

Abstract
Challenges in the selective manipulation of functional groups (chemoselectivity) in organic synthesis have historically been overcome using either reagents/catalysts that tunably interact with a substrate or through modification to shield undesired sites of reactivity (protecting groups). Although electrochemistry offers precise redox control to achieve unique chemoselectivity, this approach often becomes challenging in the presence of multiple redox-active functionalities. Historically, electrosynthesis has been performed almost solely by using direct current (DC). In contrast, applying alternating current (AC) has been known to change reaction outcomes considerably on an analytical scale but has rarely been strategically exploited for use in complex preparative organic synthesis. Here we show how a square waveform employed to deliver electric current – rapid alternating polarity (rAP) – enables control over reaction outcomes in the chemoselective reduction of carbonyl compounds, one of the most widely used reaction manifolds. The reactivity observed cannot be recapitulated using DC electrolysis or chemical reagents. The synthetic value brought by this new method for controlling chemoselectivity is vividly demonstrated in the context of classical reactivity problems such as chiral auxiliary removal and cutting-edge medicinal chemistry topics such as the synthesis of PROTACs.
Graphical Abstract
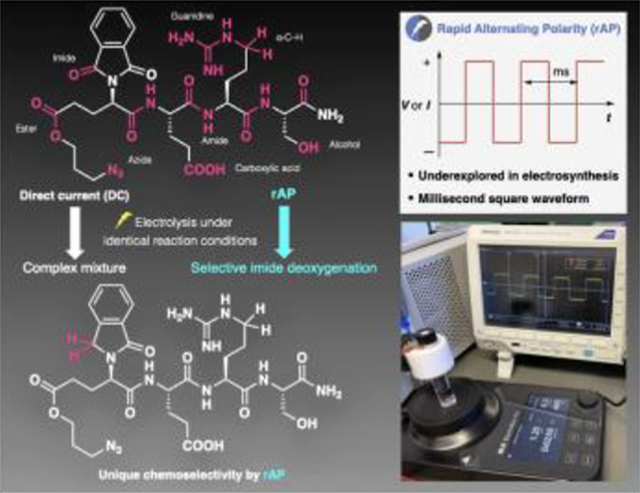
Introduction
Controlling chemoselectivity in organic synthesis is widely regarded as one of the paramount challenges of the field.1 Indeed, tuning reactions in complex settings to obtain precise position-selective outcomes is often the key bottleneck in executing synthetic pathways.2 Although computational chemistry techniques can be applied in certain contexts, identifying the right reagent or conditions to achieve such selectivity is, in most cases, an empirical endeavor. For this reason, the toolbox of organic chemistry is replete with multiple ways to achieve the same outcome, and protecting groups are still widely employed to block undesired reactivity.3,4 Amongst various ways to control a reaction outcome, precise redox control based on electron transfer is an emerging trend addressed by employing photoinduced electron transfer or direct electrochemical techniques. Although organic electrosynthesis is gaining popularity due to its ability to enable specific redox reactions to take place by adjusting the potential applied to a reaction,5 the presence of both oxidation and reduction can complicate reaction outcomes when using a simple undivided cell. Thus far, especially in the synthetic community, little attention has been paid to the effect of electron flow on the selectivity of electrochemical transformations on organic molecules. To date, nearly all synthetically useful electrochemical reactions proceed via direct current (DC)5 – where the polarity of electrodes remains unchanged such that the flow of electrons is uni-directional (Figure 1A, left). This current will drive the oxidation and reduction of molecules on anodes and cathodes respectively, with the reaction outcome being rationalized based on redox potential: a thermodynamic parameter used to describe the tendency of a molecule to accept or give electrons.6 Another mode of electric current delivery is alternating current and its varied manifestations (AC, see Supporting Information for the detailed classification)7 – wherein electrons flow back and forth between two electrodes in a reaction, most often associated with a sinusoidal waveform. In contrast to DC, reaction control using AC has been far less explored in synthetic electrochemistry (Figure 1A, right).8 This is somewhat surprising since AC electrolysis has found wide applications in electroanalysis to measure variables that cannot be easily determined by DC-based methods as well as to study reaction kinetics for electrochemical reactions.9 Besides these analytical applications, it is well-recognized that AC electrolysis has advantages outside organic synthesis such as electroplating10 and CO2 reduction.11 In fact, the potential value of AC electrolysis for organic electrosynthesis has been studied since the early 20th century,12 and different product distributions have been reported in anodic decarboxylation processes13a, the reduction of nitrobenzene,13b and propylene oxide formation.13c Thus far, the application of AC into mainstream organic synthesis has remained scarce, although recent elegant work has made inroads on the exploration of this uncharted territory.14–18 Regardless, what is undeniably missing in this arena is the discovery of unique reaction outcomes through AC as well as strategic exploitation of such an effect into complex molecule synthesis. An additional layer of difficulty stems from the lack of readily available instrumentation to generate a suitable and standardized AC method for evaluation by synthetic organic chemists.19 Herein we disclose a practical implementation of a square wave form of AC electrolysis, rapid alternating polarity (rAP), which enables precise control of specific reaction pathways and remarkable chemoselectivity in synthetically relevant reduction processes for which no known direct chemical alternative currently exists.
Figure 1.
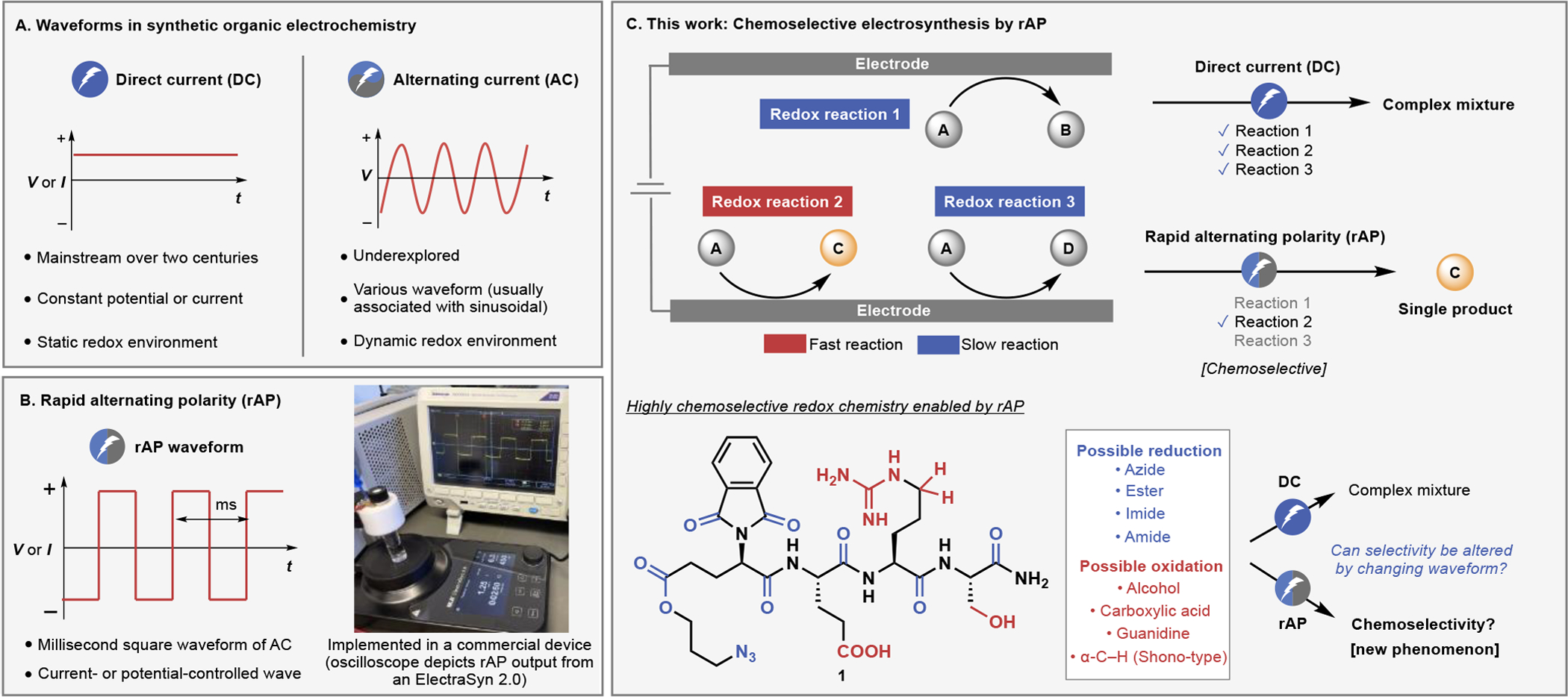
Types of waveforms in electrosynthesis and proposed reaction control enabled by rapid alternating polarity (rAP). (A) Illustration of direct current (DC) and alternating current (AC). (B) rAP waveform and implementation into a commercial device. (C) Hypothesis of achieving chemoselectivity by rAP through differentiating slow and fast electrochemical processes, and demonstration of its utility in a highly complex setting.
rAP is a type of AC featuring a square waveform (Figure 1B), achieved by alternating the polarity of an electrode in the millisecond timescale while maintaining either the current or potential constant. Such a waveform is known to be useful for improving the yield15 of electrochemical trifluromethylation20 and as a tool for mechanistic analysis.21 The underlying software to access such a waveform was recently implemented in ElectraSyn2.0, a widely employed commercial potentiostat, to facilitate widespread use of this method without any engineering barrier. rAP was developed based on the following hypothesis: if the rate of polarity switching is faster than the rate of certain processes on an electrode, a slower subset of electrochemical processes might be suppressed. This hypothesis is supported by early theoretical studies22,23 as well as recent experimental results24 suggesting that altered waveforms might influence the course of an electrochemical reaction (Figure 1C). In other words, rAP would enable differentiation of redox reactions based on their relative reaction rate. Such a method could bring a new dimension to reaction control where differentiation of multiple redox reactions and redox-active functionality are inherently challenging based only on redox potentials. An aspirational goal would therefore be to employ rAP in a functional-group-rich setting such as tetrapeptide 1, containing as many as 8 redox-active sites. Whereas fine-tuning site-specificity in a redox reaction by modulating the waveform of electric current applied may be considered possible on theoretical grounds by electrochemists, it is a tool unused and unheard of by practitioners of mainstream synthetic organic chemists. Thus, this community has yet to witness the possibility and the value brought by such a strategy.
Results and Discussion
To obtain proof-of-concept for the hypothesis, piperidine derivative 2 (Figure 2A) was selected as a simple model substrate containing both oxidizable and reducible functional groups. Specifically, this structure contains both imide carbonyl groups and activated C–H bonds, potentially susceptible to both reductive and oxidative functionalization, respectively. Figure 2 presents a detailed comparison of rAP with DC, as well as the influence of pulse duration (frequency) and waveform on the electrolysis of 2. Reaction conditions were chosen such that they could potentially lead to either reductive or oxidative outcomes (non-sacrificial electrodes, slightly acidic conditions). Each reaction was performed under identical conditions using pivalic acid as a proton source with waveform (type/frequency/amplitude) being the only variable screened. Crude 1H NMR analysis of the product distribution under standard DC electrolysis of phthalimide 2 revealed methoxy group incorporation into a large number of species resulting in a complex mixture (entry 1). Presumably, C–H oxidation (Shono oxidation) occurred along with varying degrees of imide reduction leading to this unselective and synthetically unworkable outcome.25 To exclude the possibility that this unselective reaction profile was attributed to harsh electrolysis conditions, the current and amount of electrons were reduced (entries 2 and 3) to no avail. In addition, the common tactic for maintaining clean electrode surface by switching polarity made no improvement on the outcome (entry 4).26 Throughout these experiments (entries 1–4), no product was isolated in synthetically meaningful yield. In stark contrast, rAP led to highly selective and efficient formation of either 3a or 3b, depending on the pulse duration applied (constant current rAP, entries 5–9). Remarkably, methoxy group incorporation indicative of Shono oxidation was barely observable in these entries (except entry 5, slow rAP at 200 ms). The crude 1H NMR depictions of entries 1 and 6 vividly illustrate the stark difference in product distribution. The effect is clearly beyond the reaction improvement that could be expected from common electrode cleaning by switching polarity (cf. entry 4). At shorter pulse duration (25–33 ms, 15–20 Hz), the major product was hemiaminal 3a, whereas fully reduced lactam 3b was formed at longer pulse duration (50–200 ms, 2.5–10 Hz). This product distribution suggested that current efficiency is a function of pulse duration: the longer the pulse is, the more redox reactions proceed (with an infinite pulse duration being equal to DC). The reduced current efficiency with a short pulse can be augmented by increasing the current, leading to a complete switch of product distribution from 3a to 3b [same 25 ms pulse but with 20 or 40 mA, respectively (entry 9 vs entry 10)]. The same principle could be applied to achieve a selectivity switch in the opposite direction – from 3b to 3a [same 100 ms pulse but with 20 or 10 mA, respectively (entry 6 vs entry 11)]. This dependency of current efficiency is reasonable since more energy is consumed for charging and discharging electric double layer at higher frequency. (See Supporting Information for details.) The highest yield of 3b was observed by adjusting the number of electrons (16 F/mol) at 20 mA and 50 ms pulse duration (entry 12), resulting in 76% isolated yield after 1.5 h of electrolysis. The aforementioned rAP experiments were all run using constant current, but the technique could also be applied in a constant potential mode. Thus, potential-controlled rAP at a similar cell voltage to entry 12 (alternating ±8 V) with the same pulse duration also led to efficient formation of 3b (entry 13). On the other hand, the use of a sinusoidal waveform (conventional AC) at the same terminal potential and frequency gave 3a instead of 3b, indicating a much lower current efficiency with this waveform (entry 14).15
Figure 2.
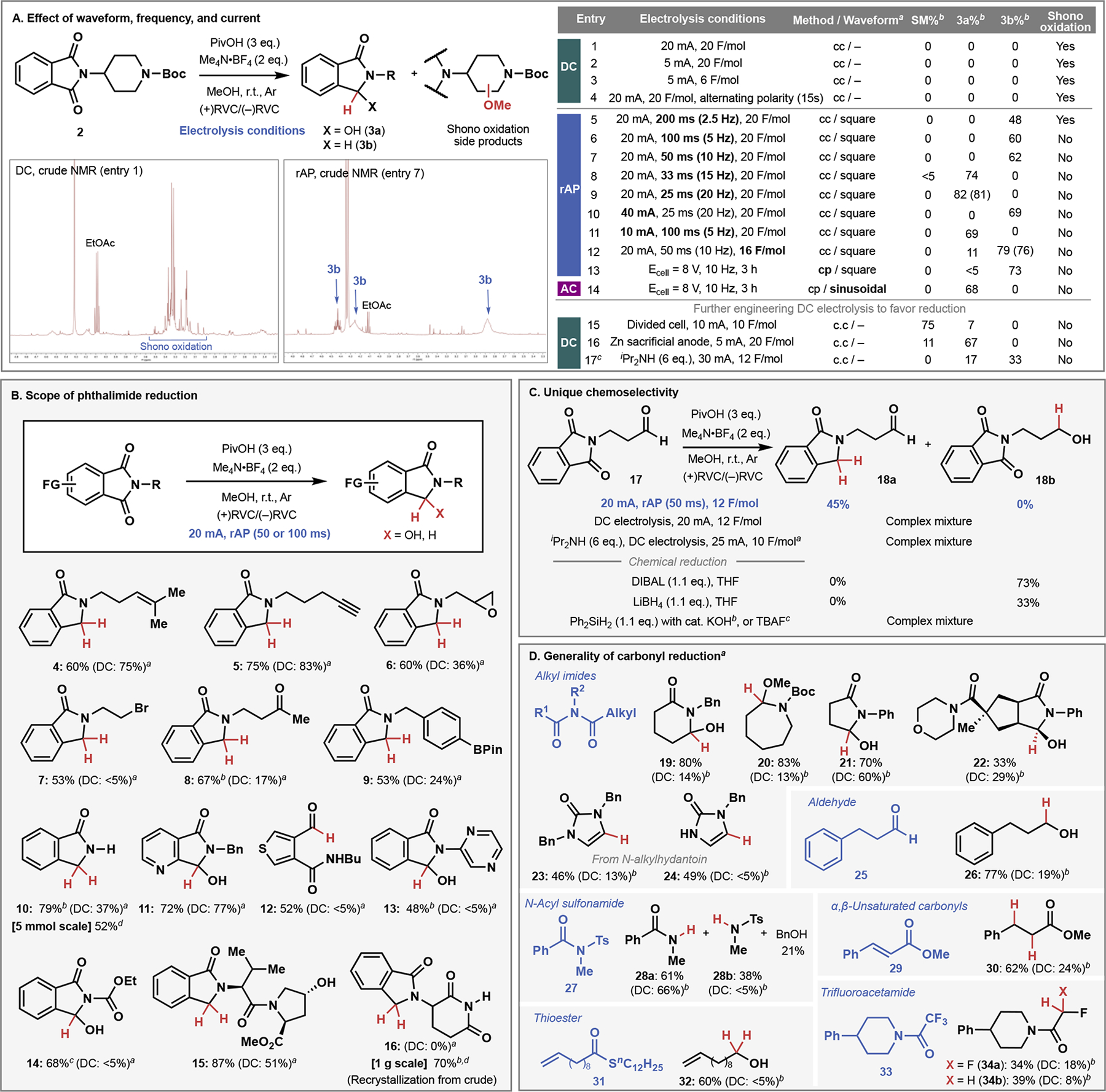
Difference between rAP and DC electrolysis in general carbonyl reduction. (A) A striking difference in reaction outcome by employing rAP with various frequencies and currents. acc = constant current, cp = constant potential. bNMR yield. Isolated yields are shown in parenthesis. cConditions adopted from reference 27. (B) Functional group tolerance in the reduction of phthalimides benchmarked with the latest DC electrolysis conditions. Reactions were performed on 0.05–0.1 mmol scale unless otherwise noted. aConditions were adopted from reference 27. bMeCN/tBuOH (1:1) was used as solvent. cMeCN was used as solvent. d100 mA was applied instead of 20 mA. (C) Discovery of chemoselectivity unachievable by known synthetic methods. aConditions were adopted from reference 27. bConditions were adopted from reference 31. cConditions were adopted from reference 30. (D) Generality of carbonyl reduction by rAP. aSee Supporting Information for reaction conditions for each substrate. bDC conditions were adopted from reference 27.
Having established rAP as a unique mode of electrolysis distinctly different from conventional DC electrolysis, one question remained: is the observed reduction unachievable by DC with further reaction engineering? In principle, Shono oxidation products could be suppressed simply using a divided cell or employing a sacrificial anode to avoid such an oxidative side reaction. Towards that end, the reaction with DC was carried out in a divided cell, furnishing 3a in low yield (entry 15). This inefficient reduction was partly accounted for the high resistance present in a divided cell setup. The reaction with a Zn sacrificial anode (entry 16) also resulted in a similar outcome (lactam 3b was not observed). Finally, employing a sacrificial electron donor (e−-donor = iPr2NH) instead of a sacrificial anode, under the conditions reported by Xiang et al., indeed afforded a 2:1 mixture of 3a and 3b (entry 17).27 As presented below, further comparison with these conditions unveiled that rAP-based reduction exhibits a dramatically expanded scope and generality in both simple and demanding settings.
To further gauge the synthetic utility of reductive rAP over conventional DC electrolysis, it was field-tested in synthetically relevant contexts (Figure 2B). Isoindolinone derivatives are an important class of molecules in both medicinal and agricultural chemistry, and routes to such molecules usually employ stepwise ring construction rather than late-stage reduction of a phthalimide.28 Reductively labile alkene (4), alkyne (5), epoxide (6), alkyl bromide (7), and ketone (8) motifs were all tolerated in addition to the oxidatively labile C–B bond (9). Efficient reduction of N-unsubstituted phthalimide ($0.1/g) afforded a valuable building block 10 ($576/g in Sigma-Aldrich). MeCN/tBuOH mixture was used instead to improve the solubility of a substrate. In the case of heteroaromatic imides, hemiaminal was obtained as a sole product (11-13). This could be explained by that dissociation of OH group is not favorable due to the presence of intramolecular hydrogen bonding (11 and 13) or causing considerable ring strain (12). N-Ethoxycarbonyl phthalimide was found to be reduced cleanly to the hemiaminal 14 by employing MeCN as solvent. This reaction was also effective for a peptidic substrate bearing an unprotected alcohol, affording 15 in high yield. The synthetic utility was further demonstrated in the successful reduction of unprotected thalidomide on gram-scale, affording deoxythalidomide 1629 in one step after simple recrystallization from the crude mixture. In addition, negligible epimerization (~3%) of the base-sensitive stereocenter was confirmed, when enantioenriched thalidomide was used (See Supporting Information). Amongst the various substrates studied, one of the most surprising selectivities was observed with phthalimide 17: selective deoxygenation of phthalimide without reducing the alkyl aldehyde (Figure 2C). The remaining mass balance was largely accounted for the presence of hemiaminal intermediate. This striking chemoselectivity was indeed unique to rAP as conventional direct electrolysis, both under the same reaction conditions as well as with e−-donor, led to extensive decomposition of 17. Well-established chemical reductants such as DIBAL and LiBH4 gave reduction of aldehyde preferentially, and reported chemical conditions specifically invented for phthalimide reduction gave a complex mixture.30,31 Finally, Figure 2D illustrates the generality of the rAP reduction across various types of carbonyl compounds. In general, electron-deficient carbonyl compounds are susceptible to this reduction, whereas simple esters, ketones, and amides are not affected. Hemiaminals were cleanly formed in the reduction of various alkyl imides, including 22 from bicyclic succinimide derivative conveniently accessible by Yu’s Pd-catalyzed C–H functionalization.32 The reduction of N-alkylhydantoins led to the formation of imidazolone instead of hemiaminal due to facile dehydration of the product hemiaminal. Although chemical reduction of fully protected hydantoins is known, reduction of mono-protected systems (to yield compounds such as 24) is challenging due to the free N–H.30 While the alkyl aldehyde was tolerated during the reduction of 17, hydrocinnamyl aldehyde 25 could be reduced under slightly modified rAP conditions by employing a longer pulse duration (see Supporting Materials for detail). Exposure of the simple N-acylsulfonamide (27) to rAP led to a mixture of desulfonylated amide 28a and sulfonamide 28b (along with benzyl alcohol in 21%). α,β-Unsaturated ester 29 and thioester 31 could also be reduced to the corresponding saturated ester (30) and alcohol (32), respectively. Reduction of trifluoroacetamide-containing piperidine (33) delivered unexpected mono- and bis-defluorinated products (34a/b) – such an outcome had no known chemical counterpart until the very recent discovery of radical-based defluorinative transformation, indicating that rAP could exhibit such interesting reactivity under simple conditions.33 Through the entirety of Figure 2, conventional DC electrolysis with a sacrificial e−-donor was run in parallel with rAP-based reduction; in nearly all cases rAP was found to be superior with more complex cases failing completely under DC (7, 12, 13, 14, 16, 17, 24, 27, 31).
The synthetic utility of the unique reactivity enabled by rAP was demonstrated in two popular contexts: chiral auxiliary removal and an emerging area of medicinal chemistry known as proteolysis targeting chimeras (PROTACs). In the first instance (Figure 3A), it is well-recognized that reductive removal of Evans auxiliary to provide an aldehyde is challenging especially in the presence of other reducible carbonyl groups. A recently reported synthesis of chiral aldehyde 37 is an emblematic example of this lingering issue.34 In order to prepare this simple aldehyde, a nine-step route involving three protecting group manipulations and five redox manipulations was required. Only one step forged a key C–C bond (allylation) that needed several corrective concession steps to install the requisite t-butyl ester group. In contrast, commencing with the same starting material (35), direct alkylation with t-butyl acrylate followed by subjecting 36 to reductive rAP cleanly delivered the hemiaminal that could be easily cleaved during workup to reveal aldehyde 37. It was confirmed that other representative chemical reductants such as DIBAL and LiBH4 led to either reduction of the ester or overreduction of the desired aldehyde (see Supporting Information). Comparable DC electrolysis with or without an e−-donor led to a complex mixture of products (37 not detected). It is worth pointing out that the alkylation of acrylates with Evans auxiliary35 is a rarely employed tactic in synthesis, perhaps due to the heretofore intractable challenge of chemoselective reduction.
Figure 3.
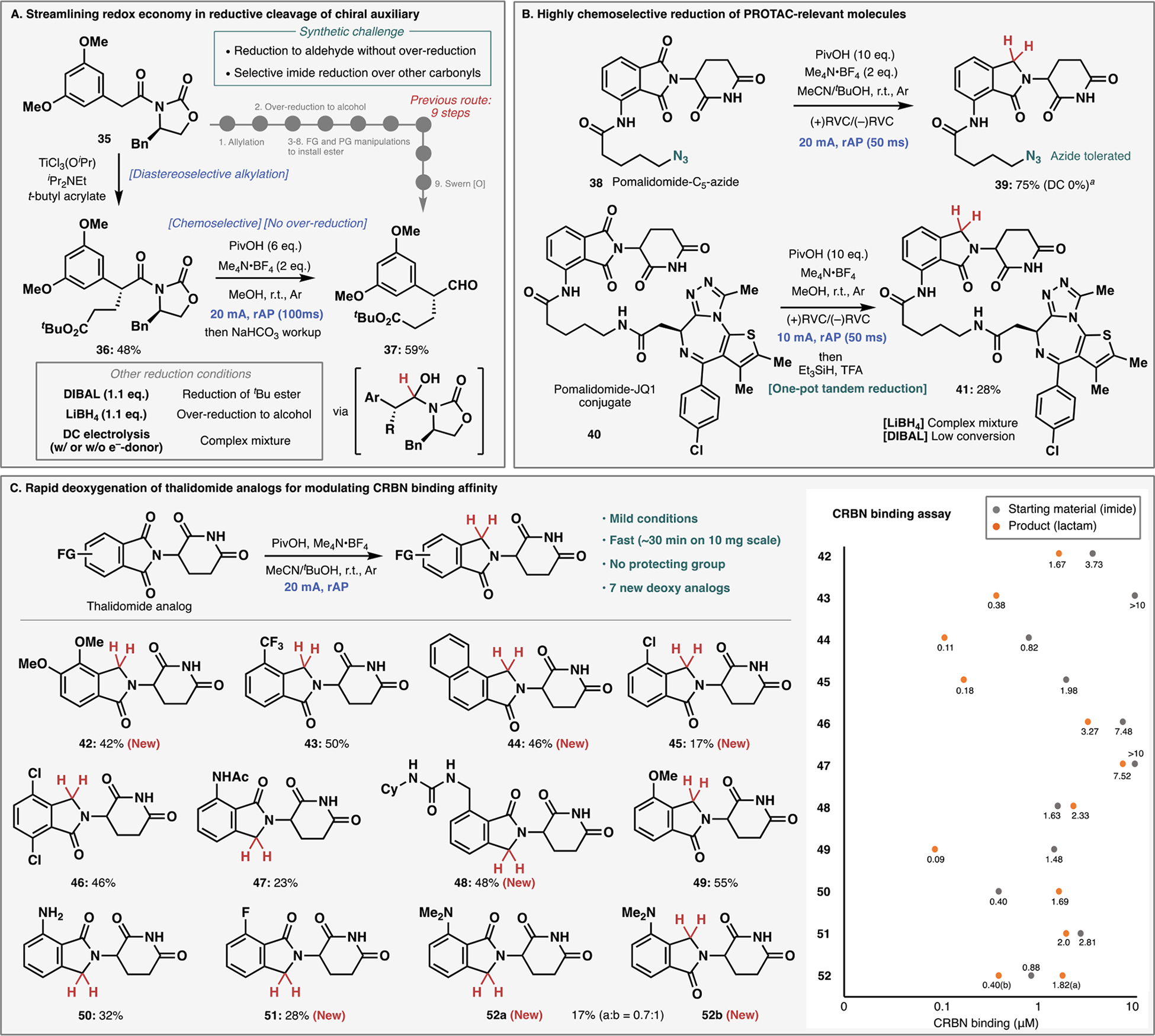
Synthetic applications of rAP. (A) Reductive removal of Evans auxiliary with high chemoselectivity among multiple carbonyl groups. (B) Highly chemoselective reduction for diversification of PROTAC molecules. (C) Rapid deoxygenation of thalidomide analogs and modulation of cereblon binding affinity.
In the second major application of reductive rAP, molecules relevant to the burgeoning field of PROTACs were investigated. Pomalidomide- (bearing a phthalimide) and lenalidomide-type (bearing an isoindolinone) scaffolds are cereblon E3 ligase binders used as standalone treatments in oncology (Pomalyst® or Revlimid®) or are incorporated into PROTAC design to elicit protein degradation. The bioactivity of PROTAC structures can vary based on the presence or absence of a single carbonyl group.36 Historically such entities are prepared through a de novo synthesis, whereas late-stage single-step reductive deoxygenations are, to our knowledge, without precedent. As shown in Figure 3B, reductive rAP deoxygenation of PROTAC building block 38 smoothly proceeded in the presence of azide functionality with no protection on the glutarimide subunit.37 The complete PROTAC molecule, pomalidomide-JQ1 conjugate 4038 was subjected to several reduction conditions, with rAP as the only method capable of reducing 40 in synthetically meaningful yield. Due to the sensitivity of the JQ1-heterocycle, a two-step tandem reduction was performed in this case.
The synthetic feasibility of this methodology was further explored by enlisting a small library of thalidomide-type analogs (Figure 3C). Under standard conditions, facile reduction was achieved to give the corresponding isoindolinone products in synthetically useful yield. Electron-donating groups were well tolerated to give products 42, 49, 50, and 52 as well as the extended aromatic 44. As suggested by products 43, 45, 46, and 51, electron-withdrawing groups were also able to withstand the reduction with dehalogenation being largely suppressed by the addition of TFA (for details, please see Supporting Information). The exquisite chemoselectivity of this transformation is highlighted by the incorporation of acyl and urea-protected amines (products 47 and 48). In both cases, no overreduction was observed with the desired products isolated in 23% and 48% yield, respectively.
With newly synthesized analogs in hand, they were analyzed in an HTRF-Tb (Homogeneous Time-Resolved Fluorescence – Terbium-based)39 cereblon-binding assay (Figure 3C, binding assay table). From this study, 9 out of 12 newly synthesized lenalidomide-type analogs showed greater binding affinity to cereblon than their thalidomide-type starting materials. Of note, 43 now shows sub-micromolar binding affinity, a drastic improvement to its parent compound having no measurable binding. Only in the cases of 48, 50, and 52a was a slight loss in affinity for cereblon observed. This exciting result underscores the benefit this new methodology may bring to the cereblon biochemistry field and, by extension, the protein degradation realm.
The unconventional outcomes observed on complex structures when employing rAP can be explained by interplay of two independent factors: chemoselectivity dictated by themodynamics (redox potential or LUMO) and reaction selection dictated by kinetics on an electrode. The former, chemoselectivity, can be rationalized by redox potentials of organic functional groups as well as frontier molecular orbital theory (Figure 4A). From the standpoint of reduction potential, the phthalimide carbonyl has the highest reduction potential (ca. −1.4 V), followed by alkyl aldehyde (ca. −1.8 V), alkyl ketone (ca. −2.4 V), and amide/ester functionalities (both <−2.5 V).6,40 Thus, these reduction potentials clearly explain the observed selectivity on substrates with multiple carbonyl groups such as 17 and 53. The difficulty in harnessing this difference using conventional chemical reductants could partly stem from the fundamental mechanistic difference in the reduction: with chemical reagents, amides and esters are generally reduced through the interaction between a carbonyl oxygen and a Lewis-acidic metal in a reagent.41 Even aldehyde, one of the most trivial functional groups to reduce by chemical reagents, was tolerated in the rAP-based reduction of 17, representing the most pronounced difference between rAP and chemical reductants. Simply stated, such selectivity is counter to textbook chemical intuition. Since the reduction potential corresponds to the LUMO (lowest unoccupied molecular orbital) based on Koopmans’ theorem, the aforementioned selectivity can be rationalized based on the calculated LUMO coefficient, which provides convenient prediction even without relying on experimental electrochemical techniques such as cyclic voltammetry. As illustrated in Figure 4A, carbonyl carbons with the largest LUMO coefficient are in good agreement with experimental reactive sites (17 and 53). When there were several carbonyl carbons with similar LUMO coefficients, mixtures of products were obtained (54, vide infra). Thus, the selectivity observed invokes a single-electron transfer (SET)-based reduction to the radical anion, followed by protonation. Experimental support for this notion was obtained using substrate 54, wherein substantial dechlorination was observed under standard conditions, indicating the intermediacy of a radical anion (Figure 4B).42 Suppression of the dechlorination by the addition of strong acid such as TFA also indicates that the intermediate is sensitive to reaction pH, supporting the existence of an anionic intermediate. The source of hydrogen could also be elucidated using substrate 56 under rAP in MeOH-d4 solvent wherein deuterium incorporation was observed on the methylene of lactam 58-d (Figure 4C). Such an inexpensive way of installing a stable-labeled isotope may find use in the synthesis of isotopically labeled materials. The fate of the carboxylic acid could also be followed in this reaction, delivering ether 59, derived from decarboxylative carbocation formation.43
Figure 4.
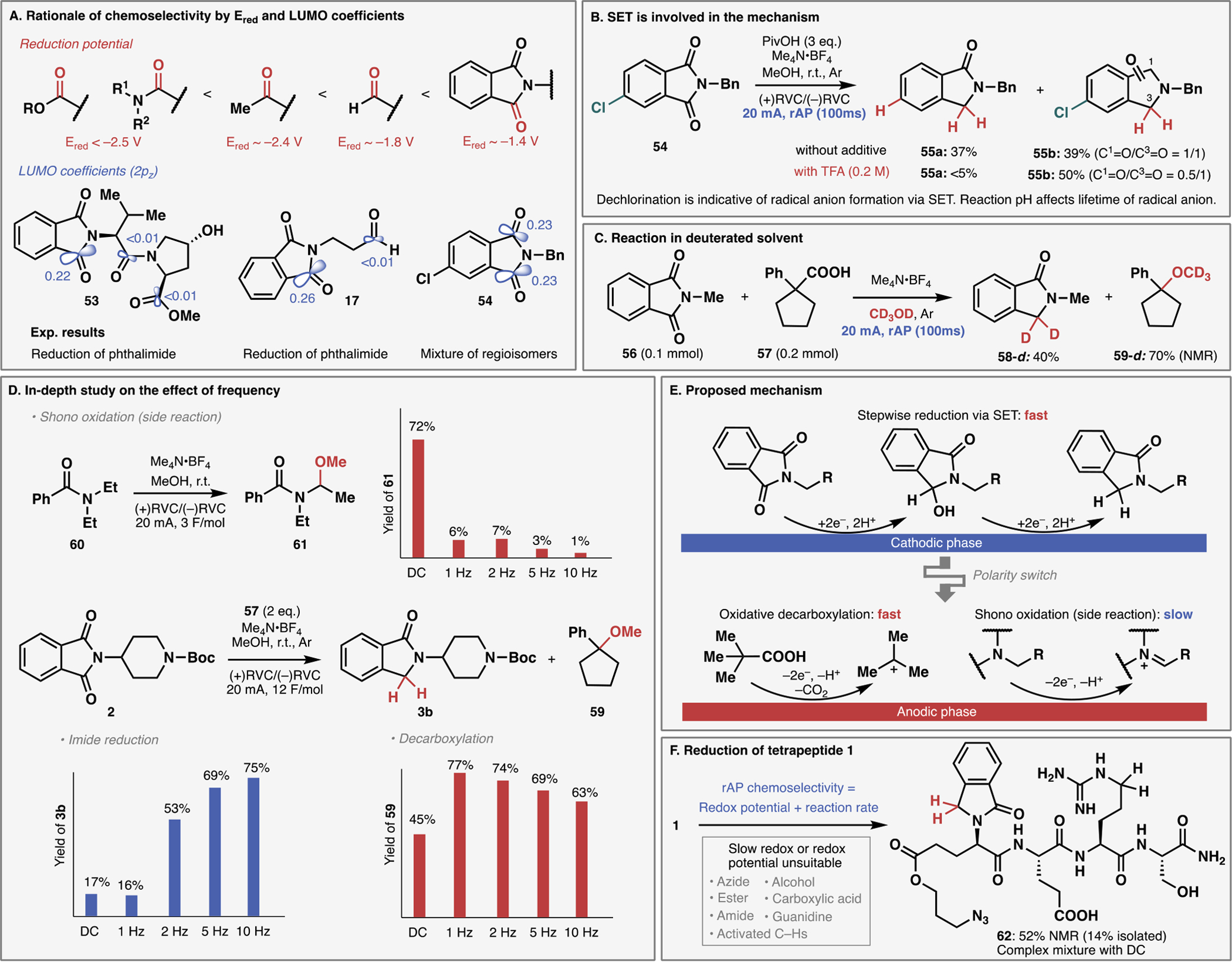
Experimental and computational rationale for unique reactivity of rAP. (A) Chemoselectivity follows reduction potential and can be predicted by FMO analysis. (B) Experimental evidence to support SET mechanism. (C) Deuterium labeling study to identify the source of proton and the fate of carboxylic acid. (D) Frequency-dependent reaction profile for carbonyl reduction, oxidative decarboxylation, and Shono oxidation. (E) Proposed mechanism based on Figure 4A–4D. (F) Demonstration of precise redox reaction based on the understanding of rAP chemoselectivity.
The most striking feature of rAP is that it enables reaction selection simply by virtue of its differing waveform, a phenomenon that has yet to be appreciated in synthetic chemistry.
The unique parameter associated with rAP, namely frequency (pulse duration), was therefore studied in more depth to confirm the unconventional reaction selection under rAP. The frequency dependence of each elementary reaction – imide reduction, decarboxylation, and Shono oxidation – was interrogated individually (Figure 4D). Since the Shono oxidation is a competing reaction in phthalimide reduction to generate multiple products, simplified substrate 60 was used to obtain the frequency-dependent profile.44 Frequency had a major impact on the progress of the Shono oxidation; the yield of 61 was significantly reduced even at a frequency as low as 1 Hz (500 ms pulse). The slow kinetics of the Shono oxidation are presumably related to intrinsically high overpotential associated with amide oxidation.45 Substrate 2 could be studied in a similar fashion with the converse observation that the yield of lactam 3b increased as frequency increased. The frequency had a relatively minor impact on the progress of oxidative decarboxylation of 57, although the yield of 59 decreases moderately as frequency increases. Taken together, a proposed mechanism consistent with these observations is depicted in Figure 4E. During the cathodic phase, reduction of the imide proceeds via SET followed by protonation. Chemoselectivity among multiple reducible functional groups during this reduction step can be predicted by redox potentials or LUMO coefficient. During the anodic phase of the pulse, the carboxylic acid is oxidatively decarboxylated, generating carbocation that is eventually trapped by solvent. Progress of the competitive Shono oxidation is considerably abrogated above a certain frequency, which accounts for reaction differentiation between rAP and conventional DC. In other words, a functional group affected by rAP is the one that is most redox-active among functional groups that have reasonably fast electrode kinetics. This rule is most emphasized in the reduction of the peptidic substrate 1 mentioned at the outset of this work (Figure 1); exposure to rAP-based reduction resulted in 52% NMR yield of lactam 62 without disrupting the numerous redox-active and epimerizable stereocenters (Figure 4F).
Conclusion
This work describes unique chemoselectivity of rAP that is unprecedented and unachievable by DC electrolysis, and provides the rational for its origin. Such control of chemoselectivity represents a unique direction for synthetic organic electrochemistry and a practical technique that is currently outside the reach of conventional chemical reagent space. As such, it warrants further study from both mechanistic and reaction discovery standpoints.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. D.-H. Huang and Dr. L. Pasternack (Scripps Research) for assistance with NMR spectroscopy; Dr. J. Chen (Automated Synthesis Facility, Scripps Research) for purification of compounds and acquisition of HRMS data; Dr. A. Ramirez (BMS) for DFT calculation; M. Bird (Scripps Research) for the synthesis of oligopeptides. We would like to acknowledge the BMS biology team for their contributions running the cereblon binding assay. We also thank Dr. S. B. J. Kan and Dr. S. Gnaim for advice and suggestions.
Funding Sources
Financial support for this work was provided by National Science Foundation Center for Synthetic Organic Electrochemistry (CHE-2002158), and the National Institutes of Health (grant number GM-118176).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, additional experimental data, the detail of DFT calculation (PDF)
There is an article concerning the asymmetric current distribution during an alternating polarity experiment using ElectraSyn2.0. (Wills, A. G.; Poole, D. L.; Alder, C. M.; Reid, M. A Mechanistic and Cautionary Case Study on the Use of Alternating Potential in Electrochemical Reactions. ChemElectroChem 2020, 7, 2771–2776.). This problem was observed only when a reference electrode was used in an alternating polarity experiment. Although this issue was already resolved by IKA, the authors declare that a reference electrode was not used in any of the experiments in this study. Accordingly, experimental results displayed in this article are not affected by such an issue.
REFERENCES
- 1.Shenvi RA; O’Malley DP; Baran PS Chemoselectivity: the mother of invention in total synthesis. Acc. Chem. Res 2009, 42, 530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sierra MA; de la Torre MC; Cossio FP; Hoffmann R More Dead Ends and Detours: En Route to Successful Total Synthesis (Wiley, Hoboken, NJ, 2013). [Google Scholar]
- 3.Yao T Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock RC Ed. (Wiley, Hoboken, NJ, ed. 3, 2018). [Google Scholar]
- 4.Wuts PGM Greene’s Protective Groups in Organic Synthesis, (Wiley, Hoboken, NJ, ed. 5, 2014). [Google Scholar]
- 5.Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- 7. Alternating polarity in organic electrosynthesis commonly refers to switching electrode polarity periodically at intervals of several seconds to minutes to alleviate electrode fouling (Please also see Reference 26 for additional references). In this work rAP is distinguished from this type of slow alternating current process. In addition, it is shown in Figure 2A that alternating polarity at every 15 s did not alter the reaction outcome significantly.
- 8.Rodrigo S; Gunasekera D; Mahajan JP; Luo L Alternating current electrolysis for organic synthesis. Current Opinion in Electrochemistry 2021, 28, 100712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bard AJ; Faulkner LR Electrochemical Methods: Fundamentals and Applications, 2nd Edition. (Wiley Global Education, 2000). [Google Scholar]
- 10.Leisner P; Zanella C; Belov I; Edström C; Wang H Influence of anodic pulses and periodic current reversion on electrodeposits. Transactions of the IMF, 2014, 92, 336–342. [Google Scholar]
- 11.(a) Xu Y; Edwards JP; Liu S; Miao RK; Huang JE; Gabardo CM; O’Brien CP; Li J; Sargent EH; Sinton D Self-Cleaning CO2 Reduction Systems: Unsteady Electrochemical Forcing Enables Stability. ACS Energy Lett 2021, 6, 809–815. [Google Scholar]; (b) Kimura KW; Casebolt R; DaSilva JC; Kauffman E; Kim J; Dunbar TA; Pollock CJ; Suntivich J; Hanrath T Selective Electrochemical CO2 Reduction during Pulsed Potential Stems from Dynamic Interface. ACS Catal, 2020, 10, 8632–8639. [Google Scholar]; (c) Kimura KW; Fritz KE; Kim J; Suntivich J; Abruña HD; Hanrath T Controlled Selectivity of CO2 Reduction on Copper by Pulsing the Electrochemical Potential. ChemSusChem 2018, 11, 1781–1786. [DOI] [PubMed] [Google Scholar]
- 12.For selected examples of pioneering investigation, see; (a) Ghosh JC Alternating Current Electrolysis, J. Am. Chem. Soc 1914, 36, 2333–2346. [Google Scholar]; (b) Shipley JW; Rogers MT The Electrolysis of Some Organic Compounds with Alternating Current. Can. J. Res 1939, 17, 147–158. [Google Scholar]
- 13.(a) Srivastava SC; Shukla SN Electrolysis of sodium salts of aliphatic acids with ac. Electrochim. Acta 1967, 12, 1049–1057. [Google Scholar]; (b) Fleischmann M; Goodridge F Anodic oxidation under pulse conditions. Discuss. Faraday Soc 1968, 45, 254–260. [Google Scholar]; (c) Smeltzer JC; Fedkiw PS Reduction of Nitrobenzene in a Parallel‐Plate Reactor: I. Differential Conversion Using Periodic Cell‐Voltage Control. J. Electrochem. Soc 1992, 139, 1358–1365. [Google Scholar]; (d) Alkire RC; Tsai JE Electrolysis of Propylene Oxide with Alternating Current. J. Electrochem. Soc 1982, 129, 1157–1158. [Google Scholar]
- 14.Schotten C; Taylor CJ; Bourne RA; Chamberlain TW; Nguyen BN; Kapur N; Willans CE Alternating polarity for enhanced electrochemical synthesis. React. Chem. Eng 2021, 6, 147–151. [Google Scholar]
- 15.Rodrigo S; Um C; Mixdorf JC; Gunasekera D; Nguyen HM; Luo L Alternating Current Electrolysis for Organic Electrosynthesis: Trifluoromethylation of (Hetero)arenes. Org. Lett 2020, 22, 6719–6723. [DOI] [PubMed] [Google Scholar]
- 16.Bortnikov EO; Semenov SN Coupling of Alternating Current to Transition-Metal Catalysis: Examples of Nickel-Catalyzed Cross-Coupling. J. Org. Chem 2021, 86, 782–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sattler LE; Otten CJ; Hilt G Alternating Current Electrolysis for the Electrocatalytic Synthesis of Mixed Disulfide via Sulfur–Sulfur Bond Metathesis towards Dynamic Disulfide Libraries. Chem. Eur. J 2020, 26, 3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee B; Naito H; Nagao M; Hibino T Alternating-Current Electrolysis for the Production of Phenol from Benzene. Angew. Chem. Int. Ed 2012, 51, 6961–6965. [DOI] [PubMed] [Google Scholar]
- 19.Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemistry: Calling All Engineers. Angew. Chem. Int. Ed 2017, 57, 4149–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien AG; Maruyama A; Inokuma Y; Fujita M; Baran PS; Blackmond DG Radical C–H Functionalization of Heteroarenes under Electrochemical Control. Angew. Chem. Int. Ed 2014, 53, 11868–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Dea JJ; Osteryoung J; Osteryoung RA Theory of Square-Wave Voltammetry for Kinetic Systems. Anal. Chem 1981, 53, 695–701. [Google Scholar]
- 22.Fedkiw PS; Scott WD Selectivity Changes in Electrochemical Reaction Sequences by Modulated Potential Control. J. Electrochem. Soc 1984, 131, 1304–1315. [Google Scholar]
- 23.Bakshi R; Fedkiw PS Optimal Time-Varying Potential Control. J. Appl. Electrochem 1993, 23, 715–727. [Google Scholar]
- 24.Blanco DE; Lee B; Modestino MA Optimizing organic electrosynthesis through controlled voltage dosing and artificial intelligence. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 17683–17689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto K; Kuriyama M; Onomura O Shono-type Oxidation for Functionalization of N-Heterocycles. Chem Rec 2021, doi: 10.1002/tcr.202100031 [DOI] [PubMed] [Google Scholar]
- 26.For selected examples of slow alternating polarity, see:; (a) Umezawa M; Kojima M; Ichikawa H; Ishikawa T; Nonaka T Electroreductive Polymerization of Dichlorocarbosilanes, Electrochem. Acta 1993, 38, 529–533. [Google Scholar]; (b) Kashimura S; Ishifune M Electroreductive Synthesis of Silicon Containing Polymers. Journal of Synthetic Organic Chemistry, Japan, 2000, 58, 966–974. [Google Scholar]; (c) Lu L; Siu JC; Lai Y; Lin S An Electroreductive Approach to Radical Silylation via the Activation of Strong Si–Cl Bond. J. Am. Chem. Soc 2020, 142, 21272–21278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai Y; Shi L; Zheng L; Ning S; Che X; Zhang Z; Xiang J Electroselective and Controlled Reduction of Cyclic Imides to Hydroxylactams and Lactams. Org. Lett 2021, 23, 2298–2302. [DOI] [PubMed] [Google Scholar]
- 28.Hu S; Yuan L; Yan H; Li Z Design, synthesis and biological evaluation of Lenalidomide derivatives as tumor angiogenesis inhibitor. Bioorg. Med. Chem. Lett 2017, 27, 4075–4081. [DOI] [PubMed] [Google Scholar]
- 29.Luzzio FA; Mayorov AV; Ng SSW; Kruger EA; Figg WD Thalidomide Metabolites and Analogues. 3. Synthesis and Antiangiogenic Activity of the Teratogenic and TNFα-Modulatory Thalidomide Analogue 2-(2,6-Dioxopiperidine-3-yl)phthalimidine 1. J. Med. Chem 2003, 46, 3793–3799. [DOI] [PubMed] [Google Scholar]
- 30.Das S; Addis D; Knöpke LR; Bentrup U; Junge K; Brückner A; Beller M Selective Catalytic Monoreduction of Phthalimides and Imidazolidine-2,4-diones. Angew. Chem. Int. Ed 2011, 50, 9180–9184. [DOI] [PubMed] [Google Scholar]
- 31.Ding G; Li C; Shen Y; Lu B; Zhang Z; Xie X Potassium Hydroxide-Catalyzed Chemoselective Reduction of Cyclic Imides with Hydrosilanes: Synthesis of ω-Hydroxylactams and Lactams. Adv. Synth. Catal 2016, 358, 1241–1250. [Google Scholar]
- 32.Park H; Yu J-Q Palladium-Catalyzed [3 + 2] Cycloaddition via Twofold 1,3-C(sp3)-H Activation. J. Am. Chem. Soc 2020, 142, 16552–16556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y-J; Zhang F-L; Peng T-Y; Wang C-L; Cheng J; Chen C; Houk KN; Wang Y-F Sequential C-F bond functionalizations of trifluoroacetamides and acetates via spin-center shifts. Science 2021, 371, 1232–1240. [DOI] [PubMed] [Google Scholar]
- 34.Guo Y-A; Zhao M; Xu Z; Ye T Total Synthesis and Stereochemical Assignment of Actinoranone. Chem. Eur. J 2017, 23, 3572–3576. [DOI] [PubMed] [Google Scholar]
- 35.Evans DA; Bilodeau MT; Somers TC; Clardy J; Cherry D; Kato Y Enantioselective Michael Reactions. Diastereoselective Reactions of Chlorotitanium Enolates of Chiral N-Acyloxazolidinones with Representative Electrophilic Olefins. J. Org. Chem 1991, 56, 5750–5752. [Google Scholar]
- 36.Petzold G; Fischer ES; Thomä NH Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 2016, 532, 127–130. [DOI] [PubMed] [Google Scholar]
- 37.Wurz RP; Dellamaggiore K; Dou H; Javier N; Lo M-C; McCarter JD; Mohl D; Sastri C; Lipford JR; Cee VJ A ‘Click Chemistry Platform’ for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation. J. Med. Chem 2018, 61, 453–461. [DOI] [PubMed] [Google Scholar]
- 38.Crew AP Crews C; Dong H; Wang J; Qian Y; Siu K; Jin M Imide-based modulators of proteolysis and associated methods of use. U. S. Patent, US2016/0058872, March 3, 2016.
- 39.Degorce F; Card A; Soh S; Trinquet E; Knapik GP; Xie B HTRF: A technology tailored for drug discovery–a review of theoretical aspects and recent applications. Curr. Chem. Genomics, 2009, 3, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griesbeck AG; Schieffer S “Electron‐Transfer Reactions of Carbonyl Compounds” in Electron Transfer in Chemistry, Balzani V, Ed. (Wiley, Hoboken, NJ, 2001), pp. 457–493. [Google Scholar]
- 41.Ashby EC; Laemmle JT Stereochemistry of Organometallic Compound Addition to Ketones. Chem. Rev 1975, 75, 521–546. [Google Scholar]
- 42.Bunnett JF Aromatic substitution by the SRN1 mechanism. Acc. Chem. Res 1978, 11, 413–420. [Google Scholar]
- 43.Leech MC; Lam K Electrosynthesis Using Carboxylic Acid Derivatives: New Tricks for Old Reactions. Acc. Chem. Res 2020, 53, 121–134. [DOI] [PubMed] [Google Scholar]
- 44.Alfonso-Súarez P; Kolliopoulos AV; Smith JP; Banks CE; Jones AM An experimentalist’s guide to electrosynthesis: the Shono oxidation. Tetrahedron Lett 2015, 56, 6863–6867. [Google Scholar]
- 45.Wang F; Stahl SS Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron−Proton Transfer Mediators. Acc. Chem. Res 2020, 53, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.