

Abstract
The effects of a vasoactive intestinal peptide (VIP) receptor antagonist on mammary carcinogenesis were investigated using the C3(1)SV40T antigen (ag) mice. Ten μg/day VIPhybrid (VIPhyb) administered daily subcutaneously increased significantly the survival of C3(1)SV40Tag mice. At 5.2 months, VIPhyb significantly reduced the mammary tumor burden in C3(1)SV40Tag mice relative to control animals. 125I-VIP bound with high affinity to mouse mammary tumor homogenate. Because (Lys15, Arg16, Leu27)VIP1–7GRF8–27 (VPAC1 selective) but not Ro25–1553 (VPAC2 selective) inhibited specific 125I-VIP binding to mammary tumor membranes with high affinity, VPAC1 receptors predominate. By RT-PCR, VPAC1 receptor mRNA was detected in mammary tumors. By Western blot, a major 60 Kdalton band was detected in mammary tumor extracts using VPAC1 receptor antisera. By immunocytochemistry, VPAC1-R immunostaining was detected in the cytosol and plasma membrane but not the nucleus of fixed mammary tumor tissue. Using laser capture microdissected tumor cells and surface enhanced laser desorption/ionization (SELDI) techniques on mammary tumor cells, the proteomic profile was altered in mice treated with VIPhyb. Because VPAC1 receptor antagonists increase the survival and reduce the tumor burden in C3(1)SV40Tag mice, they may function as chemopreventive agents in mammary cancer.
Keywords: VIP receptor antagonist, Mammary carcinogenesis, Transgenic mice, Chemoprevention, Proteomics
Introduction
Over 40,000 women in the United States will die annually from breast cancer, which is a highly diagnosed neoplasm in women (Landis et al., 1999). Breast lesions, which can be benign, in situ or invasive, and premalignant lesions, including hyperplasia, are associated with increased risk for developing breast cancer (Marshall et al., 1997). Breast carcinogenesis is associated with loss of function of tumor suppressor genes such as p53, BRCA1, BRCA2 and/or Rb (Dickson and Lippman, 1997). Oncogenes, which can be amplified in breast cancer, include erbB2, EGF receptor, c-myc and/or cyclin D1 (Ram et al., 1995; Dickson and Lippman, 1997). Many breast cancers are estrogen-receptor positive and in the breast cancer prevention trial the anti-estrogen tamoxifen causes a 49% reduction of breast cancer in high risk women (Fisher et al., 1998; Cronin et al., 1998). Also, the antiestrogen, raloxifene, reduced breast cancer incidence (Cummings et al., 1999). There is a need to develop additional agents to reduce incidence of hormone-independent breast cancer.
Animal models have been developed to study breast carcinogenesis. These include rats using 7,12-dimethylbenz(a)anthracene (DMBA) or nitrosomethylurea (NMU) to induce mammary tumors (Clarke, 1997). Retinoids such as retinyl acetate and N-(4-hydroxyphenyl)retinamide (fenretinide)reduce tumor incidence and multiplicity (Moon et al., 1976, 1983). In C3(1)SV40 Tag transgenic mice, mice treated with 50 mg/kg 9-cis-retinoic acid (9cRA) had a 20% increase in mean survival time and 67% reduction in tumor multiplicity (Wu et al., 2000). In this transgenic animal model of breast carcinogenesis, SV40Tag causes inactivation of the p53 and Rb genes resulting in spontaneous development of mammary carcinomas (Green et al., 2000; McKenzie and Sukumar, 1996; Tripathy and Benz, 1993). In C3(1)SV40.Tag female mice, mammary hyperplasia spontaneously develops by 3 months, followed by carcinoma in situ lesions and adenocarcinoma by 6 months (Green et al., 2000).
Vasoactive intestinal peptide (VIP) is a 28 amino acid peptide with sequence homology to the 27 amino acid pituitary adenylate cyclase activating polypeptide (PACAP)(Arimura, 1992; Said and Mutt, 1970). VIP binds with high affinity to the G-protein coupled receptors VPAC1 and VPAC2 (Harmar and Lutz, 1993; Ishihara et al., 1992) stimulating adenylyl cyclase. PACAP binds with high affinity to VPAC1, VPAC2 and PAC1 receptors (Pisegna and Wank, 1993; Spengler et al., 1993), the latter of which causes elevated cAMP as well as phosphatidyl inositol turnover; VIP binds with low affinity to PAC1 receptors. Human breast cancer cells bind VIP with high affinity (Gespach et al., 1988; Reubi, 1995; Waschek et al., 1995). The VIP receptor antagonist VIPhyb, inhibited the ability of VIP to elevate cAMP in breast cancer cells and inhibited their proliferation in vitro and in vivo (Gozes et al., 1991; Zia et al., 1996). These results suggest that VIP-like peptides may function as autocrine growth factors in some breast cancer cells. Here the effects of VIPhyb on mammary carcinogenesis were investigated. Our results indicate that mouse mammary tumors have VPAC1 receptors. Use of VIP receptor antagonists increased the lifetime of C3(1)SV40Tag transgenic mice, decreased tumor multiplicity and altered the tumor cell protein expression profile.
Materials and methods
Carcinogenesis
Female C3(1)SV40Tag transgenic mice were obtained from NCI(Frederick, MD) at 5 weeks of age and 20 of the mice treated subcutaneously with 100 μl of PBS/day or PBS containing 10 μg VIPhyb (neurotensin(6–11)-VIP(7–28)Phoenix Pharmaceuticals, Belmont, CA). Tumor volume was determined twice weekly using electronic calipers and the tumor volume calculated. Weights of all mice were recorded monthly. When the tumors became necrotic (2000 mm3), the animals were euthanized and each tumor dissected and used for histological analysis, mRNA analysis, protein analysis or receptor binding. The research protocol was approved by the NCI Animal Care and Use Committee.
Receptor binding
The ability of mammary tumor membranes to bind VIP-like peptides was investigated (Lee et al., 1990). Mouse mammary tumors were placed in 1 ml of cold homogenization buffer containing 50 mM Tris.HCl (pH 7.4), 0.2 mg/ml soybean trypsin inhibitor, 0.2 mg/ml benzamide and 0.5 mg/ml bacitracin (Sigma Chemical Co., St. Louis, MO). The samples were homogenized in a Polytron at speed 6 for 30 sec, and the membranes centrifuged at 1500 × g to remove nuclear and tissue debris. The supernatant was centrifuged at 20,000 × g for 20 min. The pellet, which contains plasma membranes (0.1 mg), was resuspended in homogenization buffer which contained 50 mM Tris, 0.2 mg/ml benzamide, 0.5 mg/ml bacitracin and 2 mg/ml BSA with 125I-VIP (2200 Ci/mmmol; New England Nuclear; Boston, MA) in the presence or absence of unlabeled peptide (VIP, VIPhyb, R025–1553, PACAP-27 and (Lys15,Arg16,Leu27)VIP(1–7)-GRF(8–27) were purchased from Phoenix Pharmaceuticals whereas (N-Stearyl-Nle17)VIPhyb ((SN)VIPhyb) was provided by Dr. I. Gozes. After incubation at 37 °C for 30 min, free 125I-VIP was removed by washing 3 times in receptor binding medium. The membrane pellets, which contained bound peptide, were counted in a LKB gamma counter.
Immunocytochemistry
Some of the mammary tissue was fixed in 10% formalin for 24 hr, paraffin embedded and cut into 6 mm sections. VPAC1 receptors were assessed by immunocytochemistry (Ebina et al., 1994). One section from each lung was stained with hematoxylin and eosin (H and E) for histological analysis. A rabbit polyclonal antisera was obtained from Dr. E. Goetzl (UCSF) that recognized amino acids 191–222 of the VPAC1 receptor. After removal of paraffin and blocking of endogenous peroxidase in hydrogen peroxide/methanol, the sections were blocked with 1.5% normal goat serum/0.5% bovine serum albumin (BSA), incubated overnight at 4 °C with the antisera (1:200 dilution), washed extensively and incubated with biotinylated goat anti-rabbit IgG and avidin-biotin-enzyme complex. Sections were stained with 3,3′-diaminobenzidine and hydrogen peroxide and counterstained with Mayer’s hematoxlin (Sigma Chemical Co.,St. Louis, MO).
Western blot
Mammary tumor tissue was washed twice with PBS and lysed in buffer containing 50 mM Tris.HCl (pH 7.5), 150 mM sodium chloride, 1% Triton X-100, 1% deoxycholate, 1% sodium azide, 1 mM EGTA, 0.4 M EDTA, 1.5 μg/ml aprotinin, 1.5 μg/ml leupeptin, 1 mM PMSF and 0.2 mM sodium vanadate (Sigma Chemical Co., St. Louis, MO). The lysate was sonicated for 5 sec at 4 °C and centrifuged at 10,000 × g for 15 min. Protein concentration was measured by Bio-Rad protein assay. Protein (10 μg) was analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blot. Membranes were blocked overnight at 4 °C using blotto (5% non-fat dried milk in solution containing 50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM NaCl, 0.05% Tween 20 and 0.02% sodium azide) and incubated with VPAC1 receptor antisera, followed by incubation for 2 hr at 25 °C with anti-rabbit immunoglobulinG-horseradish peroxidase conjugate. The membrane was washed for 10 min with blotto and twice for 10 min with washing solution (50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20 and 0.02% sodium azide). The blot was incubated with enhanced chemiluminescence detection reagent for 5 min and exposed to Hyperfilm ECL.
RT-PCR
Some of the mammary tumors were fixed overnight at 4 °C in ethanol, paraffin embedded, sectioned and dissected using laser capture microdissection (LCM) techniques (Bonner et al., 1997). The UV-laser microbeam (Arcturus, Mountain View, CA) used consisted of high beam precision wavelength (337 nm) coupled to an inverted PixCell microscope via the illumination path. Tumor tissue was selected, traced and photolysed by the laser beam and transferred to a thin polymer film on a microcentrifuge tube cap. After incubating the microdissected sample with 100 μl of guanidine isothiocyanate and h-mercaptoethanol at room temperature for 5 min, RNA was isolated using a RNA microisolation kit with a silica-gel matrix (Strategene, San Diego, CA). RNAse free DNAse I was added to remove genomic DNA. Phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation of the RNA was performed. RNA was eluted from the matrix in 30 μl of RNAse free water. Specific primers for the VPAC1 receptor were 5′-ATGTGCAGATGATCGAGGTG-3′(sense bases 127–146) and 5′-TGTAGCCGGTCTTCACAGAA-3′(antisense bases 431–450). To the cDNA 1 μM primer pair, 100 μM dNTP, 2.5 U Taq polymerase, 2.5 mM MgCl2 and PCR buffer was added. The PCR was performed on a Perkin–Elmer/Cetus thermal cycler; 35 cycles at 94 °C for 30 sec, 62 °C for 45 sec and 72 °C for 60 sec. Amplification products were separated by electrophoresis (2% agarose in TAE buffer) and visualized by ethidium bromide fluorescence.
Proteomics
Protein expression was evaluated by Surface enhanced laser desorption/ionization (SELDI; Wulfkuhle et al., 2001). Frozen mouse mammary tumors were sectioned and dehydrated in ethanol containing protease inhibitors followed by xylene. The sections were frozen at −80 °C until use. The mammary tumor cells were dissected using LCM techniques and the caps frozen at −80 °C until protein extraction. Two thousand microdissected cells were procured and lysed directly on an LCM cap with 1 μl of 1% NP-40. A H4 Protein ChipTM (Ciphergen Biosystems, Freemont, CA) was pretreated with 1 μl of acetonitrile. Shortly before the acetonitrile evaporated the lysate was applied to the bait surface. The analyte was concentrated by air-drying followed by 2 × 5 min washes in water (2 μl). The energy absorbing molecule (EAM, saturated 3,5-dimethoxy-4-hydrocinnaminic acid) was then added in 50% acetonitrile and 0.5% trifluoroacetic acid and allowed to crystallize. The chip was then inserted into the SELDI for laser desorption and time of flight (TOF) analysis. Routinely a signal/noise ratio of 5/1 indicated the presence of a protein.
Results
Carcinogenesis
The survival of C3(1)SV40TAg mice was determined as a function of time. All mice were viable 4 months after birth (Fig. 1). In the control group of 20 mice, tumors developed at multiple sites after 3.5 months. When the tumors became necrotic, the animals were euthanized and the first control mouse was euthanized at 4.2 months. At 5.2 months approximately half the mice had been euthanized, whereas at 5.7 months, all control mice were euthanized. One month after birth a group of mice was administered VIPhyb daily (10 μg/day, s.c.). In this group of mice, tumors were detected at 4 months and the first mouse euthanized at 4.5 months. Half of the treated mice were euthanized at 5.7 months and at 6.5 months 90% of the mice had been euthanized. The remaining mice were euthanized at 7 months. By Kaplan–Meier analysis of the survival curve, VIPhyb-treated mice were significantly different from the control (P < 0.01).
Fig. 1.
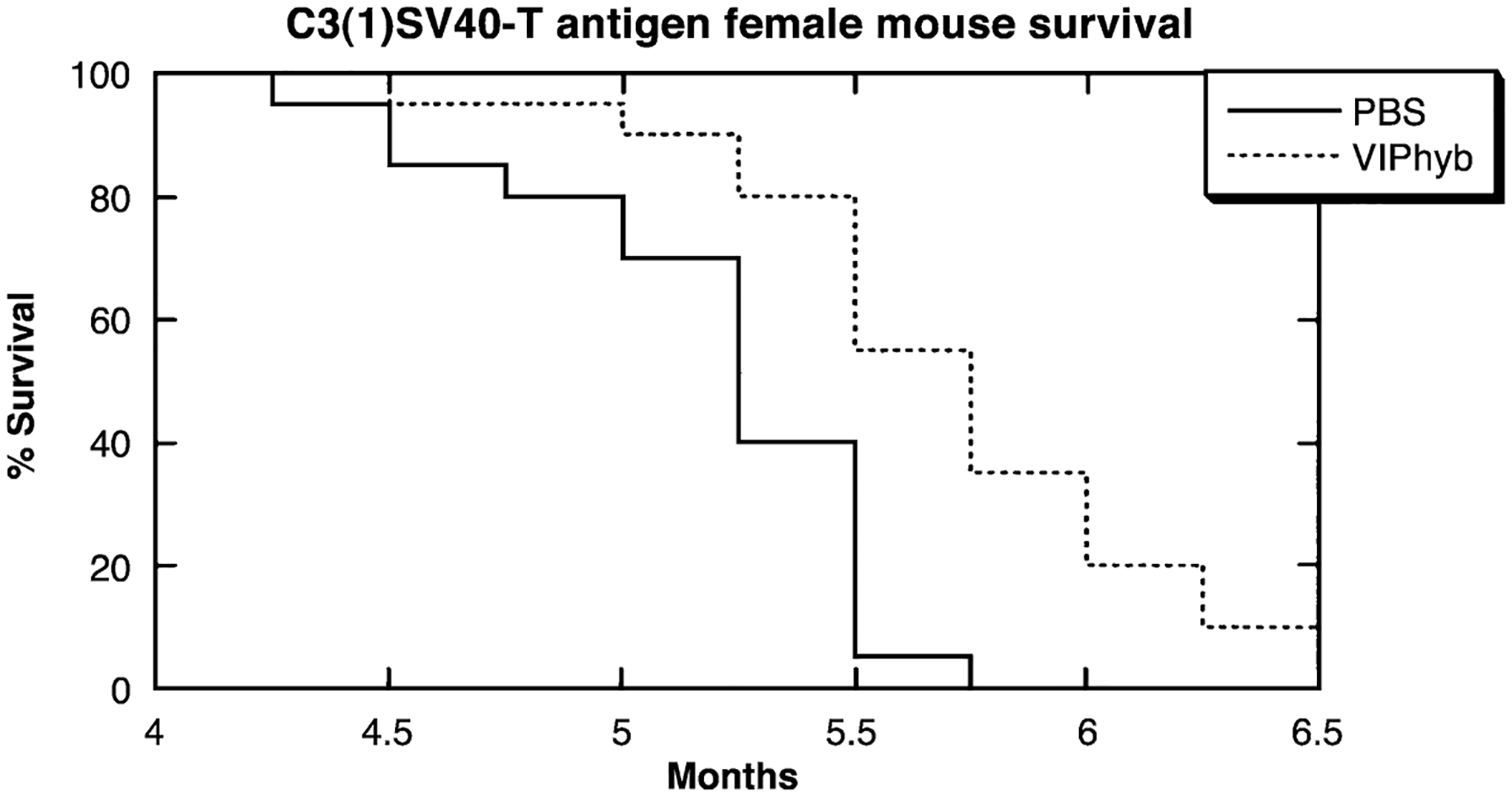
Mammary carcinogenesis. PBS (−) and 10 μg/day VIPhyb (…) were injected s.c. into C3(1)SV40T antigen mice and the survival was determined. The % survival of 20 female mice in each group is indicated. By Kaplan–Meier analysis, the curves were significantly different (p < 0.01). This experiment is representative of 2 others.
The tumor burden was determined. At 5.2 months, the control mice averaged 2.8 mammary tumors/mouse; each positive mammary gland had 1 tumor and there were 10 mammary glands/mouse (Table 1). In mice treated with VIPhyb, there was an average of 1.4 tumors/mouse and each positive mammary gland had 1 tumor. In general the tumors were larger in the control relative to VIPhyb treated mice at 5.2 months (data not shown). Using the Student’s t-test. the turmor burden was significantly less (p < 0.05) in VIPhyb treated animals than control mice.
Table 1.
Tumor burden in C3(1)SV40Tag mice
| Treatment | Tumor burden |
|---|---|
| PBS | 2.8 ± 0.5 |
| VIPhyb | 1.4 ± 0.4* |
The mean ± S.E. of 10 mice is indicated 5.2 months after birth;
p < 0.05,.
VIP receptor binding
The ability of VIP-like peptides to bind to mouse mammary tumor homogenates was investigated (Fig. 2). Specific 125I-VIP binding was strongly inhibited by 0.1 μM but not 0.0001 μM VIP. Specific 125I VIP binding was half maximally inhibited (IC50) by .002 μM VIP. Similarly PACAP and (Lys15, Arg16, Leu27)VIP1–7GRF8–27(a VPAC1 receptor agonist) inhibited specific 125I-VIP binding with high affinity (IC50 values of .003 and .003 μM respectively). (SN)VIPhyb and VIPhyb inhibited specific 125I VIP binding with IC50 values of 0.03 and 0.3 μM, respectively, whereas Ro25–1553 (a VPAC2 receptor agonist) did not inhibit specific binding (IC50 > 10 μM) Because (Lys15, Arg16, Leu27)VIP1–7GRF8–27 but not Ro25–1553 inhibited specific 125I VIP binding with high affinity, VPAC1 receptors predominate in mouse mammary tumors. Using in vitro autoradio-graphic techniques, 125I-VIP binding sites were distributed throughout the mammary tumor (data not shown).
Fig. 2.

Specificity of 125I-VIP binding to mammary tumor membranes. The % specific binding of 125I-VIP was determined as a function of unlabeled VIP (○), PACAP-27 (■), (Lys15, Arg16, Leu27)VIP1–7-GRF82–27 (□), (SN)VIPhyb (●), VIPhyb (▲) and Ro25–1553 (△) concentration. The mean value ± S.D. of 4 determinations each repeated in duplicate is indicated.
Immunocytochemistry
The mammary tumors were investigated for VPAC1 receptor immunostaining. High densities of VPAC1 receptor immunostaining were detected in the cytosol and plasma membrane of tumor cells (Fig. 3). Little VPAC1 receptor immunostaining was detected in the nucleus. Lower densities of VPAC1 receptor immunostaining were detected in adjacent normal cells such as stomal and endothelial cells. Little immunostaining was detected in control sera (data not shown). VPAC1 receptor immunostaining was present throughout the mammary tumor specimen, except for areas of necrosis.
Fig. 3.
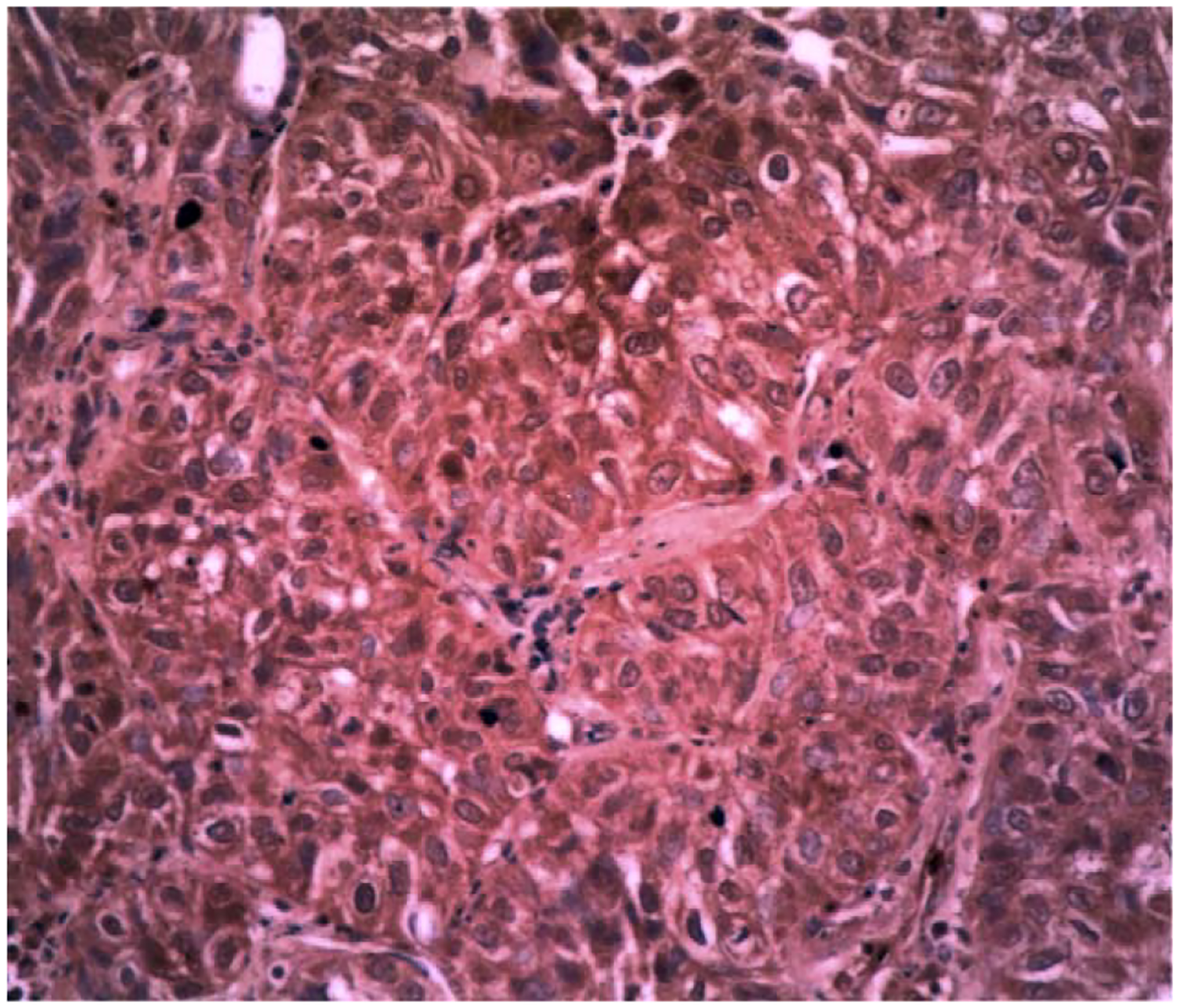
VPAC1 receptor immunocytochemistry. VPAC1 immunoreactivity (brown) is localized to the cytosol and plasma membrane of breast cancer cells but not the nucleus which is counterstained with Mayer’s hematoxlin (purple). This experiment is representative of 5 others.
Western blot
By Western blot a major 60 Kdalton protein was detected indicative of the VPAC1 receptor. Fig. 4 shows that a minor band stained at 42 Kdaltons. Major bands were not detected using mammary tumor extract and control VPAC1 receptor antisera (data not shown).
Fig. 4.
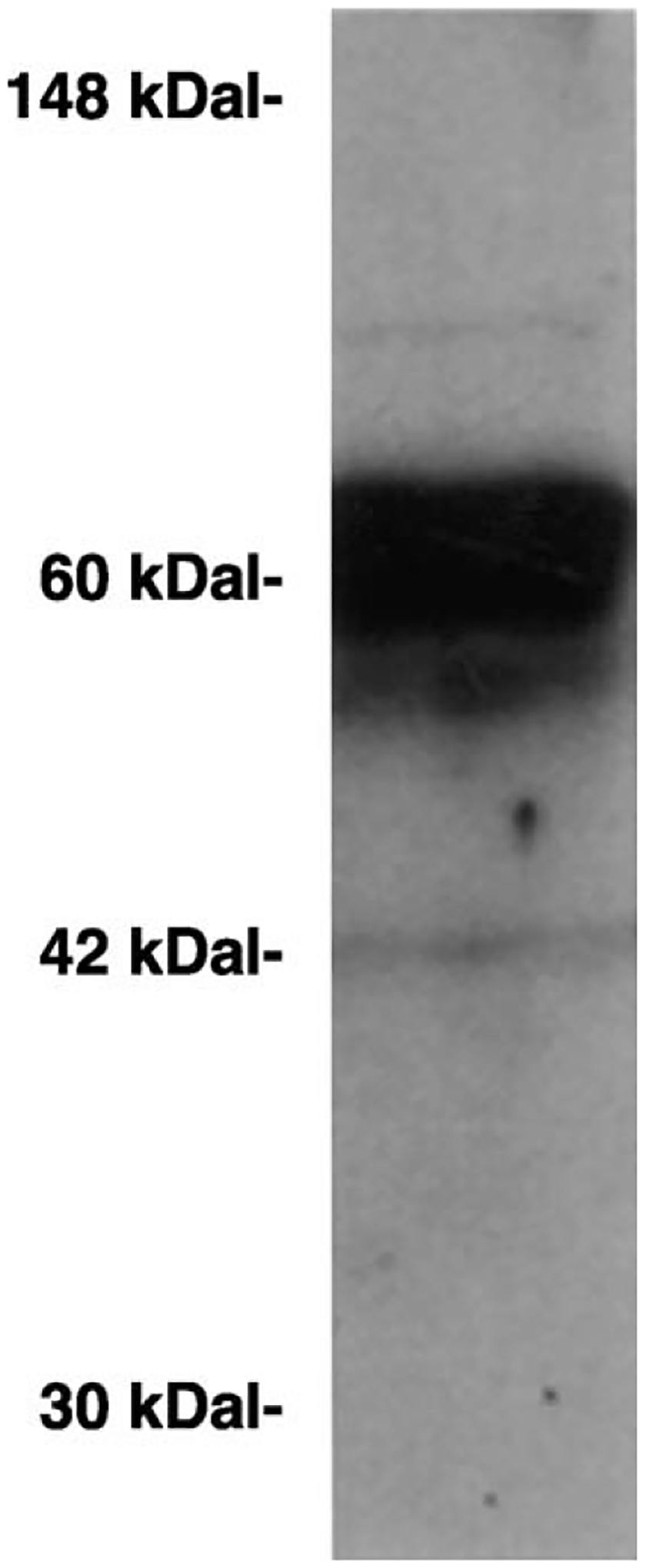
Western blot. Mammary tumor extract had a major 60 Kdalton protein band using VPAC1 receptor antibodies. This experiment is representative of 2 others.
RT-PCR
The mammary tumors were investigated for VPAC1 receptor mRNA. Fig. 5 shows that a 324 bp PCR product was present in LCM dissected mammary tumor cells derived from control and C3(1)SV40Tag mice treated with VIPhyb. These result indicate that treatment of mice with VIPhyb had little effect on VPAC1 receptor mRNA in mouse mammary tumors. Similarly, 125I-VIP bound with high affinity to tumor homogenates derived from VIP-hyb treated and control mice (data not shown). These results suggest that VIPhyb treatment had little effect on VPAC1 receptor expression.
Fig. 5.
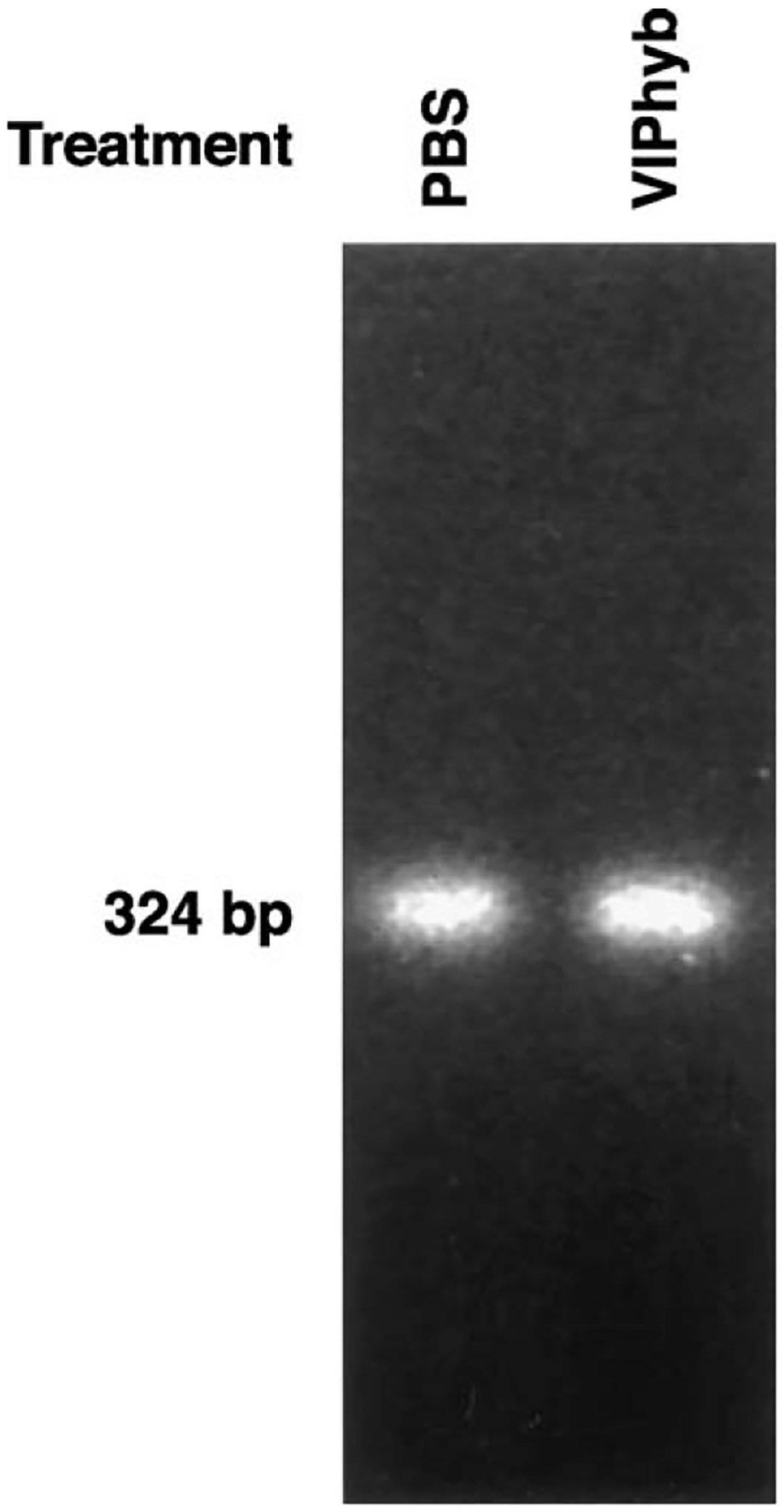
VPAC1 receptor RT-PCR. A major 324 bp PCR product indicative of the VPAC1 receptor is present in mammary tumors of mice treated with PBS or VIPhyb. This experiment is representative of 6 others.
Protein expression
Tumor cells isolated by LCM, were analyzed for proteins using SELDI techniques. Fig. 6 shows that approximately 50 proteins were detected with mass/charge ratios of 1000–40,000. Table 2 shows that major proteins with mass/charge ratios of 6902, 8931, 11,295, 12,346, 12,788 and 15,234 were detected in tumors from control animals whereas in animals treated with VIPhyb, major proteins with mass/charge ratios of 5655, 6904, 7015, 11,305, 13,797, 13,977 and 15,292 Daltons were detected. These results indicate that treatment of mice with VIPhyb alters the protein expression profile of some major C3(1)SV40Tag mouse mammary tumor proteins.
Fig. 6.
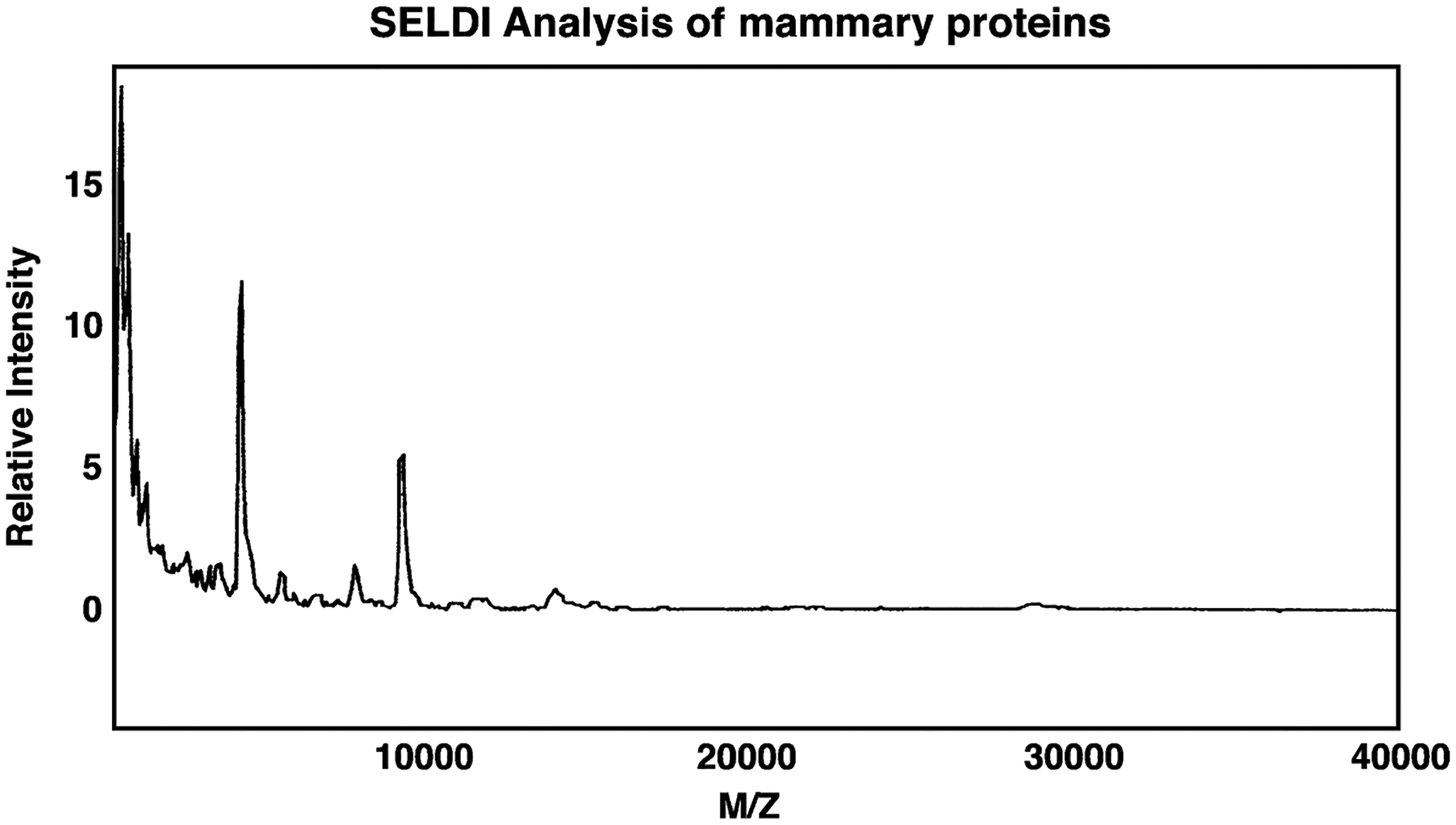
SELDI analysis of mouse mammary gland. Using a H4 chip several major proteins were observed with mass/charge (M/Z) ratios of 4000–16,000. This experiment is representative of 4 others.
Table 2.
SELDI analysis of mammary tumors
| Mouse treatment | |
|---|---|
| PBS | VIPhyb |
| 6902 | 5655 |
| 8931 | 6904 |
| 11,295 | 7015 |
| 12,346 | 11,305 |
| 12,788 | 13,797 |
| 15,234 | 13,977 |
| 15,292 | |
The protein extracts were fractionated on an H4 chip using SELDI techniques. The mass/charge ratio of major detected proteins is indicated. This experiment is representative of 2 others.
Discussion
Previously, we found that VIPhyb inhibited the growth of MCF-7 and MDA-MB231 breast cancer xenografts in nude mice (Zia et al., 1996). Also, VIP stimulated whereas VIPhyb inhibited the clonal growth of MCF-7 and MDA-MB231 cells, which are estrogen receptor positive and negative, respectively. These results indicate that VIPhyb inhibits the growth of breast cancer cells regardless of their estrogen receptor status. Here the effects of VIPhyb were investigated during mammary carcinogenesis.
C3(1)SV40TAg transgenic mice develop mammary tumors that are histologically similar to human breast cancer. The SV40Tag causes inactivation of the p53 and Rb tumor suppressor genes and female mice develop mammary hyperplasia by 3 months, followed by carcinoma in situ and invasive adenocarcinoma by 6 months (Green et al., 2000). Here we found that the mean lifetime of control mice was 5.2 months whereas the mean lifetime of mice treated with VIPhyb was significantly increased significantly to 5.7 months. Previously it was found that treatment of C3(1)-SV40-Tag mice with 9-cis retinoic acid increased lifetime significantly from 140 to 170 days (Wu et al., 2000). Also, treatment of C3(1)-SV40-Tag mice with 2-difluoromethylornithine, an ornithine decarboxylase inhibitor, or dehydroepiandrosterone, a steroid precursor to androgens and estrogens in primates, reduced tumor burden by 70 and 66% respectively (Green et al., 2001). These results indicate that 9-cis retinoic acid, 2-difluoromethylornithine, dehydroepiandrosterone and VIPhyb are chemopreventive agents for mammary carcinogenesis in C3(1)SV40Tag mice.
VIPhyb appeared to delay but not stop the carcinogenic process; by 7 months all C3(1)SV40Tag mice had mammary tumors. Previously we found that breast cancer cell lines MCF-7 and MDA-MB231 produced endogenous VIP-like peptides (Moody et al., 1998). When secreted, the VIP-like peptides may bind to cell surface VPAC1 receptors. Upon addition to breast cancer cells, exogenous VIP elevated transiently the cAMP and increased nuclear oncogene expression (c-myc and c-fos mRNA) leading to breast cancer cellular proliferation (Zia et al., 1996). Because VIP may be an autocrine growth factor for breast cancer cells, addition of receptor antagonist VIPhyb decreased cellular proliferation in vitro and in vivo. Further VIPhyb potentiated the ability of chemotherapeutic drugs such as taxol to inhibit breast cancer growth (Gelber et al., 2001; Moody et al., 2001). It remains to be determined if VIP is a promoter of mammary carcinogenesis.
VIPhyb decreased the mammary tumor burden in C3(1)SV40Tag mice. Most mice had a single tumor in 3 mammary glands 5.2 months after birth and the average tumor burden was 2.8. Mice treated with VIPhyb had a single tumor in 1 or 2 mammary glands and the average tumor burden was 1.4 at 5.2 months. Thus VIPhyb significantly reduced tumor burden by 50%. In contrast, 9-cis retinoic acid decreased tumor burden by 67% (Wu et al., 2000). While estrogen receptors are not abundant in C3(1)SV40Tag tumors, they are prevalent in Fisher rat mammary tumors (Clarke, 1997). In a rat animal model of carcinogenesis, 9-cis retinoic acid strongly prevents mammary carcinogenesis (Moon et al., 1976, 1983). Preliminary data (T. Moody, unpublished) indicate that VIPhyb prevents mammary tumors induced by NMU in Fisher rats; ras but not p53 and Rb mutations predominate in this rat model of carcinogenesis (Moon and Constantinou, 1997). These results indicate that VIPhyb prevents mammary carcinogenesis regardless of the estrogen receptor status.
VIP, which elevates the cAMP in breast cancer cells, does not appear to alter the differentiation of human breast cancer cells (Moody et al., 1998). Also, VIPhyb is a cytostatic and not cytotoxic agent. The growth inhibition caused by VIPhyb in human breast cancer cells is reversed by VIP (Zia et al., 1996). If VIPhyb injection was stopped in C3(1)SV40Tag mice, mammary carcinogenesis resumed at the normal rate (T. Moody, unpublished). It remains to be determined if PACAP(6–38), a PAC1 receptor antagonist, prevents mammary carcinogenesis in C3(1)-SV40-Tag mice. Previously we reported that PACAP(6–38) reduced breast cancer xenograft growth in nude mice (Leyton et al., 1999).
VIPhyb may be biologically active in mouse mammary tumors due to its ability to bind to VPAC1 receptors. Mouse mammary tumor homogenate binding of 125I-VIP was inhibited with high affinity by the VPAC1 receptor agonist (Lys15, Arg16, Leu27)VIP1–7GRF8–27, but not Ro25–1553, a VPAC2 receptor agonist. Similarly, VPAC1 receptor binding was demonstrated in homogenate derived from the normal mouse mammary gland but unfortunately insufficient tissue was available to assay for VPAC1 receptor binding in hyperplastic tissue (T. Moody, unpublished). It remains to be determined if the VPAC1 receptor density varies as a function of mammary carcinogenesis. Alternatively, the VIP-like peptide density could increase during mammary carcinogenesis increasing the importance of VIP as a mammary autocrine growth factor.
Also, VPAC1 receptor mRNA was present in the mouse mammary tumors. VPAC1 receptor immunostaining was abundant in the mouse mammary tumor cells. The VPAC1 receptor antibody recognized a major protein of 60 Kdaltons in mammary tumors. The mouse VPAC1 receptor, which contains 459 amino acids, has a molecular weight of 52,094 daltons, however, it can be glycosylated on Asn58, Asn 69, Asn 100, Asn104 and Asn292 leading to a higher molecular weight glycoprotein. Previously, we found that VPAC1 receptors were present in human breast cancer cell lines (Zia et al., 1996). In human biopsy specimens, VPAC1 receptors predominated in neuroblastoma as well as bladder, breast, colon, liver, lung, prostate, stomach, thyroid, uterus and prostate cancer. VPAC2 receptors predominated in stomach leimyoma cancer. PAC1 receptors were abundant in glioblastoma, adrenal and pituitary cancer (Reubi, 2000; Reubi et al., 2000).
Low molecular weight mammary cancer proteins were analyzed using SELDI techniques. Routinely 2000 cells were prepared by LCM, and extracted for total protein content. Over 50 proteins were detected with mass/charge ratios of 1000–40,000. In mammary tumors from control animals, major proteins with mass/charge ratios of 6902, 8931, 11,295, 12,346, 12,788 and 15,234 Daltons were detected. In contrast, in animals treated VIPhyb, the mammary tumors had major proteins with mass/charge ratios of 5655, 6904, 7015, 11,305, 13,797, 13,977 and 15,292 Daltons. Because SELDI has an intrinsic error of 0.1%, it is likely that the 6902 in control and 6904 in VIPhyb treated animals are identical; the 11,295 and 11,305 in VIPhyb treated animals; and the 15,234 and 15,292 proteins in VIPhyb treated animals may be identical. The 5655, 7015, 13,797 and 13,977 proteins detected in animals treated with VIPhyb may be up-regulated whereas the 8931, 12,346 and 12,788 proteins in control animals may be down- regulated by VIPhyb treatment. VIPhyb could alter the gene expression and/or post-translational modification of these proteins.
While the identity of these mouse mammary tumor proteins remains to be determined, several proteins have been identified in human ductal carcinoma in situ (DCIS) which are elevated relative to normal ductal cells (Wulfkuhle et al., 2002). Using two dimensional gels, cytoskeletal proteins such as cofilin and actin-related protein (ARP)-3 (18,592 and 47,371 Daltons respectively) were elevated in DCIS. The ARP-3 and cofilin are required for actin filament assembly. Annexin II, V and VII (38,472, 35,805 and 50,315 Daltons respectively) was elevated in DCIS relative to control tissue and may play a role in lipid and vesicular trafficking. The annexins are important in exocytosis and secretion of growth factors, which regulate breast cancer proliferation. Also peroxiredoxins 1,2 and 5 (22,110, 21,892 and 22,026 Daltons respectively) were elevated in DCIS relative to control tissue, and this may protect breast cancer cells from apoptosis associated with oxidative stress. While the molecular weight of these human breast proteins is larger than the mouse mammary proteins studied here, it is possible that some of these proteins may be metabolized by mammary proteases to smaller protein fragments.
In summary, VIPhyb significantly increased the survival of C3(1)SV40Tag mice which spontaneously develop mammary cancer, decreased tumor burden and altered the protein expression. These results indicate that VIPhyb, which antagonizes VPAC1 receptors, may be a preventive agent in breast carcinogenesis.
Acknowledgements
The authors thank Dr. J. Leyton for helpful discussions, Dr. E.Goetzl for the VPAC1 receptor antisera and Dr. I. Gozes for the (SN)VIPhyb.
References
- Arimura A, 1992. Pituitary adenylate cyclase activating polypeptide (PACAP): Discovery and current status of research. Regul. Peptides 37 (3), 287–303. [PubMed] [Google Scholar]
- Bonner RF, Emmert-Buck M, Pohida T, Chuaqui R, Goldstein S, Liotta LA, 1997. Laser capture microdissection: Molecular analysis of tissue. Science 278 (5342), 1481–1483. [DOI] [PubMed] [Google Scholar]
- Clarke R, 1997. Animal models of breast cancer: Experimental design and their use in nutrition and psychosocial research. Breast Cancer Res. Treat 46 (2–3), 117–133. [DOI] [PubMed] [Google Scholar]
- Cronin WM, Vogel V, Tovidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N, 1998. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-I study. J. Natl. Cancer Inst 90 (18), 1371–1388. [DOI] [PubMed] [Google Scholar]
- Cummings SR, Eckert S, Krueger KA, Grady D, Powldes TJ, Cauley JA, Norton L, Nickelsen T, Bjarnason NH, Morrow M, Lippman ME, Black D, Glusman JE, Costa A, Jordon VC, 1999. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. J. Am. Med. Assoc 281 (23), 1289–2197. [DOI] [PubMed] [Google Scholar]
- Dickson RB, Lippman ME, 1997. Molecular biology of breast cancer. In: DeVita VT, Hellman S, Rosenberg SA (Eds.), Cancer: Principles and Practice of Oncology, 1541–1547. [Google Scholar]
- Ebina M, Steinberg SA, Mulshine JL, Linnoila I, 1994. Relationship of p53 overexpression and up-regulation of proliferating cell nuclear antigen with the clinical course of non-small cell lung cancer. Cancer Res 54 (9), 2496–2503. [PubMed] [Google Scholar]
- Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Tovidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N, 1998. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-I Study. J. Natl. Cancer Inst 90 (18), 1371–1388. [DOI] [PubMed] [Google Scholar]
- Gelber E, Granoth R, Fridkin M, Dreznik Z, Brenneman DE, Moody TW, Gozes I, 2001. A lipophilic vasoactive intestinal peptide analog enhances the antiproliferative effect of chemotherapeutics agents on cancer cell lines. Cancer 92 (8), 2172–2180. [DOI] [PubMed] [Google Scholar]
- Gespach C, Bawab W, Decremoux P, Calvo F, 1988. Pharmacology, molecular identification and functional characteristics of vasoactive intestinal peptide receptors in human breast cancer cells. Cancer Res 48 (18), 5079–5083. [PubMed] [Google Scholar]
- Gozes I, McCune SK, Jacobson L, Warren D, Moody TW, Fridkin M, Brenneman DE, 1991. A hybrid antagonist to vasoactive intestinal peptide: Effects on cellular function in the central nervous system. J. Pharm. Exp.Ther 257 (3), 959–966. [PubMed] [Google Scholar]
- Green JE, Shibata MA, Yoshidome K, Liu M, Jorcky C, Anver MR, Wigginton J, Wiltrout R, Shibata E, Kaczmarczyk S, Wang W, Liu A, Calvo A, Couldrey C, 2000. The C3 (1)SV40 T-antigen transgenic mouse model of mammary cancer: Ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 19 (18), 1020–1027. [DOI] [PubMed] [Google Scholar]
- Green JE, Shibata MA, Shibata E, Moon RC, Anver MR, Kelloff G, Lubet R, 2001. 2-Difluoromethylornithine and dehydroepiandrosterone inhibit mammary tumor progression but not mammary or prostate tumor initiation in C3 (1)/SV40T/T-antigen transgenic mice. Cancer Res 61 (20), 7449–7455. [PubMed] [Google Scholar]
- Harmar T, Lutz E, 1993. Multiple receptors for PACAP and VIP. Trends in Pharmacological Sciences 15 (4), 97–99. [DOI] [PubMed] [Google Scholar]
- Ishihara R, Shigemoto R, Mori K, Takahashi K, Nagata S, 1992. Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron 8 (4), 811–819. [DOI] [PubMed] [Google Scholar]
- Landis S, Murray T, Bolden S, Wingo PCA, 1999. Cancer Statistics, 1999. CA Cancer, J. Clin 49 (1), 8–31. [DOI] [PubMed] [Google Scholar]
- Lee M, Jensen RT, Huang SC, Bepler G, Korman L, Moody TW, 1990. Vasoactive intestinal polypeptide binds with high affinity to non-small cell lung cancer cells and elevates cyclic AMP levels. Peptides 11 (6), 1205–1209. [DOI] [PubMed] [Google Scholar]
- Leyton J, Purdom S, Cassibang M, Gozes Y, Zia F, Coy D, Moody TW, 1999. PACAP(6–38) is a PACAP receptor antagonist for breast cancer cells. Breast Cancer Res. Treat 56 (2), 177–186. [DOI] [PubMed] [Google Scholar]
- Marshall L, Hunter D, Connolly J, Schnitt S, Byrne C, London S, Colditz C, 1997. Risk of breast cancer associated with atypical hyperplasia of lobular and ductal types. Cancer Epidemiol. Biomarkers Prev 6 (5), 297–301. [PubMed] [Google Scholar]
- McKenzie K, Sukumar S, 1996. Molecular genetics of human breast cancer. In: Huff J, Boyd J, Barrett J (Eds.), Cellular and Molecular Mechanisms of Hormonal Carcinogenesis: Environmental Influences, 183–209. [Google Scholar]
- Moody TW, Leyton J, Gozes I, Lang L, Eckelman WC, 1998. VIP and breast cancer. Ann. N.Y. Acad. Sci 865, 290–296. [DOI] [PubMed] [Google Scholar]
- Moody TW, Leyton J, Chan D, Brenneman DC, Fridkin M, Gelber E, Levy A, Gozes I, 2001. VIP receptor antagonists and chemotherapeutic drugs inhibit the growth of breast cancer cells. Breast Cancer Res. Treat 68 (1), 55–64. [DOI] [PubMed] [Google Scholar]
- Moon RC, Constantinou AI, 1997. Dietary retinoids and carotenoids in rodent models of mammary tumorigenesis. Breast Cancer Res. Treat 46 (2–3), 181–189. [DOI] [PubMed] [Google Scholar]
- Moon RC, Grubbs C, Sporn MB, 1976. Inhibition of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis by retinyl acetate. Cancer Res 36 (7PT2), 2626–2630. [PubMed] [Google Scholar]
- Moon RC, McCormick D, Mehta RG, 1983. Inhibition of carcinogenesis by retinoids. Cancer Res 43 (S5), 2469s–2475s. [PubMed] [Google Scholar]
- Pisegna JR, Wank SA, 1993. Molecular cloning and functional expression of the pituitary adenylate cyclase activating polypeptide type I receptor. Proc. Natl. Acad. Sci. USA 90 (13), 6345–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram TG, Kokeny KE, Dilts CA, Ethier SP, 1995. Mitogenic activity of neu differentiation factor/heregulin mimics that of epidermal growth factor and insulin-like growth factor I in human mammary epithelial cells. J. Cell Physiol 163 (3), 589–596. [DOI] [PubMed] [Google Scholar]
- Reubi JC, 1995. In vitro identification of vasoactive intestinal peptide receptors in human tumors: Implications for tumor imaging. J. Nucl. Med 36 (10), 1846–1853. [PubMed] [Google Scholar]
- Reubi JC, 2000. In vitro evaluation of VIP/PACAP receptors in healthy and diseases human tissues. Clinical implications. Ann. N.Y. Acad. Sci 921, 1–30. [DOI] [PubMed] [Google Scholar]
- Reubi JC, Laderach U, Waser B, Gebbers JO, Robberecht P, Laissue JA, 2000. Vasoactive intestinal peptide/pituitary adenylate cyclase activating peptide receptor subtypes in human tumors and their tissues of origin. Cancer Res 60 (11), 3105–3112. [PubMed] [Google Scholar]
- Said SI, Mutt V, 1970. Peptide with broad biological activity:Isolation from the small intestine. Science 169 (951), 1217–1218. [DOI] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeberg PH, Journot L, 1993. Differential signal transduction by five splice variants of the PACAP receptor. Nature 365 (6442), 170–175. [DOI] [PubMed] [Google Scholar]
- Tripathy D, Benz C, 1993. Activated oncogenes and putative tumor suppressor genes involved in human breast cancers. In: Benz C, Liu E (Eds.), Oncogenes and tumor suppressor genes in human malignancies, 15–60. [DOI] [PubMed] [Google Scholar]
- Waschek JL, Richards ML, Bravo DT, 1995. Differential expression of VIP/PACAP receptor genes in breast, intestinal and pancreatic lines. Cancer Lett 92 (2), 143–149. [DOI] [PubMed] [Google Scholar]
- Wu K, Kim HT, Rodriquez JL, Munoz-Medellin D, Moshsin SK, Hilsenbeck SG, Lamph WM, Gottardis MM, Shirley MA, Kuhn JG, Green JE, Brown PH, 2000. 9-cis-Retinoic acid suppresses mammary tumorigenesis in C3 (1)-Simian Virus 40 T antigen-transgenic mice. Clin. Cancer Res 6 (9), 3696–3704. [PubMed] [Google Scholar]
- Wulfkuhle JD, McLean KC, Paweletz CP, Sgroi DC, Trock BJ, Steeg PS, Petricoin EF, 2001. New approaches to proteomic analysis of breast cancer. Proteomics 1 (10), 1205–1215. [DOI] [PubMed] [Google Scholar]
- Wulfkuhle JD, Sgroi DC, Krutzsch HJ, McLean K, McGarvey K, Knowlton M, Chen S, Shu H, Sahin A, Kurek R, Wallwiener D, Merino MJ, Petricoin EF, Zhao Y, Steeg PS, 2002. Proteomics of human breast ductal carcinoma in situ. Cancer Res 62 (22), 6740–6749. [PubMed] [Google Scholar]
- Zia H, Hida T, Jakowlew S, Birrer M, Gozes Y, Reubi JC, Fridkin M, Gozes I, Moody TW, 1996. Breast cancer growth is inhibited by VIPhybrid, a synthetic VIP receptor antagonist. Cancer Res 56 (15), 3486–3489. [PubMed] [Google Scholar]