

Availability of next-generation sequencing has provided many insights into the clonal architecture and dynamics of leukemogenic events,1 but in a significant proportion (10–50%), causative molecular lesions have not yet been identified. Here we report the discovery of novel somatic mutations in the RIT1 gene in patients with myeloproliferative or mixed myelodysplastic/myeloproliferative neoplasms (MDS/MPN), in particular, chronic myelomonocytic leukemia (CMML).
Somatic mutations of the Ras gene family are present in 20–30% of all human cancers,2 including 15–20% of acute myeloid leukemia (AML), 10–50% of MDS and 11–27% of MDS/MPN cases.3,4 In hematology, certain forms of congenital rasopathies are associated with juvenile myelomonocytic leukemia.5,6 Although all Ras family members are characterized by guanosine-triphosphatase (GTPase) activity and maintain highly conserved domains (G1–G5), they possess distinct biochemical and biological activities and express both overlapping and unique functions under different conditions and in different cell types.7 Ras-like-without-CAAX-1 (RIT1) gene is a new member of the family and has been found to be critical in neuron stress-mediated survival.8 Moreover, RIT1 has been reported to be overexpressed due to amplification and occasionally mutation in a proportion (25%) of patients with hepatocellular carcinoma (HCC).9,10
We applied whole-exome sequencing to a subgroup of patients with various forms of myeloid malignancies (seven MDS and AML with myelodysplasia-related changes (AML-MRC) and eight MDS/MPN) and found two closely positioned somatic RIT1 mutations in two cases with CMML and AML-MRC (c.T244A and c.T245G) corresponding to the codon F82 (Figure 1). Sequencing of serial informative cases showed the presence of RIT1 mutation early at the initial diagnosis, suggesting its ancestral origin (Supplementary Figure 1A). Screening for RIT1 mutations was expanded to a total of 722 patients with various myeloid neoplasms (Supplementary Table 1): all together, nine mutant cases were found: six in the amino acid F82, two in E81 and one in M90. All mutations were located on the Switch II effector domain of this protein, which is conserved among species (Figure 1). Of note, the implicated residues are very close to codon Q79; this codon is analogous to the amino acid Q61 in Ras family proteins such as NRAS or KRAS, where mutations frequently occur in cancer. Moreover, the experimental Q79L mutation in RIT1 has been reported to confer constitutive activation of the protein.11
Figure 1.

Functional domains of RIT1, location of mutations and amplifications. Mutations are located in conserved sites among species. Amino acids in which a mutation was found are shown in a red rectangle. Sanger sequencing of tumor and CD3+ fraction of the two index cases with RIT1 mutations. GTP-binding domains appear in green, whereas Switch effector domains appear in blue, both with codifying amino acids. RIT1 is located at 1q22 (red bar). Blue bars show the amplified region in each patient.
Parallel analyses using SNP-array-based karyotyping and sequencing demonstrated that mutations were heterozygous with the exception of one case, which showed 1q amplification involving the RIT1 locus: in this case the mutant allele was duplicated (Supplementary Figure 2A). Complimentary DNA sequencing revealed that both mutant and wild-type alleles were equally expressed in all cases (Supplementary Figure 2B).
The E81Q somatic RIT1 mutation and alterations of amino acids located toward the N-terminus of the protein have been described in solid cancers.10 RIT1 maps to the minimal common amplified region (1q21–22) in 1q gains frequently found in HCC (~25%); RIT1 amplification is associated with overexpression of the corresponding mRNA in these cases.9 In our cohort we found 10 patients (1.8%) with 1q amplification involving the RIT1 locus (Figure 1): RIT1 mutation was found only in one of these cases and, similar to HCC, amplifications were associated with mRNA overexpression (median relative ratio 1.29 in cases with 1q+ vs 0.18 in wild-type cases, P=0.041) (Figure 2A). The clinical phenotype of cases with 1q amplification resembled those with RIT1 mutations, including CMML (n=3) and advanced MDS (n=3). The remaining four cases had myelofibrosis; three out of four were secondary to other forms of MPN (Supplementary Table 2). Similar to serial analyses of RIT1 mutant cases, sequential cytogenetic evaluation showed the presence of 1q+ in the myelofibrotic stage, but absent at the initial polycythemia vera presentation (Supplementary Figure 1), consistent with the fact that chromosomal abnormalities are rare among chronic stages of MPN, but can be acquired at the time of fibrotic evolution.12
Figure 2.
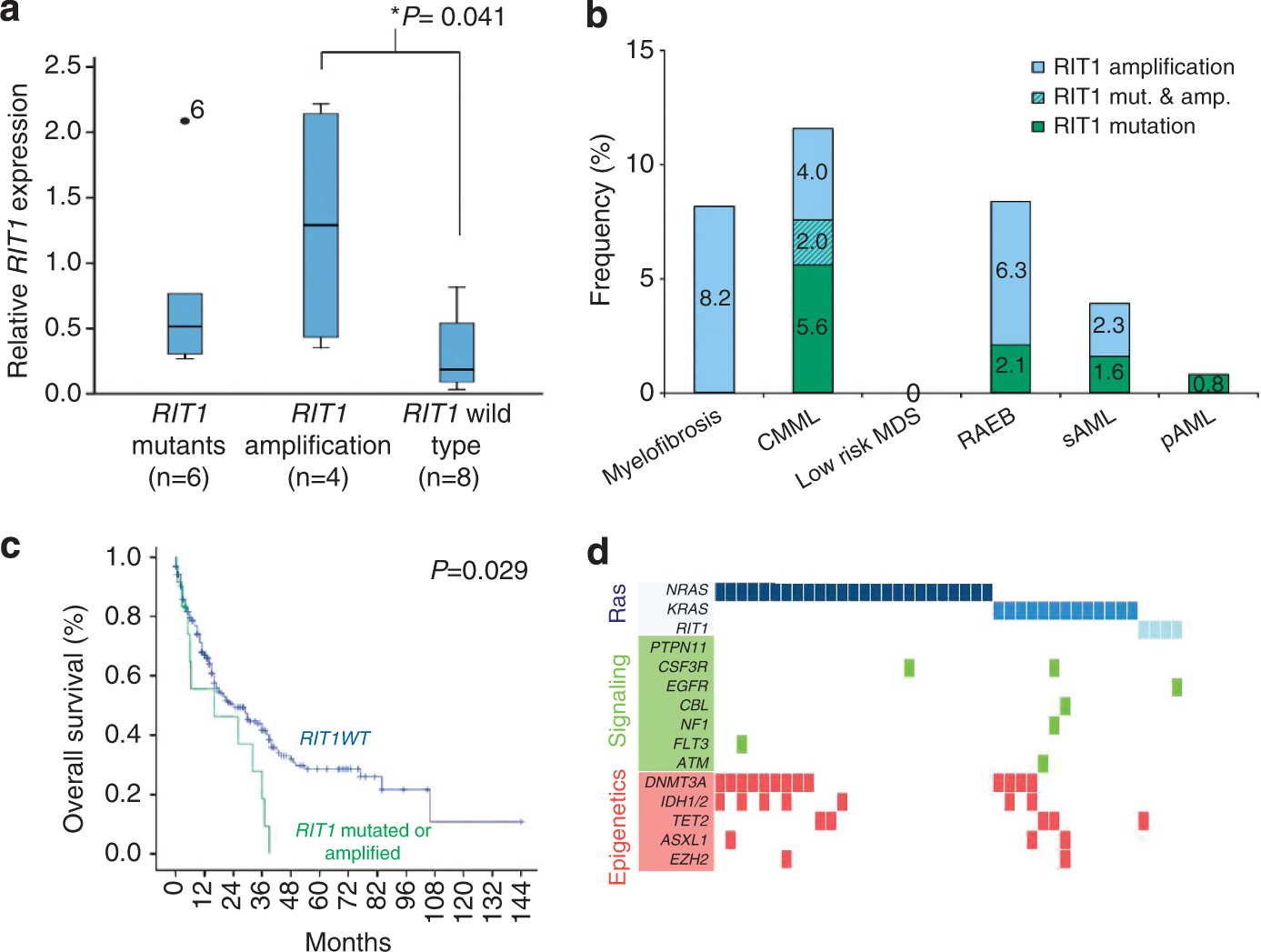
(a) Relative RIT1 expressions in patients with mutations, amplifications or wild-type gene. (b) Clinical distribution of RIT1 anomalies in myeloid neoplasms. (c) Survival curve of patients with MDS or MDS/MPN diagnosis with and without RIT1 abnormalities. (d) Molecular associations of Ras mutants in a cohort of patients studied by whole-exome sequencing, that appeared to be mutually exclusive with themselves and other mutations in genes of signaling pathways and to be related to mutations in genes involved in epigenetic regulation.
RIT1 has been shown to increase phosphorylation of AKT and thereby inhibit apoptosis and also activate proliferation through mitogen-activated protein kinase.8,13,14 Consistent with these properties, we found that BAD expression was decreased while BCL2 increased in cells with RIT1 mutations or amplifications. Similarly, MYC mRNA levels were elevated in these cases.
Comparison of RIT1 mutations with other somatic mutations in our whole-exome sequencing discovery cohort (Supplementary Table 9) demonstrated that mutations affecting Ras gene family are mutually exclusive (Figure 2d). Only in one RIT1 mutant case with 1q amplification a concomitant KRAS mutation was found. RIT1 lesions were less likely associated with FLT3 mutations (2.4% vs 18.6%, P=0.003), but occurred more commonly with DNMT3A mutations (31% vs 16%, respectively, P=0.021). In myelodysplastic subcohort (MDS, MDS/MPN and AML-MRC), Ras and RIT1 mutations were coincident with cohesin complex mutations (STAG2, SMC1A, SMC3 and RAD21; 27% vs 5%, respectively, P=0.015). Similarly, when a larger cohort of patients was analyzed by Sanger sequencing, TET2 mutant cases were coincident with RIT1 abnormalities (30% of RIT1 mutant cases vs 9% in RIT1 wild type, P=0.059) consistent with the presence of both mutations in MDS/MPN.
Because RIT1 abnormalities occur in a diverse molecular context, a distinct phenotype was difficult to discern. However, RIT1 abnormalities were significantly more frequent in CMML than other myeloid neoplasms (56% vs 9%, P=0.001) and also were associated with −7/del(7q) (33% vs 6%, P=0.017; Supplementary Table 3). RIT1 mutant and amplified cases showed a trend toward having a shorter median overall survival (19 vs 14 months, P=0.053) in both the total cohort but more pronounced when studied in patients with MDS or MDS/MPN (24 vs 16 months, P=0.029) (Supplementary Table 4). Multivariate analysis showed that the impact of RIT1 abnormalities was related to their occurrence with other high-risk features.
In conclusion, we demonstrate here that RIT1 abnormalities, including activating mutations and locus amplifications, are novel lesions in a subgroup of patients with myeloid neoplasms, particularly frequent in CMML, where the percentage rose to 11% in our series, including 7% of mutations and 4% of locus duplications.
Supplementary Material
ACKNOWLEDGEMENTS
We thank The Cancer Genome Atlas (TCGA) for access to the whole-genome sequencing results described in the text. This work was supported by the National Institutes of Health (NIH; Bethesda, MD) grants RO1HL-082983 (JPM), U54 RR019391 (JPM), K24 HL-077522 (JPM), a grant from the AA &MDS International Foundation (Rockville, MD), the Robert Duggan Charitable Fund (JPM; Cleveland, OH), and Scott Hamilton CARES grant (HM; Cleveland, OH), Grant-in-Aids from the Ministry of Health, Labor and Welfare of Japan and KAKENHI (23249052, 22134006 and 21790907) (SO; Tokyo), and the Grant ‘Rio Hortega’ from Instituto de Salud Carlos III (IGS; CM10/00321; Ministry of Science and Innovation, Spain).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C et al. Clonal diversity ofrecurrently mutated genes in myelodysplastic syndromes. Leukemia 2013; 27: 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bos J Ras oncogene in human cancer: a review. Cancer Res 1989; 49: 4682–4689. [PubMed] [Google Scholar]
- 3.Kadia TM, Kantarjian H, Kornblau S, Borthakur G, Faderl S, Freireich EJ et al. Clinical and proteomic characterization of acute myeloid leukemia with mutated RAS. Cancer 2012; 118: 5550–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dicker F, Haferlach C, Sundermann J, Wendland N, Weiss T, Kern W et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia 2010; 24: 1528–1532. [DOI] [PubMed] [Google Scholar]
- 5.Bader-Meunier B, Tchernia G, Miélot F, Fontaine JL, Thomas C, Lyonnet S et al. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr 1997; 130: 885–889. [DOI] [PubMed] [Google Scholar]
- 6.Niemeyer CM, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, Creutzig U et al. Chronic myelomonocytic leukemia inchildhood: a retrospective analysis of 110 cases. European Working Group onMyelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 1997; 89: 3534–3543. [PubMed] [Google Scholar]
- 7.Reuther GW, Der CJ. The Ras branch of small Gtpases: Ras family members don’t fall far from the tree. Curr Opin Cell Biol 2000; 12: 157–165. [DOI] [PubMed] [Google Scholar]
- 8.Shi GX, Jin L, Andres DA. A rit GTPase-p38 mitogen-activated protein kinase survival pathway confers resistance to cellular stress. Mol Cell Biol 2011; 31: 1938–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li JT, Liu W, Kuang ZH, Chen HK, Li DJ, Feng QS et al. Amplification of RIT1 in hepatocellular carcinoma and its clinical significance. Ai Zheng 2003; 22: 695–699. [PubMed] [Google Scholar]
- 10.Li JT, Liu W, Kuang ZH, Zhang RH, Chen HK, Feng QS. Mutation and amplification of RIT1 gene in hepatocellular carcinoma. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2004; 21: 43–46. [PubMed] [Google Scholar]
- 11.Shao H, Kadono-Okuda K, Finlin BS, Andres DA. Biochemical characterization of the Ras-related GTPases Rit and Rin. Arch Biochem Biophys 1999; 371: 207–219. [DOI] [PubMed] [Google Scholar]
- 12.Andrieux J, Demory JL, Caulier MT, Agape P, Wetterwald M, Bauters F et al. Karyotypic abnormalities in myelofibrosis following polycythemia vera. Cancer Genet Cytogenet 2003; 140: 118–123. [DOI] [PubMed] [Google Scholar]
- 13.Rusyn EV, Reynolds ER, Shao H, Grana TM, Chan TO, Andres DA et al. Rit, a non-lipid-modified Ras-related protein, transforms NIH3T3 cells without activating the ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene 2000; 19: 4685–4694. [DOI] [PubMed] [Google Scholar]
- 14.Cai W, Rudolph JL, Harrison SM, Jin L, Frantz AL, Harrison DA et al. An evolutionarily conserved Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress resistance. Mol Biol Cell 2011; 22: 3231–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.