

Abstract
Objective
Mechanisms leading to anti–citrullinated protein antibody (ACPA) generation in rheumatoid arthritis (RA) are hypothesized to originate in the lung. We undertook this study to understand associations between neutrophil extracellular trap (NET) formation in the lung and local ACPA generation in subjects at risk of developing RA.
Methods
Induced sputum was collected from 49 subjects at risk of developing RA, 12 patients with RA, and 18 controls. Sputum neutrophils were tested for ex vivo NET formation, and sputum‐induced NET formation of control neutrophils was measured using immunofluorescence imaging. Sputum macrophages were tested for ex vivo endocytosis of apoptotic and opsonized cells. Levels of ACPA, NET remnants, and inflammatory proteins were quantified in sputum supernatant.
Results
Spontaneous citrullinated histone H3 (Cit‐H3)–expressing NET formation was higher in sputum neutrophils from at‐risk subjects and RA patients compared to controls (median 12%, 22%, and 0%, respectively; P < 0.01). In at‐risk subjects, sputum IgA ACPA correlated with the percentage of neutrophils that underwent Cit‐H3+ NET formation (r = 0.49, P = 0.002) and levels of Cit‐H3+ NET remnants (r = 0.70, P < 0.001). Reduced endocytic capacity of sputum macrophages was found in at‐risk subjects and RA patients compared to controls. Using a mediation model, we found that sputum inflammatory proteins were associated with sputum IgA ACPA through a pathway mediated by Cit‐H3+ NET remnants. Sputum‐induced Cit‐H3+ NET formation also correlated with sputum levels of interleukin‐1β (IL‐1β), IL‐6, and tumor necrosis factor in at‐risk subjects, suggesting a causal relationship.
Conclusion
These data support a potential mechanism for mucosal ACPA generation in subjects at risk of developing RA, whereby inflammation leads to increased citrullinated protein–expressing NETs that promote local ACPA generation.
Introduction
Rheumatoid arthritis (RA) develops in multiple phases, including a “preclinical” phase of systemic autoimmunity that precedes the onset of inflammatory arthritis (IA) (1, 2). Anti–citrullinated protein antibodies (ACPAs), often characterized by anti–cyclic citrullinated peptide (anti‐CCP) antibodies, are a key autoantibody system in RA. ACPA can be pathogenic in RA‐related arthritis models (3, 4), yet it is unknown how ACPAs initially form. To study the early steps of ACPA formation, our group has focused on subjects with an elevated risk of developing RA based on familial and/or serologic RA risk factors (5, 6, 7). Herein, these subjects are described as “at risk” for RA.
While the exact mechanisms that trigger ACPA generation are unknown, data support the notion that ACPA may originate in the lung (5, 6, 7, 8, 9, 10, 11). We have previously identified anti‐CCP generation in the lung using induced sputum in RA patients as well as in a portion of subjects at risk for RA (5, 6, 7). These studies also found a strong correlation between levels of ACPA and DNA–protein remnants of neutrophil extracellular traps (NETs), including DNA–myeloperoxidase (MPO) and DNA–neutrophil elastase (NE), in the sputum of subjects at risk for RA (6, 7). NET formation, commonly termed NETosis, is a mechanism during which neutrophils decondense their nucleus and expel their chromatin in complex with intracellular proteins in response to various stimuli, particularly inflammation or bacteria (12). While NETosis is a common physiologic process, certain NET features have been linked to ACPA and RA (13, 14, 15, 16, 17). It is currently unknown whether neutrophils in the lung are inherently more prone to undergo a specific type of NETosis in RA patients or at‐risk subjects, and whether citrullinated proteins expressed on NETs are associated with ACPA in the lung.
We hypothesized that ACPAs, specifically IgA ACPA, are formed in the lungs of subjects at risk for RA as the result of increased citrullinated protein–expressing NET formation. In the present study, we specifically investigated citrullinated histone H3 (Cit‐H3) expression on NETs, because this molecule and physically associated factors are known to be externalized on a subset of NETs (18), can be a target of ACPA in RA (19), and can be readily identified using currently available antibodies. Importantly, understanding the early steps of ACPA generation, particularly in subjects at risk for RA, can improve the overall understanding of RA development, including the initial loss of tolerance to citrullinated self‐antigens and eventual development of clinically apparent IA.
Patients and Methods
Study subjects
Subjects were recruited from the Studies of the Etiologies of RA Lung cohort (5, 6, 7), which was designed to use induced sputum to study RA‐related autoimmunity in the lung during different phases of RA development.
Subjects at risk for RA
We included 49 subjects without IA who were determined to be at risk of developing RA. We defined being at risk for RA as having a first‐degree relative with RA and/or having serum ACPA positivity (CCP3.1 IgG/IgA) identified through community health fair, clinic, or research‐based blood screenings. In these 49 at‐risk subjects, 40 had a first‐degree relative with RA (of which 10 of 40 [25%] were also seropositive for ACPA), and 9 were seropositive for ACPA without a known first‐degree relative with RA.
RA patients
We included 12 patients who met the RA classification criteria based on medical chart review (20, 21). All RA patients were seropositive for ACPA (CCP3.1 IgG/IgA) and were receiving disease‐modifying antirheumatic drugs and/or biologics.
Healthy controls
We included 18 healthy controls who were recruited through local advertisement, did not have RA or IA, did not have a first‐degree relative with RA, and were seronegative for ACPA.
Sputum collection and processing
All subjects underwent induced sputum collection with nebulized hypertonic saline (10%), as previously described (5, 6, 7). Sputum samples were immediately separated into 2 equal portions and processed separately. One portion was processed to obtain the cell‐free supernatant, which included dilution with phosphate buffered saline, syringe‐based mechanical homogenization, and centrifugation, followed by storage at −80°C with protease inhibitors phenylmethylsulfonyl fluoride and EDTA. Cell‐free assays were performed to evaluate levels of ACPA, rheumatoid factor (RF), NET remnants, cytokines, chemokines, complement, and histone protein.
The other portion of the sputum sample was processed to obtain a cell‐rich sample, which included extraction of sputum plugs with tweezers, chemical homogenization with dithiothreitol, and filtration. Mechanical homogenization was not performed on this sample. The processed cellular sample was immediately used for cell‐based assays, which included ex vivo NET formation and macrophage endocytosis assays.
When sputum volume was adequate, all assays were performed. If sample volume was low, only 1 cell‐based assay was performed. A flow chart outlining the number of subjects tested for each assay is included in Supplementary Figure 1 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). For sputum cell‐free testing, all samples with adequate volume were tested for ACPA and RF, and those with remaining volume were also tested for levels of NET remnants, cytokines/chemokines, and complement.
Study visit
Blood and sputum were collected from all subjects. At‐risk and control subjects had a 66/68‐count joint examination to confirm the absence of IA. Self‐administered questionnaires were used to obtain demographic data and history of smoking and lung disease. For RA subjects, the Multidimensional Health Assessment Questionnaire (MD‐HAQ) (22) was completed to measure functional status.
ACPA and RF testing
Serum was tested for ACPA using enzyme‐linked immunosorbent assay (ELISA) (Quanta Lite CCP3.1 IgG/IgA; Inova Diagnostics). The cutoff level for positivity was based on the manufacturer’s recommendations (≥20 units). Sputum supernatant was tested for individual ACPA isotypes, IgG CCP and IgA CCP, using ELISA. IgG anti‐CCP was measured using CCP3 (IgG; Inova) with modifications including additional dilutions of the standard curve resulting in a 7‐point standard curve. IgA anti‐CCP was measured using CCP3.1 (IgG/IgA; Inova) with substitution of the anti‐human IgG/IgA conjugate for an anti‐human IgA conjugate (horseradish peroxidase–conjugated goat anti‐human IgA, no. 2050‐05; Southern Biotech) and using an in‐house standard curve generated from pooled RA patient serum. Sputum IgG anti‐CCP and IgA anti‐CCP positivity were established using a separate cohort of 100 healthy controls and setting a cutoff level for positivity at the 95th percentile. Sputum was also tested for IgA RF using ELISA (Quanta Lite RF IgA; Inova Diagnostics).
Shared epitope testing
Blood was tested for RA risk alleles containing the shared epitope using previously described methods (23).
Sputum NET remnant testing
Sputum supernatant was tested for DNA–protein complexes of NET remnants in a blinded manner using previously described methods (6, 7, 25). Briefly, we used sandwich ELISA to detect DNA–MPO, DNA–NE, and DNA–Cit‐H3 protein complexes. Detailed methods for DNA–MPO and DNA–NE testing have been previously described (7). Similar methods were used for DNA–Cit‐H3, and the antibody used to coat plates was rabbit anti‐human H3 (citrulline R2+R8+R17; Abcam). To control for plate‐to‐plate variation, 4 control samples were included on every plate and an optical density index was calculated based on the average of these healthy control samples. In addition, a negative and positive control standard were run on each plate to confirm the validity of each plate. All samples were run in duplicate.
Sputum cytokine and chemokine testing
Meso Scale Discovery assays using Custom V‐Plex Human Biomarker Panel plates were used to quantify sputum levels of interleukin‐1β (IL‐1β), IL‐6, IL‐8, IL‐10, tumor necrosis factor (TNF), monocyte chemotactic protein 1 (MCP‐1), macrophage inflammatory protein 1α (MIP‐1α), and MIP‐1β. Spike recovery testing with high and low standards was performed in coordination with the company (Meso Scale Discovery) to confirm that the biologic sample of sputum did not interfere with testing. All sputum samples were tested in duplicate, with mean levels reported in pg/ml.
Sputum complement factor testing
Complement factors Ba, soluble C5b–9, and C3a were measured by ELISA (Quidel). Complement factors C1q, C4, C2, MBL, C4b, C3, factor B, factor D, properdin, C3b, factor H, factor I, and C5a were measured by multiplex Luminex immunoassays (MilliporeSigma). Methods were optimized to measure the low concentration values in sputum, and 3 quality control samples were included in each run. All testing was performed in duplicate, with the resulting mean values reported in ng/ml, except for C5a, which was reported in pg/dl.
Ex vivo sputum NET formation assay
Detailed methods are provided in the Supplementary Materials (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Briefly, sputum cells were fixed, permeabilized, and stained with anti‐MPO and anti–Cit‐H3 antibodies. Confocal microscopy was used to quantify NET formation as previously described (25). DNA+MPO+ or DNA+MPO+Cit‐H3+ colocalization and morphologic features of DNA extrusion consistent with NET formation were used to identify neutrophils that had undergone total NET formation or Cit‐H3+ NET formation, respectively. Percentage of total or percentage of Cit‐H3+ NETs was defined as the number of neutrophils with DNA+MPO+ or DNA+MPO+Cit‐H3+ NET formation per total neutrophils.
Ex vivo sputum macrophage endocytosis assay
Detailed methods are provided in the Supplementary Materials (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Briefly, dissociated sputum cells were incubated with apoptotic and opsonized Jurkat cells for 2 hours followed by cell fixation and staining. Microscopy with manual counting was performed to quantify the endocytosis index based on the following formula:
Sputum Western blot
Sputum cell‐free supernatant from 8 at‐risk subjects (3 positive for sputum IgA anti‐CCP and 5 negative for sputum IgA anti‐CCP) was analyzed by Western blot to determine the levels of Cit‐H3 and total H3. Samples were analyzed by equal volume (3 μl sputum per well) and by total protein level (2 μg protein per well). A positive control was run with each blot that included 7 ng of in vitro–citrullinated bulk histones. Antibodies were used to detect total H3 (no. ab1791; Abcam) and Cit‐H3 (citrulline R2+R8+R17, no. ab5103; Abcam). The volume intensity of bands was calculated by densitometry using Bio‐Rad Image Lab software. Each band was normalized to the positive control.
Induction of NETosis by induced sputum
Detailed methods are provided in the Supplementary Materials (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Briefly, peripheral blood neutrophils isolated from a control donor were incubated with sputum supernatant for 2 hours followed by fixation and staining with anti‐MPO and anti–Cit‐H3 antibodies. After accounting for background, levels of NET formation were quantified using the SpectraMax iD5 (Molecular Devices).
Statistical analysis
Subject demographics, percentage NET formation, levels of sputum protein, macrophage endocytic index, and NET remnants were compared between groups using chi‐square and Wilcoxon’s rank sum tests, as appropriate. Log‐transformed levels of sputum anti‐CCP and sputum cytokine, chemokine, and complement factor levels correlated with percentage NET formation, macrophage endocytic index, and NET remnant levels, according to Spearman’s correlation. NET remnant levels correlated with MD‐HAQ scores, and sputum cytokine, chemokine, and complement levels correlated with sputum‐induced NET levels, according to Spearman’s correlation. Multivariable linear regression was used to compare sputum percentage NET formation and group status as well as NET remnant levels and log‐transformed sputum anti‐CCP levels, while adjusting for relevant covariates.
For mediation analyses, we first used established methodologies (26) to create a composite variable to represent the multiple cytokine/chemokine or complement variables using the first principal component from principal component analysis. The resulting composite score explained 79% and 61% of the total variation among the 8 cytokine/chemokines and 16 complement variables, respectively. The mediation model used the sputum composite score as the effector variable, log‐transformed sputum IgA anti‐CCP levels as the outcome variable, and sputum NET remnant levels as mediator variables. The model was run with each NET remnant mediator variable separately (DNA–MPO, DNA–NE, and DNA–Cit‐H3) to quantify the direct effect and the indirect effect in the model. Significance was determined by a false discovery rate–adjusted bootstrap P value of less than 0.05, using the Benjamini and Yekutieli method to account for multiple testing (27). The mediation models were fit using the mediation R package (28, 29).
Study approval
All study procedures were approved by the Colorado Multiple Institutional Review Board and were in compliance with the Declaration of Helsinki. Written informed consent was obtained from all subjects.
Results
Increased spontaneous sputum NET formation in at‐risk subjects and RA subjects
Subject demographic data are described in Supplementary Table 1 (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Using immunofluorescence imaging of unstimulated sputum cells (representative images in Supplementary Figure 2, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract), we found that the percentage of sputum neutrophils that underwent NET formation (DNA+MPO+) and Cit‐H3+ NET formation (DNA+MPO+Cit‐H3+) was significantly higher in at‐risk subjects and RA patients compared to controls (Figure 1A and 1B). For at‐risk subjects, RA patients, and controls, the median percentage NET formation values for total NETs were 51%, 67%, and 37% (P = 0.001), respectively, and for Cit‐H3+ NET formation were 12%, 22%, and 0% (P < 0.001), respectively. Among at‐risk subjects, sputum total NETosis was significantly higher in those who were seropositive for anti‐CCP3.1 (Figure 1C), whereas sputum Cit‐H3+ NETosis was similar between at‐risk subjects seropositive for anti‐CCP3.1 and those seronegative for anti‐CCP3.1 (Figure 1D). Increasing levels of Cit‐H3+ NETosis significantly correlated with increasing levels of sputum IgA anti‐CCP (Figure 1E) but not IgG anti‐CCP (P = 0.88) in at‐risk subjects. There was no significant correlation between total NETosis and IgA anti‐CCP or IgG anti‐CCP in sputum from at‐risk subjects (P = 0.15 and P = 0.12, respectively). There was also no significant correlation between sputum total or Cit‐H3+ NETosis and sputum IgA anti‐CCP or IgG anti‐CCP among RA patients or control subjects (data not shown).
Figure 2.
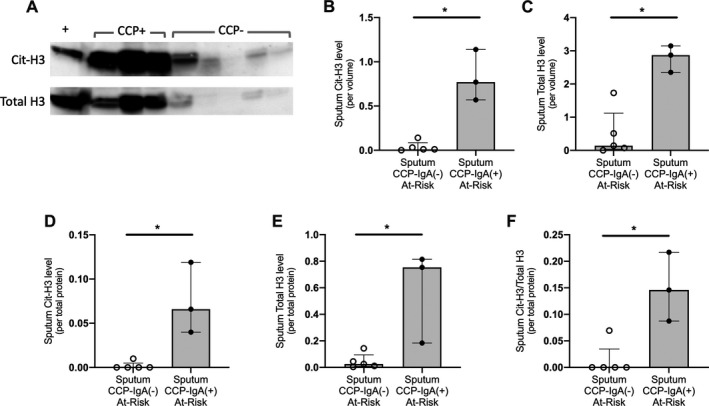
Sputum Cit‐H3 levels are associated with sputum IgA anti‐CCP levels in subjects at risk for RA. A, Western blot imaging of Cit‐H3 and total H3 in sputum supernatant from a subset of at‐risk subjects (3 subjects with IgA anti‐CCP–positive sputum and 5 subjects with IgA anti‐CCP–negative sputum). Each well was loaded with 3 μl of sputum supernatant, and the positive control lane includes in vitro–citrullinated bulk histones. B and C, Quantified levels of Cit‐H3 (B) and total H3 (C), by sputum volume (3 μl loaded per well), normalized to the positive control in at‐risk subjects positive for sputum IgA anti‐CCP and those negative for sputum IgA anti‐CCP. D–F, Quantified levels of Cit‐H3 (D), total H3 (E), and Cit‐H3:total H3 ratio (F), by sputum total protein level (2 μg loaded per well) normalized to the positive control in at‐risk subjects positive for sputum IgA anti‐CCP and those negative for sputum IgA anti‐CCP. Each band volume intensity was calculated using Bio‐Rad Image Lab software. Median levels were compared between groups using Wilcoxon’s rank sum test. Bars show the median and interquartile range. Open circles represent subjects negative for sputum IgA anti‐CCP, and solid circles represent subjects positive for sputum IgA anti‐CCP. * = P < 0.05. See Figure 1 for definitions.
Figure 1.

Sputum neutrophil extracellular trap (NET) formation (known as NETosis) and association with sputum IgA anti–cyclic citrullinated peptide (anti‐CCP). A and B, Percentage of sputum neutrophils that underwent total NETosis (DNA+MPO+) (A) and citrullinated histone H3–positive (Cit‐H3+) NETosis (DNA+MPO+Cit‐H3+) (B) in ex vivo unstimulated culture in healthy controls (n = 15), subjects at risk for rheumatoid arthritis (RA) (n = 41), and RA patients (n = 8). C and D, Percentage of sputum neutrophils that underwent total NETosis (C) and Cit‐H3+ NETosis (D) in serum anti‐CCP3.1–negative at‐risk subjects (n = 26) and serum anti‐CCP3.1–positive at‐risk subjects (n = 15). Median levels were compared between groups using Wilcoxon’s rank sum test. E, Correlation between sputum IgA anti‐CCP level and percentage Cit‐H3+ NETosis in at‐risk subjects (n = 37), by Spearman’s correlation. Bars show the median and interquartile range. Open circles represent subjects negative for sputum IgA anti‐CCP, solid circles represent subjects positive for sputum IgA anti‐CCP, and open squares represent subjects in whom sputum IgA anti‐CCP was not tested. * = P < 0.05; ** = P < 0.01; *** = P < 0.001. MPO = myeloperoxidase; NS = not significant; Ln = natural logarithm.
Of note, sputum total and Cit‐H3+ NETosis were not significantly higher based on age, sex, or history of chronic lung disease (data not shown). Because age and history of chronic lung disease differed between groups, we performed multivariable linear regression and found that sputum total and Cit‐H3+ NET formation remained significantly associated with subject group after adjusting for age and history of chronic lung disease (P < 0.01). In addition, levels of sputum total and Cit‐H3+ NETosis remained significantly higher in at‐risk and RA never‐smokers compared to controls, and sputum Cit‐H3+ NETosis remained significantly correlated with sputum IgA anti‐CCP level in at‐risk never‐smokers (Supplementary Figures 3A–C, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract).
Association of sputum Cit‐H3 levels with sputum ACPA
Proteins can be released extracellularly during NETosis (30). Using Western blot analysis, we found that median Cit‐H3 levels and total H3 protein levels were higher in at‐risk subjects with IgA anti‐CCP–positive sputum (22% median Cit‐H3+ NETosis in these samples based on ex vivo NETosis testing described above) compared to those with sputum negative for IgA anti‐CCP (1% median Cit‐H3+ NETosis in these samples) (Figures 2A–E). When blots were loaded based on total protein, we also found that the sputum Cit‐H3:total H3 ratio was higher in at‐risk subjects with sputum positive for IgA anti‐CCP (Figure 2F). Total H3 and Cit‐H3 levels also correlated with total sputum neutrophil counts (r = 0.82, P = 0.02, and r = 0.86, P = 0.01, respectively).
Sputum macrophage endocytosis diminished in at‐risk subjects and RA patients
Macrophage endocytosis is needed for effective clearance of NET remnants (31). To globally study macrophage endocytosis, we quantified both efferocytosis of apoptotic cells and phagocytosis of opsonized cells. We found that sputum macrophages from at‐risk subjects and RA patients had significant impairments in both efferocytosis (Figure 3B) and phagocytosis (Figure 3C). Among at‐risk subjects, there was no difference in endocytosis based on serum anti‐CCP3.1 positivity (Figures 3D and E). In addition, sputum macrophage endocytosis did not correlate with sputum IgA anti‐CCP level in at‐risk subjects (P = 0.24 for efferocytosis and P = 0.10 for phagocytosis).
Figure 3.
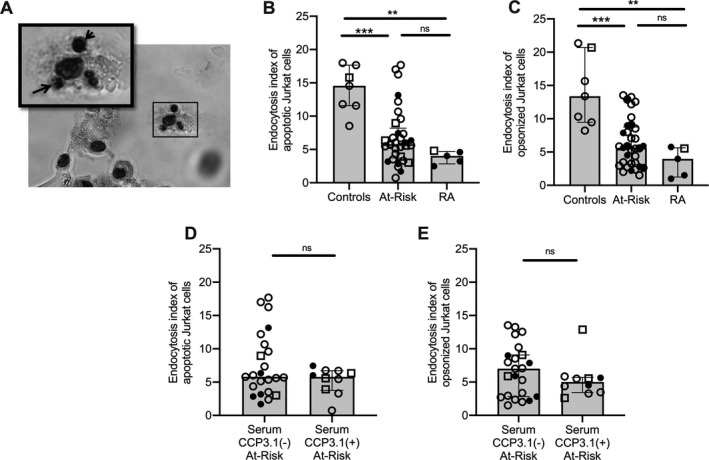
Sputum macrophage endocytosis of apoptotic and opsonized cells. A, Representative image of macrophage efferocytosis assay displaying ingestion of apoptotic Jurkat cells (arrows) by a sputum macrophage. Original magnification × 400. B and C, Endocytosis index of apoptotic (B) and opsonized (C) Jurkat cells in healthy controls (n = 7), subjects at risk for RA (n = 33), and RA patients (n = 5). D and E, Endocytosis index of apoptotic (D) and opsonized (E) Jurkat cells in at‐risk subjects with serum negative for anti–citrullinated protein antibodies (ACPA) (n = 23) and those with serum positive for ACPA (n = 10), with ACPA measured using an anti‐CCP3.1 (IgG/IgA) enzyme‐linked immunosorbent assay. Median levels were compared between groups using Wilcoxon’s rank sum test. Bars show the median and interquartile range. Open circles represent subjects negative for sputum IgA anti‐CCP, solid circles represent subjects positive for sputum IgA anti‐CCP, and open squares represent subjects in whom sputum IgA anti‐CCP was not tested. ** = P < 0.01; *** = P < 0.001. See Figure 1 for other definitions.
Sputum macrophage endocytosis was not significantly associated with age or history of chronic lung disease in any group (P > 0.05 for all). Among at‐risk subjects, macrophage efferocytosis was higher in women compared to men (P < 0.01), and there was a non‐significant trend toward lower endocytosis in ever‐smokers (P = 0.08 for efferocytosis and P = 0.09 for phagocytosis). However, when comparing only women or only never‐smokers in each group, at‐risk subjects and RA patients continued to demonstrate decreased sputum macrophage efferocytosis and phagocytosis compared to controls (P < 0.05 for all) (Supplementary Figures 3D and E, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Endocytosis also remained significantly associated with the subject group when adjusting for sex and ever‐smoking in multivariable linear regression (P < 0.01).
Correlation of sputum Cit‐H3–containing NET remnants with sputum IgA anti‐CCP in at‐risk subjects
NET remnants represent a composite of NET formation and NET clearance. Consistent with our prior data (7), sputum IgA anti‐CCP levels correlated with sputum NET remnant levels of DNA–NE (r = 0.55, P < 0.001) and DNA–MPO (r = 0.51, P = 0.003) in at‐risk subjects (Figures 4A and B). We also found that sputum DNA–Cit‐H3 protein complexes had the strongest correlation with sputum IgA anti‐CCP (r = 0.70, P < 0.001) (Figure 4C). Notably, sputum DNA–Cit‐H3 levels were not significantly higher in at‐risk subjects as a group compared to healthy controls or RA patients (P = 0.15) (Figure 4D), but among at‐risk subjects, sputum DNA–Cit‐H3 levels were significantly higher in those with sputum IgA anti‐CCP positivity (P = 0.01) (Figure 4E).
Figure 4.

Sputum IgA anti‐CCP and NET remnant levels. A–C, Correlations between levels of sputum IgA anti‐CCP and levels of sputum NET remnants, measured as DNA–neutrophil elastase (DNA–NE) (A), DNA–MPO (B), and DNA–Cit‐H3 (C) in at‐risk subjects (n = 33). Correlations were calculated using Spearman’s correlation. D, Sputum DNA–Cit‐H3 levels in controls (n = 8), at‐risk subjects (n = 33), and RA patients (n = 7). E, Sputum DNA–Cit‐H3 levels in at‐risk subjects positive for sputum IgA anti‐CCP (n = 6) and those negative for sputum IgA anti‐CCP (n = 27). Median levels were compared between groups using Wilcoxon’s rank sum test. Open circles represent subjects negative for serum anti‐CCP3.1, and solid circles represent subjects positive for serum anti‐CCP3.1. See Figure 1 for definitions.
Because IgA RF and smoking have been associated with increased NETosis in humans (6, 33, 34), we evaluated the influence of these factors. In at‐risk subjects, after adjusting for ever‐smoking and sputum IgA RF level in linear regression models, sputum IgA anti‐CCP remained significantly associated with sputum DNA–Cit‐H3 (P = 0.02) but not with DNA–MPO (P = 0.84) or DNA–NE (P = 0.06).
There was no significant correlation between sputum IgA anti‐CCP and NET remnant levels in RA subjects (data not shown). However, sputum DNA–Cit‐H3 NET remnant levels did demonstrate a significant positive correlation with MD‐HAQ score (r = 0.85, P = 0.02).
Association of sputum cytokines/chemokines with sputum IgA anti‐CCP via pathway mediated by DNA–Cit‐H3 and DNA–NE NET remnants in at‐risk subjects
Multiple inflammatory cytokines can induce NET formation (12, 14, 35). We found positive correlations between levels of sputum IgA anti‐CCP and sputum IL‐1β, IL‐6, IL‐8, IL‐10, TNF, MIP‐1α, and MIP‐1β in at‐risk subjects (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). We then performed a mediation analysis to investigate whether the relationship between a composite cytokine/chemokine score and IgA anti‐CCP was mediated by NET remnants. Our outcome of interest was the indirect effect of the model, because it quantified how much of the relationship between cytokine/chemokines and IgA anti‐CCP was mediated through NET remnants (Supplementary Figure 4, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). We found that the relationship between the sputum cytokine/chemokine score and sputum IgA anti‐CCP was significantly mediated through sputum DNA–Cit‐H3 NET remnant levels (indirect effect; P < 0.001) (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract) and to a lesser extent DNA–NE (indirect effect; P = 0.004).
As outlined in Supplementary Table 3 (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract), 86% of the relationship between the sputum cytokine/chemokine score and sputum IgA anti‐CCP was mediated by sputum DNA–Cit‐H3 levels. This relationship was maintained after adjusting for sputum IgA RF levels (P = 0.001 for DNA–Cit‐H3; P = 0.002 for DNA–NE). The contributions of individual cytokines/chemokines are outlined in Supplementary Table 4 (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract).
Sputum macrophage endocytosis index was not included in the mediation model, because it was not associated with sputum IgA anti‐CCP. Of note, when accounting for multiple comparisons, there were no significant correlations between sputum macrophage endocytosis and sputum cytokine/chemokine levels (data not shown), but there was a trend toward sputum macrophage phagocytosis negatively correlating with sputum TNF levels (r = −0.41, P = 0.03).
Association of sputum complement activation fragments with sputum IgA anti‐CCP via pathway mediated by DNA–Cit‐H3 NET remnants in at‐risk subjects
Complement activation fragments have also been found to induce NETosis (36, 37). We identified positive correlations between levels of sputum anti‐CCP‐IgA and sputum Ba, C2, C4, and factor H in at‐risk subjects (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Similar to the mediation analysis applied to cytokines/chemokines, we found a significant relationship between the composite sputum complement score and sputum IgA anti‐CCP that was mediated through sputum DNA–Cit‐H3 NET remnant levels (indirect effect; P < 0.001) (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). We found that 86% of the relationship between the sputum complement score and sputum IgA anti‐CCP was mediated by sputum DNA–Cit‐H3 levels. We did not find DNA–MPO or DNA–NE to significantly mediate this relationship. Results for individual complement proteins are listed in Supplementary Table 5 (http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). Although not included in the mediation model, when accounting for multiple comparisons, there was a non‐significant trend toward sputum macrophage phagocytosis negatively correlating with sputum levels of C3a (r = −0.51, P = 0.01), C2 (r = −0.47, P = 0.02), C1q (r = −0.44, P = 0.03), and C3 (r = −0.44, P = 0.03).
Increase in induction of NETosis due to sputum that contains elevated inflammatory cytokine levels in at‐risk subjects
Because our mediation analysis suggested that the relationship between sputum inflammatory proteins and IgA anti‐CCP was mediated by DNA–Cit‐H3 in at‐risk subjects, we evaluated the ability of the sputum from at‐risk subjects to induce Cit‐H3+ NETs in control neutrophils. We found that among at‐risk subjects, induction of Cit‐H3+ NETs significantly correlated with sputum levels of IL‐1β, IL‐6, and TNF (Figure 5 and Supplementary Table 6, http://onlinelibrary.wiley.com/doi/10.1002/art.41948/abstract). There was no significant correlation between sputum complement factor levels and NET induction. There was also no significant correlation between sputum IgA anti‐CCP, IgG anti‐CCP, or IgA RF level in at‐risk subjects and NET induction. In at‐risk subjects with IgA anti‐CCP–negative sputum, positive correlations remained between the induction of Cit‐H3+ NETs and sputum IL‐1β (P = 0.002), IL‐6 (P = 0.010), and TNF (P = 0.027), supporting the notion that sputum cytokines in at‐risk subjects can induce Cit‐H3+ NETs even in the absence of sputum IgA anti‐CCP.
Figure 5.

Sputum‐induced Cit‐H3+ NET formation and sputum cytokine levels. Correlations between levels of sputum‐induced Cit‐H3+ NET formation in control peripheral blood neutrophils measured by Cit‐H3 mean relative fluorescence intensity (RFI) and log‐transformed levels of sputum cytokines, including interleukin‐1β (IL‐1β) (A), IL‐6 (B), IL‐8 (C), and tumor necrosis factor (TNF) (D) in subjects at risk for RA (n = 33) were calculated using Spearman’s correlation. Open circles represent subjects negative for sputum IgA anti‐CCP, and solid circles represent subjects positive for sputum IgA anti‐CCP. See Figure 1 for other definitions.
Discussion
We demonstrate for the first time that sputum neutrophils from subjects at risk of developing RA and RA patients are more prone to spontaneously undergo NET formation. Of particular interest, in at‐risk subjects, IgA anti‐CCP levels in the lung were associated with increased Cit‐H3–expressing NET formation, and sputum IgA anti‐CCP most strongly correlated with Cit‐H3–containing NET remnants. Furthermore, we demonstrate that in at‐risk subjects, inflammatory cytokines, chemokines, and complement pathway proteins in the lung are associated with sputum IgA anti‐CCP through a pathway mediated by Cit‐H3–containing NET remnants, and induction of Cit‐H3+ NETosis was increased in sputum containing higher levels of IL‐1, IL‐6, and TNF. Taken together, these data suggest an important role for inflammation‐induced citrullinated protein–expressing NETs in the lungs in subjects at risk for RA.
While the exact etiology of RA remains unknown, multiple data indicate that autoimmunity and inflammation begin in the lung in a portion of individuals who ultimately develop RA (7, 9, 38, 39). Data also demonstrate that peripheral blood and synovial fluid neutrophils from RA patients are more prone to NETosis (14). Arthritogenic NET peptides can also be presented to antigen‐specific T cells in association with ACPA, suggesting that NETs can be an initial trigger or amplifier of ACPA production (39). Taking these data into account along with our current findings, we propose that neutrophils in the lungs of subjects at risk for RA are more prone to undergoing citrullinated protein–containing NETosis, and IL‐1β–, IL‐6–, and TNF‐associated inflammation, in particular, lead to an excess accumulation of citrullinated protein–containing NET remnants in the lung that can promote the local formation of ACPA. The factors that drive the inflammation in these subjects are unknown, but candidates are under investigation. We also found that sputum NET remnants in RA subjects correlated with joint disability (i.e., MD‐HAQ) and not sputum ACPA levels, suggesting that NETs in the lung may play different roles at different stages of RA development.
In the present study, we found that sputum macrophages in at‐risk subjects and RA patients had diminished endocytic function, suggesting that both increased NET formation and decreased NET clearance could contribute to the high levels of NET remnants associated with sputum IgA anti‐CCP. While we did not directly evaluate macrophage ingestion of NET remnants in this study, it is assumed that NET ingestion would also be diminished because sputum macrophages in at‐risk subjects and RA patients displayed global dysfunction of endocytosis (e.g., decreases in both efferocytosis and phagocytosis), and the receptor for advanced glycation end products can be used for both NET and apoptotic cell ingestion (39, 40). Studies have shown that NE, which can be released during NETosis, can reduce lung macrophage efferocytic capacity by cleaving phagocytic receptors on the cell surface (41). Future studies are needed to identify whether similar processes occur in subjects at risk for RA. In addition to macrophage endocytosis, NETs are cleared through degradation by DNase (31). We were unable to measure sputum DNase enzyme activity in this study, which is a limitation, but going forward, understanding differences in sputum DNase activity in at‐risk subjects will be important to determine its contribution to increased levels of sputum NET remnants and RA pathogenesis.
NETs can be divided into several subtypes. The protein cargo externalized and the inflammation triggered by NETs can differ based on the individual and the NET stimulus (14, 44, 45, 46). While smoking can induce NETosis and could play a role in increased NET formation in at‐risk and RA smokers, our findings were independent of smoking, supporting the hypothesis of involvement of other factors. Our study focused on Cit‐H3–containing NETs, but other citrullinated proteins have been identified in RA‐associated NETs (14). As such, additional studies are needed to explore the full citrullinome of NETs generated in the lung in subjects at risk for RA. In addition, we found increased Cit‐H3 and H3 protein levels in sputum associated with IgA anti‐CCP in at‐risk subjects. The source of Cit‐H3 in the sputum could be from NETosis, but other pathways could also contribute, including extracellular release of peptidylarginine deiminase during NETosis (30) and neutrophils externalizing citrullinated proteins through leukotoxic hypercitrullination or complement membrane attack complex insertion (45). Understanding each distinct pathway that could trigger ACPA through excess citrullinated protein externalization will be of interest.
There are several limitations to our study. It has been demonstrated that ACPA can trigger NET formation (13), which could result in a feed‐forward process of increased NETosis. However, we found increased sputum NETosis in at‐risk subjects without ACPA in the sputum or serum, suggesting that increased NET formation could be an initial event leading to ACPA formation and not a resultant event caused by ACPA in the lung. NETs can also induce cytokine stimulation from other nearby cells, including airway epithelial cells, and NETs can activate complement (46, 47, 48). Therefore, while our data suggest that NET remnants mediate the relationship between inflammatory factors and ACPA, we cannot exclude the possibility that elevation of sputum cytokines and/or complement results in part from the effects of increased NETosis. Figure 6 depicts our hypothesis in the context of the complexities of several possible feed‐forward loops that could contribute to further enhancement of lung inflammation and NET formation. Of note, lung inflammation and NET formation triggered by a variety of environmental factors are likely common in most individuals, regardless of their risk for RA. It is likely the generation of a sustained local and subsequently systemic adaptive immune response to citrullinated proteins is the factor that distinguishes individuals with and those at‐risk for RA from healthy controls. However, large longitudinal studies of sputum and linked systemic studies of autoimmune status are needed to fully understand the natural history and relationships of lung inflammation and ACPA generation across different sites in groups of individuals who have well‐defined outcomes.
Figure 6.
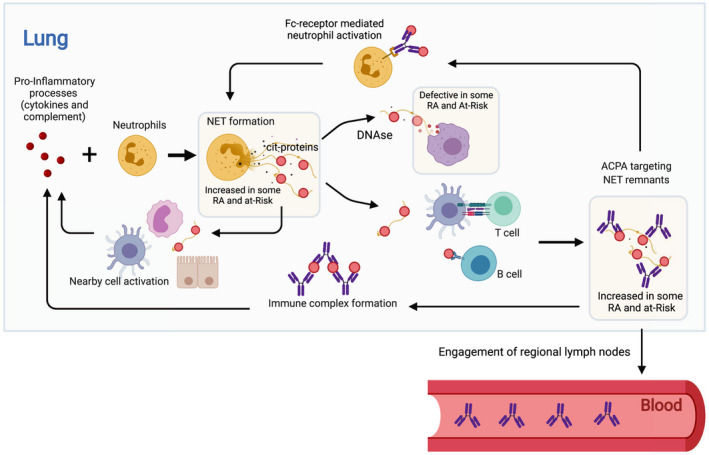
Hypothesis by which NETs in the lung can contribute to anti–citrullinated protein antibody (ACPA) generation. The figure depicts a hypothesis that proinflammatory mediators in the lung, including cytokines and complement proteins, can activate neutrophils to undergo NET formation in the lung. In addition, degradation of NET remnants can be diminished due to defective macrophage endocytosis. The resulting increased NET remnants can lead to ACPA generation in the lung via T cell and B cell activation. Following engagement of regional lymph nodes, this ACPA generation could become systemic and ultimately participate in the initiation and/or propagation of joint inflammation. There are several opportunities for feed‐forward enhancement of this process including Fc receptor–mediated NET formation by ACPA and increased inflammatory protein generation via ACPA immune complex formation/deposition or NET activation of nearby cells, including macrophages, dendritic cells, and respiratory epithelial cells that can subsequently produce inflammatory cytokines. Notably, these processes may also occur at sites other than the lung mucosa. Figure created by BioRender.com. See Figure 1 for other definitions.
In conclusion, we found increased Cit‐H3–containing NET formation and NET remnants to be associated with IgA anti‐CCP in the sputum of subjects at risk for RA. These data suggest a role for aberrant NET formation in the lung during the preclinical period of RA. They further support the importance of understanding features of sputum neutrophils in at‐risk subjects and RA patients, as NET formation and effector mechanisms may be potential future targets that abrogate ACPA generation during the preclinical period of RA.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Demoruelle had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Okamoto, Devoe, Minarchick, Rothfuss, Mohning, Thomas, Norris, Cherrington, Janssen, Kaplan, Deane, Holers, Demoruelle.
Acquisition of data
Okamoto, Devoe, Seto, Minarchick, Rothfuss, Visser, August, Charry, Fleischer, Feser, Frazer‐Abel, Cherrington, Kaplan, Deane, Demoruelle.
Analysis and interpretation of data
Okamoto, Wilson, Rothfuss, Mohning, Arbet, Kroehl, Thomas, Frazer‐Abel, Norris, Cherrington, Janssen, Kaplan, Deane, Holers, Demoruelle.
Supporting information
Supplementary Material
Supported by the NIH (grants AR‐066712, AR‐076450, AI‐101990, AI‐101981, HL‐140039, HD‐090541, AR‐51749, and GM‐103432) and by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (project ZIA‐AR‐041199).
The content of this publication is the sole responsibility of the authors and does not represent the official views of the NIH.
Dr. Norris has received a research grant from Pfizer. Dr. Deane has received consulting fees, speaking fees, and/or honoraria from Bristol Myers Squibb and Janssen (less than $10,000 each) and research grants from Pfizer and Janssen. Dr. Holers has received consulting fees, speaking fees, and/or honoraria from Bristol Myers Squibb, Janssen, and AdMIRx (less than $10,000 each) and research grants from Pfizer and Janssen, and owns stock or stock options in AdMIRx. Dr. Demoruelle has received a research grant from Pfizer. No other disclosures relevant to this article were reported.
References
- 1. Rantapää‐Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 2003;48:2741–9. [DOI] [PubMed] [Google Scholar]
- 2. Kokkonen H, Mullazehi M, Berglin E, Hallmans G, Wadell G, Rönnelid J, et al. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res Ther 2011;13:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest 2012;122:1791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest 2006;116:961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Willis VC, Demoruelle MK, Derber LA, Chartier‐Logan CJ, Parish MC, Pedraza IF, et al. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum 2013;65:2545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, et al. Antibody responses to citrullinated and noncitrullinated antigens in the sputum of subjects with rheumatoid arthritis and subjects at risk for development of rheumatoid arthritis. Arthritis Rheumatol 2018;70:516–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, et al. Anti–citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthritis Rheumatol 2017;69:1165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Demoruelle MK, Weisman MH, Simonian PL, Lynch DA, Sachs PB, Pedraza IF, et al. Airways abnormalities and rheumatoid arthritis–related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum 2012;64:1756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klareskog L, Stolt P, Lundberg K, Källberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006;54:38–46. [DOI] [PubMed] [Google Scholar]
- 10. Rangel‐Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus‐associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 2006;116:3183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reynisdottir G, Olsen H, Joshua V, Engstrom M, Forsslund H, Karimi R, et al. Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann Rheum Dis 2016;75:1722–7. [DOI] [PubMed] [Google Scholar]
- 12. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532–5. [DOI] [PubMed] [Google Scholar]
- 13. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther 2014;16:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013;5:178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis 2014;73:1414–22. [DOI] [PubMed] [Google Scholar]
- 16. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis 2016;75:1866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lloyd KA, Wigerblad G, Sahlstrom P, Garimella MG, Chemin K, Steen J, et al. Differential ACPA binding to nuclear antigens reveals a PAD‐independent pathway and a distinct subset of acetylation cross‐reactive autoantibodies in rheumatoid arthritis. Front Immunol 2018;9:3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun 2009;1:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janssen KM, de Smit MJ, Withaar C, Brouwer E, van Winkelhoff AJ, Vissink A, et al. Autoantibodies against citrullinated histone H3 in rheumatoid arthritis and periodontitis patients. J Clin Periodontol 2017;44:577–84. [DOI] [PubMed] [Google Scholar]
- 20. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 21. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 22. Pincus T, Yazici Y, Bergman M. A practical guide to scoring a Multi‐Dimensional Health Assessment Questionnaire (MDHAQ) and Routine Assessment of Patient Index Data (RAPID) scores in 10–20 seconds for use in standard clinical care, without rulers, calculators, websites or computers. Best Pract Res Clin Rheumatol 2007;21:755–87. [DOI] [PubMed] [Google Scholar]
- 23. Kolfenbach JR, Deane KD, Derber LA, O'Donnell C, Weisman MH, Buckner JH, et al. A prospective approach to investigating the natural history of preclinical rheumatoid arthritis (RA) using first‐degree relatives of probands with RA. Arthritis Rheum 2009;61:1735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lood C, Blanco LP, Purmalek MM, Carmona‐Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus‐like disease. Nat Med 2016;22:146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marder W, Knight JS, Kaplan MJ, Somers EC, Zhang X, O'Dell AA, et al. Placental histology and neutrophil extracellular traps in lupus and pre‐eclampsia pregnancies. Lupus Sci Med 2016;3:e000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song MK, Lin FC, Ward SE, Fine JP. Composite variables: when and how. Nurs Res 2013;62:45–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Statist 2001;29:1165–88. [Google Scholar]
- 28. Tingley D, Yamamoto T, Hirose K, Keele L, Imai K. mediation: R Package for Causal Mediation Analysis. J Stat Softw 2014;59:38. [Google Scholar]
- 29. R Core Team . R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2018. URL: https://www.R‐project.org/. [Google Scholar]
- 30. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol 2015;67:3135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 2013;191:2647–56. [DOI] [PubMed] [Google Scholar]
- 32. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcαRI. J Immunol 2016;197:4552–9. [DOI] [PubMed] [Google Scholar]
- 33. Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, et al. Nicotine drives neutrophil extracellular traps formation and accelerates collagen‐induced arthritis. Rheumatology (Oxford) 2017;56:644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One 2012;7:e48111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palmer LJ, Damgaard C, Holmstrup P, Nielsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J Periodontal Res 2016;51:70–6. [DOI] [PubMed] [Google Scholar]
- 36. Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, et al. Induction of genes mediating interferon‐dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 2004;279:44123–32. [DOI] [PubMed] [Google Scholar]
- 37. Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction [review]. Nat Rev Rheumatol 2018;14:542–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mankia K, Emery P. Is localized autoimmunity the trigger for rheumatoid arthritis? Unravelling new targets for prevention [review]. Discov Med 2015;20:129–35. [PubMed] [Google Scholar]
- 39. Carmona‐Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast‐neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol 2017;2:eaag3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Armstrong A, Ravichandran KS. Phosphatidylserine receptors: what is the new RAGE? EMBO Rep 2011;12:287–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, et al. Elastase‐mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest 2002;109:661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia‐Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011;3:73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skopelja S, Hamilton BJ, Jones JD, Yang ML, Mamula M, Ashare A, et al. The role for neutrophil extracellular traps in cystic fibrosis autoimmunity. JCI Insight 2016;1:e88912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017;6:e24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans–induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med 2016;8:369ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 2012;188:3522–31. [DOI] [PubMed] [Google Scholar]
- 47. Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol 2015;181:518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sabbione F, Keitelman IA, Iula L, Ferrero M, Giordano MN, Baldi P, et al. Neutrophil extracellular traps stimulate proinflammatory responses in human airway epithelial cells. J Innate Immun 2017;9:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material