

Abstract
Chaperone-mediated autophagy (CMA) is a proteolytic process whereby selected intracellular proteins are degraded inside lysosomes. Owing to its selectivity, CMA participates in the modulation of specific regulatory proteins, thereby playing an important role in multiple cellular processes. Studies conducted over the last two decades have enabled the molecular characterization of this autophagic pathway and the design of specific experimental models, and have underscored the importance of CMA in a range of physiological processes beyond mere protein quality control. Those findings also indicate that decreases in CMA function with increasing age may contribute to the pathogenesis of age-associated diseases, including neurodegeneration and cancer.
In the context of neurological diseases, CMA impairment is thought to contribute to the accumulation of misfolded/aggregated proteins, a process central to the pathogenesis of neurodegenerative diseases. CMA therefore constitutes a potential therapeutic target, as its induction accelerates the clearance of pathogenic proteins, promoting cell survival. More recent evidence has highlighted the important and complex role of CMA in cancer biology. While CMA induction may limit tumor development, experimental evidence also indicates that inhibition of this pathway can attenuate the growth of established tumors and improve the response to cancer therapeutics. Here, we describe and discuss the evidence supporting a role of impaired CMA function in neurodegeneration and cancer, as well as future research directions to evaluate the potential of this pathway as a target for the prevention and treatment of these diseases.
Keywords: chaperones, lysosomes, autophagy, protein degradation, aggregation, tumorigenesis
1. Chaperone-mediated autophagy
1.1. Introduction.
Mammalian cells depend on monitoring systems to manage protein alterations and organelle damage. One such system is autophagy, an evolutionarily conserved cellular process through which parts of the cell are degraded in the lysosomal compartment. Three types of autophagy are described to date, each differing in the manner in which cargo is delivered to the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). In macroautophagy, a portion of the cytoplasm is engulfed by a phagophore, resulting in the formation of a double-membrane organelle known as the autophagosome. After fusion of the outer autophagosomal and lysosomal membranes, lysosomal enzymes degrade the inner autophagosomal membrane and the enclosed material (De Duve and Wattiaux, 1966). Microautophagy involves the direct engulfment of cytoplasmic contents by lysosomes (Marzella et al., 1981). Finally, CMA consists of translocation of the cargo (specifically selected proteins), assisted by chaperones, across the lysosomal membrane. The most selectively regulated form of autophagy, CMA responds to environmental and physiological cues and is particularly important in the context of stress (Arias and Cuervo, 2011). In this review, we focus specifically on this unique form of selective autophagy (Fig. 1).
Figure 1. Schematic depicting the steps involved in degradation of cytosolic proteins by chaperone-mediated autophagy (CMA).
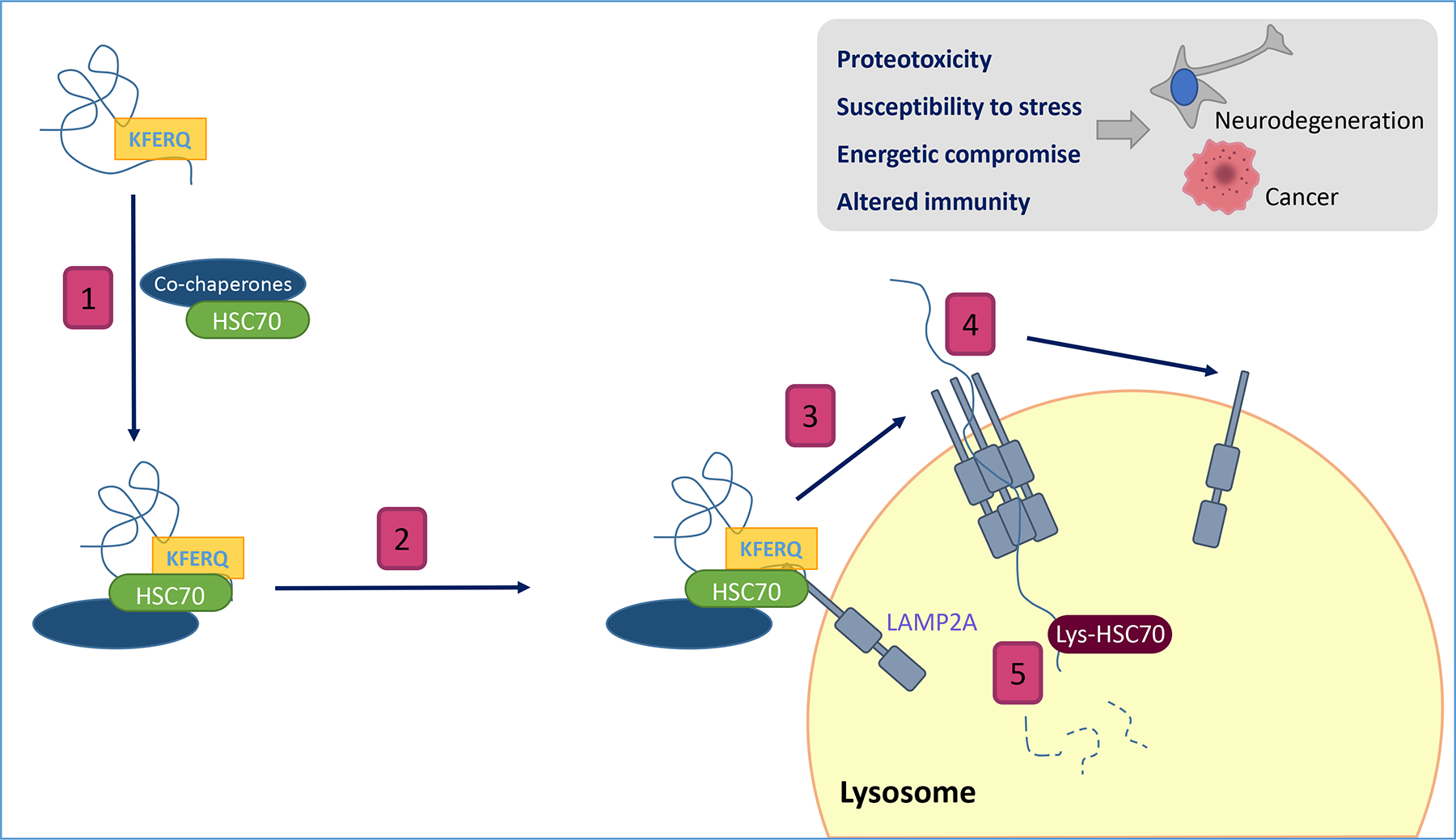
(1) Recognition of cytosolic proteins by HSC70 and its co-chaperones. (2) Binding of the chaperone/substrate complex to the receptor at the lysosomal membrane. (3) LAMP2A multimerization to form a translocation complex (4) Unfolding of the substrate protein and translocation across the lysosomal membrane. (5) Degradation in the lysosomal lumen. Insert: Consequences of CMA failure.
CMA has been ascribed a high degree of selectivity since it was first described by Dice over 30 years ago (Dice, 1990). This pathway is responsible for the degradation of around 30% of cytosolic proteins (Kirchner et al., 2019), and the peculiar mechanism by which the cargo is delivered to the lysosome involves the orchestrated action of multiple components, including chaperones, integral membrane proteins, and cytosolic proteins that transiently associate with the lysosomal membrane. A unique feature of CMA is that the substrate is limited to proteins expressing a specific amino acid sequence (Dice, 1990). This motif is recognized by cytosolic chaperone HSC70 (heat shock cognate 71 kDa protein), which targets and delivers the protein to the surface of the lysosomal membrane (Chiang et al., 1989). There, the substrate binds to the cytosolic tail of lysosome-associated membrane protein type 2A (LAMP2A), which contains a single transmembrane spanning region (Cuervo and Dice, 1996). This binding induces multimerization of LAMP2A to form a translocation complex that facilitates internalization of the substrate (Bandyopadhyay et al., 2008). Distinct chaperones, organized within (HSC70 and HSP90) and outside of (HSP90) the lysosomal membrane facilitate substrate unfolding and translocation (Bandyopadhyay et al., 2008; Cuervo et al., 1997) (Fig. 1).
LAMP2A and luminal HSC70 content have been proposed as determinants of the rate of CMA, although only levels of LAMP2A limit the rate of CMA. Consequently, quantification of LAMP2A protein levels has been used as a subordinate indicator of CMA status, and the impact of changes in CMA activity has been studied using interventions that alter lysosomal levels of LAMP2A (Schneider et al., 2015; Zhang and Cuervo, 2008). However, it is important to note that CMA activation does not require de novo LAMP2A synthesis: LAMP2A abundance at the lysosomal membrane can be altered through changes in its stability, organization, and dynamics (Bandyopadhyay et al., 2008; Cuervo and Dice, 2000b). Nonetheless, there are specific and well-established methods used to rigorously study CMA (Arias, 2017; Dong et al., 2020; Klionsky et al., 2021).
The different steps of CMA are precisely regulated by lysosome-associated proteins, including the mTORC2/AKT1/PHLPP1 axis (Arias, 2015; Arias et al., 2015). Five years ago, PHLPP1 and TORC2 were identified as endogenous stimulator and inhibitor, respectively, of CMA, and shown to act in concert to modulate basal and inducible CMA activity through their antagonist effects on AKT. Stress stimuli increase the association of AKT to the lysosomal membrane and the control of the stability of the AKT-lysosomal membrane association via the GTPase Rac1, cooperate to counterbalance the endogenous inhibitory effect of lysosomal TORC2/AKT on CMA. The GFAP/EF1α complex has been proposed as modulator of CMA (Bandyopadhyay et al., 2010). These proteins, glial fibrillary acidic protein (GFAP) and elongation factor 1α (EF1α), regulate the assembly and disassembly of LAMP2A (Bandyopadhyay et al., 2010), controlling the translocation complex that facilitates cargo internalization. GFAP binds LAMP2A at the multimeric complex and contributes to its stabilization, while phosphorylated GFAP (pGFAP), which has a lower affinity for LAMP2A, associates better with the lysosomal membrane as part of a complex with EF1α. Upon translocation of the substrate, EF1α is released and promotes the disassembly of LAMP2A as GFAP leaves the translocation complex. In addition to regulation at the lysosomal level, CMA is also modulated by signaling from the nucleus, plasma membrane, and other organelles (Table 1).
Table 1. Proteins involved in CMA.
Proteins involved in CMA are grouped based on their function (effector and modulators) and localization (lysosomal and extra-lysosomal).
| Proteins involved in CMA | Function | Refs | |
|---|---|---|---|
| Protein effectors | LAMP2A | Effector | Cuervo & Dice |
| HSC70 | Effector | Agarraberes et al. | |
| HSP90 | Effector | Agarraberes et al. | |
| HSP40 | Effector | Agarraberes et al. | |
| Lysosomal modulators | AKT1 | Inhibitor | Arias et al. |
| Cath A | Inhibitor | Cuervo et al. | |
| Rictor | Inhibitor | Arias et al. | |
| eF1a | Activator | Bandyopadhyay et al. | |
| GFAP | Activator | Bandyopadhyay et al. | |
| Humanin | Activator | Gong et al. | |
| PHLPP1 | Activator | Arias et al. | |
| RAC1 | Activator | Arias et al. | |
| Extra lysosomal modulators | NFAT | Activator | Valdor et al. |
| NRF-2 | Activator | Pajares et al. | |
| Rab11 | Inhibitor | Zhang et al. | |
| RARa | Inhibitor | Anguiano et al. |
AKT1, RAC-alpha serine/threonine-protein kinase; Cath A, lysosomal protective protein/cathepsin A; CMA, chaperone-mediated autophagy; eF1α, Elongation factor 1-alpha; GFAP, Glial fibrillary acidic protein; HSC70, Heat shock cognate 71 kDa protein; HSP40, DnaJ homolog subfamily B member 1; HSP90, Heat shock protein HSP 90; LAMP-2A, lysosome-associated membrane protein type 2A; NFAT, nuclear factor of activated T cells; NRF-2, nuclear factor erythroid 2-related factor 2; PHLPP1, PH domain leucine-rich repeat-containing protein phosphatase 1; Rab11, Ras-related protein Rab-11; RAC1, Ras-related C3 botulinum toxin substrate 1; RARα, Retinoic acid receptor alpha.
Elucidation of the key molecular components essential for CMA has led to a new era in our understanding of mammalian physiology and the pathophysiology of human diseases. Moreover, the advent of techniques to genetically modulate CMA has strengthened the association between CMA dysfunction and human disease (Bourdenx et al., 2021; Kon et al., 2011). Studies at the level of the cell and the entire organism are uncovering new functions of CMA and providing a more in-depth understanding of the physiological relevance of this autophagy pathway (Schneider et al., 2014; Valdor et al., 2014). In this review, we discuss new findings that highlight the importance of CMA in both health and disease, particularly in the context of neurodegeneration and cancer.
1.2. CMA in physiological conditions
In most tissues, basal levels of CMA in physiological conditions prevent the gradual accumulation of damaged proteins, which exert noxious effects over time, and therefore plays an important role in protein quality control (Arias and Cuervo, 2011). Identification of CMA substrates that are dysregulated in cells and tissues in which CMA is deficient is important to understand the biological role and tissue specificity of CMA. Determining the overall influence of CMA on the cellular proteome is another crucial goal: we are only beginning to understand the broad spectrum of CMA substrates and the functional implications of dysregulation of their recycling and degradation.
CMA captures and degrades specific proteins in order to sustain homeostasis and metabolism. Certain tissues, including the brain and liver (as well as other, less studied tissues), are exceptionally dependent on autophagy in general, and CMA in particular, in order to maintain homeostasis, prevent the accumulation of damaged protein/aggregates, mitigate oxidative stress, and preserve adequate metabolism. It is now known that the toxic consequences of defective CMA are due not only to the accumulation of proteins and cellular components, but also to imbalances in proteins and cellular components. These effects may be direct or indirect. For example, CMA contributes to the regulation of lipid metabolism by directly and selectively degrading lipid droplet proteins, including perilipins (Kaushik and Cuervo, 2015), thereby facilitating the initiation of both lipolysis and lipophagy (Kaushik and Cuervo, 2015), and also contributes to lipid metabolism through selective degradation of key enzymes in this pathway (and in the glucose metabolism pathway) (Schneider et al., 2014). Evidence indicates a wide range of effects of CMA on cellular homeostasis at the level of: substrate removal, maintenance of adequate metabolic function, and detoxification. The level of CMA activity and the nature of the substrates degraded or recycled via this pathway are both tissue-specific.
The metabolic role of CMA partly overlaps with that of protein and organ quality control and homeostatic processes, and further expand the impact of CMA in mammalian physiology and disease. In healthy mammalian cells CMA induction is critical to overcome starvation through the recycling of intracellular components to fuel metabolic pathways (Massey et al., 2006), as also occurs in the context of aging (Walters et al., 2018). CMA thus contributes to the maintenance of metabolic homeostasis and to cell survival, both of which are essential during nutrient deprivation. Although it is generally accepted that degradation of intracellular proteins by CMA provides metabolic support during starvation, many of the substrates involved and the metabolic pathways they support remain to be identified.
Recent findings have identified other regulatory functions of CMA, beyond the control of homeostasis and metabolic pathways. For example, CMA regulates transcriptional programs (Cuervo et al., 1998; Franch et al., 2001; Yang et al., 2009; Zhang et al., 2014), its activity has been linked to the immune response (Hu et al., 2016; Valdor et al., 2014), it is required to initiate cell cycle progression after DNA repair (Ferreira et al., 2013; Hubbi et al., 2014; Park et al., 2015) and it also influences the cellular proteome -beyond the known thirty percent of cytosolic proteins having this pathway as responsible for its degradation (Bourdenx et al., 2021; Kirchner et al., 2019). The role of CMA in cells and organs is both highly complex and tissue-dependent (Bourdenx et al., 2021; Rodriguez-Muela et al., 2013; Schneider and Cuervo, 2014). CMA deficiency is known to contribute to the pathogenicity in many diseases including neurodegenerative diseases (Bourdenx et al., 2021) and cancer (Kon et al., 2011), on which the present review is specifically focused.
2. CMA and neurodegeneration
2.1. Role of CMA in neurons
Neurons are a particularly sensitive type of cell. Given their post-mitotic status, the accumulation of damaged or misfolded proteins, aggregates, or oxidative stress products cannot be diluted by cell division. This unwanted material is usually eliminated by autophagic or proteolytic systems that work in concert (Cuervo and Wong, 2014). Such crosstalk between different pathways does not implies redundancy. Rather, these pathways can usually partially compensate for one another to ensure survival in normal conditions. However, in certain situations, such as stress, this compensation may be insufficient and can compromise viability (Cuervo and Wong, 2014). When an imbalance in proteostasis occurs, it is critical to activate control mechanisms to avoid severe cellular damage that endangers neuronal survival (Morimoto, 2008).
Protein homeostasis requires more than simple protein quality control, as it also involves RNA metabolism and processing, protein synthesis, folding, translocation, assembly/disassembly, and clearance (Balch et al., 2008). Numerous recent studies point to an important role of CMA in protecting neurons from stress and injury (Damme et al., 2015). Oxidative stress is an important player contributing to the progression neurodegenerative diseases and aging (Barber and Shaw, 2010; Behl, 1999; Browne et al., 1999; Hauser and Hastings, 2013). During oxidative stress, upregulation of CMA favors the transport of oxidized proteins into lysosomes. The increase in LAMP2A levels appears to be dependent on transcriptional regulation (Kiffin et al., 2004). Moreover, when LAMP2A expression is selectively blocked, preventing upregulation of CMA in response to stress, cells accumulate oxidative damage and cell death increases (Massey et al., 2006). CMA is also involved in cell survival after hypoxic stress. Brain ischemia results in subsequent upregulation of LAMP2A in the ischemic hemisphere. This effect has been replicated in neuronal cell culture (Dohi et al., 2012), in which modulation of CMA using chemical activators reduces hypoxia-mediated cell death, and, on the contrary, silencing of LAMP2A decreases cell survival. In mice, two key findings emphasize the importance of CMA in neurons, highlighting the role in maintaining the neuronal metastable proteome (Bourdenx et al., 2021). First, both systemic and neuron-specific LAMP2A knock-out directly impact the solubility of the neuronal proteome, leading to proteostatic collapse and functional dysregulation. Second, genetic inhibition of CMA in a mouse model of Alzheimer’s disease (AD) accelerates disease (Bourdenx et al., 2021). Aging is accompanied by a decline in CMA and LAMP2A levels that promotes the accumulation of oxidized proteins and impairs the cellular response to stress or damage. This effect is observed in most tissues, including the nervous system (Orenstein and Cuervo, 2010; Zhang and Cuervo, 2008). In conclusion, CMA helps to control proteome integrity, which is crucial to maintain biological processes and to ensure neuronal function and health (Fig. 2).
Figure 2. Impairment of CMA by pathogenic proteins in neurodegenerative diseases.
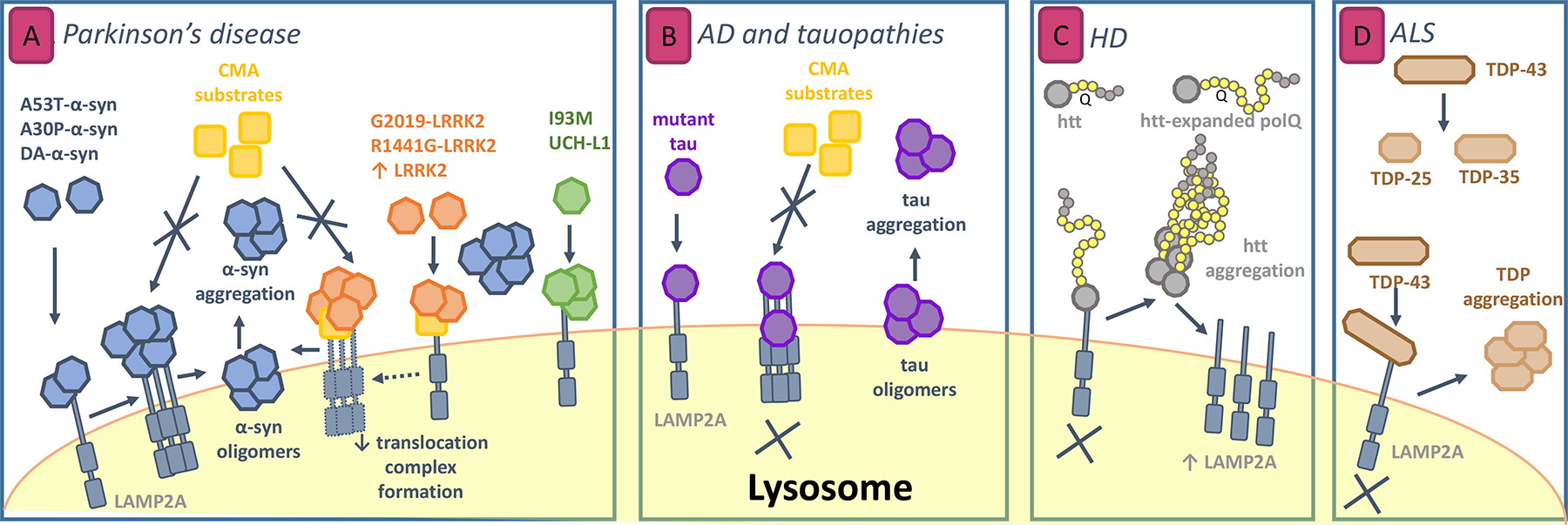
(A) Mechanisms of CMA failure in Parkinson’s disease (PD). α-synuclein and LRRK2 are degraded by CMA. Mutant forms of these proteins and of UCH-L1 bind to LAMP2A, but different alterations in binding/uptake lead to blockage of their own degradation as well as degradation of other CMA substrates. Also, abnormally high levels of wild-type LRRK2 also impair CMA translocation complex formation. Alterations by mutant LRRK2 and UCH-L1 both produce toxic effects on α-synuclein aggregation. (B) Perturbation of CMA by mutant tau in Alzheimer’s disease (AD) and tauopathies. Non translocated pathogenic variants can induce cleavage of tau into tau oligomers that may favor further tau aggregation when liberated to the lysosome. (C) CMA alteration in Huntigton’s disease (HD). Huntingtin with abnormally expanded polyQ shows slow transport across lysosomal membrane and thus accumulate in the cytosol. Aggregation of this form increases CMA by LAMP2A transcription and stabilization at the lysosomal membrane but this compensation fails with aging. (D) Role of CMA in amyotrophic lateral sclerosis (ALS). Cleavage of the protein TDP-43 is necessary prior to degradation. Non-degraded fragments could serve as seed for TDP aggregation in the cytosol.
2.2. CMA and protein aggregation in neurodegenerative diseases
Protein aggregation is a feature common to many neurodegenerative diseases. Proteins that accumulate and exert toxic effects, such as α-synuclein, amyloid beta, tau, and huntingtin, aggregate to form inclusions that are considered pathological hallmarks in neurodegenerative diseases. A common feature of neurodegenerative diseases is an inability of proteolytic systems to effectively manage the accumulation of these aggregate-prone proteins (Cuervo and Wong, 2014). Degradation of polyubiquitinated huntingtin and ataxin-3, mutant α-synuclein, and different forms of tau is strongly dependent on autophagy (Alvarez-Erviti et al., 2010; Cuervo and Dice, 2000a; Iwata et al., 2005). CMA is one of the mechanisms by which cellular homeostasis is maintained. Accordingly, CMA dysfunction has been implicated in the pathogenesis of certain human diseases, including neurological disorders (Alvarez-Erviti et al., 2010; Koga et al., 2011a; Thompson et al., 2009). The toxic effects of CMA dysregulation can be the primary cause of disease when pathogenic proteins directly interfere with components of the CMA pathway and impair the degradative capacity of CMA, thereby triggering or contributing to the progression of disease, or can be secondary when CMA is indirectly disturbed by altered or accumulated proteins affecting proteostasis (Scrivo, 2018).
Risk factors for protein aggregation and neurodegeneration due to CMA dysregulation include (i) environmental risk factors (i.e. toxins such as 6OH-DA lead in first place to increases in LAMP2A and HSC70 expression, to exert protective effects by increasing CMA activity, but persistent exposure results in a decrease in CMA) (Gao et al., 2014); (ii) genetic risk factors (pathogenic proteins carrying mutations can block CMA by binding to the lysosomal surface and preventing the degradation of other CMA substrates); and (iii) aging (the demonstrable age-associated decrease in CMA, which is mainly attributed to loss of stability of the lysosomal membrane, may contribute to pathogenesis) (Rodriguez-Navarro and Cuervo, 2012). The deterioration of CMA that occurs with aging appears to involve changes in lipidic components at the lysosomal membrane, resulting in consequent alterations in LAMP2A dynamics and stability (Kiffin et al., 2007). Lipid-rich diets can also cause small changes in lipid content, this can be exacerbated when CMA is compromised, i.e. during aging, highlighting the importance of diet and the accelerated aging effect that it can produce (Schneider et al., 2015). In the brain, lipidomic alterations (mainly in the mitochondria membrane) have been associated with aging (Gaudioso et al., 2019). Tissue-specific knock-out of CMA limiting component, LAMP2A, has furthered our understanding of the consequences of age-associated CMA impairment (Schneider et al., 2015), and studies in mice have helped elucidate the crosstalk between macroautophagy and CMA (Schneider et al., 2014; Valdor et al., 2014). Nonetheless, in some tissues this crosstalk is non-reciprocal. For example, in the retina macroautophagy cannot compensate for the age-dependent loss of CMA, and may worsen or accelerate the effects of aging (Rodriguez-Muela et al., 2013). Genetic manipulation to introduce an extra copy of LAMP2A has proven beneficial in counteracting the age-associated decrease in CMA and prolonging lifespan (Zhang and Cuervo, 2008).
Below, we will focus on the role of CMA in neurodegenerative diseases such as Parkinson’s, Alzheimer’s, and Huntington’s diseases and amyotrophic lateral sclerosis.
2.3. Exploring CMA in Parkinson’s disease
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease worldwide, after Alzheimer’s disease. It affects 6.3 million people aged over 65 years globally. The main pathological characteristics are the formation of Lewy bodies and aggregates containing α-synuclein and ubiquitin, followed by the death of tyrosine hydorxylase (TH)-positive neurons in the substantia nigra (Bourdenx et al., 2017), with major motor coordination consequences in affected patients.
Post-mortem studies of PD brains have revealed reduced levels of LAMP2A in the substantia nigra, pointing to a role of CMA in the etiology of the disease (Alvarez-Erviti et al., 2010). α-synuclein and LRRK2 (leucine-rich repeat kinase 2) are the main pathogenic proteins in both sporadic and familial PD (Cuervo et al., 2004; Orenstein et al., 2013), and both have been identified as substrates of CMA. α-synuclein bears a pentapeptide, V95KKDQ99, that is recognized by HSC70 chaperone (Alvarez-Erviti et al., 2010; Liao et al., 2021b), and LRRK2 is degraded in lysosomes by CMA, although mutations can render it resistant to CMA degradation (Orenstein et al., 2013). Moreover, decreases in LAMP2A and HSC70 protein levels have been described in the substantia nigra and amygdala of PD patients, supporting a role of impaired CMA function in the pathogenesis of PD (Alvarez-Erviti et al., 2010).
2.3.1. α-synuclein as a CMA substrate
Loss of proteostasis is directly related to the formation of protein aggregates. This suggests that different forms of autophagy and the proteasome system, which are involved in the formation and clearance of these aggregates, may be implicated in the development of PD. A 2004 study of substrate binding and uptake in intact lysosomes showed that α-synuclein is degraded by lysosomes, specifically via CMA. The mutated α-synuclein forms A53T and A30P were found to bind more strongly to the lysosomal surface than the unmodified form, but were not degraded, and therefore impaired CMA degradation of other substrates, exerting a gain of toxic function effect by favoring the aggregation of pathogenic forms and triggering cellular stress (Cuervo et al., 2004).
Post-transcriptional modifications in α-synuclein can have varying consequences on the protein’s susceptibility to degradation by CMA, and on the degradation of other CMA substrates. Monomeric, dimeric, and oligomeric forms of α-synuclein bind to the lysosomal membrane, but only monomers and dimers are translocated into the lysosomal lumen of lysosomes that are participating in CMA (Martinez-Vicente et al., 2008). Also, while the lysosomal binding capacity of oxidized α-synuclein and S129E α-synuclein, a phosphomimetic mutant, are similar to that of WT α-synuclein, S129E α-synuclein cannot be internalized in the lysosome. Similarly, decreased lysosomal uptake has been described for a phosphorylated form of α-synuclein in vitro (Martinez-Vicente et al., 2008). These modified proteins do not interfere with CMA function or the internalization of other substrates. Furthermore, other modifications such as nitration and oligomerization of α-synuclein result in reduced clearance, but have no effects on CMA performance.
Interestingly, only dopamine-reacted (+DA) and dopaminochrome-reacted (+DAC) α-synuclein affect CMA activity in lysosomes, both in cultured DA cell lines and mouse ventral midbrain (VM) primary cultures (Martinez-Vicente et al., 2008). Similar results have been reported in SH-SY5Y cells, in which dopamine-modified α-synuclein appears to be responsible for reduced CMA function and increased toxicity, rendering dopaminergic neurons particularly vulnerable (Xilouri et al., 2009). In pharmacological and genetic approaches, mice treated with paraquat, an herbicide used as Parkinsonism inducer via oxidative stress (Bastías-Candia et al., 2019), and transgenic mice overexpressing mouse α-synuclein display increased α-synuclein mRNA levels, which correlate with a marked increase in protein and mRNA levels of LAMP2A. Moreover, these animals show increased expression and colocalization of α-synuclein and LAMP2A, suggesting increased degradation of α-synuclein by CMA in these conditions (Mak et al., 2010).
In conclusion, there is evidence that accumulation of α-synuclein may be a consequence of CMA impairment, although post-transcriptional modifications in α-synuclein may also contribute to CMA dysregulation. Together, the available data point to an important role of CMA in α-synuclein clearance.
2.3.2. LRRK2
Mutations in LRRK2 are the most common cause of familial PD. LRRK2, which contains 8 putative CMA motifs, can bind to lysosomes and be actively degraded by CMA. G2019S, a LRRK2 mutant, is degraded less efficiently by CMA. Both mutant and wild-type LRRK2 inhibit CMA by blocking formation of the translocation complex (Orenstein et al., 2013). This newly described mechanism also increases LAMP2A levels in an attempt to compensate for the lack of CMA activity. The ultimate consequence is defective α-synuclein degradation (Orenstein et al., 2013). Studies in a knock-in mouse model expressing the R1441G LRRK2 mutation have shown that the presence of the mutated protein causes age-dependent accumulation of α-synuclein oligomers in the brain correlating with LAMP2A and HSC70 accumulation in the midbrain and striatum (Ho et al., 2020).
2.3.3. Other CMA substrates that contribute to the pathogenesis of Parkinson’s disease
Other proteins implicated in PD, apart from α-synuclein and LRRK2, also participate in CMA, further supporting a role of CMA dysregulation in PD progression. These proteins are described below.
MEF2D (myocyte enhancer factor 2D) is a transcription factor required for neuronal survival, and is translocated from the nucleus to the cytoplasm for lysosomal degradation in basal conditions. High levels of α-synuclein appear to impair MEF2D degradation via CMA by interfering with translocation, resulting in MEF2D accumulation in the cytoplasm. Unexpectedly this accumulated MEF2D is nonfunctional, as it loses its DNA-binding capacity (Yang and Mao, 2009). Further studies are required to elucidate the mechanism underlying this loss of DNA-binding capacity and to determine whether MEF2D accumulation exerts toxic effects.
Mutation in UCH-L1 (ubiquitin C-terminal hydrolase L1) is associated with familial PD. Setsuie and coworkers showed that transgenic mice expressing UCH-L1I93M display progressive loss of dopaminergic cells (Setsuie et al., 2007). Furthermore, the same research group has shown that UCH-L1 interacts with LAMP2A, HSC70 and Hsp90 in NIH-3T3 cells stably expressing FLAG-hemagglutinin (HA)-tagged UCH-L1 (Kabuta et al., 2008). Interestingly, the I93M mutation results in abnormal enhancement of these interactions, thus impairing CMA functioning and facilitating α-synuclein accumulation (Kabuta et al., 2008). Structural analyses have shown that the I93M mutation increases instability, leading to aberrant aggregation and abnormal interactions with CMA machinery (Andersson et al., 2011).
Mutations in the PARK7/DJ-1 gene are linked to autosomal-recessive causes of hereditary PD. PARK7/DJ-1 is an important redox reactive signaling intermediate that is activated in the presence of reactive oxygen species (ROS) in conditions of oxidative stress (Kahle et al., 2009). CMA preferentially targets oxidized and non-functional PARK7/DJ-1 to the lysosome for elimination. Thus, reductions in CMA activity impair the oxidative stress response by disrupting mitochondrial function (Wang et al., 2016)
In summary, numerous studies support an important role of CMA in the degradation of proteins implicated in PD. Given that defective CMA may contribute to the progression of PD, and that CMA is also altered as a consequence of the disease, CMA activation could constitute a useful approach to the treatment of PD.
2.4. Link between AD and other tauopathies to CMA
Extracellular deposits of the protein amyloid-β (Aβ) in the form of senile plaques (SP), and intracellular filaments of hyperphosphorylated tau protein that form neurofibrillary tangles (NFT) are the characteristic histopathological lesions in Alzheimer’s disease (AD) (Bourdenx et al., 2017). Tauopathies are a large group of diseases including AD, progressive supranuclear palsy (PSP), and frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17). Aggregation and cleavage of tau are two key pathological hallmarks of tauopathies.
Wang and colleagues developed an inducible neuronal cell model of tauopathy to study tau cleavage, aggregate-prone forms of tau, and tau clearance (Wang et al., 2010). This model consists of N2A cells expressing in an inducible manner the repeat domain (RD) of tau with a FTDP-17 mutation (TauRDΔK280). The authors proposed that tau binds to CMA active lysosomes through LAMP2A, assisted by HSC70. Subsequently, it is partially translocated into the lysosome, starting at the C-terminus. Inside the lumen, cathepsin L cleaves fragments F2 and F3, while F1 remains attached to the lysosomal surface, exerting an inhibitory effect by blocking binding/uptake of other CMA substrates and promoting formation of tau oligomers, as well as subsequent aggregation at the lysosomal surface (Wang et al., 2009). Tau oligomers can also disrupt the lysosomal membrane, causing lysosomal leakage and the release of F2 and F3 fragments into the cytosol, further promoting aggregation of TauRDΔK280 (Wang et al., 2010).
Modifications in the amino acid sequence of tau appear to affect its degradation by CMA and endosomal microautophagy (e-MI), subsequently altering the crosstalk between different autophagic pathways. These alterations may render the cells more sensitive to stress. Studies of P301L mutations, which are linked to tau aggregation in frontotemporal dementia (FTD), and of A152T, which is considered a risk factor for FTD and AD, have shown that P301L affects tau degradation by all autophagic pathways. Macroautophagy appears to constitute a compensatory pathway that is compromised in FTD and other tauopathies, thereby contributing to the impaired degradation (Caballero et al., 2018).
Tau bears two recognition motifs in its C-terminal that are required for targeting (Wang et al., 2009) and a sequence required for internalization in the lysosome located in the N-terminal. Post-translational modifications such as phosphorylation may affect degradation via CMA (Caballero et al., 2018). Tau acetylation at lysine 174 has been detected in soluble fractions from AD patients, suggesting a role of this modification in tau aggregation due to a poorly CMA degradation. Reduced CMA in a knock-out mouse of LAMP2A specific for pyramidal neurons confirmed the degradation via CMA of acetylated tau (K174). Moreover, studies in vitro and in vivo mouse studies demonstrated that CMA blockade favors rerouting of the degradative process to e-MI. These observations suggest that CMA compromise may trigger a mechanism by which extracellular tau and cell-to-cell propagation of tau are increased (Caballero et al., 2021).
A 2021 study in a mouse model of tau-mediated proteotoxicity (h-TauP301L mice) demonstrated a significant reduction in CMA in using a fluorescent KFERQ-Dendra reporter for CMA (Dong et al., 2020), suggesting that CMA may be defective in this model (Bourdenx et al., 2021). RNA-seq data from AD patients were used to analyze CMA during disease progression, and revealed neuron-specific translational downregulation of CMA in early AD. In a mouse model of AD bred with a transgenic mouse lacking L2A (and therefore deficient in CMA), attenuation of CMA was shown to exacerbate brain proteotoxicity, as evidenced by an increase in Aβ deposits and the accumulation of phosphorylated and aggregated tau, thereby accelerating disease progression (Bourdenx et al., 2021). Interestingly, pharmacological activation of CMA in two different models of tauopathies, the PS19 mouse (that exhibits phenotype indicative of frontotemporal dementia) (Yoshiyama et al., 2007) and a triple transgenic mouse model of AD (Grueninger et al., 2010), resulted in decreases in phosphorylated and pathogenic forms of tau, reduced Aβ histopathology, and amelioration of disease-related behaviors. Together, these results point to a beneficial effect of pharmacological CMA activation in AD and related pathologies (Bourdenx et al., 2021).
There are proteins, other than tau, contributing to AD as amyloid precursor protein (APP) or regulator of calcineurin 1 (RCAN1) that have been related to CMA. Studies in SH-SY5Y cells have shown that amyloid precursor protein (APP) contains a KFERQ recognition motif that is important for its processing. Deleting this motif increases the accumulation of C-terminal fragments and the secretion of soluble APP. Despite having the KFERQ recognition motif, the contribution of CMA to APP degradation needs to be further study and subsequently the potential implication in the pathogenesis of AD (Park et al., 2016). Regulator of calcineurin 1 (RCAN1), a gene encoding a region critical in Down’s syndrome, has been recently implicated in the pathogenesis of AD, as its expression is increased in the brains of patients with both diseases (Harris et al., 2007). Another study in HEK293T cells has shown that RCAN1 is degraded by CMA, and that disruption of CMA is sufficient to increase RCAN1 levels. As increased RCAN1 expression, have been linked to AD and Down syndrome, CMA impairment is potentially related to the contribution of RCAN1 to these diseases progression (Liu et al., 2009).
In summary, many studies have linked CMA dysregulation to diseases characterized by the aggregation of tau and Aβ, suggesting that defects in this autophagic pathway contribute to the pathogenesis of AD and other tauopathies.
2.5. Studies of CMA in Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant disease caused by the expansion of the polyQ tail due to a mutation in the huntingtin (Htt) gene. Mutant Htt is prone to aggregation, and the fibrils that accumulate during inclusion formation are toxic and contribute to the disease progression. Pathways that control proteostasis, including CMA, play an important role in regulating aggregate formation and degradation of huntingtin protein (Koyuncu et al., 2017; Martinez-Vicente et al., 2010).
In vitro experiments have shown that wild type and mutant Htt are degraded by both macroautophagy and CMA. To decipher the role in HD of LAMP2A and HSC70, both components of the CMA pathway, Qi and coworkers isolated lysosomes from mouse liver, which they then incubated with Htt-552 fragments bearing either 18 or 100 glutamines (Htt-552–18Q or Htt-552–100Q). CMA was capable of degrading both Htt-552–18Q and Htt-552–100Q, although clearance rates were superior for Htt-552 fragments with the shorter polyQ tail. Modulation of the levels of both LAMP2A and HSC70 by neutralizing antibodies affected Htt degradation by CMA, by decreasing degradation and therefore increasing Htt levels. The authors also demonstrated that endogenous Htt can be degraded by CMA (Qi et al., 2012). The effect of mutant Htt on CMA activity has also been studied in HEK293T cells, in which transient transfection of mutant Htt did not significantly alter LAMP2A or HSC70 expression or CMA activity when compared to normal Htt (Li and Wang, 2014), leading the authors to suggest that enhancement of CMA may constitute an effective approach to induce clearance of toxic forms of Htt.
CMA is upregulated in a variety of HD cell and mouse models, including 111QHtt knock-in mice, striatal 111QHtt knock-in cell lines, human lymphoblasts from HD patients, and striatal neuronal cultures from HD94 mice. Specifically, an increase in LAMP2A presence at the lysosomal surface is observed in these conditions. This has been demonstrated using a fluorescent CMA reporter (KFERQ-PS-CFP2) (Koga et al., 2011b) in 111QHtt versus 7QHtt cells. Increased CMA active lysosomes has also been described in HD cells (111QHtt), and increased substrate binding/uptake in isolated lysosomes (Koga et al., 2011a). Although the increased rate of CMA does not directly account for the degradation of full-length Htt, CMA appears to degrade truncated Htt forms, which are also cytotoxic. This increased proteolytic capacity is lost at 12 months of age in HD mice, which express lower levels of LAMP2A at the lysosomal surface, suggesting that the age-associated decline in CMA is accelerated in HD mice (Koga et al., 2011a).
Overexpression of LAMP2A in neuronal cells enables degradation of post-transcriptionally modified Htt by CMA, reducing intracellular protein inclusions (Thompson et al., 2009). It has been suggested that phosphorylation at Ser16 of Htt by the kinase IKK creates a KFERQ-like motif that makes Htt recognizable by HSC70. More recently, LAMP2A expression has been linked to levels of let7b miRNA in mouse striatal neurons, both of which are decreased in the presence of extended polyQ (Choi and Cho, 2021). Upregulation of let7b miRNA increases LAMP2A, and correlates with a decrease in extended polyQ. Interestingly, both let7b miRNA and LAMP2A are elevated in the striatum of pre-onset HD mice and decrease in post-onset HD, suggesting that LAMP2A-dependent CMA capacity plays an important role in HD symptom onset.
Although few studies have investigated the role of CMA in HD, early findings implicate CMA in the pathogenesis of this disease and highlight the need for further research in this area.
2.6. Relevance of CMA in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease characterized by the aggregation of misfolded proteins in motor neurons, resulting in progressive paralysis and death. The underlying causes remain unknown. TDP-43 (TAR DNA binding protein, also known as TARDBP), which binds RNA and DNA, among other functions, is one of the proteins identified in the ubiquitinated inclusion bodies (UBIs) characteristic of both frontotemporal lobar degeneration (FTLD-U) and ALS. Clearance of TDP-43 and its aggregates is a subject of particular interest in the search for therapeutic targets for ALS.
Huang and coworkers investigated whether TDP-43 could be a CMA substrate, and identified the CMA recognition motif Q134VKKD138 (Huang et al., 2014). The authors showed that TDP-43 is cleaved to generate the fragments TDP-25 and TDP-35, which can then be degraded by different cellular pathways. Interestingly, these fragments are insoluble forms that serve as seeds for subsequent formation of aggregates and inclusions (Huang et al., 2014). Other studies have examined the role of HSC70 in ALS, although not always in the specific context of CMA. In their study of a drosophila model of ALS, Coyne et al. proposed a mechanism whereby overexpressed TDP-43 sequesters hsc70–4 at the neuromuscular junction, decreasing Hsc70–4 expression. Moreover, the authors reported similar results in mice expressing mutant TDP-43 (Coyne et al., 2017). Another study investigated the potential role of LAMP2A and HSC70 in the increased TDP-43 protein levels observed in peripheral blood mononuclear cells (PBMCs) in ALS patients. Arosio et al. reported decreases in HSC70 mRNA and protein levels, and decreases in LAMP2A mRNA only in samples from patients with longer disease duration (Arosio et al., 2020). Similar effects on HSC70 were observed in lymphoblastoids from patients with sporadic ALS (sALS) and ALS patients with mutated TDP-43. The absence of any changes in TDP-43 mRNA suggests that TDP-43 protein levels are increased due to a reduction in protein clearance. Together, the findings point to a key role of HSC70 in the pathogenesis of ALS, although decreased LAMP2A levels may also play a role in advanced stages of the disease (Arosio et al., 2020). A 2020 study provided the first in vivo evidence confirming TDP-43 as a CMA substrate (Ormeño et al., 2020). This study also showed that in cell culture the aggregate-prone form of TDP-43 also interacts with HSC70, and that TDP-43 aggregation upregulates CMA activity by increasing mRNA of LAMP2A and HSC70. This aggregation also causes partial damage of lysosomes with higher activity for CMA, pointing to a general dysfunction of the pathway (Ormeño et al., 2020).
While earlier studies established a link between Hsc70 and ALS, more recent findings identifying TDP-43 as a CMA substrate will likely serve as an impetus for further research into the role of CMA in the pathogenesis of this disease.
2.7. Modulation of CMA for the treatment of neurodegenerative diseases
Misfolding of proteins, either during translation/folding or as a consequence of cellular stress, is a relatively common event. Degradative processes (like UPS, CMA, and macroautophagy) act in a coordinated manner to eliminate these misfolded proteins. In the particular case of UPS, these proteins can block the proteasomal cylinder or remain attached to the membrane of the lysosomes if they are not first unfolded by chaperones, carriers, or adaptors (Ciechanover and Kwon, 2015). Dependence on autophagy varies between tissues and is particularly important in neurons, which cannot divide after differentiation. Neurons are also highly polarized cells in which retrograde transport of autophagosomes from the distal axon to the soma is required for fusion with lysosomes and completion of degradation. Pathogenic proteins may interfere with this transport process and, consequently, with autophagy (Cuervo and Wong, 2014; Maday and Holzbaur, 2014; Scrivo et al., 2018; Volpicelli-Daley et al., 2014). As stated above a growing body of evidence supports a protective role of CMA in neurons (Cai et al., 2015).
Mice with neural tissue-specific knockout of autophagy genes can survive post-natal starvation, but develop ubiquitin-positive inclusions in neurons and display motor deficits (Mizushima et al., 2008). These findings, together with the role of autophagy, and CMA in particular, in several neurodegenerative diseases suggest that boosting or restoring CMA exerts a neuroprotective effect that may be of therapeutic value in several neurodegenerative diseases. Transgenic overexpression of LAMP2A ameliorates the loss of proteostasis and attenuates the accumulation of damaged proteins in the liver of aged mice (Zhang and Cuervo, 2008), suggesting that maintenance of LAMP2A may offset the age-associated decline in CMA function.
Xilouri et al. demonstrated that overexpression of LAMP2A by nigral injection of recombinant adeno-associated virus vectors in mice ameliorates α-synuclein-induced neurodegeneration, prolongs the survival of substantia nigra neurons, and increases the number of axons that reach the striatum (Xilouri et al., 2013). Moreover, this effect correlates with a decrease in levels of α-synuclein and potentially pathogenic α-synuclein species. This result highlights the potential of gene therapy approaches to successfully reverse α-synuclein-induced toxicity. Another 2013 study demonstrated that pharmacological modulation of CMA through inhibition of the RARα receptor using synthetic derivatives of all-trans retinoic acid effectively protected NIH3T3 mouse fibroblasts from oxidative stress and proteotoxicity (Anguiano et al., 2013). Using the same compound, the reduction in lysosomal activity that is observed in fibroblasts from LRRK2 knock-in mice expressing the LRRK2 mutant R1441G can be reversed/ameliorated in an AR-7 dependent manner (Ho et al., 2020).
Oral administration of a new version of the aforementioned CMA activating compounds has been tested in a mouse model of AD, in which it ameliorates histopathological signs, decreases amyloid and tau phenotypes, and reverses behavioral phenotypes (Bourdenx et al., 2021). This provides the first in vivo evidence supporting the therapeutic potential of CMA activation in AD.
A 2000 study was the first to confirm the therapeutic potential of downregulating expression of the mutant Htt allele in a Tet-regulated mouse model of HD (Yamamoto et al., 2000). Koga and coworkers described an early increase in CMA, considered a compensatory response to macroautophagy loss, in cells expressing mutant Htt and in a knock-in mouse model of HD (Koga et al., 2011a). However, this compensatory response was compromised with increasing age. The authors showed that overexpression of LAMP2A to increase CMA in cells expressing mutant Htt accelerated the degradation of both WT and mutant Htt. Another study demonstrated that intrastriatal delivery in R6/2 mice of a construct containing polyQ binding motifs and recognition site for HSC70A decreased mutant Htt levels without affecting those of normal Htt, and that CMA contributed to degradation of mutant Htt (Bauer et al., 2010).
In summary, the successful reversal of neurodegeneration achieved by modulating CMA suggests that this may constitute a promising approach for the treatment of neurodegenerative diseases.
3. CMA and cancer
3.1. The complex role of CMA in cancer
Cancer encompasses a broad range of subtypes and involves many different processes (abnormal cell transformation and growth, invasion of other tissues, metastasis etc.). This significantly complicates efforts to tease apart the contributions of CMA in cancer. Nonetheless, CMA is known to play an important role in cancer biology. While induction of CMA may limit tumor development, experimental evidence indicates that CMA inhibition can also limit the growth of established tumors and improve response to cancer therapeutics (Fig. 3).
Figure 3. CMA exerts anti-oncogenic effects in healthy cells and pro-oncogenic effects in cancer cells.
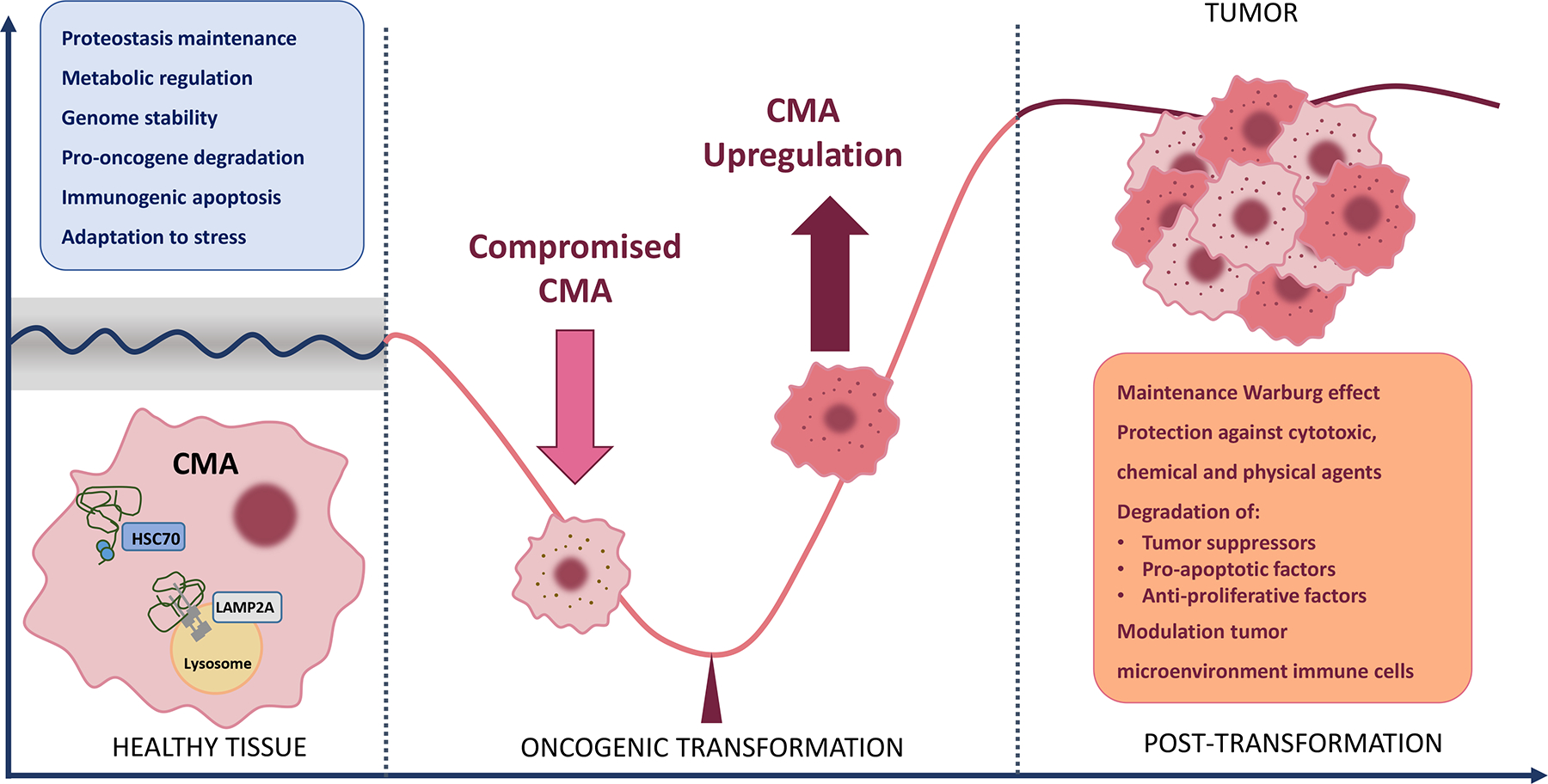
In vitro and in vivo studies have identified multiple mechanisms by which CMA exerts its anti-oncogenic effect in healthy cells (blue insert). Conditions associated with a decrease in CMA activity, such as disease and aging, favor malignant transformation. Once transformation occurs, CMA activity is upregulated. High CMA activity in cancer cells exerts a variety of pro-oncogenic effects (orange insert).
The first connection between CMA and cancer was established ten years ago (Kon et al., 2011). Since then, multiple studies have identified unique features of the interaction between CMA and cancer, as well as several key common aspects. One of the first described aspects of this interplay is a marked increase in CMA activity observed in a broad range of cancer cell lines, as well as increases in the expression of CMA components, including but not limited to LAMP2A, which is upregulated in the great majority of human tumors (Kon et al., 2011; Li et al., 2016; Saha, 2012). Survival of cancer cells, as well as tumorigenicity, is negatively affected by CMA blockade, and in xenograft models CMA blockade leads to tumor shrinkage (Kon et al., 2011). Multiple mechanisms underlie these effects of CMA in cancer, which depend on both tumor type and stage.
3.2. Anti-tumorigenic effects of CMA
The greatest risk factor for developing most forms of cancer, with a few exceptions, is age. The relationship between cancer risk and aging is well demonstrated: according to the National Cancer Institute (NCI), median patient age at the time of cancer diagnosis is 66 (https://www.cancer.gov/about-cancer/causes-prevention/risk/age). Interestingly, CMA activity decreases with age in almost all mammalian cell types and in most mammalian organs. Indeed, CMA dysfunction was first described in the context of aging. This decrease in CMA has been attributed to the reduction in LAMP2A content in lysosomes in aged cells, a consequence of altered stability of this essential CMA protein at the lysosomal membrane (Cuervo and Dice, 2000a; Kiffin et al., 2007) and age-associated changes in the lipid composition of the lysosomal membrane (Kaushik et al., 2006; Kiffin et al., 2007; Rodriguez-Navarro et al., 2012).
Below, we discuss published studies on the association between CMA and cancer and evolving ideas about the anti-oncogenic effects of CMA.
3.2.1. A player regulating oncogenic signaling
As a degradative pathway, CMA may constitute a tool used by the cell to mitigate the deleterious effects of stress. Recent findings indicate that specific oncogenic proteins can be selectively degraded either direct or indirectly by CMA, suggesting that this selective autophagy pathway may adapt to the specific conditions that mold the cancer phenotype.
The first indications of a tumor suppressing role of CMA were based on findings suggesting that CMA degrades the proto-oncogene protein MDM2 (Lu et al., 2010) and the tumor-associated protein TCTP (upon acetylation) (Bonhoure et al., 2017). Yang and colleagues recently demonstrated that 17β-estradiol (E2) stabilizes MORC2 (MORC family CW-type zinc finger 2), an emerging oncoprotein in human cancer (Yang et al., 2020). Phosphorylation of MORC2 at threonine 582 (T582) decreases its interaction with HSPA8 (heat shock protein family A [Hsp70] member 8) and LAMP2A, two of the core components of the CMA machinery, thus protecting MORC2 from lysosomal degradation by CMA. These findings point to a phosphorylation-dependent mechanism for MORC2 oncoprotein stabilization in response to estrogen and anti-estrogens through blockade of CMA-mediated lysosomal degradation. The first direct evidence of oncogene regulation by CMA to prevent malignant transformation was described for c-MYC (Gomes et al., 2017). Gomes et al. showed that inhibition of CMA in cultured fibroblasts was sufficient to increase c-MYC-induced transformation. These were acute experiments, and therefore the increase in c-MYC-induced transformation could be attributed to the elevated levels of c-MYC in CMA-deficient cells. Interestingly, c-MYC was not directly degraded by CMA. Rather, CMA facilitated its degradation via proteasomal activity by degrading the phosphatase CIP2A, which modulates site-specific phosphorylation of c-MYC and thereby controls its proteolysis through the ubiquitin-proteasome system (Gomes et al., 2017).
3.2.2. Prevention of intracellular changes that favors malignant transformation
Previous studies of CMA have revealed the important contribution of this pathway to protein quality control (Arias and Cuervo, 2011). Indeed, CMA activation appears to be a common mechanism of cell defense against proteotoxicity (Arias and Cuervo, 2011). In this section we describe recent advances in our understanding of the finely tuned incorporation of CMA activity into the proteostasis network to prevent intracellular changes that would otherwise favor malignant transformation.
Several studies have investigated the mechanisms that contribute to tumor growth in CMA-deficient tissues. More information is available for certain organs, such as the liver (Schneider et al., 2015). In this organ, deficient protein quality control in LAMP2A knockout mice leads to decreased tolerance to stress (Schneider et al., 2015) and favors malignant transformation and liver tumorigenesis (Arias and Cuervo, 2020). Liver-specific LAMP2A knockout also increases susceptibility to fibrosis caused by pro-oxidant insults (Schneider et al., 2015), similar to the increased susceptibility to fibrosis and hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease patients. In the context of liver damage, higher rates of malignant transformation can also be a consequence of underlying hepatic steatosis (Schneider et al., 2015). In fact, both, reduced protein quality control and steatosis, promote oncogenic transformation and liver tumors in CMA-deficient mice, which present areas of proteinaceous aggregates, lipidosis, and inflammation (Schneider et al., 2015). CMA is activated in response to lipid challenge (Kaushik and Cuervo, 2015), and an increased LAMP2A levels response is observed in experimental mouse models of non-alcoholic steatohepatitis (Das et al., 2013). Conversely, irregular increases in intracellular lipids inhibit CMA (Rodriguez-Navarro et al., 2012) and reduce LAMP2A expression in different models of steatosis (Cai et al., 2016; Sharma et al., 2011). This chain reaction, whereby decreased CMA promotes steatosis, which in turn inhibits CMA, may sustain the metabolic disorder and promote the progression to fibrosis and HCC. It is not known how CMA deficiency exerts a pro-fibrotic effect in the liver; while upregulation of macroautophagy is involved in stellate cells implication to liver fibrosis (Hernandez-Gea et al., 2012). Moreover, stellate cells show lower CMA activity than hepatocytes in basal conditions and similar CMA activity in response to stress(Dong et al., 2020).
Even less information is available for organs other than the liver about the processes through which CMA deficiency contributes to tumor development or growth. The results of a recent study of malignant mammary gland tumors with poor prognosis indicate that leptin, which is secreted at high levels into the blood stream of obese patients, regulates cathepsin A, which is responsible for LAMP2A degradation (Kim et al., 2020). This suggests that leptin may modulate mammary adenocarcinoma cells through cathepsin A and subsequent LAMP2A degradation.
Owing to its role in DNA repair, CMA is also part of the response to genotoxic damage (Park et al., 2015). It therefore seems likely that CMA deficiency will increase genomic instability and favor malignant transformation (Gomes et al., 2017).
3.3. Mechanisms underlying the regulatory effect of CMA in cancer
The mechanisms underlying the regulatory effect of CMA in cancer are only starting to be elucidated. A link between CMA and cancer was first demonstrated only a decade ago, when Kon et al. described the pro-carcinogenic and pro-survival effects of this pathway (Kon et al., 2011). Increased CMA was consistently identified in many different primary tumors and cancer cell lines (Kon et al., 2011), a finding that was somewhat controversial at the time, as macroautophagy, another well-known autophagy pathway, was already known to be upregulated or downregulated in cancer cells depending on tumor stage and type (White, 2012). Later studies corroborated the findings of Kon et al., identifying a constitutive increase in CMA as a common feature of cancer cells. This correlation has been ascribed prognostic value in the case of gastric (Zhou et al., 2016) and breast (Han et al., 2017) cancer. CMA has been associated with tumor size and cumulative recurrence in HCC patients (Ding et al., 2016), and with tumor growth modulation in non-small cell lung cancer (NSCLC) as well as papillary thyroid carcinoma (PTC) (Ichikawa et al., 2020; Zhou et al., 2020). Even in cancer cells with comparable expression to the match untransformed cells, LAMP2A blockade significantly inhibits cancer cell viability (Ding et al., 2016), indicating that in certain cases, basal levels of CMA activity may be sufficient to maintain carcinogenic activity. There are also many tumoral cells in which CMA exerts regulatory effects via non-conventional pathways.
Unfortunately, the mechanisms underlying CMA modulation in tumor cells remain mostly unknown, although it is described that it is a cell autonomous upregulation process, and occurs right after transformation (Kon et al., 2011). Metabolic stressors (e.g. nutrient scarcity) and oxidative stress (e.g. high ROS content, hypoxia) clearly promote increased CMA activity, together with other components of the tumor microenvironment (Cuervo et al., 1995; Dice, 1982; Finn and Dice, 2005; Hubbi et al., 2013; Kiffin et al., 2004)
3.4. Functions of CMA in cancer cells
Recent studies have helped to better define the role of CMA in cancer cells. This is currently a highly active field of research. In this section, we review some studies that have helped characterize the pro-oncogenic activity of CMA in tumoral cells. An important limitation that should be borne in mind is that many of these studies measured levels of CMA components (typically LAMP2A levels) as a readout of CMA activity, rather than performing actual functional CMA analysis, although other studies in which the latter approach has been used also support the proposed role of CMA in cancer.
3.4.1. Energetic stress and stress resistance
It is well demonstrated that cell metabolism is altered to meet the metabolic needs of cancer cells, which have the potential to proliferate uncontrollably (Pavlova and Thompson, 2016; Vander Heiden and DeBerardinis, 2017). Tumor cells use a variety of fuel sources to accomplish these goals, including CMA, which is involved in energetic homeostasis and stress resistance. Tumor cells grow within the tumor microenvironment in conditions of persistent stress. First, dysregulation of blood flow, which is adapted to the tumor’s needs, and the monitoring process of the immune system induce metabolic stress in the tumor cells and the stroma. This stress is further amplified by new signaling pressure pathways in cases in which oncological treatments are administered.
Cancer cells exhibit a marked shift toward anabolic metabolism in order to biosynthesize the essential building blocks required to maintain proliferation. Indeed, cancer cells often depend on anaerobic glycolysis for energy production, even in the presence of oxygen (Bartrons and Caro, 2007) (known as the Warburg effect) (Warburg, 1956). In melanoma and lung cancer cells, CMA blockade stabilizes p53 and decreases the transcription of glycolytic enzymes (Vousden and Ryan, 2009), an effect that contrasts with the increase in these enzymes observed in control cells upon CMA inhibition (Schneider et al., 2014). CMA activity is therefore thought to be necessary to maintain the Warburg effect in melanoma and lung cancer cells (Kon et al., 2011). Xenograft studies using HCC cells have corroborated the role of CMA in energy maintenance in these cancer cells, in which the absence of CMA markedly impacts cell viability (Ding et al., 2016).
CMA is also responsible for energetic modulation in other types of cancer through direct degradation of other glycolytic enzymes, including (i) acetylated pyruvate kinase 2, degradation of which leads to proliferation due to the accumulation of glycolytic intermediates (Lv et al., 2011); and (ii) hexokinase-II, which drives tumor cell growth (Palmieri et al., 2009) and occurs after CMA degradation depending on glucose availability in non-AML cells (Xia et al., 2015). Upregulation of CMA to deplete hexokinase-II was proposed as a means to induce metabolic crisis and cell death, although this approach has proven difficult, as phosphorylation of this enzyme at Thr473 (as described in breast cancer) prevents CMA degradation (Yang et al., 2018). However, inhibition of the kinase responsible for hexokinase-II phosphorylation results in a reduction in cancer cell growth (Yang et al., 2018).
Upregulation of CMA in breast cancer cells facilitates survival during oxidative stress, as it has been shown LAMP2A deprivation promotes apoptosis of these cells (Saha, 2012). In specific forms of lung cancer expressing misfolded N-CoR, increased CMA confers protection against ER stress. The misfolded N-CoR protein is required for activation of oncogenic survival pathways, while its degradation by CMA decreases the ER stress induced by its expression (Ali et al., 2011). Hypoxia has also been shown to activate transcription of CMA genes in several types of cancer, and CMA contributes to adaptation to hypoxia through degradation of hypoxia-inducible factor-1 alpha (HIF-1α) (Ferreira et al., 2015; Hubbi et al., 2013), further supporting a role of CMA in cancer cell survival in the context of tumor-induced hypoxia.
3.4.2. Cancer cell progression, invasion, and migration
In addition to regulating energetic homeostasis, CMA can sustain cancer cell proliferation through modulation of the cell cycle. Degradation by CMA of cell cycle-related proteins such as RND3/RhoE and HIF-1α (in this case induced by cyclin-dependent kinase 2) has been shown to be necessary for the proliferation of gastric cancer cells (Zhou et al., 2016), cervical carcinoma, and HCC (Hubbi et al., 2014; Kudo et al., 2020). Furthermore, a recent study showed that LAMP2A knockdown in NSCLC cells suppresses proliferation and colony formation (Ichikawa et al., 2020). Based on their findings, the authors proposed that intrinsic apoptosis signaling may mediate the cell death that occurs following CMA blockade, and speculated that LAMP2A may regulate malignant phenotypes of NSCLC through CMA modulation in vitro.
CMA has also been proposed to contribute to tumor metastasis in lung cancer (Kon et al., 2011): downregulation of CMA in cancer cells reduces the metastatic potential by decreasing migration and resistance to anoikis (a form of programmed cell death induced upon cell detachment from the extracellular matrix), a mechanism critical to prevent adherent-independent cell growth and attachment to an inappropriate matrix, thus preventing colonization of distant organs (Dower et al., 2018). A link between CMA and breast cancer metastasis has also been proposed (Han et al., 2017). While it remains unclear how CMA activity influences metastasis, in certain types of cancer cells degradation by CMA of the multifunctional protein HSD17B4 exerts a modulatory effect on the proliferative, invasive, and migratory properties of these cells (Huang et al., 2020; Zhang et al., 2017). A recent study reported increases in HSD17B4, which is involved in fatty acid β-oxidation and steroid metabolism, in prostate cancer (PCa) versus matched paratumor tissue, highlighting a role in PCa progression (Huang et al., 2020). The authors showed that dihydroxytestosterone (DHT) treatment increases HSD17B4 acetylation, and then promotes its degradation by CMA. Also, in the context of PCa, CMA downregulation of Atg5-dependent macroautophagy also promotes metastasis (Han et al., 2017). Furthermore, CMA decreases PPAR-γ protein expression, thereby increasing SDF1 and CXCR4 expression, enhancing tumor cell proliferation and migration, and ultimately promoting tumor growth and metastasis (Zhou et al., 2020).
3.5. Impact in the immune response
Mobilization of the immune system in response to cancer can render in sustained tumor control. This raises the question: What is the role of CMA in the anti-tumor immune response? While this issue has not been explored in depth, several research groups have already highlighted a potential tumor-protective role of CMA.
Some cancer cells have been found to modulate CMA in host cells in order to actively suppress the anti-oncogenic response. In a glioblastoma (GB) mouse model, cancer immunotolerance was induced by actively dysregulating CMA in pericytes (PC), perivascular cells that regulate capillary function and promote an immunological defense through inflammatory responses at the neurovascular unit (Valdor et al., 2019). In vivo grafting of CMA-deficient PCs into the GB mouse model reduced GB proliferation and induced a more effective immune response than that observed in mice grafted with control PCs. These findings point to abnormal upregulation of CMA as a mechanism by which GB cells harness the immunosuppressive function of PCs and stabilize GB-PC interactions to ensure tumor cell survival (Valdor et al., 2019).
CMA can also facilitate immunogenic cell death (ICD), a form of apoptosis induced by some chemotherapeutic drugs, by mediating the exposure of surface-exposed calreticulin (Garg et al., 2013). Immunogenic apoptosis of cancer cells facilitates a cooperative antitumor immune response through activation of dendritic cells, and subsequent activation of a specific T cell response (Spisek and Dhodapkar, 2007).
3.6. Tumor microenvironment in relation to CMA
Different studies have investigated the role of CMA in the tumor microenvironment. In addition to the aforementioned glioblastoma-dependent increase in CMA in pericytes, which abolishes their anti-oncogenic response (Valdor et al., 2019), an increase in LAMP2A in tumor-associated macrophages may be necessary for breast cancer progression. In fact, macrophage LAMP2A levels associated to the tumor have been proposed to be required for breast cancer progression. (Wang et al., 2019). While the specific processes by which tumor cells regulate CMA in the tumor microenvironment are unknown, proposed mechanisms include microtube-like projections (Valdor et al., 2019) and tumor-generated exosomes (Liu et al., 2019).
Cells in the tumor microenvironment can also participate in CMA regulation in cancer cells. For example, in response to chemotherapy, HCC-resident macrophages secrete IL-17, which increases levels of LAMP2A in tumor cells and likely also increases CMA activity, thus conferring resistance to chemotherapy (Guo et al., 2017), as described in greater detail in section 3.7.
An anti-oncogenic role of CMA in cells of the tumor micro-environment has also be proposed. p65 is a key component of the CMA substrate nuclear factor-kappa B (NF-κB), and reduced CMA activity in epithelial cells results in increased NF-κB signaling and contributes to progression of epithelial-mesenchymal transition and tumorigenesis (Tang et al., 2017).
3.7. Participation of CMA in cancer treatment resistance
CMA typically acts as a protective mechanism by selectively degrading proteins that are altered during cancer therapy, including proteins oxidized by pro-oxidant agents (Saha, 2012) or photodynamic therapy (Dewaele et al., 2011). However, in some cases CMA can promote resistance against anti-oncogenic interventions, as described below.
CMA prevents apoptosis and contributes to oxaliplatin resistance in hepatocellular carcinoma by degrading the apoptosis trigger cyclin D1 (Guo et al., 2017), and to irradiation resistance by degrading the antitumor protein HMGB1(Wu et al., 2017). Active degradation of the acetyltransferase p300/CBP by CMA confers resistance to 5‐fluorouracil in colorectal cancer (Du et al., 2017). Upregulation of LAMP2A by exosomes released by hepatitis B virus-associated cancer has been shown to reduce oxaliplatin-induced cell death (Liu et al., 2019). Furthermore, LAMP2A modulates the response to platinum‐based chemotherapy by regulating antiapoptotic intrinsic signaling in NSCLC patients (Ichikawa et al., 2020).
Breast tumors from patients undergoing recurrence after tamoxifen and fulvestrant treatment show increased MORC2 phosphorylation, which protects MORC2 from lysosomal degradation by CMA (Yang et al., 2020), suggesting a potential role of MORC2 in estrogen-induced resistance to anti-estrogen therapies.
Emerging evidence indicates that induction of CMA during cancer therapy may constitute a key mechanism of resistance in multiple cancer types. In patients with esophageal squamous cell carcinoma (ESCC), downregulation of CMA activity has been proposed to increase sensitivity to cisplatin (Cao et al., 2021). In HCC patients, resistance to sorafenib is a major obstacle to treatment with this drug. In conditions of hypoxia, alterations in the distribution of necroptosis-related proteins contribute to sorafenib resistance (Liao et al., 2021a). HSP90α plays a critical role in this process by binding to the necrosome complex and promoting degradation via CMA, thereby blocking necroptosis and increasing sorafenib resistance.
3.8. CMA related strategies against cancer
While research in the field of CMA continues to investigate the link between defective CMA and cancer, several factors have hindered the translation of these findings into preventive or therapeutic strategies. First, all components and modulators of CMA (e.g. signaling elements, chaperones) are involved in multiple important cell processes. This is a key obstacle to the identification of selective chemical modulators of CMA and to date no studies have identified a specific component of the CMA pathway that could serve as a target for external manipulation. Perhaps the most promising candidate is LAMP2A, although it shares a high level of homology (85%) with other spliced variants of the LAMP2 gene that mediate distinct cellular functions. To date no selective inhibitors of CMA have been generated. Selective CMA activators have been developed (Bourdenx et al., 2021): synthetic derivatives of all-trans-retinoic acid specifically neutralize the inhibitory effect of CMA through retinoic acid receptor α (RARα) (Anguiano et al., 2013).
In contrast to macroautophagy, in which contrasting effects have been described depending on the context and type of cancer, CMA appears to play a consistently pro-oncogenic role in most cases. Nonetheless, CMA is regulated very differently in tumoral cells versus matched non-transformed cells, what renders in the binary role of CMA pathway (pro-oncogenic in transformed cells, and anti-oncogenic mechanism in healthy cells). The search for CMA modulators is further complicated by the fact that the mechanism underlying the shift in CMA activity that occurs following transformation remains unknown. Regardless, current research efforts continue to search for new targets and strategies that allow the restoration and modulation of CMA as a preventive or therapeutic approach.
3.8.1. Prophylaxis
Given the well described anti-oncogenic role of CMA in healthy cells, and the results of interventions that counteract the age-associated decline in CMA, there is general consensus that restoration of CMA activity holds significant promise as a cancer prevention therapy. Zhang and Cuervo demonstrated that genetic intervention can successfully attenuate the decline in CMA that occurs in aging rodents, thereby delaying liver proteotoxicity and the accumulation of cellular damage (Zhang and Cuervo, 2008). Those authors also speculated that this intervention prevented age-associated oncogenic transformation in the liver. Whether this effect will also be observed in other organs is unclear, given the likelihood of tissue-dependent differences in the antitumoral function of CMA, and will need to be investigated in future studies.
Here it is important to highlight the tight link between CMA and nutritional status. This association suggests that nutritional interventions could be used as a tool to preserve CMA activity and protect against malignant transformation, at least in aging individuals.
3.8.2. Therapeutics
Genetic manipulation (LAMP2A knockdown) in many different types of cancer has provided the first evidence of the potential of CMA as a therapeutic target. In multiple cancer cell lines, LAMP2A knockdown results in a clear decrease in proliferation, increases susceptibility to stressors, and reduces carcinogenicity in xenografts (Dewaele et al., 2011; Han et al., 2017; Kon et al., 2011; Liu et al., 2019; Saha, 2012; Wu et al., 2017; Zhang et al., 2017; Zhou et al., 2016). Even after tumor formation in xenograft models, shRNA against LAMP2A induces regression of the primary tumor and decreases metastasis (Kon et al., 2011). Taken together, the anti-tumor effects of CMA downregulation and the resistance to cancer treatment observed after CMA upregulation supports CMA blockade as a potential cancer treatment strategy.
Upregulation of CMA to exceed the already high levels in tumor cells leads to depletion of glycolytic enzymes, ultimately resulting in energetic crisis and cell death (Xia et al., 2015), and also contributes to metabolic stress through secondary blockade of macroautophagy (Xia et al., 2015). New small molecules have been used to induce this effect in AML cells, although their selectivity for CMA and the applicability of this approach in other conditions remains to be demonstrated (Xia et al., 2015). Previously described activators of CMA could prove useful in this context (assuming RARα inhibition also govern in cancer cells) (Anguiano et al., 2013). While available data support the utility of genetic approaches to modulate CMA to treat cancer, selective inhibition of this pathway using synthetic compounds is not yet a realistic therapeutic option: the chemical alternatives described so far interfere also with other autophagy forms, as they block proteolytic activity at the lysosomal level.
Several clinical trials have investigated the effects of HCQ (hydroxychloroquine) in the treatment of cancer (clinicaltrials.gov). HCQ inhibits proteolysis in the lysosomal compartment by increasing the intralysosomal pH, and therefore affects multiple autophagic pathways. A sustained increase in lysosomal pH has a negative impact on the stability of hsc70, a luminal protein crucial for internalization of substrates through the CMA pathway, and thereby impairs CMA. However, the extent to which CMA contributes to the beneficial effect of HCQ in cancer is unknown.
4. Concluding remarks and future prospects
Although CMA is less studied and less well characterized than other autophagy subtypes, numerous studies in recent years have helped to further our understanding of this process and identify essential molecular players (Arias, 2015; Arias et al., 2015; Pajares et al., 2018). Moreover, the development of new transgenic mouse models constitutes an essential tool to help elucidate the dynamics of CMA substrate degradation in vivo (Dong et al., 2020). Research into CMA has gathered momentum, revealing new links between deficient CMA and human diseases (Arias and Cuervo, 2020; Bourdenx et al., 2021) and opening new avenues of basic and clinical research.
Studies have also established a link between neurodegeneration and decreased CMA, due either to the action of pathogenic protein aggregates or as a contributing cause for the disease. The development of new drugs that activate CMA holds significant promise for the treatment of these diseases that affect a large proportion of the aged population.
A growing body of evidence also implicates CMA in cancer. Recent findings have helped to more accurately define the role of CMA in cancer and its anti- and pro-tumorigenic action. The involvement of CMA extends beyond cancer cells themselves, as it appears to facilitate cross-talk between tumor cells and the tumor microenvironment. Further research into the role of CMA in tumor–host interactions and tumor immunity will be essential to further understand this complex relationship. Meanwhile, the effects of more potent and specific CMA modulators will need to be investigated to assess their utility as molecular tools in basic research and as clinical drug candidates.
Acknowledgements
Authors thank the members of their respective groups and colleagues in the field of autophagy for the valuable feedback and suggestions when preparing this work. We apologize to those esteemed colleagues whose work may have been omitted from this review due to space limitation.
Funding
Work in EA laboratory is supported by the National Institutes of Health: DK124308, P30DK041296 (P&F) and P30AG038072 (P&F).
RGS is recipient of the Spanish Ministry of Science and Innovation grant RTI2018-098990-J-I00.
References
- Ali AB, Nin DS, Tam J, and Khan M (2011). Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS One 6, e25268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, and Schapira AH (2010). Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67, 1464–1472. [DOI] [PubMed] [Google Scholar]
- Andersson FI, Werrell EF, McMorran L, Crone WJ, Das C, Hsu ST, and Jackson SE (2011). The effect of Parkinson’s-disease-associated mutations on the deubiquitinating enzyme UCH-L1. J Mol Biol 407, 261–272. [DOI] [PubMed] [Google Scholar]
- Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, and Cuervo AM (2013). Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nature chemical biology 9, 374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E (2015). Lysosomal mTORC2/PHLPP1/Akt axis: a new point of control of chaperone-mediated autophagy. Oncotarget 6, 35147–35148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E (2017). Methods to Study Chaperone-Mediated Autophagy. Methods in enzymology 588, 283–305. [DOI] [PubMed] [Google Scholar]
- Arias E, and Cuervo AM (2011). Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 23, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E, and Cuervo AM (2020). Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol Metab 31, 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E, Koga H, Diaz A, Mocholi E, Patel B, and Cuervo AM (2015). Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol Cell 59, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio A, Cristofani R, Pansarasa O, Crippa V, Riva C, Sirtori R, Rodriguez-Menendez V, Riva N, Gerardi F, Lunetta C, et al. (2020). HSC70 expression is reduced in lymphomonocytes of sporadic ALS patients and contributes to TDP-43 accumulation. Amyotrophic lateral sclerosis & frontotemporal degeneration 21, 51–62. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, and Kelly JW (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U, Kaushik S, Varticovski L, and Cuervo AM (2008). The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28, 5747–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, and Cuervo AM (2010). Identification of regulators of chaperone-mediated autophagy. Mol Cell 39, 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber SC, and Shaw PJ (2010). Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 48, 629–641. [DOI] [PubMed] [Google Scholar]
- Bartrons R, and Caro J (2007). Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr 39, 223–229. [DOI] [PubMed] [Google Scholar]
- Bastías-Candia S, Zolezzi JM, and Inestrosa NC (2019). Revisiting the Paraquat-Induced Sporadic Parkinson’s Disease-Like Model. Molecular neurobiology 56, 1044–1055. [DOI] [PubMed] [Google Scholar]
- Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y, et al. (2010). Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nature biotechnology 28, 256–263. [DOI] [PubMed] [Google Scholar]
- Behl C (1999). Alzheimer’s disease and oxidative stress: implications for novel therapeutic approaches. Progress in neurobiology 57, 301–323. [DOI] [PubMed] [Google Scholar]
- Bonhoure A, Vallentin A, Martin M, Senff-Ribeiro A, Amson R, Telerman A, and Vidal M (2017). Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur J Cell Biol 96, 83–98. [DOI] [PubMed] [Google Scholar]
- Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, and Tsarbopoulos A (2017). Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Progress in neurobiology 155, 171–193. [DOI] [PubMed] [Google Scholar]
- Bourdenx M, Martin-Segura A, Scrivo A, Rodriguez-Navarro JA, Kaushik S, Tasset I, Diaz A, Storm NJ, Xin Q, Juste YR, et al. (2021). Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 184, 2696–2714 e2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE, Ferrante RJ, and Beal MF (1999). Oxidative stress in Huntington’s disease. Brain pathology (Zurich, Switzerland) 9, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B, Bourdenx M, Luengo E, Diaz A, Sohn PD, Chen X, Wang C, Juste YR, Wegmann S, Patel B, et al. (2021). Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun 12, 2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, Mandelkow EM, Mandelkow E, and Cuervo AM (2018). Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Jogasuria A, Yin H, Xu MJ, Hu X, Wang J, Kim C, Wu J, Lee K, Gao B, et al. (2016). The Detrimental Role Played by Lipocalin-2 in Alcoholic Fatty Liver in Mice. Am J Pathol 186, 2417–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zeng W, Tao K, E Z, Wang B, and Yang Q (2015). Chaperone-mediated autophagy: roles in neuroprotection. Neuroscience bulletin 31, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D, Shan D, Yan W, Zhang Z, Song Q, Jiang Y, Zhang X, Zhang Z, Wang Z, Wang Y, et al. (2021). Chaperone-mediated autophagy affects tumor cell proliferation and cisplatin resistance in esophageal squamous cell carcinoma. Thorac Cancer 12, 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HL, Terlecky SR, Plant CP, and Dice JF (1989). A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385. [DOI] [PubMed] [Google Scholar]
- Choi SH, and Cho K (2021). LAMP2A-mediated autophagy involved in Huntington’s disease progression. Biochem Biophys Res Commun 534, 561–567. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, and Kwon YT (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Experimental & molecular medicine 47, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, Lorenzini I, Chou CC, Torvund M, Rogers RS, Starr A, Zaepfel BL, Levy J, Johannesmeyer J, Schwartz JC, et al. (2017). Post-transcriptional Inhibition of Hsc70–4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell reports 21, 110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (1996). A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (2000a). Age-related decline in chaperone-mediated autophagy. J Biol Chem 275, 31505–31513. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (2000b). Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–583. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF, and Knecht E (1997). A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 272, 5606–5615. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Hu W, Lim B, and Dice JF (1998). IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell 9, 1995–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Knecht E, Terlecky SR, and Dice JF (1995). Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 269, C1200–1208. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, and Sulzer D (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, and Wong E (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Research 24, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damme M, Suntio T, Saftig P, and Eskelinen EL (2015). Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta neuropathologica 129, 337–362. [DOI] [PubMed] [Google Scholar]
- Das S, Seth RK, Kumar A, Kadiiska MB, Michelotti G, Diehl AM, and Chatterjee S (2013). Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 305, G950–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Duve C, and Wattiaux R (1966). Functions of lysosomes. Annu Rev Physiol 28, 435–492. [DOI] [PubMed] [Google Scholar]
- Dewaele M, Martinet W, Rubio N, Verfaillie T, de Witte PA, Piette J, and Agostinis P (2011). Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J Cell Mol Med 15, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice JF (1982). Altered degradation of proteins microinjected into senescent human fibroblasts. J Biol Chem 257, 14624–14627. [PubMed] [Google Scholar]
- Dice JF (1990). Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15, 305–309. [DOI] [PubMed] [Google Scholar]
- Ding ZB, Fu XT, Shi YH, Zhou J, Peng YF, Liu WR, Shi GM, Gao Q, Wang XY, Song K, et al. (2016). Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int J Oncol 49, 2367–2376. [DOI] [PubMed] [Google Scholar]
- Dohi E, Tanaka S, Seki T, Miyagi T, Hide I, Takahashi T, Matsumoto M, and Sakai N (2012). Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem Int 60, 431–442. [DOI] [PubMed] [Google Scholar]
- Dong S, Aguirre-Hernandez C, Scrivo A, Eliscovich C, Arias E, Bravo-Cordero JJ, and Cuervo AM (2020). Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat Commun 11, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dower CM, Wills CA, Frisch SM, and Wang HG (2018). Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 14, 1110–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Huang D, Peng Y, Yao Y, Zhao Y, Yang Y, Wang H, Cao L, Zhu WG, and Gu J (2017). 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett 400, 183–193. [DOI] [PubMed] [Google Scholar]
- Ferreira JV, Fofo H, Bejarano E, Bento CF, Ramalho JS, Girao H, and Pereira P (2013). STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9, 1349–1366. [DOI] [PubMed] [Google Scholar]
- Ferreira JV, Soares AR, Ramalho JS, Pereira P, and Girao H (2015). K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci Rep 5, 10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn PF, and Dice JF (2005). Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem 280, 25864–25870. [DOI] [PubMed] [Google Scholar]
- Franch HA, Sooparb S, Du J, and Brown NS (2001). A mechanism regulating proteolysis of specific proteins during renal tubular cell growth. J Biol Chem 276, 19126–19131. [DOI] [PubMed] [Google Scholar]
- Gao L, She H, Li W, Zeng J, Zhu J, Jones DP, Mao Z, Gao G, and Yang Q (2014). Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxid Redox Signal 20, 2936–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, Dudek AM, and Agostinis P (2013). Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis 4, e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudioso A, Garcia-Rozas P, Casarejos MJ, Pastor O, and Rodriguez-Navarro JA (2019). Lipidomic Alterations in the Mitochondria of Aged Parkin Null Mice Relevant to Autophagy. Front Neurosci 13, 329–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LR, Menck CFM, and Cuervo AM (2017). Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 13, 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueninger F, Bohrmann B, Czech C, Ballard TM, Frey JR, Weidensteiner C, von Kienlin M, and Ozmen L (2010). Phosphorylation of Tau at S422 is enhanced by Abeta in TauPS2APP triple transgenic mice. Neurobiol Dis 37, 294–306. [DOI] [PubMed] [Google Scholar]
- Guo B, Li L, Guo J, Liu A, Wu J, Wang H, Shi J, Pang D, and Cao Q (2017). M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 8, 44465–44476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Deng Y, Chen S, Chen R, Yang M, Zhang Z, Sun X, Wang W, He Y, Wang F, et al. (2017). Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep 7, 4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CD, Ermak G, and Davies KJ (2007). RCAN1–1L is overexpressed in neurons of Alzheimer’s disease patients. FEBS J 274, 1715–1724. [DOI] [PubMed] [Google Scholar]
- Hauser DN, and Hastings TG (2013). Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiology of Disease 51, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, and Friedman SL (2012). Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PW, Leung CT, Liu H, Pang SY, Lam CS, Xian J, Li L, Kung MH, Ramsden DB, and Ho SL (2020). Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 16, 347–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, Yin L, and Shu HB (2016). Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 45, 555–569. [DOI] [PubMed] [Google Scholar]
- Huang CC, Bose JK, Majumder P, Lee KH, Huang JT, Huang JK, and Shen CK (2014). Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci 127, 3024–3038. [DOI] [PubMed] [Google Scholar]
- Huang H, Liu R, Huang Y, Feng Y, Fu Y, Chen L, Chen Z, Cai Y, Zhang Y, and Chen Y (2020). Acetylation-mediated degradation of HSD17B4 regulates the progression of prostate cancer. Aging (Albany NY) 12, 14699–14717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbi ME, Gilkes DM, Hu H, Kshitiz, Ahmed I, and Semenza GL (2014). Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc Natl Acad Sci U S A 111, E3325–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, and Semenza GL (2013). Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem 288, 10703–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa A, Fujita Y, Hosaka Y, Kadota T, Ito A, Yagishita S, Watanabe N, Fujimoto S, Kawamoto H, Saito N, et al. (2020). Chaperone-mediated autophagy receptor modulates tumor growth and chemoresistance in non-small cell lung cancer. Cancer Sci 111, 4154–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, and Kopito RR (2005). Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A 102, 13135–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabuta T, Furuta A, Aoki S, Furuta K, and Wada K (2008). Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 283, 23731–23738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ, Waak J, and Gasser T (2009). DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med 47, 1354–1361. [DOI] [PubMed] [Google Scholar]
- Kaushik S, and Cuervo AM (2015). Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17, 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Massey AC, and Cuervo AM (2006). Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J 25, 3921–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffin R, Christian C, Knecht E, and Cuervo AM (2004). Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15, 4829–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, Martinez-Vicente M, and Cuervo AM (2007). Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci 120, 782–791. [DOI] [PubMed] [Google Scholar]
- Kim JW, Mahiddine FY, and Kim GA (2020). Leptin Modulates the Metastasis of Canine Inflammatory Mammary Adenocarcinoma Cells Through Downregulation of Lysosomal Protective Protein Cathepsin A (CTSA). Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner P, Bourdenx M, Madrigal-Matute J, Tiano S, Diaz A, Bartholdy BA, Will B, and Cuervo AM (2019). Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol 17, e3000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17, 1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, and Cuervo AM (2011a). Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci 31, 18492–18505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, and Cuervo AM (2011b). A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun 2, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, and Cuervo AM (2011). Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 3, 109ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu S, Fatima A, Gutierrez-Garcia R, and Vilchez D (2017). Proteostasis of Huntingtin in Health and Disease. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo Y, Sugimoto M, Arias E, Kasashima H, Cordes T, Linares JF, Duran A, Nakanishi Y, Nakanishi N, L’Hermitte A, et al. (2020). PKClambda/iota Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 38, 247–262 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, Zhang X, and Ye L (2016). Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene 35, 4048–4057. [DOI] [PubMed] [Google Scholar]
- Li X, and Wang J (2014). Maintenance of chaperone-mediated autophagy activity in cultured cells expressing mutant huntingtin. Biomedical reports 2, 529–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Yang Y, Pan D, Ding Y, Zhang H, Ye Y, Li J, and Zhao L (2021a). HSP90alpha Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia. Cancers (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Wang B, Liu W, Xu Q, Hou L, Song J, Guo Q, and Li N (2021b). Dysfunction of chaperone-mediated autophagy in human diseases. Mol Cell Biochem 476, 1439–1454. [DOI] [PubMed] [Google Scholar]
- Liu DX, Li PP, Guo JP, Li LL, Guo B, Jiao HB, Wu JH, and Chen JM (2019). Exosomes derived from HBV-associated liver cancer promote chemoresistance by upregulating chaperone-mediated autophagy. Oncol Lett 17, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang P, Song W, and Sun X (2009). Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J 23, 3383–3392. [DOI] [PubMed] [Google Scholar]
- Lu TL, Huang GJ, Wang HJ, Chen JL, Hsu HP, and Lu TJ (2010). Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem Biophys Res Commun 398, 26–31. [DOI] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. (2011). Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, and Holzbaur EL (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Developmental cell 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, and Di Monte DA (2010). Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem 285, 13621–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzella L, Ahlberg J, and Glaumann H (1981). Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B Cell Pathol Incl Mol Pathol 36, 219–234. [DOI] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, and Cuervo AM (2006). Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103, 5805–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI (2008). Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & development 22, 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein SJ, and Cuervo AM (2010). Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 21, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormeño F, Hormazabal J, Moreno J, Riquelme F, Rios J, Criollo A, Albornoz A, Alfaro IE, and Budini M (2020). Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Frontiers in molecular neuroscience 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajares M, Rojo AI, Arias E, Diaz-Carretero A, Cuervo AM, and Cuadrado A (2018). Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 14, 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri D, Fitzgerald D, Shreeve SM, Hua E, Bronder JL, Weil RJ, Davis S, Stark AM, Merino MJ, Kurek R, et al. (2009). Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol Cancer Res 7, 1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Suh Y, and Cuervo AM (2015). Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 6, 6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Kim DH, and Yoon SY (2016). Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep 49, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, and Qin ZH (2012). The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One 7, e46834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, and Boya P (2013). Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, and Cuervo AM (2012). Dietary lipids and aging compromise chaperone-mediated autophagy by similar mechanisms. Autophagy 8, 1152–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, and Cuervo AM (2012). Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A 109, E705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha T (2012). LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 8, 1643–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JL, and Cuervo AM (2014). Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol 11, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JL, Suh Y, and Cuervo AM (2014). Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20, 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, and Cuervo AM (2015). Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 14, 249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrivo A, Bourdenx M, Pampliega O, and Cuervo AM (2018). Selective autophagy as a potential therapeutic target for neurodegenerative disorders. The Lancet. Neurology 17, 802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setsuie R, Wang YL, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, Li H, Furuta A, Sano Y, Sun YJ, et al. (2007). Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem Int 50, 119–129. [DOI] [PubMed] [Google Scholar]
- Sharma S, Mells JE, Fu PP, Saxena NK, and Anania FA (2011). GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One 6, e25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spisek R, and Dhodapkar MV (2007). Towards a better way to die with chemotherapy: role of heat shock protein exposure on dying tumor cells. Cell Cycle 6, 1962–1965. [DOI] [PubMed] [Google Scholar]
- Tang J, Zhan MN, Yin QQ, Zhou CX, Wang CL, Wo LL, He M, Chen GQ, and Zhao Q (2017). Impaired p65 degradation by decreased chaperone-mediated autophagy activity facilitates epithelial-to-mesenchymal transition. Oncogenesis 6, e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O’Rourke JG, Khashwji H, et al. (2009). IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol 187, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R, Garcia-Bernal D, Riquelme D, Martinez CM, Moraleda JM, Cuervo AM, Macian F, and Martinez S (2019). Glioblastoma ablates pericytes anti-tumor immune function through aberrant upregulation of chaperone-mediated autophagy. . Proc Nat Acad Sci USA E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, and Macian F (2014). Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 15, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, and Lee VM (2014). Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 25, 4010–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, and Ryan KM (2009). p53 and metabolism. Nat Rev Cancer 9, 691–700. [DOI] [PubMed] [Google Scholar]
- Walters RO, Arias E, Diaz A, Burgos ES, Guan F, Tiano S, Mao K, Green CL, Qiu Y, Shah H, et al. (2018). Sarcosine Is Uniquely Modulated by Aging and Dietary Restriction in Rodents and Humans. Cell Rep 25, 663–676 e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, Feng D, Gao G, and Yang Q (2016). Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 12, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Liu Y, Liu L, Chen M, Wang X, Yang J, Gong Y, Ding BS, Wei Y, and Wei X (2019). Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 40, 118–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, and Mandelkow E (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18, 4153–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, and Mandelkow E (2010). Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 6, 182–183. [DOI] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- White E (2012). Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JH, Guo JP, Shi J, Wang H, Li LL, Guo B, Liu DX, Cao Q, and Yuan ZY (2017). CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol 23, 2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T, et al. (2015). Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol 210, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, and Stefanis L (2013). Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain : a journal of neurology 136, 2130–2146. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, and Stefanis L (2009). Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 4, e5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, and Hen R (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66. [DOI] [PubMed] [Google Scholar]
- Yang F, Xie HY, Yang LF, Zhang L, Zhang FL, Liu HY, Li DQ, and Shao ZM (2020). Stabilization of MORC2 by estrogen and antiestrogens through GPER1- PRKACA-CMA pathway contributes to estrogen-induced proliferation and endocrine resistance of breast cancer cells. Autophagy 16, 1061–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, and Mao Z (2009). The complexity in regulation of MEF2D by chaperone-mediated autophagy. Autophagy 5, 1073–1074. [DOI] [PubMed] [Google Scholar]
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, and Mao Z (2009). Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323, 124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Ren C, Qiao P, Han X, Wang L, Lv S, Sun Y, Liu Z, Du Y, and Yu Z (2018). PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 37, 5997–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, and Lee VM (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- Zhang C, and Cuervo AM (2008). Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Sun Y, Fei M, Tan C, Wu J, Zheng J, Tang J, Sun W, Lv Z, Bao J, et al. (2014). Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 10, 1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu YY, Yao CB, Li JT, Zhao XN, Yang HB, Zhang M, Yin M, Chen J, and Lei QY (2017). Acetylation targets HSD17B4 for degradation via the CMA pathway in response to estrone. Autophagy 13, 538–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xie X, Chen Y, Lin Y, Cai Z, Ding L, Wu Y, Peng Y, Tang S, and Xu H (2020). Chaperone-mediated Autophagy Governs Progression of Papillary Thyroid Carcinoma via PPARgamma-SDF1/CXCR4 Signaling. J Clin Endocrinol Metab 105. [DOI] [PubMed] [Google Scholar]
- Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J, Zhang S, Shang Y, Jiang X, Guo H, et al. (2016). Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 12, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]