

Abstract
The adenosine (ADO) A1 receptor (A1R) is a promising therapeutic target for potential non-opioid analgesics to treat neuropathic pain [1, 2]. However, development of analgesic orthosteric A1R agonists has failed because of a lack of sufficient on-target selectivity as well as off-tissue adverse effects [3]. Here, we demonstrate that [2-amino-4-(3,5-bis(trifluoromethyl)phenyl)thiophen-3-yl)(4-chlorophenyl)methanone] (MIPS521), a positive allosteric modulator (PAM) of the A1R, displays in vivo analgesic efficacy through modulation of elevated endogenous ADO that occurs in rat spinal cord in neuropathic pain states. We also report the structure of the A1R co-bound to ADO, MIPS521 and a Gi2 heterotrimer, revealing a novel extrahelical lipid/detergent-facing allosteric binding pocket involving transmembrane helixes 1, 6 & 7. Molecular dynamics simulations and ligand kinetic binding experiments support a molecular mechanism whereby MIPS521 stabilises the ADO-receptor-G protein complex. This study provides proof of concept for structure-based allosteric drug design of disease context-specific, non-opioid analgesics.
Introduction
Chronic pain remains a widespread global health burden. The paucity of current therapeutic options has led to an over-reliance on opioids analgesics, which provide limited relief in patients with chronic (particularly neuropathic) pain while exhibiting severe adverse effects, such as respiratory depression and addiction [4, 5]. These limitations contribute to the current global opioid crisis, highlighting the urgent need for non-opioid analgesics that are safe and effective for the treatment of pathological pain.
The purine nucleoside, ADO, is an important cellular protective molecule with the potential to modulate different types of pain, particularly through the A1R, a member of the G protein-coupled receptor (GPCR) superfamily that preferentially couples to Gi/o proteins [2, 6] and is present on central terminals of peripheral nociceptive neurons [7, 8]. Indeed, prototypical agonists acting via the A1R are antinociceptive, but have not progressed into clinical development due to on-target limitations [3]. Due to high sequence and structural conservation within the orthosteric binding site of the four ADO GPCRs, A1R subtype-selectivity is difficult to achieve [3]. Additionally, since A1R is widely expressed not only in the central nervous system but in the heart and adipose tissue; A1R agonists may elicit dose-limiting side effects such as bradycardia [9].
The limitations of orthosteric A1R activators may, however, be overcome through targeting allosteric binding sites. Structurally, allosteric sites can exhibit higher selectivity between GPCR subtypes due to greater sequence variability [10, 11]. Mechanistically, allosteric drugs offer an additional advantage due to the reciprocity of communication between the two binding domains, i.e., the direction and magnitude of effect that an allosteric ligand exerts on the endogenous orthosteric agonist will also be exerted by the agonist on the modulator [11]. This bi-directional communication is pharmacologically quantified as the ‘cooperativity’ between two ligands, and can potentially support tissue- or context-based allosteric drug selectivity i.e., where the desired site of therapeutic action (e.g., spinal cord) exhibits localised changes in concentration (tone) of the endogenous agonist as a consequence of disease.
The A1R was the first GPCR for which synthetic, small molecule PAMs were described, predominantly represented by 2-amino-3-benzoylthiophenes (2A3BTs) [12]. Some A1R PAMs have efficacy in reducing chronic mechanical allodynia in rodent pain models [13–15], but 2A3BT scaffolds are suboptimal for drug development as metabolism of the thiophene core can lead to the formation of reactive metabolites [16]. Despite many years of research, the location of allosteric sites on the A1R remains unknown, although prior mutagenesis studies have proposed a pocket comprising the extracellular face of the transmembrane (TM) bundle, including extracellular loop (ECL) 2, similar to that of many other class A GPCRs [17–19]. Recent structural biology advances provide promising approaches for identification of allosteric sites within GPCRs that may also be used to guide the development of novel allosteric analgesics [20].
Here, we provide mechanistic validation that in vivo analgesic efficacy of A1R PAMs is driven by on-target positive cooperativity with ADO that is elevated at the site of action in an animal model of neuropathic pain, and report the cryo-electron microscopy (cryo-EM) structure of this PAM in complex with ADO-bound A1R-Gi2 protein. All-atom simulations using a robust Gaussian accelerated molecular dynamics (GaMD) method [21] indicate that the PAM stabilises the active ADO-A1R-G protein complex. We reveal a hitherto unappreciated allosteric binding pocket and propose a molecular mechanism of allostery that expands the scope for structure-based design of non-opioid allosteric GPCR analgesics.
Allosteric cooperativity drives A1R efficacy in treating neuropathic pain
Previously, we demonstrated that sciatic nerve-injury-induced neuropathic pain elevates endogenous ADO tone in the rat spinal dorsal horn [22]. We also described a recently-characterised A1R PAM, VCP171 [(2-amino-4-(3-(trifluoromethyl)phenyl)thiophen-3-yl)(phenyl)methanone] [22] (Ext Data Fig 1a), which reduced electrically-evoked, AMPA receptor-mediated, excitatory postsynaptic currents (eEPSCs) in spinal cord nociceptive neurons [22]; at the time, VCP171 was the highest affinity A1R PAM arising from our structure-activity based medicinal chemistry program [23, 24].
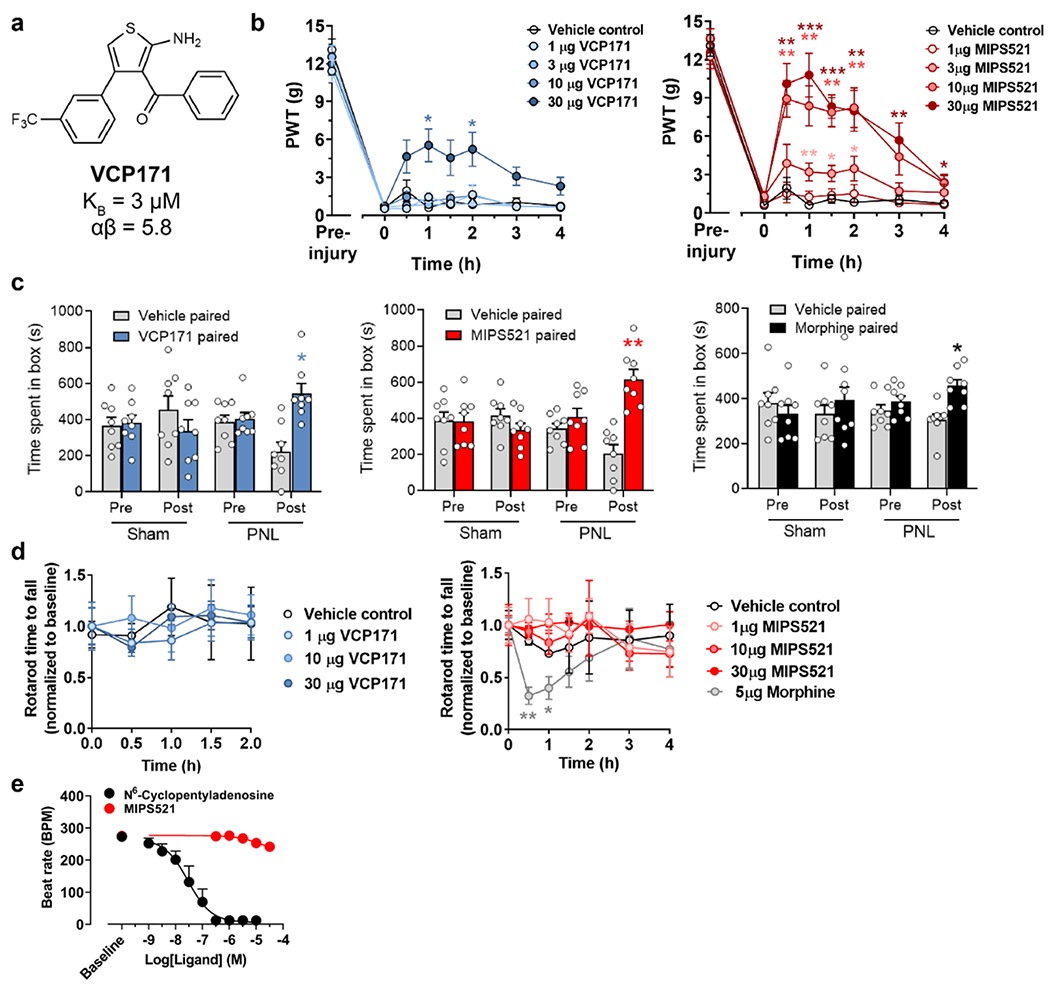
To determine whether the cellular effects of VCP171 in spinal cord translated to analgesic efficacy in vivo, we measured mechanical allodynia in nerve-injured rats following direct intrathecal administration of the PAM. VCP171 had no effect on paw withdrawal threshold between 1-10 μ g, but gave a small reduction of allodynia at 30 μg (Fig. 1d; Ext. Fig. 1b). At this highest dose, VCP171 also reduced spontaneous pain as measured in conditioned place preference experiments (Ext Fig 1c). As VCP171 was administered intrathecally and is stable in human and rat plasma for at least 4 h (Supp Table 1), we hypothesised that the weak in vivo effect was not a result of poor target engagement, but due to limited positive cooperativity (αβ between the PAM and endogenous ADO (αβ = 5.8, Fig 1a). Although VCP171 is also able to partially activate the A1R directly when tested in overexpressed recombinant CHO lines (LogτB = 0.29 ± 0.12; τB = 1.9; Fig 1a), this effect is absent in native cells of the spinal cord upon treatment with adenosine deaminase [20] and thus does not play a role in the actions of the PAM under physiological conditions. To test the cooperativity hypothesis, we re-evaluated A1R PAMs that were synthesised as part of our earlier focussed structure-activity medicinal chemistry campaign. Relative to VCP171, MIPS521 (compound 13a in [23]) (Fig. 1a) had lower affinity for the A1R allosteric site (pKB = 4.95 ± 0.40; KB = 11 μM), slightly higher signaling efficacy (Log τB = 0.96 ± 0.34; τ B = 9.12), but substantially higher positive cooperativity (Log αβ = 1.81 ± 0.53; αβ = 64.6) with ADO when assessed in a recombinant cell-based assay of A1R-mediated inhibition of forskolin-stimulated cAMP (Fig. 1a). In electrophysiological recordings of dorsal horn neurons, MIPS521 was more potent than VCP171 in reducing eEPSCs in spinal cord from nerve-injured animals, with a pEC50 of 6.9 ± 0.4 compared to 5.6 ± 0.3 for VCP171 (n=4-12; Fig 1c). Importantly, the maximal MIPS521-induced decrease in synaptic current amplitude was significantly greater in nerve-injured animals compared with sham surgery controls (Fig 1c), indicating a tissue- and context-specific sensitivity to differences in localised ADO tone in the disease model relative to control animals. Reversal of the ADO tone using the A1R antagonist DPCPX gave an increase of 19.7 ± 6.1% in eEPSC amplitude from baseline in nerve-injured animals and 7.89 ± 2.7% in sham controls. Both compounds reduced amplitude frequency of spontaneous EPSCs in nerve injured rats (Ext Data Fig 2). It is possible that the A1R PAMS also act on postsynaptic A1Rs in lamina II of the dorsal horn, contributing to the analgesic effect. However, any postsynaptic effects are likely to be subtle compared to presynaptic A1R activation due to sparse postsynaptic A1R expression [25], and thus beyond the scope of the current study. Irrespective, when tested in vivo, MIPS521 reversed the mechanical hyperalgesia at lower concentrations than VCP171, promoting a robust antinociceptive effect (Fig 1d, Ext Data Fig 1b). MIPS521 also significantly reduced spontaneous pain in a conditioned place preference model at a lower dose than VCP171 (Ext Fig 1c). For the in vivo studies, it should be noted that the DMSO/saline ratio was kept at 60:40 for all doses and vehicle control, as this was the concentration of DMSO required for the highest doses prior to administration; as with many early-stage drug discovery programs, the current series of tool PAMs are not yet optimized for aqueous solubility. Although such high concentrations of DMSO can be pro-nociceptive, the DMSO/saline vehicle control used in our von Frey experiments did not produce any significantly different results relative to nerve-injured rats that had been injected with pure saline (data not shown; see Methods for further details).
Figure 1 |. MIPS521 reduces spinal nociceptive signalling and mechanical allodynia in an animal model of neuropathic pain.
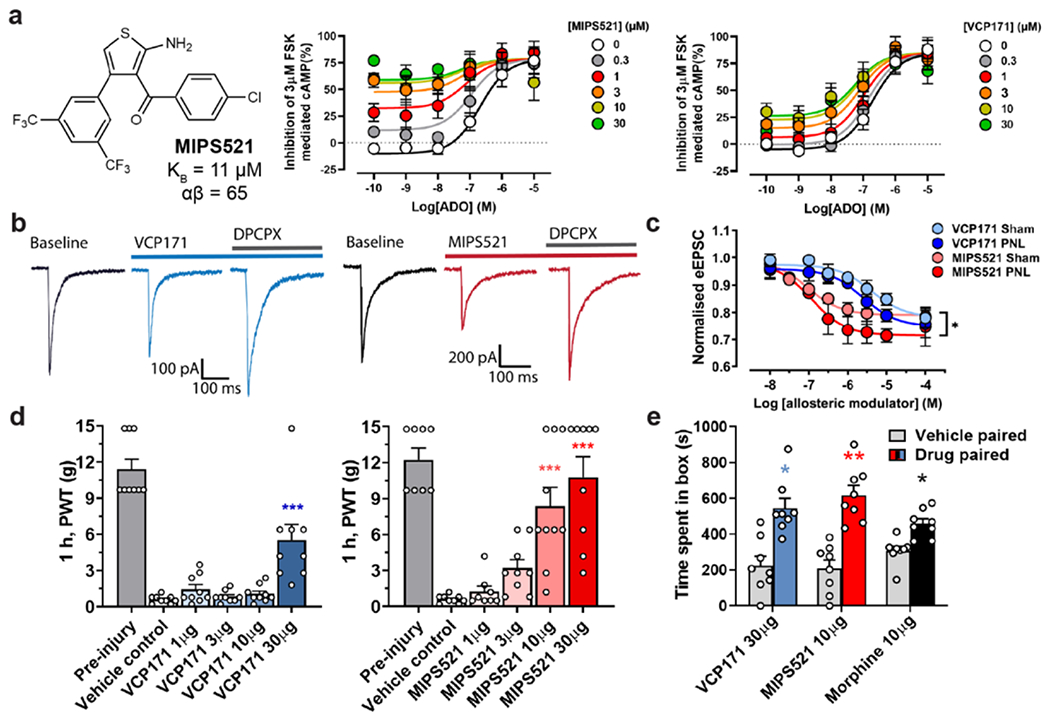
a, MIPS521 and VCP171 produce a concentration-dependent potentiation of signalling by the endogenous agonist, adenosine (ADO), in an inhibition of cAMP assay (shown as inhibition of 3μM forskolin mediated cAMP) mediated by the human A1R expressed in CHO cells. An operational model of allosterism and agonism yielded estimated MIPS521 and VCP171 affinities of pKB = 4.95 ± 0.40 & 5.50 ± 0.29, cooperativity of Logαβ = 1.81 ± 0.53 & 0.76 ± 0.25 and direct allosteric agonism of LogτB = 0.96 ± 0.34 & −0.23 ± 0.12, respectively; data shows mean ± SEM n =3, duplicate readings. b, Examples of dorsal root eEPSCs in neurons of the superficial laminae (I-II) of the spinal dorsal horn at baseline and following superfusion of 1 μM VCP171 or MIPS521, which is reversed by the antagonist, DPCPX in nerve-injured rats (partial nerve ligation; PNL). c, MIPS521 is more potent than VCP171 at reducing eEPSC amplitude in dorsal horn neurons. Effects of MIPS521, but not VCP171, were increased in the nerve-injured group compared to the sham surgery control group (n = 4-12 per data point); Data are presented as mean +/− SEM. d, Paw withdrawal threshold (PWT) to mechanical stimulus in nerve-injured rats 1-hour post-intrathecal administration of VCP171 (blue, n = 9 per group) or MIPS521 (red, n = 8-10 per group). Data is displayed as mean +SEM (bars) and individual values (circles). Significant differences were determined using a one-way ANOVA, * P < 0.05, ** P < 0.01, *** P < 0.001. e, The effect of intrathecal VCP171 (30 μg, blue), MIPS521 (10 μg, red) or morphine (10 μg, black) on spontaneous pain was determined using conditioned place preference tests in nerve injured rats (n = 8 per group); Data are presented as mean values +/− SEM.
Opioid analgesics such as morphine can cause acute motor impairment through effects on the ventral horn of the spinal cord [26]. Although this region also expresses A1Rs [27], neither MIPS521 nor VCP171 affected motor function, in contrast to morphine (Ext Data Fig 1d). Moreover, a major limitation of traditional orthosteric agonists of the A1R as analgesics is the potential to produce off-tissue adverse effects, in particular cardiovascular events such as bradycardia. Encouragingly, MIPS521 had minimal effect on rat atrial beat rate, in contrast to the orthosteric A1R agonist N6-Cyclopentyladenosine (CPA) (Ext. Fig. 1e).
Similar to VCP171, MIPS521 was stable in human and rat plasma over a 4-hour incubation period (Supp Table 1). Collectively, our findings are thus consistent with the degree of A1 PAM analgesic efficacy being determined by the extent of on-target positive cooperativity on the elevated ADO tone that occurs in the spinal cord upon the development of neuropathic pain. Thus, MIPS521 is an important proof-of-concept tool for the development of tissue- and disease selective-allosteric A1R analgesics. A possible limitation of our current study is the lack of definitive validation of the site of action of our selective PAMs being the A1R through the use of A1R knockout (KO) mice. It should be noted, however, that even such studies are prone to difficulties in interpretation because A1R KO mice exhibit increased hyperalgesia and are susceptible to epilepsy [28]. Previous studies that have already identified the A1R as a promising analgesic target, together with our pharmacological validation using the A1R-selective antagonist, DPCPX, nonetheless strongly support the A1R as the primary target of our novel PAMs.
Given the lack of detailed information regarding the location of the allosteric binding site occupied by A1R PAMs, however, and the relative paucity of chemotypes beyond the 2A3BT scaffold, we next sought to determine the active-state structure of the A1R bound to ADO, MIPS521 and a cognate heterotrimeric G protein partner where stability of the complex was enabled by incorporation of dominant-negative mutations in the Gαi2 [29, 30].
Cryo-EM structure of a PAM-bound ADO-A1R-Gi complex
We expressed and purified the A1R-Gi2 complex bound to ADO and MIPS521 as described previously [29, 30] where high concentrations of both ADO and MIPS521 were present throughout the purification procedure (Ext Data Fig 3a–d). The structure of the MIPS521-ADO-A1R-Gi2 complex was determined using cryo-EM to a nominal resolution of 3.2 Å reconstructed from 689,000 particle projections [Ext Data Fig 4a]. The cryo-EM density map exhibited well-resolved side chains, allowing confident rotamer placements for most amino acids [Ext Data Fig 4c]. Both orthosteric and allosteric pockets were well-resolved, with local resolution of ~3.3-3.4 Å for both regions. We also re-processed the original dataset for our previously determined ADO-A1R-Gi2 structure [30] to take advantage of recent improvements in cryo-EM software. This resulted in a new map with a local resolution of 3.3 Å [Ext Data Fig 4b]. We used this map to create an improved model for the ADO-A1R-Gi2 complex in the absence of allosteric modulator for all subsequent comparisons (Fig 2a). Protein chains were well resolved in both structures with the exception of ICL3 in the receptor, flexible N- and C- termini in Gβ and Gγ subunits, and the highly mobile ɑ-helical domain of Gɑi2, which was masked out during map reconstructions.
Figure 2 |. Comparison of the structures of the A1R-Gi2 complex in the presence and absence of the positive allosteric modulator, MIPS521.
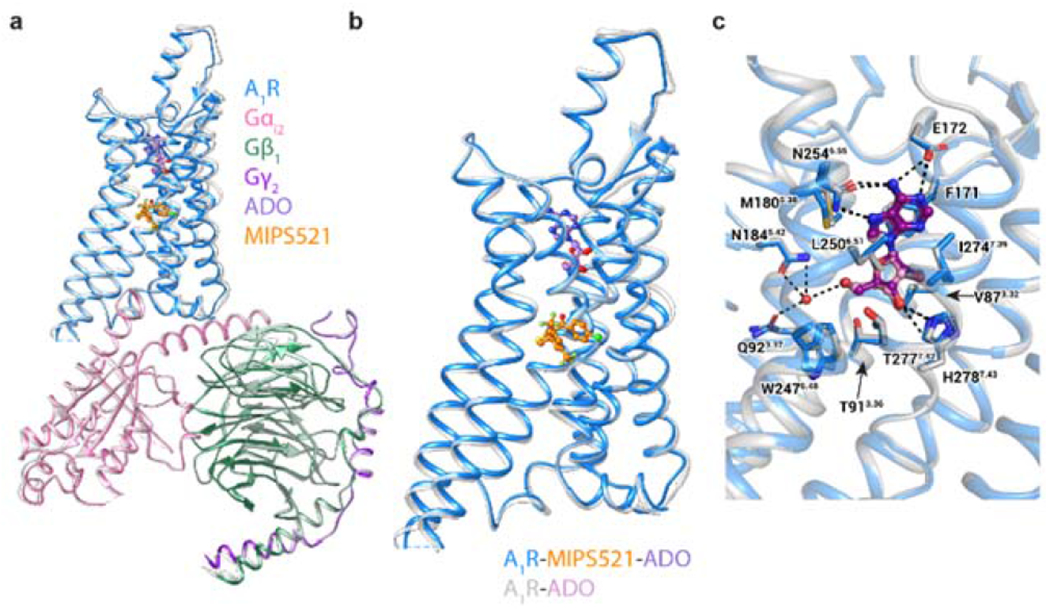
a, b, Overlay of the A1R-Gi2 complex in the presence and absence of MIPS521, showing (a) the whole complex and (b) receptor alone (in MIPS521-ADO-A1R-Gi2 the receptor is blue and the heterotrimeric Gi2 pink, cyan and dark purple for α, β and γ, respectively, ADO in purple and MIPS521 in orange; the ADO-A1R-Gi2 complex is coloured grey, with ADO in plum), c, The orthosteric binding site of the A1R-Gi2 complex in the presence and absence of MIPS521 is highly conserved. Water molecule shown as red sphere. ADO is shown in ball and stick representation and residues of the orthosteric binding pocket in stick representation, coloured by heteroatom.
Superposition of ADO-A1R-Gi2 and MIPS521-ADO-A1R-Gi2 structures showed near-identical conformations for the receptor and the G protein (root mean square deviation of 0.46 Å, Fig 2a,b). The placement of the orthosteric agonist ADO and the rotamers of the binding pocket side chains are similar between the two structures. A stable water molecule was also evident in both structures, coordinated by Q92 and N184, stabilising the bound ADO through hydrogen bonds (Fig 2c).
A unique extrahelical allosteric binding pocket in the A1R
Surprisingly, in the PAM-bound complex, there was no density within the extracellular vestibule that was previously predicted to be a binding site for 2A3BT scaffolds [17–19]. However, we observed robust density at an extrahelical position outside of TM1, 6 and 7 that was not present in the cryo-EM map for the ADO-A1R-Gi2 complex [30], and thus we attributed this to MIPS521. The distinctly non-planar and extrahelical binding pose of MIPS521 was unexpected and to our knowledge is a novel allosteric binding pocket not observed previously for other GPCRs.
Inspection of this novel allosteric binding site reveals that the PAM occupies a shallow pocket formed by sidechains of L2426.43, L2456.46, S2466.47, L2767.41, M2837.48, F2757.40, V221.45, L181.41, I191.42 and is exposed to the lipid/detergent interface (Fig 3a). Consistent with the relatively low affinity of MIPS521, H-bonds are only observed between the 2-amino substituent of the thiophene ring of MIPS521 and S2466.47 and the backbone of L2767.41 of the A1R. The rest of the contacts are weak van der Waals interactions with the 4-chlorophenyl group and the core thiophene ring of MIPS521. To accommodate MIPS521, small conformational variances in receptor side chains were evident relative to the non-PAM bound structure, including pivoting of M2837.48 and subtle movements of L242, L245, L18, I19 and V22 (Fig 3b).
Figure 3 |. Identification of an extrahelical lipid-facing allosteric binding pocket involving TMs 1,6 & 7 on the A1R.
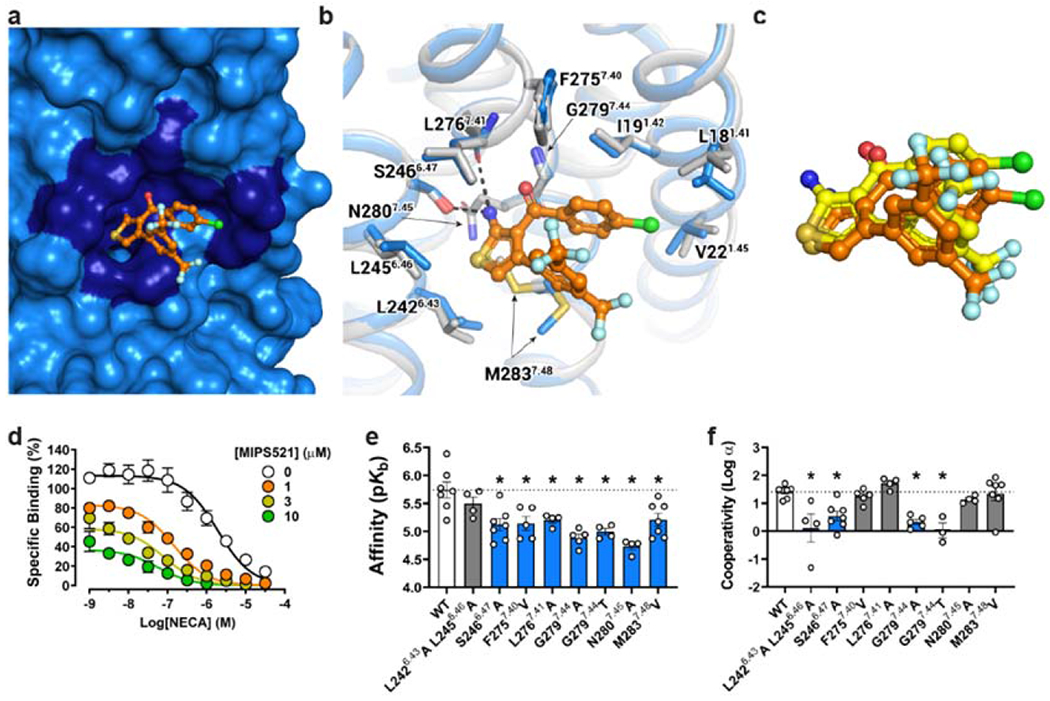
a, Surface rendering of the MIPS521 binding site located in an extrahelical position between TM6 & 7. b, Extrahelical allosteric binding pocket of the A1R in the presence and absence of MIPS521, with a pivoting of M2837.48 to accommodate MIPS521 (Pocket residues in stick representation coloured by heteroatom; MIPS521-ADO-A1R-Gi2 complex (blue), MIPS521 (orange); ADO-A1R-Gi2 complex (grey), c, Comparison of the binding pose of MIPS521 from the cryo-EM structure (orange) and in situ docking (yellow), d, Allosteric modulation of orthosteric ligand affinity at the WT A1R demonstrated in a [3H]-DPCPX radioligand interaction binding assay in the presence of the orthosteric agonist NECA and MIPS521. Data are presented as mean +/− SEM, n=6. e, f, Changes in MIPS521 affinity (pKB) (e) or binding cooperativity (log a) with the orthosteric agonist NECA (f) following mutation of residues proposed to form the allosteric pocket identified in the cryo-EM structure. Parameter estimates are the mean ± SEM determined from n=3-6 (white circles) performed in duplicate. *P < 0.05 (compared with wild type; one-way ANOVA, Dunnett’s post hoc test).
We next performed in situ docking and site-directed mutagenesis to further validate this extrahelical allosteric binding pocket. The docked MIPS521 pose is very similar to the cryo-EM structure (RMSD 1.7 Å), consistent with our modelling of MIPS521 into the cryo-EM map, despite the discontinuous density in this region (Fig 3c, Ext Data Fig 4c). Further, in both GaMD and conventional MD (cMD) simulations, MIPS521 formed stable hydrogen bonds with receptor residues Ser6.47 and Leu7.41 (Ext Data Fig 5).
For the mutagenesis, we performed whole-cell [3H]DPCPX saturation binding assays and interaction binding assays in the absence and presence of the orthosteric agonist NECA, with and without MIPS521, in cell lines expressing WT and mutant A1Rs (Fig 3d–f, Supp Table 2, Supp Table 3). Interaction binding data were fitted to an allosteric ternary complex model, allowing estimates of allosteric ligand affinity (pKB) and binding cooperativity (log α) between the allosteric ligand (MIPS521 or VCP171) and orthosteric agonist (NECA) [31]. None of these point mutations significantly altered the affinity of the radioligand itself or receptor expression relative to wild-type A1R (Ext Fig 6a–b, Supp Table 2). However, there were several mutations within the extrahelical binding site (L2426.43A/L2456.46A, S2466.47A, and N2807.45A) that significantly enhanced the affinity of the orthosteric agonist NECA, while the G2797.44A mutant reduced NECA affinity (Ext Data Fig 6c, Supp Table 3).
Consistent with the extended weak interactions between MIPS521 and the receptor, most binding site mutations resulted in a significant reduction in MIPS521 affinity and/or cooperativity (Fig 3b, d–f, Supp Table 3). S2466.47A resulted in a significant decrease in both PAM affinity and cooperativity, likely due to the loss of a hydrogen bond with the 2-amino substituent of the thiophene of MIPS521 (Fig 3b, d–f, Supp Table 3). The decrease in affinity for F2757.40V is likely a result of a decreased number of van der Waals contacts due to the smaller side chain of V275. Consistent with the role of M2837.48 in shaping of the allosteric pocket, M2837.48V significantly reduced the affinity of MIPS521 (Figure 3b, d–f, Supp Table 3). Introduction of bulkier side chains into position 7.44 (the G2797.44) resulted in significant loss of both modulator affinity and cooperativity, likely due to steric hindrance of MIPS521 binding (Figure 3b, d–f, Supp Table 3). L2767.41A significantly decreased MIPS521 affinity, likely due to a loss of this hydrophobic interaction with the smaller side chain of A276. In contrast, alanine mutation of both L2426.43 and L2456.46 caused a significant decrease in PAM cooperativity but had no effect on affinity, likely due to the weak nature of the hydrophobic interactions with MIPS521. Collectively, the mutagenesis data support the location of the novel extrahelical allosteric binding pocket at the A1R identified in the cryo-EM structure.
Molecular mechanism of positive allosteric modulation at the A1R
While an extrahelical allosteric site at TMs 1, 6 & 7 is unique to the A1R, alternative lipid-facing allosteric binding pockets have been identified at other Class A GPCRs [20]. These include binding sites for the negative allosteric modulators, BPTU (P2Y1 receptor), ORG27569 (CB1), NDT9513727 (C5aR) and AZ3451 (PAR2) [32–35], and the PAMs, AP8 (GPR40), LY3154207 (D1R) and compound-6FA (α2-adrenergic receptor) [36–38] (Ext Data Fig. 7). In each of these previous instances, the allosteric ligands appear to affect the ability of the receptor to transition between active and inactive states. To gain insights into molecular mechanisms underlying the effects of MIPS521 as a PAM at the A1R, we performed GaMD simulations to interrogate how MIPS521 alters dynamics of both the receptor and the G protein.
Using the MIPS521-ADO-A1R-Gi2 and ADO-A1R-Gi2 cryo-EM structures as starting points, we performed GaMD and cMD simulations of receptor-G protein complexes (cryo-EM models) or receptors alone (cryo-EM models with omitted G protein heterotrimers). MIPS521 underwent high fluctuations in the ADO-A1R-MIPS521 system and could even dissociate in the GaMD simulations [Ext Data Fig 8], while it remained bound to the ADO-A1R-Gi2 complex during both the GaMD and cMD simulations [Ext Data Fig 8]. We then focused on dynamics of the agonist, ADO. In the absence of G protein, ADO sampled a large conformational space in the orthosteric pocket and exhibited higher flexibilities in simulations with and without MIPS521 (Fig 4a, b, Ext Data Fig 9a, b, o, p). The presence of G protein in the simulations decreased the conformational dynamics of ADO (Fig 4a, c Ext Data Fig 9a, c, m, n), consistent with a ternary complex model where the G protein allosterically stabilises agonist binding in the orthosteric pocket [39]. In the presence of MIPS521, ADO was stabilised even further when Gi2 was present in the A1R complex (Fig 4b, d Ext Data Fig 9b, d, m, n). Next, we examined whether the effect of MIPS521 on ADO stability in the orthosteric pocket resulted from changes in receptor and G protein dynamics in the simulations. In the absence of G protein, the active receptor in the ADO-A1R GaMD simulations relaxed toward the inactive structure, for which the Arg1053.50–Glu2296.30 distance could decrease to ~8.3 Å (Fig 4e) [19]. Simulations in the presence of G protein, MIPS521, or both, revealed TM3-6 distances consistent with the A1R in the active conformation [Fig 4f–h, Ext Data Fig 9f–h], suggesting a direct stabilisation of the A1R in a ‘G protein-bound-like’ conformation by MIPS521 post removal of Gi2. Moreover, there was reduced mobility between G protein and receptor (measured as the receptor NPxxY–Gα distance) in the MIPS521-ADO-A1R-Gi2 complex compared to the complex in the absence of PAM [Ext Data Fig 9i–I]. Taken together, the GaMD simulations provide mechanistic insight into the positive cooperativity of MIPS521 that most likely occurs by stablisation of the A1R-Gi2 complex via effects to facilitate formation of, and retard relaxation from, TM6 and TM7 in the ‘G protein-bound-like’ conformation. This mechanism could potentially also explain why several mutations in the allosteric site significantly affected the affinity of the orthosteric agonist, NECA (Ext Data Fig 6); through an indirect perturbation of the ability of the receptor to adopt the active conformation.
Figure 4 |. MIPS521 stabilises the A1R-Gi2 ternary complex.
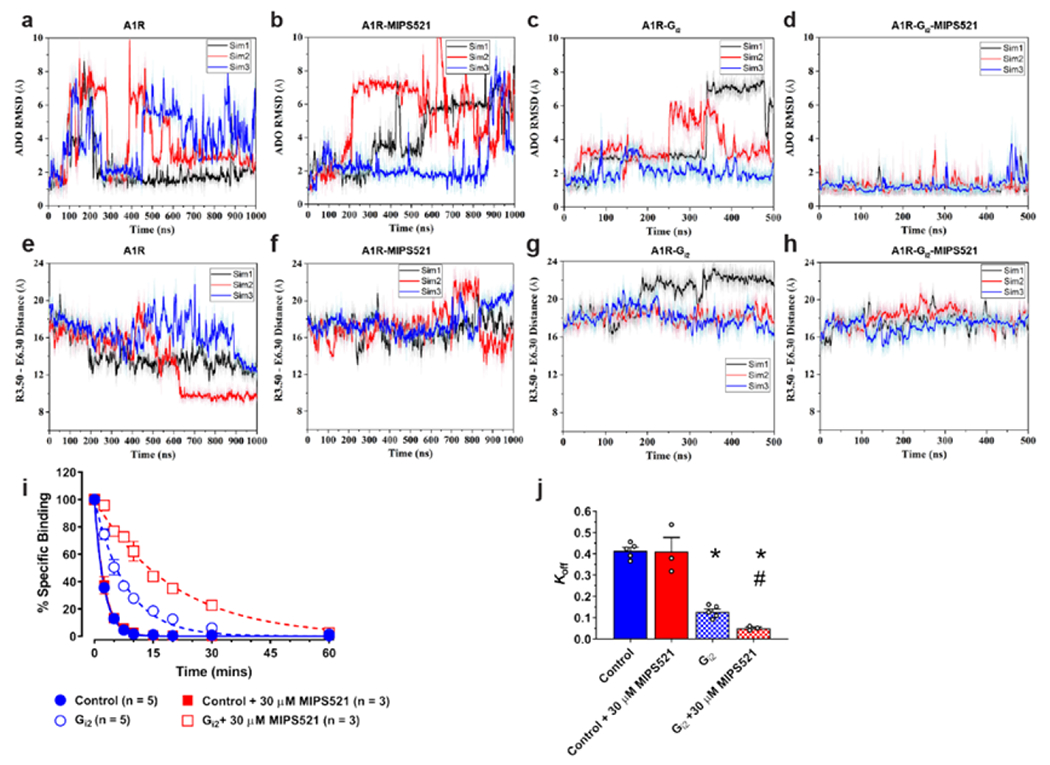
a-d, RMSD (Å) of ADO from GaMD simulations completed in the (a) absence or (b) presence of MIPS521, (c) Gi2, or (d) both Gi2 and MIPS521. e-h, Distance between the intracellular ends of TM3 and TM6 (measured as the distance in Å between Arg1053.50 and Glu2296.30) in the (e) absence or (f) presence of MIPS521, (g) Gi2, or (h) both Gi2 and MIPS521. Each condition represents three GaMD simulations, with each simulation trace displayed in a different colour (black, red, blue). The lines depict the running average over 2 ns. i, MIPS521 has no effect on the dissociation rate of [3H]DPCPX, promoted by isotopic dilution with excess antagonist, SLV320 (1 μM; Control), at the A1R alone reconstituted in rHDL. Addition of a saturating concentration of Gi results in a retardation of [3H]DPCPX dissociation that is reduced by co-addition of MIPS521. Data is mean ± SEM, n = 3-5 j, Quantification of dissociation rates from traces. Data is mean ± SEM, n ≥ 3 experiments performed in duplicate. *P < 0.05 (compared to control; one-way ANOVA, Tukey’s post hoc test). #P < 0.05 (compared to control in the presence of Gi2; one-way ANOVA, Tukey’s post hoc test).
Finally, to validate the allosteric mechanism proposed from the GaMD simulations, we reconstituted purified A1R and heterotrimeric Gi2 proteins in high-density lipoprotein (rHDL) particles and determined the effects of MIPS521 on the ability of a pre-bound orthosteric ligand to dissociate from the complex. In the absence of G protein, MIPS521 had no effect on the dissociation of pre-equilibrated [3H]DPCPX at A1R-rHDL particles (Fig 4i, j, Supp Table 4). Upon the addition of a saturating concentration of Gi2, a significant slowing of the dissociation of [3H]DPCPX was observed (Fig 4i, j, Supp Table 4), consistent with previous studies of G protein effects on antagonist dissociation at the β2 adrenergic receptor [39]. Although the active state promoted by the G protein would dramatically reduce radiolabelled antagonist association and affinity (and hence the overall proportion of receptors bound to antagonist), it would also “trap” any pre-equilibrated radioligand through a contraction of the extracellular domains of the GPCR, hence the observed reduction in the [3H]DPCPX dissociation rate constant. Strikingly, when these experiments were repeated in the presence of MIPS521, an even more profound retardation of [3H]DPCPX dissociation was observed (Fig 4i, j, Supp Table 4), supporting a synergistic mechanism of active-state stabilisation by the PAM and the G protein.
In conclusion, we present a novel non-opioid analgesic PAM of the A1R, MIPS521, as proof of concept that disease-context-specific selectivity can be achieved through exploiting the cooperativity between an allosteric GPCR modulator and cognate endogenous agonist. The solution of the MIPS521- and ADO-bound A1R-Gi2 cryo-EM structure revealed a hitherto unappreciated detergent/lipid facing allosteric binding pocket at the extrahelical interface of TM 1,6 and 7 of the GPCR and a mechanism of positive cooperativity consistent with stabilisation of the dynamics of the GPCR-G protein ternary complex. Our findings have broad implications for understanding the molecular basis of GPCR allosteric modulation and can facilitate rational, structure-based drug design of improved novel PAMs at this important therapeutic target.
Methods
Neuropathic pain model:
A partial nerve ligation (PNL) or sham surgery was performed to injure the left sciatic nerve of 7-12 week old male and female Sprague-Dawley rats (n = 223 for electrophysiology and behavioural studies), as described previously [40]. Briefly, rats were anaesthetized with isoflurane and the sciatic nerve proximal to its trifurcation was surgically exposed and a single suture was tied around one third to one half of the nerve. Rats were assessed for mechanical allodynia two weeks post-PNL surgery using a von Frey assay and used for electrophysiology and behavioural experiments between 2-3.5 weeks post-surgery. Age-matched Sham surgery rats with all other procedures, but no nerve ligation, were used as controls (n = 71). Rats were housed in a temperature-controlled environment 22±2 °C with a 12 hour light/dark cycle in in groups of 3-4 with the exception of those with intrathecal catheters, which were housed individually to prevent catheter removal. All experiments involving animals were approved by the Monash University Animal Ethics Committee and every precaution was taken to prevent animal suffering during these experiments.
Mechanical allodynia assessment:
Paw withdrawal threshold (PWT) was used to assess the development of mechanical allodynia prior to behavioral or electrophysiology experiments. Animals were acclimatized to clear Perspex containers with a steel mesh floor twice per day for two days before the day of experiment and for around 15 minutes prior to testing. A series of von Frey filaments (0.52-14.8 g) were presented using an up-down paradigm to calculate paw withdrawal threshold, which were converted to bending force [41]. Mechanical PWT was tested prior to surgery on day 0 and 14 days following surgery. A reduction in von Frey threshold from a pre-surgery baseline of ≥9.7g to ≤2.8 g days after surgery was used as a threshold criterion that neuropathic pain had developed [42].
Intrathecal drug administration and in vivo pain behavioural assays:
Twelve days post PNL or sham surgery, chronic polyethylene lumbar intrathecal catheters were surgically inserted into male and female Sprague-Dawley rats (9-14 weeks) to allow intrathecal drug administration into the lumbar spinal region in conscious animals for von Frey studies (n = 89), rotarod (n=31) and conditioned place preference studies (n = 53) [43]. Rats were randomly assigned to treatment groups of 8-10 animals and group sizes were based on previous similar studies by the investigators. Under isoflurane anesthesia, a skin incision was made in the midline over the lumbar spine and the intrathecal space between the fourth and fifth lumbar vertebrae was punctured using a 21 g needle. A catheter was introduced through the needle and advanced 3-4cm in the intrathecal space and the other end tunneled underneath the skin and exteriorized at the neck, flushed with normal saline and sutured in place with silk suture. Two days after catheter placement, drug injections and behavioral testing were conducted. Following baseline mechanical allodynia measurements, VCP171, MIPS521 or DMSO/saline vehicle (blinded to the experimenter) were injected in a 10 μL volume, which is rapidly diluted in cerebrospinal fluid within the intrathecal cavity once injected, followed by 25 μL of saline flush; collectively ensuring that the effective DMSO concentration (at the site of action) is reduced below levels that may be pro-nociceptive in their own right. Catheter patency and placement was confirmed after each experiment by administration of intrathecal lignocaine (20 μL, 1%) which causes brief bilateral hind limb paralysis. Rats that did not develop paralysis were excluded from the data set (n = 8). Each rat was used only once for behavioral testing. For von Frey assays, four animals were tested per session, as this was the maximum that could be surgically prepared in one day and tested at the planned time points by one experimenter.
Conditioned place preference:
The effects on MIPS521 and VCP171 on spontaneous pain were investigated using methods described by King et al, 2009 [44]. Briefly, rats (n = 53, 24 males and 24 females tested, 10-16 weeks) underwent pre-conditioning training in a 3-chamber conditioned place preference (CPP) box, with full access to all chambers, starting 10 days post PNL or sham surgery following intrathecal catheter surgery. On the third training day, behaviour was recorded for 15 minutes and animals that spent more than 80% of their time were eliminated from the study (n = 5). Rats received vehicle control (DMSO/Saline or saline) paired with one chamber in the morning, followed by drug treatment in the opposite chamber 4 hours later. Chamber pairings alternated for each animal in the group and were counterbalanced. The following day (20 hours post drug-pairing), rats were recorded in the CPP box for 15 minutes with access to all chambers.
Rotarod:
An accelerating rotarod (Ugo Basile) was set so speed increased from 6 to 80 rpm over 170 seconds. Naive male and female Sprague Dawley rats (n=31, groups of 3-4), 7-9 weeks of age were trained on the rotarod twice daily for two days (≥2 trials per session) until performance times were stable. On the day of the experiment, three baseline trials were recorded. Compounds were administered through lumbar implanted intrathecal catheters. The control group received vehicle (DMSO/saline) and the positive control group intrathecal morphine (5 μg). Latency to fall (seconds) was measured in triplicate at 0.5, 1, 1.5. 2, 3 and 4 hours post drug administration.
Spinal cord slice electrophysiology:
Adult Sprague-Dawley rats (9-15 weeks) (n=91 PNL rats and sham controls) were anaesthetized with isoflurane, decapitated and the lumbar region of the spinal cord was removed. Parasagittal slices (340 μm thick) of spinal cord were cut on a vibrotome in oxygenated ice-cold sucrose-based artificial CSF (sACSF) that contained (mM): 100 sucrose, 63 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 25 glucose, and 25 NaHCO3. Slices were transferred to a submerged chamber containing NMDG-based recovery ACSF (rACSF) for 15 minutes at 34°C, equilibrated with 95% O2 and 5% CO2 and composed of (mM): 93 NMDG, 2.5 KCI, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 Glucose, 5 Na ascorbate, 2 thiourea, 3 Na pyruvate, 10 MgSO4 and 0.5 CaCl2, and adjusted to pH 7.4 with HCI. Following the recovery incubation slices were transferred to normal oxygenated ACSF where they were allowed to recover for 1 hour at 34° C and maintained at room temperature. Normal ACSF had the following composition (mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 25 glucose, and 11 NaHCO3 and was equilibrated with 95% O2 and 5% CO2. Slices were transferred to a recording chamber and superfused continuously at 2 ml/min with oxygenated normal ACSF which was maintained at 34°C with an inline heater and monitored by a thermister in the slice chamber. Dodt-contrast optics were used to identify laminae I and II neurons in the superficial dorsal horn. Effects of VCP171 on synaptic currents were measured from 46 neurons from sham control rats and 53 neurons from PNL rats. Effects of MIPS521 were measured from 35 neurons from sham control rats and 46 neurons from PNL rats. A Cs+-based internal solution was used to record electrically evoked excitatory post-synaptic currents (eEPSCs) and spontaneous excitatory post-synaptic currents (sEPSCs), which contained (mM): 140 CsCl, 10 EGTA, 5 HEPES, 2 CaCl2, 2 MgATP, 0.3 NaGTP, 5 QX-314.CI and 0.1% biocytin (osmolarity 285-295 mosmol l−1). Moderate expression of adenosine A1R has previously been detected on all C- and A-fibre sensory neuron types [45], which suggests pre-synaptic effects were not limited to one type of nociceptor and may also affect other sensory functions such as mechanosensation and proprioception. However, if this were the case, this would be detected in our rotarod experiments, but we did not see any such changes in sensory functions mediated by the A1R PAMs.
Patch electrodes had a resistance of between 3-5 MΩ. Tungsten electrodes placed in the dorsal roots were used to elicit eEPSCs using a stimulus strength sufficient to evoke reliable submaximal currents, including both C and A fibre input. Synaptic currents were measured in whole-cell voltage-clamp (−70 mV) from superficial laminae neurons. Post-synaptic effects were eliminated with QX-314 in the patch electrodes and Cs to inhibit postsynaptic A1R effects mediated by potassium channels. All eEPSCs were recorded in the presence of gabazine (10 μM) and strychnine (0.5 μM). Following a stable baseline recording, VCP171 or MIPS521 were superfused onto slices at a rate of 2ml/min and reversed using 1 μM DPCPX in normal oxygenated ACSF at 34°C.
Native tissue and in vivo data analysis:
Pooled values are presented as mean ± SEM, or drug effect normalized to the baseline. Statistical tests between a treatment group and vehicle control at specific time points were made using a two-tailed unpaired t test assuming unequal variance. All treatment groups were compared to vehicle control using a one-way ANOVA with Dunnett post hoc test to correct for multiple comparisons. Normalized eEPSC concentration-response data was pooled and fitted with a logistic function using Prism 6 software. Significance was set at * P < 0.05, ** P < 0.01, *** P < 0.001 levels.
Isolated rat atrial experiments:
Animals were humanely killed by CO2 inhalation, or with either 0.5ml of 325mg/ml sodium pentobarbitone or decapitation under isoflurane inhalation anaesthetic. The chest cavity of the rat was opened to fully expose the heart, which was rapidly removed and placed in Krebs-Henseleit solution. The right and left atria were isolated (still attached to each other but detached from the ventricles). The heart was cut transversely below the right and left atria to remove the ventricles and the remaining ventricles were trimmed carefully [24]. Atria were then mounted in an organ bath at 37°C, bubbled with 5% CO2/95% O2 and allowed to contract spontaneously [24]. The rate of atrial contraction was measured using a force-transducer connected to a PowerLab data acquisition system. Concentration-response curves to the prototypical A1R orthosteric agonist N6-Cyclopentyladenosine (CPA), or the PAM MIPS521 were then constructed as in [24]. All experiments involving animals were approved by the Monash University Animal Ethics Committee.
Protein purification constructs:
For structural studies, human A1R in the pVL1392 vector was modified to include an N-terminal Flag tag epitope and a C-terminal 8×histidine tag; both tags are removable by 3C protease cleavage. A dominant-negative Gαi2 construct was generated previously by site directed mutagenesis to incorporate mutations that reduce nucleotide binding, stabilize the G0 state and increase interactions with the βγ subunits [29, 30]
A1R-DNGi2-MIPS521-ADO complex purification:
The expression and purification of the A1R-DNGi2 complex were performed as previously reported [30]. Briefly, A1R and DNGi2 were expressed in HighFive insect cells (Expression Systems, LLC), solubilised separately in 20 mM HEPES pH 7.4, 100 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace), 0.05% (w/v) cholesteryl hemisuccinate (CHS, Anatrace) supplemented with cOmplete Protease Inhibitor Cocktail tables (Roche). Solubilized A1R and heterotrimeric Gi were combined and complex formation was initiated by addition of 1 mM ADO (Sigma), 100nM MIPS521 and apyrase (25 mU ml–1, NEB); followed by 2 h incubation at 4 °C. Insoluble material was removed by centrifugation at 30,000g for 30 min and the solubilized complex was immobilized by batch binding to M1 anti-Flag affinity resin in the presence of 5 mM CaCl2. The resin was packed into a glass column and washed with 20 column volumes of 20 mM HEPES pH 7.4, 100 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 1mM ADO, 0.01% (w/v) LMNG and 0.001% (w/v) CHS supplemented with 1 mM ADO (Sigma), 100 nM MIPS521 before bound material was eluted in buffer containing 10 mM EGTA and 0.1 mg ml-1 Flag peptide. The complex was concentrated using an Amicon Ultra Centrifugal Filter (MWCO 100 kDa) and subjected to size-exclusion chromatography on a Superdex 200 Increase 10/300 column (GE Healthcare) pre-equilibrated with 20 mM HEPES pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM ADO, 100 nM MIPS521, 0.01% (w/v) L-MNG and 0.001% (w/v) CHS to separate complex from contaminants. Eluted fractions consisting of receptor and G-protein complex were pooled, spiked with 100 μM MIPS521 and concentrated. Final yield of purified complex was approximately 0.2 mg l–1 insect cell culture.
SDS–PAGE and western blot analysis:
Samples collected from each purification step were analysed by SDS–PAGE and western blot as previously described [30].
Cryo-EM sample preparation and data acquisition:
Samples were prepared on Quantifoil R1.2/1.3 Cu/Rh 200 mesh grids (Quantifoil, Germany) washed in advance with acetone on a piece of filter paper. The grids were glow discharged less than an hour before sample vitrification in low pressure air for 60 s with 10 mA current in a PIB-10 Ion Bimbarder (JEOL, Japan). The samples were vitrified on a Vitrobot Mark IV (Thermo Fisher Scientific, USA) set to 4 °C and 100 %RH. 3 μl of the sample was applied on the grid, blotted for 10 s and plunge-frozen in liquid ethane kept at close-to-liquid-nitrogen temperature.
Data was collected on Titan Krios (Thermo Fisher Scientific, USA) 300 kV electron microscope equipped with GIF Quantum energy filter and K3 detector (Gatan, USA). The acquisition was performed in EFTEM NanoProbe mode, indicated magnification x105k, spot 4, C2 aperture 50 μm, beam diameter 1.4 μm, zero-loss slit 25 eV, K3 counted mode (2x hardware binning), pixel size 0.83 Å, exposure rate 13.47 counts/pixel/s, exposure time 3.495 s, total exposure 68.34 e/Å2, 70 frames, target defocus range 0.8 – 1.5 μm. The data was collected automatically with homemade scripts for SerialEM [46] performing a 9-hole beam-image shift acquisition scheme with one exposure in the centre of each hole. In total, 4222 movies were acquired with an average throughput of 218 movies/hour.
Data processing:
MIPS521-ADO-A1R-Gi2 dataset - A total of 4222 movies were collected and subjected to the correction of beam-induced motion using MotionCor2 [47]. CTF estimation was performed using Gctf [48] on non-dose-weighted micrographs. The 4006 micrographs, with CTF fit resolution below 4 Å, were selected for further examination. All future processing was performed in Relion 3.0. The 4,975,220 particles were autopicked using templates from a different GPCR dataset, following by 2D and 3D classifications. The 3D refinement of the resulting dataset of 990,833 particles yielded a 3.75 Å map. This dataset was subjected to several rounds of CTF refinement, particle polishing, and further 3D classifications with local angular sampling to yield the final set of 683,928 particles that were used for the final reconstruction. The nominal resolution of the final map was 3.2 Å, based on the gold standard Fourier shell correlation cut-off of 0.143. The final map was sharpened with a B-factor of −80 Å2. Local resolution was determined using the internal local resolution procedure in Relion, using half-reconstructions as input maps.
ADO-A1R-Gi2 dataset - We reprocessed the Volta Phase Plate dataset published in [30] using the new advances in Relion 3.0. The data processing routine was almost identical to that of the MIPS521-ADO-A1R-Gi2 complex. Briefly, 3220 movies were processed using MotionCor2 and Gctf. Template-based picking resulted in ~2.5M particles, which were reduced to 838K following 2D and 3D classification, yielding a 3.6 Å map. Multiple rounds of particle polishing and 3D classification improved the map resolution to 3.46 Å. To further increase the map quality we subtracted the signal from the lipid micelle and the ɑ-helical domain of Gɑi2 subunit. This led to the final 3D reconstruction at 3.3 Å from 716K particles. The final map was sharpened with a B-factor of −80 Å2.
Model building:
Previously deposited coordinates of the ADO-A1R-Gi2 complex (PDB 6D9H) were used as an initial model for building the receptor and Gɑ subunit [30]. The Gβ and Gγ coordinates were taken from the high-resolution x-ray structure of a heterotrimeric G-protein in complex with an antibody fragment (PDB 6CRK) [49]. All models were rigid-body fitted into the cryo-em map, following by iterative model adjustment and rebuilding in COOT [50] and real-space refinement in PHENIX [51]. Restraints for the agonists, ADO and MIPS521, were generated using the GRADE server, https://www.globalphasing.com (v.12.13). Model validation was performed in MolProbity [52]. Figures were prepared in USCF Chimera [53] or PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC).
cAMP assay, ADO vs VCP171/MIPS521.
Inhibition of cAMP accumulation was assessed as described previously [54]. Briefly, FlpIN-CHO cells stably expressing the WT A1R were grown to were grown to 90% confluence and maintained in Dulbecco’s modified eagle medium (DMEM) containing 20 mM HEPES, 5% fetal bovine serum (FBS) and 500 μg/mL of hygromycin B at 37°C in a humidified incubator containing 5% CO2: 95% O2. Cells were then harvested by trypsinization followed by centrifugation (300 g, 5 min). Cells were then seeded into 96-well plates at a density of 20,000 cells/well and incubated overnight at 37°C. Interaction assays were performed at 37°C in stimulation buffer (140 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 0.2 mM Na2HPO4, 0.44 mM KH2PO4, 1.3 mM CaCl2, 5.6 mM D-glucose, 5 mM HEPES, 0.1% bovine serum albumin, and 10 mM rolipram, pH 7.45). Cells were washed and incubated in stimulation buffer for 30 minutes, followed by a 10-minute exposure to either VCP171 or MIPS521, followed by addition of 3 μM forskolin in the absence and presence of increasing concentrations of ADO. After a 30-minute incubation at 37°C, the reaction was terminated by rapid removal of buffer and the addition of 50 mL of ice-cold ethanol. After ethanol evaporation, detection was performed as outlined previously [54]. All data was expressed as a percentage of the forskolin mediated cAMP accumulation.
Whole-cell [3H]-DPCPX saturation and interaction binding assays in whole cells:
Generation of Chinese hamster ovary (CHO) FlpIN cell lines, stably expressing a 3x haemagglutinin (HA)-tagged wildtype human A1R or mutant A1R, [3H]DPCPX saturation binding, and interaction binding in the absence and presence of the orthosteric agonist NECA, with and without MIPS521, were all performed as described previously [55]. Point mutations were generated using the oligonucleotides listed in Supplementary Table 5. All other details were as previously described [17, 30, 55]. Cells were routinely tested and confirmed to be free from mycoplasma contamination.
Molecular docking calculations:
The AutoDockTools package and Autodock4.2.6 [56] were used to prepare and perform molecular docking to the A1R cryo-EM structure. A grid with 48x42x48 points and a spacing of 0.375 Å centered on the binding site was generated using AutoGrid4. The docking calculation was performed using the Lamarckian genetic algorithm with an initial population of 300 individuals, a maximum of 10x106 energy evaluations and 27000 generations. In total, 1000 independent runs were performed, and the lowest energy pose was selected.
Gaussian accelerated molecular dynamics (GaMD):
GaMD is an enhanced sampling method that works by adding a harmonic boost potential to reduce the system energy barriers[21]. When the system potential drops below a reference energy E, the modified potential of the system is calculated as:
| (1) |
where k is the harmonic force constant. The two adjustable parameters E and k can be determined based on three enhanced sampling principles. First, for any two arbitrary potential values and found on the original energy surface, if , ΔV should be a monotonic function that does not change the relative order of the biased potential values; i.e., . Second, if , the potential difference observed on the smoothed energy surface should be smaller than that of the original; i.e., . By combining the first two criteria and plugging in the formula of and , we obtain
| (2) |
Where Vmin and Vmax are the system minimum and maximum potential energies. To ensure that Eq. 2 is valid, k has to satisfy: . Let us define , then 0 < k0 ≤ 1. Third, the standard deviation (SD) of ΔV needs to be small enough (i.e., narrow distribution) to ensure accurate energetic reweighting: , where Vavg and σV are the average and SD of ΔV with σ0 being a user-specified upper limit (e.g., 10kBT) for accurate reweighting. When E is set to the lower bound E = Vmax according to Eq. 2, k0 can be calculated as
| (3) |
Alternatively, when the threshold energy E is set to its upper bound , k0 is calculated as:
| (4) |
If is found to be between 0 and 1. Otherwise, k0 is calculated using Eq. 3.
System Setup.
The MIPS521-ADO-A1R-Gi2 and ADO-A1R-Gi2 cryo-EM structures were used for setting up simulation systems. The initial structures of ADO-bound A1R-MIPS521 and A1R were obtained by removing the Gi2 and Gi2/MIPS521 from the MIPS521-ADO-A1R-Gi2 cryo-EM structure. According to previous findings, intracellular loop (ICL) 3 is highly flexible and removal of ICL3 does not appear to affect GPCR function[57, 58], The ICL3 missing in the cryo-EM structures was thus omitted as in the current GaMD simulations [19], Similar as to previous study[59], the helical domain of the Gi2 protein missing in the cryo-EM structures was not included in the simulation models. This was based on earlier simulation of the β2AR-Gs complex, which showed that the helical domain fluctuated substantially[57]. Six missing residues (LAEDEE) in the s4h3 of Gi2 were added using VMD[60]. All chain termini were capped with neutral groups (acetyl and methylamide). All the disulphide bonds in the complexes (i.e., Cys803.25-Cys169ECL2 and Cys2606.61-Cys263ECL3 in the A1R) that were resolved in the cryo-EM structures were maintained in the simulations. Using the psfgen plugin in VMD[60], missing atoms in protein residues were added and all protein residues were set to the standard CHARMM protonation states at neutral pH. For each of the complex systems, the receptor was inserted into a palmitoyl-oleoyl-phosphatidyl-choline (POPC) bilayer with all overlapping lipid molecules removed using the membrane plugin in VMD[60]. The system charges were then neutralized at 0.15M NaCl using the solvate plugin in VMD[60]. The simulation systems were summarized in Supplementary Table 6.
Simulation Protocol.
The CHARMM36 parameter set[61–63] was used for the proteins and lipids. Force field parameters of the agonist ADO and PAM MIPS521 were obtained from the CHARMM ParamChem web server[64, 65]. GaMD simulations of these systems followed a similar protocol used in previous studies of GPCRs[59, 66, 67]. For each of the complex systems, initial energy minimization, thermalization, and 20ns cMD equilibration were performed using NAMD2.12[68]. A cutoff distance of 12 Å was used for the van der Waals and short-range electrostatic interactions and the long-range electrostatic interactions were computed with the particle-mesh Ewald summation method[69]. A 2-fs integration time step was used for all MD simulations and a multiple-time-stepping algorithm was used with bonded and short-range non-bonded interactions computed every time step and long-range electrostatic interactions every two timesteps. The SHAKE algorithm[70] was applied to all hydrogen-containing bonds. The NAMD simulation started with equilibration of the lipid tails. With all other atoms fixed, the lipid tails were energy minimized for 1,000 steps using the conjugate gradient algorithm and melted with a constant number, volume, and temperature (NVT) run for 0.5 ns at 310 K. The four systems were further equilibrated using a constant number, pressure, and temperature (NPT) run at 1 atm and 310 K for 10 ns with 5 kcal/(mol· Å2) harmonic position restraints applied to the protein and ligand atoms. Final equilibration of each system was performed using a NPT run at 1 atm pressure and 310 K for 0.5 ns with all atoms unrestrained. After energy minimization and system equilibration, conventional MD simulations were performed on each system for 20 ns at 1 atm pressure and 310 K with a constant ratio constraint applied on the lipid bilayer in the X-Y plane.
With the NAMD output structure, the system topology and CHARMM36 force field files, the ParmEd tool in the AMBER package was used to convert the simulation files into the AMBER format[71]. The GaMD module implemented in the GPU version of AMBER18[21, 71] was then applied to perform the simulations. GaMD simulations of A1R-Gi2 and A1R-Gi2-MIPS521 included an 8-ns short cMD simulation used to collect the potential statistics for calculating GaMD acceleration parameters, a 64-ns equilibration after adding the boost potential, and finally three independent 500-ns GaMD production simulations with randomized initial atomic velocities. The average and SD of the system potential energies were calculated every 800,000 steps (1.6 ns). GaMD simulations of A1R and A1R-MIPS521 with smaller system sizes (Supplementary Table 6) included a 2.5-ns short cMD simulation used to collect the potential statistics for calculating GaMD acceleration parameters, a 20-ns equilibration after adding the boost potential, and finally three independent 1000-ns GaMD production simulations with randomized initial atomic velocities. The average and SD of the system potential energies were calculated every 250,000 steps (0.5 ns). All GaMD simulations were run at the “dual-boost” level by setting the reference energy to the lower bound. One boost potential was applied to the dihedral energetic term and the other to the total potential energetic term. The upper limit of the boost potential SD, σ0 was set to 6.0 kcal/mol for both the dihedral and the total potential energetic terms. Similar temperature and pressure parameters were used as in the NAMD simulations. In addition, cMD simulations of the same lengths were also performed on the A1R systems for comparison.
Simulation Analysis.
CPPTRAJ[72] and VMD[60] were used to analyze the GaMD simulations. The root-mean square deviations (RMSDs) of the agonist ADO and PAM MIPS521 relative to the simulation starting structures and the distance between the receptor TM3 and TM6 intracellular ends were selected as reaction coordinates. The distance between the conserved NPxxY motif in the TM7 intracellular end of the A1R and the C terminus of the Gα α5 helix was used to characterize the receptor-G protein interactions. Particularly, distances were calculated between the Cα atoms of receptor residues Arg1053.50 and Glu2296.30, the center-of-mass (COM) distance between the receptor NPxxY motif and the last 5 residues of the Gα α5 helix. Time courses of these reaction coordinates obtained from the GaMD simulation are plotted in Fig 5, Ext Data Fig 5, Ext Data Fig 8 and Ext Data Fig 9. Root-mean-square fluctuations (RMSFs) were calculated for the protein residues and ligands, averaged over three independent GaMD simulations and color-coded for schematic representation of each complex system (Ext Data Fig 10).
A1R nanodisc dissociation experiments
The A1R was inserted into rHDL/nanodisc particles, as previously described [73]. In brief, purified A1R was mixed with MSP1D1 and a lipid mixture (POPC:POPG = 3:2) at a molar ratio of 1:10:800 and the mixture was incubated or 1 h at 4°C. Bio-beads SM2 (0.5 g/mL; Bio-Rad) were added into the mixture and incubated overnight at 4°C. The reconstitution mixture was spun down and the supernatant was loaded onto an M1 anti-Flag affinity column to remove empty rHDL/nanodiscs.
For nanodisc dissociation experiments, the A1R-rHDL particles +/− saturating concentration of Gi2 heterotrimeric protein (1:50 molar ratio) were incubated with 2nM [3H]DPCPX for 1 hour in the presence or absence of 30 μM MIPS521. Rebinding of [3H]DPCPX was prevented by addition of SLV320, and treatments consisted of 10 μM SLV320 and either vehicle, 30 μM MIPS521 or 30 μM MIPS521 and Gi2 heterotrimeric protein. The experiment was stopped by filtration over Whatman GF/C filters. Filters were washed with ice-cold 2% NaCl, dried, and subjected to liquid scintillation counting on a MicroBeta2 plate counter (Perkin-Elmer, MA). Bound ligand never exceeded 10% of the total ligand added. Grouped values are presented as mean ± SEM of n = 3-5 independent experiments performed in duplicate. Normalized group data was curve-fitted to a mono-exponential decay function using Prism 8.2 (GraphPad). All conditions were compared to each other using a one-way ANOVA with Tukey’s post hoc test. Significance was set at * P < 0.05.
Extended Data
Extended Data Figure 1. Physiological effects of VCP171 and MIPS521,
a, Chemical structure of VCP171. b, Time courses of paw withdrawal threshold (PWT) to mechanical stimulus by von Frey filaments in nerve-injured rats post-intrathecal administration of VCP171 (blue) or MIPS521 (red). Significance to vehicle control was determined using Greenhouse-Geisser correction for multiple comparisons, corrected via Dunnett’s post hoc test, * P < 0.05, ** P < 0.01, *** P < 0.001. Data are shown as mean +/− SEM (n=8-10 rats per data group). c, Single trial place preference conditioning with intrathecal VCP171 (30 μg, blue), MIPS521 (10 μg, red) and morphine (10 μg, black) increased the time nerve-injured rats spent in the drug paired chamber, with a corresponding decrease in the vehicle paired chamber. Sham surgery rats showed no chamber preference. Empty circles show individual data points, and bars show mean +SEM (n = 8 per group). Significance was determined using a two-tailed unpaired t test assuming unequal variance, * P < 0.05, ** P < 0.01, compared to vehicle control. d, Rotarod latency in rats following intrathecal administration of VCP171 (blue) or MIPS521 (red) is not significantly different to vehicle controls, whereas intrathecal administration of morphine reduces rotarod latency to fall. Data is shown as mean +/− SEM (n = 3-4 per group). Significance was determined using a two-tailed unpaired t test assuming unequal variance, * P < 0.05, ** P < 0.01, compared to vehicle control. e, Effect of CPA (black; n = 4) or MIPS521 (solid red; n = 6) on rate of atrial contraction. Data represent mean ± SD.
Extended Data Figure 2.
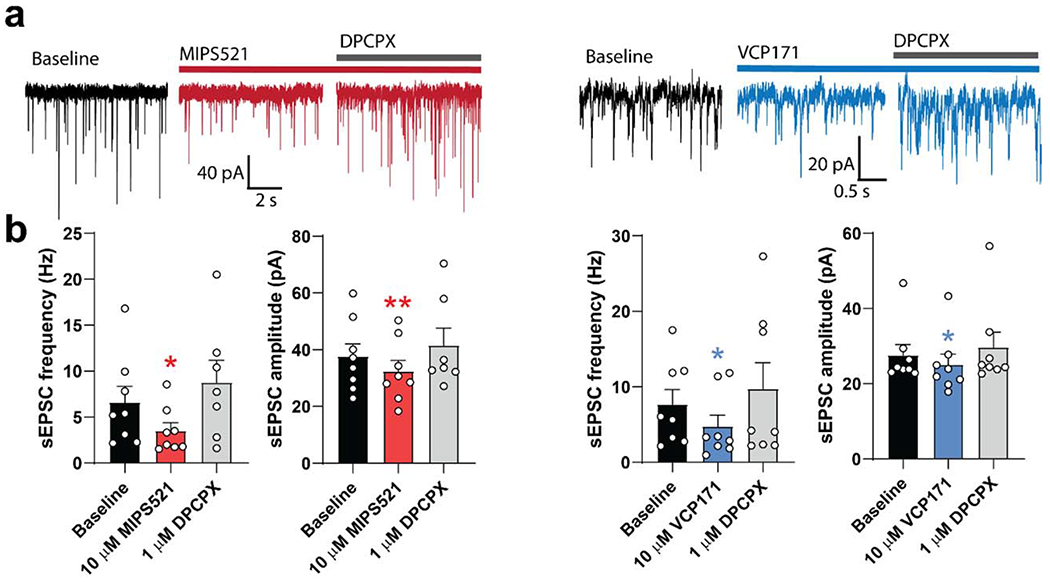
a, Examples of spontaneous excitatory postsynaptic potentials (sEPSCs) recorded from neurons of the superficial laminae of the spinal dorsal horn of nerve-injured rats. b, sEPSC frequency and amplitude were reduced following superfusion of VCP171 or MIPS521, which is reversed by the antagonist, DPCPX (n=8 per group); Data are presented as mean values +/− SEM. Significance compared to baseline was determined using a two-tailed paired t test, * P < 0.05, ** P < 0.01.
Extended Data Figure 3 |. Expression and purification of the MIPS521-ADO–A1R–Gi2 complex.
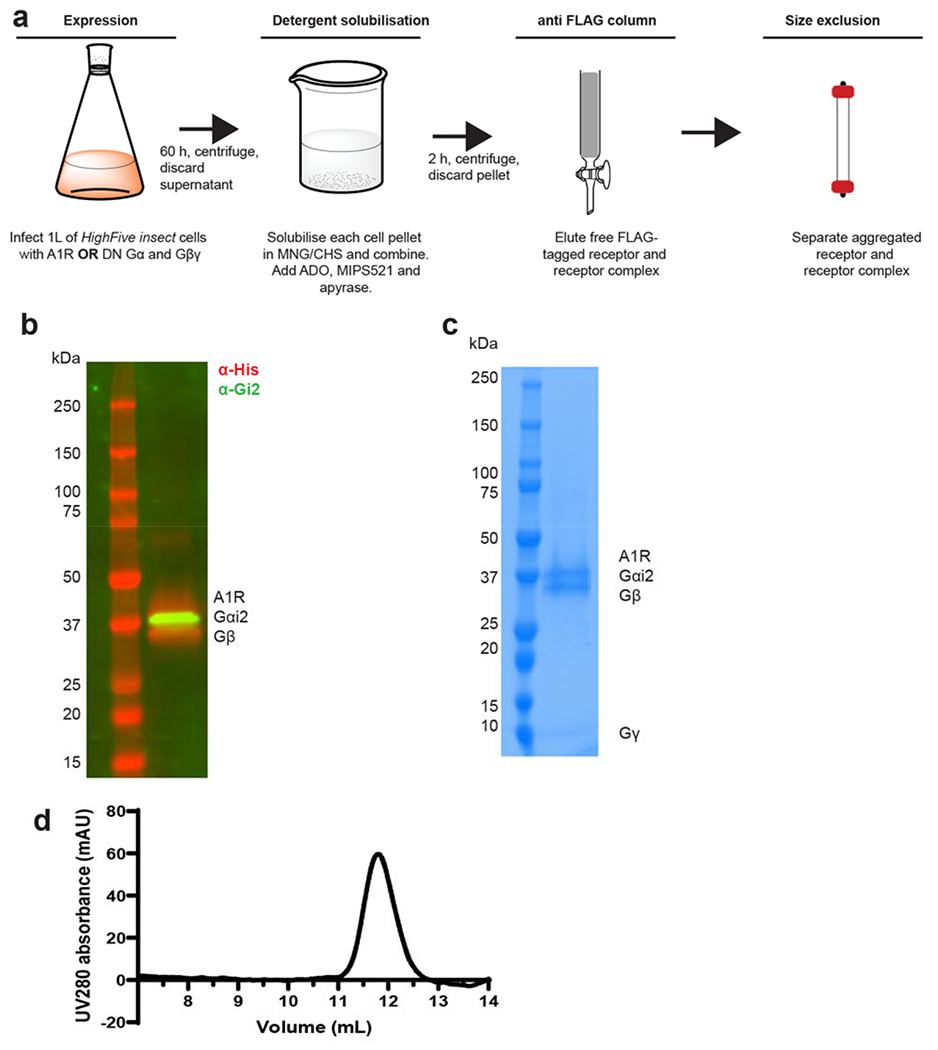
a, Expression and purification flowchart for the A1R–Gi2 complex. A1R and the Gi2 heterotrimer with Gβ1γ2 were expressed separately in insect cell membranes. Addition of ADO (1 mM) and MIP521 (100 nM) initiated complex formation, which was solubilised with 0.5% (w/v) lauryl maltose neopentyl glycol and 0.05% (w/v) cholesteryl hemisuccinate. Solubilised A1R and A1R –Gi2 complex was immobilised on Flag antibody resin. Flag-eluted fractions were purified by size-exclusion chromatography (SEC). b, Representative SDS–PAGE/western blot of the purified A1R–Gi2 complex. An anti-His antibody was used to detect Flag–A1R-His and Gβ1-His (red) and an anti-Gi2 antibody was used to detect Gαi2 (green). Experiment was performed three times with similar results c, Representative SDS–PAGE/Coomassie blue stain of the purified complex concentrated from the Superdex 200 Increase 10/30 column. Experiment was performed three times with similar results d, Representative elution profile of Flag-purified complex on Superdex 200 Increase 10/30 SEC. Experiment was performed three times with similar results
Extended Data Figure 4 |. Cryo-EM data processing for the MIPS521-ADO-A1R-Gi2 (a) and ADO-A1R-Gi2 (b) complexes.
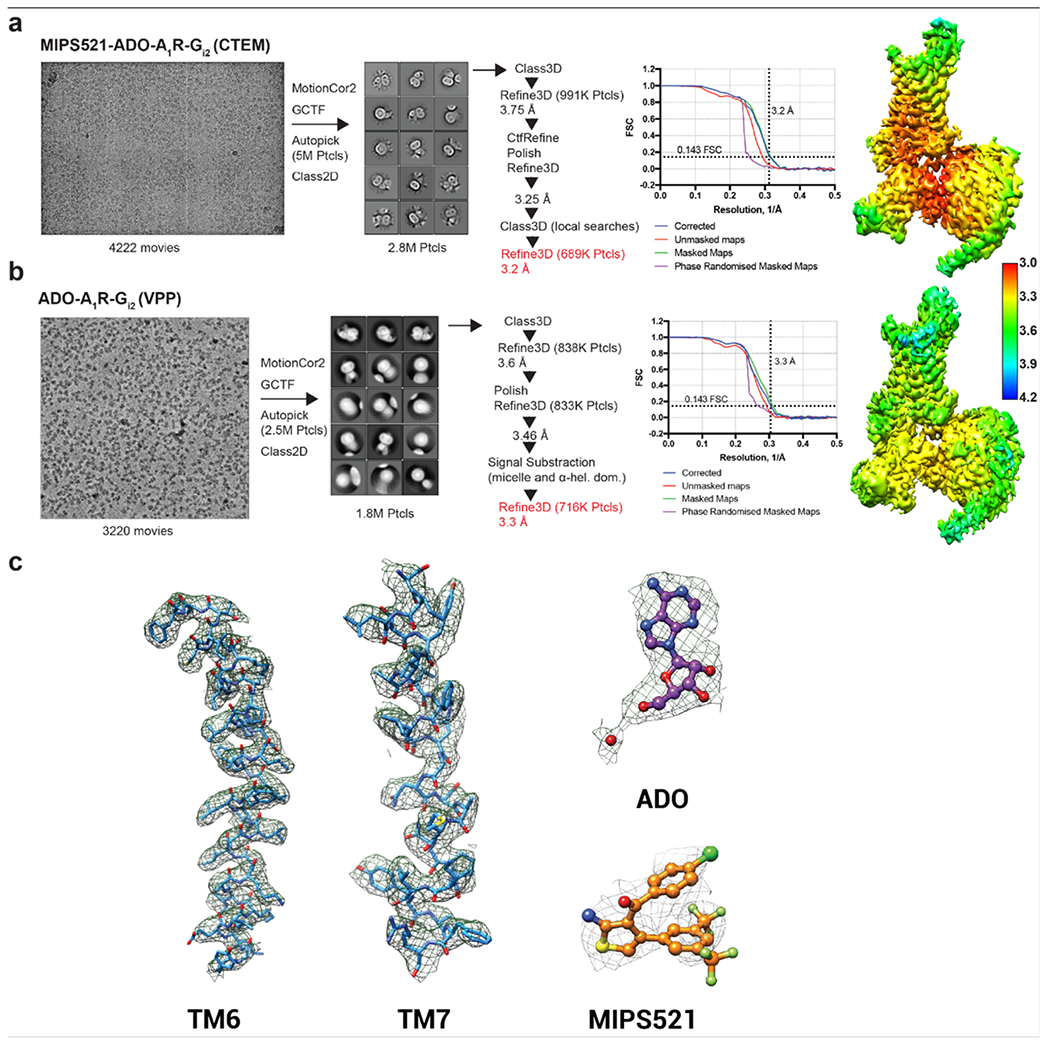
Representative cryo-EM micrographs of each of the complexes. Reference-free 2D class averages of the complexes in LMNG and CHS detergent micelles. Gold-standard Fourier shell correlation (FSC) curves, showing the overall nominal resolution of 3.2 Å and 3.3 Å, respectively, at FSC 0.143. Corresponding 3D cryo-EM maps coloured according to local resolution estimation (Å) in Relion. c, Atomic resolution model of representative regions from the MIPS521-ADO-A1R-Gi2 structure of the A1R transmembrane domain, ADO, and MIPS521. The molecular model is shown in ball and stick representation, coloured by heteroatom, and the cryo-EM map displayed in mesh contoured at 0.02.
Extended Data Figure 5 |. Stable hydrogen bonds formed between residue S6.47/L7.41 in A1R and MIPS521 in A1R-Gi2-MIPS521:
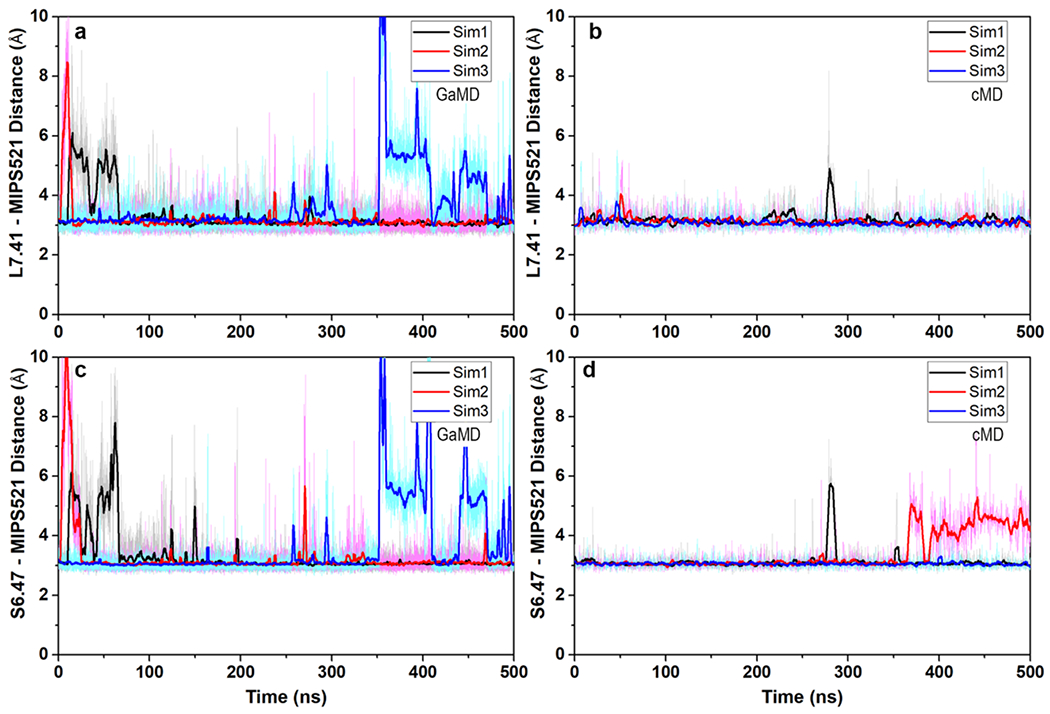
(a and c) GaMD and (b and d) cMD simulations. Each simulation trace is displayed in a different colour (black, red, blue). The lines depict the running average over 2 ns.
Extended Data Figure 6 |. Affinity of orthosteric ligands at mutations of the MIPS521 extrahelical allosteric binding pocket.
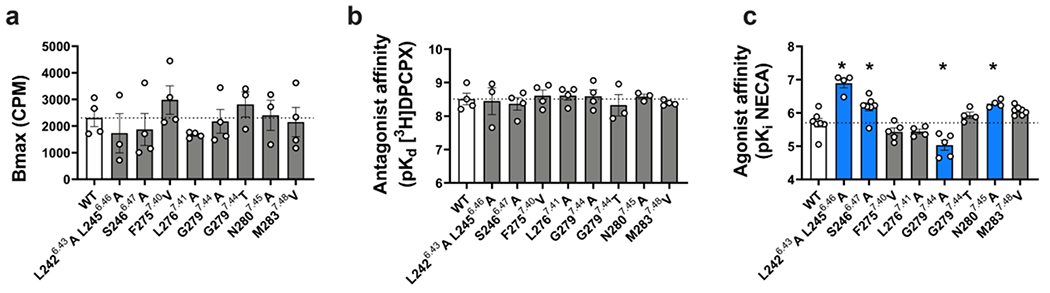
a, c, The affinity of (a) [3H]DPCPX and (c) NECA for wildtype and mutant A1Rs performed in FlpInCHO cells. b, Bmax; determined by [3H]DPCPX radioligand saturation binding studies. Data are the means + S.E.M. of 3-7 independent experiments (shown as circles) performed in duplicate. *P < 0.05 (compared with WT; one-way analysis of variance, Dunnett’s post hoc test).
Extended Data Figure 7 |. Extrahelical binding sites for allosteric modulators of class A GPCRs.
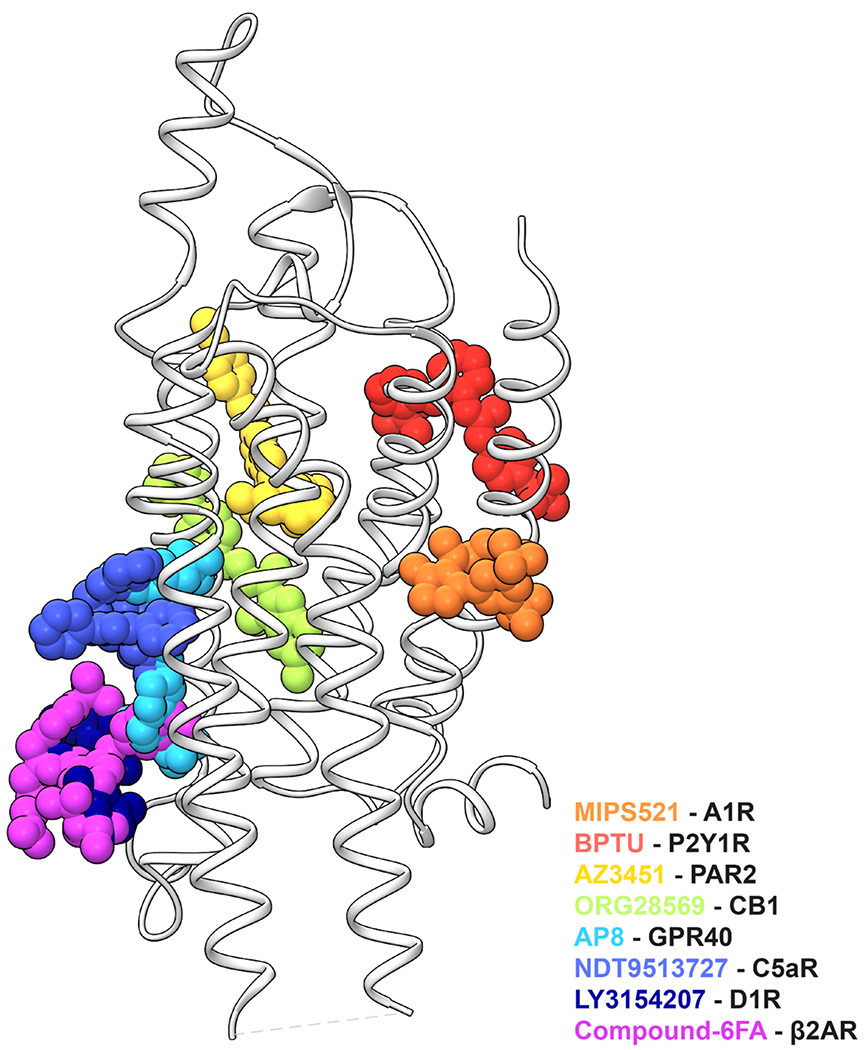
The unique extrahelical binding pose of MIPS521 in the A1R (orange) compared to previously reported extrahelical allosteric binding pockets for class A GPCRs in P2Y1R (BPTU, red; PDB 4XNV), PAR2 (AZ3451, yellow; PDB 5NDZ), CB1 (ORG28569, green; 6KQI), GPR40 (AP8, cyan; PDB 5TZY), C5aR (NDT9513727, blue; PDB 5O9H), D1R (LY3154207, navy; PDB 7LJD), and β2AR (Compound-6FA, pink; PDB 6N48).
Extended Data Figure 8 |. Stability of MIPS521 at the allosteric binding site of A1R is enhanced by Gi2 protein coupling to the receptor.
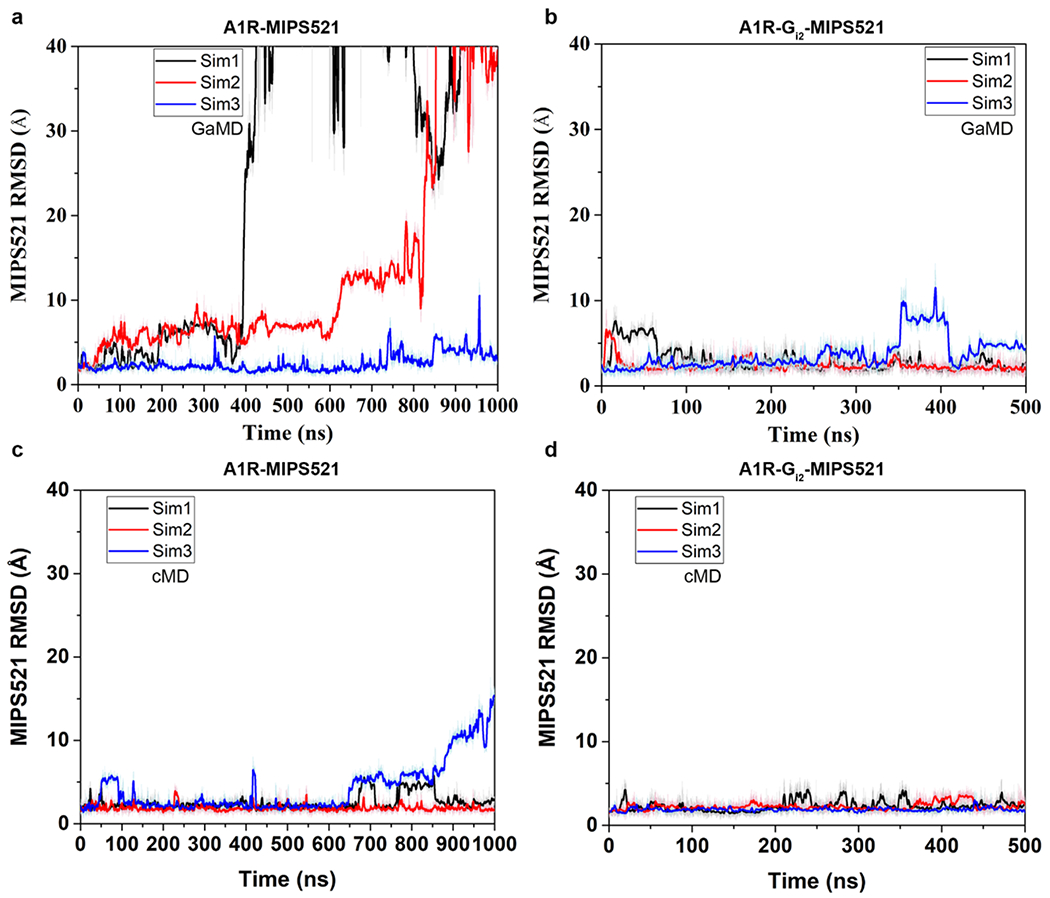
a, b, RMSD (Å) of MIPS521 relative to the starting cryo-EM conformation obtained from GaMD simulations in the (a) absence and (b) presence of Gi2. c, d, RMSD (Å) of MIPS521 relative to the starting cryo-EM conformation obtained from cMD simulations in the (c) absence and (d) presence of Gi2. Each condition represents three GaMD/cMD simulations, with each simulation trace displayed in a different colour (black, red, blue). Lines depict the running average over 2 ns.
Extended Data Figure 9 |. MIPS521 stabilises the A1R-Gi2 ternary complex.
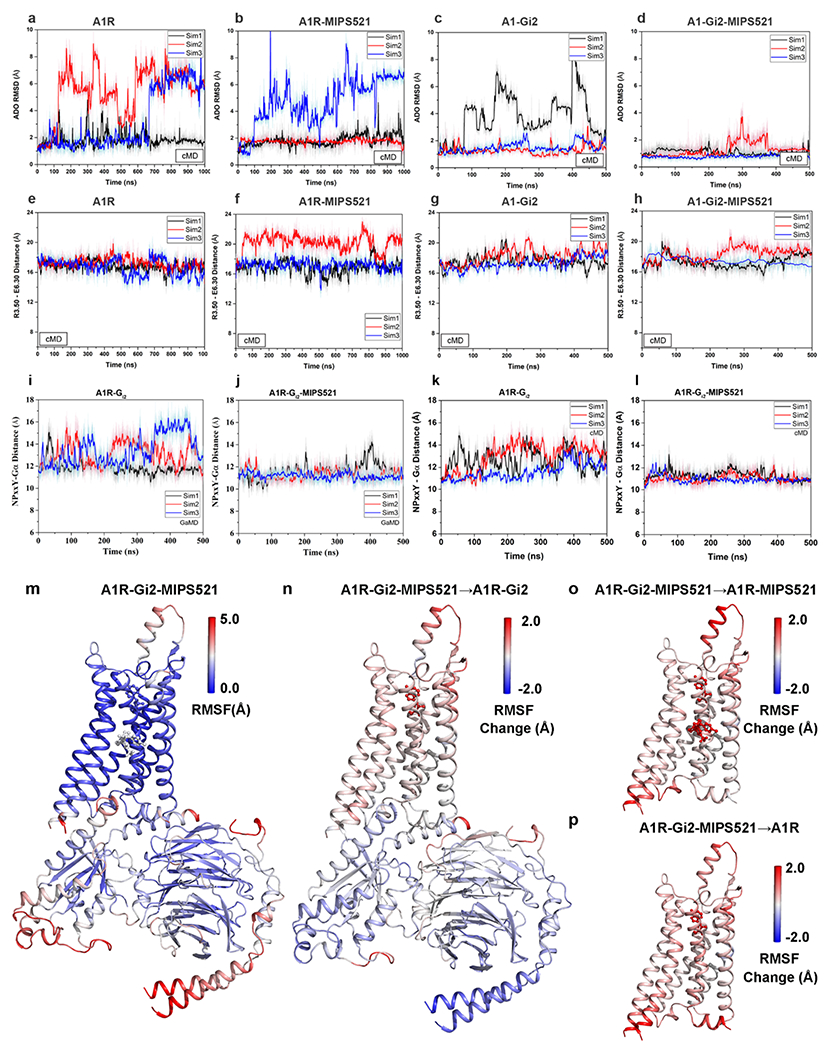
a-d, RMSD (Å) of ADO from cMD simulations completed in the (a) absence or (b) presence of MIPS521, (c) Gi2, or (d) both Gi2 and MIPS521. e-h, Distance between the intracellular ends of TM3 and TM6 (measured as the distance in Å between Arg1053.50 and Glu2296.30) in the (e) absence or (f) presence of MIPS521, (g) Gi2, or (h) both Gi2 and MIPS521. Each condition represents three cMD simulations, with each simulation trace displayed in a different colour (black, red, blue). The lines depict the running average over 2 ns. i,j, Distance between A1R and Gi2 (measured as the distance in Å between the NPxxY motif of A1R and the C terminus of the Gα α5 helix) from GaMD simulations in the (i) absence and (j) presence of MIPS521. k,l, Distance between A1R and Gi2 from cMD simulations in the (k) absence and (l) presence of MIPS521. Each condition represents three GaMD/cMD simulations, with each simulation trace displayed in a different colour (black, red, blue). Thick lines depict the running average over 2 ns. m-p, Flexibility change upon removal of PAM and/or Gi2 protein from the ADO-bound A1R obtained from GaMD simulations. (m) Root-mean-square fluctuations (RMSFs) of the A1R-Gi2-MIPS521. A colour scale of 0.0 Å (blue) to 5.0 Å (red) was used. (n) Change in the RMSFs of the A1R-Gi2 when MIPS521 was removed from A1R-Gi2-MIPS521. (o) Change in the RMSFs of the A1R and MIPS521 when the Gi2 was removed from A1R-Gi2-MIPS521. (p) Change in the RMSFs of the A1R when the Gi2 and MIPS521 were removed from A1R-Gi2-MIPS521 system. A colour scale of −2.0 Å (blue) to 2.0 Å (red) was used for n, o and p.
Extended Data Figure 10 |.
Cryo-EM data collection, refinement and validation statistics
Supplementary Material
Acknowledgements
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) project grants 1145420 and 1147291, NHMRC program grant 1050083, American Heart Association grant 17SDG33370094 and the National Institutes of Health grant R01GM132572. P.M.S., W.L.I. and D.W. are NHMRC Senior Principal Research, Career Development and Senior Research Fellows, respectively. C.J.D-J., A.G. and D.M.T. are Australian Research Council Discovery Early Career Research Fellows. L.T.M. is an Australian Heart Foundation Future Leaders Fellow. J.C. acknowledges support from the Swedish Research Council (2017-04676). The authors are grateful to Prof. Susan Charman and Dr. Karen White, from the Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, for performing the VCP171 and MIPS521 plasma and liver microsome stability studies. The authors acknowledge use of facilities within the Monash Ramaciotti Cryo-EM platform. This work was supported by the MASSIVE HPC facility (www.massive.org.au) and the Extreme Science and Engineering Discovery Environment supercomputing award TG-MCB180049.
Footnotes
The authors declare no competing financial interests.
Data availability
Cryo-EM coordinates are deposited in the PDB under the accession codes 7LD3 (MIPS521 & ADO bound A1R-Gi2 complex), 7LD4 (ADO bound A1R-Gi2 complex); the corresponding EM maps are deposited in the EMDB under accession codes EMD-23280, EMD-23281. Source data are provided with this paper.
References
- 1.Nakamura I, Ohta Y, and Kemmotsu OM, Characterization of Adenosine Receptors Mediating Spinal Sensory Transmission Related to Nociceptive Information in the Rat. Anesthesiology, 1997. 87(3): p. 577–584. [DOI] [PubMed] [Google Scholar]
- 2.Poon A and Sawynok J, Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain, 1998. 74(2-3): p. 235–45. [DOI] [PubMed] [Google Scholar]
- 3.Zylka MJ, Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends in molecular medicine, 2011. 17(4): p. 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King A, Analgesia without opioids. Nature, 2019. 573(7773): p. S4. [Google Scholar]
- 5.Busse JW, et al. , Opioids for Chronic Noncancer Pain: A Systematic Review and Meta-analysis. JAMA, 2018. 320(23): p. 2448–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribeiro JA, Sebastião AM, and de Mendonça A, Adenosine receptors in the nervous system: pathophysiological implications. Prog Neurobiol, 2002. 68(6): p. 377–92. [DOI] [PubMed] [Google Scholar]
- 7.Choca JI, Proudfit HK, and Green RD, Identification of A1 and A2 adenosine receptors in the rat spinal cord. J Pharmacol Exp Ther, 1987. 242(3): p. 905–10. [PubMed] [Google Scholar]
- 8.Choca JI, Green RD, and Proudfit HK, Adenosine A1 and A2 receptors of the substantia gelatinosa are located predominantly on intrinsic neurons: an autoradiography study. J Pharmacol Exp Ther, 1988. 247(2): p. 757–64. [PubMed] [Google Scholar]
- 9.Yang Z, et al. , Cardiac overexpression of A1-adenosine receptor protects intact mice against myocardial infarction. Am J Physiol Heart Circ Physiol, 2002. 282(3): p. H949–55. [DOI] [PubMed] [Google Scholar]
- 10.Christopoulos A and Kenakin T, G Protein-Coupled Receptor Allosterism and Complexing. Pharmacological Reviews, 2002. 54(2): p. 323–374. [DOI] [PubMed] [Google Scholar]
- 11.May LT, et al. , Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol, 2007. 47: p. 1–51. [DOI] [PubMed] [Google Scholar]
- 12.Bruns RF and Fergus JH, Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol, 1990. 38(6): p. 939–49. [PubMed] [Google Scholar]
- 13.Li X, et al. , Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain, 2002. 97(1-2): p. 117–25. [DOI] [PubMed] [Google Scholar]
- 14.Childers SR, et al. , Allosteric modulation of adenosine A1 receptor coupling to G-proteins in brain. J Neurochem, 2005. 93(3): p. 715–23. [DOI] [PubMed] [Google Scholar]
- 15.Vincenzi F, et al. , TRR469, a potent A(1) adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology, 2014. 81: p. 6–14. [DOI] [PubMed] [Google Scholar]
- 16.Gramec D, Peterlin Mašič L, and Sollner Dolenc M, Bioactivation Potential of Thiophene-Containing Drugs. Chemical Research in Toxicology, 2014. 27(8): p. 1344–1358. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen AT, et al. , Role of the Second Extracellular Loop of the Adenosine A1 Receptor on Allosteric Modulator Binding, Signaling, and Cooperativity. Mol Pharmacol, 2016. 90(6): p. 715–725. [DOI] [PubMed] [Google Scholar]
- 18.Miao Y, et al. , Structural Basis for Binding of Allosteric Drug Leads in the Adenosine A1 Receptor. Sci Rep, 2018. 8(1): p. 16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glukhova A, et al. , Structure of the Adenosine A(1) Receptor Reveals the Basis for Subtype Selectivity. Cell, 2017. 168(5): p. 867–877.e13. [DOI] [PubMed] [Google Scholar]
- 20.Thal DM, et al. , Structural insights into G-protein-coupled receptor allostery. Nature, 2018. 559(7712): p. 45–53. [DOI] [PubMed] [Google Scholar]
- 21.Miao Y, Feher VA, and McCammon JA, Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput, 2015. 11(8): p. 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imlach WL, et al. , A positive allosteric modulator of the adenosine A1 receptor selectively inhibits primary afferent synaptic transmission in a neuropathic pain model. Molecular pharmacology, 2015. 88(3): p. 460–468. [DOI] [PubMed] [Google Scholar]
- 23.Aurelio L, et al. , Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem, 2009. 52(14): p. 4543–7. [DOI] [PubMed] [Google Scholar]
- 24.Valant C, et al. , Separation of on-target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. Proc Natl Acad Sci U S A, 2014. 111(12): p. 4614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulte G, et al. , Distribution of antinociceptive adenosine A1 receptors in the spinal cord dorsal horn, and relationship to primary afferents and neuronal subpopulations. Neuroscience, 2003. 121(4): p. 907–16. [DOI] [PubMed] [Google Scholar]
- 26.Wu Z-Y, et al. , Endomorphin-2 Decreases Excitatory Synaptic Transmission in the Spinal Ventral Horn of the Rat. Frontiers in neural circuits, 2017. 11: p. 55–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geiger J, LaBella F, and Nagy J, Characterization and localization of adenosine receptors in rat spinal cord. The Journal of Neuroscience, 1984. 4(9): p. 2303–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johansson B, et al. , Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proceedings of the National Academy of Sciences, 2001. 98(16): p. 9407–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang YL, et al. , Dominant Negative G Proteins Enhance Formation and Purification of Agonist-GPCR-G Protein Complexes for Structure Determination. ACS Pharmacol Transl Sci, 2018. 1(1): p. 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Draper-Joyce CJ, et al. , Structure of the adenosine-bound human adenosine A(1) receptor-G(i) complex. Nature, 2018. 558(7711): p. 559–563. [DOI] [PubMed] [Google Scholar]
- 31.Leach K, et al. , Molecular mechanisms of action and in vivo validation of an M 4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology, 2010. 35(4): p. 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang D, et al. , Two disparate ligand-binding sites in the human P2Y 1 receptor. Nature, 2015. 520(7547): p. 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng RK, et al. , Structural insight into allosteric modulation of protease-activated receptor 2. Nature, 2017. 545(7652): p. 112. [DOI] [PubMed] [Google Scholar]
- 34.Robertson N, et al. , Structure of the complement C5a receptor bound to the extrahelical antagonist NDT9513727. Nature, 2018. 553(7686): p. 111–114. [DOI] [PubMed] [Google Scholar]
- 35.Shao Z, et al. , Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat Chem Biol, 2019. 15(12): p. 1199–1205. [DOI] [PubMed] [Google Scholar]
- 36.Lu J, et al. , Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nature structural & molecular biology, 2017. 24(7): p. 570. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, et al. , Mechanism of β2AR regulation by an intracellular positive allosteric modulator Science, 2019. 364(6447): p. 1283–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhuang Y, et al. , Mechanism of dopamine binding and allosteric modulation of the human D1 dopamine receptor. Cell Research, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeVree BT, et al. , Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature, 2016. 535(7610): p. 182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Method References
- 40.Seltzer Z, Dubner R, and Shir Y, A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain, 1990. 43(2): p. 205–18. [DOI] [PubMed] [Google Scholar]
- 41.Bonin RP, Bories C, and De Koninck Y, A simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments. Mol Pain, 2014. 10: p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imlach WL, et al. , Glycinergic dysfunction in a subpopulation of dorsal horn interneurons in a rat model of neuropathic pain. Sci Rep, 2016. 6: p. 37104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Størkson RV, et al. , Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods, 1996. 65(2): p. 167–72. [DOI] [PubMed] [Google Scholar]
- 44.King T, et al. , Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci, 2009. 12(11): p. 1364–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Usoskin D, et al. , Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci, 2015. 18(1): p. 145–53. [DOI] [PubMed] [Google Scholar]
- 46.Schorb M, et al. , Software tools for automated transmission electron microscopy. Nature Methods, 2019. 16(6): p. 471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng SQ, et al. , MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods, 2017. 14(4): p. 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang K, Gctf: Real-time CTF determination and correction. J Struct Biol, 2016. 193(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maeda S, et al. , Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nature Communications, 2018. 9(1): p. 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, et al. , Features and development of Coot. Acta Crystallogr D Biol Crystallogr, 2010. 66(Pt 4): p. 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams PD, et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr, 2010. 66(Pt 2): p. 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen VB, et al. , MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr, 2010. 66(Pt 1): p. 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pettersen EF, et al. , UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem, 2004. 25(13): p. 1605–12. [DOI] [PubMed] [Google Scholar]
- 54.Baltos JA, et al. , Quantification of adenosine A(1) receptor biased agonism: Implications for drug discovery. Biochem Pharmacol, 2016. 99: p. 101–12. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen AT, et al. , Extracellular Loop 2 of the Adenosine A1 Receptor Has a Key Role in Orthosteric Ligand Affinity and Agonist Efficacy. Mol Pharmacol, 2016. 90(6): p. 703–714. [DOI] [PubMed] [Google Scholar]
- 56.Morris GM, et al. , AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry, 2009. 30(16): p. 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dror RO, et al. , Structural basis for nucleotide exchange in heterotrimeric G proteins. Science, 2015. 348(6241): p. 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dror RO, et al. , Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA, 2011. 108(46): p. 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J and Miao Y, Mechanistic Insights into Specific G Protein Interactions with Adenosine Receptors. J. Phys. Chem. B, 2019. 123(30): p. 6462–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humphrey W, Dalke A, and Schulten K, VMD: Visual molecular dynamics. J. Mol. Graph , 1996. 14(1): p. 33–38. [DOI] [PubMed] [Google Scholar]
- 61.Vanommeslaeghe K and MacKerell AD, CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta. Gen. Subj , 2015. 1850(5): p. 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang J, et al. , CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods, 2016. 14: p. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klauda JB, et al. , Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B, 2010. 114(23): p. 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vanommeslaeghe K and MacKerell AD, Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model, 2012. 52(12): p. 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanommeslaeghe K, Raman EP, and MacKerell AD, Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model , 2012. 52(12): p. 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miao Y and McCammon JA, Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. USA, 2016. 113(43): p. 12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miao Y and McCammon JA, Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. Proc. Natl. Acad. Sci. USA, 2018. 115(12): p. 3036–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phillips JC, et al. , Scalable molecular dynamics with NAMD. J. Comput. Chem, 2005. 26(16): p. 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Darden T, York D, and Pedersen L, Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys, 1993. 98: p. 10089. [Google Scholar]
- 70.Ryckaert J-P, Ciccotti G, and Berendsen HJ, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys, 1977. 23(3): p. 327–341. [Google Scholar]
- 71.Bernstein N, et al. , QM/MM simulation of liquid water with an adaptive quantum region. Physical Chemistry Chemical Physics, 2012. 14(2): p. 646–656. [DOI] [PubMed] [Google Scholar]
- 72.Roe DR and Cheatham TE, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput, 2013. 9(7): p. 3084–3095. [DOI] [PubMed] [Google Scholar]
- 73.Whorton MR, et al. , A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A, 2007. 104(18): p. 7682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM coordinates are deposited in the PDB under the accession codes 7LD3 (MIPS521 & ADO bound A1R-Gi2 complex), 7LD4 (ADO bound A1R-Gi2 complex); the corresponding EM maps are deposited in the EMDB under accession codes EMD-23280, EMD-23281. Source data are provided with this paper.