

Abstract
KRAS mutations hinder therapeutic efficacy of epidermal growth factor receptor (EGFR)-specific monoclonal antibodies cetuximab and panitumumab-based immunotherapy of EGFR+ cancers. Although cetuximab inhibits KRAS-mutated cancer cell growth in vitro by natural killer (NK) cell-mediated antibody-dependent cellular cytotoxicity (ADCC), KRAS-mutated colorectal carcinoma (CRC) cells escape NK cell immunosurveillance in vivo. To overcome this limitation, we used cetuximab and panitumumab to redirect Fcγ chimeric receptor (CR) T cells against KRAS-mutated HCT116 colorectal cancer (CRC) cells. We compared four polymorphic Fcγ-CR constructs including CD16158F-CR, CD16158V-CR, CD32131H-CR, and CD32131R-CR transduced into T cells by retroviral vectors. Percentages of transduced T cells expressing CD32131H-CR (83.5 ± 9.5) and CD32131R-CR (77.7 ± 13.2) were significantly higher than those expressing with CD16158F-CR (30.3 ± 10.2) and CD16158V-CR (51.7 ± 13.7) (p < 0.003). CD32131R-CR T cells specifically bound soluble cetuximab and panitumumab. However, only CD16158V-CR T cells released high levels of interferon gamma (IFNγ = 1,145.5 pg/ml ± 16.5 pg/ml, p < 0.001) and tumor necrosis factor alpha (TNFα = 614 pg/ml ± 21 pg/ml, p < 0.001) upon incubation with cetuximab-opsonized HCT116 cells. Moreover, only CD16158V-CR T cells combined with cetuximab killed HCT116 cells and A549 KRAS-mutated cells in vitro. CD16158V-CR T cells also effectively controlled subcutaneous growth of HCT116 cells in CB17-SCID mice in vivo. Thus, CD16158V-CR T cells combined with cetuximab represent useful reagents to develop innovative EGFR+KRAS-mutated CRC immunotherapies.
Keywords: Fc gamma CR T cells, polymorphisms, immunotherapy, KRAS-mutated CRC, anti-EGFR mAb
Introduction
Epidermal growth factor receptor (EGFR) is overexpressed in several solid tumors.1 Upon binding by epidermal growth factor (EGF), EGFR triggers a series of signaling pathways supporting invasion and metastasis.1 The important role of EGFR in promoting cancer progression has provided the rationale for the development of EGFR-targeted monoclonal antibody (mAb)-based treatments.2
EGFR-specific cetuximab is a chimeric IgG1 mAb preventing EGFR dimerization by stimulating its internalization and degradation. EGFR-specific panitumumab is a human IgG2 mAb interfering with EGF binding to its receptor. Cancer cells incubated with cetuximab or panitumumab undergo cell cycle arrest and apoptosis.2 However, a variety of EGFR+ cancer cells, including colorectal carcinoma (CRC) cells, are insensitive to EGFR-specific mAbs since they carry RAS gene mutation(s) downstream of EGFR. Because of these mutations, cancer cells can bypass antitumor activities of both cetuximab and panitumumab. Lack of sensitivity of KRAS-mutated CRC cells to EGFR-specific mAb has serious clinical consequences since in these cases both cetuximab and panitumumab have either no effect on tumor growth or worsen CRC clinical course.3,4
Increasing evidence suggests that this limitation can be overcome, at least for cetuximab, by taking advantage of its ability to mediate ADCC since EGFR+ cells, opsonized with cetuximab, undergo ADCC by activating the CD16 receptor expressed on natural killer (NK) cells.5 Nevertheless, cancer progression in patients is not arrested.3,4
Failure to control cancer growth might be due to the ability of cancer cells to escape recognition by innate and adaptive immune cells. In patients with cancer, both NK cells, the major ADCC effector cells, and T cells are characterized by a variety of functional defects.6,7 However, T-cell infiltration into the tumor microenvironment is more frequently detectable and is associated with favorable prognosis.8–11 In contrast, NK cell infiltration in solid tumors is rare and usually devoid of prognostic significance.12
To restore the sensitivity of KRAS-mutated cancer cells to EGFR-specific mAbs, we investigated different strategies based on the generation of extracellular CD16-(chimeric receptor) CR linked to intracellular signaling and activating molecules.13–17 Because of NK cell limitations,6–10,12,18 we decided to use T cells, as effectors, since they easily infiltrate the tumor microenvironment and effectively protect hosts from cancer progression.19 Selected CD16-CR constructs were transduced into T cells to redirect them by EGFR-specific mAbs toward EGFR+ cancer cells.
Other than NK cells, myeloid cells also mediate effector functions including proinflammatory cytokine production20,21 and cytotoxic activity including ADCC.20,22 Unlike NK cells, myeloid cells have the exclusive property to recognize Fc fragments of IgG2 antibodies complexed with the corresponding antigens on target cells, utilizing the Fc receptor CD32 and triggering ADCC activation.23 Both CD16 and CD32 are polymorphic and their polymorphisms influence their binding to IgG Fc fragments.24 It is still unknown whether CD32 and CD16 polymorphisms impact the antitumor activity of Fcγ-CR T cells against KRAS-mutated CRC cells.
The goal of this study is to compare the ability of polymorphic CD16-CR and CD32-CR to inhibit KRAS-mutated CRC cell proliferation and tumor progression in vitro and in vivo.
Materials and Methods
Antibodies, reagents, and cell lines
Fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD3 (cat. 555332), FITC-conjugated-annexin V (cat. 556420), phycoerythrin (PE)-conjugated mouse anti-CD16 (cat. 555407), PE-conjugated mouse anti-human CD32 (cat. 550586), propidium Iodide (PI) (cat. 556463), allophycocyanin (APC)-conjugated mouse anti-human CD3 (cat. 555335), mouse anti-human CD3 (cat. 555329), CD28 (cat. 555725), CD32 (8.26) (cat. 557333), and CD16 (3g8) (cat. 556617) were purchased from BD Bioscience (San Jose, CA). Cetuximab (Erbitux 5 mg/ml) and panitumumab (Vectibix 20 mg/ml) were purchased from Merck Serono (Darmstadt, Germany) and Amgen (Thousand Oaks, CA), respectively. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich (Saint Louis, MO) and GeneJuice® Transfection Reagent (Novagen) from Millipore (Burlington, MA). Human recombinant interleukin-7 (IL-7) and interleukin-15 (IL-15) were purchased from PeproTech (London, UK) and Lipofectamine 2000 from Life Technologies (Carlsbad, CA). Retronectin (Recombinant Human Fibronectin) was purchased from Takara Bio (Saint-Germain-en-Laye, France). Dulbecco’s modified Eagle’s medium, Iscove’s modified Dulbecco’s medium, RPMI 1640 medium, fetal bovine serum (FBS), l-glutamine, and penicillin/streptomycin were purchased from Thermo Fisher Scientific (Waltham, MA). Complete media (CM) were supplemented with 10% FBS, 2 mM l-glutamine, 0.1 mg/ml streptomycin, and 100 U/ml penicillin. Mycoplasma-free, KRAS-mutated HCT116 cells25 (RRID:CVCL_0291) were maintained in RPMI 1640, CM. Mycoplasma-free, KRAS-mutated, non-small-cell-lung cancer cell line, A549 (RRID:CVCL_0023) was kindly provided by Dr. Antonio Rossi (Institute of Translational Pharmacology, CNR, Rome, Italy). The cells were authenticated on November 21, 2018, by PCR-single-locus-technology (Eurofins, Ebersberg, Germany). The cells were kept in culture for a maximum of four to eight passages.
Fcγ chimeric receptors
Generation of CD16158F-CD8α-CD28-CD3ζ CR has previously been described.16 The extracellular region of CD32A131R was amplified by reverse-transcriptase polymerase chain reaction (RT-PCR) from RNA extracted from peripheral blood mononuclear cells (PBMCs) utilizing the following primers: forward 5′-GAGAATTCACCATGACTATGGAGACCCAAATG-3′ and reverse 5′-CGTACGCCCCATTGGTGAAGAGCTGCC-3′ (Thermo Fisher Scientific, Waltham, MA). PCR product was fused by restriction enzyme-compatible ends with the CD8α-CD28-CD3ζ domain contained in the pcDNA3.1/V5-His TOPO TA (Invitrogen, Carlsbad, CA). CD16158F-CR and CD32131R-CR were subcloned into NcoI and MluI sites of the SFG retroviral vector. CD16158V and CD32A131H were assembled by using synthetic oligonucleotides. Fragments were separately inserted into SFG vector and sequenced by GeneArt Gene Synthesis team (Invitrogen-Thermo Fisher Scientific, Regensburg, Germany).
Retrovirus production and T-cell transduction
Retroviral supernatants were obtained by transient transfection of 293T cells, with Peg-Pam plasmid encoding the Moloney murine leukemia virus gag and pol genes, and RDF plasmid encoding the RD114 envelope and the CD32131R-, CD32131H-, CD16158F-, or CD16158V-CR SFG retroviral vectors using the GeneJuice reagent. Forty-eight and 72 hr post-transfection, retrovirus-containing supernatants were harvested, filtered, snap frozen, and stored at −80°C until use. To generate Fcγ-CR T cells, PBMCs (0.5 × 106 PBMCs/ml) were cultured for 3 days in nontissue culture treated 24-well plates precoated with 1 μg/ml anti-CD3 and 1 μg/ml anti-CD28 mAbs in the presence of 10 ng/ml IL-7 and 5 ng/ml IL-15. Viral supernatants were placed on retronectin-coated nontissue culture-treated 24-well plates and spun for 1.5 hr at 2000g. Activated T cells were seeded into retrovirus-loaded plates, spun for 10′, and incubated for 72 hr at 37°C in 5% CO2 atmosphere. After transduction, T cells were expanded in RPMI 1640 CM supplemented with 10 ng/ml IL-7 and 5 ng/ml IL-15.
In vitro tumor cell viability assays
The antitumor activity of Fcγ-CR T cells in vitro was evaluated by MTT assays. Tumor target cells (7 × 103/well) were seeded in 96-well plates, and Fcγ-CR T cells (35 × 103/well) were added in the presence or absence of (3 μg/ml) cetuximab or panitumumab. Following a 48–72 hr incubation at 37°C, nonadherent T cells were removed and 100 μl/well of fresh medium supplemented with 20 μl of MTT (5 mg/ml) were added to adherent cells. Incubation was prolonged for 3 hr at 37°C. Supernatants were then removed and 100 μl of dimethyl sulfoxide were added to each well. Absorbance was measured at 570 nm.
Cytokine release
Twofold dilutions of a Fcγ-CR T cell suspension (1×105/100 μl/well) were added to 96-well plates in triplicates. Then, EGFR+, KRAS-mutated HCT116 cells (2 × 104/100 μl/well) were added at a 5:1 E:T ratio in the presence or absence of 3 μg/ml cetuximab or panitumumab. Supernatants were collected after 48 hr incubation at 37°C and IFNγ, and TNFα levels were measured by ELISA (Thermo Fisher Scientific, Waltham, MA).
Flow cytometry and cytotoxicity assay
Fcγ-CR expression levels on engineered T cells were assessed by flow cytometry upon incubation for 30 min at 4°C with FITC-conjugated mouse anti-human CD3, PE-conjugated mouse anti-human CD32, or PE-conjugated mouse anti-human CD16 mAbs.
To assess cytotoxic potential of transduced lymphocytes, KRAS-mutated HCT116 cells or A549 cells (0.12 × 106) were incubated overnight at 37°C in 5% CO2 in the presence or absence of CD16158V-CR T cells (0.6 × 106), with or without anti-EGFR mAbs (10 μg/ml) while nontransduced T cells (0.6 × 106) were used as control. Cultures were then harvested and cells were stained with FITC-Annexin V, APC-conjugated anti-CD3, and PI. HCT116 and A549 cells were identified by gates posted on CD3 negative and FSC-Hhigh cells. The cells were analyzed by a 2-laser BD FACSCalibur (Becton Dickinson, S. Jose, CA) flow cytometer. Results were evaluated utilizing the Tree Star Inc. FlowJo software.
Xenograft mouse model
In vivo experiments were performed in accordance with the guidelines and regulations of the European Directive 2010/63/EU. The Italian Ministry of Health approved animal handling and procedures (authorization code: 186/2016-PR). Antitumor activity of CD16158V-CR T cells with or without cetuximab was assessed using 8-week-old male CB17-SCID mice (CB17/lcr-PrkdcSCID/lcrlcoCrl, Charles River Laboratories, Lecco, Italy, Cat. CRL:236, RRID: IMSR CRL:236), 12–18 g body weight, engrafted with KRAS-mutated HCT116 CRC cells. Mice were housed in temperature-controlled rooms with 12 hr light/dark cycle and free access to sterile water and autoclaved standard chow diet (4RF25; Mucedola, Milan, Italy). Endogenous NK cell activity was suppressed by intra-peritoneal injection of 20 μl rabbit anti-asialo-GM1 antibody (Wako, Chemicals, Richmond, VA, Cat. 986–10001). Mice received anti-asialo-GM1 antibody on days −3, 0, +14, and +21 since tumor cell engraftment. On day 0, mice were grafted subcutaneously in the right flank, with 1 × 106 HCT116 cells and then randomly separated into four groups (5 mice per group). Group 1 received only HCT116, group 2 received cetuximab (150 μg), group 3 HCT116 and CD16158V-CR T cells, and group 4 HCT116 CD16158V-CR T cells and cetuximab (150 μg). Effector cells were administered at a 5:1 E:T ratio. Tumor volumes were measured every 3 days with caliper and calculated using the formula: TV (cm3) = 4/3pr3, where r = (length + width)/4. Mice were sacrificed when tumor volume reached 2 cm3.
Statistical analysis
Results were analyzed by paired-t-test, Mann–Whitney test, and two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple-comparison correction, as necessary. Disease-free survival (DFS) was evaluated by log-rank-(Mantel-Cox) test. Differences were considered significant with p-values <0.05.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Results and Discussion
To enhance antitumor potential of EGFR-specific mAbs, we generated polymorphic CD16 and CD32-CR vectors (Fig. 1a). We successfully expressed all indicated Fcγ-CR into transduced T cells (Fig. 1b). However, T cell transduction of CD32131H-CR (83.5% ± 9.5%) and CD32131R-CR (77.7% 13.2%) was significantly more effective than that of CD16158V-CR (51.7% ± 13.7%) and CD16158F-CR (30.3% ± 10.2%) (p < 0.003 Fig. 1c). Considering that all vectors are packaged using the same methodology and viral titers are similar, these results may suggest that in human T lymphocytes polymorphic CD32-CR are more stably expressed as compared to polymorphic CD16-CR, although underlying mechanism(s) remain to be explored. Notably however, Cheeseman et al. provided evidence that hematopoietic cells tend to express CD32 more efficiently than CD16 on their surfaces.26
Figure 1.
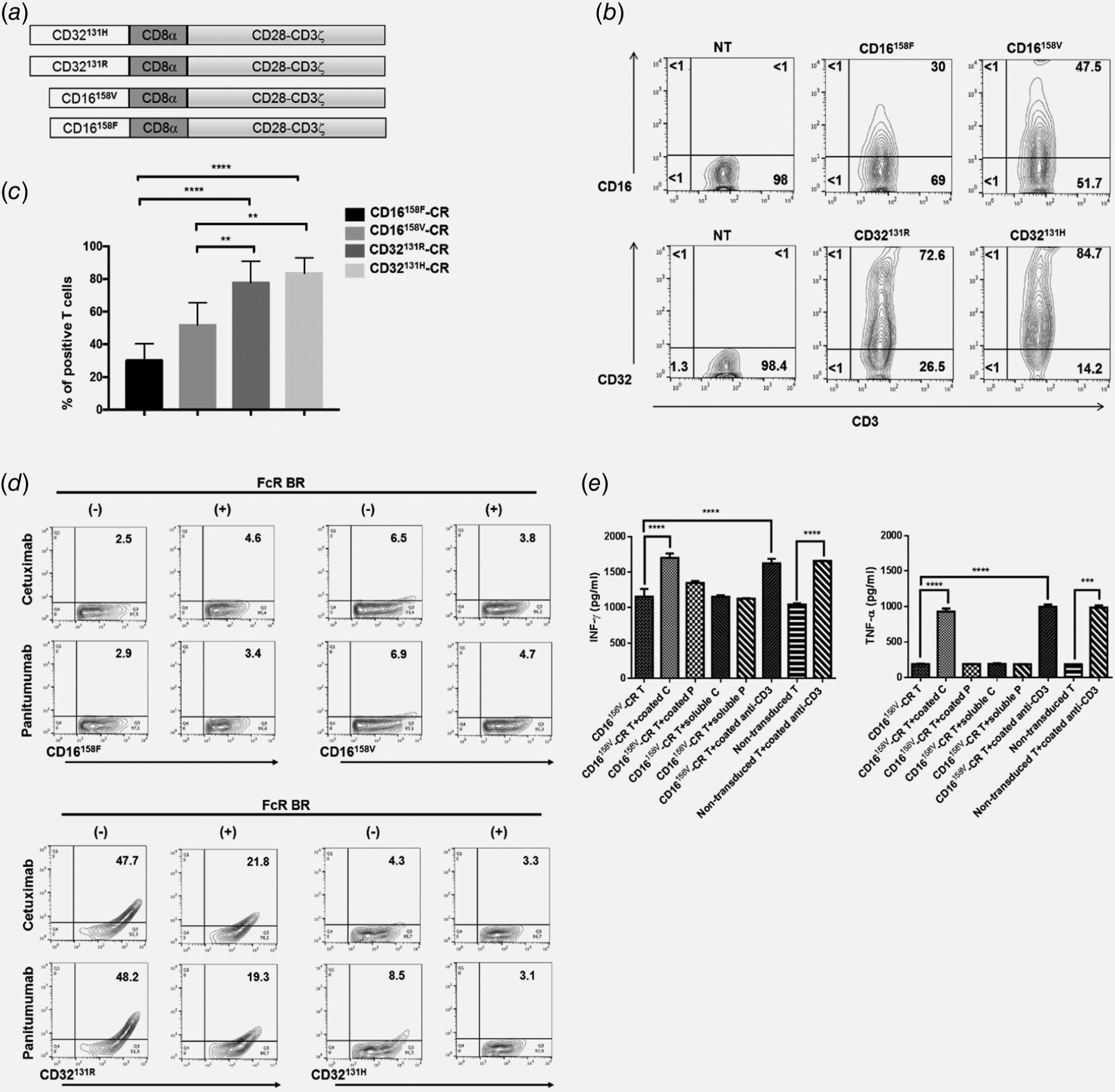
Molecular structure and functional features of Fcγ-CRs. Panel A shows a schematic representation of the gene constructs encoding the indicated Fcγ-CRs. The extracellular domain of high- and low-affinity CD32-CRs and CD16-CRs incorporate the transmembrane CD8α and CD28/CD3ζ chain. Panel B reports a representative experiment showing the expression of Fcγ-CRs on the surface of T cells 48 hr after retroviral transduction. Percentages of cells expressing the transgenes are shown in the quadrants. Panel C shows cumulative percentages of positive T lymphocytes for each Fcγ-CR 48 hr following viral infection of cells from five healthy donors. ****p < 0.0001, **p < 0.003. Panel D shows the binding of soluble cetuximab and panitumumab on the surface of Fcγ-CR T cells. T lymphocytes expressing each CD16-CR (upper panel) and CD32-CR (lower panel) polymorphisms were incubated with 10 μg/ml of either cetuximab or panitumumab for 30 min at 4°C with or without FcR BR. The cells were then washed and stained with a FITC-conjugated mouse anti-human IgG antibody. Binding of cetuximab and panitumumab was then evaluated by flow cytometry. FcR BR: Fc receptor blocking reagent. Panel E shows ELISA assays of IFNγ and TNFα release in the culture supernatants, of CD16158V-CR T cells cultured, in 24-well plates, for 72 hr at 37°C, in the presence or absence of 10 μg/ml of the following mAbs: soluble or immobilized cetuximab and soluble or immobilized panitumumab while an anti-CD3 mAb (10 μg/ml) was utilized as a positive control. ***p < 0.0007, ****p < 0.0001.
CD16-CR has also been produced in other laboratories, whereas, to the best of our knowledge, CD32-CR has not. CD16 and CD32 binding affinity for IgG is known to be influenced by their polymorphisms. The presence of valine instead of phenylalanine at position 158 of CD16 (CD16158V) and the presence of histidine instead of arginine at position 131 of CD32 (CD32131H) enhance the IgG binding affinity of these receptors.24,27 Notably, CD16158V has preferentially been utilized for in vitro and in vivo studies.13–15,28
To evaluate Fcγ-CR T cell antibody-binding capacity, polymorphic CD32-CR and CD16-CR T cells were incubated with cetuximab or panitumumab, for 30 min, at 4°C. CD32131R-CR T cells efficiently bound both cetuximab and panitumumab, whereas CD32131H-CR T cells only displayed a minimal binding for panitumumab (Fig. 1d, lower panel). In contrast, CD16158V-CR T cells and CD16158F-CR T cells failed to bind both cetuximab and panitumumab (Fig. 1d, upper panels). Binding of cetuximab and panitumumab to CD32-CR T cells was highly specific and promptly inhibited in the presence of Fc receptor blocking reagent (FcR BR) (Fig. 1d, lower panels).
Although in our study, neither CD16158V-CR nor CD16158F-CR showed significant binding affinity for soluble cetuximab (IgG1) or panitumumab (IgG2), Kudo et al.15 previously reported that CD16158V-CR has a significantly higher affinity for rituximab (IgG1) than that of CD16158F-CR. Notably, however, in accord with our data, Snyder et al. observed that NK92 cells expressing the high-affinity CD16A176V variant do not bind trastuzumab (IgG1).29 A possible explanation of these discrepancies might reside in structural differences in the stimulatory domains of the transduced chimeras. Nevertheless, our data clearly indicate that multivalent presentation of immobilized cetuximab, but not panitumumab, effectively induces CD16158V-CR stimulation, leading to significant enhancement of IFNγ and TNFα release, thereby suggesting that CD16-CR T successfully bind immobilized cetuximab (Fig. 1e).
We next asked whether polymorphic Fcγ-CR T cells recognize KRAS-mutated HCT116 cells opsonized with EGFR-specific mAb and affect their viability. Transduced and nontransduced T cells were incubated, for 72 hr, at 37°C with KRAS-mutated HCT116 cells with or without cetuximab or panitumumab. Production of IFNγ and TNFα was measured in culture supernatants. Only CD16158V-CR T cells, combined with cetuximab but not panitumumab, produced levels of both IFNγ (1,145.5 ± 16.5 pg/ml) and TNFα (614 ± 21 pg/ml) significantly higher than those released by the other Fcγ-CR T cells or nontransduced T cells (Fig. 2a).
Figure 2.
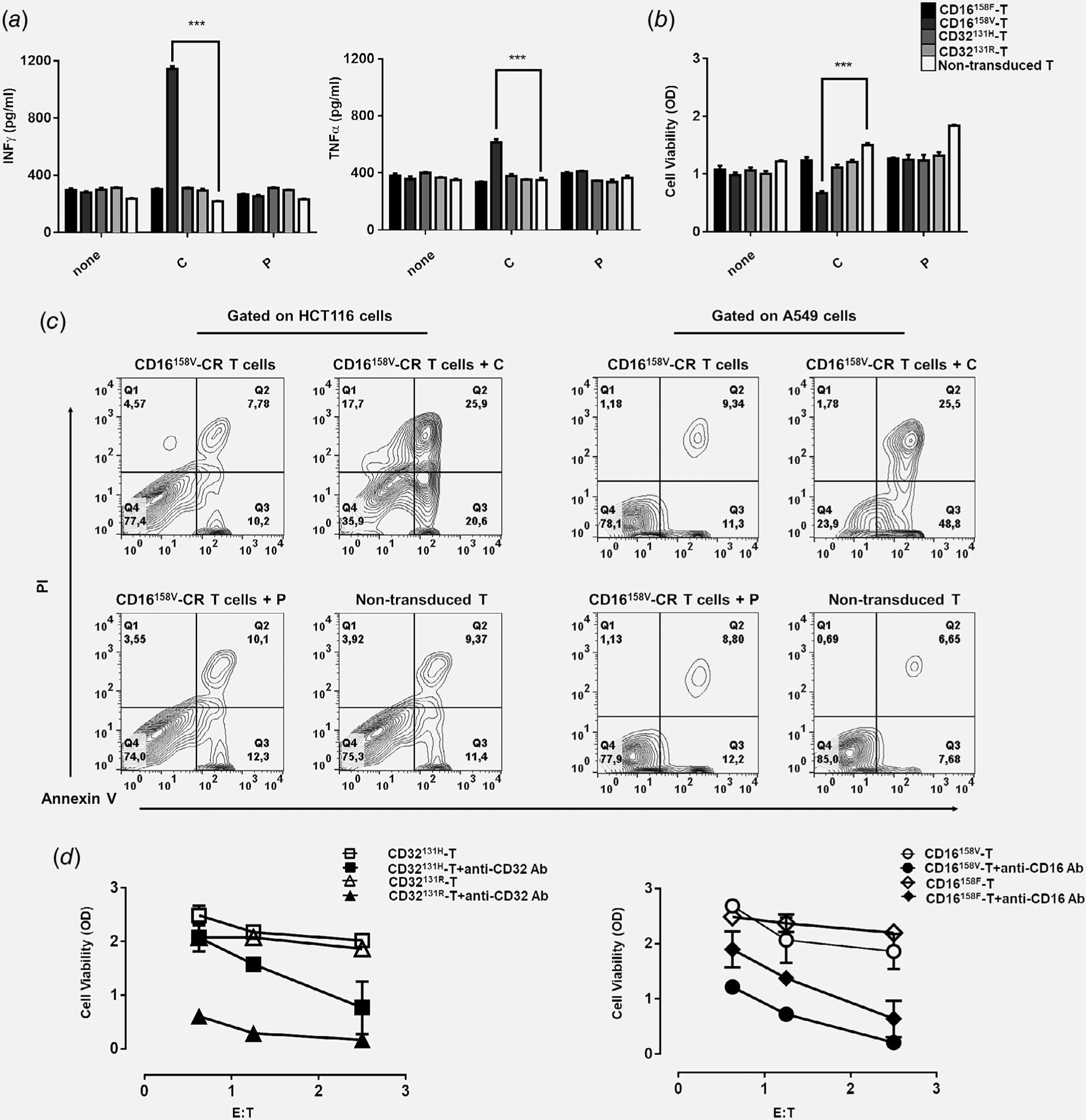
Recognition of HCT116 CRC cells by CD16158V-CR T cells in combination with cetuximab leads to proinflammatory cytokine production and HCT116 cell elimination. Panel (a) shows IFNγ and TNFα levels in supernatants of HCT116 cells incubated for 48 hr at 37°C with the indicated Fcγ-CR T cells in the presence or absence of cetuximab (3 μg/ml) or panitumumab (3 μg/ml), both at a E:T ratio of 5:1. Panel (b) shows viability, as evaluated by MTT assays of HCT116 cells incubated for 48 hr at 37°C with the indicated Fcγ-CR T cells with or without cetuximab or panitumumab at a E:T cell ratio of 5:1. C, cetuximab; P, panitumumab; white bars, nontransduced T cells. Asterisks indicate a p-value <0.001. The figure reports cumulative data, with mean ± SD values, of HCT116 cell viability obtained by using effector cells from five different donors in independent experiments. Panel (c) shows CD16158V-CR T cells killing of KRAS-mutated cell lines (HCT116 and A549) with or without cetuximab or panitumumab at an E:T ratio of 5:1. Nontransduced T cells were used as a control. After 16 hr, incubation cells were harvested, stained with APC-anti-CD3, FITC-annexin V and propidium iodide (PI), and analyzed by flow cytometry. Data are representative of five experiments independently performed. Panel (d) shows the results of redirected assays on the viability of stably transfected CD32 + HCT116 cells, as measured by the MTT assay. Fcγ-CR T cells were incubated for 3 days, at 37°C with CD32 + HCT116 in the presence or absence of anti-CD16 mAb (3 μg/ml) (right panel) with or without anti-CD32 mAb (3 μg/ml) (left panel) at the indicated E:T cell ratio. Viability of HCT116 target cells was then measured as described in the Methods section. Asterisks indicate p < 0.001.
Then, we tested whether polymorphic Fcγ-CR engineered T cells could impair the viability of KRAS-mutated HCT116 cell opsonized with cetuximab or panitumumab, by utilizing an ADCC mechanism. Figure 2b shows that KRAS-mutated HCT116 cell viability, expressed as optical density (OD), was significantly reduced following a 48 hr incubation at 37°C with CD16158V-CR T cells and cetuximab (0.67-OD ± 0.03-OD) as compared to nontransduced T cells (1.5-OD ± 0.03-OD). In contrast, no change in viability was detected when cetuximab was replaced with panitumumab. Both polymorphic CD32-CR T cells and CD16158F-CR T cells with or without anti-EGFR mAbs, failed to impair KRAS-mutated HCT116 cell viability.
To further validate the cytotoxic activity of CD16158V-CR, HCT116, and A549 cells, also characterized by KRAS mutations and EGFR expression were incubated overnight at 37°C in 5% CO2, with or without CD16158V-CR T cells, in the presence or absence of cetuximab. In these assays, we observed that cetuximab-opsonized HCT116 and A549 cells clearly undergo apoptosis and necrosis in the presence of CD16158V-CR T (Fig. 2c). These data indicate that efficient recognition of cetuximab-opsonized cancer cells by CD16158V-CR T cells leads to the activation of effector functions including production of proinflammatory cytokines and impairment of KRAS-mutated HCT116 and A549 cell viability. Therefore, CD16158V-CR T cells restore the ability of cetuximab to target KRAS-mutated HCT116 CRC cells by an immune-mediated mechanism in vitro.
Since these data challenged the effector potential of CD16158F- and CD32-CR T cells, we tested them in redirected ADCC assays, using, as targets, KRAS-mutated HCT116 cells stably transfected with CD32. Although to different extents, all CR-engineered T cells, at different E:T ratios, significantly reduced the viability of FcγR+HCT116 cells in the presence of 3g8 (anti-CD16) or 8.26 (anti-CD32) mAb (Fig. 2d). These data indicate that all engineered T cells are fully competent effector cells.
Thus, although transgenic CD32 binds soluble mAbs and is able to provide a cytotoxic signal, CD32-CR T cells are unable to produce proinflammatory cytokines in co-culturing with opsonized KRAS-mutated HCT116 and to impair their viability. Therefore, soluble mAb binding and redirected killing do not appear to represent effective surrogate assays to test the ability of transduced T cells to elicit mAb-mediated effector functions targeting tumor cells. Notably, affinity of IgG1 cetuximab for CD32 is low and panitumumab mediates ADCC only in the presence of myeloid cells.23
The in vivo antitumor potential of CD16158V-CR T cells was then assessed. Transduced cell ability to impair KRAS-mutated HCT116 viability was tested prior to their administration to experimental animals. CD16158V-CR T cells were incubated at 37°C with target cells and cetuximab or panitumumab, while nontransduced T cells were used as a negative control. Following 72 hr incubation, KRAS-mutated HCT116 viability was assessed by the MTT assay. Data reported in Figure 3a confirm that co-culture with CD16158V-CR T cells, in combination with cetuximab, significantly affects HCT116 cell viability. Instead, CD16158V-CR T alone or together with panitumumab or nontransduced T cells were completely ineffective.
Figure 3.
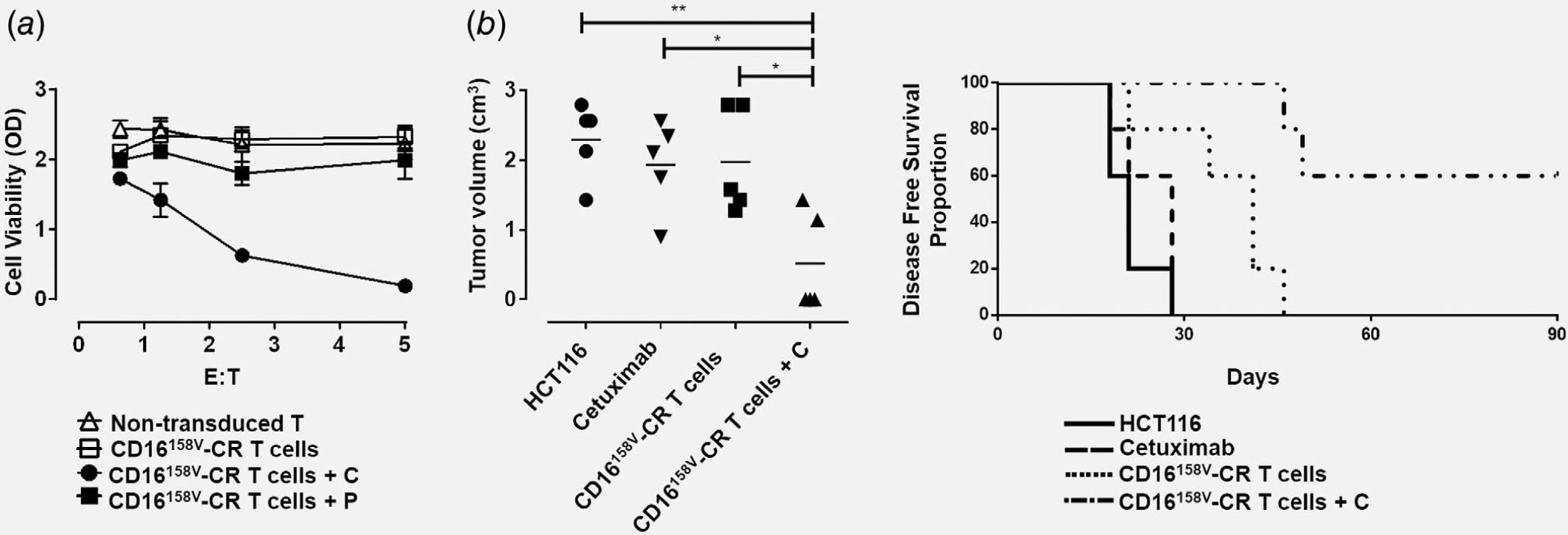
CD16158V-CR T cells in combination with cetuximab protect CB17 SCID mice from subcutaneous growth of KRAS-mutated HCT116 cancer cells. Panel (a) shows HCT116 cells incubated for 72 hr, at 37°C, in the presence or absence of CD16158V-CR T cells at the indicated E:T ratios with or without cetuximab (C) or panitumumab (P) both at the concentration of 3 μg/ml. Nontransduced T cells were used as a control. HCT116 cell viability was evaluated by MTT assay. Values are expressed as mean ± SD. Data are analyzed by a two-way ANOVA test. Panel (b) shows four groups of CB17 SCID mice (N = 5 per group) injected with HCT116 cells (1 × 106) subcutaneously, in the right flank. Following HCT116 injection, three groups of mice were injected, in the area adjacent to HCT116 injection, with 150 μg of cetuximab (group 2), 5 × 106 CD16158V-CR T cells (group 3), and 5 × 106 CD16158V-CR T cells plus 150 μg of cetuximab (group 4). HCT116 cell growth was then monitored. Left panel shows a scatterplot analysis of tumor volumes resulting from subcutaneous injection of KRAS-mutated HCT116 cells 64-days postinjection. Right panel shows DFS analysis, as evaluated by the log-rank (Mantel-Cox) test. *p < 0.02, **p < 0.001.
We then engrafted CB17-SCID mice subcutaneously with KRAS-mutated HCT116 cells. One hour later, CD16158V-CR T cells, with or without cetuximab, were injected in proximity to the injection site of HCT116 cells. Figure 3b shows volumes of HCT116 tumors on day 64 postinjection. CD16158V-CR T cells combined with cetuximab significantly protected mice from tumor growth. DFS of treated animals is reported in Figure 3c. Interestingly, tumor growth was also significantly delayed in the group of mice receiving CD16158V-CR only.
SCID mice are characterized by an efficient NK cell compartment. Therefore, in vivo antitumor effects might be attributed to resident NK cells rather than to exogenously administered CD16158V-CR T cells. However, no antitumor activity was detectable upon cetuximab administration in the absence of CD16158V-CR T cells. Moreover, although cetuximab can mediate ADCC in vitro,5 it has no impact on progression of KRAS-mutated colorectal cancers.3,4 Furthermore, our experiments were performed upon repeated administration of anti-asialo-GM1Abs, resulting in NK cell depletion.
This is the first study demonstrating the ability of CD16158V-CR combined with cetuximab to control KRAS-mutated cancer cell growth in vitro and in vivo. Our results, obtained by using the same target cells, document the superior antitumor potential of CD16158V, as compared to the other CR under investigation, despite an equivalent reverse ADCC capacity. CD16158V-CR superiority may reflect its optimal interaction with cetuximab Fc fragment.5
Taken together, these data contribute to a repositioning of currently available anti-EGFR therapeutic mAbs in the treatment of insensitive tumors, and pave the way toward innovative immunotherapies targeting KRAS-mutated cancers.
What’s new?
KRAS mutations downstream the epidermal growth factor receptor (EGFR) represent the major limitation in monoclonal antibody-targeted therapy of EGFR+ colorectal cancers. To restore the sensitivity of KRAS-mutated cancer cells to EGFR-specific mAbs, here the authors explore different strategies based on the generation of extracellular CD16-chimeric receptors linked to intracellular signalling and subsequent transduction into T cells. They demonstrate that T cells engineered with CD16158V-chimeric receptor can be redirected by EGFR-specific mAbs toward EGFR+ cancer cells, overcoming the limitation conferred by KRAS mutations in vitro and in vivo. This study might help develop innovative and effective treatments of EGFR+ KRAS-mutated cancers.
Acknowledgements
We thank Marta Coccia, Antonio Rossi, Matilde Paggiolu, and Pamela Papa for technical assistance.
Grant sponsor: Associazione Italiana per la Ricerca sul Cancro; Grant number: IG17120; Grant sponsor: Foundation for the National Institutes of Health; Grant numbers: CA216114, CA231766
Abbreviations:
- ADCC
antibody-dependent cellular cytotoxicity
- CM
complete media
- CR
chimeric receptor
- CRC
colorectal carcinoma
- DFS
disease-free survival
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FcR BR
Fc-receptor blocking reagent
- FITC
fluorescein isothiocyanate
- IL-15
interleukin-15
- IL-7
interleukin-7
- INFγ
interferon gamma
- mAb
monoclonal antibody
- OD
optical density
- PBMCs
peripheral blood mononuclear cells
- RT-PCR
reverse-transcriptase polymerase chain reaction
- TNFα
tumor necrosis factor alpha
Footnotes
Conflict of Interest: The authors declare no potential conflict of interest.
References
- 1.Rocha-Lima CM, Soares HP, Raez LE, et al. EGFR targeting of solid tumors. Cancer Control 2007;14: 295–304. [DOI] [PubMed] [Google Scholar]
- 2.Martinelli E, De Palma R, Orditura M, et al. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin Exp Immunol 2009;158:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757–65. [DOI] [PubMed] [Google Scholar]
- 4.Douillard J-Y, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369:1023–57. [DOI] [PubMed] [Google Scholar]
- 5.Veluchamy JP, Spanholtz J, Tordoir M, et al. Combination of NK cells and cetuximab to enhance antitumor responses in RAS mutant metastatic colorectal cancer. PLoS One 2016;11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balsamo M, Scordamaglia F, Pietra G, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A 2009;106:20847–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pietra G, Manzini C, Rivara S, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res 2012;72:1407–15. [DOI] [PubMed] [Google Scholar]
- 8.Sconocchia G, Spagnoli GC, Del Principe D, et al. Defective infiltration of natural killer cells in MICA/B-positive renal cell carcinoma involves β 2-integrin-mediated interaction. Neoplasia 2009; 11:662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sconocchia G, Zlobec I, Lugli A, et al. Tumor infiltration by FcγRIII (CD16)+ myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer 2011;128: 2663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sconocchia G, Arriga R, Tornillo L, et al. Melanoma cells inhibit NK cell functions. Cancer Res 2012;72:5428–9. [DOI] [PubMed] [Google Scholar]
- 11.Pagès F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 2005;353:2654–66. [DOI] [PubMed] [Google Scholar]
- 12.Desbois M, Rusakiewicz S, Locher C, et al. Natural killer cells in non-hematopoietic malignancies. Front Immunol 2012;3:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clemenceau B, Congy-Jolivet N, Gallot G, et al. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood 2006;107: 4669–77. [DOI] [PubMed] [Google Scholar]
- 14.Ochi F, Fujiwara H, Tanimoto K, et al. Gene-modified human a/b-T cells expressing a chimeric CD16-CD3z receptor as adoptively transferable effector cells for anticancer monoclonal antibody therapy. Cancer Immunol Res 2014;2:249–62. [DOI] [PubMed] [Google Scholar]
- 15.Kudo K, Imai C, Lorenzini P, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res 2014;74:93–103. [DOI] [PubMed] [Google Scholar]
- 16.D’Aloia MM, Caratelli S, Palumbo C, et al. T lymphocytes engineered to express a CD16-chimeric antigen receptor redirect T-cell immune responses against immunoglobulin G-opsonized target cells. Cytotherapy 2016;18:278–90. [DOI] [PubMed] [Google Scholar]
- 17.Marei HE, Althani A, Caceci T, et al. Recent perspective on CAR and Fcγ-CR T cell immunotherapy for cancers: preclinical evidence versus clinical outcomes. Biochem Pharmacol 2019;166:335–46. [DOI] [PubMed] [Google Scholar]
- 18.Coppola A, Arriga R, Lauro D, et al. NK cell inflammation in the clinical outcome of colorectal carcinoma. Front Med 2015;2:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weixler B, Cremonesi E, Sorge R, et al. OX40 expression enhances the prognostic significance of CD8 positive lymphocyte infiltration in colorectal cancer. Oncotarget 2015;6:37588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sconocchia G, Campagnano L, Adorno D, et al. CD44 ligation on peripheral blood polymorphonuclear cells induces interleukin-6 production. Blood 2001;97:3621–7. [DOI] [PubMed] [Google Scholar]
- 21.Lande R, Urbani F, Di Carlo B, et al. CD38 ligation plays a direct role in the induction of IL-1b, IL-6, and IL-10 secretion in resting human monocytes. Cell Immunol 2002;220:30–8. [DOI] [PubMed] [Google Scholar]
- 22.Shaw GM, Levy PC, Lobuglio AF. Human monocyte antibody-dependent cell-mediated cytotoxicity to tumor cells. J Clin Invest 1978;62:1172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider-Merck T, van Bueren JJL, Berger S, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol 2010;184:512–20. [DOI] [PubMed] [Google Scholar]
- 24.Bruhns P, Iannascoli B, England P, et al. Specificity and affinity of human fc gamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009;113:3716–25. [DOI] [PubMed] [Google Scholar]
- 25.Alves S, Castro L, Fernandes MS, et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015; 6:30787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheeseman HM, Olejniczak NJ, Rogers PM, et al. Broadly neutralizing antibodies display potential for prevention of HIV-1 infection of mucosal tissue superior to that of nonneutralizing antibodies. J Virol 2017;91:1762–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, Zhao L, Zhang Y, et al. CD16 and CD32 gene polymorphisms may contribute to risk of idiopathic thrombocytopenic purpura. Med Sci Monit 2016;18:2086–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rataj F, Jacobi SJ, Stoiber S, et al. High-affinity CD16-polymorphism and fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br J Cancer 2019;120:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snyder KM, Hullsiek R, Mishra HK, et al. Expression of a recombinant high affinity IgG fc receptor by engineered NK cells as a docking platform for therapeutic mAbs to target cancer cells. Front Immunol 2018;9:2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.