

SUMMARY
Neurons in the cerebral cortex connect through descending pathways to hindbrain and spinal cord to activate muscle and generate movement. Although components of this pathway have been previously generated and studied in vitro, the assembly of this multi-synaptic circuit has not yet been achieved with human cells. Here, we derive organoids resembling the cerebral cortex or the hindbrain/spinal cord and assemble them with human skeletal muscle spheroids to generate 3D cortico-motor assembloids. Using rabies tracing, calcium imaging, and patch-clamp recordings, we show that corticofugal neurons project and connect with spinal spheroids, while spinal-derived motor neurons connect with muscle. Glutamate uncaging or optogenetic stimulation of cortical spheroids triggers robust contraction of 3D muscle, and assembloids are morphologically and functionally intact for up to 10 weeks post-fusion. Together, this system highlights the remarkable self-assembly capacity of 3D cultures to form functional circuits that could be used to understand development and disease.
Graphical Abstract
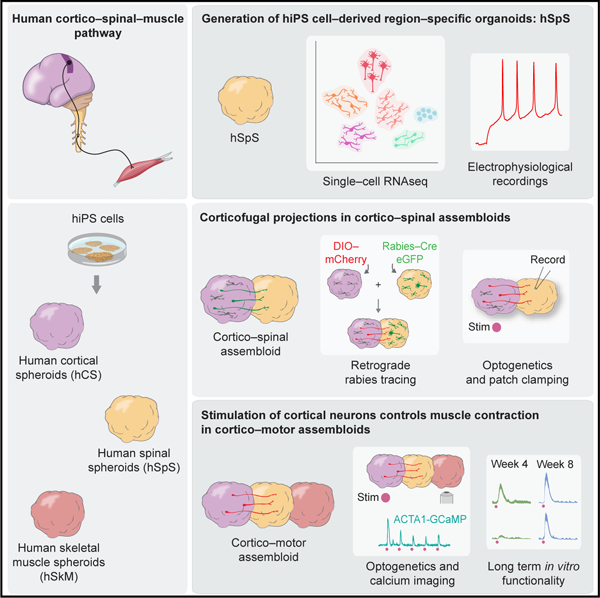
In Brief
The assembly of 3D cultures derived from hiPS cells resembling cerebral cortex, hindbrain/spinal cord, and skeletal muscle form neural circuits that can be readily manipulated to model cortical control of muscle contraction in vitro long term.
INTRODUCTION
The main function of the cortico-motor pathway is the regulation of neural activity in the hindbrain and spinal cord to generate coordinated movement (Lemon, 2008). Corticofugal glutamatergic neurons in the cerebral cortex send long-range axons to modulate networks in the hindbrain and spinal cord to ultimately induce muscle contraction (Kiehn, 2016; Lodato and Arlotta, 2015; Shim et al., 2012; ten Donkelaar et al., 2004) (Figure 1A). Damage or degeneration of the cortico-motor circuit following traumatic injury, amyotrophic lateral sclerosis, or autoimmune disorders result in severe motor dysfunction (Blesch and Tuszynski, 2009). Recent evidence points at primate-specific reorganization and connectivity of the cortico-spinal pathway, including the presence of direct monosynaptic cortico-spinal connections, which may be related to corticalization of motor control and the evolution of dexterity (Gu et al., 2017; Lemon, 2008; Sousa et al., 2017). These species-specific differences are further highlighted by challenges in translating findings from animal models (Kaiser and Feng, 2015; Philips and Rothstein, 2015; van der Worp et al., 2010). Therefore, direct access to human cortico-spinal-muscle circuits has the potential to accelerate our understanding of motor disorders.
Figure 1. Generation of hSpS from hiPS Cells.
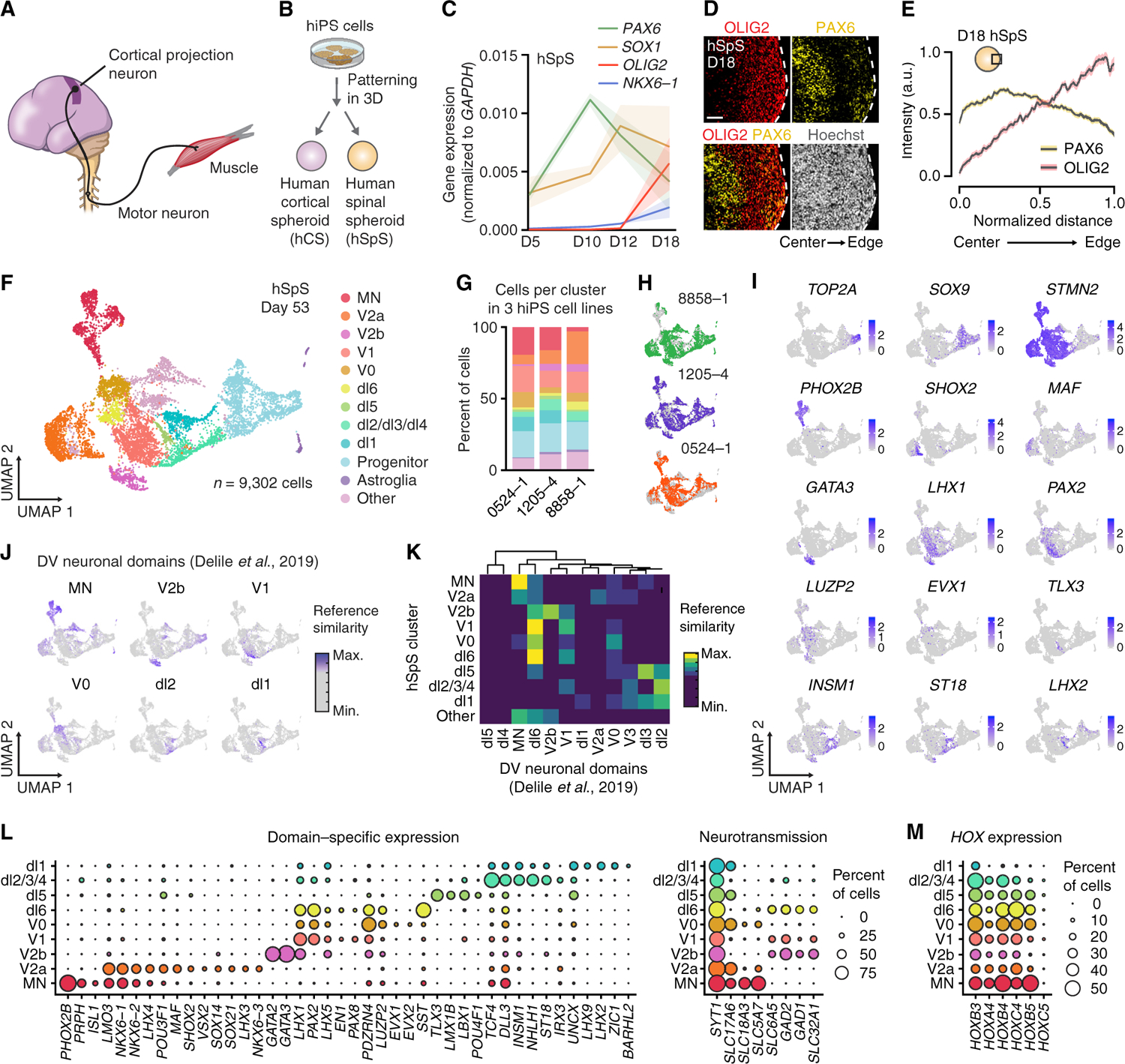
(A) Schematic illustrating the main cellular components of the cortico-motor system.
(B) Schematic illustrating the generation of human cortical spheroids (hCS) and human spinal spheroids (hSpS) from hiPS cells.
(C) Gene expression (RT-qPCR) of spinal cord-related gene markers at 5, 10, 12, and 18 days in hSpS. Data represent mean ± SEM (n = 3 hiPS cell lines from 1 differentiation; two-way ANOVA, interaction F[9,32] = 3.29, p = 0.005).
(D) Representative immunocytochemistry images of OLIG2 and PAX6 in hSpS at day 18. Scale bar, 50 µm.
(E) Fluorescence intensity analysis showing expression of OLIG2 and PAX6 plotted versus the distance from the center (left) to the edge (right) of hSpS at day 18. Black traces represent the mean and shaded bars represent the SEM (n = 20 hSpS derived from 3 hiPS cell lines from 2 separate differentiations, with 2–4 cryosections quantified per hSpS).
(F) UMAP visualization of single cell gene expression of hSpS at day 53 (n = 9,302 cells from 3 hiPS cell lines).
(G) Graph showing the percentage of cells in each of the three hiPS cell lines used for this experiment belonging to each cluster in hSpS.
(H) UMAP plot showing cells separately colored by the hiPS cell line they were derived from.
(I) UMAP plots showing gene expression of selected hSpS cluster-specific markers. Colored scale shows normalized gene expression data, log (counts per 10,000).
(J) hSpS UMAP plots colored by reference similarity spectrum (RSS) to selected single-cell RNA sequencing (RNA-seq) clusters from mouse spinal cord from Delile et al. (2019).
(K) Hierarchical clustering showing RSS analysis of hSpS clusters to single-cell RNA-seq clusters from mouse developing spinal cord neuronal clusters from Delile et al. (2019).
(L and M) Dot plots showing the expression of selected domain-specific and neurotransmitter identity-related genes (L), and expression of HOX genes (M) in each of the neuronal clusters in hSpS. The size of the circle represents the percent of cells expressing each gene per cluster.
See also Figures S1, S2, and S3 and Table S1.
Induced pluripotent stem cells hold promise in building personalized disease models. Although components of the cortico-spinal-motor pathway have been generated or studied in isolation (Ogura et al., 2018; Sances et al., 2016; Shi et al., 2018; Steinbeck et al., 2016), the in vitro derivation and assembly of this three-part system have not yet been achieved with human cells. We have previously developed an approach to model complex cellular interactions during human brain development by specifying region-specific spheroids or organoids and then fusing them in vitro to generate three-dimensional (3D) assembloids (Birey et al., 2017; Paşca, 2019; Sloan et al., 2018). Long-range neural projections have been observed in a cortico-thalamic assembloid (Xiang et al., 2019), in organotypic mouse co-cultures (Takuma et al., 2002) and in a preparation that used sliced cortical organoids and rodent spinal cord explants (Giandomenico et al., 2019). However, in vitro derivation and assembly of a functional multi-synaptic circuit from human cells have not been achieved.
Here, we leverage a modular assembloid approach to separately generate and then functionally integrate the components of a cortico-motor circuit. More specifically, we find that human-induced pluripotent stem (hiPS) cell-derived region-specific spheroids form physiologically relevant connections when assembled together. Spheroids resembling the cerebral cortex form long-range functional connections with spheroids resembling the hindbrain/cervical spinal cord, and these in turn project and connect with human skeletal muscle. Importantly, these cortico-motor assembloids can be used to trace anatomical and functional connectivity, can be maintained long term in vitro to capture cellular and functional changes, and can be manipulated optogenetically or pharmacologically at multiple levels to control human muscle contraction.
RESULTS
Generation of Hindbrain/Cervical Spinal Cord Spheroids from Pluripotent Stem Cells
We first generated spheroids resembling the human hindbrain/cervical spinal cord (hSpS) starting from hiPS cells (Figure 1B; Table S1 summarizes the color-coded use of n = 9 hiPS cell lines in various experiments). Specification of the progenitor domains in the hindbrain and spinal cord relies on signaling molecules like WNT and retinoic acid (RA) secreted along the rostro-caudal axis, and sonic hedgehog (SHH) along the dorso-ventral axis (Alaynick et al., 2011; Goulding, 2009; Jessell, 2000; Kiehn, 2006) (Figures S1A and S1B). A number of groups have developed protocols to generate spinal motor neurons by modulating these pathways in vitro (Amoroso et al., 2013; Butts et al., 2019; Gouti et al., 2014; Wichterle et al., 2002). To establish hSpS, we designed a combinatorial approach (12 conditions) to test FGF-2, RA, WNT, and SHH modulators at varying concentrations following neural ectoderm specification (Figure S1C). Briefly, hiPS cell colonies were lifted using the enzyme dispase, moved to ultra-low attachment plates to form spheroids, and exposed to dual SMAD inhibition (Paşca et al., 2015; Sloan et al., 2018). Starting on day 4, spheroids were exposed to the WNT activator CHIR-99021 (3 µM) and to 20 ng/mL epidermal growth factor (EGF) from day 6 onward. Spheroids also received a combination of RA (0.01 or 0.1 µM) and FGF-2 (0, 10, or 20 ng/mL) from day 6, and the SHH activator smoothened agonist (SAG; 0, 0.01, 0.1 or 1 µM) from day 11. To determine the fate of the resulting spheroids in each condition, we assessed gene expression at day 20 (Figures S1D–S1F; primers listed in Table S1). Exposure to high levels of RA and low levels of FGF-2 results in more rostral fates (Figure S1D; HOXA2, p = 0.002), whereas exposure to low levels of RA and high levels of FGF-2 yields more caudal fates (Figure S1D; HOXC9, p = 0.002). Moreover, high SAG exposure is associated with higher expression of ventral marker genes (Figures S1E and S1F; OLIG2, p = 0.001; NKX6–1, p = 0.002; CHX10, p = 0.007; and ISL1, p = 0.01). Ventral and dorsal identities were also confirmed by immunocytochemistry in hSpS cryo-sections (Figure S1G; antibodies listed in Table S1). Based on the overall diversity of domain-specific markers expressed, we chose to pursue condition 8 for further experiments (0.1 µM RA, 0.1 µM SAG, and 10 ng/mL FGF-2) (Figure S1H). Other conditions, such as condition 12, showed higher expression of motor neuron markers, but had lower expression of interneuron markers, which are essential for hindbrain/spinal cord circuitry. In condition 8 (hSpS), the neuroectodermal markers SOX1 and PAX6 peaked at day 10, followed by an increase in the expression of OLIG2 and NKX6–1 from day 12 (Figure 1C; interaction p = 0.005, gene p < 0.0001, time p = 0.01).
In vivo, hindbrain/spinal cord progenitors are organized in a stereotypical pattern (Dasen and Jessell, 2009). To explore organization in day 18 hSpS, we used immunohistochemistry for the ventral marker OLIG2 and the medial marker PAX6. We found that OLIG2 was more highly expressed toward the edge of the hSpS, whereas PAX6 showed the opposite pattern, with higher expression toward the center (Figures 1D and 1E). We next further characterized OLIG2+ progenitors, which give rise to motor neurons (Figures S1I–S1L), and found that ~40% of OLIG2+ cells co-expressed the marker NKX2.2 and ~15% co-expressed NKX6.1 (Figure S1L). This pattern of expression suggests the presence of hindbrain and spinal cord motor neuron progenitor populations in hSpS (Guthrie, 2007).
Next, we tested whether the presence of the Notch pathway modulator DAPT (Borghese et al., 2010) changed the proportions of cell types present in hSpS (Figures S2A and S2B). For this, we performed single-cell transcriptional profiling (Fan et al., 2015) in day 45 hSpS that were differentiated in the presence or absence of DAPT from day 19 to day 24 (Figure S2C) (n = 7,888 cells; BD Rhapsody). Analysis of cells using the t-distributed stochastic neighbor embedding (t-SNE) approach (van der Maaten and Hinton, 2008) identified several domains, including groups of GABAergic, glycinergic and glutamatergic interneurons (4, 5, 7, 8, and 9), as well as a mixed neuronal cluster (6), and a motor neuron cluster (10) (Figures S2C and S2D; Table S1). When differentiated in the presence of DAPT, although overall similar to the unexposed condition (hSpS(–)DAPT; R2 = 0.85, p < 0.0001), hSpS contained a ~3-fold increase in the proportion of motor neurons as well as a 4-fold decrease in the proportion of an undefined mixed neuronal population (Figures S2E and S2F). We further validated the presence of these neuronal and glial populations by performing real-time qPCR for a set of domain-specific markers in day 30 hSpS and hSpS–DAPT derived from 5 hiPS cell lines in 4 separate differentiation experiments (Figures S2G–S2I), and confirmed expression of some of these genes by immunocytochemistry in cryo-sections (Figures S2J and S2K). Based on these data, we chose to use the hSpS condition that included DAPT exposure.
One of the challenges of using organoids for studying development and disease is the reproducibility of differentiation between hiPS cell lines. We have previously demonstrated high reliability in the generation of organoids resembling the dorsal forebrain (Yoon et al., 2019), and to investigate this aspect in hSpS, we performed droplet-based single-cell transcriptomics at day 53 (10x Genomics Chromium) in three different hiPS cell lines that were differentiated in parallel (n = 9,302 cells). We first used the uniform manifold approximation and projection (UMAP) dimensionality reduction technique to visualize cells and determine their identity (Figure 1F; Figure S3A shows t-SNE reduction; Table S1 shows the top 50 differentially expressed genes per cluster). We found that cells grouped into 12 main clusters, and when we compared the proportions of cells in each cluster across lines, we found that cell diversity was overall similar (Figures 1G, 1H, and S3B). We also compared expression of all genes across lines and found that they were highly correlated (8858–1 versus 0524–1 R2 = 0.98, 8858–1 versus 1205–4 R2 = 0.99, and 1205–4 versus 0524–1 R2 = 0.98, Figure S3C). Similarly, we found that gene expression in each domain was highly correlated across lines (Figure S3D).
Next, we explored hSpS cell cluster identity and found they included progenitors expressing SOX9 and TOP2A, astroglia, and several neuronal domains including a dorsal group of cells expressing LHX2, INSM1, ST18, and TLX3 (clusters dl1, dl2/3/4, dl5, and dl6), four ventral clusters that showed expression of LHX1, PAX2, EVX1, SHOX2 and GATA3 (V0, V1, V2a, and V2b), and a group of motor neurons expressing ISL1, PRPH, and PHOX2B (MN) (Figures 1I and 1L). We did not find a clear V3 domain with this analysis, and we found no non-ectodermal cells, however, we identified a group of neural cells that we could not define based on known marker genes. One hiPS cell line (8858–1) had a higher proportion of V2a neurons at the expense of motor neurons (Figure 1G). The balance between V2a interneurons and motor neurons is known to be linked (Thaler et al., 1999), and loss of motor neurons in Olig1/2 knockout mice results in a dramatic increase of Chx10+ V2a interneurons (Zhou and Anderson, 2002). Real-time qPCR for ISL1 and CHX10 showed that out of 16 samples from 5 lines in 4 differentiations, 2 had higher expression of CHX10 compared to ISL1 (Figure S2G). Variation in the state of hiPS cell lines can lead to divergent neuronal biases (Micali et al., 2020), and real-time qPCRs for these two markers at ~day 30 in hSpS could be implemented as a quality control step.
To further validate the cluster assignment, we compared hSpS to annotated neuronal clusters from the developing mouse spinal cord (Delile et al., 2019) using reference similarity spectrum (RSS) analysis (Kanton et al., 2019). We found that, overall, cluster annotation in hSpS matched previously annotated spinal cord clusters (Figures 1J and 1K) (Delile et al., 2019; Rosenberg et al., 2018; Sathyamurthy et al., 2018). Some dorsal clusters appeared to be less correlated compared to ventral clusters, which may be explained by the lack of dorsalizing signals in hSpS. We also looked at neurotransmitter identity in the neuronal clusters and we found that it broadly matched that described in the literature (Delile et al., 2019) and included glutamatergic (SLC17A6+), GABAergic (GAD1+, GAD2+ or SLC32A1+), glycinergic (SLC6A5+), and cholinergic (SLC5A7+ or SLC18A3+) cells (Figure 1L). Genes encoding cell adhesion and neuronal guidance molecules, such as PCDH7, NRXN3, and ROBO3, were also expressed in a domain-specific manner (Figure S3E).
To explore rostro-caudal identity of hSpS, we examined the expression of HOX genes in hSpS and found expression of HOXB4, HOXC4, and HOXB5, suggestive of caudal hindbrain/cervical spinal cord identity (Figures 1M and S3F). Moreover, RSS analysis comparing hSpS to different CNS regions in the mouse (Zeisel et al., 2018) showed that hSpS was most closely correlated to hindbrain cell types, further validating the identity of hSpS (Figures S3G and S3H). However, direct comparison to human developing hindbrain/spinal cord in the future could better address these questions.
Next, to further characterize motor neurons in hSpS, we re-clustered the MN cluster from the single cell gene expression analysis. We found that MN subdivided into at least 6 groups (Figures S3I–S3K). Most of these cells expressed the motor neuron marker PHOX2B, but did not express the visceral motor neuron marker TBX20, suggesting motor neurons in hSpS have a largely somatic identity (Figure S3M). MN cluster 6 was negative for PHOX2B and positive for ISL1/2 and HB9 (MNX1), indicating a limb-innervating lateral motor column-like identity (Figures S3I–S3M).
We also confirmed the presence of cholinergic motor neurons in hSpS by immunocytochemistry for the enzyme that catalyzes the biosynthesis of the neurotransmitter acetylcholine (CHAT), the vesicular acetylcholine transporter VACHT, the neurofilament SMI-32, and the transcription factors HB9, ISL1, and PHOX2B (Figures 2A, 2B, and S4A–S4F), and by electrophysiological recordings using an Hb9::GFP reporter (Figures 2C and 2D). We also observed the presence of ventral neuronal domains in hSpS by immunohistochemistry at day 45 (Figures 2E, 2F, and S4G), as well as the presence of astrocytes and oligodendrocytes at day 75 (Figure 2G). Whereas spinal cord progenitors are organized in a well-defined laminar pattern during development, post-mitotic neurons in the spinal cord do not appear to maintain this organization at later stages (Lai et al., 2016).
Figure 2. Characterization of hSpS.
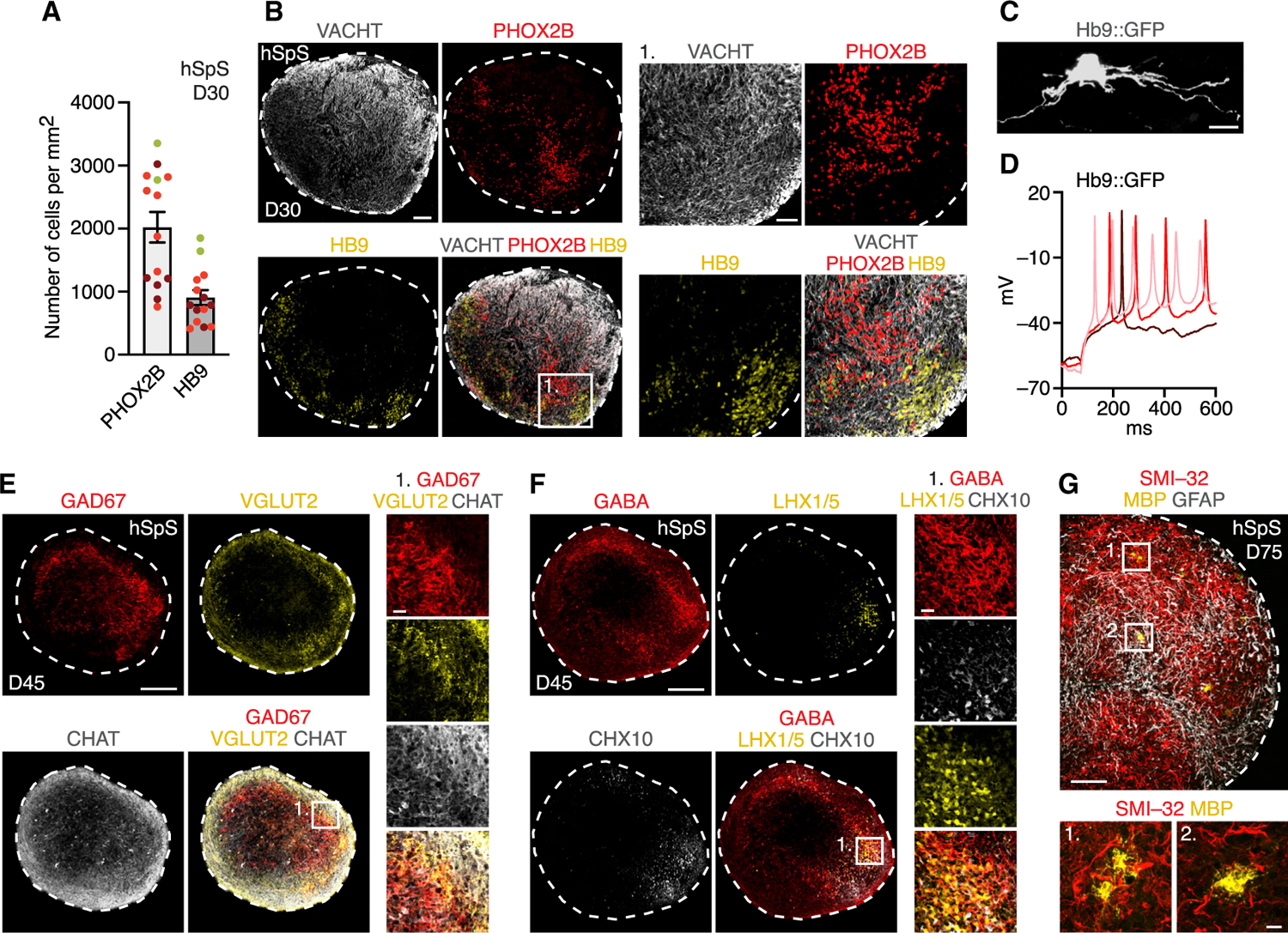
(A) Quantification of the total number of cells expressing PHOX2B or HB9 per area (mm2) in 16 µm hSpS cryosections at day 30. Data represent mean ± SEM (n = 14 hSpS derived from 3 hiPS cell lines from 1–2 differentiations, with 2–4 cryosections quantified per hSpS).
(B) Representative immunohistochemistry images of day 30 hSpS.
(C) Immunocytochemistry showing a lenti-Hb9::GFP+ neuron in an hSpS cryosection.
(D) Whole-cell current-clamp recording from an Hb9::GFP cell showing action potential generation in response to depolarizing current injections.
(E and F) Representative immunohistochemistry images of neuronal domains with diverse neurotransmitter identities on day 45 hSpS.
(G) Immunocytochemistry in day 75 hSpS showing expression of the astrocyte marker GFAP and the oligodendrocyte marker MBP.
Scale bars, 10 µm (C), inset in (G), 20 µm (insets in E and F), 50 µm (inset in B), 100 µm (B and G), and 200 µm (E and F).
Generation of Cortico-Spinal Assembloids
We previously described the generation of 3D spheroids resembling the cerebral cortex (human cortical spheroids [hCS]) (Birey et al., 2017; Paşca et al., 2015; Sloan et al., 2017, 2018), which contain deep and superficial glutamatergic neurons. We verified expression of cortico-spinal-associated markers in hCS, including FEZF2, BCL11B (CTIP2), and SOX5, using single-cell profiling of hCS (Birey et al., 2017) and found a population of cells co-expressing these genes in the glutamatergic neuron cluster but not in a subpallial-derived GABAergic cluster (Figure S5A). Moreover, we confirmed the expression of some these genes by real-time qPCR from day 45 to day 130 (Figure S5C), as well as by immunocytochemistry with antibodies that we validated in slices of human cortical tissue at post-conception week 17 (PCW17) (Figures S5D and S5E). Importantly, we found no expression of cholinergic markers CHT1 and VACHT in hCS (Figure S5B).
To generate cortico-spinal assembloids, we fused hSpS with an hCS that had been infected with an AAV-hSYN1::eYFP reporter (Figure 3A). Live imaging at 5 days after fusion (daf) showed processes derived from cells in the hCS extending into hSpS (Figure 3B; Video S1). At 30 daf, immunocytochemistry for YFP showed extensive hCS-derived projections in hSpS (Figure 3C). Quantification of the eYFP+-covered area in hSpS in intact hCS-hSpS assembloids showed a progressive increase of eYFP over time (5 daf versus 10 daf p = 0.009; 5 daf versus 20 daf p < 0.0001; Figures 3D, S5F, and S5G for examples of hCS-hSpS assembloids at 20 daf), and this was not observed to the same extent in hCS-hCS assembloids (5 daf versus 10 daf p > 0.9; 5 daf versus 20 daf p = 0.06; Figures 3E, S5H, and S5I). Immunohistochemistry in hCS-hSpS assembloids at 28 daf showed that hCS-derived eYFP processes often project toward ISL1+ cell clusters (Figure 3F). We also generated hSpS using a fluorescently labeled CAG::EGFP hiPS cell line to examine reciprocal hSpS interactions with hCS and found that hSpS cells sometimes projected into hCS but seldom migrated (Figure S5J). Similarly, we did not observe hSpS-derived Hb9 cells migrating into hCS in hCS-hSpS assembloids where hSpS had been infected with Hb9::mCherry (Figure S5K).
Figure 3. Generation of hCS-hSpS Assembloids and Characterization of Cortico-Spinal Projections.
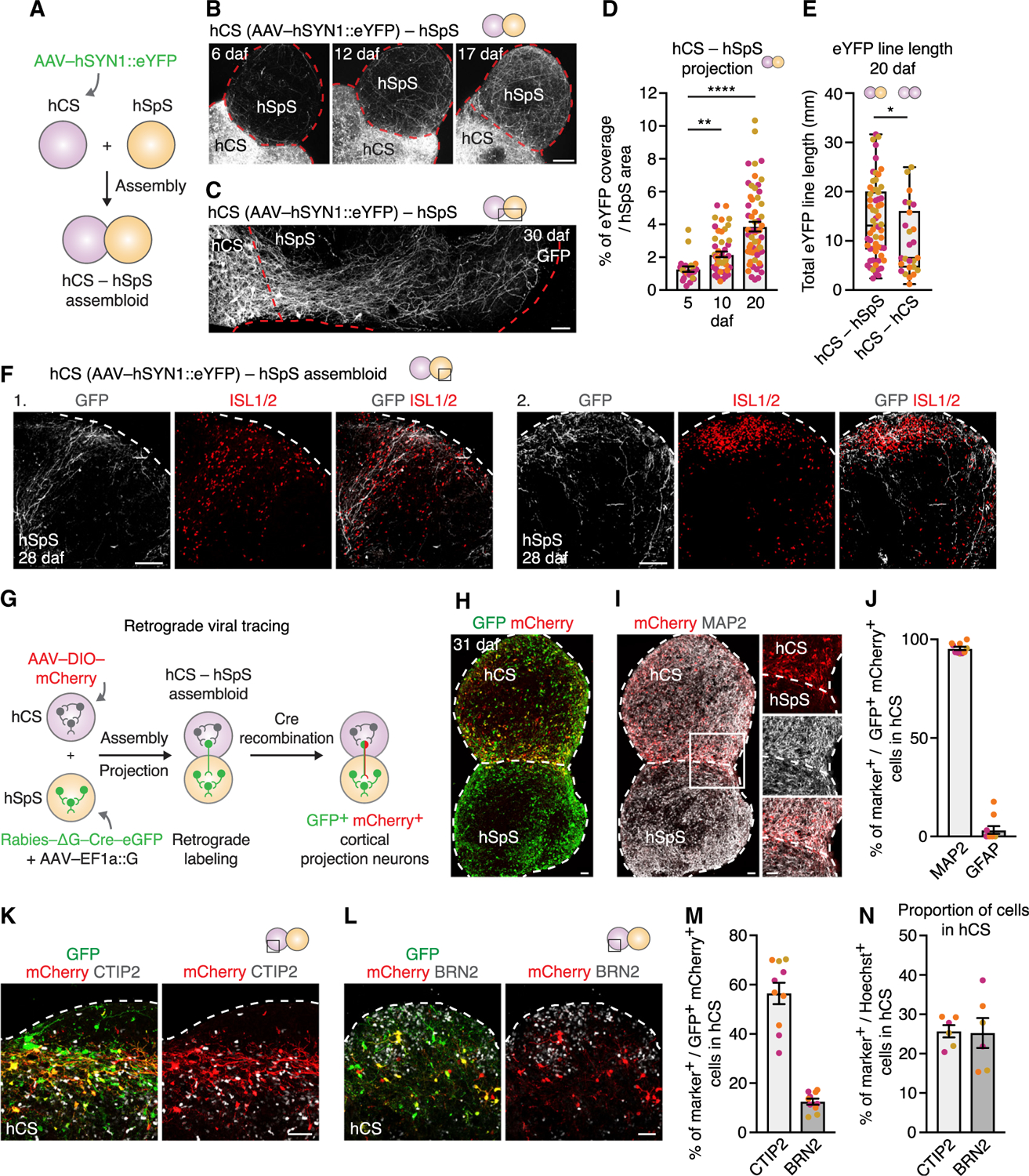
(A) Schematic illustrating the fusion of hCS and hSpS to form hCS-hSpS assembloids.
(B) Representative images of intact hCS-hSpS assembloid showing hCS-derived hSYN1::eYFP projections 6, 12, and 17 days after fusion (daf). See Video S1 for live imaging of AAV-hSYN1::eYFP projection at 5 daf.
(C) Immunocytochemistry of hCS-hSpS assembloid 30 days after fusion (daf).
(D) Quantification of hCS-derived eYFP coverage in hSpS area at 5, 10, and 20 daf in hCS-hSpS assembloids (n = assembloids from 3 hiPS cell lines from 3 separate differentiations, Kruskal-Wallis test p < 0.0001 with Dunn’s multiple test comparison: **p = 0.009 for 10 daf versus 5 daf, ****p < 0.0001 for 20 daf versus 5 daf).
(E) Quantification of total eYFP line length on the projection side of hCS-hSpS or hCS-hCS assembloids at 20 daf (n = 3 hiPS cell lines from 1–2 differentiations; Mann-Whitney test: *p = 0.01). Boxplot shows median and 75th and 25thth percentiles, and whiskers show minimum (min.) and maximum (max.) values.
(F) Immunohistochemistry images in hCS-hSpS assembloids at 28 daf showing eYFP projections overlapping with ISL1+ cell clusters.
(G) Schematic detailing retrograde viral tracing experiment in hCS-hSpS assembloids.
(H) Immunocytochemistry of hCS-hSpS assembloid at 31 daf showing co-expression of GFP and mCherry on the hCS side.
(I) Immunocytochemistry of hCS-hSpS assembloid at 31 daf showing expression of mCherry and MAP2.
(J) Quantification of the percentage (%) of GFP+ and mCherry+ cells on the hCS side that co-express MAP2 or GFAP (n = 10 assembloids from 3 hiPS cell lines from 1–2 separate differentiations, with 2–3 cryosections quantified per assembloid).
(K and L) Representative immunocytochemistry image for CTIP2 (K) or BRN2 (L) on the hCS side of hCS-hSpS assembloids at 31 daf.
(M) Quantification of the percentage (%) of GFP+ and mCherry+ cells on the hCS side that co-express either CTIP2 or BRN2 (n = 10 assembloids derived from 3 hiPS cell lines from 1–2 separate differentiations, with 2–3 cryosections quantified per assembloid).
(N) Quantification of the percentage (%) of CTIP2+ or BRN2+ among all Hoechst+ cells in hCS (n = 6 assembloids derived from 3 hiPS cell lines from 1–2 separate differentiations, with 2–3 cryosections quantified per assembloid).
Data represent mean ± SEM unless otherwise specified. Scale bars, 50 µm (C, H, I, K, L, and inset in I), 100 µm (F), and 200 µm (B).
Viral Tracing, Optogenetics, and Calcium Imaging in Cortico-Spinal Assembloids
To characterize the specificity of hCS-derived projections, we used a retrograde rabies tracing approach (Figure 3G). We separately infected hSpS with a ∆G-rabies virus carrying Cre-eGFP and with an AAV carrying the rabies glycoprotein (G) required for trans-synaptic spread (Coulon et al., 1983; Etessami et al., 2000), and hCS with an AAV encoding mCherry under a double-floxed inverse orf (DIO-mCherry). After 6–7 days of infection, day ~80 hCS and days ~40–50 hSpS were assembled and expression of GFP and mCherry was examined at 31 daf. We predicted infected neurons in hSpS would express GFP from the rabies-Cre virus, and hCS neurons would co-express GFP and mCherry following rabies-Cre retrograde transport and recombination. We observed extensive expression of GFP in hSpS and cells co-expressing GFP and mCherry in hCS (Figures 3H and S5L). We found that ~95% of the GFP+/mCherry+ cells in hCS co-expressed the neuronal marker MAP2 and fewer than 4% expressed the glial lineage marker GFAP (Figures 3I, 3J, S5M, and S5N). Corticofugal projection neurons, which include cortico-spinal projecting cells, express CTIP2 and are mainly located in deep layers of the cerebral cortex, whereas neurons located in superficial layers express BRN2 and project to the contra-lateral cortex (Greig et al., 2013). To verify the projection identity of GFP+/mCherry+ cells, we quantified the proportion of cells that co-expressed either CTIP2 or BRN2. We found that almost 60% of all GFP+/mCherry+ cells co-expressed the corticofugal marker CTIP2, and only ~12% co-expressed BRN2 (Figures 3K–3M and S5O), although the relative proportions of these cell types were not different in hCS at this in vitro stage (Figure 3N).
We next used optogenetics and calcium imaging to probe whether hCS-derived projections formed functional connections. We used AAV viruses to separately deliver, before assembly, the light-sensitive opsin Chrimson (Chrim, AAV-hSYN1-ChrimsonR-tdT) (Klapoetke et al., 2014) into hCS and the genetically encoded calcium indicator GCaMP7s (AAV-hSYN1-jGCaMP7s) into hSpS (Figures 4A and 4B). We then imaged calcium transients (10 Hz) at 29–46 daf, while delivering short (100 ms) pulses of red light (625 nm) onto hCS using a fiber optic. We found that neurons in hSpS in 4 out of 10 assembloids responded with calcium spikes that followed the stimulus after ~200 ms (Figures 4C and 4D). To confirm the relationship between stimulation and calcium responses, we compared the stimulus-triggered change in amplitude (ΔF/F) to random time-locked ΔF/F in the same cell. We found that the median amplitude of the response was ~5 times higher for real stimulation compared to randomly selected time points (n = 16 cells, p = 0.0005); Figure 4E). These responses were blocked by application of the ionotropic glutamate receptor blockers NBQX (20 µM) and APV (50 µM) (Figures 4D and 4F), indicating glutamatergic transmission from hCS to hSpS.
Figure 4. Functional Connectivity in hCS-hSpS Assembloids.
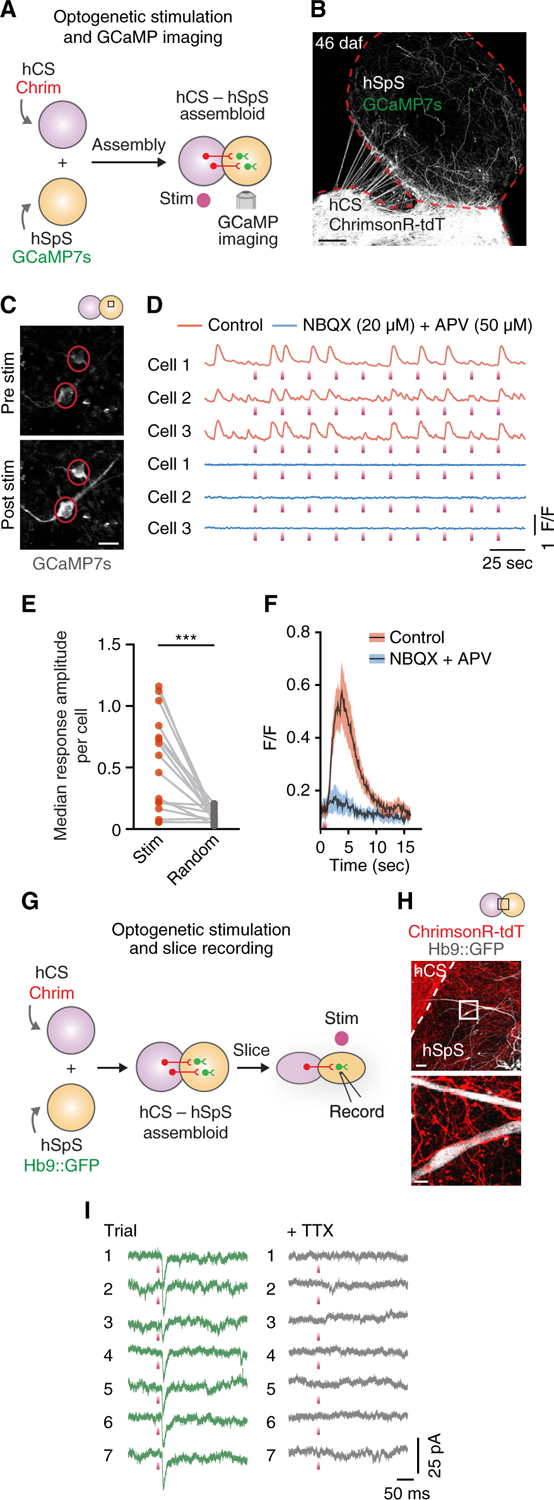
(A) Schematic detailing optogenetic stimulation coupled with GCaMP imaging in hCS-hSpS assembloids.
(B) Representative picture of intact hCSAAV-Chrim-hSpSGCaMP7s assembloid 46 days after fusion (daf).
(C) Representative images of two GCaMP7s-infected cells in an hCS-hSpS assembloid before and after optogenetic stimulation (625 nm, 100 ms each).
(D) Representative ΔF/F traces showing spontaneous and light-evoked calcium responses in three GCaMP7s-infected cells before and after adding NBQX (20 µM) and APV (50 µM). Optogenetic stimulation (625 nm, 100 ms each) is indicated with a purple rectangle. ΔF/F indicates the fluorescence intensity over baseline fluorescence.
(E) Quantification of the stimulation-triggered ΔF/F amplitudes, shown in comparison to randomized-triggered amplitudes (n = 16 cells from 4 assembloids derived from 2 hiPS cell lines from 1–2 differentiations; two-tailed paired t test: ***p = 0.0005).
(F) Stimulation-triggered average of the calcium response to optogenetic stimulation in paired cells with and without NBQX and APV (n = 16 cells from 4 assembloids derived from 2 hiPS cell lines). Black traces represent the mean and shaded bars represent the SEM.
(G) Schematic detailing optogenetic stimulation coupled with patch clamping slice recording in hCS-hSpS assembloids.
(H) Example images of an hCSAAV-Chrim-hSpSHb9::GFP assembloid.
(I) Chrimson-triggered EPSCs in an Hb9::GFP+ neuron voltage clamped to –70 mV, (left) and lack of EPSCs following TTX application (right).
Scale bars, 10 µm (inset in H), 20 µm (C), 50 µm (H), and 200 µm (B).
See also Table S1.
To verify that cortical neurons in hCS are synaptically connected to motor neurons in hSpS, we performed voltage clamp recordings in slices of hCS-hSpS assembloids. We infected hCS with AAV1-Syn1::Chrimson-mCherry and used the Hb9::GFP reporter in hSpS to visually identify motor neurons (Figures 4G and 4H). We found that consecutive light stimulation to the area surrounding Hb9+ cells could result in reliable post-synaptic currents (Figure 4I; 2 out of 30 recorded cells in 4 assembloids). These responses could be blocked by application of TTX, indicating that they have been triggered by action potentials in axons of hCS neurons by mono- or di-synaptic connections. Together, these assays indicate that corticofugal neurons in hCS synaptically connect with neurons in hSpS.
Functional Assembly of Spinal Spheroids with Muscle
The functional output of the cortico-motor circuit is muscle contraction through motor neuron activity. To probe the ability of hSpS to mediate muscle contraction, we dissected mouse limb buds at embryonic day 11.5 (E11.5) before spinal cord axonal innervation and directly assembled them with hSpS derived from a TUBA1B-mEGFP hiPS cell line. We observed extensive projections from the hSpS into the limb bud upon assembly (Figure 5A). Moreover, limb buds displayed spontaneous contractions when assembled with hSpS, but not when kept in isolation or when assembled with hCS (χ2 test, p = 0.02; Figure 5B). Activity in hSpS-limb assembloids persisted for at least 2 weeks in vitro and was completely blocked by addition of the acetylcholine receptor antagonist curare (100 µM; χ2 test, p = 0.02, Figure 5B; Video S2, sequence 1).
Figure 5. hSpS Control of Muscle Activity.
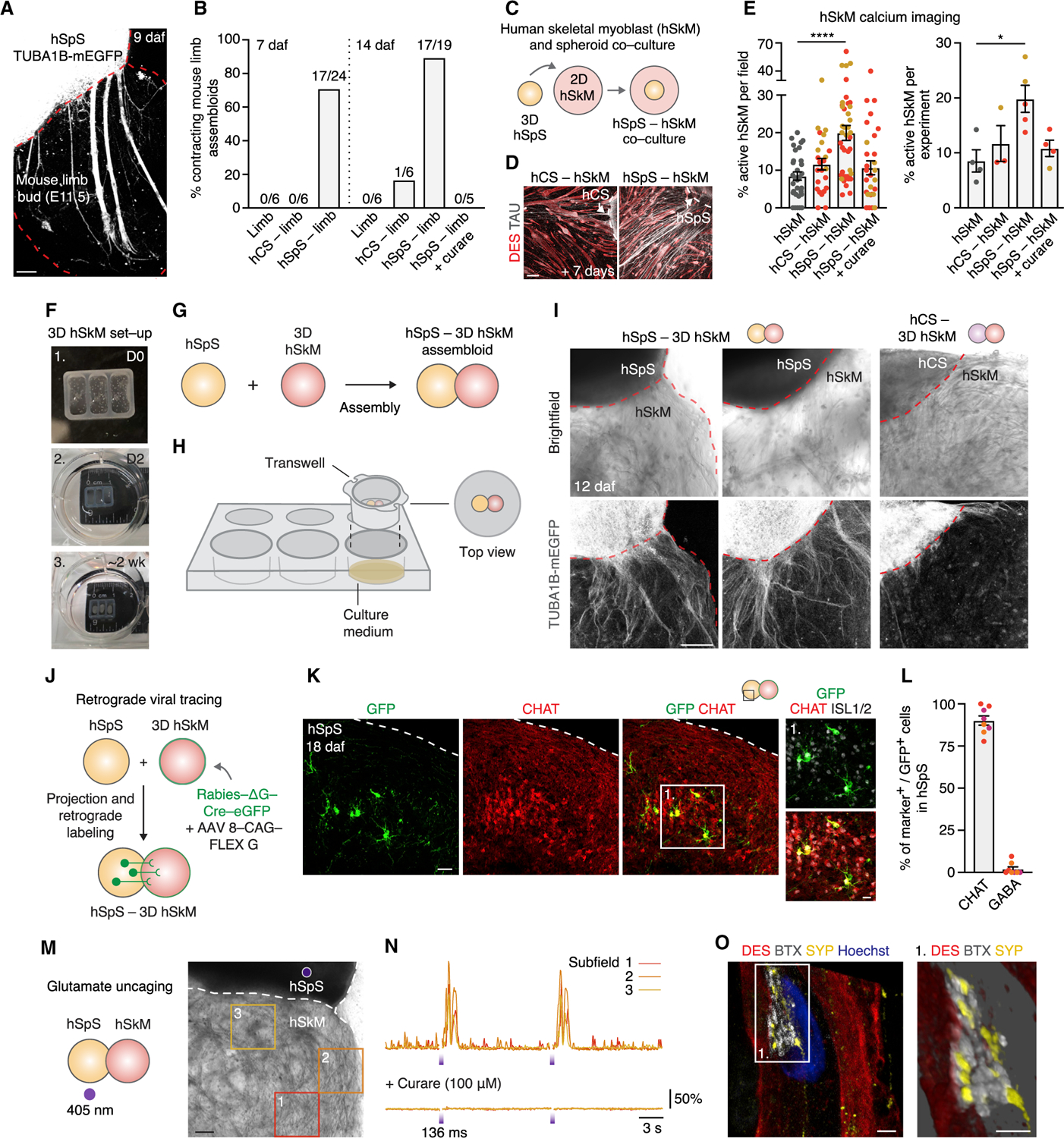
(A) Image of intact assembloid showing hSpS derived from TUBA1B-mEGFP projecting into mouse limb. This image was generated by manual stitching of individual images.
(B) Quantification of the proportion of contracting limb, hCS-limb or hSpS-limb assembloids 7 and 14 days after fusion (daf; n = 3 hiPS cell lines from 1 differentiation; χ2 test, p = 0.02), in the absence or presence of curare (100 µM; n = 3 hiPS cell lines; χ2 test, p = 0.02). See Video S2, sequence 1, for live imaging of a contracting hSpS-limb assembloid with or without curare.
(C) Schematic showing the co-culture of 2D human skeletal myoblasts (hSkM) and spheroids (hCS or hSpS).
(D) Immunocytochemistry of 2D hSkM 7 days after co-culture with hCS or hSpS.
(E) Quantification of spontaneous calcium activity in hSkM (Cal-590 AM) in either hSkM alone or after co-culture with hCS, hSpS or hSpS + curare (100 µM). Graph on the left shows % of active hSkM per field recorded (n = 2 hiPS cell lines from 1–2 differentiations; Kruskal-Wallis test: p < 0.0001, with Dunn’s multiple comparison test ****p < 0.0001). Graph on the right shows the percentage (%) of active hSkM per co-culture experiment (fields imaged in one experiment combined; n = 2 hiPS cell lines from 1–2 differentiations; Kruskal-Wallis test: p = 0.02, with Dunn’s multiple comparison test *p = 0.01). See Video S2, sequence 2 for calcium activity of hSkM and hSpS-hSkM.
(F) Images showing the generation of 3D hSkM and the assembly with hCS and hSpS. (1) Dissociated hSkM are resuspended in Geltrex and placed in silicone wells. (2) Silicone wells are placed in ultra-low attachment plates in hSkM growth medium. (3) hSkM growth medium is switched to differentiation medium.
(G and H) Schematics showing hSpS-hSkM assembloid set-up.
(I) Representative images of 3D hSkM that have been assembled with hCS or hSpS derived from a TUBA1B-mEGFP hiPS cell line.
(J) Schematic detailing retrograde viral tracing experiment in hSpS-hSkM assembloid.
(K) Representative immunohistochemistry image for rabies-derived GFP, CHAT, and ISL1 on the hSpS side of hSpS-hSkM assembloids at 18 daf.
(L) Quantification of the percentage (%) of GFP+ cells on the hSpS side of hSpS-hSkM assembloids that co-express either CHAT or GABA (n = 8 assembloids derived from 3 hiPS cell lines from 1 differentiation, with 3–6 cryosections quantified per assembloid).
(M and N) Glutamate uncaging in hSpS-hSkM assembloid. UV light (405 nm, 136 ms) uncages glutamate on hSpS (M). Displacement normalized to baseline over time is shown for 3 subfields in the presence or absence of curare (100 µM; N). See Video S3 for an example of glutamate uncaging in an hSpS-hSkM assembloid. Table S1 shows details of all stimulation experiments.
(O) Representative immunohistochemistry showing a neuromuscular junction (NMJ) with synaptophysin 1 (SYP) and bungarotoxin (BTX) on a desmin+ (DES) myofiber. Inset: 3D rendering of the NMJ.
Data represent mean ± SEM. Scale bars, 2 µm (O), 20 µm (inset in K), 50 µm (K), 100 µm (A), and 200 µm (D, I, and M).
See also Figure S6.
Next, we probed the ability of hSpS to modulate the activity of human muscle cells. To achieve this, we used human skeletal myoblasts (hSkM) (Figure S6A) derived from adult muscle biopsies. Differentiated myotubes expressed markers of mature skeletal muscle, such as desmin (DES), titin (TTN), and the heavy chain myosin (MyHC), and became multinucleated (Figures S6B and S6C). We placed hSpS on top of hSkM grown in 2D adherent cultures (Figure 5C), and within a week, hSpS projected out to cover muscle cells (Figure 5D). Using live imaging with the calcium indicator Cal-590, we found that the proportion of active hSkM doubled in hSpS-hSkM in comparison to hSkM in isolation or co-cultured with hCS (hSpS-hSkM versus hSkM p = 0.01; Figures 5E and S6D; Video S2, sequence 2). This effect was observed mainly in hSkM within 1 mm of hSpS (hSkM versus hSpS-hSkM <1 mm, p < 0.0001; Figures S6E and S6F) and was blocked by curare (Figure 5E). Immunocytochemistry of hSkM after co-culture with hSpS revealed binding of bungarotoxin (BTX) (Figure S6G), a peptide that binds with high affinity to nicotinic acetylcholine receptors at neuromuscular junctions (NMJs).
We then generated 3D hSkM by combining dissociated proliferative hSkM in an extracellular matrix within a silicone well (Figure 5F; see STAR Methods for details). We placed an hSpS and a 3D hSkM in close proximity on top of a 6-well transwell insert to generate hSpS-hSkM assembloids (Figures 5G, 5H, and S6H). Using spheroids derived from the TUBA1B-mEGFP hiPS cell line, we noticed that by 12 daf hSpS sent out abundant projections to the 3D hSkM (Figure 5I). To verify the specificity of these projections, we next performed retrograde rabies tracing. We delivered the ∆G-rabies-Cre-eGFP virus together with an AAV-G to differentiated hSkM that were assembled with day 37 hSpS (Figure 5J). We examined expression of GFP in hSpS 18 days after assembly and found that 90% of GFP+ cells were CHAT+, while less than 5% of these cells were GABA+ (Figures 5K, 5L, S6I, and S6J). Further quantification showed that ~45% of these cells were also positive for the motor neuron-related transcription factor ISL1 but not for CHX10, PROX1, or FOXP2 (Figures S6K–S6M).
Next, we investigated whether hSpS cholinergic projections were functionally connected to hSkM using glutamate uncaging. We used 405 nm photostimulation in the presence of MNI-caged glutamate in the medium to rapidly and locally release glutamate (Figure S6N; Table S1 includes details of all stimulation experiments). Photostimulation of hSpS in hSpS-hSkM assembloids resulted in robust hSkM contraction in the presence, but not in the absence, of MNI-caged glutamate (Figures 5M, 5N, S6O, and S6P; Video S3), and addition of curare stopped hSkM contractions (100 µM; Figure 5N). Last, we used immunocytochemistry to verify the presence of neuromuscular junctions by labeling Desmin+ myofibers with the pre-synaptic marker synaptophysin and BTX (Figures 5O and S6Q–S6S).
Functional Applications in Cortico-Motor Assembloids
To model higher-order control of motor output, we implemented the tools described above to generate and characterize a functional cortico-motor unit in vitro. We fused intact hCS with hSpS and 3D hSkM on top of transwell inserts (Figures 6A and 6B). We then verified the presence of spontaneous contractions in hSkM. We looked at displacement of pixels over time in imaging fields (subdivided into 16 subfields; Figure S7A). We found that hSpS-hSkM and hCS-hSpS-hSkM assembloids displayed ~5× more spontaneous contractions compared to hSkM alone or hCS-hSkM assembloids (p = 0.03 for hSpS-hSkM versus hSkM; p = 0.0003 for hCS-hSpS-hSkM versus hSkM) (Figure 6C; Video S4). Moreover, we found that spontaneous activity in hSpS-hSkM and hCS-hSpS-hSkM assembloids was more coordinated across fields as assessed by analysis of the covariance between subfields in an imaging field (p = 0.01 for hSpS-hSkM versus hSkM, p = 0.03 for hCS-hSpS-hSkM versus hSkM) (Figures 6D, 6E, S7B, and S7C). However, we found no significant difference in the number of contractions or the covariance between hSpS-hSkM and hCS-hSpS-hSkM (p = >0.99).
Figure 6. Cortical Activity Modulates Muscle Function in hCS-hSpS-hSkM Assembloids.
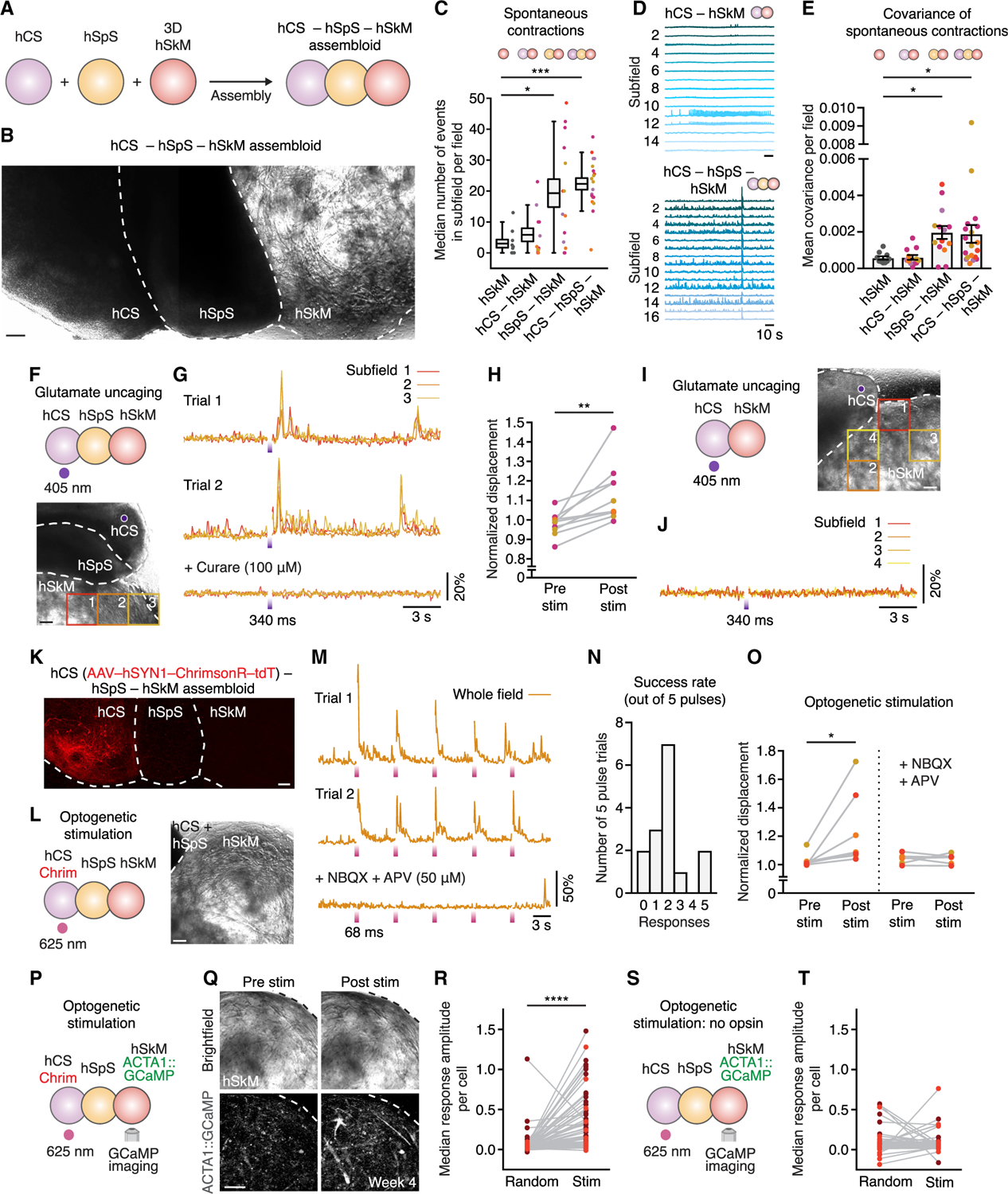
(A) Schematic showing hCS-hSpS-hSkM assembloid set-up.
(B) Representative image showing intact hCS-hSpS-hSkM assembloid. This image was generated by manual stitching of individual images.
(C) Quantification of spontaneous contractions over a 2-min period showing the median number of events in subfields per field (n = 10 fields from 5 assembloids for hSkM, n = 12 fields from 6 assembloids from 1–2 differentiations for hCS-hSkM, n = 14 fields from 7 assembloids from 1–2 differentiations for hSpS-hSkM, n = 19 fields from 11 assembloids from 1–2 differentiations for hCS-hSpS-hSkM; Kruskal-Wallis test p < 0.0001, with Dunn’s multiple comparison test: *p = 0.03 for hSpS-hSkM versus hSkM, ***p = 0.0003 for hCS-hSpS-hSkM versus hSkM). Boxplot shows mean ± SEM and whiskers show 90th and 10th percentiles. See Video S4 for an example of spontaneous contractions in an hCS-hSpS-hSkM assembloid.
(D) Representative spontaneous contraction traces in subfields of hCS-hSkM or hCS-hSpS-hSkM assembloids.
(E) Correlation of displacements between subfields in a field quantified using covariance analysis. Data represent mean ± SEM (n = 10 fields from 5 assembloids for hSkM, n = 12 fields from 6 assembloids for hCS-hSkM, n = 14 fields from 7 assembloids for hSpS-hSkM, n = 19 fields from 11 assembloids for hCS-hSpS-hSkM; Kruskal-Wallis test p = 0.001, with Dunn’s multiple comparison test: *p = 0.01 for hSpS-hSkM versus hSkM, *p = 0.03 for hCS-hSpS-hSkM versus hSkM).
(F and G) Glutamate uncaging in hCS-hSpS-hSkM assembloid. UV light (405 nm) uncages glutamate specifically on hCS (F). Displacement normalized to baseline over time is shown for 3 subfields and two trials (G).
(H) Quantification of displacement normalized to baseline in different glutamate uncaging experiments. Values of the last frame before stimulation (Pre stim) and the highest of the first 3 frames or 204 ms after stimulation (Post stim) are plotted per field (subfields per field are averaged; n = 10 fields from 7 assembloids derived from 3 hiPS cell lines from 1–2 differentiations; Wilcoxon test **p = 0.002).
(I and J) Glutamate uncaging of hCS in hCS-hSkM assembloid (I). Displacement normalized to baseline over time is shown for 4 subfields in the presence of caged glutamate (J). Similar results were obtained in n = 4 hCS-hSkM assembloids derived from 2 hiPS cell lines.
(K) Representative image showing an intact hCS-hSpS-hSkM assembloid in which hCS was infected with AAV-hSYN1-ChrimsonR-tdT prior to assembly. This image was generated by manual stitching of individual images.
(L and M) Optogenetic stimulation in hCS-hSpS-hSkM assembloids. Five consecutive pulses of light (625 nm, 68 ms in duration each and 6.8 s apart) were delivered (L). Traces of whole-field muscle displacement are shown after normalization to the pre-stimulation baseline in the absence or presence of NBQX (50 µM) and APV (50 µM) (M). See Video S5, sequence 1 for an example of optogenetic stimulation in a hCS-hSpS-hSkM assembloid.
(N) Histogram illustrating the success rate of optogenetic stimulation (out of 5 consecutive pulses for each assembloid; n = 15 trials of 5 pulses in 7 assembloids from 1–2 differentiations).
(O) Quantification of displacement (normalized to pre-stimulation baseline) per assembloid in the presence or absence of NBQX and APV (50 µM). Pre stim represents the highest value of displacement in the 20 frames (1.36 s) before stimulation. Post stim represents the average across 5 pulses of the highest value in the 20 frames immediately following stimulation (left: n = 7 assembloids derived from 3 hiPS cell lines from 1–2 differentiations; Wilcoxon matched paired t test *p = 0.01; right: n = 6 assembloids derived from 3 hiPS cell lines from 1–2 differentiations; two-tailed paired t test p = 0.94). Table S1 includes details of stimulation experiments.
(P) Optogenetic stimulation coupled with calcium imaging in hCSAAV-Chrim-hSpS-hSkMACTA1::GCaMP6s. Five consecutive pulses of light (625 nm, 100 ms in duration each and 10 s apart) were delivered.
(Q) Representative bright-field and ACTA1::GCaMP6s images of hSkM in hCS-hSpS-hSkM assembloid before and after stimulation. See Video S5, sequence 2 for an example of optogenetic stimulation coupled with calcium imaging in a hCS-hSpS-hSkM assembloid.
(R) Quantification of the stimulation-triggered ΔF/F amplitudes, shown in comparison to randomized-triggered amplitudes in hSkM cells. The median ΔF/F amplitude of the five pulses delivered per cell is shown (n = 82 cells from 8 fields in 6 assembloids derived from 2 hiPS cell lines and 2 separate differentiations; Wilcoxon matched paired t test: ****p < 0.0001).
(S) Optogenetic stimulation in hCS-hSpS-hSkMACTA1::GCaMP6s assembloid (no opsin; five consecutive 100-ms pulses).
(T) Quantification of stimulation-triggered ΔF/F amplitudes, shown in comparison to randomized-triggered amplitudes in hSkM cells of hCS-hSpS-hSkM assembloids without Chrimson. The median ΔF/F amplitudes of the five pulses delivered per cell are shown (n = 91 cells from 6 fields in 4 assembloids derived from 2 hiPS cell lines and 1 differentiation; Wilcoxon matched paired t test: p = 0.27).
Scale bars, 200 µm (B, F, I, K, L, and Q).
See also Figures S7 and S8.
Next, we probed the functional connectivity by selective stimulation of hCS. We used glutamate uncaging to photostimulate hCS in hCS-hSpS-hSkM assembloids (405 nm, 340 ms). This resulted in robust muscle contraction (p = 0.002) (Figures 6F–6H) suggesting functional assembly of a cortico-spinal-muscle functional unit. The contraction was blocked by treatment with 100 µm curare (Figures 6G, S7D, and S7E) and was not related to UV exposure (Figures S7F and S7G). Moreover, glutamate uncaging of hCS in hCS-hSkM did not elicit a response (Figures 6I and 6J), showing that muscle contraction upon cortical stimulation is dependent on the presence of the hSpS.
To validate cortical activation of muscle in hCS-hSpS-hSkM assembloids and achieve broader stimulation, we used optogenetics. We delivered Chrimson into hCS before assembly (Figures 6K and S7H) and used light stimulation (68 ms at 625 nm, 5 pulses, 100 frames or 6.8 s apart each) at 20–30 days after hCS-hSpS-hSkM assembly. Activation of Chrimson-expressing hCS induced global contractions in hSkM in 85.7% of tested assembloids (p = 0.01) (Figures 6L and 6M; Video S5, sequence 1) and 40% of pulses (Figure 6N shows the distribution of successful stimulation and Figures S7I and S7J show an example where muscle contraction is induced in only 2 of the 5 light pulses; Table S1), while stimulation of Chrimson-expressing hCS in hCS-hSkM did not trigger a response (Figures S7K and S7L). To test whether this response was mediated by glutamatergic transmission, we added the AMPA and NMDA receptor blockers NBQX (50 µM) and APV (50 µM), which completely abolished light-induced responses (Figures 6M and 6O).
Next, to test whether hCS-hSpS-hSkM assembloids could be entirely derived from hiPS cells, we generated hiPS cell-derived skeletal myoblasts using a protocol adapted from Chal et al. (2016) (Figure S8A). Following myoblast differentiation, sub-culturing and fluorescence-activated cell sorting (FACS) purification (Figures S8B–S8F), we generated 3D hiPS cell-derived hSkM (ihSkM) as we did for primary myoblasts in Figure 5F. We found that these 3D ihSkM can also be assembled with hSpS and hCS to generate three-part assembloids (Figure S8G) that display contraction following optogenetic stimulation (Figures S8H and S8I).
Because quantification of contractions and displacement depends on changes in pixel intensities, and this could vary from preparation to preparation and over time, we next implemented the use of a calcium indicator expressed under a muscle-specific promoter to monitor cellular activity in hSkM. For this, we delivered Chrimson into hCS as well as lenti-ACTA1::GCaMP6s into hSkM, and used light stimulation (100 ms at 625 nm, 5 pulses, 100 frames or 10 s apart each) at 4 weeks after hCS-hSpS-hSkM assembly (Figure 6P). Activation of Chrim-expressing hCS induced calcium spikes in hSkM fibers (Figures 6Q and 6R; Video S5, sequence 2), and these were dependent on the presence of Chrimson, as light stimulation of hCS-hSpS-hSkM assembloids in the absence of the opsin did not elicit any responses (Figures 6S and 6T).
Long-Term Culture and Functionality of Cortico-Motor Assembloids
Conventional motor neuron and muscle co-cultures are not amenable to long-term experiments, as cells tend to detach over time (Thomson et al., 2012). We found that hCS-hSpS-hSkM can be kept in culture for up to 10 weeks post-assembly (~16–18 weeks from the initiation of hSpS differentiation). To characterize cortico-motor assembloids over time, we first performed electron microscopy 8 weeks after assembly (Figures 7A–7F and S7M–S7S). We found that assembloids at this time present intact axons and dendrites (Figure S7N), mature synapses (Figures 7B, 7C, S7O, and S7P), organized skeletal muscle fibers (Figures 7D and S7Q), and neuromuscular contacts (Figures 7E, 7F, S7R, and S7S). Moreover, we used immunohistochemistry in hCS-hSpS-hSkM assembloids at 10 weeks (Figure 7G) and found the presence of hCS-derived projections into hSpS (Figures 7H and S7U), motor neurons (Figure 7I, S7U, and S7V), non-reactive astrocytes (Figures 7H and S7V), myelinating oligodendrocytes (Figures 7H and 7I) and neuromuscular junctions (Figure 7J). Together, these data indicate that hCS-hSpS-hSkM assembloids can be maintained in culture for up to 10 weeks without structural disintegration.
Figure 7. Long-Term Culture and Functionality of hCS-hSpS-hSkM Assembloids.
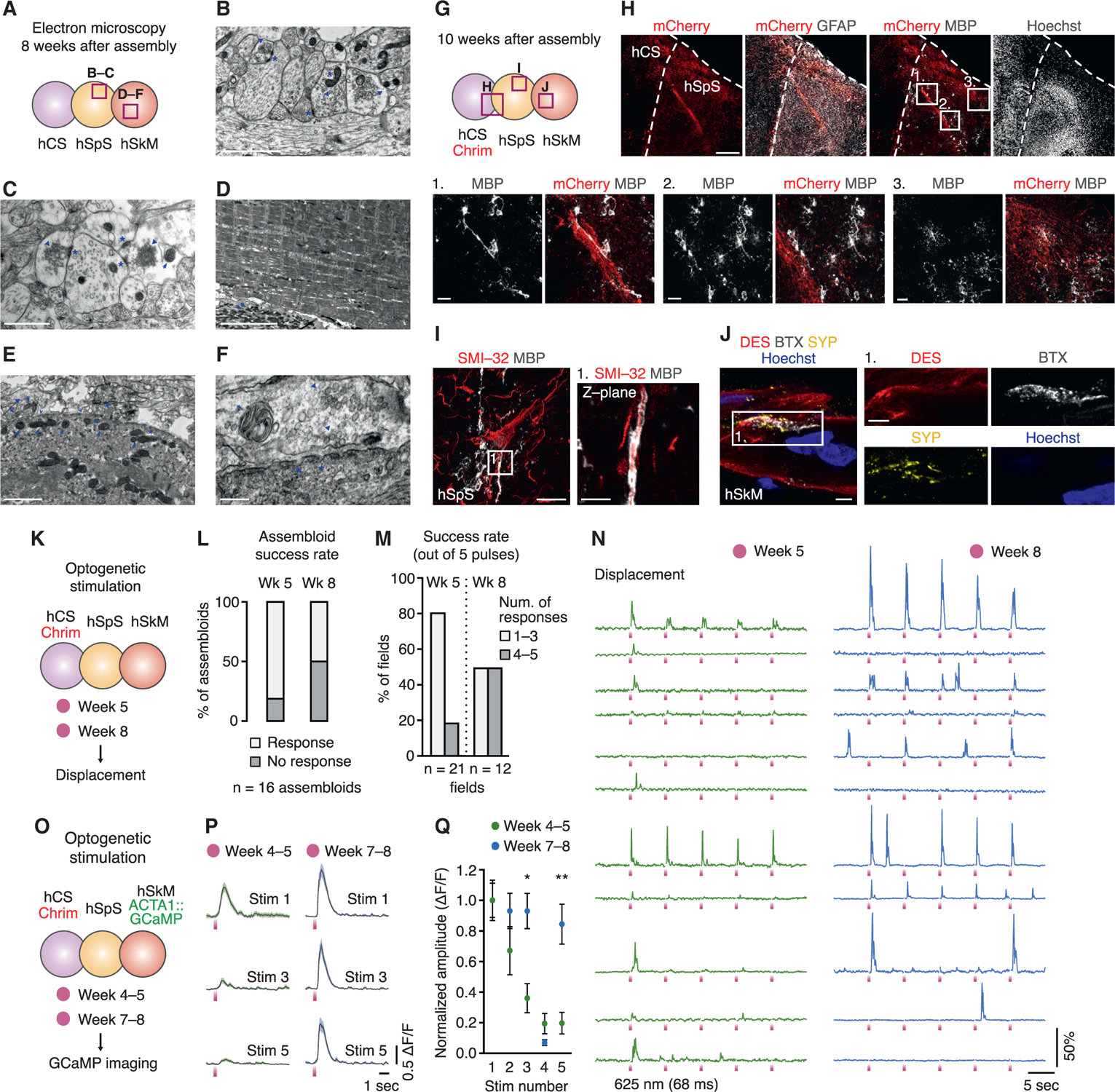
(A) Schematic detailing location of electron microscopy images within hCS-hSpS-hSkM assembloids.
(B and C) Representative images within hSpS in hCS-hSpS-hSkM assembloids showing dendrites and synapses with synaptic vesicles (arrowheads), post-synaptic densities (asterisks), and mitochondria (arrows).
(D) Representative images within hSkM in hCS-hSpS-hSkM assembloids showing skeletal muscle fibers with actin and myosin filaments and mitochondria (arrows).
(E and F) Representative images within hSkM in hCS-hSpS-hSkM assembloids showing points of contact between hSpS neurons and hSkM. Neuron terminals are vesicle-laden (arrow heads) and present mitochondria (arrows). hSkM are surrounded by a basal lamina (hollow arrowheads) and present small invaginations on the membrane (asterisks, F).
(G) Schematic detailing location of images within hCSAAV-Chrim-hSpS-hSkM assembloids.
(H–J) Representative immunohistochemistry images in cryosections of hCSAAV-Chrim-hSpS-hSkM assembloids showing oligodendrocytes (H), motor neurons (I) and neuromuscular junctions (J). Insets in (H) show oligodendrocytes in hSpS, and inset in (I) shows a single Z plane. Optogenetic stimulation in hCS-hSpS-hSkM assembloid (five consecutive 68 ms pulses) at 5 and 8 weeks post-assembly. Displacement in hSkM was quantified.
(L) Success rate per assembloid following optogenetic stimulation at 5 and 8 weeks post-assembly. Two separate fields per assembloid were imaged at each time-point (n = 16 assembloids for both time-points from 2 hiPS cell lines and 1 differentiation; χ2 test, p = 0.06).
(M) Success rate out of 5 consecutive light pulses per responding field following optogenetic stimulation at 5 and 8 weeks post-assembly (n = 21 and 12 fields for weeks 5 and 8, respectively, from 13 or 8 assembloids derived from 2 hiPS cell lines and 1 differentiation; χ2 test, p = 0.06).
(N) Representative traces of whole-field muscle displacement shown after normalization to the pre-stimulation baseline in hCS-hSpS-hSkM assembloid (five consecutive 68-ms pulses) at 5 and 8 weeks post-assembly.
(O) Optogenetic stimulation coupled with calcium imaging in hCSAAV-Chrim-hSpS-hSkMACTA1::GCaMP6s at 4–5 and 7–8 weeks post-assembly. Five consecutive pulses of light (625 nm, 100 ms in duration each and 10 s apart) were delivered.
(P) Average ACTA1::GCaMP6s signal aligned to the time of the pulse in stimulations of numbers 1, 3, and 5 in hCS-hSpS-hSkM assembloids at 4–5 and 7–8 weeks after assembly.
(Q) GCaMP6s ΔF/F amplitudes plotted for stimulation numbers 1–5 normalized to the first stimulation (n = 22 cells from 4 assembloids from 1 differentiation at 4– 5 weeks, and 13 cells from 3 assembloids from 1 differentiation at 7–8 weeks; two-way repeated-measures ANOVA with Tukey multiple comparisons: *p = 0.1, **p = 0.003).
Scale bars, 200 nm (F), 1 µm (C and E), 2 µm (B), 5 µm (D, J, and inset in I), 20 µm (I and insets in H), and 200 µm (H)
Next, we probed the ability of hCS-hSpS-hSkM assembloids to maintain functional connections over 8 weeks post-assembly. For this, we generated hCSAAV-Chrim-hSpS-hSkM assembloids and performed optogenetic stimulations at 5 and 8 weeks (Figure 7K). We found that hSkM contracted after light stimulation at both time points, although the success rate for contracting assembloids at 8 weeks tended to be decreased (50% versus 81% success rate at 8 and 5 weeks, respectively; p = 0.06; Figure 7L). Interestingly, however, assembloids at 8 weeks tended to be more likely to repeatedly contract following five consecutive stimulations (Figures 7M and 7N; p = 0.06), suggesting a more robust response after long-term culture. To investigate this further, we imaged calcium activity using GCaMP6s in hSkM fibers following optogenetic stimulation at 4–5 or at 7–8 weeks (Figure 7O). Light-induced hCS activity elicited calcium spikes in hSkM at both 4–5 and 7–8 weeks, but although hSkM fibers in assembloids respond to the first stimulation at both ages, only at the later time point cells are able to maintain a robust response after 5 stimulations (Figures 7P and 7Q; p = 0.003 for Stim 5).
Taken together, these results demonstrate that the assembly of intact 3D human cultures resembling the cerebral cortex, hindbrain/spinal cord and skeletal muscle enables the formation of neural circuits that can be readily manipulated over several weeks in vitro to model cortical control of muscle contraction.
DISCUSSION
Cell reprograming and the subsequent derivation of human neurons from patients holds great promise for studying brain disorders (Amin and Paşca, 2018; Dolmetsch and Geschwind, 2011; Han et al., 2011). However, modeling of complex cell-cell interactions and circuit assembly in vitro remains a challenge (Paşca, 2018, 2019). We previously showed the potential of using region-specific spheroids as a modular approach to study human interneuron migration and identify disease phenotypes (Birey et al., 2017). Here, we leveraged this approach to generate an intact, three-component cortico-motor circuit in which cortical neural activation controls muscle contraction via activation of motor neurons. This represents a significant advance beyond prior models in which individual components of motor circuits have been generated in 3D in isolation or co-cultured in 2D (Duval et al., 2019; Ogura et al., 2018; Sances et al., 2016; Shi et al., 2018; Steinbeck et al., 2016). First, we used human-derived components that are assembled in a self-organizing 3D preparation, and we achieved this reliably using multiple hiPS cells lines. Second, we built assembloids from intact, 3D cultures that have been patterned to resemble specific regions of the central nervous system. Specifically, we have generated hSpS that contain a large diversity of spinal neuronal domains and shown they resemble in vivo neuronal populations in mouse. This allows greater cell diversity while leveraging the ability of specific neural populations to form human circuits. Third, we showed the generation, from three components, of a functional human neural circuit that contains at least two synaptic contacts. Moreover, we implemented a series of neuroscience tools and showed that this system can be used for tracing connectivity using a rabies virus, as well as for probing and manipulating neural activity using calcium imaging, glutamate uncaging, and optogenetic stimulation. Finally, we showed that this three-part assembloid can be maintained functionally and morphologically intact for up to 10 weeks in vitro. This feature may allow for further refinement and maturation of connections, as well as myelination, which could facilitate the study of cellular phenotypes of neurodegenerative disorders.
Moving forward, there are a number of potential applications for this cellular platform that could be used to gain insights into the evolution, development, and disorders of the cortico-spinal-muscle circuit. For instance, primates possess direct monosynaptic cortico-spinal connections that control fine motor movements (Lemon, 2008; Sousa et al., 2017), and a better understanding of the underlying developmental mechanisms of assembly could bring insights into the unique primate aspects of this circuit. Moreover, patient-derived cells could be used to dissect cell-autonomous effects and cell-specific vulnerabilities in the context of amyotrophic lateral sclerosis or spinal muscular atrophy (Garden and La Spada, 2012). Co-culture with autologous immune cells may reveal the cellular cross-talk underlying multiple sclerosis and other autoimmune conditions. We also envision that, once assembled, this three-way system could be physically perturbed at various levels (cortical, hindbrain-cervical) to develop a human cellular model of spinal cord injury (SCI) (Thiry et al., 2020) and to investigate regeneration at early stages of development. In the context of SCI, these studies could also explore damage affecting the phrenic nerve that controls the thin diaphragmatic skeletal muscle. In addition, assembly with other brain regions such as dorsal root ganglia neurons or midbrain spheroids, could be used to model sensory input (Koch et al., 2018; Levine et al., 2012) or rubrospinal tract connectivity (Cregg et al., 2020). Ultimately, assembloids of various parts of the CNS could bring insights into understanding assembly of different types of human circuits and into identifying therapeutic strategies.
Limitations of Study
These applications would greatly benefit from further improvements such as the generation of more caudal spinal cord cell types (Faustino Martins et al., 2020; Gouti et al., 2014), the presence of organizing centers or implementation of other strategies to improve progenitor domain organization as illustrated by Duval et al. (2019) and Ogura et al. (2018), and the assessment of ascending projections. Moreover, further optimization of optogenetic approaches with cell-type-specific reporters could better capture connectivity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sergiu P. Paşca (spasca@stanford.edu).
Materials Availability
This study did not generate unique reagents.
Data and Code availability
Processed gene expression data are available in the Gene Expression Omnibus under accession number GEO: GSE123722.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Culture of hiPS cells
The hiPS cell lines used in this study were validated using standard methods as previously described (Paşca et al., 2011; Sloan et al., 2018). A total of nine hiPS cell lines derived from fibroblasts collected from eight healthy subjects were used for experiments (see Table S1 for details of the hiPS cell lines used for each experiment). hiPS cell lines TUBA1B-mEGFP and LMNB1-mEGFP were derived by the Allen Institute and obtained from Coriell. The CAG::EGFP hiPS cell lines was genetically engineered in the lab using the line #CW30261 obtained from Coriell. The sex of each of the hiPS cell lines is indicated in Table S1. Cultures were tested and maintained mycoplasma free. hiPS cells were cultured on inactivated mouse embryonic fibroblast feeders (EmbryoMax PMEF) in hiPS cell medium containing DMEM/F12, knockout serum (20%), non-essential amino-acids (1:100, Life Technologies), GlutaMax (1:200, Life Technologies), β-mercaptoethanol (0.1mM, Sigma-Aldrich), penicillin and streptomycin (1:100, Life Technologies), and supplemented with FGF-2 (10 ng ml−1; R&D Systems). hiPS cell lines used for the generation of skeletal myoblasts and the CAG::EGFP hiPS cell line were maintained and passaged as previously described (Yoon et al., 2019). Briefly, hiPS cells were maintained in 6-well plates coated with recombinant human vitronectin (VTN-N, Life Technologies, A14700) in Essential 8 medium (Life technologies, A1517001). To passage hiPS colonies, cells were incubated with 0.5 mM EDTA for 7 minutes at room temperature, resuspended in Essential 8 medium and distributed in new 6-well plates. Approval for using these lines was obtained from the Stanford IRB panel and informed consent was obtained from all subjects.
Human primary brain tissue
Human brain specimens were obtained under a protocol approved by the Research Compliance Office at Stanford University. PCW17 forebrain tissue was delivered overnight on ice and immediately processed after arrival. Cortical tissue was fixed overnight in 4% PFA, washed three times with PBS and embedded as described below.
METHOD DETAILS
Variability terminology
There are three levels of variability that we accounted for in our study. These are outlined below, together with the steps we have taken to describe it in this study:
Line-to-line variability: hiPS cell lines derived from different individuals; graphs are color-coded so that each cell line used in the study is represented by one color.
Experiment-to-experiment (or differentiation) variability: a differentiation experiment represents a batch of hiPS cells that are differentiated in parallel; graphs are separated per differentiation experiment.
Spheroid-to-spheroid or assembloid-to-assembloid variability: values for individual spheroids or assembloids are shown in graphs, unless specified otherwise.
Table S1 shows details on how many and which hiPS cell lines were used for each experiment, as well as the number of differentiations the data is generated from, and from how many spheroids or assembloids. This information can also be found in the corresponding Figure legends. RT-qPCR data is generated from 2–3 spheroids pooled per differentiation.
Generation of hCS and hSpS from hiPS cells
The generation of hCS from hiPS cells was performed as previously described (Birey et al., 2017; Paşca et al., 2015; Sloan et al., 2018). Briefly, hiPS cell colonies were lifted from the plates using dispase (0.35 mg ml−1) and transferred into ultra-low attachment plastic dishes (Corning) in hiPS cell medium supplemented with the two SMAD inhibitors dorsomorphin (5 µM; Sigma-Aldrich, P5499) and SB-431542 (10 µM; Tocris, 1614). This medium was replaced daily for the first five days. On the sixth day in suspension, neural spheroids were transferred to neural medium containing neurobasal-A (Life Technologies, 10888022), B-27 supplement without vitamin A (Life Technologies, 12587010), GlutaMax (1:100, Life Technologies), penicillin and streptomycin (1:100, Life Technologies) and supplemented with the growth factors EGF (20 ng ml−1; R&D Systems, 236-EG) and FGF-2 (20 ng ml−1; R&D Systems, 233-FB) until day 24. From day 25 to 42, the neural medium was supplemented with the growth factors BDNF (20 ng ml−1; Peprotech, 450–02) and NT3 (20 ng ml−1; Peprotech, 450–03) with medium changes every other day. From day 43 onward hCS were maintained in neural medium with medium changes every four to six days.
To generate hSpS, hiPS cell medium was supplemented with dual SMAD inhibitors until day 5 and the WNT activator CHIR 99021 (3 µM; Selleckchem, S1263) from day 4 to day 18. On day 6, spheroids were transferred to neural medium supplemented with RA (0.1 µM; Sigma-Aldrich, R2625), EGF (20 ng ml−1; R&D Systems) and FGF-2 (10 ng ml−1; R&D Systems), with addition of the SHH modulator smoothened agonist (SAG, 0.1 µM; Millipore, 566660) from day 11. From day 7, the medium was changed every other day. On day 19, hSpS were transferred to neural medium supplemented with N-2 supplement (Life Technologies, 17502048), BDNF (20 ng ml−1, Peprotech), IGF-1 (10 ng ml−1; Peprotech, 100–11), L-Ascorbic Acid (AA, 200 nM; Wako, 321–44823) and cAMP (50 nM; Sigma-Aldrich, D0627). For hSpS, the Notch pathway inhibitor DAPT (2.5 µM; STEMCELL technologies, 72082) was added on days 19, 21 and 23. From day 43 onward, the medium was changed every four to five days. For the generation of hSpS with the CAG::EGFP hiPS cell line, we used AggreWell 800 (STEMCELL Technologies, 34815) containing 300 microwells as previously described (Yoon et al., 2019). From days 1 to 6 Essential 6 medium supplemented with dorsomorphin (2.5 µM, Sigma-Aldrich) and SB-431542 (10 µM, Tocris) was used, with all other steps performed as described above. A schematic detailing the hSpS recipe is shown in Figure S2A.
For the combinatorial growth factor matrix, small molecules were added on the same days as described above, and the concentrations for dual SMAD inhibitors, CHIR 99021 and EGF were the same as above. Concentrations tested for RA, FGF-2 and SAG are shown in Figure S1C. From day 7, the neural medium was changed every other day until day 20, when spheres were collected. No DAPT was added for this experiment.
Generation of CAG::EGFP hiPS cell line
The parental hiPS cell line was obtained from Coriell (#CW30261) and was maintained in 6-well plates using StemFlex medium (Life Technologies, A3349401). Cas9, gRNA and donor plasmids were obtained from Addgene (plasmids #42230, #41818 and #52344, respectively). For nucleofection 3 μg Cas9, 1 μg gRNA and 1 μg donor plasmids were used. On the day of nucleofection, hiPS cells were washed with DPBS and incubated with 1 mL Accutase at 37°C for 10 min, after which 9 mL Essential 8 Medium was added to the well for resuspension. After cell counting, the single cell suspension containing 3 ×106 cells was centrifuged. The cell pellet was used for nucleofection using the P3 Primary Cell 4D-NucleofectorTM X Kit L (Lonza, V4XP-3024), a 4D-nucleofector core unit and X unit (Lonza) using the nucleofection program DC100. After nucleofection, the cells were immediately seeded into a well of a 6-well plate that was precoated with vitronectin and contained pre-warmed Essential 8 Medium supplemented with the ROCK inhibitor Y-27632 (10 µM; Selleckchem, S1049). While the nucleofected cells recovered to reach 70%–80% confluency, 1 μg ml−1 of puromycin was applied for 5 days, after which the media was switched back to StemFlex media. Puromycin-resistant clones became visible after 7 days. Clones were pooled together, expanded, cryopreserved and later sorted into 96-well plates to ensure single clone formation by seeding one cell per well. The sorting was performed on a BD Aria II (Stanford Shared FACS Facility). The clone used in this study was selected based on its morphology and uniform expression of GFP. Validation of hiPS genome integrity was performed by high density SNP arrays.
Generation of hCS-hSpS assembloids
To generate cortico-spinal (hCS-hSpS) assembloids, hCS and hSpS were generated separately, and later assembled by placing them in close proximity with each other in 1.5 mL microcentrifuge tubes for 3 days in an incubator. The neural medium used for assembly was supplemented with BDNF (20 ng ml−1; Peprotech), NT3 (20 ng ml−1; Peprotech), L-Ascorbic Acid (AA, 200 nM; Wako) and cAMP (50 nM; Sigma-Aldrich). Media was carefully changed on day 2 after assembly, and placed in a 24-well ultra-low attachment plate (Corning, 3473) using a cut P1000 pipette tip on the third day. Medium was changed every 3–4 days thereafter. Assembly was performed between days (D) 60 and D120 of hCS and between D30 and D50 of hSpS. For hCS-hCS assembloids, one hCS was D60-D75 and the second hCS was D45 (to match the hSpS age).
Culture of hSkM
Human skeletal myoblasts (hSkM) were obtained from Thermo Fisher Scientific (A12555, Lot# 1837192) and maintained in an undifferentiated state with Skeletal Muscle Cell Growth Medium (ready to use; Promocell, C23060) in 10-cm plates (Primaria Cell Culture Dish, Corning). Medium was changed every 2–3 days, and hSkM were passaged using Trypsin (Trypsin-EDTA, 0.25%, phenol red; Life Technologies) when they reached ~80% confluency. hSkM from passages 1 to 4 were used for experiments. For analysis of hSkM differentiation ability, hSkM were plated on wells of 24-well plates (Corning) that had been coated with Geltrex™ (1:50 diluted in DMEM/F12, 1 hour at 37°C; Life Technologies, A1413202). 30,000 hSkM were plated on day 0 in Skeletal Muscle Cell Growth Medium. Medium was replaced the day after plating and every other day after that. When hSkM reached ~90% confluency (2–3 days after plating), Skeletal Muscle Cell Growth Medium was replaced with Skeletal Muscle Cell Differentiation Medium (ready to use; Promocell, C23061). On days 0, 5 and 15, hSkM were washed with PBS once and fixed for 10 minutes with 4% paraformaldehyde (PFA).
Generation of 3D hSkM
For the generation of 3D hSkM cultures, hSkM were dissociated using Trypsin (Trypsin-EDTA, 0.25%, phenol red; Life Technologies) and resuspended in Geltrex™ (Life Technologies) at a density of 3,000 hSkM per μl. Fifty ml of this viscous cell suspension were aliquoted into silicone wells (Ibidi, 80369) located inside 6-well tissue culture plates (Corning), and incubated for 30 minutes at 37°C to allow Geltrex™ gelling, at which point 4 mL of Skeletal Muscle Cell Growth Medium was added. The next day, silicone wells containing hSkM were placed into 6-well ultra-low attachment plates, and medium was changed every 2–3 days. After 7–10 days, medium was changed to Skeletal Muscle Cell Differentiation Medium to allow for differentiation of hSkM with medium changes every 2–3 days. For some experiments, including ACTA1::GCaMP6s imaging, smaller 3D hSkM were generated by resuspending 24,000 cells in 10 μL of Geltrex™ and aliquoting them in small silicone wells (Ibidi, 80409). 3D hSkM were used for assembloid generation 10 to 25 days after the switch to differentiation medium. Figures 5F and S6H show pictures of the 3D hSkM set-up.
Generation of ihSkM and 3D ihSkM
Generation of ihSkM was performed as described in Chal et al., 2016 with some modifications. On day –2, hiPS cells at 65%–80% confluency were detached and plated at a density of 1 ×105 cells per well in 12-well plates coated with Matrigel (1:25; Corning, 354230) and Essential 8 medium (Life technologies, A1517001) supplemented with Y-27632 (10 µM; Selleckchem, S1049). From day 0–3, DMEM/F12 medium (Life Technologies, 11330032) supplemented with 1% Non-Essential Amino Acids (NEAA; Life Technologies), 1% Insulin-Transferrin-Selenium (ITS; Life Technologies, 25–800-CR), 1% penicillin and streptomycin (Life Technologies), and small molecules CHIR 99021 (3 µM; Selleckchem) and LDN-193189 (500 nM; Selleckchem, S7507) was changed daily. From day 3–6, FGF-2 (20 ng ml−1; R&D Systems) was added in addition to CHIR 99021 and LDN-193189 as described above. From day 6 until sub-culturing, DMEM/F12 with 15% knockout serum (KSR, Life Technologies), 1% penicillin and streptomycin (Life Technologies), 1% Non-Essential Amino Acids (NEAA; Life Technologies), and β-mercaptoethanol (0.1 µM; Sigma-Aldrich) was used. From day 6–9 the basal medium was supplemented daily with LDN-193189 (500 nM; Selleckchem), recombinant FGF-2 (20 ng ml−1; R&D Systems), recombinant HGF (10 ng ml−1; Peprotech, 315–23) and recombinant IGF-1 (2 ng ml 1; Peprotech). From day 9–11 cultures were supplemented only with 2 ng ml−1 IGF-1. From day 12 onward, media was supplemented with 2 ng ml−1 IGF-1 and 10 ng ml−1 HGF every other day. Cultures were sub-passaged or sub-cultured at day 33–34. For sub-culture, four wells of a 12-well plate were washed with DPBS twice and then incubated with 1 mL of TrypLE Express Enzyme each (GIBCO, 12604013) for 9–10 minutes at 37°C. Following incubation, cells were detached, transferred to a 50 mL conical tube with 20 mL of DMEM/F12 with 10% FBS and centrifuged. Cells were then resuspended in Skeletal Muscle Cell Growth Medium (Promocell) and passed through a 70 µm strainer. Cells were resuspended in Skeletal Muscle Cell Growth Medium supplemented with 10 µM Y-27632 and plated at a density of 2.5 ×105 per well in Matrigel-coated 12-well plates. Medium was changed the following day. These cells, referred as P2, could be maintained and expanded in Skeletal Muscle Cell Growth Medium with daily medium changes and passages every 2–4 days. Starting at P4 cells could be cryopreserved or used for FACS.
The FACS purification of myoblasts was performed as described in Pakula et al. (2019). Briefly, cells were detached using TrypLE Express, resuspended in Hank’s Balanced Saline Solution (HBSS; Life Technologies) supplemented with 5% FBS, and counted. The cell suspensions were incubated with APC anti-CD56 and/or PE anti-CD82 at a concentration of 5 mL per 1×106 cells (BioLegend, 318309 and 342103, respectively) for 30 minutes on ice, washed with HBSS/FBS and pipetted through the strainer cap of a 5 mL round-bottom FACS tube. Samples were then sorted on a BD Aria II instrument (Stanford Shared FACS Facility) into Skeletal Muscle Cell Growth Medium, and plated on Matrigel-coated 12-well plates for further expansion.
The generation of 3D ihSkM was performed as described above for hSkM. Briefly, 2.5–3.5 ×105 ihSkM were seeded into silicone wells (Ibidi, 80369) with 50 μL of Geltrex™ (Life Technologies) and maintained in Skeletal Muscle Cell Growth Medium with medium changes every 1–2 days. After 7–10 days, medium was changed to Skeletal Muscle Cell Differentiation Medium with medium changes every 2–3 days.
Generation of hSpS-hSkM and hCS-hSpS-hSkM
To generate neural-muscle assembloids, 3D hSkM that had been in differentiation medium for at least 10 days (see above) were removed from the silicone wells and placed on top of cell culture inserts (0.4 µm pore size; Corning, 353090) that were positioned in 6-well plates containing 2 mL of DMEM/F12 medium supplemented with 1% Non-Essential Amino Acids (NEAA; Life Technologies), 1% Insulin-Transferrin-Selenium (ITS; Life Technologies), 1% penicillin and streptomycin (Life Technologies), L-Ascorbic Acid (AA, 200 nM; Wako) and cAMP (50 nM; Sigma-Aldrich). Next, spheroids (either hSpS or hCS) were placed on the inserts containing 3D hSkM (or ihSkM) and arranged so that they were in contact with one another and were allowed to interact. For hCS-hSpS-hSkM assembloids, hSpS-hSkM was assembled first, and hCS was added 1–2 days later. For this combination, sometimes more than one (1–2) hSpS were added. hSpS tend to be smaller in size than hCS, and adding more than one hSpS avoids hCS being in direct contact with hSkM. Only one assembloid was maintained per insert, and half medium changes were performed every other day. Figure 5H shows a schematic detailing this set-up, and Figures 5F and S6H show pictures of the generation of 3D hSkM and the insert set-up.
Imaging of 3D hSkM spontaneous contractions was performed under environmentally controlled conditions (37°C, 5% CO2) using a 5x objective in a confocal microscope (Leica SP8). Assembloids, still in transwells, were incubated in the environmentally controlled chamber for 20–30 minutes before imaging, and they were imaged for 2 minutes at a frame rate of 14.7 frames/sec. 1–2 fields were imaged per assembloid.
Co-culture of mouse limb and spheroids
For mouse co-culture experiments, timed-pregnant female mice were sacrificed at E11.5, embryos were collected, and limb buds dissected (both forelimbs and hindlimbs were used for this experiment). Limbs and spheroids (at D25) were then assembled together by placing them in close proximity in a 1.5 mL microcentrifuge tube for 3 days in an incubator. One limb and one spheroid were placed per tube. On day 2 medium was carefully changed. Neural medium supplemented with N-2 supplement (Life Technologies, 17502048) was used. After assembly, mouse-spheroid cultures were placed in 24-well ultra-low attachment plates (Corning), and medium was changed every other day. For contraction quantification, assembloids were visualized using brightfield illumination in an EVOS FL Cell Imaging System (Life Technologies), and they were deemed to be contracting if they moved within a time-window of 30 s. Approval for mouse experiments was obtained from the Stanford University’s Administrative Panel on Laboratory Animal Care (APLAC).
Viral labeling and rabies-∆G tracing
Viral labeling of neural spheroids was performed as previously described (Sloan et al., 2018). In brief, spheroids were placed in a 1.5 mL microcentrifuge tube containing 250 mL neural medium with the desired virus and incubated overnight. Fresh medium was added the following day, and spheroids were transferred to ultra-low attachment plates (Corning) the next day. The viruses used for this study are: AAV-DJ-hSYN1::eYFP, lenti-Hb9::GFP or lenti-Hb9::mCherry (Nakano et al., 2005), lenti-ACTA1::GCaMP6s (generated by VectorBuilder) (Brennan and Hardeman, 1993), rabies-∆G-Cre-eGFP, AAV-DJ-EF1a-CVS-G-WPRE-pGHpA (Addgene, Plasmid #67528) (Wertz et al., 2015), AAV-DJ-DIO-mCherry, AAV-1-hSYN1-ChrimsonR-tdT (Addgene, #59171-AAV1), AAV-1-syn-jGCaMP7s-WPRE (Addgene, Plasmid #104487-AAV1, gift from Douglas Kim & Genie Project) and AAV-8-CAG-FLEX-Rabies G. Lentivirus was generated in-house by transfecting HEK293T cells with Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) and concentrating the supernatant with Lenti-X concentrator (Clontech, 631232) 72 h later. AAVs were generated at the Stanford Gene Vector and Virus Core at Stanford University School of Medicine or acquired from Addgene. Rabies-∆G viruses were obtained from the Salk institute Viral Vector Core.
For viral tracing experiments with rabies-∆G in hCS-hSpS assembloids, ~d82–88 hCS were labeled with AAV-DJ-DIO-mCherry and ~d46–54 hSpS separately labeled with both rabies-∆G-Cre-eGFP and AAV-DJ-EF1a-CVS-G-WPRE-pGHpA. Six to seven days after viral infection, hCS and hSpS were thoroughly washed with neural medium, assembled, and maintained in culture with media changes every 3–4 days. For viral tracing experiments with rabies-∆G in hSpS-hSkM assembloids, 3D hSkM were infected with rabies-∆G-Cre-eGFP and AAV-8-CAG-FLEX-Rabies G, thoroughly washed, assembled one week later with d37 hSpS, and maintained in culture with half medium changes every other day. After 31 days of fusion (hCS-hSpS) or 18 days of fusion (hSpShSkM), assembloids were fixed with 4% paraformaldehyde and processed for immunocytochemistry as described below. Only assembloids with at least 10 GFP+ cells were included in the analysis. Widespread infection of 3D hSkM with rabies-∆G-Cre-eGFP and AAV-8-CAG-FLEX-Rabies G was challenging, which resulted in some assembloids having fewer GFP cells for quantification.
Projection imaging in intact hCS-hSpS
The projection of hCS-derived AAV-DJ-hSYN1::eYFP into hSpS was imaged under environmentally controlled conditions (37°C, 5% CO2) in intact, assembled hCS-hSpS using a confocal microscope with a motorized stage (Leica SP8). Assembloids were transferred to a glass-bottom 96-well plate (Corning) with 200 mL of neural medium, and incubated in the environmentally controlled chamber for 20–30 minutes before imaging. Images were taken using a 10x objective to capture the entire hSpS side at a depth of 50–150 µm. For long-term live imaging of hCS-derived AAV-DJ-hSYN1::eYFP, the same set-up was used, and hCS-hSpS were imaged for 8–12 hours at a rate of 10 min per frame.
hSkM Cal-590 calcium imaging
For calcium imaging co-culture experiments, hSkM were plated on 24-well plates as described above and differentiated with Skeletal Muscle Cell Differentiation Medium. After 6–7 days of exposure to differentiation medium, hSkM were co-cultured with hSpS or hCS. Skeletal Muscle Cell Differentiation Medium was replaced with DMEM/F12 supplemented with 1% Non-Essential Amino Acids (NEAA; Life Technologies), 1% Insulin-Transferrin-Selenium (ITS; Life Technologies), 1% penicillin and streptomycin (Life Technologies), L-Ascorbic Acid (AA, 200 nM; Wako) and cAMP (50 nM; Sigma-Aldrich). hSpS or hCS were placed in the middle of the 24-well plate, taking care not to disrupt the hSkM. One hSpS or hCS were placed per well. hSkM-spheroid co-cultures were left undisturbed for two days, and half medium was carefully replaced every other day thereafter. Calcium imaging was performed after 6–7 days of co-culture. Cultures were incubated with Cal-590 AM (10 µM; AAT Bioquest, 20510) and PowerLoad (1:100; Invitrogen, P10020) for 30 minutes at 37°C, washed once for 10 minutes with full medium and then imaged. A Leica SP8 confocal microscope with a resonant scanner was used for imaging. Spontaneous calcium activity was recorded for 2 min (10 frames per second) in 6–9 fields per well, and for each field the distance from the spheroid was measured. (+)-tubocurarine chloride pentahydrate (curare; Sigma-Aldrich, 93750) was used at a final concentration of 100 µM.
Optogenetics and GCaMP imaging in hCS-hSpS
For optogenetic stimulation in hCS-hSpS assembloids, intact hCS-hSpS were placed in a 35 µm Glass bottom dish with a 20 µm micro-well #0 cover glass (Cellvis) and imaged using a 10x objective in a confocal microscope (Leica SP8) under environmentally controlled conditions (37°C, 5% CO2). GCaMP7s was imaged at a frame rate of 10 frames/sec. A typical stimulation consisted of ten pulses of light (625 nm, 100 ms in duration each and 100–200 s apart) delivered using an optical fiber-coupled LED (400 µm-diameter, 14 mW/mm2; Thorlabs, M625F2) that was directed toward the hCS. Pulses were generated by a CYCLOPS LED driver coupled with the Leica SP8. NBQX (Tocris, 0373) and APV (Tocris, 0106) were used at a final concentration of 20 µM and 50 µM each.
Glutamate uncaging and optogenetics in hCS-hSpS-hSkM
Intact assembloids were imaged under environmentally controlled conditions (37°C, 5% CO2) using a 5x or 10x objective in a confocal microscope (Leica SP8). Assembloids, still in transwells, were incubated in the environmentally controlled chamber for 20–30 minutes before imaging. For glutamate uncaging experiments, MNI-caged-L-glutamate (Tocris, 1490) was used at a final concentration of 3.3 µM in culture medium (see above). The FRAP software module of the Leica SP8 confocal microscope was used to uncage glutamate using UV light (405 nm). At a frame rate of 14.7 frames/sec, a typical stimulation experiment consisted of 500 frames acquired during pre-stimulation, 2–5 frames of UV stimulation (in specified region of interest, ROI) and 200 frames acquired during post-stimulation. For optogenetic stimulation, five consecutive pulses of light (625 nm ± 5 nm, 68 or 100 ms in duration each and ~6.8 or ~10 s apart) were delivered using an optical fiber-coupled LED (400 µm-diameter, 14 mW/mm2; Thorlabs) that was directed toward the hCS. Pulses were generated by a CYCLOPS LED driver (Open Ephys) coupled with the Leica SP8. (+)-tubocurarine chloride pentahydrate (curare; Sigma-Aldrich) was used at a final concentration of 100 µM. NBQX (Tocris) and APV (Tocris) were used at a final concentration of 50 µM each.
We note that optogenetic stimulation experiments depend on at least two factors: (i) the duration of light pulses, and (ii) the efficiency of opsin delivery. Experiments in this manuscript were performed with stimulations of 68–100 ms for increased sampling speed and to avoid any light-induced toxicity, yet longer stimulations may yield higher success rates. Moreover, because low opsin expression may result in reduced stimulation success, inspection of hCS AAV-1-hSYN1-ChrimsonR-tdT expression before assembly is recommended.
Cryopreservation and immunohistochemistry
Cryopreservation and immunocytochemistry in hCS and hSpS was performed as previously described (Sloan et al., 2018). Briefly, neural spheroids were fixed in 4% paraformaldehyde (PFA in PBS, Electron Microscopy Sciences) for 2 hours. Early spheroids (>25 days) were fixed for 30 minutes. Fixation was followed by three PBS washes, sucrose cryopreservation (30% sucrose in PBS for 24–48 hours or until samples sank to the bottom), embedding in 1:1, 30% sucrose: OCT (Tissue-Tek OCT Compound 4583, Sakura Finetek) and freezing. For immunocytochemistry, 16 µm thick sections were cut using a cryostat (Leica). PCW17 cryosections were 30 µm thick. Cryosections were then washed with PBS to remove excess OCT, blocked for 1 h at room temperature (10% normal donkey serum (NDS), 0.3% Triton X-100 diluted in PBS), and incubated overnight at 4°C with primary antibodies in blocking solution (Table S1 shows the primary antibodies used in this study). Next day, cryosections were washed with PBS and then incubated with secondary antibodies for 1 h at room temperature. Alexa Fluor secondary antibodies (Life Technologies) diluted in blocking solution at 1:1,000 were used. For neuromuscular junction staining, cryosections were incubated for 30 minutes with anti-bungarotoxin (BTX) conjugated to Alexa Fluor-647 in blocking solution (1:500) after secondary antibody incubation. Following washes with PBS, nuclei were visualized with Hoechst 33258 (Life Technologies). Finally, slides were mounted for microscopy with cover glasses (Fisher Scientific) using Aquamount (Polysciences) and imaged on a Zeiss M1 Axioscope, Keyence fluorescence microscope or Leica TCS SP8 confocal microscope. Images were processed in ImageJ (Fiji) (Schindelin et al., 2012). 3D rendering of neuromuscular junction was performed using the software Imaris. The same procedure was followed for immunocytochemistry of 2D hSkM.
Transmission electron microscopy
hCS-hSpS-hSkM were fixed in 2% glutaraldehyde and 4% PFA in 0.1 M sodium cacodylate buffer (pH 7.4) for 15 minutes at room temperature. After fixation, samples were then moved to 4°C for processing. The fixative was removed and replaced with 1% OsO4 and samples were then allowed to warm to room temperature rotating for 2 hours. Samples were then washed 3 times with ultra-filtered water. After the 3rd rinse, samples were stained in 1% uranyl acetate for 2 hours while rotating. Samples were then dehydrated in a series of ethanol washes for 20 minutes each at room temperature: 50% ethanol, 70% ethanol (at 4°C overnight), 95% ethanol (while allowing to warm at room temperature) and, lastly 100% ethanol. A 2nd 20-minute rinse in 100% ethanol was performed followed by a 15 minute rinse in propylene oxide (PO) at room temperature. Samples were then infiltrated with resin mixed 1:2, 1:1, and 2:1 with PO for 2 hours each, leaving samples in 2:1 resin with PO overnight rotating at room temperature. The following day, the samples were placed in 100% EMbed-812 for 2–4 hours and then placed into molds with fresh resin and placed at 65°C overnight. Sections were picked up on formvar/Carbon coated slot Cu grids, stained for 30 s in 3% uranyl acetate in 50% acetone followed by staining in Sato’s Lead Citrate for 3–5 minutes. Sections were inspected using a JEOL JEM-1400 120kV, and images were taken using a Gatan Orius 832 4k X 2.6k digital camera (9 µm pixel).
Real-time quantitative PCR - qPCR
For qPCR analysis of spheroids, at least 2–3 spheroids were pooled per sample. mRNA was isolated using the RNeasy Mini kit and RNase-Free DNase set (QIAGEN), and template cDNA was prepared by reverse transcription using the SuperScript III First-Strand Synthesis SuperMix for qRT–PCR (Life Technologies). qPCR was performed using SYBR Green (Roche) on a ViiA7 machine (Applied Biosystems, Life Technologies). Primers used are listed in Table S1.
Single cell dissociation
Dissociation of hSpS into single cells for transcriptomics analysis was performed as previously described (Birey et al., 2017; Paşca et al., 2015; Sloan et al., 2017, 2018). Briefly, 8–10 hSpS per sample were chopped and incubated in 40 U/ml papain enzyme solution at 37°C for 90 minutes. After digestion, samples were washed with a protease inhibitor solution and gently triturated to achieve a single cell suspension and strained using a 70 µm Flowmi strainer (Bel-Art).
Single cell gene expression - BD Rhapsody
To capture single cell transcriptomic information of hiPS cell derived hSpS, we used the BD Rhapsody system (formerly known as BD Resolve) (BD Biosciences) as previously reported (Birey et al., 2017; Fan et al., 2015). hSpS with or without DAPT exposure were dissociated enzymatically into single cells at day 45 of differentiation and processed on the same day. Ten spheroids for each condition were combined, the proportion of live cells was estimated using a fluorescent assay (~90%) and all cells were used for further processing. Single-cell suspension of ~10,000 cells were captured from all isolated cells, without selection, on an array of > 200,000 microwells through a limited dilution approach. Beads with oligonucleotide barcodes were added to saturation so that a bead was paired with a cell in a microwell. After exposure to cell lysis buffer, poly-adenylated RNA molecules hybridized to the beads. Beads were retrieved into a single tube for reverse transcription. Upon cDNA synthesis, each cDNA molecule was tagged on the 5′ end (that is, the 3′ end of a mRNA transcript) with a molecular index and cell label indicating its cell of origin. Whole transcriptome libraries were prepared from 40% of the captured cells by subsampling the Rhapsody beads that were then subject to second strand cDNA synthesis, adaptor ligation, and universal amplification using twenty-two cycles of PCR. The rest of the beads were archived. Sequencing libraries were prepared using random priming PCR of the whole-transcriptome amplification products to enrich the 3′ end of the transcripts linked with the cell label and molecular indices. The libraries were sequenced on HiSeq4000 (Illumina) using 101 × 2 chemistry.
The BD Rhapsody analysis pipeline was used to process sequencing data (.fastq files). Cell labels and molecular indices were identified, and gene identity was determined by alignment against the gencode comprehensive hg19 reference. A table containing molecule counts per gene per cell was the output. Gene expression profiles of 4,175 and 3,173 cells were recovered for hSpS with or without DAPT, respectively, with an average number of reads of ~17,000, ~2,400 molecules and ~1,400 number of genes detected per cell with average molecular index coverage (that is, the number of times a molecule was sequenced) of 4.8.
Analysis of the single cell transcriptome profiles was performed with BD™ Data View as we previously described (Birey et al., 2017). Cells with mitochondrial gene (with a gene symbol starting with MT) content > 30%, were discarded, retaining a total of 7,888 cells from both samples. We extracted the expression profiles of the 1,278 genes (Table S1) that define the 10 populations in hSpS and conducted tSNE projection on the filtered data.
Single cell gene expression - 10x Genomics
Dissociated cells were resuspended in ice-cold PBS containing 0.02% BSA and loaded onto a Chromium Single cell 3′ chip (with an estimated recovery of 6,000 cells per channel) to generate gel beads in emulsion (GEMs). scRNA-seq libraires were prepared with the Chromium Single cell 3′ GEM, Library & Gel Bead Kit v3 (10x Genomics, PN: 1000075). Libraries from different samples were pooled and sequenced by Admera Health on a NovaSeq S4 (Illumina) using 150 × 2 chemistry. Demultiplexing, alignment, barcode and UMI counting and aggregation were performed using Cell Ranger (v3.0 with default settings). This provided us with a feature by count matrix. Further analysis was performed using the R package Seurat (v3.0) from the Satija Lab (Butler et al., 2018). We excluded cells with more than 6,000 or less than 1,000 detected genes, as well as those with a mitochondrial content higher than 18%. We excluded genes that were not expressed in at least three cells. Due to sex differences between hiPS cell lines we also removed all genes on the X and Y chromosomes from the count matrix. Gene expression was then normalized using a global-scaling normalization method (normalization.method = ‘‘LogNormalize,’’ scale.factor = 10000, in Seurat), and the 1,500 most variable genes were then selected (selection.method = ‘‘vst,’’ in Seurat) and scaled (mean = 0 and variance = 1, for each gene, as recommended by Seurat) prior to principal component analysis (PCA). The top 18 principal components were utilized to do the clustering with a resolution of 1.1, implemented using the FindNeighbors and FindClusters functions in Seurat. We identified clusters based on expression of known markers (Delile et al., 2019). In some cases, using expression of known markers, we grouped together clusters that were originally separate based on the Seurat clustering, namely the ‘MN’, ‘V2a’, ‘astroglia’ and ‘other’ clusters were composed of 2–4 seurat clusters. Motor neuron subclustering was performed using Seurat as described above using 800 variable genes and the top 10 principal components.
Reference similarity spectrum (RSS) analysis (Kanton et al., 2019) was performed to compare hSpS single cell transcriptomics data to adult mouse brain regions (Zeisel et al., 2018) and developing mouse spinal cord (Delile et al., 2019). Briefly, processed reference data and cell cluster annotations were downloaded from http://mousebrain.org/downloads.html (Zeisel et al., 2018) and https://github.com/juliendelile/MouseSpinalCordAtlas/tree/master/dataset (Delile et al., 2019). Analysis was restricted to non-progenitor cell clusters, neuronal cell clusters, and clusters from central nervous system tissues. For reference datasets, gene expression was averaged for each cell cluster and the top 2000 highly variable genes were identified using the FindVariableFeatures function of the Seurat R package (Stuart et al., 2019) with default parameters. Human ortholog mapping of mouse gene symbols was performed as described in Skene et al. (2018) with the following annotation file: https://github.com/NathanSkene/EWCE/blob/master/data/mouse_to_human_homologs.rda. Pearson correlation between each reference cell cluster and hSpS data (either cell clusters or individual cells) was performed across matched highly variable genes. z-Transformation was then applied to get the normalized similarities.
Electrophysiology
Sections of hSpS (day 45–75) for electrophysiology were obtained as previously described (Sloan et al., 2018). In brief, spheroids were incubated in bicarbonate-buffered aCSF at 23 °C and equilibrated with a mixture of 95% O2 and 5% CO2. The aCSF solution contained: 126 mM NaCl, 26 mM NaHCO3, 10 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgSO4 and 2 mM CaCl2. Slicing was performed using a Leica VT1200 vibratome. Immediately after sectioning, slices were moved to a circulation chamber containing oxygenated aCSF at room temperature.
Patch-clamp recordings were performed from cells expressing the Hb9::GFP fluorescent reporter using an upright microscope (Slicescope, Scientifica). Recording electrodes of borosilicate glass had a resistance of 7–10 MΩ when filled with internal solution. The internal solution contained: 145 mM K-gluconate, 0.1 mM CaCl2, 2.5 mM MgCl2, 10 mM HEPES, 0.2 mM EGTA, 4 mM Na-phosphocreatine. 4/7 cells were able to fire repetitive action potentials.
To evoke APs in hCS axon terminals while recording from Hb9::GFP cells, we performed whole field illumination via the microscope’s 40x objective (Olympus), using a broad-spectrum green-red LED (560 ± 50 nm, CoolLed). Hb9::GFP cells were selected based on the proximity to Chrimson+ fibers. The stimulation duration was set to 5 ms and the stimulation frequency was 0.1 Hz. Chrimson-triggered EPSCs were blocked by bath application of TTX (1 µM; Tocris, 1069).
Data were collected using a 1550A digitizer (Molecular Devices), a 700B patch-clamp amplifier (Molecular Devices) and acquired with pClamp 10.7 software (Molecular Devices). Data were low-pass filtered at 10 kHz and digitized at 20 kHz. Data averaging, digital subtraction of null traces, and current peak detection were performed using clampfit (Molecular Devices).
QUANTIFICATION AND STATISTICAL ANALYSIS
Fluorescence intensity quantification
Fluorescence intensity in cryosections was obtained using the Plot Profile command in ImageJ (Fiji). For each cryosection 3–8 regions were measured.
Projection quantification
hCS-derived AAV-hSYN1::eYFP projections were quantified using ImageJ (Fiji). ROIs were manually drawn to cover the area on the hSpS or hCS to be measured in max projection confocal stacks. Both the brightfield and fluorescent channels were used to draw the ROIs. Following background subtraction (50 rolling ball radius), FeatureJ Hessian filter and contrast enhancement (0.4 saturated), the percentage of YFP+ pixels over total area of hSpS or hCS was calculated in binary images. For the quantification of line length, the ridge detection plugin of ImageJ was used in binary images.
hSkM Cal-590 calcium imaging
Calcium imaging data was processed using ImageJ (Fiji) and custom MATLAB routines. ROIs corresponding to hSkM fibers were automatically generated using the analyze particles plugin on average intensity projections (300 frames) using ImageJ (Fiji). A total number of 798 (hSkM condition), 652 (hCS + hSkM condition), 1006 (hSpS + hSkM condition) and 727 (hSpS + hSkM + curare condition) hSkM fibers were analyzed. Following ROI registration, raw time-series movies were transformed to relative changes in fluorescence: ΔF/F(t) = (F(t)-F0)/F0, where F0 represented the 5th percentile value of the time series of each ROI. To remove slow fluctuations originating from the summation of multiple events, we first high-passed filtered the ROI’s ΔF/F(t) functions (ΔF/F(t)’. Calcium candidate events were detected whenever the ROI’s ΔF/F(t) crossed a threshold of 7 median absolute deviations (MAD). Active hSkM generated at least 1 calcium event over a 2-minute period. Calcium events are typically characterized by a sharp rise followed by slower decay. To capture these features, we only considered events that follow this behavior. Event time was set to the time the event crossed the threshold.
hSkM GCaMP6s analysis
GCaMP6s imaging data was processed using ImageJ (Fiji) and custom MATLAB routines as described above. ROIs corresponding to GCaMP6s positive hSkM fibers were generated using standard deviation projections.
GCaMP7s analysis
GCaMP7s imaging data was processed using ImageJ (Fiji) and custom MATLAB routines. For the purpose of this experiment (comparison of stimulation-triggered responses before and after application of NBQX + APV), only fields where cells appeared to be active at baseline were used. We found four out of ten assembloids to have clear responding cells. A cell’s ΔF/F amplitude was calculated as the maximum ΔF/F value inside a 6 s window after the LED stimulation time, minus the mean of the baseline 1 s before the stimulation. To eliminate shape-dependent artifacts, the median was used instead of the mean. To demonstrate that responses were time-locked to the stimulation and decouple them from the spontaneous firing, we compared the time-locked ΔF/F response to that of random time-locked ΔF/F.
Contraction analysis
Muscle contraction of 3D hSkM was quantified using the automated, open-source ImageJ plugin MUSCLEMOTION (Sala et al., 2018) (https://github.com/l-sala/MUSCLEMOTION). MUSCLEMOTION quantifies movement by subtracting the summed, absolute changes in pixel intensity between a reference frame and the frame of interest. Because each imaging field consists of a large area containing multiple muscle fibers, several fibers may be moving simultaneously and summation of pixel intensities in these cases may result in non-changing summed values. Therefore, to reduce the chance of subtraction of pixel values, each imaging field (1.8 mm by 1.8 mm in size) was divided into 16 subfields and the analysis was performed in each of the subfields individually (Figure S7A) . For the analysis of spontaneous contractions, event detection was performed using custom MATLAB routines. Events over 5 median absolute deviations (MAD) were counted as a contraction event. Correlation between subfields in a field was calculated in MATLAB by computing a non-normalized covariance calculation. The mean covariance per field was plotted.
For the quantification of stimulation experiments of assembloids, pixel intensity analysis was performed with MUSCLEMOTION as described above. Displacement over time was calculated by normalizing all values to 500 frames preceding stimulation. If different fields (i.e., areas) were stimulated per assembloid, then these were plotted separately. If the same field was stimulated more than once, values were averaged and plotted as one point. For optogenetic stimulation, only the first trial per field was used for quantification, and data values resulting from each of the five pulses of light were averaged.
Supplementary Material
(A and B) Schematics illustrating marker gene expression along the rostro-caudal (A) and dorso-ventral (B) axes of the hindbrain and spinal cord.
(C) Schematic detailing the 12 conditions in the combinatorial growth factor matrix and the molecules and concentrations they receive.
(D–F) Gene expression analysis of genes expressed along the rostro-caudal (D) and dorso-ventral (E), (F) axes in the 12 conditions of the combinatorial matrix at day 20 of in vitro differentiation. Each condition shows 3 colored bars representative of n = 3 hiPS cell lines from 1 differentiation (Kruskal-Wallis test p = 0.002 for HOXA2, p = 0.008 for HOXB4, p = 0.003 for HOXC9, p = 0.002 for HOXC9, p = 0.0008 for PAX7, p = 0.02 for PAX6, p = 0.001 for OLIG2, p = 0.002 for NKX6–1,p= 0.0006 for LHX1, p = 0.007 for CHX10, p = 0.06 for HB9; one-way ANOVA p = 0.002 for HOXC5, p < 0.0001 for FOXA2, p < 0.0001 for EVX1, p = 0.01 for ISL1).
(G) Immunocytochemistry in cryosections of hSpS conditions #4 and #8 showing dorsal-like and ventral-like identities. This image was generated by automatic stitching of individual images.
(H) Schematic detailing differentiation conditions used for deriving hSpS.
(I and J) Representative immunocytochemistry images in cryosections of hSpS showing expression of OLIG2 and NKX2.2 (I) or NKX6.1 (J) at day 18 of in vitro differentiation.
(K) Quantification of the total number of OLIG2 cells per area (mm2) in day 18 hSpS (n = 10 hSpS derived from 3 hiPS cell lines from 1 differentiation, with 2 cryosections quantified per hSpS).
(L) Quantification of the percentage (%) of OLIG2 positive cells that co-express either NKX2.2 or NKX6.1 in day 18 hSpS (n = 10 hSpS derived from 3 hiPS cell lines from 1 differentiation, with 2–3 cryosections quantified per hSpS).
Data represent mean ± s.e.m. Scale bars, 50 µm (insets in (I), (J), 100 µm (G), 200 µm (I), (J).
(A) Schematic detailing differentiation conditions used for deriving hSpS with or without DAPT.
(B) Schematic illustrating hindbrain/spinal cord neuronal domains, their corresponding marker genes and their neurotransmitter identities.
(C) t-SNE visualization of single cell gene expression of hSpS with or without DAPT at day 45 of in vitro differentiation (n = 7,888 cells; BD™ Rhapsody system) showing 10 main clusters that include a spinal motor neuron (MN) cluster. List of genes for each cluster is included in Table S1.
(D) Boxplots for neurotransmitter-related genes enriched in each of the single cell clusters (VGAT also called SLC32A1, GLYT2 also called SLC6A5, CHT1 also called SLC5A7, VACHT also called SLC18A3, VGLUT2 also called SLC17A6).
(E) Distribution of cells in conditions hSpS and hSpS (– DAPT) in t-SNE plot. R2 value shows the correlation between the two hSpS conditions (R2 = 0.85, p < 0.0001).
(F) Proportion of cells per condition in each of the ten clusters (χ2 test, hSpS versus hSpS (–DAPT); p < 0.0001).
(G) Gene expression analysis of neuronal cell type markers in hSpS at day 30 of in vitro differentiation in 4 separate differentiations that were either pooled (left) or separated (right; n = samples from 5 hiPS cell lines; two-tailed t test: ****p = < 0.0001 for HB9, ***p = 0.0003 for EVX1; Mann-Whitney test: **p = 0.0014 for SIM1, ****p < 0.0001 for ISL1, ****p = < 0.0001 for GATA3, **p = 0.0045 for CHX10, ****p = < 0.0001 for EN1).
(H) Gene expression analysis of neurotransmitter identity markers in hSpS at day 30 of in vitro differentiation in 4 separate differentiations that were either pooled (left) or separated (right; n = samples from 5 hiPS cell lines; two-tailed t test: ***p < 0.0002 for CHAT, p = 0.7 for VGLUT2; Mann-Whitney test: p = 0.01 for GAD1, ***p = 0.0001 for GLYT2).
(I) Gene expression analysis of glial progenitor markers in hSpS at day 30 of in vitro differentiation in 4 separate differentiations that were either pooled (left) or separated (right; n = samples from 5 hiPS cell lines; Mann-Whitney test: ****p < 0.0001 for FABP7, p = 0.1 for SOX10).
(J) Immunocytochemistry in hSpS and hSpS (– DAPT) cryo-sections at day 45 of in vitro differentiation showing expression of different neuronal domain marker genes.
(K) Immunocytochemistry in hSpS and hSpS (– DAPT) cryo-sections at day 45 of in vitro differentiation showing expression of different neuronal and neurotransmitter identity markers.
Data represent mean ± s.e.m. Scale bars, 50 µm (J), 200 µm (K).
(A) t-SNE visualization of single cell gene expression of hSpS at day 53 of in vitro differentiation (n = 9,302 cells; 10x Genomics) showing 12 main clusters that include a spinal motor neuron (MN) cluster. See Figure 1F for visualization using UMAP.
(B) UMAP showing cells in hSpS colored by the hiPS cell line they were derived from.
(C) Plots showing the Pearson correlation of the normalized average gene expression, log (counts per 10,000) between each of the three hiPS cell lines used for this experiment. R2 shows the Pearson correlation value for each.
(D) Heatmap showing the Pearson correlation of the ventral, dorsal, progenitor, astroglia and other domains between hiPS cell lines used for this experiment. Hierarchical clustering of sample correlations is displayed with a dendogram.
(E) Dot plot showing expression of cell adhesion and neuronal guidance genes in each of the neuronal clusters in hSpS. The size of the circle represents the percent of cells expressing each gene per cluster.
(F) UMAP plots showing average gene expression of selected HOX genes.
(G) Hierarchical clustering showing RSS analysis of hSpS clusters to single-cell RNA-seq clusters from adult mouse central nervous system (CNS) regions in Zeisel et al. (2018). CB, cerebellum; HBCHOL3, hindbrain cholinergic neuron, HBGLU2: hindbrain excitatory neuron; HBINH2, hindbrain inhibitory neuron.
(H) hSpS UMAP plots colored by RSS to selected single cell RNaseq clusters from adult mouse CNS from Zeisel et al. (2018).
(I and J) Schematic illustrating hindbrain/spinal cord motor columns (I) and their corresponding marker gene expression (J). SAC, spinal accessory column; PMC, phrenic motor column; LMC, lateral motor column; MMC, medial motor column; PGC, preganglionic column; HMC, hypaxial motor column. Figure adapted from Dasen and Jessell (2009) and Stifani (2014).
(K) UMAP plot showing sub-clustering of the motor neuron cluster.
(L) Heatmap showing the top 20 differentially expressed genes in each motor neuron cluster (MN 1–6).
(M) UMAP plots showing average gene expression of selected markers, including neurotransmission-related genes, column-specific genes, cell adhesion genes and HOX genes.
(A) Representative immunohistochemistry images in cryosections of day 25 hSpS showing expression of post-mitotic neurons expressing MAP2 at the edge of hSpS and proliferating progenitors expressing SOX2 and KI67 in the center.
(B) Representative immunohistochemistry images in cryosections of day 25 hSpS showing expression of post-mitotic motor neurons expressing PHOX2B at the edge of hSpS and medial-dorsal progenitors expressing PAX6 in the center.
(C and D) Immunocytochemistry in cryosections of hSpS showing motor neuron markers at day 30 of in vitro differentiation.
(E and F) Representative immunohistochemistry images in cryosections of day 45 hSpS showing expression of motor neuron markers ISL1, HB9 (F) and PHOX2B (G).
(G) Representative immunohistochemistry images in cryosections of day 45 hSpS showing localization of spinal neuronal domain V2a represented by CHX10 and LHX3 glutamatergic cells.
Scale bars, 10 µm (C), 20 µm (A), (B), (D), insets in (E), (F), (G), 200 µm (E), (F), (G).
(A) t-SNE visualization of single cell gene expression of hCS and hSS (human subpallial spheroids) at day 105 of differentiation (n = 11,838 cells; BD™ Resolve system, from Birey et al. [2017]) showing the distribution of expression of a set of genes associated with cortico-spinal neuronal identity (SOX5, FEZF2, BCL11B or CTIP2, BHLHE22, LDB2, CRIM1 and OTX1).
(B) t-SNE visualization of single cell gene expression of day 105 hCS and hSS showing expression of CHT1 (also called SLC5A7) and VACHT (also called SLC18A3).
(C) Gene expression analysis of cortico-spinal-related genes in hCS over time (n = 4–6 hiPS cell lines from 1–2 differentiations; one-way ANOVA interaction: p = 0.001 for FEZF2, p < 0.0001 for BCL11B, p = 0.5 for OTX1; Kruskal-Wallis test interaction: p = 0.002 for SOX5).
(D and E) Immunocytochemistry in hCS cryosections at day 130 of in vitro differentiation showing expression of cortico-spinal markers CTIP2 and OTX1 (D). Antibodies were validated in slices of human cortical tissue at post-conception week 17 (PCW17, E).
(F) Quantification of hCS-derived eYFP fluorescence coverage in hSpS areas in hCS-hSpS assembloids (at 20 daf) with samples separated by differentiation (n = 3 hiPS cell lines; one-way ANOVA interaction: p = 0.3). Figure 3D shows data combined.
(G) Images of intact hCS-hSpS assembloids 20 days after fusion (daf) showing hCS-derived hSYN1::eYFP projections.
(H and I) Representative image of intact hCS-hCS assembloid (H) and quantification of the extent of projection of hSYN1::eYFP (I) (n = 3 hiPS cell lines from 1–2 differentiations; Kruskal-Wallis test interaction p = 0.01, with Dunn’s multiple comparison test: p > 0.9 for 10 versus 5 daf, p = 0.06 for 20 versus 5 daf). See Figure 3D for quantification of the projection in hCS-hSpS assembloids.
(J) Representative images of intact hCS-hSpSCAG::EGFP assembloids showing some reciprocal projections from hSpS to hCS and limited cell migration.
(K) Representative image of intact hCS-hSpS assembloid where hCS was infected with AAV-hSYN1::eYFP and hSpS with lenti-Hb9::mCherry. No hSpS-derived Hb9::mCherry projections are observed in hCS (similar results were observed in 15 assembloids from 2 differentiations and 3 hiPS cell lines).
(L) Representative immunocytochemistry image of hCS-hSpS assembloid in retrograde viral tracing experiment at 31 daf showing GFP expression in hSpS and colocalization of GFP and mCherry in hCS. mCherry projections from hCS to hSpS can also be seen.
(M) Representative immunocytochemistry image of GFP, mCherry and the glial marker GFAP on the hCS side of hCS-hSpS assembloid at 31 daf.
(N and O) Quantification of the percentage (%) of GFP+ and mCherry+ cells on the hCS side of hCS-hSpS assembloids that co-express either the neuronal marker MAP2 or the glial marker GFAP (O) or the cortical layer-specific markers CTIP2 or BRN2 (N) separated by differentiation (n = 10 assembloids from 3 hiPS cell lines, with 2–3 cryosections quantified per assembloid). Figures 3J and 3M show quantification with both differentiations combined.
Data represent mean ± s.e.m. Scale bars, 50 µm (D), 100 µm (L), (M), 200 µm (G), (H), (J), (K), 500 µm (E).
(A and B) Characterization of human skeletal myoblast (hSkM) differentiation. hSkM differentiate upon removal of FBS from the culture medium (A). Immunocytochemistry images for desmin (DES), titin (TTN) and the heavy myosin chain (MyHC) show differentiation of myoblasts over time (B).
(C) Distribution and mean number of Hoechst+ nuclei in desmin cells at day 0 and day 15 of hSkM differentiation. Data represent mean ± s.e.m. (n = 1,031 desmin+ cells at day 0, and n = 190 desmin+ cells at day 15; data was collected in each case from 5 fields within a culture well in 2 hSkM differentiation replicates; Mann-Whitney test: ****p < 0.0001).
(D) Representative calcium imaging for hSkM and hSpS-hSkM. Images show average (avg) intensity projections of Cal-590 AM in a field (average intensity projections were used to delineate cells for quantification). Traces show examples of active and inactive hSkM cells in either hSkM alone or hSpS-hSkM cultures. ΔF/F indicates the fluorescence intensity over baseline fluorescence. Arrows show cells analyzed.
(E) Schematic detailing hSkM analyzed for calcium imaging experiment. Spontaneous calcium activity was recorded in 6–9 fields per culture well. Of these fields some were within 1 mm of the spheroid, and some were further than 1 mm from the spheroid.
(F) Quantification of spontaneous calcium imaging activity in hSkM over a period of 2 minutes. Fields were separated according to their distance from the spheroid (hCS or hSpS). Data represent mean ± s.e.m. (n = 2 hiPS cell lines from 1–2 differentiations; one-way ANOVA interaction: p = < 0.0001, with Dunnett’s multiple comparison test ****p < 0.0001).
(G) Representative immunocytochemistry image showing BTX binding in hSkM that had been co-cultured with hSpS.
(H) Step number 4 in the generation and assembly of hSpS-3D hSkM. After 2–3 weeks in differentiation medium, 3D hSkM can be co-cultured with hSpS and/or hCS. For this, 3D hSkM and spheroids are placed on insert wells with 2 mL of medium per well.
(I and J) Representative immunohistochemistry images in cryosections for rabies-derived GFP and the cholinergic markers CHAT (I) and GABA (J) on the hSpS side of hSpS-hSkM assembloids at 18 daf.
(K) Quantification of the percentage (%) of GFP positive (+) cells on the hSpS side of hSpS-hSkM assembloids that co-express either ISL1/2, CHX10, FOXP2 or PROX1 (n = 8 to 9 assembloids derived from 3 hiPS cell lines and 1 differentiation, with 3–6 cryosections quantified per assembloid).
(L and M) Representative immunohistochemistry images in cryosections for rabies-derived GFP and either GABA and FOXP2 (L) or PROX1 and CHX10 (M) on the hSpS side of hSpS-hSkM assembloids at 18 daf.
(N) Schematic illustrating the glutamate uncaging approach. Caged glutamate is added to the culture medium and uncaged upon UV light stimulation (405 nm).
(O and P) Glutamate uncaging of hSpS in hSpS-hSkM assembloid (H). Displacement normalized to baseline over time is shown for 3 subfields in the presence (I) or absence (J) of caged glutamate in the medium.
(Q and R) Representative immunohistochemistry in cryosections of hSpS-hSkM assembloids 31 days after fusion showing desmin (DES), BTX, synaptophysin 1 (SYP) and Hoechst.
(S) Single Z-plane and orthogonal views of projection in (R) showing YZ (right) and XZ (bottom) planes of stack and indicating apposition of BTX and SYP. Yellow lines indicate the point in the stack being visualized.
Data represent mean ± s.e.m. Scale bars, 10 µm (Q), (R), (S), insets in (G), 50 µm (I), (J), (L), (M), 100 µm (G), 200 µm (B), (D), (O), 400 µm (A).
(A) Imaging fields (1.8 mm x 1.8 mm in size) are divided into 16 subfields for analysis. Only subfields containing hSkM are analyzed.
(B and C) Representative spontaneous contraction traces in subfields of hSkM or hSpS-hSkM assembloids (B). The correlation of displacements between subfields in a field is quantified using covariance analysis. Heatmaps show representative examples of covariance between subfields in a field of hSkM, hCS-hSkM, hSpS-hSkM or hCS-hSpS-hSkM assembloids (C). Video S4 shows live imaging of spontaneous contractions in an hCS-hSpS-hSkM assembloid.
(D and E) Glutamate uncaging in hCS-hSpS-hSkM assembloids. UV light (405 nm) uncages glutamate specifically on hCS (D). Displacement normalized to baseline over time is shown for 3 subfields without or with addition of curare (100 µM, E).
(F and G) Glutamate uncaging of hCS in hCS-hSpS-hSkM assembloid. Displacement normalized to baseline over time is shown for 3 subfields in the absence of caged glutamate in the medium (see Figures 6F and 6G for glutamate uncaging in this field in the presence of caged glutamate, and see Table S1 for details of all stimulation experiments).
(H) Representative brightfield image showing an intact hCS-hSpS-hSkM assembloid. Figure 6K shows AAV-hSYN1-ChrimsonR-tdT in hCS.
(I and J) Representative example of optogenetic stimulation in hCS-hSpS-hSkM assembloids (five consecutive 68 ms pulses). hCS were infected with AAV-hSYN1-ChrimsonR-tdT (Chrim). Traces of displacement in the whole field in (I) are shown normalized to the pre-stimulation baseline. Two trials for the same assembloid are shown. Addition of NBQX (50 µM) and APV (50 µM) abolished muscle contraction upon light-induced hCS stimulation (J).
(K and L) Optogenetic stimulation in hCS-hSkM assembloid (five consecutive 68 ms pulses). hCS were infected with AAV-hSYN1-ChrimsonR-tdT (K). Displacement over time normalized to pre-stimulation baseline is shown for the whole field in (L). Similar results were obtained in n = 5 hCS-hSkM assembloids derived from 2 hiPS cell lines.
(M) Schematic detailing location of electron microscopy images within hCS-hSpS-hSkM assembloids.
(N–P) Representative images showing axons and synapses in hCS-hSpS-hSkM assembloids with synaptic vesicles (arrow heads), post-synaptic densities (asterisks) and mitochondria (arrows). Inset in (P) shows an axo-axonic synapse.
(Q) Representative images within hSkM in hCS-hSpS-hSkM assembloids showing skeletal muscle fibers with actin and myosin filaments and mitochondria (arrows).
(R and S) Representative images within hSkM in hCS-hSpS-hSkM assembloids showing points of contact between hSpS neurons and hSkM. Neuron terminals are vesicle-laden (arrow heads) and present mitochondria (arrows). hSkM are surrounded by a basal lamina (hollow arrowhead).
(T) Schematic detailing location of images within hCSAAV-Chrim-hSpS-hSkM assembloids.
(U and V) Representative immunohistochemistry images in cryosections of hCSAAV-Chrim-hSpS-hSkM assembloids showing the presence of SMI-32+ motor neurons in hSpS with hCS-derived mCherry-positive projections (U) and astrocytes (V).
Scale bars, 200 nm (inset in P), 1 µm (O), (P), (S), 2 µm (N), (Q), (R), 20 µm (U), (V), 200 µm (A), (F), (H), (I), (K).
(A) Schematic detailing the differentiation condition used for generating hiPS cell-derived skeletal myoblasts. Protocol adapted from Chal et al. (2016).
(B) Schematic indicating marker genes expressed during the in vitro generation of myoblasts from hiPS cells.
(C) Gene expression analysis of skeletal myogenesis markers at days 5/6, 20 and 33/34 of in vitro myoblast differentiation (n = 3 hiPS cells lines from 2–3 differentiations).
(D) Representative images of myoblasts derived from 3 hiPS cell lines 2 to 4 days after subculture at day 33 of in vitro differentiation.
(E) FACS plots showing purification of the CD56+ / CD82+ myoblast population.
(F) Representative images of CD56+ / CD82+ myoblasts under either growth conditions or differentiation conditions.
(G) Representative image of an hSpS-3D ihSkM assembloid.
(H and I) Optogenetic stimulation in hCS-hSpS-ihSkM assembloids. hCS were infected with AAV-hSYN1-ChrimsonR-tdT (Chrim) before assembly. Five consecutive pulses of light (625 nm, 200 ms in duration each and 20 s apart) were delivered using a fiber-coupled LED directed toward the hCS (H). Traces of whole-field muscle displacement are shown after normalization to the pre-stimulation baseline.
Scale bars, 200 µm (D), (G), (H), 210 µm (F).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| All antibodies used for immunohistochemistry in this study, the concentrations used, their source and their identifier are listed on Table S3 | N/A | N/A |
| APC anti-CD56 | BioLegend | Cat# 318309 |
| PE anti-CD82 | BioLegend | Cat# 342103 |
| Bacterial and Virus Strains | ||
| AAV-DJ-hSYN1::eYFP | Stanford Gene Vector and Virus Core | N/A |
| G-deleted Rabies Cre-eGFP | Salk Institute Viral Vector Core | N/A |
| AAV-DJ-EF1a-CVS-G-WPRE-pGHpA | Stanford Gene Vector and Virus Core (Wertz et al., 2015) | Addgene plasmid #67528 |
| AAV-DJ-DIO-mCherry | Stanford Gene Vector and Virus Core | N/A |
| AAV-1-hSYN1-ChrimsonR-tdT | Addgene | Addgene; #59171-AAV1 |
| AAV-1-syn-jGCaMP7s-WPRE | Addgene | Addgene; #104487-AAV1 |
| AAV-8-CAG-FLEX-Rabies G | Stanford Gene Vector and Virus Core | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| B-27 supplement without vitamin A | Life Technologies | Cat# 12587010 |
| N-2 supplement | Life Technologies | Cat# 17502048 |
| Human recombinant FGF-2 | R&D Systems | Cat# 233-FB |
| Human recombinant EGF | R&D Systems | Cat# 236-EG |
| Human recombinant BDNF | Peprotech | Cat# 450–02 |
| Human recombinant NT3 | Peprotech | Cat# 450–03 |
| Human recombinant IGF-1 | Peprotech | Cat# 100–11 |
| L-Ascorbic Acid | Wako | Cat# 321–44823 |
| cAMP | Sigma-Aldrich | Cat# D0627 |
| Dorsomorphin | Sigma-Aldrich | Cat# P5499 |
| SB-431542 | Tocris | Cat# 1614 |
| CHIR 99021 | Selleckchem | Cat# S1263 |
| SAG | Millipore | Cat# 566660 |
| Retinoic Acid (RA) | Sigma-Aldrich | Cat# R2625 |
| DAPT | STEMCELL Technologies | Cat# 72082 |
| Y-27632 | Selleckchem | Cat# S1049 |
| LDN-193189 | Selleckchem | Cat# S7507 |
| Recombinant murine HGF | Peprotech | Cat# 315–23 |
| Geltrex LDEV-Free Reduced Growth Factor Basement Membrane Matrix | Life Technologies | Cat# A1413201 |
| (+)-tubocurarine chloride pentahydrate (curare) | Sigma-Aldrich | Cat# 93750 |
| NBQX | Tocris | Cat# 0373 |
| D-AP5 (APV) | Tocris | Cat# 0106 |
| Tetrodotoxin citrate (TTX) | Tocris | Cat# 1069 |
| MNI-caged-L-glutamate | Tocris | Cat# 1490 |
| Critical Commercial Assays | ||
| P3 Primary Cell 4D-NucleofectorTM X Kit L | Lonza | V4XP-3024 |
| Chromium Single cell 3’ GEM, Library & Gel Bead Kit v3 | 10x Genomics | PN: 1000075 |
| Deposited Data | ||
| Processed single cell RNA-seq data; hSpS d53 and hSpS +/− DAPT | This study | GEO: GSE123722 |
| Experimental Models: Cell Lines | ||
| Human iPSC line 2242–1 | Paşca lab | N/A |
| Human iPSC line 8858–1 | Paşca lab | N/A |
| Human iPSC line 0307–1 | Paşca lab | N/A |
| Human iPSC line 0524–1 | Paşca lab | N/A |
| Human iPSC line 1205–4 | Paşca lab | N/A |
| Human iPSC line 8119–1 | Paşca lab | N/A |
| Human iPSC line TUBA1B-eGFP | Coriell | AICS-0012 cl.105 |
| Human iPSC line LMNB-eGFP | Coriell | AICS-0013 cl.210 |
| Human iPSC line CAG::EGFP | Coriell; modified in the Paşca lab | CW30261 |
| Human skeletal myoblasts (hSkM) | Thermo Fisher Scientific | A12555, Lot# 1837192 |
| EmbryoMax PMEF | Millipore | Cat# PMEF-N-K |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| All primers used for this study are listed on Table S2 | N/A | N/A |
| Recombinant DNA | ||
| lenti-ACTA1::GCaMP6s | Vector Builder (Brennan and Herdeman, 1993) | N/A |
| lenti-Hb9::GFP | Vector Builder (Nakano et al., 2015) | N/A |
| lenti-Hb9::mCherry | Vector Builder (Nakano et al., 2015) | N/A |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | Plasmid# 42230 |
| gRNA_AAVS1-T2 | Addgene | Plasmid# 41818 |
| AAVS1-CAG-hrGFP | Addgene | Plasmid# 52344 |
| Software and Algorithms | ||
| ImageJ (Fiji) | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Matlab_R2018a | MathWorks | https://www.mathworks.com/help/matlab/ref/rand.html |
| Cell Ranger v3.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat v3.0 | Satija Lab (Butler et al., 2018) | https://satijalab.org/seurat/ |
| Reference Similarity Spectrum (RSS) | Kanton et al., 2019 | https://github.com/quadbiolab/primate_cerebral_organoids |
| Human ortholog mapping of mouse genes | Skene et al., 2018 | https://github.com/NathanSkene/EWCE/blob/master/data/mouse_to_human_homologs.rda |
| MUSCLEMOTION | Sala et al., 2018 | https://github.com/l-sala/MUSCLEMOTION |
| Other | ||
| 10 cm ultralow attachment plates | Corning | Cat# 3262 |
| 24-well ultralow attachment plates | Corning | Cat# 3473 |
| Silicone wells | Ibidi | Cat# 80369 and 80409 |
| Cell culture inserts, 0.4 μm pore size | Corning | Cat# 353090 |
| Glass-bottom 96-well plate | Corning | Cat# 4580 |
| Optical fiber-coupled LED | Thorlabs | Cat# M625F2 |
| CYCLOPS LED driver 3.6 with M8 connector | Open Ephys | Cat# f2 |
Highlights.
Generation of hiPS cell-derived 3D spheroids resembling the hindbrain/spinal cord
Synaptically connected corticofugal projections in fused cortico-spinal assembloids
Stimulation of cortical neurons controls skeletal muscle in three-part assembloid
Cellular and functional changes in cortico-motor assembloids maintained long term
ACKNOWLEDGMENTS
We thank K.M. Hennig, S.J. Yoon, M.L. Fabian, F. Birey, R.M. Agoglia, M. Li, A.E. Trevino, and the Paşca lab for advice and support. We also thank J. Perrino (Stanford Cell Sciences Imaging Facility) for support with electron microscopy. Cell sorting was performed in the Stanford Shared FACS Facility. The rabies virus work was supported by the GT3 Core Facility of the Salk Institute with funding from NIH NCI CCSG P30 014195, an NINDS R24 Core Grant, and funding from NEI. Electron microscopy work was supported, in part, by ARRA Award Number 1S10RR026780–01 from the National Center for Research Resources (NCRR). This work was supported by grants from National Institute of Mental Health (NIMH) (BRAINS Award R01MH107800), the Chan Zuckerberg Initiative Ben Barres Investigator Award, the NYSCF Robertson Stem Cell Investigator Award, the Stanford Brain Organogenesis Progam in the Wu Tsai Neurosciences Institute and the Big Idea Grant, Stanford Bio-X, the Kwan Research Fund, V. Coates Foundation, the Senkut Funds (to S.P.P.), the Idun Berry Postdoctoral Fellowship (to J.A.), the Stanford Medicine Dean’s Fellowship (to J.A. and Y.M.), and the Stanford Maternal & Child Health Research Institute (MCHRI) Fellowship (to O.R. and Y.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.11.017.
DECLARATION OF INTERESTS
Stanford University has filed a provisional patent application that covers the generation and assembly of region-specific cortico-spinal-muscle spheroids. H.C.F. and E.M.W. were employees of BD Genomics during this study.
REFERENCES
- Alaynick WA, Jessell TM, and Pfaff SL (2011). SnapShot: spinal cord development. Cell 146, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin ND, and Paşca SP (2018). Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 100, 389–405. [DOI] [PubMed] [Google Scholar]
- Amoroso MW, Croft GF, Williams DJ, O’Keeffe S, Carrasco MA, Davis AR, Roybon L, Oakley DH, Maniatis T, Henderson CE, and Wichterle H (2013). Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J. Neurosci 33, 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler KRC, Panagiotakos G, Thom N, et al. (2017). Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, and Tuszynski MH (2009). Spinal cord injury: plasticity, regeneration and the challenge of translational drug development. Trends Neurosci 32, 41–47. [DOI] [PubMed] [Google Scholar]
- Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B, Koch P, Edenhofer F, Hampl A, and Brüstle O (2010). Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 28, 955–964. [DOI] [PubMed] [Google Scholar]
- Brennan KJ, and Hardeman EC (1993). Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J. Biol. Chem 268, 719–725. [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butts JC, Iyer N, White N, Thompson R, Sakiyama-Elbert S, and McDevitt TC (2019). V2a interneuron differentiation from mouse and human pluripotent stem cells. Nat. Protoc 14, 3033–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chal J, Al Tanoury Z, Hestin M, Gobert B, Aivio S, Hick A, Cherrier T, Nesmith AP, Parker KK, and Pourquié O (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc 11, 1833–1850. [DOI] [PubMed] [Google Scholar]
- Coulon P, Rollin PE, and Flamand A (1983). Molecular basis of rabies virus virulence. II. Identification of a site on the CVS glycoprotein associated with virulence. J. Gen. Virol 64, 693–696. [DOI] [PubMed] [Google Scholar]
- Cregg JM, Leiras R, Montalant A, Wanken P, Wickersham IR, and Kiehn O (2020). Brainstem neurons that command mammalian locomotor asymmetries. Nat. Neurosci 23, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasen JS, and Jessell TM (2009). Hox networks and the origins of motor neuron diversity. Curr. Top. Dev. Biol 88, 169–200. [DOI] [PubMed] [Google Scholar]
- Delile J, Rayon T, Melchionda M, Edwards A, Briscoe J, and Sagner A (2019). Single cell transcriptomics reveals spatial and temporal dynamics of gene expression in the developing mouse spinal cord. Development 146, dev173807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R, and Geschwind DH (2011). The human brain in a dish: the promise of iPSC-derived neurons. Cell 145, 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval N, Vaslin C, Barata TC, Frarma Y, Contremoulins V, Baudin X, Nedelec S, and Ribes VC (2019). BMP4 patterns Smad activity and generates stereotyped cell fate organization in spinal organoids. Development 146, dev175430. [DOI] [PubMed] [Google Scholar]
- Etessami R, Conzelmann KK, Fadai-Ghotbi B, Natelson B, Tsiang H, and Ceccaldi PE (2000). Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: an in vitro and in vivo study. J. Gen. Virol 81, 2147–2153. [DOI] [PubMed] [Google Scholar]
- Fan HC, Fu GK, and Fodor SP (2015). Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 347, 1258367. [DOI] [PubMed] [Google Scholar]
- Faustino Martins JM, Fischer C, Urzi A, Vidal R, Kunz S, Ruffault PL, Kabuss L, Hube I, Gazzerro E, Birchmeier C, et al. (2020). Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 26, 172–186. [DOI] [PubMed] [Google Scholar]
- Garden GA, and La Spada AR (2012). Intercellular (mis)communication in neurodegenerative disease. Neuron 73, 886–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, Sutcliffe M, Boulanger J, Tripodi M, Derivery E, et al. (2019). Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci 22, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding M (2009). Circuits controlling vertebrate locomotion: moving in a new direction. Nat. Rev. Neurosci 10, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M, Tsakiridis A, Wymeersch FJ, Huang Y, Kleinjung J, Wilson V, and Briscoe J (2014). In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol 12, e1001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, and Macklis JD (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci 14, 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kalambogias J, Yoshioka S, Han W, Li Z, Kawasawa YI, Pochareddy S, Li Z, Liu F, Xu X, et al. (2017). Control of species-dependent corticomotoneuronal connections underlying manual dexterity. Science 357, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie S (2007). Patterning and axon guidance of cranial motor neurons. Nat. Rev. Neurosci 8, 859–871. [DOI] [PubMed] [Google Scholar]
- Han SS, Williams LA, and Eggan KC (2011). Constructing and deconstructing stem cell models of neurological disease. Neuron 70, 626–644. [DOI] [PubMed] [Google Scholar]
- Jessell TM (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet 1, 20–29. [DOI] [PubMed] [Google Scholar]
- Kaiser T, and Feng G (2015). Modeling psychiatric disorders for developing effective treatments. Nat. Med 21, 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, Guijarro P, Sidow L, Fleck JS, Han D, et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. [DOI] [PubMed] [Google Scholar]
- Kiehn O (2006). Locomotor circuits in the mammalian spinal cord. Annu. Rev. Neurosci 29, 279–306. [DOI] [PubMed] [Google Scholar]
- Kiehn O (2016). Decoding the organization of spinal circuits that control locomotion. Nat. Rev. Neurosci 17, 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SC, Acton D, and Goulding M (2018). Spinal Circuits for Touch, Pain, and Itch. Annu. Rev. Physiol 80, 189–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HC, Seal RP, and Johnson JE (2016). Making sense out of spinal cord somatosensory development. Development 143, 3434–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon RN (2008). Descending pathways in motor control. Annu. Rev. Neurosci 31, 195–218. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Lewallen KA, and Pfaff SL (2012). Spatial organization of cortical and spinal neurons controlling motor behavior. Curr. Opin. Neurobiol 22, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, and Arlotta P (2015). Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell Dev. Biol 31, 699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micali N, Kim SK, Diaz-Bustamante M, Stein-O’Brien G, Seo S, Shin JH, Rash BG, Ma S, Wang Y, Olivares NA, et al. (2020). Variation of Human Neural Stem Cells Generating Organizer States In Vitro before Committing to Cortical Excitatory or Inhibitory Neuronal Fates. Cell Rep 31, 107599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Windrem M, Zappavigna V, and Goldman SA (2005). Identification of a conserved 125 base-pair Hb9 enhancer that specifies gene expression to spinal motor neurons. Dev. Biol 283, 474–485. [DOI] [PubMed] [Google Scholar]
- Ogura T, Sakaguchi H, Miyamoto S, and Takahashi J (2018). Three-dimensional induction of dorsal, intermediate and ventral spinal cord tissues from human pluripotent stem cells. Development 145, dev162214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakula A, Spinazzola JM, and Gussoni E (2019). Purification of Myogenic Progenitors from Human Muscle Using Fluorescence-Activated Cell Sorting (FACS). Methods Mol. Biol 1889, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca SP (2018). The rise of three-dimensional human brain cultures. Nature 553, 437–445. [DOI] [PubMed] [Google Scholar]
- PPaşca SP (2019). Assembling human brain organoids. Science 363, 126–127. [DOI] [PubMed] [Google Scholar]
- Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasxca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med 17, 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, et al. (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, and Rothstein JD (2015). Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol 69, 5.67.1–5.67.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala L, van Meer BJ, Tertoolen LGJ, Bakkers J, Bellin M, Davis RP, Denning C, Dieben MAE, Eschenhagen T, Giacomelli E, et al. (2018). MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ. Res 122, e5–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, Livesey MR, Lowry E, Macklis JD, Rushton D, et al. (2016). Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci 19, 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, and Levine AJ (2018). Massively Parallel Single Nucleus Transcriptional Profiling Defines Spinal Cord Neurons and Their Activity during Behavior. Cell Rep 22, 2216–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Kwan KY, Li M, Lefebvre V, and Sestan N (2012). Cis-regulatory control of corticospinal system development and evolution. Nature 486, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, Giusti-Rodriguez P, Hodge RD, Miller JA, Muñoz-Manchado AB, et al. ; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium (2018). Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan SA, Darmanis S, Huber N, Khan TA, Birey F, Caneda C, Reimer R, Quake SR, Barres BA, and Pasca SP (2017). Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 95, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan SA, Andersen J, Paşca AM, Birey F, and Paşca SP (2018). Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc 13, 2062–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AMM, Meyer KA, Santpere G, Gulden FO, and Sestan N (2017). Evolution of the Human Nervous System Function, Structure, and Development. Cell 170, 226–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck JA, Jaiswal MK, Calder EL, Kishinevsky S, Weishaupt A, Toyka KV, Goldstein PA, and Studer L (2016). Functional Connectivity under Optogenetic Control Allows Modeling of Human Neuromuscular Disease. Cell Stem Cell 18, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stifani N (2014). Motor neurons and the generation of spinal motor neuron diversity. Front. Cell. Neurosci 8, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H, Sakurai M, and Kanazawa I (2002). In vitro formation of corticospinal synapses in an organotypic slice co-culture. Neuroscience 109, 359–370. [DOI] [PubMed] [Google Scholar]
- ten Donkelaar HJ, Lammens M, Wesseling P, Hori A, Keyser A, and Rotteveel J (2004). Development and malformations of the human pyramidal tract. J. Neurol 251, 1429–1442. [DOI] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, and Pfaff SL (1999). Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23, 675–687. [DOI] [PubMed] [Google Scholar]
- Thiry L, Hamel R, Pluchino S, Durcan T, and Stifani S (2020). Characterization of Human iPSC-derived Spinal Motor Neurons by Single-cell RNA Sequencing. Neuroscience, S0306–4522(20)30275-X. [DOI] [PubMed]
- Thomson SR, Wishart TM, Patani R, Chandran S, and Gillingwater TH (2012). Using induced pluripotent stem cells (iPSC) to model human neuromuscular connectivity: promise or reality? J. Anat 220, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, and Macleod MR (2010). Can animal models of disease reliably inform human studies? PLoS Med. 7, e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz A, Trenholm S, Yonehara K, Hillier D, Raics Z, Leinweber M, Szalay G, Ghanem A, Keller G, Ró zsa, B., et al. (2015). PRESYNAPTIC NETWORKS. Single-cell-initiated monosynaptic tracing reveals layer-specific cortical network modules. Science 349, 70–74. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Tanaka Y, Cakir B, Patterson B, Kim KY, Sun P, Kang YJ, Zhong M, Liu X, Patra P, et al. (2019). hESC-Derived Thalamic Organoids Form Reciprocal Projections When Fused with Cortical Organoids. Cell Stem Cell 24, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SJ, Elahi LS, Paşca AM, Marton RM, Gordon A, Revah O, Miura Y, Walczak EM, Holdgate GM, Fan HC, et al. (2019). Reliability of human cortical organoid generation. Nat. Methods 16, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, and Anderson DJ (2002). The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109, 61–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A and B) Schematics illustrating marker gene expression along the rostro-caudal (A) and dorso-ventral (B) axes of the hindbrain and spinal cord.
(C) Schematic detailing the 12 conditions in the combinatorial growth factor matrix and the molecules and concentrations they receive.
(D–F) Gene expression analysis of genes expressed along the rostro-caudal (D) and dorso-ventral (E), (F) axes in the 12 conditions of the combinatorial matrix at day 20 of in vitro differentiation. Each condition shows 3 colored bars representative of n = 3 hiPS cell lines from 1 differentiation (Kruskal-Wallis test p = 0.002 for HOXA2, p = 0.008 for HOXB4, p = 0.003 for HOXC9, p = 0.002 for HOXC9, p = 0.0008 for PAX7, p = 0.02 for PAX6, p = 0.001 for OLIG2, p = 0.002 for NKX6–1,p= 0.0006 for LHX1, p = 0.007 for CHX10, p = 0.06 for HB9; one-way ANOVA p = 0.002 for HOXC5, p < 0.0001 for FOXA2, p < 0.0001 for EVX1, p = 0.01 for ISL1).
(G) Immunocytochemistry in cryosections of hSpS conditions #4 and #8 showing dorsal-like and ventral-like identities. This image was generated by automatic stitching of individual images.
(H) Schematic detailing differentiation conditions used for deriving hSpS.
(I and J) Representative immunocytochemistry images in cryosections of hSpS showing expression of OLIG2 and NKX2.2 (I) or NKX6.1 (J) at day 18 of in vitro differentiation.
(K) Quantification of the total number of OLIG2 cells per area (mm2) in day 18 hSpS (n = 10 hSpS derived from 3 hiPS cell lines from 1 differentiation, with 2 cryosections quantified per hSpS).
(L) Quantification of the percentage (%) of OLIG2 positive cells that co-express either NKX2.2 or NKX6.1 in day 18 hSpS (n = 10 hSpS derived from 3 hiPS cell lines from 1 differentiation, with 2–3 cryosections quantified per hSpS).
Data represent mean ± s.e.m. Scale bars, 50 µm (insets in (I), (J), 100 µm (G), 200 µm (I), (J).
(A) Schematic detailing differentiation conditions used for deriving hSpS with or without DAPT.
(B) Schematic illustrating hindbrain/spinal cord neuronal domains, their corresponding marker genes and their neurotransmitter identities.
(C) t-SNE visualization of single cell gene expression of hSpS with or without DAPT at day 45 of in vitro differentiation (n = 7,888 cells; BD™ Rhapsody system) showing 10 main clusters that include a spinal motor neuron (MN) cluster. List of genes for each cluster is included in Table S1.
(D) Boxplots for neurotransmitter-related genes enriched in each of the single cell clusters (VGAT also called SLC32A1, GLYT2 also called SLC6A5, CHT1 also called SLC5A7, VACHT also called SLC18A3, VGLUT2 also called SLC17A6).
(E) Distribution of cells in conditions hSpS and hSpS (– DAPT) in t-SNE plot. R2 value shows the correlation between the two hSpS conditions (R2 = 0.85, p < 0.0001).
(F) Proportion of cells per condition in each of the ten clusters (χ2 test, hSpS versus hSpS (–DAPT); p < 0.0001).
(G) Gene expression analysis of neuronal cell type markers in hSpS at day 30 of in vitro differentiation in 4 separate differentiations that were either pooled (left) or separated (right; n = samples from 5 hiPS cell lines; two-tailed t test: ****p = < 0.0001 for HB9, ***p = 0.0003 for EVX1; Mann-Whitney test: **p = 0.0014 for SIM1, ****p < 0.0001 for ISL1, ****p = < 0.0001 for GATA3, **p = 0.0045 for CHX10, ****p = < 0.0001 for EN1).
(H) Gene expression analysis of neurotransmitter identity markers in hSpS at day 30 of in vitro differentiation in 4 separate differentiations that were either pooled (left) or separated (right; n = samples from 5 hiPS cell lines; two-tailed t test: ***p < 0.0002 for CHAT, p = 0.7 for VGLUT2; Mann-Whitney test: p = 0.01 for GAD1, ***p = 0.0001 for GLYT2).
(I) Gene expression analysis of glial progenitor markers in hSpS at day 30 of in vitro differentiation in 4 separate differentiations that were either pooled (left) or separated (right; n = samples from 5 hiPS cell lines; Mann-Whitney test: ****p < 0.0001 for FABP7, p = 0.1 for SOX10).
(J) Immunocytochemistry in hSpS and hSpS (– DAPT) cryo-sections at day 45 of in vitro differentiation showing expression of different neuronal domain marker genes.
(K) Immunocytochemistry in hSpS and hSpS (– DAPT) cryo-sections at day 45 of in vitro differentiation showing expression of different neuronal and neurotransmitter identity markers.
Data represent mean ± s.e.m. Scale bars, 50 µm (J), 200 µm (K).
(A) t-SNE visualization of single cell gene expression of hSpS at day 53 of in vitro differentiation (n = 9,302 cells; 10x Genomics) showing 12 main clusters that include a spinal motor neuron (MN) cluster. See Figure 1F for visualization using UMAP.
(B) UMAP showing cells in hSpS colored by the hiPS cell line they were derived from.
(C) Plots showing the Pearson correlation of the normalized average gene expression, log (counts per 10,000) between each of the three hiPS cell lines used for this experiment. R2 shows the Pearson correlation value for each.
(D) Heatmap showing the Pearson correlation of the ventral, dorsal, progenitor, astroglia and other domains between hiPS cell lines used for this experiment. Hierarchical clustering of sample correlations is displayed with a dendogram.
(E) Dot plot showing expression of cell adhesion and neuronal guidance genes in each of the neuronal clusters in hSpS. The size of the circle represents the percent of cells expressing each gene per cluster.
(F) UMAP plots showing average gene expression of selected HOX genes.
(G) Hierarchical clustering showing RSS analysis of hSpS clusters to single-cell RNA-seq clusters from adult mouse central nervous system (CNS) regions in Zeisel et al. (2018). CB, cerebellum; HBCHOL3, hindbrain cholinergic neuron, HBGLU2: hindbrain excitatory neuron; HBINH2, hindbrain inhibitory neuron.
(H) hSpS UMAP plots colored by RSS to selected single cell RNaseq clusters from adult mouse CNS from Zeisel et al. (2018).
(I and J) Schematic illustrating hindbrain/spinal cord motor columns (I) and their corresponding marker gene expression (J). SAC, spinal accessory column; PMC, phrenic motor column; LMC, lateral motor column; MMC, medial motor column; PGC, preganglionic column; HMC, hypaxial motor column. Figure adapted from Dasen and Jessell (2009) and Stifani (2014).
(K) UMAP plot showing sub-clustering of the motor neuron cluster.
(L) Heatmap showing the top 20 differentially expressed genes in each motor neuron cluster (MN 1–6).
(M) UMAP plots showing average gene expression of selected markers, including neurotransmission-related genes, column-specific genes, cell adhesion genes and HOX genes.
(A) Representative immunohistochemistry images in cryosections of day 25 hSpS showing expression of post-mitotic neurons expressing MAP2 at the edge of hSpS and proliferating progenitors expressing SOX2 and KI67 in the center.
(B) Representative immunohistochemistry images in cryosections of day 25 hSpS showing expression of post-mitotic motor neurons expressing PHOX2B at the edge of hSpS and medial-dorsal progenitors expressing PAX6 in the center.
(C and D) Immunocytochemistry in cryosections of hSpS showing motor neuron markers at day 30 of in vitro differentiation.
(E and F) Representative immunohistochemistry images in cryosections of day 45 hSpS showing expression of motor neuron markers ISL1, HB9 (F) and PHOX2B (G).
(G) Representative immunohistochemistry images in cryosections of day 45 hSpS showing localization of spinal neuronal domain V2a represented by CHX10 and LHX3 glutamatergic cells.
Scale bars, 10 µm (C), 20 µm (A), (B), (D), insets in (E), (F), (G), 200 µm (E), (F), (G).
(A) t-SNE visualization of single cell gene expression of hCS and hSS (human subpallial spheroids) at day 105 of differentiation (n = 11,838 cells; BD™ Resolve system, from Birey et al. [2017]) showing the distribution of expression of a set of genes associated with cortico-spinal neuronal identity (SOX5, FEZF2, BCL11B or CTIP2, BHLHE22, LDB2, CRIM1 and OTX1).
(B) t-SNE visualization of single cell gene expression of day 105 hCS and hSS showing expression of CHT1 (also called SLC5A7) and VACHT (also called SLC18A3).
(C) Gene expression analysis of cortico-spinal-related genes in hCS over time (n = 4–6 hiPS cell lines from 1–2 differentiations; one-way ANOVA interaction: p = 0.001 for FEZF2, p < 0.0001 for BCL11B, p = 0.5 for OTX1; Kruskal-Wallis test interaction: p = 0.002 for SOX5).
(D and E) Immunocytochemistry in hCS cryosections at day 130 of in vitro differentiation showing expression of cortico-spinal markers CTIP2 and OTX1 (D). Antibodies were validated in slices of human cortical tissue at post-conception week 17 (PCW17, E).
(F) Quantification of hCS-derived eYFP fluorescence coverage in hSpS areas in hCS-hSpS assembloids (at 20 daf) with samples separated by differentiation (n = 3 hiPS cell lines; one-way ANOVA interaction: p = 0.3). Figure 3D shows data combined.
(G) Images of intact hCS-hSpS assembloids 20 days after fusion (daf) showing hCS-derived hSYN1::eYFP projections.
(H and I) Representative image of intact hCS-hCS assembloid (H) and quantification of the extent of projection of hSYN1::eYFP (I) (n = 3 hiPS cell lines from 1–2 differentiations; Kruskal-Wallis test interaction p = 0.01, with Dunn’s multiple comparison test: p > 0.9 for 10 versus 5 daf, p = 0.06 for 20 versus 5 daf). See Figure 3D for quantification of the projection in hCS-hSpS assembloids.
(J) Representative images of intact hCS-hSpSCAG::EGFP assembloids showing some reciprocal projections from hSpS to hCS and limited cell migration.
(K) Representative image of intact hCS-hSpS assembloid where hCS was infected with AAV-hSYN1::eYFP and hSpS with lenti-Hb9::mCherry. No hSpS-derived Hb9::mCherry projections are observed in hCS (similar results were observed in 15 assembloids from 2 differentiations and 3 hiPS cell lines).
(L) Representative immunocytochemistry image of hCS-hSpS assembloid in retrograde viral tracing experiment at 31 daf showing GFP expression in hSpS and colocalization of GFP and mCherry in hCS. mCherry projections from hCS to hSpS can also be seen.
(M) Representative immunocytochemistry image of GFP, mCherry and the glial marker GFAP on the hCS side of hCS-hSpS assembloid at 31 daf.
(N and O) Quantification of the percentage (%) of GFP+ and mCherry+ cells on the hCS side of hCS-hSpS assembloids that co-express either the neuronal marker MAP2 or the glial marker GFAP (O) or the cortical layer-specific markers CTIP2 or BRN2 (N) separated by differentiation (n = 10 assembloids from 3 hiPS cell lines, with 2–3 cryosections quantified per assembloid). Figures 3J and 3M show quantification with both differentiations combined.
Data represent mean ± s.e.m. Scale bars, 50 µm (D), 100 µm (L), (M), 200 µm (G), (H), (J), (K), 500 µm (E).
(A and B) Characterization of human skeletal myoblast (hSkM) differentiation. hSkM differentiate upon removal of FBS from the culture medium (A). Immunocytochemistry images for desmin (DES), titin (TTN) and the heavy myosin chain (MyHC) show differentiation of myoblasts over time (B).
(C) Distribution and mean number of Hoechst+ nuclei in desmin cells at day 0 and day 15 of hSkM differentiation. Data represent mean ± s.e.m. (n = 1,031 desmin+ cells at day 0, and n = 190 desmin+ cells at day 15; data was collected in each case from 5 fields within a culture well in 2 hSkM differentiation replicates; Mann-Whitney test: ****p < 0.0001).
(D) Representative calcium imaging for hSkM and hSpS-hSkM. Images show average (avg) intensity projections of Cal-590 AM in a field (average intensity projections were used to delineate cells for quantification). Traces show examples of active and inactive hSkM cells in either hSkM alone or hSpS-hSkM cultures. ΔF/F indicates the fluorescence intensity over baseline fluorescence. Arrows show cells analyzed.
(E) Schematic detailing hSkM analyzed for calcium imaging experiment. Spontaneous calcium activity was recorded in 6–9 fields per culture well. Of these fields some were within 1 mm of the spheroid, and some were further than 1 mm from the spheroid.
(F) Quantification of spontaneous calcium imaging activity in hSkM over a period of 2 minutes. Fields were separated according to their distance from the spheroid (hCS or hSpS). Data represent mean ± s.e.m. (n = 2 hiPS cell lines from 1–2 differentiations; one-way ANOVA interaction: p = < 0.0001, with Dunnett’s multiple comparison test ****p < 0.0001).
(G) Representative immunocytochemistry image showing BTX binding in hSkM that had been co-cultured with hSpS.
(H) Step number 4 in the generation and assembly of hSpS-3D hSkM. After 2–3 weeks in differentiation medium, 3D hSkM can be co-cultured with hSpS and/or hCS. For this, 3D hSkM and spheroids are placed on insert wells with 2 mL of medium per well.
(I and J) Representative immunohistochemistry images in cryosections for rabies-derived GFP and the cholinergic markers CHAT (I) and GABA (J) on the hSpS side of hSpS-hSkM assembloids at 18 daf.
(K) Quantification of the percentage (%) of GFP positive (+) cells on the hSpS side of hSpS-hSkM assembloids that co-express either ISL1/2, CHX10, FOXP2 or PROX1 (n = 8 to 9 assembloids derived from 3 hiPS cell lines and 1 differentiation, with 3–6 cryosections quantified per assembloid).
(L and M) Representative immunohistochemistry images in cryosections for rabies-derived GFP and either GABA and FOXP2 (L) or PROX1 and CHX10 (M) on the hSpS side of hSpS-hSkM assembloids at 18 daf.
(N) Schematic illustrating the glutamate uncaging approach. Caged glutamate is added to the culture medium and uncaged upon UV light stimulation (405 nm).
(O and P) Glutamate uncaging of hSpS in hSpS-hSkM assembloid (H). Displacement normalized to baseline over time is shown for 3 subfields in the presence (I) or absence (J) of caged glutamate in the medium.
(Q and R) Representative immunohistochemistry in cryosections of hSpS-hSkM assembloids 31 days after fusion showing desmin (DES), BTX, synaptophysin 1 (SYP) and Hoechst.
(S) Single Z-plane and orthogonal views of projection in (R) showing YZ (right) and XZ (bottom) planes of stack and indicating apposition of BTX and SYP. Yellow lines indicate the point in the stack being visualized.
Data represent mean ± s.e.m. Scale bars, 10 µm (Q), (R), (S), insets in (G), 50 µm (I), (J), (L), (M), 100 µm (G), 200 µm (B), (D), (O), 400 µm (A).
(A) Imaging fields (1.8 mm x 1.8 mm in size) are divided into 16 subfields for analysis. Only subfields containing hSkM are analyzed.
(B and C) Representative spontaneous contraction traces in subfields of hSkM or hSpS-hSkM assembloids (B). The correlation of displacements between subfields in a field is quantified using covariance analysis. Heatmaps show representative examples of covariance between subfields in a field of hSkM, hCS-hSkM, hSpS-hSkM or hCS-hSpS-hSkM assembloids (C). Video S4 shows live imaging of spontaneous contractions in an hCS-hSpS-hSkM assembloid.
(D and E) Glutamate uncaging in hCS-hSpS-hSkM assembloids. UV light (405 nm) uncages glutamate specifically on hCS (D). Displacement normalized to baseline over time is shown for 3 subfields without or with addition of curare (100 µM, E).
(F and G) Glutamate uncaging of hCS in hCS-hSpS-hSkM assembloid. Displacement normalized to baseline over time is shown for 3 subfields in the absence of caged glutamate in the medium (see Figures 6F and 6G for glutamate uncaging in this field in the presence of caged glutamate, and see Table S1 for details of all stimulation experiments).
(H) Representative brightfield image showing an intact hCS-hSpS-hSkM assembloid. Figure 6K shows AAV-hSYN1-ChrimsonR-tdT in hCS.
(I and J) Representative example of optogenetic stimulation in hCS-hSpS-hSkM assembloids (five consecutive 68 ms pulses). hCS were infected with AAV-hSYN1-ChrimsonR-tdT (Chrim). Traces of displacement in the whole field in (I) are shown normalized to the pre-stimulation baseline. Two trials for the same assembloid are shown. Addition of NBQX (50 µM) and APV (50 µM) abolished muscle contraction upon light-induced hCS stimulation (J).
(K and L) Optogenetic stimulation in hCS-hSkM assembloid (five consecutive 68 ms pulses). hCS were infected with AAV-hSYN1-ChrimsonR-tdT (K). Displacement over time normalized to pre-stimulation baseline is shown for the whole field in (L). Similar results were obtained in n = 5 hCS-hSkM assembloids derived from 2 hiPS cell lines.
(M) Schematic detailing location of electron microscopy images within hCS-hSpS-hSkM assembloids.
(N–P) Representative images showing axons and synapses in hCS-hSpS-hSkM assembloids with synaptic vesicles (arrow heads), post-synaptic densities (asterisks) and mitochondria (arrows). Inset in (P) shows an axo-axonic synapse.
(Q) Representative images within hSkM in hCS-hSpS-hSkM assembloids showing skeletal muscle fibers with actin and myosin filaments and mitochondria (arrows).
(R and S) Representative images within hSkM in hCS-hSpS-hSkM assembloids showing points of contact between hSpS neurons and hSkM. Neuron terminals are vesicle-laden (arrow heads) and present mitochondria (arrows). hSkM are surrounded by a basal lamina (hollow arrowhead).
(T) Schematic detailing location of images within hCSAAV-Chrim-hSpS-hSkM assembloids.
(U and V) Representative immunohistochemistry images in cryosections of hCSAAV-Chrim-hSpS-hSkM assembloids showing the presence of SMI-32+ motor neurons in hSpS with hCS-derived mCherry-positive projections (U) and astrocytes (V).
Scale bars, 200 nm (inset in P), 1 µm (O), (P), (S), 2 µm (N), (Q), (R), 20 µm (U), (V), 200 µm (A), (F), (H), (I), (K).
(A) Schematic detailing the differentiation condition used for generating hiPS cell-derived skeletal myoblasts. Protocol adapted from Chal et al. (2016).
(B) Schematic indicating marker genes expressed during the in vitro generation of myoblasts from hiPS cells.
(C) Gene expression analysis of skeletal myogenesis markers at days 5/6, 20 and 33/34 of in vitro myoblast differentiation (n = 3 hiPS cells lines from 2–3 differentiations).
(D) Representative images of myoblasts derived from 3 hiPS cell lines 2 to 4 days after subculture at day 33 of in vitro differentiation.
(E) FACS plots showing purification of the CD56+ / CD82+ myoblast population.
(F) Representative images of CD56+ / CD82+ myoblasts under either growth conditions or differentiation conditions.
(G) Representative image of an hSpS-3D ihSkM assembloid.
(H and I) Optogenetic stimulation in hCS-hSpS-ihSkM assembloids. hCS were infected with AAV-hSYN1-ChrimsonR-tdT (Chrim) before assembly. Five consecutive pulses of light (625 nm, 200 ms in duration each and 20 s apart) were delivered using a fiber-coupled LED directed toward the hCS (H). Traces of whole-field muscle displacement are shown after normalization to the pre-stimulation baseline.
Scale bars, 200 µm (D), (G), (H), 210 µm (F).
Data Availability Statement
Processed gene expression data are available in the Gene Expression Omnibus under accession number GEO: GSE123722.