

Abstract
Cytochrome P450 3A4 (CYP3A4) is the most important drug-metabolizing enzyme in humans and has been associated with harmful drug interactions. The activity of CYP3A4 is known to be modulated by several compounds and by the electron transfer partner, cytochrome P450 reductase (CPR). The underlying mechanism of these effects, however, is poorly understood. We have used hydrogen–deuterium exchange mass spectrometry to investigate the impact of binding of CPR and of three different substrates (7-benzyloxy-4-trifluoromethyl-coumarin, testosterone, and progesterone) on the conformational dynamics of CYP3A4. Here, we report that interaction of CYP3A4 with substrates or with the oxidized or reduced forms of CPR leads to a global rigidification of the CYP3A4 structure. This was evident from the suppression of deuterium exchange in several regions of CYP3A4, including regions known to be involved in protein–protein interactions (helix C) and substrate binding and specificity (helices B′ and E, and loop K/β1). Furthermore, the bimodal isotopic distributions observed for some CYP3A4-derived peptides were drastically impacted upon binding to CPR and/or substrates, suggesting the existence of stable CYP3A4 conformational populations that are perturbed by ligand/CPR binding. The results have implications for understanding the mechanisms of ligand binding, allostery, and catalysis in CYP enzymes.
Graphical Abstract
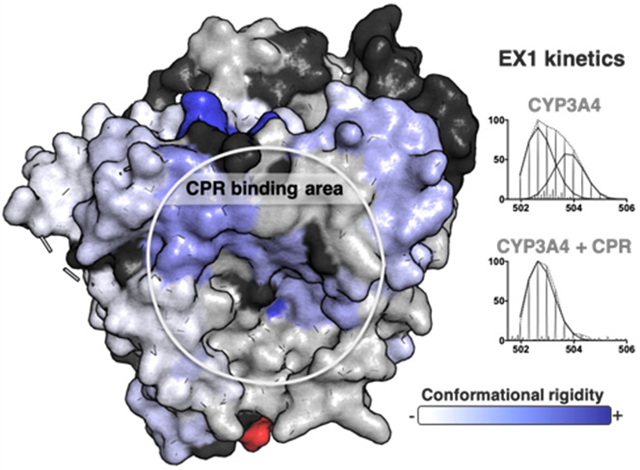
Cytochrome P450 enzymes (CYPs) make up a large family of heme-containing monooxygenases that share a similar three-dimensional architecture. Most CYPs utilize O2 and electrons donated by a redox partner to catalyze substrate monooxygenation. Cytochrome P450 reductase (CPR) is the primary electron transfer partner of mammalian CYPs. Importantly, polymorphisms in the human cpr gene are associated with metabolic disorders, such as defects in steroid metabolism.1 It has been suggested that the cpr mutations may differentially affect the ability of the mutant CPR proteins to functionally interact with their endogenous CYP partners.1 CPR is a diflavin reductase that contains one molecule each of flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN). The flavin cofactors enable CPR to shuttle electrons from NADPH (an obligate two-electron donor) one at a time to CYP to support oxygen activation and, ultimately, substrate oxidation. During these electron transfer steps, CPR undergoes large conformational changes from a closed form to an open form.2–4,56 In addition, various studies have reported that CPR binding may induce conformational changes in CYPs that promote interprotein electron transfer, oxygen activation, and substrate binding.6,7 However, the current understanding of CYP conformational changes induced by redox partner binding is based mostly on the bacterial P450cam-putidaredoxin (Pdx) system. The extent to which these findings apply to other CYPs remains to be investigated.8–10
Human CYP3A4 is central to the metabolism of approximately 50% of all pharmaceuticals and is often implicated in drug interactions and chemoresistance.11,12 This enzyme is well-known to exhibit complex ligand-dependent allosteric and cooperative behaviors.13 Whereas many studies have focused on elucidating the allosteric effects of substrates,14–17 effectors,15,18,19 other CYPs,20,21 and the membrane on CYP3A4,22,23 very little attention has been paid to understanding the allosteric effects of CPR binding and its underlying impact on the conformational dynamics of CYP3A4, which could have profound effects on the catalytic properties of the enzyme.
It is well accepted that a negatively charged surface of the FMN-binding domain of CPR interacts with a positively charged concave surface on the proximal face of CYPs (Figure 1).24 While the exact CPR binding mode remains elusive, residues in helix C of CYP3A4, part of the positively charged patch, have been associated with redox partner binding in many CYP isoforms.7 Because CYP–CPR interactions may be modulated by substrates, effectors, and other CYP enzymes,25–27 a better understanding of the structural basis for this phenomenon will improve our understanding of CYP3A4 cooperativity and our ability to predict drug metabolism and interactions.
Figure 1.
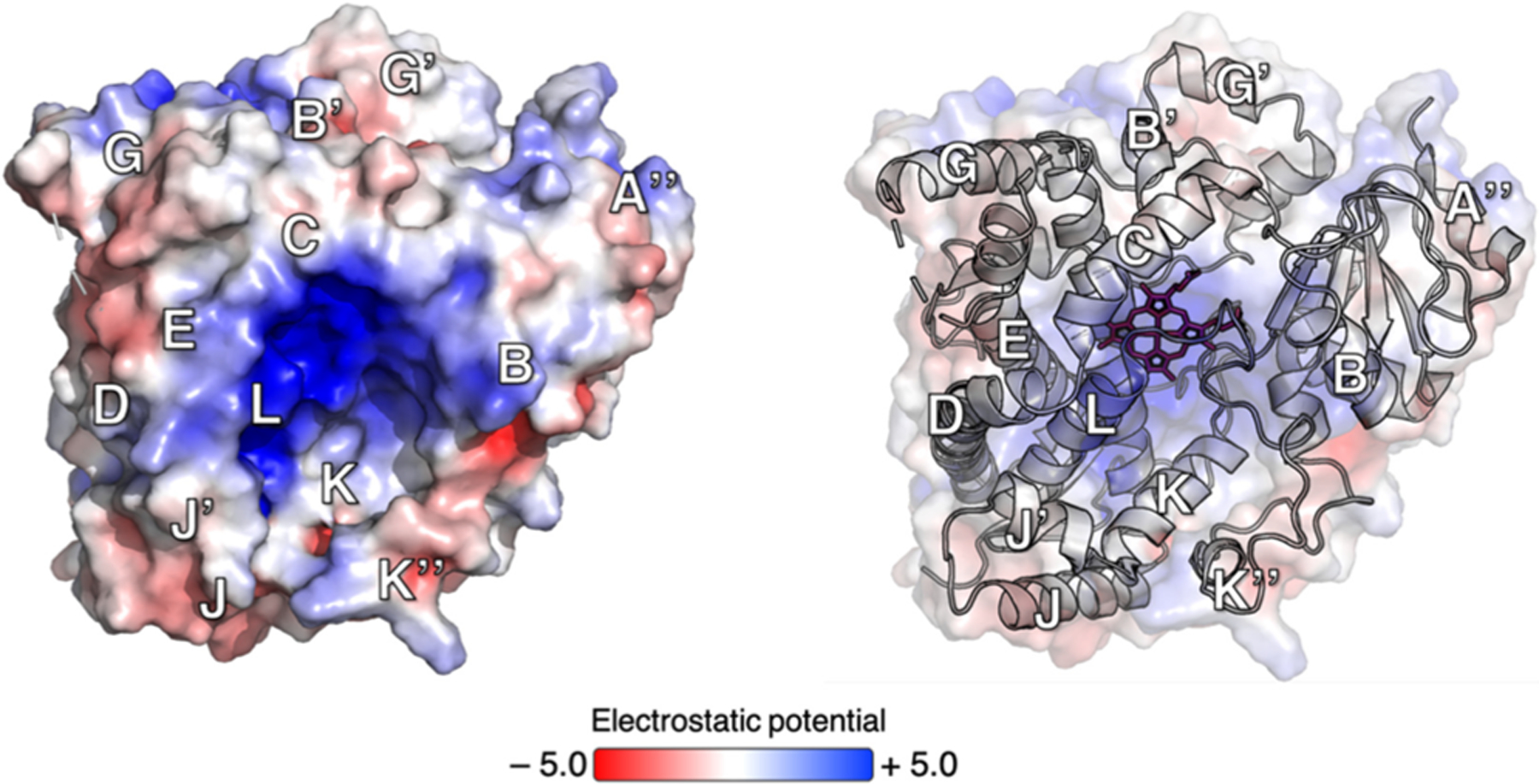
Electrostatic potential (volts) of the CYP3A4 proximal surface relative to the heme group (red wireframe). Blue denotes regions displaying a positive potential, and red regions displaying a negative potential. The positively charged concave surface involves helices B, C, E, K, and L.
In this study, we employ hydrogen–deuterium exchange mass spectrometry (HDX-MS) to characterize the conformational flexibility of CYP3A4 in the presence of CPR and/or its substrates, 7-benzyloxy-4-trifluoromethyl-coumarin (BFC), testosterone (TST), and progesterone (PRG). HDX-MS probes the dynamics of structural elements in proteins by monitoring the time-dependent exchange of deuterium atoms between the solvent and backbone amide groups.28 The rate of deuterium uptake is highly sensitive to the local hydrogen bonding environment and, as such, can be used to deduce ligand binding interactions and/or to infer the existence of conformational changes under different conditions (e.g., ligand-bound and/or unbound states).29 A decrease in the level of deuterium exchange typically reflects a structural organization, whereas an increase in the rate of deuterium uptake suggests enhanced flexibility.
As presented herein, our HDX-MS results reveal an overall increase in the rigidity of CYP3A4 upon interaction with CPR or substrates. Interestingly, the structural elements affected by CPR and substrates are largely shared, suggesting that binding of CYP3A4 to either CPR or the substrate may stabilize enzyme conformations poised for catalysis. This conclusion is further supported by the coalescence of bimodal isotopic distributions of CYP3A4 peptides derived from helices B′, C, and E, and sheet β1, suggesting a shift in the CYP3A4 conformational ensemble to a more homogeneous population of structures upon substrate and/or CPR binding. Finally, the dynamic structural elements of CYP3A4 perturbed by CPR are not restricted to the CYP–CPR interface but extend to regions associated with substrate binding and specificity. Cumulatively, these data build on the growing body of evidence for the functional relevance of conformational dynamics in CYP3A4.30–32
METHODS
Protein Expression and Purification.
The N-terminally truncated Δ3–13 CYP3A4 was expressed and purified as previously described.18 Truncation of the transmembrane helix is commonly employed to improve the protein expression yield and solubility and does not significantly impact enzymatic function under in vitro conditions.33,34 The protein was stored at −80 °C in 0.1 M potassium phosphate buffer (pH 7.4) containing 10% glycerol. Rat cytochrome P450 reductase (CPR) was expressed and purified as previously reported35 except for one important modification: the liquid growth medium was supplemented with a filter-sterilized, fully dissolved riboflavin solution (0.2 mg/mL, 50:50 acetonitrile/H2O, pH 11), which was added at a volume ratio of 5 mL/L to the growth medium at the time of inoculation. Amino acid sequences of CYP3A4 and CPR are listed in Figure S1.
BFC Hydroxylation Assay.
The activity of CYP3A4 toward BFC was determined by measuring the initial rate of the enzyme-catalyzed debenzylation of BFC to yield 7-hydroxy-4-(trifluoromethyl)coumarin (HFC) using a micro-titer plate reader with fluorescence detection. The assay was performed in potassium phosphate (KPi) buffer (0.1 M, pH 7.4, 10% glycerol) containing CYP3A4 (0.5 μM), CPR (2 μM), BFC (100 μM from an initial stock of 30 mM in DMSO), and NADPH (1 mM from a 25 mM stock in deionized water) in a total reaction volume of 150 μL. The final DMSO concentration in the assay was <1%. Prior to initiation of the reaction, CYP3A4 and CPR were incubated in assay buffer for 1 h at room temperature to allow proper interaction. BFC was then added to the solution, before being transferred to individual plate wells. The reaction was initiated by the addition of NADPH. HFC production was monitored for 2 h (excitation and emission at 410 and 530 nm, respectively) at 37 °C, and the measured signal was converted to micromolar per minute using an HFC calibration curve. Initial rates were measured at different BFC concentrations (10–100 μM), and HFC formation was monitored for 90 min. Slopes were calculated from the initial linear portions of the kinetic curves. The kinetic data were fitted with the Michaelis–Menten and Hill equations using GraphPad Prism 7.0. Better fits (higher R2 values) were obtained with the Hill equation.
TST and PRG Hydroxylation Assays.
The enzyme activity of CYP3A4 with the substrates TST and PRG in the presence of either CPR/NADPH or the cofactor surrogate cumene hydroperoxide (CHP) was monitored by LC-MS. The reaction mixtures contained CYP3A4 (0.5 μM), PRG or TST (10–150 μM from an initial stock of 1–15 mM in DMSO), CPR (2 μM), and NADPH (1 mM from a 25 mM stock in deionized water), or CHP (0.4 mM) in 0.1 M KPi buffer. When CPR was used, CPR and CYP3A4 were first preincubated together in KPi buffer at room temperature for 1 h, and the reactions were initiated with the addition of NADPH and allowed to proceed at 37 °C for 75 min. When CHP was used, the reactions were initiated with CHP and the total reaction time was 6 min. All reactions were performed in triplicate. For each substrate concentration, reaction rates were obtained from the linear portion of the kinetic curve containing three time points (CHP, 2, 4, and 6 min; CPR, 45, 60, and 75 min). At the appropriate time point, 150 μL of the reaction mixture was quenched by addition of 0.5 mL of DCM. The substrates and products were extracted in DCM (2 × 0.5 mL), and the combined organic layers were dried under N2. The dried samples were redissolved in acetonitrile (0.1 mL) before LC-UV-MS analysis. The samples were eluted on a C-18 Omega Polar HPLC column (Phenomenex, 250 mm × 4.6 mm, 5 mm particle size) using a 50:50 acetonitrile/H2O solvent for 3 min, followed by a linear increase to 80% acetonitrile over 9 min, and another linear increase to 95% acetonitrile over 8 min. The products were detected by their absorption at 244 nm, and their corresponding masses were confirmed by MS detection in positive ion mode. Hydroxylated TST products eluted between 7 and 10 min, and TST eluted at 15 min. Hydroxylated PRG products eluted between 12 and 16 min, and PRG eluted at 21 min. Calibration curves of TST and PRG were used to convert the absorption signals of the product peaks into concentration (micromolar).
Equilibrium Titrations.
Titration of CYP3A4 with TST and PRG was conducted at room temperature in 0.1 M potassium phosphate buffer (pH 7.4) supplemented with 10% glycerol. Substrates were dissolved in DMSO and added to a 1.5 μM protein solution in small aliquots, keeping the total volume of the added titrant to <2% of the final volume. After each addition, the reaction mixture was allowed to equilibrate for at least 20 min. Ultraviolet–visible (UV–vis) absorption spectra were recorded when the signal was stable. Titration plots were constructed from the difference spectra and fit to the Hill equation to derive dissociation constants (KD) and Hill coefficients (nH).
Hydrogen–Deuterium Exchange Protocol.
Separately, the CYP3A4 and CPR solutions were concentrated to ~100 μM using Spin-X concentrators (30 kDa molecular weight cutoff, 0.5 mL, Corning). All deuterium exchange reaction mixtures consisted of 1 μM CYP3A4 in deuterated potassium phosphate buffer [0.1 M KPi/D2O (pD 7.0)] to ensure a final D2O concentration of >98% (D2O/H2O), which is critical for maximizing deuterium uptake. As needed, CPR (4 μM), NADPH (1 mM), and/or substrate (100 μM BFC, TST, or PRG) was added to the reaction mixture. Stock solutions were prepared for NADPH (60 mM in Milli-Q) and the substrates (20 mM in DMSO). Prior to the hydrogen–deuterium exchange reaction, when appropriate, the concentrated stocks of CYP3A4 and CPR were preincubated together for 20 min to allow for optimal interaction between the two proteins. The substrate stock solutions were premixed in deuterated buffer before addition to the enzyme solution. HDX reactions were initiated by the addition of the enzyme(s) to deuterated buffer (with or without substrate). At the desired time points (0.5–60 min), aliquots of the reaction mixture (50 μL) were immediately quenched with pH 1.7 buffer (100 μL, 100 mM KPi) to yield a final pH of 2.5, thus ensuring minimal amide H/D back exchange. The quenched samples were immediately flash-frozen in liquid nitrogen and stored at −80 °C. Each reaction was performed in triplicate on the same day using the same stock solutions.
MS Data Acquisition.
All HDX-MS experiments were performed on a Waters Synapt G2-Si instrument with HDX technology, and MS data acquisition was performed as previously reported.36 One at a time, the quenched reaction samples (150 μL, stored at −80 °C in 1.5 mL Eppendorf tubes) were thawed for 70 s in a water bath at 32 °C and then loaded into a 40 μL injection loop of the HDX manager. The samples were injected into the HDX manager exactly 2 min after they were removed from the freezer. In the HDX manager, the protein was cleaved by elution (100 μL/min flow rate of 0.1% formic acid in Milli-Q water) through an online immobilized pepsin column for 3 min at 15 °C. The resulting peptic peptides were trapped on a C18 guard column held at 0.4 °C before reverse-phase chromatographic separation using a Waters BEH C18 UPLC column (1 mm × 100 mm). Prior to the loading of the peptides, the column was equilibrated in a 97:3 (v/v) Milli-Q water/acetonitrile solvent containing 0.1% formic acid. The peptides were eluted at 0.4 °C with a linear gradient from 3% to 100% acetonitrile containing 0.1% formic acid applied over 10 min. The samples were ionized by electrospray ionization (ESI) with a capillary voltage of 2.8 kV, a sampling cone of 30 V, a source offset of 30 V, and a desolvation temperature of 175 °C. Data were collected in positive ion and resolution modes. The ionized peptides were then further separated in the gas phase by traveling wave ion mobility using a wave velocity of 650 m/s, a wave height of 40 V, a bias voltage of 3 V, and a nitrogen pressure of 3.1 mbar. After exiting the ion mobility region, the peptides were subjected to two alternating collision energy regimes applied over 0.4 s intervals: a low collision energy of 6 V and a high-collision energy ramp of 21–44 V. The high-energy regime fragments ions by collision-induced dissociation (CID), while the low-energy regime conserves the peptide precursor masses. This process allows the matching of peptide fragments with their respective parent ions and enables peptide identification with a high degree of confidence. A [glu-1]-fibrinopeptide B (Glu-Fib) external lock mass standard was always run in parallel with all samples to enable correction of the peptide m/z values. An average variance in the determination of deuterium uptake values for this specific workflow was previously determined to be 0.087 Da with a standard deviation of 0.095 Da.36 These values are similar to the previously reported peptide-level continuous exchange, bottom-up HDX-MS deuterium uptake differences.37,38
MS Data Processing.
A peak list of the CYP3A4 peptides reproducibly detected by our workflow was generated. This list was determined by analyzing reference samples for CYP3A4 prepared in triplicate in protiated potassium phosphate buffer (100 mM, pH 2.5) and subjected to the HDX-MS workflow described above. The peptide reference lists were generated by uploading the raw MS data into the Protein Lynx Global Server (PLGS) software (Waters) as previously described.36 PLGS scans the raw data and associates the detected peptide peaks with expected masses. Spectral assignments were scored by a variety of user-defined criteria: parts per million error, difference in chromatographic retention time, difference in ion mobility drift time, and number of MS/MS ion matches. The PLGS output was uploaded into DynamX 3.0 (Waters) for additional thresholding and data analysis. The final peptide list was restricted to include only peptides with m/z values within 5 ppm of the theoretical mass, which produced a minimum of 0.2 product (fragment) ion per amino acid, that were detected in all three replicates. This resulted in a final list of 147 peptides covering 89.1% of the CYP3A4 sequence with an amino acid redundancy of 4.25.
MS Data Analysis.
DynamX 3.0 was used to quantify the level of deuterium uptake for each peptide under all conditions tested. DynamX 3.0 calculates a centroid mass from the isotopic distribution of deuterated peptide m/z signals. The centroid mass is then compared to the similarly determined centroid mass for the nondeuterated reference peptides. The deuterium uptake is quantified on the basis of this comparison and is reported either as relative uptake (in daltons) or as relative fractional uptake (RFU, percent), where the absolute deuterium uptake is normalized by the total number of amide protons in the peptide. Data were curated manually to ensure that all peptide assignments by the software were correct. Differential HDX was used to compare two states by subtracting the deuterium uptake data for each peptide for the two states of interest. We ensured that all compared peptides were from the same charge state and that the reference and deuterated peptide had matching chromatographic retention times, ion mobility drift times, and m/z values. A summary of the conditions tested is provided in Table S1. Bimodal isotope distributions were fitted with HX-express version 2.39
Assessing HDX-MS Variance.
The overall reproducibility of the workflow was established as previously described.36 All difference data reported in this work represent a sum of the HDX difference values for each peptide at each of the analyzed time points. The significant deuterium uptake difference between compared states was determined using the Deuteros version 2.0 developed by Politis and co-workers.40 The hybrid statistical test was used to calculate the significance of deuterium uptake differences between the compared HDX profiles. The RFU difference for the differential profiles of interest was mapped onto the CYP3A4 structure [Protein Data Bank (PDB) entry 1W0F]. The RFU normalizes the total level of deuterium uptake to the theoretical uptake maximum, based on the number of exchangeable backbone amides in the peptide.
RESULTS
Impact of CPR on the Functional Cooperativity and Substrate Binding Affinity of CYP3A4.
The kinetics of BFC, TST, and PRG oxidation were measured using either CPR (4-fold excess) and NADPH or the hydrogen peroxide donor cumene hydroperoxide (CHP) as a surrogate redox partner (Figure S10). CHP is known to sustain the activity of CYP3A4 and to bypass the electron transfer steps normally catalyzed by CPR in the native system.41 Both CPR/NADPH and CHP concentrations were adjusted to achieve maximum catalytic activity to enable a more optimal comparison between the two systems (Figure S5). The results revealed that the functional positive cooperativity (sigmoidal behavior) for these substrates42 was generally more pronounced in the presence of CPR, suggesting a direct contribution of CPR to this phenomenon. In agreement with our previous results,41 the CYP3A4 activity was significantly higher with all substrates when CHP was used as the redox system, likely as a result of bypassing the rate-limiting electron transfer steps in the CPR/NADPH system.
Spectrophotometric equilibrium titrations were next performed to evaluate the impact of CPR binding on the dissociation constants (Kd) and binding cooperativity of TST and PRG, which induce a type I spectral shift (blue shift) in the λmax of the Soret band of heme absorption (Figure S11). BFC binding is not associated with a spectral shift and was therefore not investigated in this experiment. We found that the presence of CPR decreased the extent of the PRG-induced spectral shift and reduced the dissociation constant (Kd) for PRG by nearly 2-fold, from 35 to 19 μM. In contrast, CPR enhanced the TST-dependent spectral shift and slightly weakened the binding affinity for TST from 46 to 55 μM. Thus, the binding of CPR to CYP3A4 clearly influences binding of the substrate yet has distinct effects on different substrates. The Hill coefficients (nH) for binding of both substrates were only modestly affected by CPR, suggesting that CPR binding does not significantly affect the binding cooperativity of TST and PRG. These interesting results prompted us to investigate how binding of substrates and/or CPR alters the structural dynamics of CYP3A4, as this may provide insight into mechanisms leading to functional cooperativity.
Overview of the HDX Workflow.
To investigate the structural dynamics of CYP3A4, a continuous-labeling, bottom-up HDX-MS approach was utilized (Table S1). Deuterium exchange on the backbone amide groups of CYP3A4 was achieved by exposing the protein to a deuterated buffer for various periods of time ranging from 0.5 to 60 min, in the presence or absence of CPR (oxidized or reduced), BFC, TST, or PRG. The workflow resulted in the reproducible detection of 56–65 peptides, providing a CYP3A4 sequence coverage of 75–81%. Regions that were incompletely covered included helix A″ and small portions of helices D, G′, G, and I, for which the raw data were searched manually to locate additional peptides. To determine the significance in deuterium uptake changes across the CYP3A4 structure, we used the hybrid statistical test as implemented in Deuteros version 2.0,40 with confidence intervals varying between 95% and 99.9%. This statistical approach combines a Welch’s t test with a globally estimated threshold calculated from the variance of all data points.43 All significant peptides detected in our study are detailed in Figures S2 and S3, and comparisons between results obtained under different conditions can be visualized in butterfly plots (Figure S4).
The significant uptake changes over time were summed to capture more subtle changes in HDX. CYP3A4 is known to oligomerize in solution.44,45 Thus, we previously carried out a control experiment to ensure that CYP3A4 aggregation was not interfering with the deuterium uptake observed under our conditions.30
Impact of the Binding of Oxidized CPR on the Structural Dynamics of CYP3A4.
To determine the impact of CPR binding on the conformational dynamics of CYP3A4, we first compared the dynamics of the enzyme in the presence and absence of oxidized CPR (oxCPR). The oxidation state of CPR was determined spectroscopically (Figure S6). To be consistent with our kinetic and binding studies, a 4-fold excess of CPR to CYP3A4 was used in these experiments. Of 65 CYP3A4 peptic peptides compared with and without oxCPR, 26 peptides showed a significant difference [±0.60 Da, confidence interval (CI) of 99.9%] in deuterium uptake (Figures S2 and S7). The respective sum of relative fractional uptake (RFU, percent) differences for these peptides at all D2O exposure times (0.5, 5, and 30 min) is mapped onto the CYP3A4 structure in Figure 2. Interestingly, changes in structural dynamics associated with oxCPR binding were distributed throughout the CYP3A4 structure rather than localized at the putative CPR–CYP interface near helix C of CYP3A4. Furthermore, binding of oxCPR results in an overall decrease in the rate of deuterium uptake, which suggests a global reduction in the conformational flexibility of CYP3A4. As expected, a substantial deuterium uptake difference was observed at the binding interface between CYP3A4 and CPR (helix C), with a total decrease in uptake of −3.2 Da upon CPR binding. Large decreases in the rate of deuterium uptake were also observed for helices B′ and E (−5.5 and −3.4 Da, respectively) and loop β1/K (−3.0 Da) upon oxCPR binding. These regions are associated with substrate binding and specificity, suggesting that interaction of CYP3A4 with CPR may help to define substrate-binding sites. Notably, the proximal loop K″/L involved in the regulation of the heme redox potential also becomes more rigid in the presence of oxCPR. In contrast to the rigidification observed throughout most of the CYP3A4 structure, helix J undergoes substantial loosening upon oxCPR binding (+7.7 Da). The functional relevance of this change is not clear.
Figure 2.
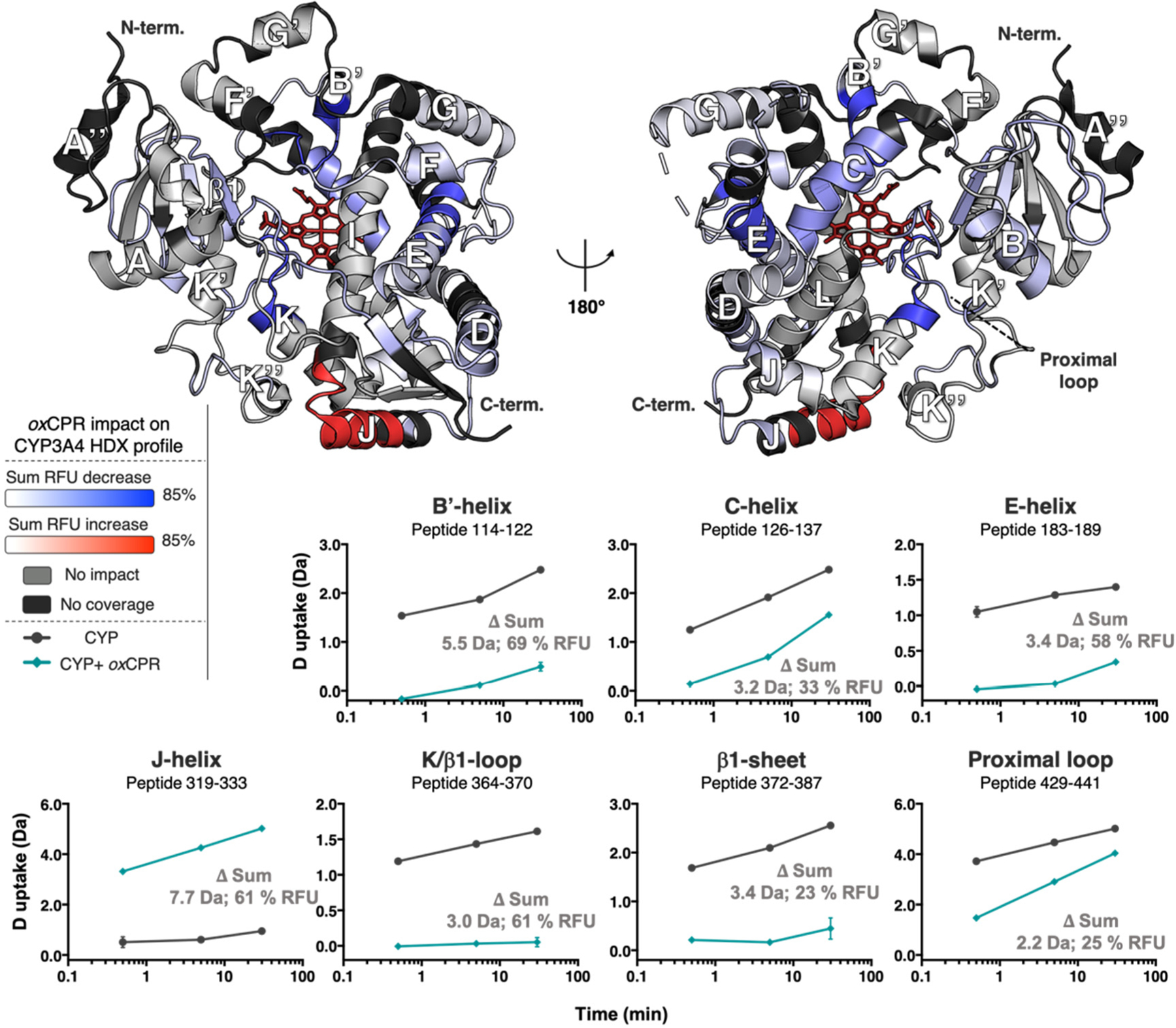
Impact of oxCPR binding on CYP3A4 dynamics represented by the deuterium uptake difference of the free enzyme, [CYP], minus the enzyme:CPR complex, [CYP:oxCPR]. The significant sum RFU difference of three time points (0.5, 5, and 30 min) is mapped onto the CYP3A4 structure (PDB entry 1W0F), which is shown in both distal (left) and proximal (right) views relative to the heme group (red wireframe). Blue denotes regions that undergo a decrease in the rate of deuterium uptake upon interaction with oxCPR, whereas red designates regions that undergo an increase in the rate of deuterium uptake. The light gray color shows regions unaffected by oxCPR binding, and the regions shown in black were not covered by the MS analysis. Deuterium uptake time courses are shown for the most impacted peptides in the bottom panels. The sum of deuterium uptake and RFU difference is shown for each peptide (CI of 99.9%: ±0.60 Da). The plots for CYP3A4 are colored black, and those for the [CYP3A4:oxCPR] complex are colored teal. Error bars represent the standard deviations of triplicate measurements.
Impact of the Binding of Reduced CPR on the Structural Dynamics of CYP3A4.
CPR is believed to undergo important conformational changes upon reduction.2–6 The reduced form of CPR (redCPR) was also reported to have an affinity for CYPs higher than that of oxCPR.46 Therefore, we hypothesized that binding of redCPR might affect the structural dynamics of CYP3A4 differently than those of oxCPR and could potentially reveal CYP3A4 conformational changes that are more specifically relevant to the electron transfer process. To investigate this, we first generated a differential deuterium exchange profile comparing free CYP3A4 with the [CYP3A4:redCPR] complex. Reduction of CPR was achieved with the addition of 1 mM NADPH and was confirmed by UV–vis absorption spectroscopy (Figure S6). The deuterium exposure times (0.5, 5, and 30 min) were selected to ensure that CPR remained reduced across all time points in the HDX reaction (Figure S6). The resulting full differential HDX profile is shown in Figure 3, and the complete list of significant peptides is provided in Figure S2. All deuterium uptake plots containing significant peptides are reported in Figure S9.
Figure 3.
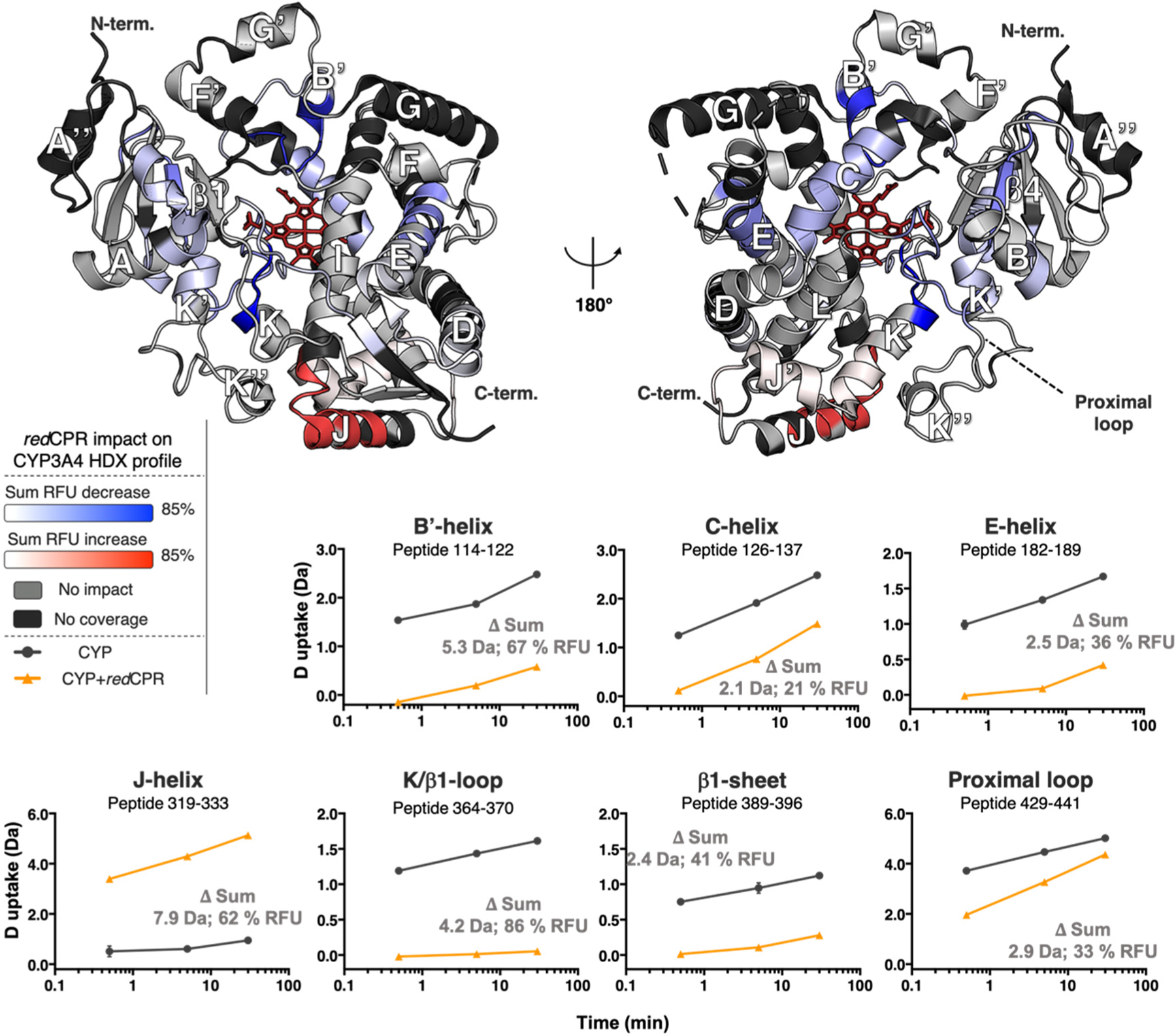
Impact of redCPR binding on CYP3A4 dynamics represented by the deuterium uptake difference of the free enzyme, [CYP], minus the enzyme:CPR complex, [CYP:redCPR]. The significant sum RFU difference of the three time points (0.5, 5, and 30 min) is mapped onto the CYP3A4 structure (PDB entry 1W0F), which is shown in both distal (left) and proximal (right) views relative to the heme group (red wireframe). Blue denotes regions that undergo a decrease in the rate of deuterium uptake upon interaction with redCPR, whereas red designates regions that undergo an increase in the rate of deuterium uptake. The light gray color shows regions unaffected by redCPR binding, and the black regions were not covered by the MS analysis. Deuterium uptake time courses are shown for the most impacted peptides in the bottom panels. The sum of deuterium uptake and RFU difference is shown for each (CI of 99.9%, ±0.69 Da). The plots for CYP3A4 are colored black, and those for the [CYP3A4:redCPR] complex are colored orange. Error bars represent the standard deviations of triplicate measurements.
Overall, the effect of redCPR binding on the conformational dynamics of CYP3A4 was found to be very similar to that of oxCPR binding and was characterized by an obvious rigidification of the CYP3A4 structure relative to the free enzyme. Comparison of the [CYP:oxCPR] and [CYP:redCPR] states, however, revealed six significantly different peptides (±0.34 Da, CI of 99%), many of which were slightly more flexible in the [CYP:redCPR] complex than in the [CYP:oxCPR] complex (Figure 4). These regions include loop D/E (+0.4 Da), loop J/J′ (+0.6 Da), and loop H/I (+0.6 Da). Interestingly, proximal loop K″/L bearing the heme iron Cys442 ligand also becomes significantly more dynamic in the presence of redCPR (+0.5 Da) compared to oxCPR. Strikingly, the middle portion of helix I, which contains the conserved Thr309 residue critical for proton relay during oxygen activation, is more flexible in the [CYP:redCPR] complex. The only region found to undergo a decrease in the rate of deuterium uptake in the [CYP:redCPR] complex relative to the [CYP:oxCPR] state was helix K′. The pertinency of the rigidification of helix K′ is not immediately obvious.
Figure 4.
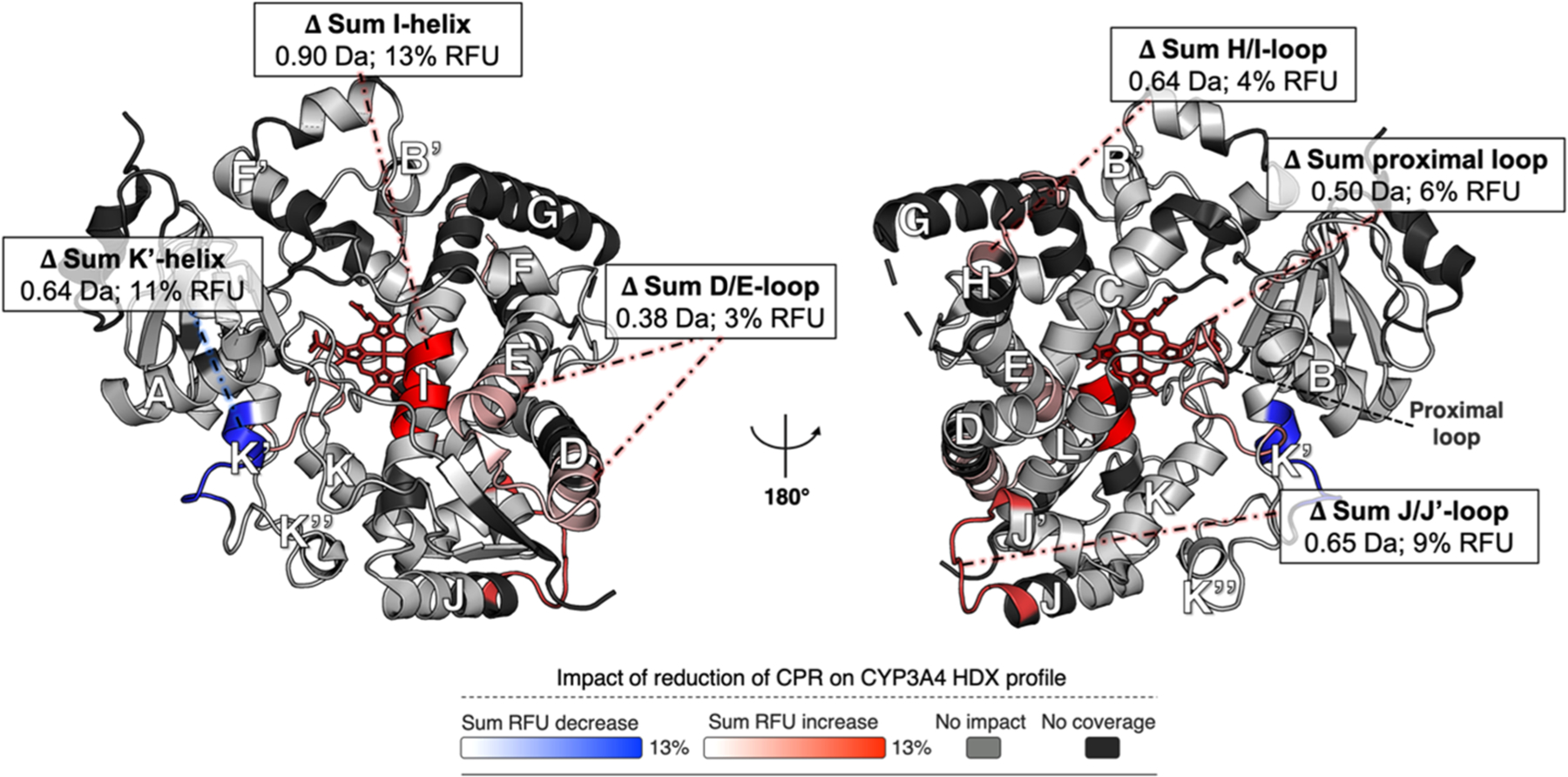
Impact of the binding of redCPR compared to that of oxCPR on the HDX profile of CYP3A4. The RFU differences were calculated as the uptake of the [CYP:oxCPR] state minus the uptake of the [CYP:redCPR] state. The sum RFU difference of three time points (0.5, 5, and 30 min) is mapped onto the CYP3A4 structure (PDB entry 1W0F), which is shown in both distal (left) and proximal (right) views relative to the heme group (red wireframe). Blue denotes regions that undergo less deuterium uptake upon interaction with redCPR than with oxCPR, whereas red designates regions that undergo more deuterium uptake. The light gray color shows regions for which there is no difference between oxCPR and redCPR binding, and the black regions were not covered by the MS analysis. Overall, the CYP3A4 structure is more dynamic in the presence of redCPR. The sum of deuterium uptake and RFU difference is shown for all statistically significant changes (±0.34 Da, CI of 99%).
Impact of Substrate Binding on the Structural Dynamics of CYP3A4.
We next looked at the structural dynamics of CYP3A4 in complex with each of three different substrates, BFC, TST, and PRG. Although substrates are expected to be located on the distal side of the heme group during catalysis (Figure S12 shows the six conserved substrate recognition sites, or SRSs, of P450 enzymes), details of the productive binding mode of the substrates used here are unavailable. The differential HDX profiles of CYP3A4 in the presence or absence of BFC, TST, and PRG were obtained for two D2O exposure time points (5 and 60 min), which revealed 16, 17, and 12 peptides, respectively, for which deuterium exchange was differentially affected by substrate binding (±0.34–0.45 Da, CI of 99%). The substrate concentration (100 μM) was selected to be higher than the respective dissociation constants (by 2–5-fold) without exceeding the substrate solubility limit in phosphate buffer. Overall, the differential HDX profiles acquired with each of the three substrates were very similar and comparable to the profiles obtained with CPR binding, i.e., characterized by a general rigidification of the CYP3A4 structure (Figure 5 and Figure S8). As a representative example, the uptake changes associated with TST binding are shown in Figure 5. The CYP3A4 regions most affected by binding of each substrate include helices B′, E, and K′. Interestingly, helix C, involved in CPR binding, also becomes more rigid upon BFC or PRG binding. These data suggest that CPR and substrates may act synergistically on the dynamic structure of CYP3A4 to modulate each other’s binding affinities.
Figure 5.
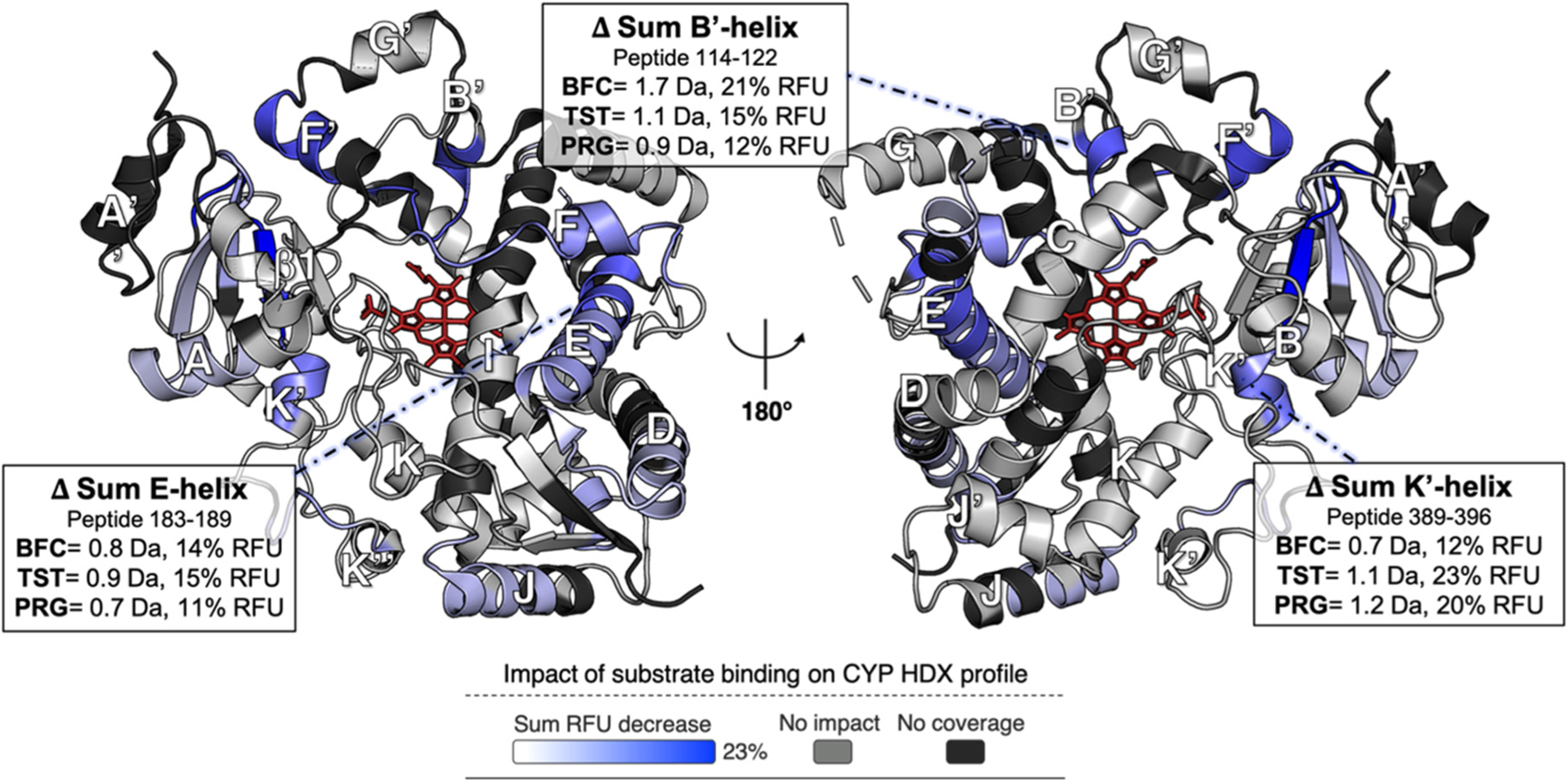
Deuterium uptake difference of [CYP] minus the [CYP:TST] complex at 100 μM TST. The sum RFU difference of two time points (5 and 60 min) is mapped onto the CYP3A4 structure (PDB entry 1W0F), which is shown in both distal (left) and proximal (right) views relative to the heme group (red wireframe). Blue denotes regions that undergo a decrease in the rate of deuterium uptake upon interaction with TST. Light gray denotes regions unaffected by TST binding, and the black regions were not covered by the MS analysis. Binding of BFC or PRG resulted in similar uptake changes relative to the free enzyme (Figure S8). The sum of deuterium uptake and RFU difference is shown for all statistically significant changes, using a CI of 99%, i.e., ±0.36 Da for TST, ±0.34 Da for BFC, and ±0.45 Da for PRG. The three regions showing the most important changes in structural dynamics for all three substrates are indicated with annotations in the boxes.
Despite the high degree of similarity in the overall structural dynamic changes induced by substrate binding, several regions of CYP3A4 exhibited subtle differences in substrate-dependent uptake (Figure S13). These regions included the end portion of helix E and the helix F region, as well as loop D/E and helices B′ and K′. Although these regions are widely separated on the polypeptide chain, most of them are close to each other in the three-dimensional structure, suggesting that the conformational dynamics in these CYP3A4 elements may be coupled to one another. These subtle differences in the dynamics of substrate-bound CYP3A4 may reflect the multiple binding modes of substrates in the large active site of this enzyme. Despite the small magnitude of these uptake differences between the various substrate-bound states, the values are still significant, illustrating the power of HDX-MS to reveal slight structural variations between biochemical states of an enzyme.
Comparison of the relative impact of CPR and substrate binding on deuterium uptake by CYP3A4 reveals a more extensive rigidification induced by CPR binding (Figure 6). Moreover, most regions rigidified by CPR binding are also rigidified by substrate binding, which include helices B′ and C–F and the sheet β1 region. The only structural motif of CYP3A4 that exhibited opposite changes in deuterium uptake was helix J, which became more flexible in the presence of CPR but slightly more rigid upon substrate binding. The potential relevance of the change in dynamics of helix J is not immediately clear, but helix J dynamics could impact the position of helix I relative to the heme. Movement of helix I is required to make space for dioxygen and substrate binding.7
Figure 6.
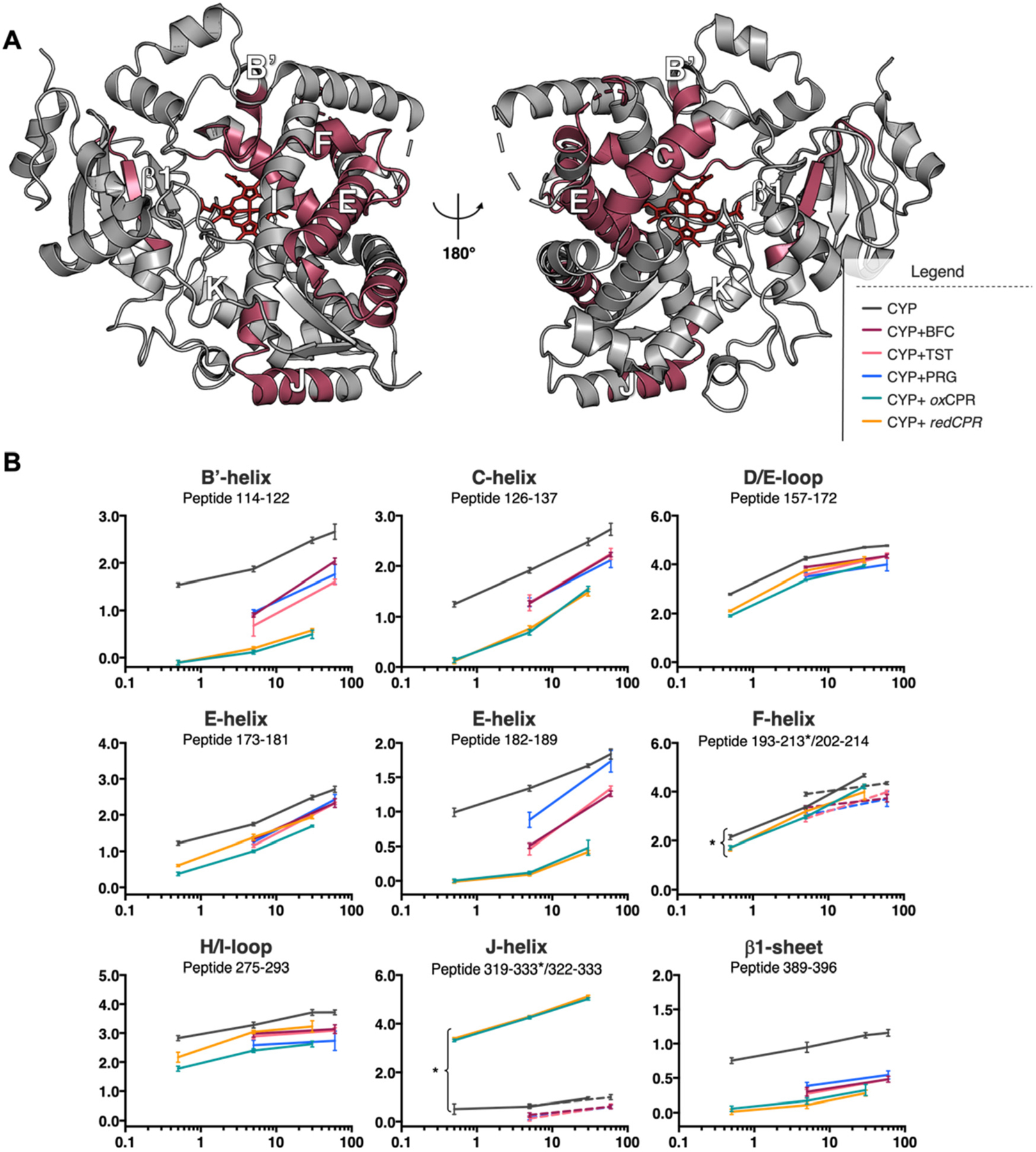
CYP3A4 regions that exhibit significant deuterium uptake differences upon binding to either CPR or a substrate. (A) The location of the peptides is colored raspberry on the CYP3A4 structure (PDB entry 1W0F) showing distal (left) and proximal (right) views relative to the heme group (red wireframe). (B) Deuterium uptake plots of these common regions showing uptake data for all biochemical states tested in this study. Uptake plots are colored black for CYP, purple for [CYP:BFC], pink for [CYP:TST], blue for [CYP:PRG], teal for [CYP:oxCPR], and orange for [CYP:redCPR]. Slightly different coverage was obtained for helices F and J in the CPR- and substrate-bound states. In these cases, two different peptides are displayed on the deuterium uptake plots. Data for peptides 193–213 and 319–333 are plotted as solid lines and designated with an asterisk. Data for peptides 202–214 and 322–333 are plotted as dotted lines.
Impact of CPR and Substrate Binding on the Bimodal Isotopic Distribution.
Under native conditions, most proteins take up deuterium in the EX2 kinetic regime, where the protein samples unfolded conformations that refold at rates that are higher than the rate of deuterium exchange. This results in peptide mass spectra with a unimodal isotope distribution.29 Careful analysis of the mass spectrum of each peptic peptide revealed a few spectra that deviated from this common EX2 behavior, showing instead a bimodal isotope pattern characteristic of the EX1 kinetic regime. Such an isotope distribution results from peptides that sample long-lived open conformational states that fully exchange with solvent deuterium before refolding into the closed state. A total of five peptides from free CYP3A4 showed a clear bimodal distribution pattern (Figure 7).30 These include peptides derived from helices B′ and C, the end portion of helix E, helix F, and the sheet β1 region. With the exception of the sheet β1 peptide, these regions show a significant decrease in the rate of deuterium uptake upon CPR or substrate binding (Figures 1, 2, and 4 and Figures S2–S4). Interestingly, the uptake difference in the sheet β1 region did not significantly change upon binding of the CPR or substrate, but the bimodal distribution was almost completely gone in the CYP–CPR and CYP–substrate complexes. This suggests that CPR binding and substrate binding induce a more rapid conformational equilibrium in sheet β1, leading to exchanges in the EX2 regime. In all cases, except for the helix F region, a large decrease in the relative abundance of the highly exchanged population of the free enzyme was observed upon ligand binding, consistent with the general rigidification of the enzyme upon CPR or substrate binding. The effect was again more pronounced upon CPR binding. CPR or substrate binding apparently shifts the equilibrium of CYP3A4 conformations toward a more structured form of the enzyme that samples the open (rapidly exchanging) conformation less often. The interaction of CPR with CYP3A4 almost completely suppressed the highly exchanged population of helices B′, C, and E and the sheet β1 region. In contrast, substrate binding decreased the abundance of the highly exchanged population of helix C (believed to directly interact with CPR) and sheet β1 (located on the distal face near the heme group) but did not significantly affect the bimodal isotope distribution of the helix E peptide. Interestingly, the bimodal distribution of helix B′ (which is involved in substrate binding and specificity) seemed to be more sensitive to the identity of the bound substrate. No drastic changes were observed in the bimodal distribution of helix F, except that in all cases the slowly exchanged populations appear to take up less deuterium. These data illustrate once more the ability of HDX-MS to reveal subtle changes in structural dynamics induced by different ligand binding modes.
Figure 7.
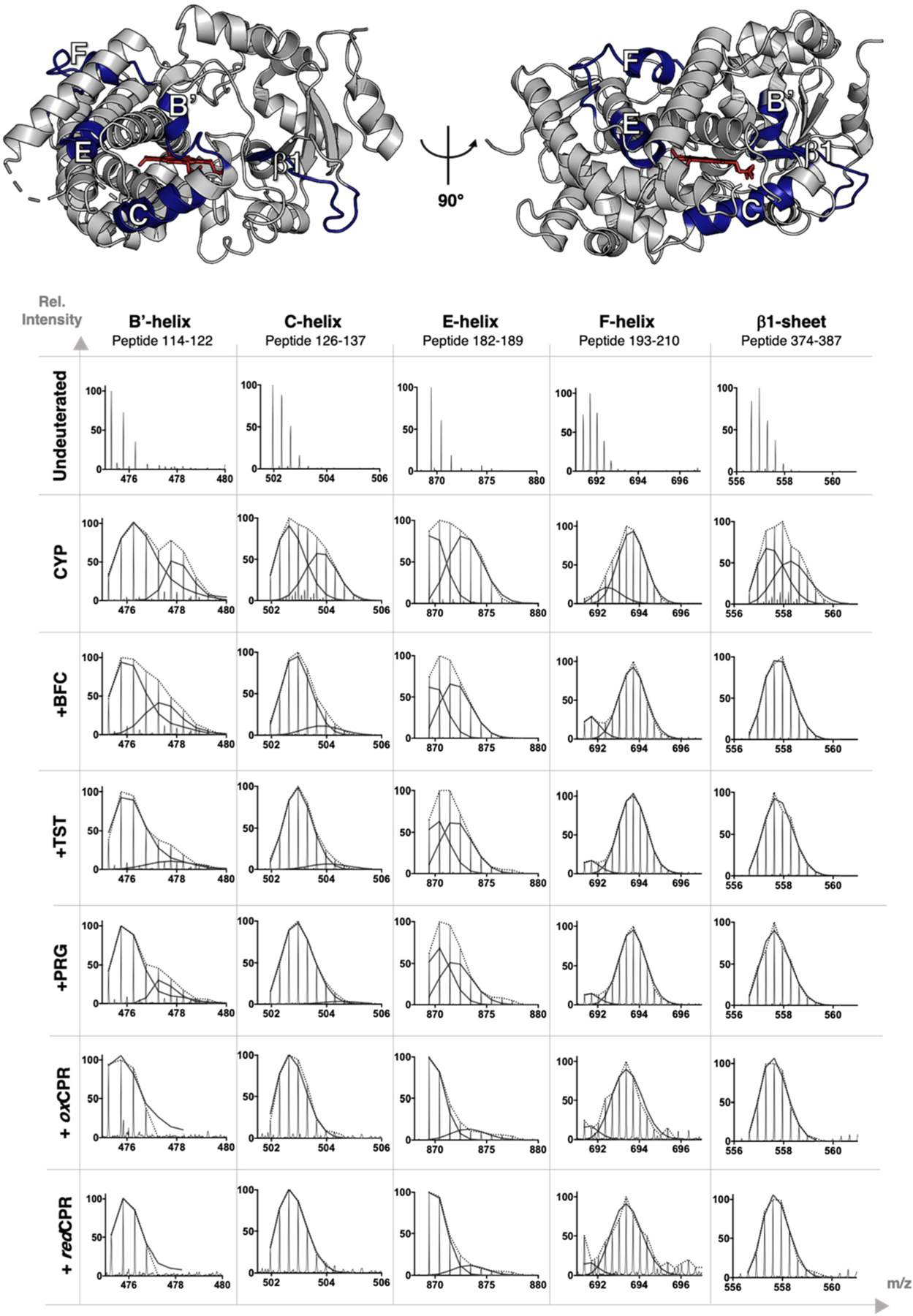
Mass spectra for the five HDX peptides of CYP3A4 that displayed a bimodal isotope distribution. Helices B′, C, E, and F and sheet β1 are mapped onto the CYP3A4 structure (PDB entry 1W0F) in deep blue. The mass spectra of these peptides are compared for CYP3A4 alone or in the presence of BFC, TST, PRG, oxCPR, or redCPR. The data are presented for the highest D2O exposure time point, i.e., 60 min, for the [CYP:substrate] complexes and 30 min for the [CYP:CPR] complexes. The lighter-colored lines indicate the peptide mass envelope, and the solid lines represent the fitted deconvolution of the isotope distribution to a sum of two binomial populations.
DISCUSSION
In this study, we investigated the effects of CPR and substrate binding on the conformational dynamics of human CYP3A4, an enzyme that exhibits remarkably relaxed substrate specificity and complex allosteric behavior. Our approach utilizes HDX-MS to provide peptide-level, localized information about protein conformational dynamics. Human CPR is encoded by a single gene and serves as a redox partner to support the activity of various enzymes, including many CYPs and heme oxygenases. CYPs share a common fold and likely possess a similar CPR-binding surface. In particular, mutagenesis and bioconjugation studies support a role for the basic residues of helix C of human CYP2B1, CYP2B4, and CYP2B6 in CPR binding.47–49 Both the deuterium uptake levels and isotope distribution data presented here are consistent with a direct interaction between helix C of CYP3A4 and CPR. Namely, rigidification of helix C (decreased rate of deuterium uptake) supports the involvement of this CYP element in protein–protein interactions with CPR. Suppression of the highly exchanged EX1 population (as indicated by the bimodal mass distribution of peptides from helix C) can be interpreted as a perturbation in the CYP3A4 conformational ensemble toward a more structured form upon CPR binding.
Crystal structures of CPR have revealed important conformational changes upon flavin reduction.2,4,50 Molecular dynamics simulations and nuclear magnetic resonance studies also support the ability of CPR to sample both open and closed conformations.5,51–55 The closed form enables intraprotein electron transfer between the two flavin coenzymes of CPR, whereas the open form of CPR mediates interprotein electron transfer from CPR to CYPs.5 This implies that the CYP–CPR complex may dissociate between the two distinct single-electron transfer steps required for oxygen activation.5,24 Our HDX-MS results reveal a significant overall rigidification of CYP3A4 upon CPR binding, regardless of the oxidation state of the reductase. These data suggest that both oxCPR and redCPR may bind to similar CYP3A4 conformations and/or induce comparable conformational perturbation in CYP3A4 upon interaction. While the overall HDX profiles of the [CYP:oxCPR] and [CYP:redCPR] complexes are quite similar, reduction of CPR leads to an increase in the flexibility of loop D/E, proximal loop K″/L, and helix I relative to the oxCPR state. Loop K″/L has been implicated in the regulation of the heme redox potential, whereas helix I bears the conserved Thr309 residue that mediates proton transfer during dioxygen activation.34 Conformational flexibility in helix I was also observed in P450cam upon association with Pdx and is thought to be important for oxygen activation.9,56–58 These data illustrate the potential for CPR binding to modulate the structural dynamics of CYP3A4 with a finely tuned regioselectivity that depends on the redox state of the reductase.
Structural evidence that supports a mechanism by which redox partner binding may cause conformational changes in the CYP active site and even at the distal surface of the protein is accumulating.24 This was first explored with P450cam, whose association with the electron donor (Pdx) was found to cause a shift from a closed to an open conformation of the P450 enzyme via motions of helices F and G.9,10 A putative communication pathway has been proposed for the Pdx-induced conformational change via coupled motions of helices B′, C, I, D, and F,56 which is consistent with our results for the CYP3A4–CPR pair (Figure 1 and Figure S7). Among all regions of CYP3A4 impacted by CPR binding, the structural rigidification was strongest in helix B′, which defines two major substrate access channels, and active site loop K/β1, which participates in substrate binding. A moderate change in the dynamic properties of helix F was also observed. This region is important for achieving the open and closed conformations of P450cam and CYP3A410,59 and is also implicated in substrate access and binding affinity. Together with our spectral and enzyme activity results (Figures S10 and S11), the HDX data suggest that association with CPR alters the ligand binding properties at the active site of CYP3A4.60–62
All three substrates tested were found to similarly influence the structural dynamics of CYP3A4 (Figure 5). This is of particular interest as it was suggested earlier that binding of the ligand to CYPs could induce conformational changes that favor and/or disfavor redox partner association.41–45 For example, Zhang et al.8 recently showed that binding of the substrate androstenedione decreased the affinity of human aromatase for CPR and that the presence of CPR increased the rate of substrate binding. Our HDX-MS data suggest that both substrate and CPR binding may be shifting the CYP3A4 conformational ensemble to a structurally similar active state. Thus, both substrate and CPR may synergistically affect each other’s binding to CYP3A4 through a structural dynamic change of the protein that, in turn, could contribute to the well-known positive kinetic cooperativity of this system.
Previous HDX-MS investigations with truncated (Δ3–12) CYP3A4 revealed small but significant changes in deuterium uptake when the lipid nanodisc-incorporated and detergent-solubilized enzymes were compared.32 Overall, the deuterium uptake properties were similar, except for the dynamics of helices F and G, which are expected to interact directly with the membrane mimic. These regions were found to be more rigid in the presence of the membrane nanodiscs. More recently, the same group reported the effect of binding of midazolam (MDZ) to CYP3A4 in lipid nanodiscs.31 Under the conditions used (60 μM midazolam, 13.5 min D2O exposure time), the largest impact of ligand binding was observed for helices F and G (increased flexibility) and loop K/β1 (rigidification). This is consistent with our HDX results, as the same regions were impacted by BFC, TST, or PRG. However, we observed rigidification of the helix F region, which contrasts with the increased flexibility for this structural element reported by Atkins and co-workers.31 This difference may arise from the distinct experimental settings, e.g., lipid-free versus membrane environment, and/or the utilization of distinct substrates. Nevertheless, our study provides the basis for future investigations of the membrane-incorporated [CYP3A4:CPR] complex that could help to resolve some of these discrepancies.63–66
Recently, Chou et al. reported the use of double electron–electron resonance (DEER) spectroscopy to investigate the flexibility of the CYP3A4 helix F/G region in the presence of a substrate (MDZ) and two inhibitors (ketoconazole and ritonavir).67 On the basis of the distance distributions measured between spin-labels located on helices A, G′, and G, the authors reported that the binding of MDZ, but not of the inhibitors, increased the number of structural states, which were attributed to different conformations of the helix G′/G region. In contrast, our results suggest only minor (insignificant) dynamical changes in the helix G′/G region, although our peptide coverage of this region is incomplete (one to three peptides per state). This finding could indicate that the multiple conformational states of the helix G/G′ region detected by Chou et al. have similar protein backbone dynamics that do not produce resolvable HDX signals in the peptides detected in our study. Alternatively, the multiple structural states detected by Chou may be in rapid equilibrium such that any deuterium uptake differences become averaged out over the time scale of the H/D exchange reaction. Better coverage of helices G and G′ with additional HDX peptides may help to further reconcile these phenomena. Nevertheless, these data emphasize the complexity of the conformational dynamics of CYP3A4 and underscore the value of investigating the system with different experimental approaches.
In summary, this is the first HDX-MS study that examined the effects of CPR binding on the structural dynamics of CYP3A4 and provides new information about redox partner interactions and substrate binding in CYP enzymes. Our data suggest that (i) both oxCPR and redCPR trigger a similar conformational reorganization in CYP3A4, most notably in helices B′, C, and E and the loop K/β1 region, and (ii) CPR modulates the substrate binding properties and catalytic activity of CYP3A4 in a substrate-dependent manner. The fact that the dynamics of similar regions are impacted by both CPR and substrate binding could also represent a structural basis for functional cooperativity. Furthermore, important similarities between the regions undergoing conformational changes upon association of redox partners with CYP3A4 and P450cam were noted and may suggest a common allosteric mechanism for dioxygen binding/activation stemming from redox partner binding. Together, our results help to improve our understanding of the structural dynamics of CYP3A4 and its potential role in modulating catalytic function.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. J. R. Halpert for providing the CYP3A4 plasmid and Dr. C. B. Kasper for the CPR plasmid. The authors are also grateful for the generous support provided by Dr. M. Guttman in the use of HX-express version 2.
Funding
This research was funded by National Science and Engineering Research Council of Canada (NSERC) Grant RGPIN-2017-04107 and FRQNT-funded Center in Green Chemistry and Catalysis (CGCC) Grant FRQNT-2020-RS4-265155-CCVC (K.A.) and by National Institutes of Health Grant ES025767 (I.F.S.). J.D. was supported by scholarships from the CGCC and the FRQNT.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.1c00178.
Amino acid sequences of human CYP3A4 and CPR, HDX experimental conditions, detailed differential HDX uptake data, CPR- and CHP-dependent activity plots for CYP3A4, and spectral confirmation of CPR reduction by NADPH (PDF)
Accession Codes
Cytochrome P450 3A4 (CYP3A4), P08684.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.1c00178
Contributor Information
Julie Ducharme, Department of Chemistry, McGill University, Montreal, Quebec, Canada H3A 0B8.
Irina F. Sevrioukova, Department of Molecular Biology and Biochemistry, University of California, Irvine, California 92697, United States;.
Christopher J. Thibodeaux, Department of Chemistry, McGill University, Montreal, Quebec, Canada H3A 0B8;.
Karine Auclair, Department of Chemistry, McGill University, Montreal, Quebec, Canada H3A 0B8;.
REFERENCES
- (1).Pandey AV, and Flück CE (2013) NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther 138, 229–254. [DOI] [PubMed] [Google Scholar]
- (2).Wang M, Roberts DL, Paschke R, Shea TM, Siler Masters BS, and Kim J-JP (1997) Three-dimensional structure of NADPH – cytochrome P450 reductase : Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. U. S. A 94, 8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yasukochi Y, and Siler Masters BS (1976) Some properties of a detergent solubilized NADPH cytochrome c (cytochrome P 450) reductase purified by biospecific affinity chromatography. J. Biol. Chem 251, 5337–5344. [PubMed] [Google Scholar]
- (4).Djordjevic S, Roberts DL, Wang M, Shea T, Camitta MGW, Masters BSS, and Kim JJP (1995) Crystallization and preliminary x-ray studies of NADPH-cytochrome P450 reductase. Proc. Natl. Acad. Sci. U. S. A 92, 3214–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sugishima M, Sato H, Higashimoto Y, Harada J, Wada K, Fukuyama K, and Noguchi M (2014) Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. U. S. A 111, 2524–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kandel SE, and Lampe JN (2014) Role of protein-protein interactions in cytochrome P450-mediated drug metabolism and toxicity. Chem. Res. Toxicol 27, 1474–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Poulos TL, Johnson EF, Waskell L, Kim J-JP, Denisov IG, Sligar SG, Ortiz de Montellano PR, Almira Correia M, Hollengberg PF, McLean KJ, Leys D, Munro AW, Schuler MA, Girhard M, Bakkes PJ, Mahmoud O, Urlacher VB, Guengerich PF, Gotoh S, Ohno M, Yoshinari K, Negishi M, Kawajiri K, Waxman DJ, Chang TKH, Miller WL, Auchus RJ, Edin ML, Cheng J, Gruzdev A, Hoopes SL, and Zeldin DC (2015) in Cytochrome P450 - Structure, Mechanism and Biochemistry (Ortiz de Montellano PR, Ed.) 4th ed., Springer International Publishing, Cham, Switzerland. [Google Scholar]
- (8).Zhang C, Catucci G, Di Nardo G, and Gilardi G (2020) Effector role of cytochrome P450 reductase for androstenedione binding to human aromatase. Int. J. Biol. Macromol 164, 510–517. [DOI] [PubMed] [Google Scholar]
- (9).Chuo SW, Wang LP, Britt RD, and Goodin DB (2019) An Intermediate Conformational State of Cytochrome P450cam-CN in Complex with Putidaredoxin. Biochemistry 58, 2353–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hollingsworth SA, Batabyal D, Nguyen BD, and Poulos TL (2016) Conformational selectivity in cytochrome P450 redox partner interactions. Proc. Natl. Acad. Sci. U. S. A 113, 8723–8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, and Sligar SG (2015) Mechanism of Drug-Drug Interactions Mediated by Human Cytochrome P450 CYP3A4Monomer. Biochemistry 54, 2227–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Doehmer J, Goeptar AR, and Vermeulen NPE (1993) Cytochromes P450 and drug resistance. Cytotechnology 12, 357–366. [DOI] [PubMed] [Google Scholar]
- (13).Denisov IG, and Sligar SG (2012) A novel type of allosteric regulation: functional cooperativity in monomeric proteins. Arch. Biochem. Biophys 519, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hosea NA, Miller GP, and Guengerich FP (2000) Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry 39, 5929–5939. [DOI] [PubMed] [Google Scholar]
- (15).Davydov DR, Rumfeldt JAO, Sineva EV, Fernando H, Davydova NY, and Halpert JR (2012) Peripheral Ligand-Binding Site in Cytochrome P450 3A4 Located with Fluorescence Resonance Energy Transfer (FRET). J. Biol. Chem 287, 6797–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Okada Y, Murayama N, Yanagida C, Shimizu M, Guengerich FP, and Yamazaki H (2009) Drug Metab. Dispos 37, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Harrelson JP, Atkins WM, and Nelson SD (2008) Biochemistry 47, 2978–2988. [DOI] [PubMed] [Google Scholar]
- (18).Ducharme J, Polic V, and Auclair K (2019) A Covalently Attached Progesterone Molecule Outcompetes the Binding of Free Progesterone at an Allosteric Site of Cytochrome P450 3A4. Bioconjugate Chem 30, 1629–1635. [DOI] [PubMed] [Google Scholar]
- (19).Hutzler JM, Wienkers LC, Wahlstrom JL, Carlson TJ, and Tracy TS (2003) Activation of cytochrome P450 2C9-mediated metabolism: mechanistic evidence in support of kinetic observations. Arch. Biochem. Biophys 410, 16–24. [DOI] [PubMed] [Google Scholar]
- (20).Fernando H, Davydov DR, Chin CC, and Halpert JR (2007) Role of subunit interactions in P450 oligomers in the loss of homotropic cooperativity in the cytochrome P450 3A4 mutant L211F/D214E/F304W. Arch. Biochem. Biophys 460, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Davydov DR (2016) Molecular organization of the microsomal oxidative system: a new connotation for an old term. Biochem. Suppl. Ser. B Biomed. Chem 10, 10–21. [DOI] [PubMed] [Google Scholar]
- (22).Denisov IG, Shih AY, and Sligar SG (2012) Structural differences between soluble and membrane bound cytochrome P450s. J. Inorg. Biochem 108, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hackett JC (2018) Membrane-embedded substrate recognition by cytochrome P450 3A4. J. Biol. Chem 293, 4037–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ortiz de Montellano PR (2015) in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano PR, Ed.) 4th ed., Springer International Publishing, Cham, Switzerland. [Google Scholar]
- (25).Davydov DR (2011) Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions. Expert Opin. Drug Metab. Toxicol 7, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Backes WL, and Kelley RW (2003) Organization of multiple cytochrome P450s with NADPH-cytochrome P450 reductase in membranes. Pharmacol. Ther 98, 221–233. [DOI] [PubMed] [Google Scholar]
- (27).Hazai E, Bikádi Z, Simonyi M, and Kupfer D (2005) Association of cytochrome P450 enzymes is a determining factor in their catalytic activity. J. Comput.-Aided Mol. Des 19, 271–285. [DOI] [PubMed] [Google Scholar]
- (28).Konermann L, Pan J, and Liu YH (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev 40, 1224–1234. [DOI] [PubMed] [Google Scholar]
- (29).Ferraro DM, Lazo ND, and Robertson AD (2004) EX1 Hydrogen Exchange and Protein Folding. Biochemistry 43, 587–594. [DOI] [PubMed] [Google Scholar]
- (30).Ducharme J, Polic V, Thibodeaux C, and Auclair K (2021) Combining small-molecule bioconjugation and hydrogen-deuterium exchange mass spectrometry (HDX-MS) to expose allostery: the case of human cytochrome P450 3A4. ACS Chem. Biol 16, 882–890. [DOI] [PubMed] [Google Scholar]
- (31).Redhair M, Hackett JC, Pelletier RD, and Atkins WM (2020) Dynamics and Location of the Allosteric Midazolam Site in Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 59, 766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Treuheit NA, Redhair M, Kwon H, McClary WD, Guttman M, Sumida JP, and Atkins WM (2016) Membrane Interactions, Ligand-Dependent Dynamics, and Stability of Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 55, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gillam EMJ (2008) Engineering Cytochrome P450 Enzymes. Chem. Res. Toxicol 21, 220–231. [DOI] [PubMed] [Google Scholar]
- (34).Poulos TL (2014) Heme Enzyme Structure and Function. Chem. Rev 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Polic V, and Auclair K (2017) Allosteric Activation of Cytochrome P450 3A4 via Progesterone Bioconjugation. Bioconjugate Chem 28, 885–889. [DOI] [PubMed] [Google Scholar]
- (36).Habibi Y, Uggowitzer KA, Issak H, and Thibodeaux CJ (2019) Insights into the Dynamic Structural Properties of a Lanthipeptide Synthetase using Hydrogen-Deuterium Exchange Mass Spectrometry. J. Am. Chem. Soc 141, 14661–14672. [DOI] [PubMed] [Google Scholar]
- (37).Houde D, Berkowitz SA, and Engen JR (2011) The Utility of Hydrogen/Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J. Pharm. Sci 100, 2071–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Burkitt W, and O’Connor G (2008) Assessment of the repeatability and reproducibility of hydrogen/deuterium exchange mass spectrometry measurements. Rapid Commun. Mass Spectrom 22, 3893–3901. [DOI] [PubMed] [Google Scholar]
- (39).Guttman M, Weis DD, Engen JR, and Lee KK (2013) Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J. Am. Soc. Mass Spectrom 24, 1906–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lau AM, Claesen J, Hansen K, and Politis A (2021) Deuteros 2.0: Peptide-level significance testing of data from hydrogen deuterium exchange mass spectrometry. Bioinformatics 37, 270–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chefson A, Zhao J, and Auclair K (2006) Replacement of natural cofactors by selected hydrogen peroxide donors or organic peroxides results in improved activity for CYP3A4 and CYP2D6. ChemBioChem 7, 916–919. [DOI] [PubMed] [Google Scholar]
- (42).Domanski TL, He Y-AA, Khan KK, Roussel F, Wang Q, and Halpert JR (2001) Phenylalanine and tryptophan scanning mutagenesis of CYP3A4 substrate recognition site residues and effect on substrate oxidation and cooperativity. Biochemistry 40, 10150–10160. [DOI] [PubMed] [Google Scholar]
- (43).Hageman TS, and Weis DD (2019) Reliable Identification of Significant Differences in Differential Hydrogen Exchange-Mass Spectrometry Measurements Using a Hybrid Significance Testing Approach. Anal. Chem 91, 8008–8016. [DOI] [PubMed] [Google Scholar]
- (44).Baas BJ, Denisov IG, and Sligar SG (2004) Arch. Biochem. Biophys 430, 218–228. [DOI] [PubMed] [Google Scholar]
- (45).Sevrioukova IF, and Poulos TL (2015) Anion-Dependent Stimulation of CYP3A4Monooxygenase. Biochemistry 54, 4083–4096. [DOI] [PubMed] [Google Scholar]
- (46).Barnaba C, Taylor E, and Brozik JA (2017) Dissociation Constants of Cytochrome P450 2C9/Cytochrome P450 Reductase Complexes in a Lipid Bilayer Membrane Depend on NADPH: A Single-Protein Tracking Study. J. Am. Chem. Soc 139, 17923–17934. [DOI] [PubMed] [Google Scholar]
- (47).Bumpus NN, and Hollenberg PF (2010) Cross-linking of human cytochrome P450 2B6 to NADPH-cytochrome P450 reductase: Identification of a potential site of interaction. J. Inorg. Biochem 104, 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Prade E, Mahajan M, Im SC, Zhang M, Gentry KA, Anantharamaiah GM, Waskell L, and Ramamoorthy A (2018) A Minimal Functional Complex of Cytochrome P450 and FBD of Cytochrome P450 Reductase in Nanodiscs. Angew. Chem., Int. Ed 57, 8458–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Omata Y, Dai R, Smith SV, Robinson RC, and Friedman FK (2000) Synthetic peptide mimics of a predicted topographical interaction surface: The cytochrome P450 2B1 recognition domain for NADPH-cytochrome P450 reductase. J. Protein Chem 19, 23–32. [DOI] [PubMed] [Google Scholar]
- (50).Hamdane D, Xia C, Im SC, Zhang H, Kim J-JJP, and Waskell L (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem 284, 11374–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Laursen T, Singha A, Rantzau N, Tutkus M, Borch J, Hedegård P, Stamou D, Møller BL, and Hatzakis NS (2014) Single molecule activity measurements of cytochrome P450 oxidoreductase reveal the existence of two discrete functional states. ACS Chem. Biol 9, 630–634. [DOI] [PubMed] [Google Scholar]
- (52).Sündermann A, and Oostenbrink C (2013) Molecular dynamics simulations give insight into the conformational change, complex formation, and electron transfer pathway for cytochrome P450 reductase. Protein Sci 22, 1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Xia C, Hamdane D, Shen AL, Choi V, Kasper CB, Pearl NM, Zhang H, Im SC, Waskell L, and Kim JJP (2011) Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem 286, 16246–16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Vincent B, Morellet N, Fatemi F, Aigrain L, Truan G, Guittet E, and Lescop E (2012) The closed and compact domain organization of the 70-kDa human cytochrome P450 reductase in its oxidized state as revealed by NMR. J. Mol. Biol 420, 296–309. [DOI] [PubMed] [Google Scholar]
- (55).Jenner M, Ellis J, Huang WC, Lloyd Raven E, Roberts GCK, and Oldham NJ (2011) Detection of a protein conformational equilibrium by electrospray ionisation-ion mobility-mass spectrometry. Angew. Chem., Int. Ed 50, 8291–8294. [DOI] [PubMed] [Google Scholar]
- (56).Liou SH, Chuo SW, Qiu Y, Wang LP, and Goodin DB (2020) Linkage between Proximal and Distal Movements of P450cam Induced by Putidaredoxin. Biochemistry 59, 2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Wei JY, Pochapsky TC, and Pochapsky SS (2005) Detection of a high-barrier conformational change in the active site of cytochrome P450cam upon binding of putidaredoxin. J. Am. Chem. Soc 127, 6974–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).OuYang B, Pochapsky SS, Dang M, and Pochapsky TC (2008) A Functional Proline Switch in Cytochrome P450cam. Structure 16, 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Benkaidali L, André F, Moroy M, Tangour B, Maurel F, and Petitjean M (2019) Four Major Channels Detected in the Cytochrome P450 3A4: A Step toward Understanding Its Multi-specificity. Int. J. Mol. Sci 20, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wen B, Atkins WM, Patricia Campbell A, Cameron MD, Nelson SD, and Roberts AG (2007) Cooperative Binding of Acetaminophen and Caffeine within the P450 3A4 Active Site. Chem. Res. Toxicol 20, 1434–1441. [DOI] [PubMed] [Google Scholar]
- (61).Torimoto N, Ishii I, Hata M, Nakamura H, Imada H, Ariyoshi N, Ohmori S, Igarashi T, and Kitada M (2003) Direct Interaction between Substrates and Endogenous Steroids in the Active Site May Change the Activity of Cytochrome P450 3A4. Biochemistry 42, 15068–15077. [DOI] [PubMed] [Google Scholar]
- (62).Kawakami N, Shoji O, and Watanabe Y (2011) Use of perfluorocarboxylic acids to trick cytochrome P450BM3 into initiating the hydroxylation of gaseous alkanes. Angew. Chem., Int. Ed 50, 5315–5318. [DOI] [PubMed] [Google Scholar]
- (63).Gentry KA, Zhang M, Im SC, Waskell L, and Ramamoorthy A (2018) Substrate mediated redox partner selectivity of cytochrome P450. Chem. Commun 54, 5780–5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Scott EE, Wolf CR, Otyepka M, Humphreys SC, Reed JR, Henderson CJ, McLaughlin LA, Paloncyova M, Navratilova V, Berka K, Anzenbacher P, Dahal UP, Barnaba C, Brozik JA, Jones JP, Estrada DF, Laurence JS, Park JW, and Backes WL (2016) The Role of Protein-Protein and Protein-Membrane Interactions on P450 Function. Drug Metab. Dispos 44, 576–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Scott EE, White MA, He YA, Johnson EF, Stout CD, and Halpert JR (2004) Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9-Å resolution: Insight into the range of P450 conformations and the coordination of redox partner binding. J. Biol. Chem 279, 27294–27301. [DOI] [PubMed] [Google Scholar]
- (66).Agrawal V, Choi JH, Giacomini KM, and Miller WL (2010) Substrate-specific modulation of CYP3A4 activity by genetic variants of cytochrome P450 oxidoreductase. Pharmacogenet. Genomics 20, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chuo SW, Liou SH, Wang LP, Britt RD, Poulos TL, Sevrioukova IF, and Goodin DB (2019) Conformational Response of N-Terminally Truncated Cytochrome P450 3A4 to Ligand Binding in Solution. Biochemistry 58, 3903–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.