronzo F, Vendemiale G, Altomare E, Sabba C, et al. Effect of tauroursodeoxycholic acid on serum liver enzymes and dyspeptic symptoms in patients with chronic active hepatitis. Current Therapeutic Research 1993;53(5):521‐32. [Google Scholar]
Rolandi 1991 {published data only}
- Rolandi E, Franceschini R, Cataldi A, Cicchetti V, Carati L, Barreca T, et al. Effects of ursodeoxycholic acid (UDCA) on serum liver damage indices in patients with chronic active hepatitis. European Journal of Clinical Pharmacology 1991;40(5):473‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Song 1998 {published data only}
- Song Z, Song J, Li Y. The effect of UDCA on 42 chronic hepatitis patients with persisting high level of gamma GT [(original title in Chinese)]. Chinese Journal of Intergretive Traditional and Western Medicine on Liver Diseases 1998;8(2):106‐7. [Google Scholar]
Zhu 1997 {published data only}
- Zhu Y, Liang J, Lu Q. Combination of UDCA, Bing Du Zuo, and Ju Jibao for the treatment on 53 chronic hepatitis C patients (Chinese) [(original title in Chinese)]. Bulletin of Jiamusi Medical College 1997;20(1):44‐5. [Google Scholar]
Additional references
Alam 1995
- Alam JJ. Interferon‐beta treatment of human disease. Current Opinion in Biotechnology 1995;6(6):688‐91. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Armstrong 1982
- Armstrong MJ, Carey MC. The hydrophobic‐hydrophilic balance of bile salt. Inverse correlation between reversephase high performance liquid chromatographic mobilities and micellar cholesterol‐solubilizing capacities. Journal of Lipid Research 1982;23(1):70‐80. [MEDLINE: ] [PubMed] [Google Scholar]
Batta 1989
- Batta AK, Salen G, Arora R, Shefer S, Tint GS, Abroon J, et al. Effect of ursodeoxycholic acid on bile acid metabolism in primary biliary cirrhosis. Hepatology 1989;10(4):414‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Boucher 1995
- Boucher E, Jouanolle H, Andre P, Ruffault A, Guyader D, Moirand R, et al. Interferon and ursodeoxycholic acid combined therapy in the treatment of chronic viral hepatitis: results from a controlled randomized trial in 80 patients. Hepatology 1995;21(2):322‐7. [MEDLINE: ] [PubMed] [Google Scholar]
Brechot 1996
- Brechot C. Hepatitis B and C viruses and primary liver cancer. Baillieres Clinical Gastroenterology 1996;10(2):335‐73. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Brok 2005
- Brok J, Gluud LL, Gluud C. Ribavirin plus interferon versus interferon for chronic hepatitis C. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD005445] [DOI] [PubMed] [Google Scholar]
Calmus 1990
- Calmus Y, Gane P, Rouger P, Poupon R. Hepatic expression of class I and class II major histocompatibility complex molecules in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology 1990;11(1):12‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chen 2001
- Chen W, Liu JP, Gluud C. Bile acids for viral hepatitis (Protocol for a Cochrane Review). Cochrane Database of Systematic Reviews 2001, Issue 3. [Google Scholar]
Chen 2002
- Chen W, Gluud C. Bile acids for primary sclerosing cholangitis (Protocol for a Cochrane Review). Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI] [PubMed] [Google Scholar]
Chen 2003
- Chen W, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD003626] [DOI] [PubMed] [Google Scholar]
Chretien 1989
- Chretien Y, Poupon R, Gherardt MF, Chazouilleres O, Labbe D, Myara A, et al. Bile acid glycine and taurine conjugates in serum of patients with primary biliary cirrhosis: effect of ursodeoxycholic treatment. Gut 1989;30(8):1110‐5. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
DeMets 1987
- DeMets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ 1997;315(7109):629‐34. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fallon 1997
- Fallon HT. Chronic hepatitis C: how to recognize, when to treat. Consultant 1997;37(10):2596‐604. [Google Scholar]
Farrell 1999
- Farrell GC, Omata M, Sarin SK, Shiratori Y, Chutaputti A, Lu ZM, et al. Management of hepatitis C. Gastroenterology and Hepatology 1999;Suppl:4(i‐viii). [Google Scholar]
Fattovich 1995
- Fattovich G, Giustina G, Schalm SW, Hadziyannis S, Sanchez‐Tapias J, Almasio P, et al. Occurrence of hepatocellular carcinoma and decompensation in Western European patients with cirrhosis type B. Hepatology 1995;21(1):77‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fried 2002
- Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology 2002;36(5 Suppl 1):S237‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fuchs 1999
- Fuchs M, Stange EF. Metabolism of bile acids. In: Johannes Bircher, Jean‐Pierre Benhamou, Neil McIntyre, Mario Rizzetto, Juan Rodes editor(s). Oxford Textbook of Clinical Hepatology. 2. Vol. 1, Oxford: Oxford University Press, 1999:223‐56. [Google Scholar]
Gallo 1993
- Gallo V, Micheli AG, Chiandussi L. Tauro‐ursodeoxycholic acids vs ursodeoxycholic acid in the dissolution of biliary calculi. Result of a single blind study. La Clinica Terapeutica 1993;143(5):421‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Gluud 2001a
- Gluud C. Alcoholic hepatitis: no glucocorticosteroids?. Steatohepatitis (NASH and ASH)‐Falk Symposium 121. Lancaster: Kluwer Academic Publisher, 2001:322‐42. [Google Scholar]
Gluud 2001b
- Gluud C, Christensen E. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD000551] [DOI] [PubMed] [Google Scholar]
Gluud 2007
- Gluud C, Brok J, Gong Y, Koretz R. Hepatology may have problems with putative surrogate outcome measures. Journal of Hepatology 2007;46:734‐2. [DOI] [PubMed] [Google Scholar]
Goulis 1999
- Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic‐acid therapy for primary biliary cirrhosis: a meta‐analysis. Lancet 1999;354(9184):1053‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Guicciardi 2002
- Guicciardi M, Gores GJ. Ursodeoxycholic acid cytoprotection: dancing with death receptors and survival pathways. Hepatology 2002;35(4):971‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hahn 2000
- Hahn S, Williamson PR, Hutton JL, Garner P, Flynn EV. Assessing the potential for bias in meta‐analysis due to selective reporting of subgroup analyses within studies. Statistics in Medicine 2000;19(24):3325‐36. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. The Cochrane Library, Issue 3. Chichester, UK: John Wiley & sons, Ltd, 2005. [Google Scholar]
Hoffnagle 1997
- Hoffnagle JH, Bisceglie AM. The treatment of chronic viral hepatitis. The New England Journal of Medicine 1997;336(5):347‐56. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. Code of Federal Regulations & International Conference on Harmonization Guidelines. Media: Parexel Barnett, 1997. [Google Scholar]
Jaeschke 2002
- Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicological Sciences 2002;65(2):166‐76. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodological quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Knodell 1981
- Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1981;1(5):431‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Larghi 1997
- Larghi A, Crosignani A, Battezzati PM, Valle G, Allocca M, Invernizzi P, et al. Ursodeoxycholic and tauro‐ursodeoxycholic acids for the treatment of primary biliary cirrhosis: a pilot crossover study. Alimentary Pharmacology & Therapeutics 1997;11(2):409‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Liaw 1999
- Liaw YF, Guan R, Lau GKK, Leung N, Merican I, Stace NH, et al. Management of chronic hepatitis B. Gastroenterology and Hepatology 1999;Supplement:2 (i‐viii). [Google Scholar]
Liu 2002
- Liu J, Kjaergard LL, Gluud C. Misuse of randomization: a review of Chinese randomized trials of herbal medicines for chronic hepatitis B. American Journal of Chinese Medicine 2002;30(1):173‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lok 2001
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2001;34(6):1225‐41. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Maddrey 1980
- Maddrey WC. Drug‐related acute and chronic hepatitis. Clinical Gastroenterology 1980;9(1):213‐24. [MEDLINE: ] [PubMed] [Google Scholar]
Manns 2001
- Manns MP, Cornberg M, Wedemeyer H. Current and future treatment of hepatitis C. Indian Journal of Gastroenterology 2001;20(Suppl 1):C47‐51. [MEDLINE: ] [PubMed] [Google Scholar]
Mihm 1997
- Mihm S, Fayyazi A, Hartmann H, Ramadori G. Analysis of histopathological manifestations of chronic hepatitis C virus infection with respect to virus genotype. Hepatology 1997;25(3):735‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervetion efficacy reported in meta‐analyses. Lancet 1998;352(9128):609‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Myers 2001a
- Myers RP, Regimbeau C, Thevenot T, Leroy V, Mathurin P, Opolon P, et al. Interferon for acute hepatitis C. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD000369] [DOI] [PMC free article] [PubMed] [Google Scholar]
Myers 2002b
- Myers RP, Regimbeau C, Thevenot T, Leroy V, Mathurin P, Opolon P, et al. Interferon for interferon naive patients with chronic hepatitis C. Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI: 10.1002/14651858.CD000370] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nakagawa 1990
- Nakagawa M, Colombo C, Setchell KDR. Comprehensive study of the biliary bile acid composition of patients with cystic fibrosis and associated liver disease before and after UDCA administration. Hepatology 1990;12(2):322‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Palmer 1972
- Palmer RH. Bile acids, liver injury and liver disease. Archives of Internal Medicine 1972;130(4):606‐17. [MEDLINE: ] [PubMed] [Google Scholar]
Paumgartner 2002
- Paumgartner G, Beuers U. Ursodeoxycholiic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36(3):525‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Podda 1996
- Podda M, Crosignani A, Battezzati PM, Quagliuolo M, Valsania C, Invernizzi P, et al. Tauroursodeoxycholic acid for the treatment of chronic hepatitis: a dose‐response study (IASL Abstract). Hepatology 1996;23(1):I‐70. [Google Scholar]
Poynard 1997
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC Groups. Lancet 1997;349(9055):825‐32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Qiao 2002
- Qiao L, Yacoub A, Studer E, Gupta S, Pei XY, Grant S, et al. Inhibition of the MAPK and PI3K pathways enhances UDCA‐induced apoptosis in primary rodent hepatocytes. Hepatology 2002;35(4):779‐89. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Renault 1989
- Renault PF, Hoofnagle JH. Side effects of alpha interferon. Seminar in Liver Disease 1989;9(4):273‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rizzetto 1986
- Rizzetto M, Verme G, Gerin JL, Purcell RH. Hepatitis delta virus disease. Progress in Liver Disease 1986;8:417‐31. [MEDLINE: ] [PubMed] [Google Scholar]
Rodrigues 2000
- Rodrigues CM, Brites D, Serejo F, Costa A, Ramalho F, Moura MC. Apoptotic cell death does not parallel other indicators of liver damage in chronic hepatitis C patients. Journal of Viral Hepatitis 2000;7(3):175‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes R, Altman D. Empirical evidence of bias. JAMA 1995;273(5):408‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Seeff 2002
- Seeff LB, Hoofnagle JH. National Institutes of Health Consensus Development Conference: management of hepatitis C: 2002. Hepatology 2002;36(5 Suppl 1):S1‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sholmerich 1984
- Sholmerich J, Becher MS, Schmidt KH, Schubert R, Kremer B, Felhaus S, et al. Influence of hydroxylation and conjugation of bile salts on their membrane damaging properties. Hepatology 1984;4(4):661‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stapleton 1994
- Stapleton JT, Lemon SM. Hepatitis A and Hepatitis E. In: Hoeprich PD, Jordan MC, Ronald AR editor(s). Infectious Disease. Fifth. Philadelphia: Lippincott Co, 1994:790‐7. [Google Scholar]
Wetterslev 2007
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trials sequential analyses of six Cochrane Neonatal Group meta‐analyses. Journal of Clinical Epidemiology 2007;(in press). [Google Scholar]
WHO 2000a
- WHO. Hepatitis B. Fact Sheet WHO/204, www.who.int/inf‐fs/en/fact204.html October 2000.
WHO 2000b
- WHO. Hepatitis C. Fact Sheet WHO/164, www.who.int/inf‐fs/en/fact164.html October 2000.
Wolfram 1999
- Wolfram H, Gerlich, Thomssen R. The viruses of hepatitis. In: Bricher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J editor(s). Oxford Textbook of Clinical Hepatology. 2. Vol. 1, Oxford: Oxford University Press, 1999:827‐954. [Google Scholar]
Zuckermann 1993
- Zuckermann AR, Thomas HC. Viral Hepatitis. 1. London: Churchill Livingstone, 1993. [Google Scholar]