

Abstract
Deep-learning methods for computational pathology require either manual annotation of gigapixel whole-slide images (WSIs) or large datasets of WSIs with slide-level labels and typically suffer from poor domain adaptation and interpretability. Here we report an interpretable weakly supervised deep-learning method for data-efficient WSI processing and learning that only requires slide-level labels. The method, which we named clustering-constrained-attention multiple-instance learning (CLAM), uses attention-based learning to identify subregions of high diagnostic value to accurately classify whole slides and instance-level clustering over the identified representative regions to constrain and refine the feature space. By applying CLAM to the subtyping of renal cell carcinoma and non-small-cell lung cancer as well as the detection of lymph node metastasis, we show that it can be used to localize well-known morphological features on WSIs without the need for spatial labels, that it overperforms standard weakly supervised classification algorithms and that it is adaptable to independent test cohorts, smartphone microscopy and varying tissue content.
Advances in digital pathology and artificial intelligence have presented the potential to analyse gigapixel whole-slide images (WSIs) for objective diagnosis, prognosis and therapeutic-response prediction[1, 2]. Apart from the immediate clinical benefits[3, 4, 5, 6], computational pathology has demonstrated promise in a variety of different tasks for quantifying the tissue microenvironment[7, 8, 9, 10, 11, 12], conducting integrative image-omic analysis[13, 14, 15, 16, 17, 18, 19], identifying morphological features of prognostic relevance[20, 21] and associating morphologies with response and resistance to treatment[22].
Although deep learning[23, 24] has revolutionized medical imaging by solving many image classification and prediction tasks[25, 26, 27, 28, 29, 30], whole-slide imaging is a complex domain with several unique challenges. Deep-learning-based computational pathology approaches require either manual annotation of gigapixel WSIs in fully supervised settings or large datasets with slide-level labels in a weakly supervised setting. Given that slide-level labels may only correspond to tiny regions of each large gigapixel image, most approaches have relied on pixel, patch or region-of-interest (ROI)-level annotations to saliently localize these ‘needles in a haystack’[31, 32, 33, 34]. Although promising results have been reported by assigning the same label to every patch in a WSI[35], this approach suffers from noisy training labels and is not applicable to problems that may have limited tumour content (for example, micro-metastasis). Furthermore, if only a subset of tissue regions in WSIs are sampled for training at the ROI or patch-level, the model may not generalize well at test time or provide useful slide-level interpretability. Recent work has demonstrated exceptional clinical-grade performance using slide-level labels for training binary classifiers for patient stratification in a weakly supervised setting[36] based on variants of multiple-instance learning (MIL). However, this methodology was reported to require thousands of WSIs to achieve comparable performance to fully supervised and ROI-level classifiers. Such large datasets, although important and beneficial for capturing the immense diversity and heterogeneity present in histology, are difficult to curate for rare diagnoses where only a handful of examples may exist or for clinical trials where it may be useful to predict the outcome from a small cohort of patients. Moreover, to produce a slide-level prediction from ROI or patch-level predictions, weakly supervised whole-slide classification methods commonly require the selection of a fixed, predefined aggregation function (for example, max-pooling or averaging over ROIs) and may not be suitable for both binary tumour versus normal classification and multi-class tissue subtyping problems, where normal tissue slides are not available. In addition, the performance of deep-learning diagnostic models, when trained using patch-level supervision, has been shown to suffer when tested on data from different sources and imaging devices[35, 36]. Such methods also need to be interpretable, with the capability to saliently localize regions used to make predictive determinations. In summary, for the broader adaptation of computational pathology in both clinical and research settings, there is a need for methods that do not require manual ROI extraction, pixel/patch-level labelling or naive sampling, which are still data efficient, interpretable, adaptable and generally applicable to both binary classification and multi-class subtyping problems.
In this Article, we propose clustering-constrained-attention multiple-instance learning (CLAM) as a high-throughput deep-learning framework that aims to address the key challenges with the whole-slide-level computational pathology outlined above. In three separate analyses (renal-cell-carcinoma (RCC) and non-small-cell-lung-cancer (NSCLC) subtyping and the detection of lymph node metastasis) using both publicly available datasets as well as independent test cohorts, we show that our approach is data efficient and can achieve high performance across different tasks while using a systematically decreasing number of training labels. We demonstrate the adaptability of CLAM by showing that models trained on tissue resection WSIs can be directly applied to biopsy WSIs as well as photomicrographs taken with a consumer-grade smartphone using data from independent test cohorts. We also demonstrate that CLAM can generalize to multi-class classification and subtyping problems in addition to the binary tumour versus normal classification tasks typically studied in weakly supervised settings. Our study presents a computational pathology framework that extends attention-based multiple-instance aggregation[37] to general multi-class weakly supervised WSI classification without requiring any pixel-level annotation, ROI extraction or sampling. We make this possible by first using transfer learning and a convolutional neural network (CNN) encoder with pre-trained parameters for dimensionality reduction, which also has the benefit of drastically increasing the speed of model training. Through the use of attention-based learning, CLAM is able to produce interpretable heatmaps that allow clinicians to visualize, for each slide, the relative contribution and importance of every tissue region to the predictions of the model without using any pixel-level annotations during training. These heatmaps show that our models are capable of identifying well-known morphological features used by pathologists to make diagnostic determinations, and show that the models are capable of distinguishing between tumour and adjacent normal tissue without any normal slides or ROIs used during training. CLAM is publicly available as an easy-to-use Python package over GitHub (https://github.com/mahmoodlab/CLAM), and whole-slide-level attention maps can be viewed in our interactive demo (http://clam.mahmoodlab.org).
CLAM is a deep-learning-based weakly supervised method that uses attention-based learning to automatically identify subregions of high diagnostic value to accurately classify the whole slide, while also enabling the use of instance-level clustering over the representative regions identified to constrain and refine the feature space. Under the standard MIL formulation and the weakly supervised learning paradigm in general, one major challenge in developing high-performance machine-learning classifiers for computational pathology is the suboptimal usage of labelled WSI data. For example, when only the slide-level labels are known, despite having access to many (up to hundreds of thousands) instances or patches per WSI, the standard MIL algorithm uses max-pooling and thus uses the gradient signal from only a single instance in each slide to update the learning parameters of the neural network model. This drawback might partly explain why, empirically, a deep-learning model trained using MIL would require the observation of a large number of example WSIs that are annotated at the slide level to achieve high performance for relatively simple binary classification tasks[36]. On the other hand, although assigning the slide-level label to each and every patch in the slide and treating them as independent training examples maximizes the number of labelled data points, it might not benefit model performance as a result of the use of noisy labels.
For whole-slide-level learning without annotation, CLAM uses an attention-based pooling function to aggregate patch-level features into slide-level representations for classification. At a high level, during both training and inference, the model examines and ranks all patches in the tissue regions of a WSI, assigning an attention score to each patch, which informs its contribution or importance to the collective slide-level representation for a specific class (Fig. 1). This interpretation of the attention score is reflected in the slide-level aggregation rule of attention-based pooling, which computes the slide-level representation as the average of all patches in the slide weighted by their respective attention score. Unlike the standard MIL algorithm[36, 37, 38], which was designed and widely used for weakly supervised positive/negative binary classification (for example, cancer versus normal), CLAM is designed to solve generic multi-class classification problems. A CLAM model has N parallel attention branches that together calculate N unique slide-level representations, where each representation is determined from a different set of highly attended regions in the image viewed by the network as strong positive evidence for the one of N classes in a multi-class diagnostic task (Fig. 1b,c). Each class-specific slide representation is then examined by a classification layer to obtain the final probability score predictions for the whole slide.
Fig 1. Overview of the CLAM conceptual framework, architecture and interpretability.
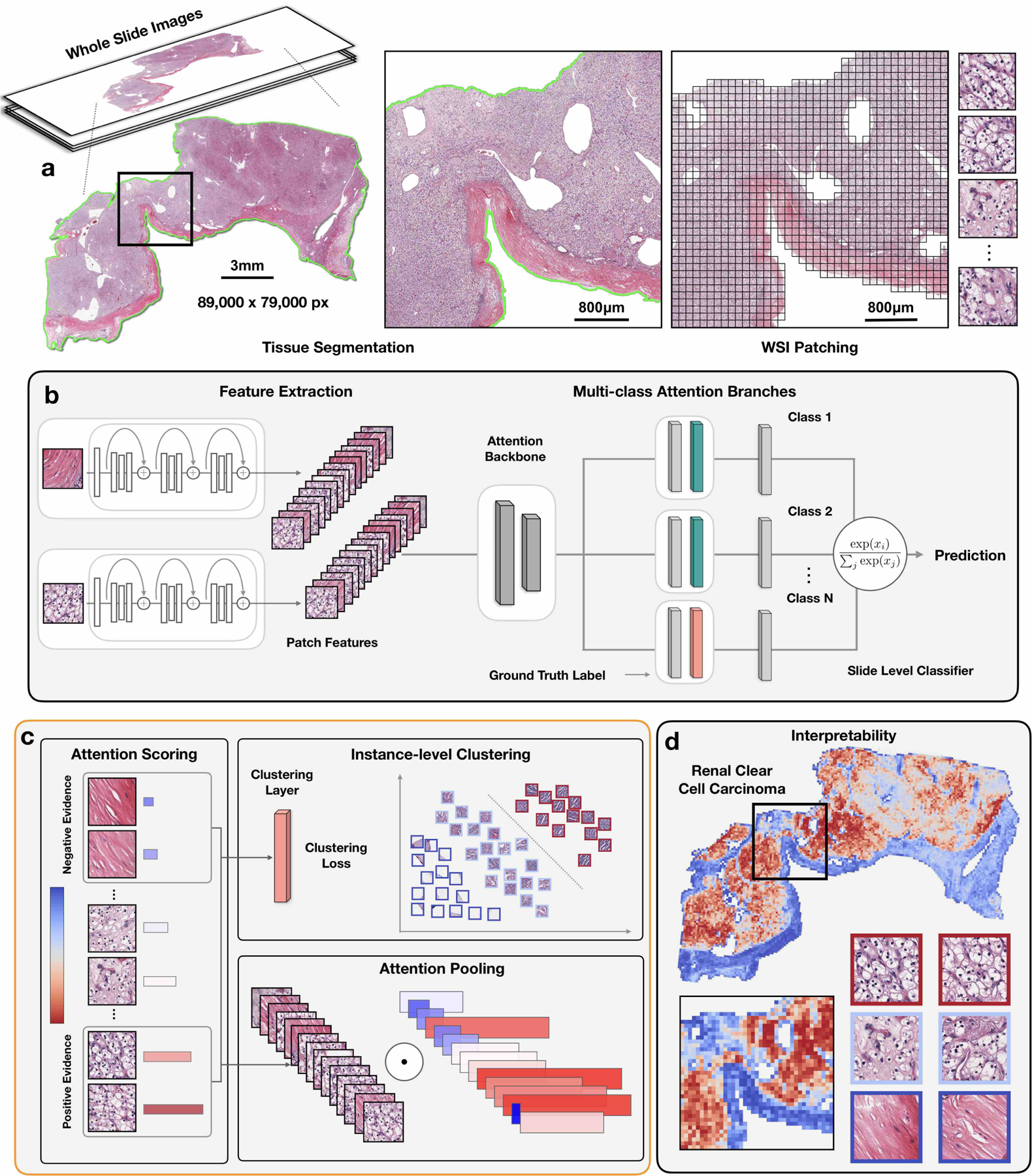
a, Following segmentation (left), image patches are extracted from the tissue regions of the WSI (right). b, Patches are encoded once by a pre-trained CNN into a descriptive feature representation. During training and inference, the extracted patches in each WSI are passed to a CLAM model as feature vectors. An attention network is used to aggregate patch-level information into slide-level representations, which are used to make the final diagnostic prediction. c, For each class, the attention network ranks each region in the slide and assigns an attention score based on its relative importance to the slide-level diagnosis (left). Attention pooling weighs patches by their respective attention scores and summarizes patch-level features into slide-level representations (bottom right). During training, given the ground-truth label, the strongly attended (red) and weakly attended (blue) regions can additionally be used as representative samples to supervise clustering layers that learn a rich patch-level feature space separable between the positive and negative instances of distinct classes (top right). d, The attention scores can be visualized as a heatmap to identify ROIs and interpret the important morphology used for diagnosis.
Beyond adopting the attention-based pooling[37] aggregation rule in favour of max-pooling, we explored additional means to address the data inefficiency in existing weakly supervised learning algorithms for computational pathology. Namely, we make use of the slide-level ground-truth label and the attention scores predicted by the network to generate pseudo labels for both highly and weakly attended patches as a technique to increase the supervisory signals for learning a separable patch-level feature space. During training, the network learns from an additional supervised learning task of separating the most- and least-attended patches of each class into distinct clusters. In addition, it is possible to incorporate domain knowledge into the instance-level clustering to add further supervision. For example, cancer subtypes are often mutually exclusive or assumed to be mutually exclusive during classification. If the mutual exclusivity assumption is adopted, in addition to supervising the attention branch for which the ground-truth class is present, the attention network branches corresponding to the remaining classes can be supervised by clustering their highly attended instances as ‘false positive’ (that is, negative) evidence for their respective classes. In practice, if one were to assume that only morphology corresponding to a single class is present in a given slide, one can also choose to use a simpler framework of having a single attention module instead of multiple branches by always treating the high-attention patches from the attention module as positive evidence for the ground-truth class and as false positive evidence for the remaining classes when computing the clustering loss.
To make CLAM a high-throughput pipeline that researchers can readily adopt and utilize without requiring dedicated high-performance compute clusters, we also propose and make available an open source easy-to-use WSI processing and learning toolbox. Our pipeline first automatically segments the tissue region of each slide and divides it into many smaller patches (for example, 256 × 256 pixels) so they can serve as direct inputs to a CNN (Fig. 1a). Next, using a CNN for feature extraction, we convert all tissue patches into sets of low-dimensional feature embeddings (Fig. 1b). Following this feature extraction, both training and inference can occur in the low-dimensional feature space instead of the high-dimensional pixel space. The volume of the data space is decreased nearly 200-fold and we can drastically reduce the subsequent computation required to train supervised deep-learning models. We found that working with a low-dimensional feature space enables training models on thousands of gigapixel-sized resection slides within hours on modern workstations with consumer-grade Graphics Processing Units (GPUs).
In the proceeding sections, we demonstrate the data efficiency, adaptability and interpretability of CLAM on three different computational pathology problems: (1) RCC subtyping, (2) NSCLC subtyping and (3) the detection of breast-cancer lymph node metastasis. We also show that CLAM models trained on WSIs are adaptable to smartphone microscopy images and biopsy slides.
Results
Dataset-size dependent, cross-validated model performance.
We evaluated the slide-level classification performance of CLAM for the three clinical diagnostic tasks mentioned above using 10-fold Monte Carlo cross-validation. For each cross-validated fold, we randomly partitioned each public WSI dataset into a training set (80% of cases), a validation set (10% of cases) and a test set (10% of cases), stratified by each class. In the event that a single case has multiple slides, all of them are sampled together into the same set. In each fold, the performance of the model on the validation set is monitored during training and used for model selection while the test set is held out and referred to just once after training is complete to evaluate the model. On the Cancer Genome Atlas (TCGA) RCC dataset (Fig. 2a), the model achieved a 10-fold macro-averaged one-versus-rest mean test area under the curve (AUC) ± s.d. of 0.991 ± 0.004 for the three-class RCC subtyping of papillary (PRCC), chromophobe (CRCC) and clear cell RCC (CCRCC) at ×20 magnification. For the per subtype one-versus-rest AUC, see Supplementary Fig. 1. For the two-class NSCLC subtyping of lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) on the combined TCGA and Clinical Proteomic Tumor Analysis Consortium (CPTAC) NSCLC dataset, at ×20 magnification, the model achieved an average test AUC of 0.956 ± 0.020 (Fig. 2b). On the combined CAMELYON16 and CAMELYON17 dataset for breast-cancer-metastasis detection in axillary lymph nodes, the model achieved an average test AUC of 0.953 ± 0.029 at ×40 magnification (Fig. 2c). Additional performance metrics are reported in Supplementary Tables 1–3. All of our training data are from publicly available sources, which, although they represent some of the largest public WSI datasets, are 5–10× smaller than the proprietary labelled datasets studied in several recent workscite[5, 36]. However, despite the moderate sizes of the datasets used (884, 1,967 and 899 total slides, respectively, of which only approximately 80% are used for training in each fold), the high performance (>0.95 AUC) on all three tasks indicates that our method can be effectively applied to solve both conventional positive-versus-negative cancer detection binary classification and more general multi-class cancer subtyping problems across a variety of tissue types.
Fig 2. Performance, data efficiency and comparative analysis.
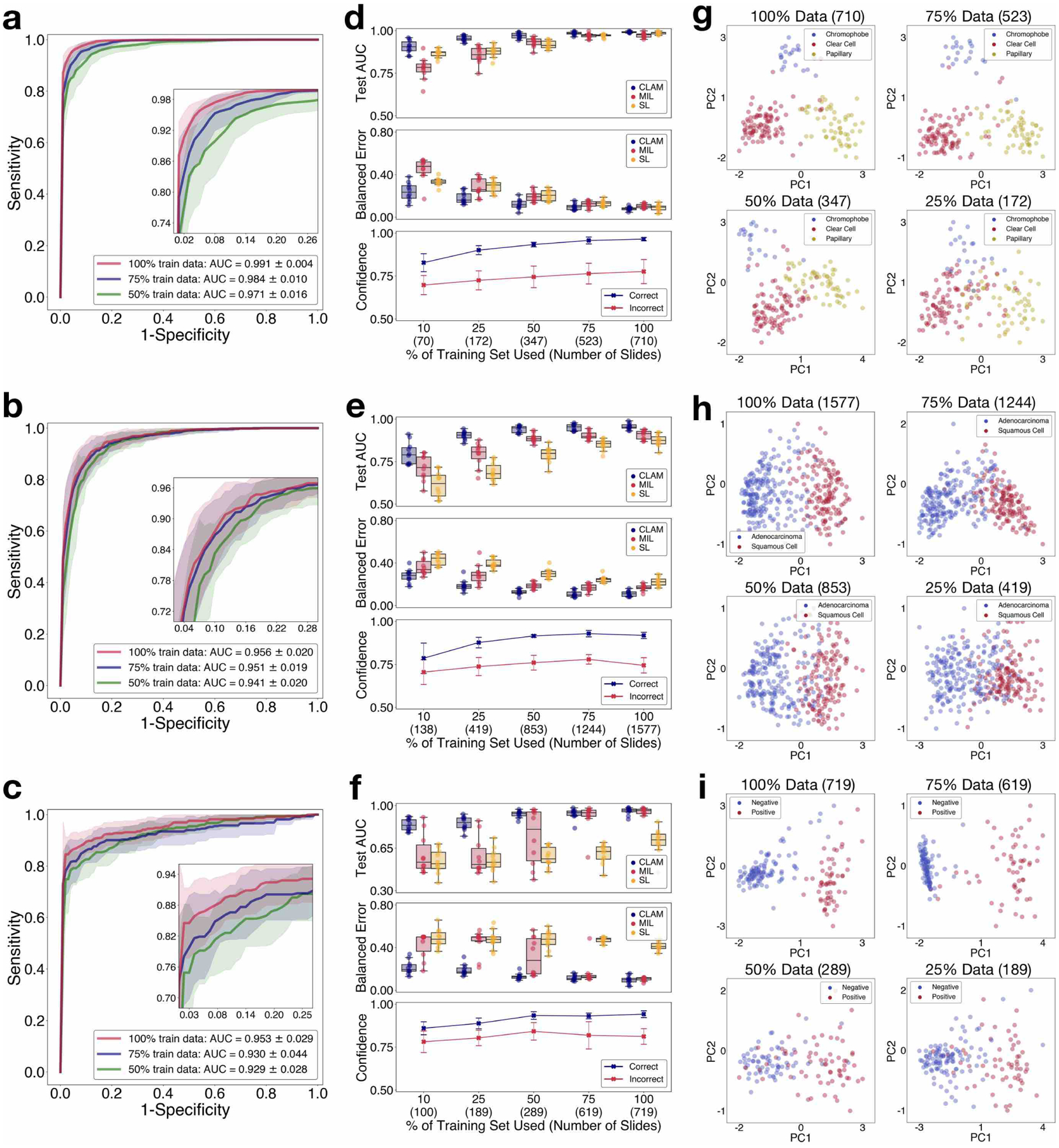
a–i, The 10-fold Monte Carlo cross-validation prediction results and test performance of CLAM models are analysed for RCC subtyping (a,d,g; n = 86), NSCLC subtyping (b,e,h; n = 196) and the detection of lymph node metastasis (c,f,i; n = 89). a–c, Mean test AUC ± s.d. of CLAM models using 100, 75 and 50% of cases in the training set. The confidence band shows ±1 s.d. for the averaged receiver-operating-characteristic curve. For multi-class RCC subtyping, the macro-averaged curve and AUC is reported. Insets: zoomed-in view of the curves. d–f, The dataset-size-dependent performance of various weakly supervised classification algorithms, in terms of the 10-fold test AUCs (top) and balanced error scores (middle) is shown using box plots for each training-set size (100, 75, 50, 25 and 10% of cases). The boxes indicate the quartile values and the whiskers extend to data points within 1.5× of the interquartile range. Mean confidence (± 1 s.d.) of the predictions made by the CLAM models for correctly and incorrectly classified slides (bottom). g–i, Visualization of the learned slide-level feature space for CLAM models; following PCA, the final slide-level feature representation used for the prediction of the model is plotted for each slide in both the validation and test set for a single cross-validated fold. PC, principal component. d–i, The number of slides used for each training-set size is shown in parentheses.
Labelled WSI data are often difficult to acquire, and it may not be feasible to collect thousands of slides in the context of rare diseases (for example, CRCC), unusual findings or clinical trials. In light of these limitations, to investigate the data efficiency of our models, we sequentially sampled subsets of training data equal to 75, 50, 25 and 10% of the total number of cases in each training set created during cross-validation. For each subsampled training set, its corresponding test set was kept the same to investigate the dependency of the performance of the model on the amount of training data available. We also kept each corresponding validation set constant to avoid the introduction of the model selection criteria as an additional confounding variable to the test performance of a model. When supervising CLAM models with the smaller sampled subsets of training data, we observed that the number of slides required to achieve satisfactory performance (AUC > 0.9) varies depending on the classification task. For example, merely 25% of the total available training cases (which represents an average of approximately 170 slides in each cross-validated fold) is sufficient to achieve an average test AUC above 0.94 on RCC subtyping, whereas 25% of the lung training set (419 slides) and 50% of the lymph-node-metastasis dataset (289 slides) might be needed for NSLCC subtyping and the detection of lymph node metastasis, respectively. Finally, to investigate the value of attention pooling over max-pooling, we compared the performance of CLAM with MIL and the other popular weakly supervised method of naively assuming the same slide-level label for every patch, denoted as ‘same label’ (SL). We implemented a multi-class variant of MIL for three-class RCC subtyping, which we denote mMIL (see Methods for technical details). In our comparative study we found that CLAM consistently outperforms the max-pooling-based algorithms for all tasks and training-set sizes (Fig. 2d–f). The AUC difference between CLAM, max-pooling-based algorithms and SL are more pronounced when fewer slides are used for training. For example, SL demonstrates a reasonable performance for RCC subtyping at 100 and 75% of training data, probably because of the high tumour content present in the TCGA RCC dataset, which means most of the training labels used by SL will be correct when assigning the slide-level diagnosis to all regions in each WSI. On the other hand, SL performs poorly in the detection of lymph node metastasis, given that the areas of metastasis can be small and sparse, which leads to a high amount of label noise when naively assigning the slide-level label to every location of tissue in each slide. Overall, we note that CLAM is data efficient, as it is often able to achieve test AUC > 0.9 using only several hundred slides for training. To investigate whether the additional task of instance-level clustering in CLAM contributes to the increased data efficiency, we conducted ablation studies for all disease models across training sets of different sizes and observed that the additional instance-level supervision improves model performance over using bag-level supervision alone when the training-set size is small (Supplementary Table 4).
We also conducted experiments to assess the performance of different algorithms under data constraint using 60/10/30 and 40/10/50 partitions instead of 80/10/10 train/validate/test partitions, which allows for model evaluation on larger test sets (Supplementary Table 5). To enable comparisons with future studies, we conducted additional experiments using the publicly available TCGA, CPTAC and CAMELYON datasets (see Supplementary Table 6 for details). Furthermore, we analysed the performance of CLAM in the context of the larger body of related works (Supplementary Table 7) evaluated on the public datasets that we used for the three different diagnostic tasks. First, we applied CLAM to the public CAMELYON16 lymph-node-metastasis detection challenge. We trained on the official training set (without using any of the pixel-level annotation provided) after splitting the 270 WSIs into approximately 85% training and 15% validation. Our best model achieved a test AUC of 0.936 (95% confidence interval (CI): 0.890– 0.983) on the official test set of 129 WSIs. This is an encouraging result given that no pixel-level labels were used during training. Similarly, we trained a CLAM model to perform NSCLC subtyping on just TCGA diagnostic WSIs, where 15% of cases (80 LUAD and 81 LUSC WSIs) were held out as the test set and the remaining data were divided into 85% training and 15% validation. This model achieved a test AUC of 0.963 (95% CI: 0.937–0.990).
Generalization to independent test cohorts.
Due to differences in institutional standards and protocols for tissue processing, slide preparation and digitization, WSIs can vary greatly in image appearance. Therefore, it is important to validate that models trained under the CLAM weakly supervised framework using publicly available data sources of a moderate size are robust to data-specific variables and generalize to real-world clinical data from scanners and staining protocols that are not encountered during training. We collected and scanned a total of 135 RCC (CRCC, 43; CCRCC, 46; and PRCC, 46), 131 NSCLC (LUAD, 63; and LUSC, 68) and 133 lymph node (negative, 66; and positive, 67) whole slides at the Brigham and Women’s Hospital (BWH) as independent test cohorts to evaluate the generalization performance of our trained models (further explained in Methods and Supplementary Table 8). For each task and training-set size, the ten models trained during cross-validation on our public datasets were directly evaluated on the completely held-out independent test set. We observed that for smaller denominations of the training set, the variance in the cross-validation performance of different models were often much higher, in which case testing using a single best-performing model may give the illusion of data efficiency although the performance of the algorithm on the independent test set would be inconsistent and vary highly across models developed using different random splits of training data. To accommodate this, we used the average performance of all ten models (instead of a single selected model) to estimate the performance of our algorithm for each training-set size. When testing on independent test cohorts, the 10-fold cross-validated CLAM models trained using 100% of the training set achieved an average one-versus-rest AUC (macro-averaged) of 0.972 ± 0.008 on RCC subtyping, and an average AUC of 0.975 ± 0.007 for NSCLC subtyping and 0.940 ± 0.015 for the detection of axillary lymph node metastasis (Fig. 3a–c). In addition, we observed that even CLAM models trained on the smaller subsets of the full training set can achieve respectable performance (test AUC > 0.9) on data from independent sources after learning from just hundreds of slides (Fig. 3d–f). When compared with mMIL/MIL and SL, CLAM delivered improved performance across all tasks and training-set sizes (Fig. 3d–f, top and middle) and especially when constrained by limited training data. For example, when trained with 25% of the full training set, CLAM outperformed MIL/mMIL by 14.2, 5.77 and 29.2% in average test AUC on RCC subtyping, NSCLC subtyping and the detection of lymph node metastasis, respectively, and similarly outperformed SL by 7.32, 16.6 and 29.7% in these same respective experiments (for comparisons using additional classification metrics, see Supplementary Table 9–11). In addition, we observed that CLAM models became on average less confident as the size of the training set was reduced (Fig. 3d–f, bottom), which is in general more desirable than having inaccurate but overly confident models that severely and erroneously overfit on the small training set that they observe.
Fig 3. Adaptability to independent test cohorts.
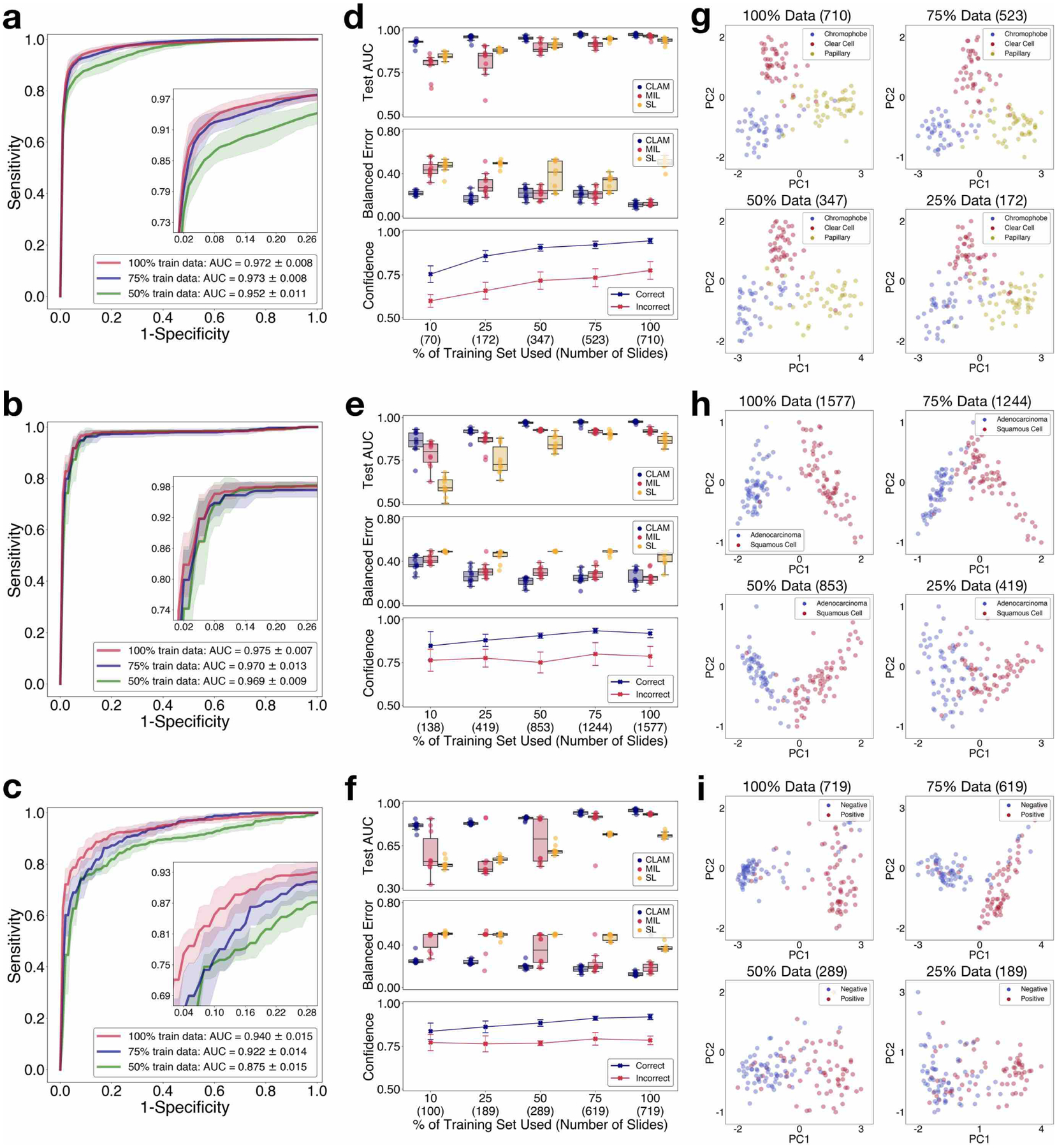
a–i, Independent test cohorts from BWH for RCC (a,d,g), NSCLC (b,e,h) and lymph node metastasis (c,f,i) are used to assess and analyse the capability of CLAM models trained on public datasets to generalize to new data sources that are not encountered during training. a–c, Performance of the CLAM model in terms of 10-fold mean test AUCs ± s.d. for RCC subtyping (n = 135), NSCLC subtyping (n = 131) and the detection of lymph node metastasis (n = 133). Insets: zoomed-in view of the curves. d–f, For each training-set size, the test AUCs (top) and balanced error scores (middle) of ten models are reported for CLAM, MIL (mMIL for RCC subtyping) and SL using box plots. The boxes indicate the quartile values and the whiskers extend to data points within 1.5× of the interquartile range. The results demonstrate that CLAM models can generalize to new data sources after training on a limited number of labelled slides and outperform other weakly supervised baselines with high consistency. Mean confidence (±1 s.d.) of CLAM model predictions for correctly and incorrectly classified slides (bottom). In general, CLAM models become less confident when trained using fewer data. g–i, Visualization of the slide-level feature space in two dimensions for select models from different training-set sizes. d–i, The number of slides used for each training-set size is shown in parentheses.
For NSCLC and RCC subtyping, the models trained on the public datasets of TCGA and CPTAC must also adapt to the different micrometre-per-pixel (m.p.p.) resolution produced by the in-house Hamamatsu scanner compared with the Aperio scanners used to digitize the training data. Whereas the vast majority of WSIs from TCGA RCC and NSCLC and CPTAC NSCLC had a ×20 equivalent m.p.p. close to 0.5, the in-house WSIs had a ×20 equivalent m.p.p. of 0.44. On the in-house NSCLC lung dataset, we also tested a mechanism to standardize the resolution at test time by downscaling the image patches to an approximate m.p.p. of 0.5 before they were embedded by the CNN encoder during data processing. However, we only observed a small improvement in mean test AUC to 0.979 ± 0.005 when using this technique. To further investigate the impact of variability introduced by different scanner hardware, we digitized all of the in-house lung-resection slides using an additional 3DHistech MiraxScan 150 scanner, which produces an m.p.p. of 0.328. We found that our models were able to achieve an average test AUC of 0.910 ± 0.022 when evaluating on the native scanning resolution of the new scanner despite the drastic difference in the m.p.p. resolution of the 3DHistech scanner in comparison to the Aperio scanners used to digitize the public training data (Supplementary Fig. 2). On the other hand, by standardizing the image patches from the 3DHistech scans to an m.p.p. of 0.5, we improved the test AUC to 0.965 ± 0.006. These results reasonably demonstrate that our proposed weakly supervised learning framework is quite robust to variation in scanner hardware but also illustrates the potential importance of m.p.p. standardization when evaluating on slides from new data sources, especially when the m.p.p. difference between the training data and test data is large.
Overall, the results from our study are highly encouraging and serve as supporting evidence that using CLAM, datasets of a moderate size curated from multiple institutions (with source-specific variability) and a diverse patient distribution (for example, TCGA) are sufficient to develop accurate, weakly supervised computer-aided diagnostic models capable of generalization. For best performance during real-world clinical deployment, we additionally propose to ensemble the diagnostic predictions from multiple models instead of selecting a single model. This is computationally inexpensive to accomplish as we only have to perform feature extraction on our data once, unlike methods that require tuning a feature encoder for each model. The ensemble performance (with 95% CI) of trained CLAM models on all independent test cohorts is demonstrated in Supplementary Fig. 3 and Supplementary Tables 12–14.
Interpretability and whole-slide attention visualization.
Human-readable interpretability of the trained weakly supervised deep-learning classifier can validate that the predictive basis of the model aligns with well-known morphology used by pathologists and can also be used to analyse failure cases. In addition, whole-slide-level heatmaps can be used for artificial-intelligence assisted human-in-the-loop clinical diagnoses. A CLAM model makes its slide-level prediction by first identifying and aggregating regions in the WSI that are of high diagnostic importance (high attention score) while ignoring regions of low diagnostic relevance (low attention score). To visualize and interpret the relative importance of each region in the WSI, we can generate an attention heatmap by converting the attention scores for the predicted class of the model into percentiles and mapping the normalized scores to their corresponding spatial location in the original slide. Fine-grained attention heatmaps can be created using overlapping patches (for example, 95% overlap) and averaging the attention scores in the overlapped regions (see Supplementary Fig. 4 for a discussion on the visual quality of heatmaps for different degrees of overlap). Although pixel-level or patch-level annotation was never used during training to explicitly inform the model whether each region is tumour tissue (and, if so, which subtype of tumour), we observed that through weakly supervised learning using slide-level labels only, trained CLAM models are generally capable of delineating the boundary between tumour and normal tissue (Fig. 4a–c; see the interactive demo at http://clam.mahmoodlab.org for high-resolution heatmaps). This is an especially welcoming property given that for RCC and NSCLC subtyping, all training data collected from the TCGA are positive cases and contain tumour regions. The finding demonstrates that CLAM has the potential to be used towards meaningful whole-slide-level interpretability and visualization in cancer subtyping problems for clinical or research purposes, without the need to observe negative cases during training (which would require either collecting slides from adjacent normal tissue or manual annotation of negative regions in positive slides). Of equal importance, high-attention regions generally correspond with morphology already established and recognized by pathologists for all of the three classification tasks studied (Fig. 4a–c). For example, the CLAM model trained for NSCLC subtyping highlights prominent intercellular bridges and keratinization, and uses them as strong evidence (high attention) for LUSC (Fig. 4b), in concordance with human pathology expertise. In addition, we examined the attention heatmap of the model with corresponding cytokeratin (AE1/AE3) immunohistochemical staining to further validate its predictive basis for a representative case of lymph node metastasis (Supplementary Fig. 5). These heatmaps can also be used to analyse and investigate misclassified slides. We observed challenging cases in our in-house independent test data in which the high-attention patches selected by the model for prediction failed to clearly indicate the correct class due to poor differentiation in the tumour cells or the limited presence of contextual cues to delineate the tumour architecture (Supplementary Fig. 6). For the detection of lymph node metastasis, false positive predictions typically highlighted large epithelioid histiocytes that mimic tumour cells to some degree, whereas false negatives tended to result from small isolated clusters of tumour cells in micro-metastases and isolated tumour cells. Despite their practical usefulness, caution should be taken to not overly rely on the attention heatmaps with the expectation that they can serve as pixel-perfect segmentation masks; intuitively, the attention scores for each region in the slide are relative and simply represent the interpretation by the model of which regions are more important (relative to others) in determining the slide-level prediction. Nonetheless, this simple and intuitive interpretability and visualization technique can provide researchers insight into the morphological patterns driving the predictions of the model; after further quantitative investigation, we also found that the attention heatmaps exhibit a high level of agreement with the pathologist annotations of tumour regions across all tasks when evaluated on our in-house resection slides (Supplementary Fig. 7).
Fig 4. Interpretability and visualization.
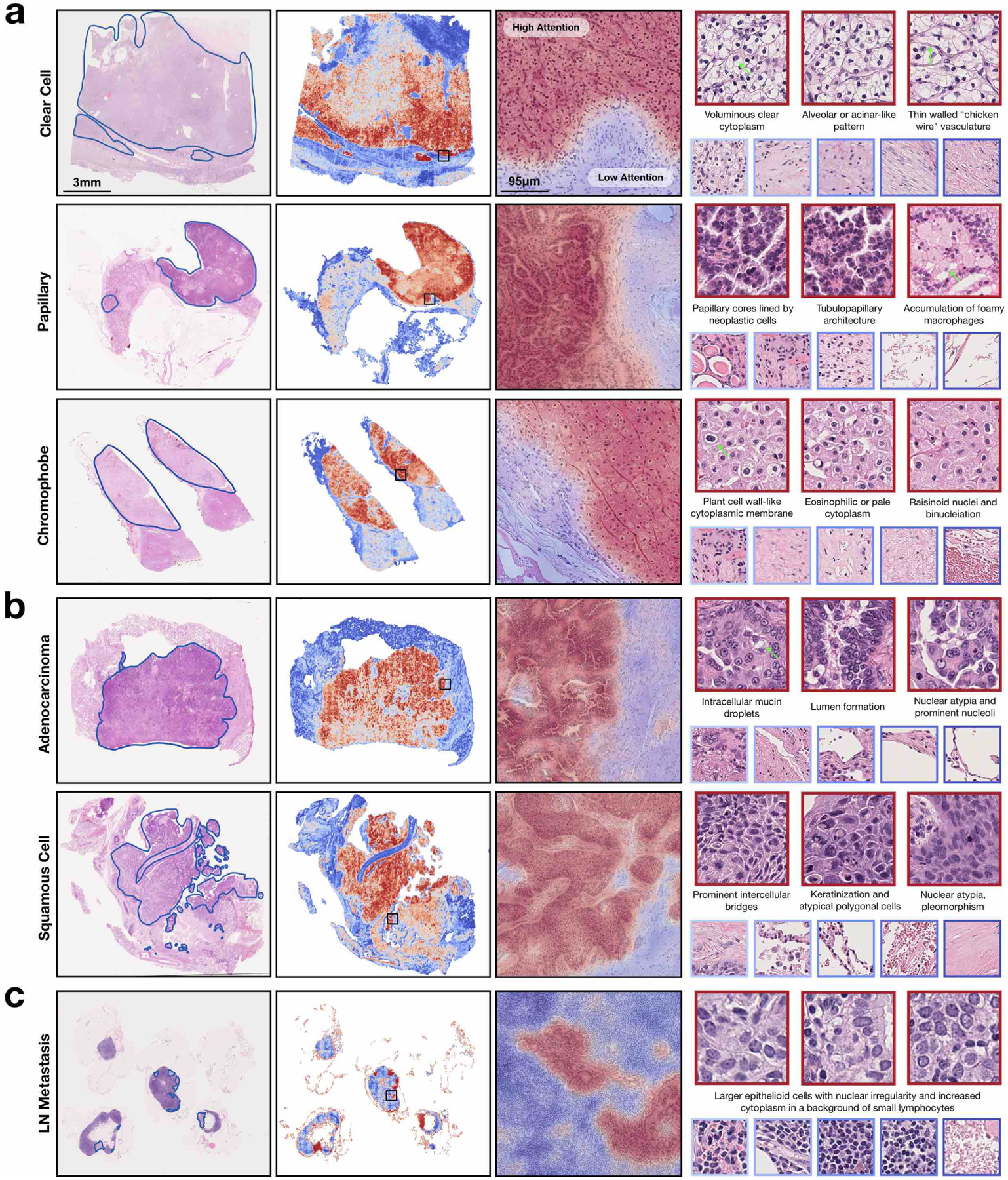
a,b, For RCC (a) and NSCLC (b) subtyping, a representative slide from each subtype was annotated by a pathologist (left), who roughly highlighted the tumour tissue regions. c, Similarly, regions of metastasis are highlighted for a case of lymph node metastasis (left). a–c, A whole-slide attention heatmap corresponding to each slide was generated by computing the attention scores for the predicted class of the model over patches tiled with a spatial overlap of 25% (second column); the fine-grained ROI heatmap, which highlights parts of the tumour normal boundary, was generated using a 95% overlap and overlaid onto the original H&E image (third column; zoomed-in view of the regions in the black squares in the images to its left). Patches of the most highly attended regions (red border) generally exhibit well-known tumour morphology and low-attention patches (blue border) include normal tissue among different background artefacts (right). Green arrows highlight specific morphology corresponding to the textual description. High-resolution WSIs and heatmaps corresponding to these slides may be viewed in our interactive demo (http://clam.mahmoodlab.org).
As a means of enhanced interpretability, we further investigated the patch-level feature space learned by the CLAM models. We randomly sampled a subset of patches from each slide in the independent test cohorts, reduced their learned instance-level 512-dimensional feature representations into two dimensions using principal component analysis (PCA) and examined their class predictions assigned by the clustering layers of the network (Supplementary Fig. 8). For the RCC and NSCLC slides, patches of different predicted classes are separated into distinct clusters in the feature space and exhibit morphology characteristic of their respective subtype. For the detection of axillary lymph node metastasis, sampled patches predicted as the positive cluster include tumour tissue, whereas negative (agnostic) patches capture a wide array of morphologies, including normal tissue and dense immune cell populations.
Adaptability to smartphone microscopy images.
We also explored the ability of our models (which are trained exclusively on WSIs) to directly adapt to microscopy images captured using a smartphone camera (commonly known as photomicrographs). In resource-constrained areas with limited access to pathologist expertise, consult cases are often imaged using a smartphone attached to a conventional microscope[39]. Training a deep-learning classifier specifically based on smartphone microscopy images would probably require the time-consuming and laborious process of manually curating a large set of labelled ROIs. These ROIs should not only be representative of the underlying pathological conditions but also capture a wide range of tissue-site and patient-specific appearances and artefacts to ensure that the model can adapt to the heterogeneity inherently found in histopathology slides and WSIs. A robust model trained on WSIs that is capable of directly adapting to cellphone images (CPIs) and deliver accurate automated diagnosis is therefore of tremendous value to the wider adoption of telepathology. As part of our model adaptability study, 4–8 fields of view (FOVs) from each slide in our independent test cohorts were captured using a consumer-grade iPhone X smartphone camera and patches from all FOV ROIs were collectively used by the model to predict the slide-level label. A variable number of FOVs were selected from each slide to cover the necessary tissue area relevant for diagnosis. CLAM achieved an average test AUC of 0.873 ± 0.025 on the NSCLC CPI dataset and an average one-versus-rest macro-averaged AUC of 0.921 ± 0.023 on the RCC CPI dataset (Fig. 5b,c and Supplementary Tables 15 and 16). The drop in performance compared with testing on WSIs (Fig. 5d) can probably be attributed to the imperfect conditions under which CPIs are captured (poor focus, non-uniform illumination, noise artefacts, vignetting, colour shift, magnification changes and so on). Although some of these adversities can potentially be reduced through the use of both conventional and deep-learning-based image-processing techniques (for example, stain normalization based on deep convolutional adversarial generative modelling[40]), we did not attempt to correct or normalize the images so as to test the robustness and adaptability of our models and keep the processing time and computational cost low to potentially allow inference directly on smartphone hardware. Despite these challenging variables, we found that in most cases, the model still accurately attends to regions in the FOV that exhibit well-known morphology characteristics of each cancer subtype (Fig. 5e,f). Furthermore, different classes are still visibly separated into distinct clusters in the feature space that the model has learned from WSIs (Fig. 5g,h). These results instil confidence with regards to the potential wider applicability of our weakly supervised learning framework to the telepathology domain.
Fig 5. Adaptability to smartphone microscopy images.
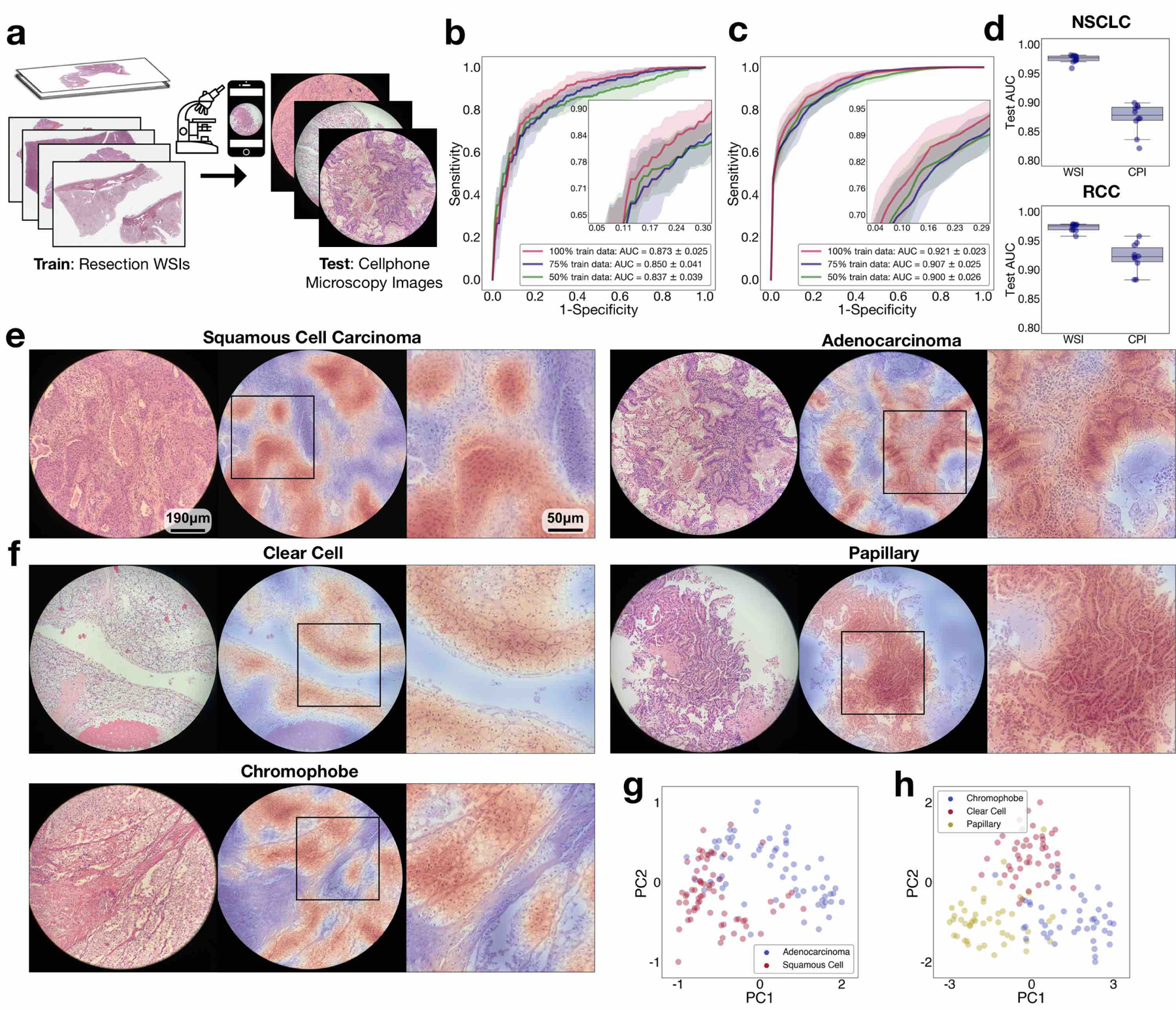
a, CLAM models trained on WSIs are adapted to CPIs taken with a consumer-grade smartphone camera without domain adaptation, stain normalization or further fine-tuning. b,c, An average test AUC of 0.873 ± 0.025 and 0.921 ± 0.023 was achieved for the BWH NSCLC (b; n = 131) and BWH RCC (c; n = 135) independent test sets, respectively. For each slide, patches extracted from all FOVs are collectively used by the CLAM model to inform the slide-level diagnosis. Insets: zoomed-in view of the curves. d, A drop in performance is expected when directly adapting models trained on data from one imaging modality (WSIs) to another (CPIs). We noted a decrease of 0.102 and 0.051 in the mean test AUC (relative to the performances on the corresponding WSI independent datasets) for NSCLC (top) and RCC (bottom) subtyping, respectively, when evaluating CLAM models (using 100% of the training set) on our CPI datasets. The boxes indicate the quartile values and the whiskers extend to data points within 1.5× of the interquartile range. e,f, The attention heatmaps (shown for NSCLC (e) and RCC (f) subtyping) help make model predictions interpretable by highlighting the discriminative regions in each FOV used by the model to make the slide-level diagnostic prediction. We observed that the model attends strongly to tumour regions and largely ignores normal tissue and background artefacts, as expected. However, due to the circular-shaped cutout of each FOV, patches near the border inevitably encapsulate varying degrees of black space in addition to the tissue content, which can mislead the model towards assigning weaker attention to those regions than it would otherwise. Zoomed-in views of the boxed regions are shown on the right. g,h, As additional validation that CLAM models trained on WSIs are directly applicable to the classification of CPIs, we visualized the attention-pooled feature representation of each set of CPIs and observed that there is visible separation between distinct classes in both the NSCLC (g) and RCC (h) smartphone datasets.
Adapting networks trained on resections to biopsies.
The publicly available WSIs that we used for training in our study are all resections. Compared with resected tissue, core-needle-biopsied tissue is generally substantially smaller in size. The limited tissue content as well as the presence of cell distortion due to crush artefact can challenge the diagnostic ability of the model. Accordingly, given that we did not use biopsy slides during training, it was important to investigate whether models trained solely on resections can adapt directly to biopsy slides and make accurate diagnostic predictions. We collected 110 lung (55 LUAD and 55 LUSC) and 92 kidney biopsy slides (53 CCRCC, 26 PRCC and 13 CRCC) at BWH as our independent test cohorts and directly tested our models that had been trained on the publicly available resection data. Each slide contains a variable number of embedded biopsy specimens, ranging from one to six for lung-biopsy WSIs and one to five for kidney-biopsy WSIs (Supplementary Table 17). For each WSI, tissue regions from all biopsy specimens embedded in the slide are provided to the model as input to make a single prediction for evaluation at the WSI level. On the lung-biopsy test set, CLAM achieved an average AUC of 0.902 ± 0.016 and on the kidney-biopsy test set, the average one-versus-rest macro-averaged test AUC was 0.951 ± 0.011 (Fig. 6b,c and Supplementary Tables 18,19). These results are highly encouraging because many biopsy slides, especially in the case of the lung-biopsy dataset, contained poorly differentiated tumours, which make them extremely difficult or impossible for pathologists to accurately diagnose based on the haematoxylin and eosin (H&E) stains alone (without immunohistochemistry). In addition, to assess the applicability of our models to potential real-world fully automated computer-aided diagnosis, when testing on biopsy slides, we did not manually select ROIs that contain high tumour content to avoid exposing the model to non-tumour features (blood vessels, inflammation, necrotic regions and so on)[35] that might lead to misclassification. We also did not perform any pre-processing techniques such as stain normalization on our test set and used the entire tissue region of each slide during evaluation. Using the same visualization and interpretability technique as before, we generated attention heatmaps for each subtype (Fig. 6d,e). We continued to observe a high similarity between the strongly attended regions highlighted by the trained CLAM models and the tumour regions annotated by the pathologist despite the tumours generally occupying smaller and more sparse tissue regions than in the resection slides.
Fig 6. Adaptability to biopsy slides.
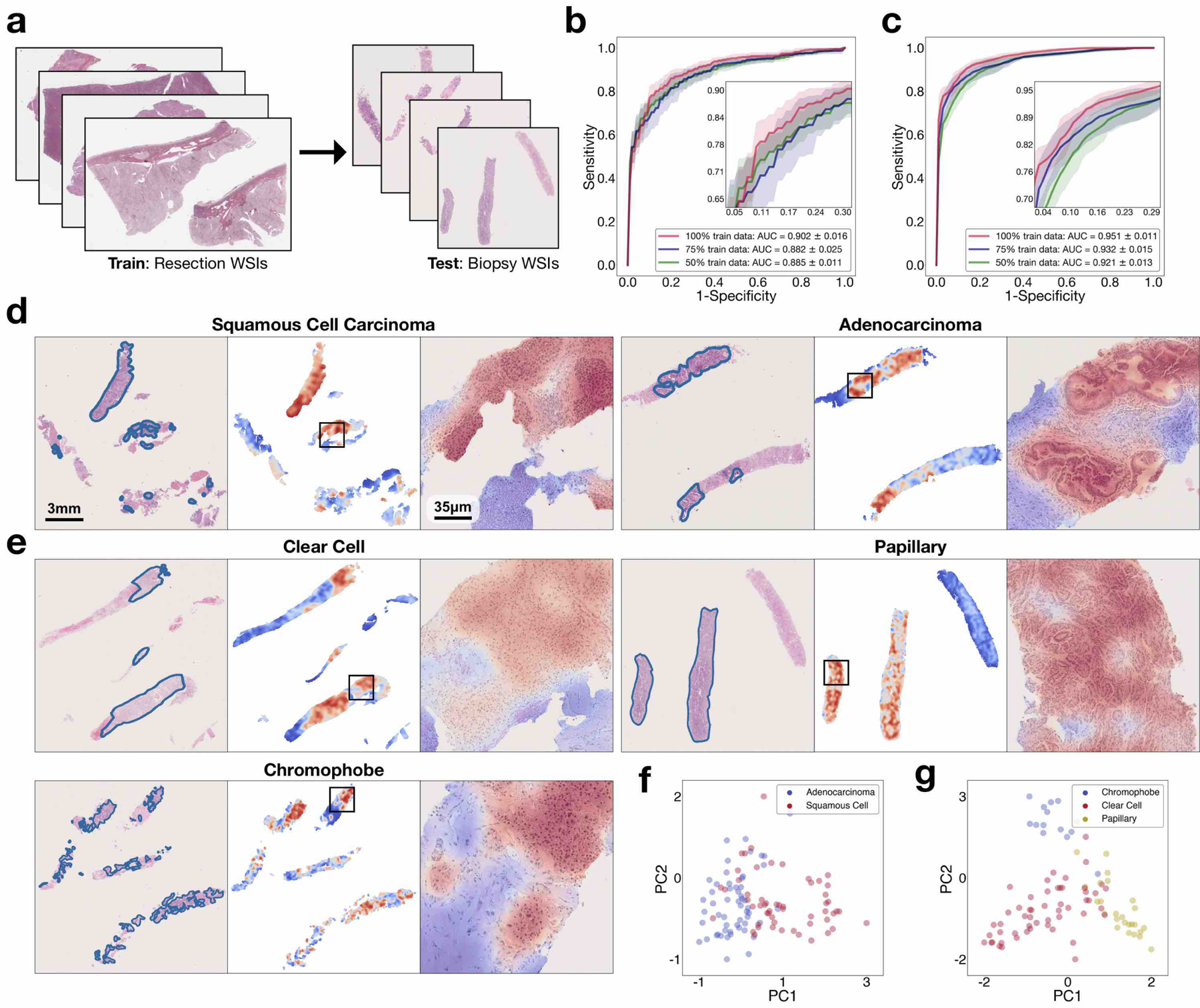
a, Compared with resection WSIs, biopsy WSIs generally contain a much lower tissue content (for example, the average number of patches extracted from the tissue regions of each slide is 820 in our BWH lung biopsy dataset compared with 24,714 in the lung-resection dataset). The presence of crush artefacts as well as poorly differentiated and sparsely distributed tumour cells can further challenge accurate diagnosis. b,c, We observed that CLAM models trained on resections are directly adaptable to biopsy WSIs, achieving a respectable average test AUC of 0.902 ± 0.016 and 0.951 ± 0.011 on our NSCLC (b; n = 110) and RCC (c; n = 92) biopsy independent test cohorts, respectively, without further fine-tuning or ROI extraction. Insets: zoomed-in view of the curves. d,e, Attention heatmap visualization for NSCLC (d) and RCC (e) biopsy slides. H&E slide with annotation by the pathologist for tumour regions (left). Heatmap for patches tiled with a 95% overlap (middle). Zoomed-in view of tumour regions attended by the CLAM model (right). Consistent with our findings on the resection and smartphone datasets, the regions that were most strongly attended by the model consistently correspond to tumour tissue. The attention heatmaps also tend to clearly highlight the tumour–normal tissue boundaries, despite the fact that no patch-level or pixel-level annotation was required or used during training. f,g, The slide-level feature representations of the biopsy datasets are visualized in two dimensions using PCA. We observed that the feature space learned by the CLAM model from resections remains visibly separable among the distinct subtypes when it is adapted to biopsy slides for both NSCLC (f) and RCC (g). A high-resolution version of these biopsy whole slides and heatmaps may be viewed our interactive demo (http://clam.mahmoodlab.org).
Discussion
Altogether, we showed that CLAM addresses several key challenges in computational pathology. Specifically, our analysis demonstrated that CLAM can be used to train interpretable, high-performance deep-learning models for both binary and multi-class WSI classification using only slide-level labels without any additional annotation. We are encouraged to note that our approach overcomes the barrier of time-costly annotation while also being more data efficient; we showed that it achieves a strong performance and also has the ability to generalize to independent test cohorts, smartphone microscopy and varying tissue content using a reasonable number of slides for training. Using CLAM, we are also able to showcase high-resolution interpretability heatmaps for the entire WSI, which may be used as an interpretability tool in research applications to identify morphological features associated with response and resistance to treatment or as a visualization tool for a secondary opinion in anatomic pathology to highlight ROIs. Although the use of attention-based pooling in CLAM provides the model with the flexibility of selectively aggregating information from multiple relevant ROIs to inform the slide-level diagnosis, a limitation of CLAM and MIL-based approaches in general for weakly supervised classification is that they typically treat different locations in the slide as independent regions and do not learn potential nonlinear interactions between instances, which may help the model become more context-aware. One line of future work will focus on extending the proposed weakly supervised framework to additional problems in computational pathology and developing more context-aware approaches. In addition, while fine-tuning the feature encoder in an end-to-end manner and using extensive data augmentation will probably to lead to further improvement in performance, end-to-end training that involves working with the original data space of image pixels is expected to drastically increase the total training time and computational resources required. In contrast with such a resource-hungry undertaking, the use of low-dimensional feature representations enables large-scale experimentation and allows us to conduct a detailed analysis of the data efficiency of different weakly supervised learning algorithms using extensive 10-fold cross-validation across a variety of tasks. However, this leaves room for future methods that will be able to flexibly strike a balance between end-to-end training that seeks to maximize the expressiveness of the model (especially when large diverse datasets are available to curb overfitting) and computationally efficient usage of feature representations for the weakly supervised learning on gigapixel WSIs. Last, other challenges that remain to be addressed and investigated in future studies include developing data-efficient weakly supervised methods for survival prediction, learning in the presence of noisy labels, poorly differentiated cases, mixed cancer subtypes and from extremely limited number of labelled data (for example, fewer than ten cases), predictions with uncertainty estimates and human in-the-loop decision-making.
Weakly supervised computational pathology is closer to clinical adaption because it only requires slide- or patient-level labels that are collected for clinical purposes. The improvement in data efficiency brought forth by our approach helps reduce the trade-off between weak supervision and the number of labelled whole slides required for training. While large diverse datasets are valuable assets for capturing as much heterogeneity within the data distribution as possible, data- efficient whole-slide training is essential to enable the applicability of computational pathology for classification in rare conditions as well as patient stratification for clinical trials where it is valuable to predict the response or resistance to treatment from a small cohort of existing patient cases. Within the context of our study, we found that CLAM is indeed capable of stratifying patients into predominant and relatively rare classes (for example, CCRCC versus CRCC). As we look forward to validating CLAM on a wider array of problems, we are also optimistic about the potential utility of CLAM in applications beyond the classification of WSI resections. For instance, we have found that models trained using CLAM and weak supervision are highly adaptable to independent data sources, biopsy slides, different scanning hardware and smartphone microscopy images without using any form of domain adaptation or fine-tuning. These important properties should allow researchers to develop models using resection slides (average tissue coverage: 142 mm2, 11,182 patches), which maximizes the diversity of tissue content encountered during training, with the flexibility to later adapt to biopsies (average tissue coverage: 15.6 mm2, 1,225 patches). Similarly, CLAM models trained on WSIs covering large tissue volume can adapt to CPIs with a limited FOV and have the potential to enable the routine use of telepathology in remote resource-constrained settings with limited anatomic pathology expertise, where consult cases are often imaged via a consumer-grade smartphone attached to brightfield microscopes. Overall, we hope our study and method will provide researchers with new ways to solve diagnostic and research problems using whole-slide images of routine histology specimens, thereby improving clinical care and facilitating knowledge discovery in computational pathology.
Methods
CLAM.
CLAM is a high-throughput deep-learning empowered toolbox designed to solve weakly supervised classification tasks in computational pathology, in which each WSI in the training set is a single data point with a known slide-level diagnosis but for which no class-specific information or annotation is available for any pixel or region in the slide. CLAM builds on the MIL framework, which views each WSI (known as a bag) as a collection comprised of many (up to hundreds of thousands) smaller regions or patches (known as instances). The MIL framework typically restricts its scope to binary classification problems of a positive and a negative class based on the assumption that if at least one patch belongs to the positive class, then the entire slide should be classified as positive, whereas a slide should be classified as negative if all patches are of the negative class. This assumption is reflected in the rigid non-trainable aggregation function of max-pooling, which simply uses the patch with the highest predicted probability for the positive class for the slide-level prediction, rendering MIL unsuitable for both multi-class classification and binary classification problems in which no intrinsic positive/negative assumption can be made. Besides max-pooling, although other aggregation functions such as the mean operator, generalized mean, log-sum-exp, the quantile function, noisy-or and noisy-and[41, 42, 43] can be used, they suffer from limited flexibility for problem and data-specific tuning and do not offer a simple, intuitive mechanism for model interpretability. In contrast, CLAM is generally applicable to multi-class classification and is built around the trainable and interpretable attention-based pooling function[37] to aggregate slide-level representations from patch-level representations for each class. In our design of multi-class attention pooling, the attention network predicts N distinct sets of attention scores corresponding to the N classes in a multi-class classification problem. This enables the network to unambiguously learn which morphological features should be considered as positive evidence (characteristic of the class) versus negative evidence (non-informative, absent of class-defining characteristics) for each class and summarize N unique slide-level representations. Specifically, for a WSI represented as a bag of K instances (patches), we denote the instance-level embedding corresponding to the kth patch as zk. In CLAM, the first fully connected layer further compresses each fixed patch-level representation to a 512-dimensional vector hk = W1zk (for simplicity, all bias terms are implied and not explicitly written). The attention network consists of several stacked fully connected layers; if we consider the first two layers of the attention network and and W1 collectively as part of the attention backbone shared by all classes, the attention network then splits into N parallel attention branches . Similarly, N parallel, independent classifiers, Wc,1, … Wc,N are built to score each class-specific slide-level representation. Accordingly, the attention score of the kth patch for the ith class, denoted ai,k, is given by equation (1) [37] and the slide-level representation aggregated per the attention score distribution for the ith class, denoted , is given by equation (2):
| (1) |
| (2) |
The corresponding unnormalized slide-level score sslide,i is given via the classifier layer by sslide,i = Wc,ihslide,i. We use dropout (P = 0.25) after each layer in the model’s attention backbone for regularization. For inference, the predicted probability distribution over each class is computed by applying a softmax function to the slide-level prediction scores sslide.
Instance-level clustering.
To further encourage the learning of class-specific features, we introduce an additional binary clustering objective during training. For each of N classes, we place a fully-connected layer after the first layer, W1. If we denote the weights of the clustering layer that corresponds to the ith class as , the cluster assignment scores predicted for the kth patch, denoted by pi,k, is given as:
| (3) |
Given we do not have access to patch-level labels, we use the outputs of the attention network to generate pseudo labels for each slide in each iteration of training to supervise the clustering. Instead of clustering all the patches in the slide, we only optimize the objective over the subset of most and least attended regions. Let the entire label set be , to avoid confusion, for a given slide, with ground truth class label , we refer to the attention branch that corresponds with this ground truth class (Wa,Y) as ‘in-the-class’, and the remaining N − 1 attention branches as ‘out-of-the-class’. If we denote the sorted list of in-the-class attention scores (in ascending order) as , we take the B patches with the lowest attention scores and assign them the negative cluster label (yY,b = 0, 1 ≤ b ≤ B) while the B patches with the highest in-the-class attention scores receive the positive cluster label (yY,b = 1, B + 1 ≤ b ≤ 2B). Intuitively, because each attention branch is supervised by the slide-level label during training, the B patches with high attention scores (hence the positive cluster) are expected to be strong positive evidence for class Y, while the B patches with low attention scores (hence the negative cluster) are expected to be strong negative evidence for class Y. Therefore, the clustering task can be intuitively interpreted as constraining the patch-level feature space hk such that the strong characterizing evidence of each class is linearly separable from its negative evidence. For cancer subtyping problems, all classes are often assumed to be mutually exclusive (i.e. they cannot be present in the same slide), as we cluster the most attended and least attended patches of the in-the-class attention branch into positive and negative evidence respectively, it makes sense to also impose additional supervision on the N − 1 out-of-the-class attention branches. Namely, given the ground truth slide label Y, , the B patches with the highest attention scores cannot be positive evidence for class i provided that we assume none of the patches on the slide is of class i (due to the mutual exclusivity). As a result, in addition to clustering the 2B patches selected from the in-the-class attention branch, we assign the negative cluster label to the top B attended patches in all out-of-the-class attention branches since they are assumed to be “false positive” evidence. On the other hand, if the mutual exclusivity assumption does not hold (e.g. cancer vs. no cancer problem, where a slide can contain patches from both tumor tissue and normal tissue), then we do not supervise the clustering of highly attended patches from out-of-the-class branches since we do not know if they are false positives or not. Using the aforementioned notations, the full instance-level clustering algorithm is summarized below in Algorithm 1.
Smooth SVM loss.
For the instance-level clustering task, we chose to use the smooth top1 SVM loss[44], which is based on the well-established multiclass SVM loss[45]. In a general N-class classification problem, neural network models output a vector of prediction scores s, where each entry in s corresponds to the model’s prediction for a single class. Given the set of all possible ground truth labels and ground truth label , the multiclass SVM loss penalizes the classifier linearly in the difference between the prediction score for the ground truth class and the highest prediction score for remaining classes only if that difference is greater than a specified margin α (equation (4)). The smoothed variant (equation (5)) adds a temperature scaling τ to the multiclass SVM loss, with which it has been shown to be infinitely differentiable with non-sparse gradients and suitable for the optimization of deep neural networks when the algorithm is implemented efficiently[44]. The smooth SVM loss can be viewed as a generalization of the widely used cross-entropy classification loss for different choices of finite values for the margin and different temperature scaling.

The introduction of a margin to the loss function has been empirically shown to reduce over-fitting when the data labels are noisy or when data are limited. During training, the pseudo labels we create to supervise the instance-level clustering task are expected to be noisy. Namely, the top attended patches might not necessarily correspond to the ground truth class and likewise, the least attended patches are also not guaranteed to be actual negative evidence of the class. Therefore, instead of the widely used cross-entropy loss (which is used for the slide-level classification task), we apply the binary top-1 smooth SVM loss to the outputs of the clustering layers of the network. In all our experiments, α and τ are both set to 1.0.
| (4) |
| (5) |
Training details.
During training, slides are randomly sampled and provided to the model using a batch size of 1. The multinomial sampling probability of each slide is inversely proportional to the frequency of its ground truth class (i.e. slides from underrepresented classes are more likely to be sampled relative to others) in order to mitigate class imbalance in the training set. Weights and bias parameters of the attention module are initialized randomly and trained end-to-end with the rest of the model using the slide-level labels since no ground truth attention is available. The total loss for a given slide is the sum of both the slide-level classification loss and the instance-level clustering loss with optional scaling via scalar c1 and c2:
| (6) |
To compute , sslide is compared against the ground truth slide-level label using the standard cross-entropy loss and to compute the instance-level clustering prediction scores pk for each sampled patch are compared against their corresponding pseudo cluster labels using the binary smooth SVM loss (recall for non-subtyping problems there are a total of 2B patches sampled from the in-the-class branch while for subtyping problems there are 2B patches sampled from the in-the-class branch and B patches sampled via each of N − 1 out-of-the-class attention branches). Unless otherwise specified, for each disease model, we tuned for B ∈ {8, 16, 32, 64, 128} on a single random validation fold by training on a subset of the training data (50% of the full training set). We considered c1 + c2 = 1 and similarly, tuned for c1 ∈ {0.3, 0.5, 0.7, 0.9} for the chosen B. Specifically, for the main 10-fold experiments, B = 8 was used for RCC subtyping and B = 32 for NSCLC subtyping and LN metastasis detection and c1 = 0.7 was used for all three tasks. We however did not observe a drastic difference in the validation performance for different values of B and c1 (Supplementary Fig. 9). The model parameters are updated via the Adam optimizer with a learning rate of 2 × 10−4 and weight decay of 1 × 10−5. For all experiments, default coefficient values for computing running averages of the first and second moment of the gradient were used (β1 = 0.9 and β2 = 0.999) and ϵ term (for numerical stability) was set to 1 × 10−8 (the default value). A commonly used technique that may help improve model generalization when training data is limited is data augmentation. However, to investigate the data efficiency of CLAM we did not attempt to perform data augmentation.
Model selection.
All models are trained for at least 50 epochs and up to a maximum of 200 epochs if the early stopping criterion is not metastasis Namely, the validation loss is monitored each epoch and when it has not decreased from the previous low for over 20 consecutive epochs, early stopping is used. The saved model, which has the lowest validation loss, is then tested on the test set.
Computational hardware and software
We used multiple hard drives to store the raw files of digitized whole slides. Segmentation and patching of WSIs were performed on Intel Xeon CPUs (central processing units) and feature extraction using a pre-trained neural network model was accelerated through data batch parallelization across multiple NVIDIA P100 GPUs on Google Cloud Compute instances or 2080 Ti GPUs on local workstations. All weakly supervised deep-learning models were trained with a total of ten local, consumer workstation-grade NVIDIA 2080 Ti GPUs by streaming extracted features from fast local solid-state-drive storage. Our whole-slide processing pipeline is implemented in Python (version 3.7.5) and takes advantage of image-processing libraries, such as openslide (version 3.4.1), opencv (version 4.1.1) and pillow (version 6.2.1). For loading data and training deep-learning models using CLAM, we used the Pytorch (version 1.3.1) deep-learning library. Based on our consumer-grade hardware, we also analysed the run time of CLAM for performing streamlined inference on our in-house WSI data. On a single local workstation and using two NVIDIA 2080 Ti GPUs, on average, using non-overlapping patches, CLAM requires 106.26 s (41.46 s for inference and 64.8 s for generating and saving a heatmap) for a ×20 resection WSI and 15.65 s (4.42 s for inference and 11.23 s for heatmap generation) for a ×20 biopsy WSI. Note that the inference speed includes the time to perform tissue segmentation, extract patches, extract features and perform classification, and heatmaps are generated and saved at the ×10 magnification. High-overlap (95%) and high-resolution (×10) WSI heatmaps shown in our interactive demo require multiple runs divided into many mini-batches of patches and are created and saved in 5,445 s per ×20 resection slide and 279 s per ×20 biopsy slide. The high compute time associated with generating high-resolution heatmaps based on a large number of overlapping patches can probably be substantially reduced using production-grade hardware and more efficient software parallelization.
All plots were generated using matplotlib (version 3.1.1) and seaborn (version 0.8.1). The AUC of the receiver-operating-characteristic curve was estimated using the Mann–Whitney U statistic, for which the algorithmic implementation is provided in the scikit-learn scientific computing library (version 0.22.1). The 95% confidence intervals of the true AUC were computed using DeLong’s method implemented by pROC (version 1.16.2) in R (version 3.6.1).
WSI datasets.
A summary of all of the datasets used are included in Supplementary Table 8. For the in-house test data, the BWH pathology archives were queried and cases were randomly sampled and requested from in-house pathology archives (2016–2019). We requested 150 resection cases for each problem, and 110 biopsy cases each for both NSCLC and RCC subtyping. We received slides based on their on-site availability at the time of study, and scanned slides with substantial markings covering the tissue area, damaged slides as well as slides that did not contain tumour (for RCC and NSCLC) were excluded before testing performance on our models; no other slides were excluded. Further details about each cohort are given in the following subsections. For model development and evaluation on public datasets using 10-fold Monte Carlo cross-validation, random train/validate/test dataset partitions are created where slides from the same patient case are sampled together to ensure that, for example, different slides from the same case are not sampled into both the training and test set. The number of slides available for each patient case can differ, which means that although all ten folds always have the same number of cases in their train/validate/test set, the exact number of slides might differ. For brevity, when we refer to the number of slides in the training or test set for the cross-validation folds, we refer to the average number of slides across all folds.
Public RCC WSI dataset.
Our public RCC dataset consists of a total of 884 diagnostic WSIs from the TCGA RCC repository under the Kidney Chromophobe (TCGA-KICH), Kidney CCRCC (TCGA-KIRC) and Kidney Renal Papillary Cell Carcinoma (TCGA-KIRP) projects. There are 111 CRCC slides from 99 cases, 489 CCRCC slides from 483 cases and 284 PRCC slides from 264 cases. The mean number of patches extracted per slide at ×20 magnification is 13,907.
Independent BWH RCC WSI dataset.
Our internal RCC dataset consists of a total of 135 WSIs from 133 cases, of which 43 slides are CRCC, 46 are CCRCC and 46 are PRCC. The mean number of patches extracted per slide at ×20 magnification is 20,394. Our RCC biopsy dataset consists of a total of 92 WSIs from 79 cases, of which 13 slides are CRCC, 53 are CCRCC and 26 are PRCC. The sample sizes for the CRCC biopsies were limited by the availability of patient cases for this rare condition (represents approximately 5% of all RCC cases with only a few biopsy cases). The mean number of patches extracted per slide at ×20 magnification is 1,709. Our RCC smartphone dataset comprises 4–8 FOVs per slide for each of the 135 slides. The mean number of patches extracted for each set of FOVs is 419. All slides were collected and processed at the BWH between 2016 and 2019.
Public NSCLC WSI dataset.
Our public NSCLC dataset consists of 993 diagnostic WSIs from the TCGA NSCLC repository under the TCGA-LUSC and TCGA-LUAD projects. There are 507 LUAD slides from 444 cases and 486 LUSC slides from 452 cases. In addition, we collected a total of 1,526 WSIs from the TCIA CPTAC Pathology Portal at the time of study that have lung as the topological site. From these WSIs, 668 slides from 223 cases are labelled as LUAD and 306 slides from 108 cases are labelled as LUSC. The remaining 552 slides are labelled as normal tissue and were excluded. Accordingly, our public lung dataset contains a total of 1,967 WSIs (1,175 LUAD slides from 667 cases and 792 LUSC cases from 560 patients). The mean number of patches extracted per slide at ×20 magnification is 9,958.
Independent BWH NSCLC WSI dataset.
Our internal NSCLC dataset consists of a total of 131 resection (63 LUAD and 68 LUSC) and 110 biopsy (55 LUAD and 55 LUSC) slides. Each slide comes from a unique case. The mean number of patches extracted per biopsy slide and per resection slide at ×20 magnification is 820 and 24,714, respectively. All slides were collected and processed at the BWH between 2016 and 2019. Our lung smartphone dataset comprises 4–8 FOVs per slide for each of the 131 resection slides. The mean number of patches extracted for each set of FOVs is 406. In addition, lung resection slides were scanned with a 3DHistech MiraxScan 150 to investigate adaptability to different scanning hardware and varying m.p.p.
Public lymph node WSI dataset.
CAMELYON16 and CAMELYON17 [46] are two of the largest publicly available, annotated breast-cancer lymph-node-metastasis detection datasets. CAMELYON16 consists of 270 annotated whole slides for training and another 129 slides as a held-out official test set collected at the Radboud University Medical Center and the University Medical Center Utrecht in the Netherlands. On the other hand, CAMELYON17 consists of a total of 1,000 slides from five different medical centres in the Netherlands. Because slide-level labels for the 500 slides in the official test set of CAMELYON17 were not yet publicly available, we used just the training portion of CAMELYON17, which consists of 500 slides (with corresponding slide-level diagnosis) for 100 cases. We combined CAMELYON16 and CAMELYON17 into a single dataset with a total of 899 slides (591 negative and 308 positive) from 499 cases. The mean number of patches extracted per slide at ×40 magnification is 41,802.
Independent BWH lymph node metastasis (breast cancer) WSI dataset.
Our internal breast-cancer lymph node metastasis dataset consists of a total of 133 WSIs from 131 cases (66 negative slides and 67 positive slides). The mean number of patches extracted per slide at ×40 magnification is 51,426. These slides were collected at BWH between 2017 and 2019.
Public Lymph Node WSI Dataset.
Camelyon16 and Camelyon17[46] are two of the largest publicly available, annotated breast cancer lymph node metastasis detection datasets. Camelyon16 consists of 270 annotated whole slides for training and another 129 slides as a held-out, official test set collected at the Radboud University Medical Center and the University Medical Center Utrecht in the Netherlands. On the other hand, Camelyon17 consists of a total of 1000 slides from 5 different medical centers in the Netherlands. Because slide-level labels for the 500 slides in the official test set of Camelyon17 were not yet publicly available, we used just the training portion of Camelyon17, which consists of 500 slides (with corresponding slide-level diagnosis) for 100 cases. We combined Camelyon16 and Camelyon17 into a single dataset with a total of 899 slides (591 negative and 308 positive) from 499 cases. The mean number of patches extracted per slide at 40 magnification is 41802.
Independent BWH Lymph Node metastasis (Breast Cancer) WSI Dataset.
Our internal breast cancer lymph node metastasis dataset consists of a total of 133 WSIs from 131 cases (66 negative slides and 67 positive slides). The mean number of patches extracted per slide at 40× magnification is 51426. These slides were collected at BWH between year 2017 and 2019.
WSI processing
Segmentation.
For each digitized slide, our pipeline begins with automated segmentation of the tissue regions. The WSI is read into memory at a downsampled resolution (for example, 32× downscale), converted from RGB to the HSV colour space. A binary mask for the tissue regions (foreground) is computed based on thresholding the saturation channel of the image after median blurring to smooth the edges and is followed by additional morphological closing to fill small gaps and holes. The approximate contours of the detected foreground objects are then filtered based on an area threshold and stored for downstream processing while the segmentation mask for each slide is made available for optional visual inspection. A human-readable text-file is also automatically generated, which includes the list of files processed along with editable fields containing the set of key segmentation parameters used. Although the default set of parameters are generally sufficient for reliable tissue segmentation, they can also be easily manually edited for any individual slide should the user find its segmentation results unsatisfactory.
Patching.
After segmentation, for each slide, our algorithm exhaustively crops 256 × 256 patches from within the segmented foreground contours at the user-specified magnification and stores stacks of image patches along with their coordinates and the slide metadata using the hdf5 hierarchical data format. Depending on the size of each WSI and the specified magnification, the number of patches extracted from each slide can range from hundreds (biopsy slide patched at ×20 magnification) to hundreds of thousands (large resection slide patched at ×40 magnification).
Feature extraction.
Following patching, we use a deep CNN to compute a low-dimensional feature representation for each image patch of each slide. Namely, we take a ResNet50 model pre-trained on ImageNet[47] and use adaptive mean-spatial pooling after the third residual block of the network to convert each 256 × 256 patch into a 1,024-dimensional feature vector using a batch size of 128 per GPU across multiple GPUs. The benefits of using extracted features as inputs to deep-learning models for supervised learning include a drastically faster training time and lower computational cost. This enables us to train a deep-learning model on thousands of WSIs in a matter of a few hours once the features have been extracted. Compared with using raw pixels, using low-dimensional features also makes it feasible to fit all patches in a slide (up to 150,000 or more) into memory simultaneously on a single consumer-grade GPU, thus avoiding the need for sampling patches and using noisy labels.
Visualization.
Visualizing slide-level feature space.
For each public WSI dataset, a model trained on one of the ten training sets created for cross-validation was used to compute a 512-dimensional slide-level feature representation for every slide in its corresponding validation and test set for the slide-level prediction of the model. The resulting set of slide-level feature vectors were reduced to two-dimensional space for visualization through transformation via PCA and each point was shaded by its ground-truth slide-level label. We then repeated this procedure for the models trained on 25, 50 and 75% of the same training set. We also performed the same analysis on the slides in each independent test cohort using the best-performing model for each training-set size.
Interpreting model prediction via attention heatmap.
To interpret the relative importance of different regions in a slide to the final slide-level prediction of the model, we computed and saved the unnormalized attention scores (before they were converted to probability distribution by applying the softmax function) for all of the patches extracted from the slide, using the attention branch that corresponded to the predicted class of the model. These attention scores were converted to percentile scores and scaled to between zero and 1.0 (with 1.0 being most attended and zero being least attended). The normalized scores were converted to RGB colours using a diverging colourmap and displayed on top of their respective spatial locations in the slide to visually identify and interpret regions of high attention displayed in red (positive evidence, high contribution to the prediction of the model relative to other patches) and low attention displayed in blue (low contribution to prediction of the model relative to other patches). To create more fine-grained heatmaps, we tiled the slides or smaller ROIs (for example, 8,000 × 8,000) into 256 × 256 patches using an overlap and calculated the raw attention score for each patch. We then followed a similar procedure and used the same colourmap as above to convert the raw score of each patch in the ROI to RGB colours. To ensure that the normalized attention scores computed for patches produced with an overlap were directly comparable to those for the set of non-overlapping patches used by the model for prediction, we referred to the set of unnormalized attention scores over the entire slide (without overlap) when calculating the percentile score of each patch. The ROI heatmaps were overlaid over the original WSI with a transparency value of 0.5 to simultaneously visualize the underlying morphological structures in the original H&E slide. Biopsy and ROI heatmaps were produced with an overlap of 95%. To produce fine-grained heatmaps for CPIs, a 95% overlap was used and attention scores were normalized over each image.
Visualizing patch-level feature space.
For each slide in the independent test cohort, we uniformly randomly sampled 2% of its tissue patches and recorded their clustering probability predictions, made by each of the N clustering branches in addition to their 512-dimensional feature representations after the first fully connected layer. For subtyping problems, patches for which all clustering branches predicted a positive probability of less than 0.5 (in other words, the clustering branch of every class considers them as negative evidence for its respective class) were labelled as class-agnostic, whereas the remaining patches were labelled with the class for which its positive probability is the highest. For metastasis detection in axillary lymph nodes, the clustering branch corresponding to the positive class was used to label patches as positive (positive probability greater than or equal to 0.5) and class-agnostic (positive probability less than 0.5). Using the same technique above for visualizing the slide-level feature space, we reduced each patch-level feature vector to two dimensions using PCA.
Quantitative evaluation of attention heatmaps.
While the attention heatmaps generated from CLAM models trained in a weakly supervised learning fashion are not designed or intended to perform pixel-level annotation of ROIs, to assess the possibility of using the heatmaps as an assistive annotator in a clinical or research setting as well as the correctness of attention, we evaluated the predicted attention heatmaps produced by a single CLAM model against the pathologist annotations using quantitative metrics including the Dice score, intersection over union and Cohen’s κ for each disease model. For all resection slides in the in-house dataset of each disease model, two anatomic pathologists were asked to independently and exhaustively annotate the tumour regions in all slides using the annotation tool Automated Slide Analysis Platform (ASAP). No time constraints were placed and for the annotation of metastasis in axillary lymph nodes, AE1/AE3 immunohistochemistry were used to assist in the annotation and ensure small tumour regions (micro-metastasis) were not missed. For evaluation, all heatmaps were generated by tiling patches at a 75% overlap. Binary masks were produced from heatmaps after dynamic thresholding, in concordance with the probable real-world scenario where a human operator can freely adjust the display threshold for the desired range to identify contiguous and dense regions of high attention. Each heatmap was thresholded without assistance from the pathologist annotations. Following binarization, simple post processing techniques including morphological closing and opening are applied to reduce fragmentation, close small cavities and suppress small artefacts. We did not apply closing and opening for lymph node metastasis, due to the presence of micro-metastasis, which can make up pixel-islands of extremely small area that are easily destroyed by such operations. We instead slightly dilated the foreground to connect neighbouring fragments and filter out pixel-islands for which all pixels have an attention of less than 0.95. Finally, despite extensive thoroughness, it is not possible to exclude all negative pixels that are present inside regions of tumour, thus we apply a tissue segmentation algorithm to detect large cavities inside the tissue and exclude such regions from the evaluation of the heatmaps. However, we note that this cannot automatically identify all regions of cavity, especially if they are small, and also does not take into account small areas of normal tissue inside an annotated tumour region. The results for both sets of pathologist annotations for all disease models are summarized in Supplementary Fig. 7.
Comparative analysis using MIL.
The most well-known MIL decision rule involves a diagnostic model making a prediction for every patch in a whole slide and the patch with the highest predicted probability for the positive class is selected to both inform the final diagnostic decision for the entire slide as well as gradient signals during training. In addition to using MIL, which simply takes the highest probability patch, the authors of a recent study[36] introduced a recurrent-neural-network-based aggregation that sequentially passes the top S patches ranked on the basis of their predicted probability for the positive class through a recurrent neural network to obtain the final slide-level prediction. However, on three different large datasets (prostate cancer, skin cancer basal cell carcinoma and lymph node metastasis detection), they noted a test AUC ranging from marginal improvement to no improvement using recurrent-neural-network-based aggregation. In light of these findings, we used the widely adopted max-pooling MIL formulation as our baseline for comparison. For each slide, during training, feature embeddings of all patches in the slide are read into memory at once, which corresponds to an input into the MIL network of shape K × 1,024. K is the number of patches (known as the bag size), which varies from slide to slide, and each patch is described by a fixed 1,024-dimensional vector representation zk, produced previously in the feature-extraction step using a pre-trained ResNet50 model. The MIL network has one fully connected layer with 512 hidden units and is followed by rectified linear unit (ReLU) activation and the classification layer. If we denote the weights and bias of each layer as , and , , respectively, the unnormalized prediction score sk, 1 ≤ k ≤ K for each patch can therefore be defined as:
| (7) |
According to the max-pooling aggregation rule, the patch whose predicted probability score for the positive class is the highest is then selected to represent the final slide-level prediction.
As previously mentioned, this MIL algorithm was designed specifically for binary classification. To compare the performance of CLAM against MIL in the multi-class setting, we also implemented a multi-class variant of MIL, which we call mMIL; mMIL has a fully connected layer of the same dimension as our binary MIL network but we adjust the binary classification layer to instead be to predict the N-class probability distribution of every patch in the slide. Similar to performing max-pooling in the binary case, based on the raw scores, we select the patch with the highest single class probability score across all classes as the slide-level prediction. For both the MIL and mMIL models, we used dropout (P = 0.25) after the model hidden layer for regularization.
Training details.
During training, for each slide, scores of the patch selected via max-pooling are passed to the cross-entropy loss function and the model parameters are optimized via stochastic gradient descent using a batch size of one and the Adam optimizer with the same hyperparameters as CLAM. Namely, we use a learning rate of 2 × 10−4, weight decay of 1 × 10−5, with β1 set to 0.9, β2 set to 0.999 and ϵ set to 1 × 10−8. Similarly, we use the same mini-batch sampling strategy and early stopping and model selection criteria for MIL/mMIL as for CLAM. For inference, the predicted probability distribution over each class is computed by normalizing the raw predicted scores of the max-pooled patch using the softmax function.
Comparative analysis using the slide-level label assigned to every patch.
Another weakly supervised learning framework used in computational pathology when pixel- or ROI-level annotations are not available is to simply sample patches from the tissue regions of each WSI and assign the slide-level label to each and every patch retrieved from that slide. We refer to this technique as SL in this study. By following this procedure, patches sampled from all WSIs in the training set can simply be treated as independent labelled data points during training. Without any annotation to guide the sampling process, this procedure implies that it is possible to infer the slide-level label from every patch sampled from that slide, which cannot be reasonably substantiated in most classification problems performed on WSIs and results in noisy labels. For example, in a positive lymph node containing a micro-metastasis, only a tiny fraction of the patches sampled from the slide would contain tumour and hence be responsible for the slide-level label, while all remaining negative patches will be mislabelled as positive for the purpose of training. Therefore, one would expect the performance of SL to be limited by the level of label noise, which is closely related to the signal-to-noise ratio of patches in each WSI. We used the 1,024-dimensional feature vector representation for each patch in our datasets as per CLAM and MIL/mMIL. The SL models consist of a stack of fully connected layers and (with the same dimensions as those in the MIL/mMIL networks) for mapping each patch embedding into N-class probability scores following softmax activation. Consistent with the CLAM and MIL/mMIL models, we used dropout (P = 0.25) after the hidden layer of the SL model.
Training details.
During training, patches are randomly sampled from slides in the training set using a batch size of 512. For inference during validation and test time, to get the slide-level prediction, we followed a previous study[35] by using the model to first make a prediction for every patch in the slide and then averaging their probability scores. We validate the model after every 100,000 patches and use early stopping on the model when the validation loss does not decrease for 20 consecutive validation epochs. The model checkpoint with the lowest validation loss is used for evaluation on the test set, which is consistent with the model selection criteria we use for MIL/mMIL and CLAM. Similarly, we use the cross-entropy loss function, and the model parameters are optimized via stochastic gradient descent using the Adam optimizer with a learning rate of 2 × 10−4 and weight decay of 1 × 10−5, with β1 = 0.9, β2 = 0.999 and an ϵvalue of 1 × 10−8.
Data availability
The TCGA diagnostic whole-slide data (NSCLC, RCC) and corresponding labels are available from the NIH genomic data commons (https://portal.gdc.cancer.gov). The CPTAC whole-slide data (NSCLC) and the corresponding labels are available from the NIH cancer imaging archive (https://cancerimagingarchive.net/datascope/cptac). Metastatic-lymph-node data are publicly available from the CAMELYON16 and CAMELYON17 website (https://camelyon17.grand-challenge.org/Data). We included links to all public data in Supplementary Table 20. All reasonable requests for academic use of in-house raw and analysed data can be addressed to the corresponding author. All requests will be promptly reviewed to determine whether the request is subject to any intellectual property or patient-confidentiality obligations, will be processed in concordance with institutional and departmental guidelines and will require a material transfer agreement.
Code availability
All code was implemented in Python using PyTorch as the primary deep learning library. The complete pipeline for processing whole slide images and training and evaluating deep learning models is available at https://github.com/mahmoodlab/CLAM and can be used to reproduce the experiments of this paper. All source code is provided under the GNU GPLv3 free software license.
Supplementary Material
Acknowledgements
The authors would like to thank Alexander Bruce for scanning internal cohorts of patient histology slides at BWH; Jingwen Wang, Katerina Bronstein, Lia Cirelli and Sharifa Sahai for querying the BWH slide database and retrieving archival slides; Martina Bragg, Sarah Zimmet and Terri Mellen for administrative support; and Zahra Noor for developing the interactive demo website. This work was supported in part by internal funds from BWH Pathology, NIH National Institute of General Medical Sciences (NIGMS) R35GM138216A (to F.M.), Google Cloud Research Grant and the Nvidia GPU Grant Program. R.J.C. was additionally supported by the NSF Graduate Research Fellowship and NIH National Human Genome Research Institute (NHGRI) T32HG002295. The content is solely the responsibility of the authors and does not reflect the official views of the National Institute of Health, National Institute of General Medical Sciences, National Human Genome Research Institute and the National Science Foundation.
Footnotes
Competing interests
The authors declare that they have no competing financial interests.
Ethics oversight
The study was approved by the Mass General Brigham (MGB) IRB office under protocol 2020P000233.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
- [1].Bera K, Schalper & Madabhushi, A. Artificial intelligence in digital pathology—new tools for diagnosis and precision oncology. Nature Reviews Clinical Oncology 16, 703–715 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Niazi MKK, Parwani AV & Gurcan MN Digital pathology and artificial intelligence. The Lancet Oncology 20, e253–e261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hollon TC et al. Near real-time intraoperative brain tumor diagnosis using stimulated raman histology and deep neural networks. Nature Medicine 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kather JN et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nature medicine 25, 1054–1056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bulten W et al. Automated deep-learning system for gleason grading of prostate cancer using biopsies: a diagnostic study. The Lancet Oncology (2020). [DOI] [PubMed] [Google Scholar]
- [6].Ström P et al. Artificial intelligence for diagnosis and grading of prostate cancer in biopsies: a population-based, diagnostic study. The Lancet Oncology (2020). [DOI] [PubMed] [Google Scholar]
- [7].Schapiro D et al. histocat: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nature methods 14, 873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Moen E et al. Deep learning for cellular image analysis. Nature methods 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mahmood F et al. Deep adversarial training for multi-organ nuclei segmentation in histopathology images. IEEE transactions on medical imaging (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Graham S et al. Hover-net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Medical Image Analysis 58, 101563 (2019). [DOI] [PubMed] [Google Scholar]
- [11].Saltz J et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell reports 23, 181–193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Javed S et al. Cellular community detection for tissue phenotyping in colorectal cancer histology images. Medical Image Analysis 101696 (2020). [DOI] [PubMed] [Google Scholar]
- [13].Mobadersany P et al. Predicting cancer outcomes from histology and genomics using convolutional networks. Proceedings of the National Academy of Sciences 115, E2970–E2979 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Heindl A et al. Microenvironmental niche divergence shapes brca1-dysregulated ovarian cancer morphological plasticity. Nature communications 9, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yuan Y et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Science translational medicine 4, 157ra143–157ra143 (2012). [DOI] [PubMed] [Google Scholar]
- [16].Lazar AJ et al. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171, 950–965 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fu Y et al. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nature Cancer 1, 800–810 (2020). [DOI] [PubMed] [Google Scholar]
- [18].Kather JN et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nature Cancer 1, 789–799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen RJ et al. Pathomic fusion: An integrated framework for fusing histopathology and genomic features for cancer diagnosis and prognosis. IEEE Transactions on Medical Imaging (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Beck AH et al. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Science translational medicine 3, 108ra113–108ra113 (2011). [DOI] [PubMed] [Google Scholar]
- [21].Yamamoto Y et al. Automated acquisition of explainable knowledge from unannotated histopathology images. Nature Communications 10, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pell R et al. The use of digital pathology and image analysis in clinical trials. The Journal of Pathology: Clinical Research 5, 81–90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].LeCun Y, Bengio Y & Hinton G Deep learning. nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- [24].Esteva A et al. A guide to deep learning in healthcare. Nature medicine 25, 24–29 (2019). [DOI] [PubMed] [Google Scholar]
- [25].Esteva A et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature 542, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Poplin R et al. Prediction of cardiovascular risk factors from retinal fundus photographs via deep learning. Nature Biomedical Engineering 2, 158 (2018). [DOI] [PubMed] [Google Scholar]
- [27].McKinney SM et al. International evaluation of an ai system for breast cancer screening. Nature 577, 89–94 (2020). [DOI] [PubMed] [Google Scholar]
- [28].Mitani A et al. Detection of anaemia from retinal fundus images via deep learning. Nature Biomedical Engineering 4, 18–27 (2020). [DOI] [PubMed] [Google Scholar]
- [29].Shen L, Zhao W & Xing L Patient-specific reconstruction of volumetric computed tomography images from a single projection view via deep learning. Nature biomedical engineering 3, 880–888 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tellez D, Litjens G, van der Laak J & Ciompi F Neural image compression for gigapixel histopathology image analysis. IEEE Transactions on Pattern Analysis and Machine Intelligence (2019). [DOI] [PubMed] [Google Scholar]
- [31].Bejnordi BE et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. Jama 318, 2199–2210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen P-HC et al. An augmented reality microscope with real-time artificial intelligence integration for cancer diagnosis. Nature medicine 25, 1453–1457 (2019). [DOI] [PubMed] [Google Scholar]
- [33].Nagpal K et al. Development and validation of a deep learning algorithm for improving gleason scoring of prostate cancer. NPJ digital medicine 2, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang S et al. Rmdl: Recalibrated multi-instance deep learning for whole slide gastric image classification. Medical image analysis 58, 101549 (2019). [DOI] [PubMed] [Google Scholar]
- [35].Coudray N et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nature medicine 24, 1559–1567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Campanella G et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nature medicine 25, 1301–1309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ilse M, Tomczak J & Welling M Attention-based deep multiple instance learning. In International Conference on Machine Learning, 2132–2141 (2018). [Google Scholar]
- [38].Maron O & Lozano-Pérez T A framework for multiple-instance learning. In Advances in neural information processing systems, 570–576 (1998). [Google Scholar]
- [39].Schaumberg AJ et al. Interpretable multimodal deep learning for real-time pan-tissue pan-disease pathology search on social media. Modern Pathology 1–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].BenTaieb A & Hamarneh G Adversarial stain transfer for histopathology image analysis. IEEE transactions on medical imaging 37, 792–802 (2017). [DOI] [PubMed] [Google Scholar]
- [41].Couture HD, Marron JS, Perou CM, Troester MA & Niethammer M Multiple instance learning for heterogeneous images: Training a cnn for histopathology. In International Conference on Medical Image Computing and Computer-Assisted Intervention, 254–262 (Springer, 2018). [Google Scholar]
- [42].Kraus OZ, Ba JL & Frey BJ Classifying and segmenting microscopy images with deep multiple instance learning. Bioinformatics 32, i52–i59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang C, Platt JC & Viola PA Multiple instance boosting for object detection. In Advances in neural information processing systems, 1417–1424 (2006). [Google Scholar]
- [44].Berrada L, Zisserman A & Kumar MP Smooth loss functions for deep top-k classification. In International Conference on Learning Representations (2018). [Google Scholar]
- [45].Crammer K & Singer Y On the algorithmic implementation of multiclass kernel-based vector machines. Journal of machine learning research 2, 265–292 (2001). [Google Scholar]
- [46].Litjens G et al. 1399 h&e-stained sentinel lymph node sections of breast cancer patients: the camelyon dataset. GigaScience 7, giy065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Russakovsky O et al. Imagenet large scale visual recognition challenge. International journal of computer vision 115, 211–252 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TCGA diagnostic whole-slide data (NSCLC, RCC) and corresponding labels are available from the NIH genomic data commons (https://portal.gdc.cancer.gov). The CPTAC whole-slide data (NSCLC) and the corresponding labels are available from the NIH cancer imaging archive (https://cancerimagingarchive.net/datascope/cptac). Metastatic-lymph-node data are publicly available from the CAMELYON16 and CAMELYON17 website (https://camelyon17.grand-challenge.org/Data). We included links to all public data in Supplementary Table 20. All reasonable requests for academic use of in-house raw and analysed data can be addressed to the corresponding author. All requests will be promptly reviewed to determine whether the request is subject to any intellectual property or patient-confidentiality obligations, will be processed in concordance with institutional and departmental guidelines and will require a material transfer agreement.