

Abstract
O-GlcNAcylation is a post-translational modification occurring on serine/threonine residues of nuclear and cytoplasmic proteins, mediated by the enzymes OGT and OGA which catalyze the addition or removal of the UDP-GlcNAc moieties, respectively. Structural changes brought by this modification lead to alternations of protein stability, protein-protein interactions, and phosphorylation. Importantly, O-GlcNAcylation is a nutrient sensor by coupling nutrient sensing with cellular signaling. Elevated levels of OGT and O-GlcNAc have been reported in a variety of cancers and has been linked to regulation of multiple cancer signaling pathways. In this review, we discuss the most recent findings on the role of O-GlcNAcylation as a metabolic sensor in signaling pathways and immune response in cancer.
Keywords: OGT, O-GlcNAc, cancer, signaling, metabolism, transcription
Introduction
The Hexosamine Biosynthetic Pathway (HBP) utilizes metabolic inputs such as glucose, glutamine, nucleotides and fatty acids to generate uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), a sugar moiety conjugated in an O- or N-linked manner onto endoplasmic reticulum (ER) or Golgi proteins or utilized for O-GlcNAcylation, which is a single sugar moiety conjugation on nuclear or cytoplasmic proteins [1, 2]. O-linked β-N-acetylglucosamine modification or O-GlcNAcylation is a dynamic post-translational modification (PTM) occurring on serine (Ser) and threonine (Thr) residues of nuclear and cytoplasmic proteins [3]. OGlcNAcylation is exclusively under the control of two enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), which catalyze the addition and removal of O-GlcNAc moieties, respectively [4]. At the molecular level, O-GlcNAcylation is similar to and often in competition with phosphorylation for altering protein stability, function, protein-protein interaction, and modulating various cellular signaling pathways in multiple biological processes such as transcription, translation, metabolism and autophagy [2, 5–8]. Exploration of the role of OGT and O-GlcNAc in adult biology is starting to attract interest from the perspective of different fields that have elucidated the role of OGT/O-GlcNAc in development, immunity and cardiac health [9–11]. Aberrant O-GlcNAcylation has been implicated with pathological complications including neurodegenerative disorders, cardiovascular diseases, and diabetes [12, 13]. In particular, an increase in HBP flux and O-GlcNAcylation has been characterized in a variety of tumors [5, 12, 14–18] where the cellular mechanisms of O-GlcNAc in promoting tumorigenesis are being widely explored [19–22]. Here, we summarize the most recent understanding of mechanisms by which O-GlcNAcylation regulates cancer cell signaling with focus on transcriptional pathways, metabolic, and cancer immune regulation.
Transcriptional Pathways
HIPPO pathway
The HIPPO Pathway, originally discovered from the mutation of the Warts (Wts) gene in D. melanogaster [mammalian large tumor suppressor kinase ½ (LATS1/2)] as a regulator of organ size, [23, 24] is a tumor suppressor kinase-cascade-signaling pathway deregulated in cancer [25, 26]. Regulation of the Hippo pathway is complex because it is dependent on multiple signals such as cell-cell contact [27], cell adhesion and polarity [28], [29], mechano-transduction cues [30–34] hormonal signals, cellular energy stress and nutrients acting through G-protein coupled receptors [35–41] and signaling pathways such as Wnt, epidermal growth factor receptor- mitogen-activated protein kinase (EGFR-MAPK), phosphoinositide-3-kinase (PI3K), and AMP-activated protein kinase (AMPK) [40, 42–46]. In response to signals that impede cell proliferation, the HIPPO pathway is activated, where upstream kinase mammalian sterile-20-like protein kinase (MST1/2) phosphorylates and activates large tumor suppressor (LATS1/2) [47] which in turn phosphorylates Yes-associated protein (YAP) and its transcriptional co-activator with PDZ-binding motif (TAZ) (Figure 1). Specifically, phosphorylation of YAP at Serine (Ser127) creates a binding site for protein 14–3-3 leading to cytoplasmic retention of YAP/TAZ [27]. Additionally, YAP phosphorylation at Ser381 by LATS1/2 results in a subsequent phosphorylation by casein kinase 1 (CK1) and the ultimate recruitment of SCF beta-TRCP E3 ligase [48] which degrades YAP/TAZ (Figure 1). On the contrary, unphosphorylated YAP/TAZ translocate to the nucleus where it interacts with the transcription enhanced associate domain (TEAD 1–4) family of transcription factors [49] to bind DNA and activate genes that promote proliferation and inhibit apoptosis [50]. By regulating gene transcription, YAP is the biological mediator of the HIPPO pathway and has been identified as an oncogene [50–52]. Until 2017 no link between the HBP and HIPPO in cancer had been reported, but recent studies have elucidated a clear regulation of the HIPPO pathway by O-GlcNAcylation.
Figure 1. Increased O-GlcNAc levels induces transcription of proliferative genes.

Graphical representation of transcriptional pathways that regulate and are regulated by O-GlcNAc to promote cell proliferation. Non-voltage dependent Ca2+ channel TRPM7 regulates O-GlcNAc levels in an unknown mechanism and increased O-GlcNAc serve to O-GlcNAcylate and stabilize Calveolin 1 and c-MYC. O-GlcNAc levels are also regulated by a mechanism in which RANBP2 SUMOylates CEBP-a, inducing its degradation and blocking OGA transcription which leads to increased O-GlcNAc levels. Increased O-GlcNAc increases NANOG, aldehyde dehydrogenase (ALDH), and Kruppel-like factor 8 (KLF8) levels, and O-GlcNAcylates SOX2 at Ser246 promoting its nuclear translocation. All of these transcription factors are considered to be Cancer Stem Cell (CSC) markers that promote tumor initiation/proliferation and epithelial to mesenchymal transition (EMT). Simultaneously, increased O-GlcNAc promotes O-GlcNAcylation of transcription factor SMAD4 which induces TGF-b production that also promotes EMT. In parallel, increased O-GlcNAc levels create an O-GlcNAc driven highly proliferative chromatin region which overlaps with markers of active promoters and super-enhancers, particularly c-MYC and host cell factor 1 (HCF-1) both of which are O-GlcNAcylated and regulate transcription of Cyclin B and polo-like kinase 1 (PLK1) which drive proliferation. Metastasis associated protein 1 (MTA1) is also O-GlcNAcylated which promotes its interaction with gene promoters involved in chemoresistance only under this modification. Finally, under increased OGT levels, YAP/TAZ is O-GlcNAcylated at Ser 109 and Thr 241 both of which inhibit phosphorylation of YAP/TAZ at Ser127 consequently blocking its cytoplasmic retention. O-GlcNAcylated YAP/TAZ translocates to the nucleus where it binds its transcriptional co-activator TEAD and promotes transcription of genes involved in cell survival and replication.
YAP O-GlcNAcylation has been reported in three different findings in pancreatic, liver, and thyroid cancer. Peng et al. reported that YAP is O-GlcNAcylated at Ser109, Ser334 and Thr83, with Ser109 being the primary site [53], a finding validated in thyroid cancer cells by Li et al. [54] (Figure 1). Glucose starvation and OGT knockdown or inhibition trigger the phosphorylation of YAP at Ser127, decrease YAP O-GlcNAcylation and localize YAP in the cytoplasm. Specifically, OGT was reported to directly interact with and O-GlcNAcylate YAP [53, 54] and mutated YAP-Serine-109-Alanine O-GlcNAcylated site exhibited increased Ser127 phosphorylation, LATS1/2 and 14–3-3 binding, weakened TEAD binding and increased cytoplasmic localization (Figure 1). Importantly, unlike wild type, YAP-Ser-109-Ala O-GlcNAcylated mutant could not rescue the growth deficiency induced by knockdown of YAP in a pancreatic cancer xenograft tumor model [53]. Interestingly, YAP-TEAD may directly regulate OGT transcription and therefore YAP O-GlcNAcylation as the OGT promoter region contains a TEAD-binding site, YAP knockout cells contain reduced OGT RNA and protein levels, and OGT overexpression leads to increased levels of YAP downstream target genes [53, 55].
Similar findings of OGT upregulating YAP activity were reported in liver and thyroid cancer cells. These studies indicate that increased O-GlcNAcylation and OGT overexpression stimulate reduction of YAP Ser127 phosphorylation, increased TEAD transcriptional activity and increased YAP stability by promoting dissociation of YAP and its E3 ligase beta-TRCP [54, 55]. Similar to Peng et al. findings, Zhang et al. confirm that YAP is O-GlcNAcylated but report important O-GlcNAcylation site Thr241 which competes with Ser127 phosphorylation of YAP by reducing LATS1/2-YAP interaction, thus promoting YAP activity (Figure 1). The O-GlcNAcylated Thr241 is increased in liver cancer tissue and mutated Thr241 site had reduced tumorigenic characteristics and shorter half-life. In conclusion, these recent findings suggest that OGT mediates increase of YAP activity that is essential for liver and pancreatic tumorigenesis in vivo.
c-MYC dependent pathways
O-GlcNAc and c-MYC seem to be tightly interconnected as c-MYC can be directly O-GlcNAcylated in cancer cells [56]and c-MYC can increase OGT and O-GlcNAc levels in cancer cells[57]. Recent studies have added to the interconnection between c-MYC and O-GlcNAc in cancer cells. Itkonen et al. recently determined that reduction of O-GlcNAcylation through OGT inhibition induces a loss in O-GlcNAc chromatin marks, one conserved in different cancer cell lines which overlaps with markers of active promoters, including super enhancers (SE), a specific type of enhancers of highly transcribed genes and c-MYC binding sites. Specifically, this O-GlcNAc-driven highly transcriptionally active chromatin region is populated by the interaction between c-MYC and host cell factor 1 (HCF-1), which are both O-GlcNAcylated and whose interaction is disrupted upon reduced O-GlcNAc (Figure 1). Additionally, O-GlcNAcylation levels controlled by OGT activity regulate MYC-dependent transcriptome and proteome of mitotic genes Cyclin B and polo-like kinase 1 (PLK1) (Figure 1), which drive proliferation, thus elucidating a pathway in which increased O-GlcNAcylation promotes prostate cancer proliferation through MYC driven proliferation [58].
In non-small cell lung cancer (NSCLC), increased O-GlcNAcylation regulates cancer progression through transient receptor potential melastatin 7 (TRPM7), a non-voltage activated calcium (Ca2+) channel, previously associated with aggressive cancer behaviors [59–62]. Luanpitpong et al. show that silencing or inhibition of TRPM7 reduced O-GlcNAcylation in NSCLC blocking cell motility, which reveals a direct role of O-GlcNAc in regulation of cell migration [63]. Mechanistically, TRPM7 regulates O-GlcNAc levels which are required for O-GlcNAcylation and stability of transcription factor c-MYC and Calveolin-1 (Cav-1) (Figure 1), while knockdown of TRPM7 reduces their protein and mRNA expression, rescued by increased O-GlcNAc, thus elucidating a novel mechanism that regulates O-GlcNAc levels alternatively to glycolytic based regulation.
Cancer stem cell transcription factors
O-GlcNAcylation has also been implicated with regulation of cancer stem cell (CSCs) populations which rely on transcription factors such as Sry-boc 2 (SOX2), Nanog homeobox (NANOG), and POU5F1 (OCT4) for maintenance of pluripotency. It is specifically the property, presence and upregulation of self-renewal transcription factors in the tumor heterogeneous population that correlate with disease reoccurrence which are thought to be responsible for tumor relapse and drug resistance [64]. Many of these self-renewal transcription factors have been shown to be O-GlcNAcylated in cancer cells [5]. For example, O-GlcNAcylation of SOX2 at Ser246 enhances protein stability and promotes nuclear localization favoring tumor progression in pancreatic adenocarcinoma (PDAC), while decreased O-GlcNAcylation through OGT inhibition delays tumor initiation and lessens tumor burden in vivo [65]. Akella et al. have identified that increased O-GlcNAc levels promote breast cancer cell mammosphere formation, an assay that selects for stem cell-like properties to form spherical structures, suggesting that increased O-GlcNAc levels may increase CSC population. Increased O-GlcNAcylation induces an increased expression of CSCs factors CD44H/CD24L, NANOG+ and ALDH+ in vitro and OGT overexpression increases tumor initiation in vivo. This regulation may involve Kruppel-like factor 8 (KLF8), identified in OGT overexpressing mammospheres, whose expression correlates with O-GlcNAc levels providing a novel insight and therapeutic target on the role of O-GlcNAc in regulating tumor-initiation and progression (Figure 1) [66].
Other transcriptional regulators
Various cancers overexpress transcription factor specificity protein 1 (Sp1) which regulates the expression of tumor promoting genes involved in angiogenesis, apoptotic resistance, and cell growth [67]. Notably, O-GlcNAcylation of Sp1 in the cytosol protects the protein from proteasomal degradation and O-GlcNAcylation of Sp1 also promotes nuclear localization. Once the Sp1 has entered the nucleus, OGA removes the O-GlcNAc moiety, allowing Sp1 to be phosphorylated and bind to DNA and promote transcription of the target genes [5].
Chromatin modifier metastasis-associated protein 1 (MTA1) promotes the expression of target genes involved in transformation, metastasis, growth, and invasion in response to PTM [68]. A recent study in breast cancer showed the O-GlcNAcylation of MTA1. This modification promotes MTA1 interaction with gene promoter regions of MTA1 target genes, especially genes involved in chemoresistance of breast cancer cells (Figure 1) [69]. MTA1 is a member of the nucleosome remodeling and histone deacetylase complex [70]. Interestingly, OGT has been found to interact with multiple members of the nucleosome remodeling and deacetylase (NuRD) complex in cancer cells including histone deacetylase complex ½ (HDAC1/2), MTA1/2/3, and chromodomain helicase DNA binding protein 4 (CHD4) [71]. However, the functional role of this association in cancer cells remains unclear.
In hepatocellular carcinoma (HCC), hyper-O-GlcNAcylation of transcription coactivator peroxisome proliferative-activated receptor gamma coactivator 1 alpha (PGC1α), contributes largely to hepatocarcinogenesis [72, 73]. Recently, the RAN-binding protein 2 (RANBP2), a small ubiquitin like modifier (SUMO) in the nuclear pore complex, was identified as an enriched marker in HCC patient tissues and correlated with lower patient survival rate, higher neoplastic grade, tumor size and pT status [74]. RANBP2 SUMOylates transcription factor CCAAT/enhancer binding protein alpha (CEBPα) at Lysine 169 inducing its degradation. CEBPα negatively regulates O-GlcNAcylation by promoting transcription of OGA (Figure 1), and RNAi silencing of RANBP2 reduced proliferation, invasive ability, tumor growth in vivo and promoted apoptosis all of which were rescued upon RNAi silencing of CEBPα, demonstrating that RANBP2 promotes CEBPα dependent O-GlcNAcylation. Interestingly, PGC1α, an HCC marker of mitochondrial biogenesis, tumor transformation and metabolism previously known to be stabilized by O-GlcNAcylation[75], was shown to also interact with OGA and decreased O-GlcNAcylation upon RANBP2 silencing were detected, speculating that RANBP2 silencing may increase OGA-RANBP2 interaction, contributing to its destabilization which leads to poor tumorigenic outcome, thus determining a novel mechanism for OGT/OGA balance maintenance and reconfirming the increased O-GlcNAcylation as a driver of tumorigenesis. [74].
Additionally, findings suggest that O-GlcNAcylation of SMAD4 at Thr63, a transcriptional complex component, stabilizes (mothers against decapentaplegic homolog 4) SMAD4 protein levels by impeding SMAD-glycogen synthase kinase 3 (GSK-3B) interaction which in turn blocks degradation of SMAD 4. SMAD4 mediates regulation of transforming growth factor-β (TGF-β) signaling, thus increased O-GlcNAc induces TGF-β production promoting EMT and metastasis (Figure 1) [76]. Overall, these findings provide novel mechanistic insights on the cross talk between transcription factors and O-GlcNAcylation in promoting tumorigenesis.
Metabolic Pathways
Metabolic sensor
The role of O-GlcNAcylation as a metabolic sensor has been widely explored [77–79]. Recent findings support regulation of cellular bioenergetics through O-GlcNAc modifications. Although not extensively studied, the mitochondrial isoform of OGT can also O-GlcNAcylate proteins in the mitochondria. It was recently shown that mitochondrial OGT (mOGT) expression and O-GlcNAc levels in breast cancer cells are glucose dependent and that overexpression of mOGT levels leads to elevated mitochondrial membrane potential and respiration leading to an increase in reactive oxygen species (ROS) and decrease of cellular ATP. O-GlcNAcylated proteins in cells overexpressing mOGT were found in the mitochondrial matrix and inner membrane, and these identified proteins play a role in mitochondrial respiration, fatty acid metabolism, transport, translation, and apoptosis reflecting the possible role of O-GlcNAcylation in cellular bioenergetics and mitochondrial homeostasis and function [80]. Increased O-GlcNAc is also observed in Diabetes Mellitus 2 (DM2) patients majorly characterized by a hyperglycemic condition which favors increased risk of developing breast cancer [81, 82] with poor survival outcomes [83]. In a syngeneic diabetes-induced breast cancer mouse model, tumors from diabetic mice had increased levels of O-GlcNAc compared to tumors in non-diabetic mice and exhibited higher malignant tumor features such as vascularization, proliferation, aligned collagen and an M2 Tumor Associated Macrophage (TAM) profile, which favors a protumor environment. These phenotypes, including O-GlcNAc levels were reduced upon treatment with metformin, an antihyperglycemic drug [84]. The higher O-GlcNAc levels in tumors from diabetic mice reflect the role of O-GlcNAc as a metabolic sensor of the nutrient state of the organism that may contribute to a more aggressive tumor phenotype induced by hyperglycemia.
Despite elevated O-GlcNAc levels being maintained in many cancer cells, a homeostatic response to O-GlcNAc fluctuations is also active. Homeostasis of cellular O-GlcNAc levels is maintained by feedback regulation of OGT and OGA whose expression is modulated in response drastic changes in O-GlcNAc levels, which has been demonstrated in neuroblastoma, leukemia, cervical cancer and colon cancer cell lines [85], [86]. A recent study suggests that upon induced increased or decreased levels of O-GlcNAc, OGA expression is downregulated at the transcriptional level and in an epigenetic dependent mechanism regulated by histone acetylation[87]. Conversely, increased O-GlcNAc induces 4E-BP1 O-GlcNAcylation on Ser5 and Ser6 which acts as a sensor of O-GlcNAc levels to upregulate OGT protein levels in a feedback loop in lung cancer cells [87].
Modulation of signaling metabolic pathways
Increased glucose uptake being shunted into glycolysis is a hallmark of cancer cells originally observed by Otto Warburg [88]. Recently, the cancer cell dependence for glycolysis instead of oxidative phosphorylation was reported to involve increased O-GlcNAcylation. Specifically, the phosphoglycerate kinase 1 (PGK1), the first ATP-producing enzyme in glycolysis is O-GlcNAcylated at Thr255, promoting its mitochondrial translocation where it inhibits pyruvate dehydrogenase and thus reduces oxidative phosphorylation in colon cancer cells [89]. Another example of the glycolytic-HBP interrelation regulating tumor cell proliferation includes the increased O-GlcNAc levels in pancreatic cancer promoting O-GlcNAcylation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) on Ser172 in hypoxic conditions. This provides competition for phosphorylation at this site by ERK and promotes nuclear translocation of the O-GlcNAcylated PFKFB3, preventing p27-hypoxia induced accumulation and leading to cell cycle progression [90].
Another study suggested control of O-GlcNAc levels from glucose utilization occurring from regulation of HBP enzymes. Glutamine-fructose-6-phosphate transaminase 2 (GFPT2) was reported to induce an increase in O-GlcNAc levels which may contribute to epithelial-mesenchymal transition (EMT) in serous ovarian cancer (SOC) [91].
Besides the evident role of OGT and O-GlcNAcylation in regulating tumorigenesis, the role of its opposing partner, OGA in cancer is studied to a lesser extent. Singh et al. show that overexpression of OGA induced increased tumor progression in mice through its non-intrinsic acetyl transferase activity. Specifically, OGA promotes acetyltransferase activity on pyruvate kinase muscle isoform 2 (PKM2), the enzyme that controls the conversion of phosphoenolpyruvate (PEP) to pyruvate, by inducing its acetylation which in turn promotes increased O-GlcNAcylation, correlating with increased tumorigenesis promoted by overexpression of OGA. This regulation is glucose dependent and inhibits the catalytic activity of PKM2 by blocking its tetramerization, promoting aerobic glycolysis and tumor growth [92]. Altogether, these findings enforce the dependency of O-GlcNAcylation on nutrient availability and its potential in influencing metabolic rewiring that supports cancer progression.
O-GlcNAcylation has also been linked to regulation of lipogenesis in adipocytes and lipogenic cancers. Studies have shown that in adipocytes O-GlcNAc levels, regulated by glutamine fructose-6-phosphate amidotransferase 1 (GFAT1), the first rate-limiting enzyme in HBP, are increased during adipogenesis and that reduction of UDP-GlcNAc which reduces O-GlcNAcyaltion levels results in a reduction of fatty acid synthase (FASN) and other lipid accumulation proteins as well as a blockade in adipogenesis [93]. This regulation is thought to be directed by O-GlcNAcylation of (CCAAT/enhancer binding protein β) C/EBPβ and (peroxisome proliferator-activated receptor γ) PPARγ [93]. Moreover, increased O-GlcNAc levels in obese mice livers correlated with increased FASN levels and interestingly, FASN was found to be O-GlcNAcylated which reduces its proteosomal degradation by promoting an interaction with the deubiquitinating enzyme nucleolar and spindle-associated protein 1 (NUSAP1) in normal liver cells lines and hepatocarcinoma lines. [94]. In addition, Sodi et al. have shown that O-GlcNAc regulates the master lipid regulator SREBP1 and its transcriptional targets including FASN, ACLY, ACC and others via AMPK regulation in breast cancer cells and lipogenic tissue, and that SREBP1 can overcome growth defects conferred by reduced O-GlcNAcylation in breast tumors [95].
Raab et al. have recently shown that OGT and FASN are co-localized in hepatocellular carcinoma HepG2 lines, O-GlcNAcylation of FASN is dependent on glucose levels and FASN expression depends on OGT upon serum stimulation in a cell-cycle dependent manner [96]. Importantly, their study reveals regulation of FASN in light of a previously studied link between O-GlcNAc and another fundamental energy sensing pathway such as mammalian target of rapamycin (mTOR) [57, 97]. FASN expression correlated with mTOR activation and increased O-GlcNAcylation in two mouse models with obesity and chronically activated insulin and mTOR, and importantly inhibition of FASN reduced O-GlcNAcylation and mTOR activation which suggest a dual regulation of FASN by O-GlcNAc and mTOR but also a feedback loop between these three nutrient dependent pathways. O-GlcNAc can also regulate de novo lipogenesis in breast cancer cell lines by O-GlcNAcylation of serine/arginine-rich protein kinase 2 (SRPK2) at a nuclear localization signal which triggers its nuclear translocation through importin-α and allows for phosphorylation of proteins that promote splicing of lipogenic pre-mRNAs [98]. In hepatocellular carcinoma (HCC), acyl-CoA ligase 4 (ACSL4) levels, that mediates fatty acid synthesis, are increased and ACSL4 promotes HCC proliferation through activation of mTOR, glucose transporter (GLUT1) and increased O-GlcNAc and conversely, O-GlcNAcylation increases ACSL4 expression [99] exhibiting yet another example of O-GlcNAc regulation by feedback loops. Thus O-GlcNAc plays a key role in regulating lipid metabolism in cancer cells.
Modulation of other signaling pathways
In recent years, increased O-GlcNAcylation in various cancers was shown to promote cancer progression through multiple signaling pathways. Chen et al. report that increased O-GlcNAc levels in bladder cancer promote increased NUSAP1 protein levels, a microtubule binding protein that promotes proliferation and inhibits apoptosis in bladder cancer cells, while decreased O-GlcNAc levels induce downregulation of NUSAP1 expression and block cancer progression [100]. Yu et al. report that increased O-GlcNAc levels in colorectal cancer are associated with an increase in integrin 5α (ITGA5) levels and show that ITAG5 is O-GlcNAcylated which blocks its degradation to promote tumorigenesis [101].
The Laman group demonstrated a link between F-box protein (FBP) encoded by FBXL17 and global increases of O-GlcNAcylation in breast cancer. FBXL17 protein typically functions in the ubiquitination pathway; in breast cancer, FBXL17 is often rearranged, causing a loss in ubiquitination activity of the FBXL17 protein. FBXL17 can interact with UAP1, the last enzyme in the HBP, UDP-N-acetylhexosamine pyrophosphorylase 1 (UAP1) catalyzes the conversion of UTP and GlcNAc-1-P into UDP-GlcNAc. FBXL17 can regulate UAP1 activity via binding leading to an inhibition of UAP1 phosphorylation activity, while knockdown of FBXL17 leads to unregulated UAP1 and elevated levels of O-GlcNAcylation in breast cancer cells [102].
Another role of O-GlcNAc in liver cancer was identified through a link between X-active-specific transcript (XIST) and OGT, both of which are upregulated in liver cancer and correlate with a poor prognosis in patients [103] and miR-424–5p, a microRNA which was shown to have a tumor suppressor role in HCC [104–106] and to be a regulator of OGT [107]. In liver cancer cells silencing of XIST reduced migration, invasion and increased apoptosis and epithelial to mesenchymal transition (EMT) markers, which were revered upon miR-424–5p silencing. XIST was shown to bind to miR-424–5p, whose expression is inversely correlated to OGT and liver tumorigenesis in patient tissues and miR-424–5p was found to bind to OGT and negatively regulate O-GlcNAcylation which is required for RAF1 proto-oncogene serine/threonine kinase stabilization [108], thus providing a novel insight where miR-424–5p dependent downregulation of O-GlcNAc levels reduces the pro-oncogenic effect of RAF1 [109].
Cancer Immune Regulation
The immune system is an intricate matrix of cells and proteins which get activated to provide a pro-inflammatory response to infection or tissue damage through innate or adaptive immunity [110]. Activation of components of the adaptive immune system, such as T-cell development, differentiation, proliferation, and activation are controlled by a metabolic profile that is marked by increased glycolysis, glutamine and acetyl-CoA. Direct links to regulation of immune activation by O-GlcNAcylation have been identified even in non-cancerous environments. [111–114]. For example, lymphocyte activation and activation of T-cells through T-cell receptor (TCR) depend on and lead to increased O-GlcNAcylation [115–117]. Differentiation of CD4+T-cells into T helper cell 17 (TH17) and regulatory T-cells (Tregs) requires O-GlcNAcylation of NF-kB, which regulates production of pro-inflammatory cytokines interleukin (IL) IL-17A, IL-17F, IL-21, and IL-22 by Th17 cells [118–121] and O-GlcNAcylation of nuclear factor of activated T-cells (NFAT), signal transducer and activator or transcription STAT3, STAT5 and forkhead box protein 3 (FOXP3) can promote T-cell differentiation, production of IL-6 and IL-10 and maintains lineage identity and Tregs function [119, 122, 123]. Additionally, increased levels of O-GlcNAc increase IL-17A production which is regulated by the O-GlcNAcylation of acetyl-CoA carboxylase 1 (ACC1) that promotes production of RAR-related orphan receptor γ t variant (RORγt) ligands, linking inflammation to excess nutrition [118]. Conversely, another study suggests that OGT suppresses innate immune response through O-GlcNAcylation of receptor interacting serine/threonine kinase 3 (RIPK3) [124]. In addition, O-GlcNAc, acting as a nutrient sensor of the metabolic state of the cell has been predicted to be a mediator of macrophage polarization in innate immunity, as influx of metabolites such as glucosamine and glutamine routing toward UDP-GlcNAc is increased in macrophage M2 polarization [125–128]. Considering such evidence, the study of increased O-GlcNAc in cancer immunology has become pertinent.
Recent studies have elucidated the role of O-GlcNAc in tumor microenvironment (TME) by monitoring the tumor associated macrophage (TAM) polarization, an event that favors antitumor properties through M1-like macrophage population, which secrete pro-inflammatory molecules such as IL-1, IL-12, tumor necrosis factor α (TNF-α), or protumor properties through M2-like macrophage population, which secrete anti-inflammatory factors such as IL-10, C-C motif chemokine ligand 2 (CCL2) and stromal cell-derived factor 1 (SDF-1) that suppress anti-tumor immunity [129–133]. Elevated O-GlcNAc levels has been marked as a characteristic of M2 polarization [126] and further investigated in cancer models. In an OGT transgenic mouse model transplanted with melanoma cells, infiltrated M1-like macrophages and pro-inflammatory cytokine production was reduced in the larger tumors of OGT-transgenic mouse compared to control mice, suggesting a correlation between increased O-GlcNAcylation and progression of tumor due to suppression of antitumor immunity through p38 MAPK activity and upregulation of the extracellular signaling regulated kinase ½ (ERK1/2) signaling pathway (Figure 2) [134]. Alternatively, inhibition of Hedgehog signaling in TAMs of mammary tumors reduced flux through the UDP-GlcNAc leading to reduced STAT6 O-GlcNAcylation which damped the M2 profile, inducing a macrophagic phenotype reminiscent of M1 with a switch in metabolism from fatty acid oxidation to glycolysis (Figure 2) [135]. In addition, it has been shown that OGT expression in macrophages is dependent on cullin 3 (CUL3) whose deletion regulates nuclear factor E2–related factor-2 (Nrf2) Ogt promoter binding and STAT3 phosphorylation and inflammation driven tumorigenesis (Figure 2) [123]. Furthermore, increased O-GlcNAcylation, resulting from enhanced glucose flow in hyperglycemic diabetes mellitus (DM) condition is responsible for macrophage polarization which favors a decrease of M1 markers CD86, major histocompatibility complex class II (MHC-II) and nitric oxide synthase 2 (NOS2) and an increase in M2 markers CD206, Arginase1. This phenotype is reversed upon OGT inhibition, while in vivo, inhibition of O-GlcNAcylation through inhibition of HBP by 6-diazo-5-oxo-l-norleucin (DON) reduced tumor growth in an acute hyperglycemic mouse model, reducing pro-tumorigenic phenotype of macrophages and increasing M1 macrophage markers [136]. On the other hand, a novel hypothesis suggests that decreased O-GlcNAc levels were detected during M1 like polarization in human macrophages and that increased O-GlcNAc signaling in an overnutrition mouse model suppresses M1macrophage pro-inflammatory activation. Increased O-GlcNAcylation inhibits mTOR/S6K1 pathway by directly O-GlcNAcylating S6K1 at Ser489 which blocks its phosphorylation and mTORC1 signaling leading to suppression of pro-inflammatory macrophage polarization, although this finding was not recapitulated in a cancer model [137].
Figure 2. OGT promotes anti-inflammatory tumor microenvironment.
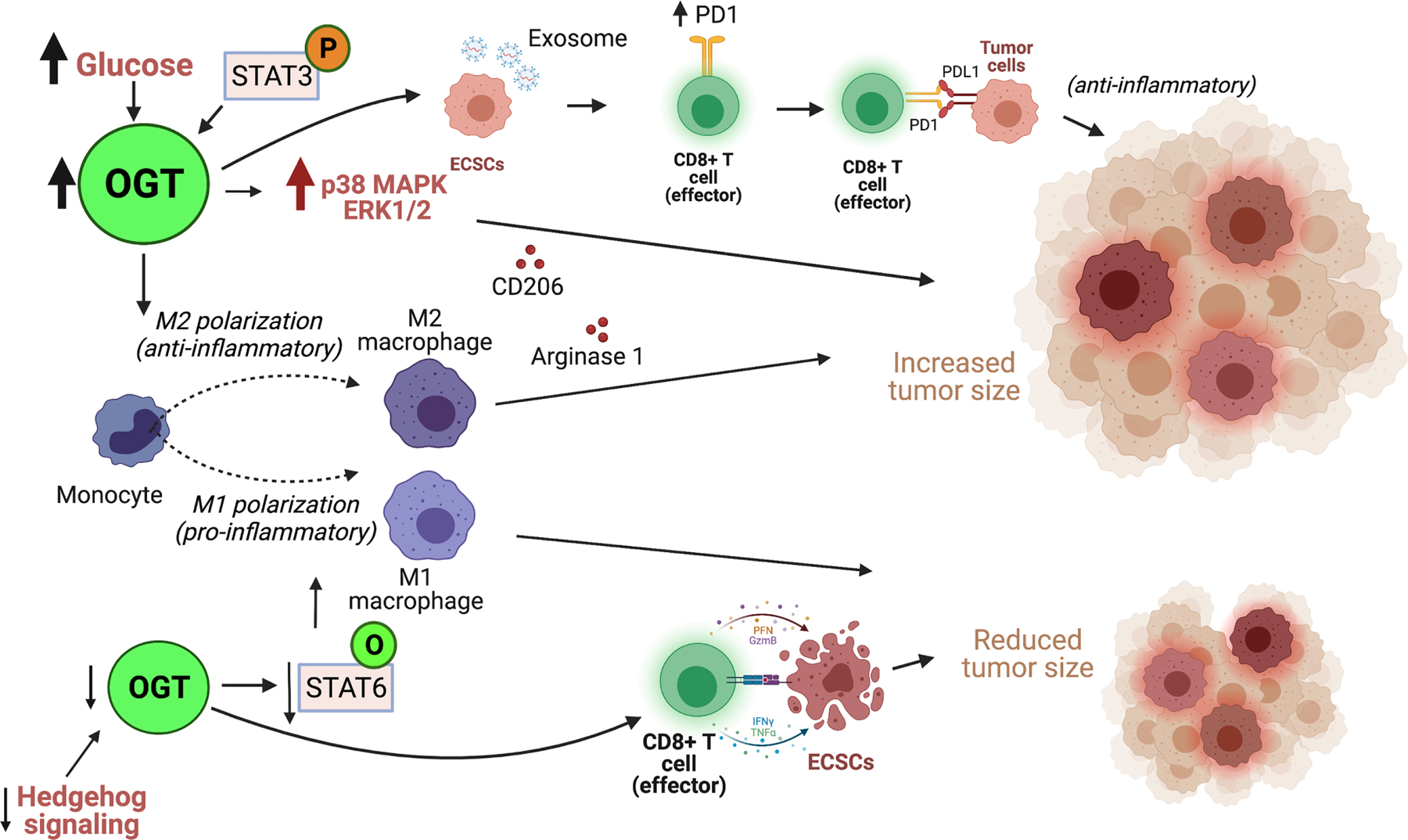
Increased OGT levels mediate a monocyte polarization towards and M2 profile which is associated with an anti-inflammatory response that promotes tumor growth. Increased OGT, induced by increased glucose levels or STAT3 phosphorylation directly promotes an M2 macrophage profile through release of anti-inflammatroy cytokines Arginase 1 and CD206 that contribute to increased tumor size. Alternatively, increased OGT increased p38 MAPK ERK1/2 which also contributes to increased tumor size. In addition, under increased OGT levels exosomes released by esophageal cancer stem cells (ECSCs) promote increased PD1 expression on CD8+ T-cells surface which binds to PDL-1 of tumor cells to promote an anti-inflammatory response and increased tumor growth. Under decreased OGT levels induced by decreased Hedgehog signaling, a decrease in O-GlcNAcyaltion of STAT6 mediates an M1 macrophage polarization profile, associated with pro-inflammatory response and reduced tumor size. Similarly, under decreased OGT/O-GlcNAc levels, CD8+ T-cells release granzymeB which recognized and mediates apoptosis of ECSCs contributing to reduced tumor size.
The role of elevated OGT and O-GlcNAcylation in tumorigenesis through different mechanisms [2], [19] has been investigated even with respect to changes in immune activation through cellular differentiation. Malaker et al. identified 36 O-GlcNAcylated MHC class I–associated peptides in primary leukemia samples which serve as potential neoantigens able to elicit memory T-cell responses in healthy donors [138]. Recently, it was shown that treatment of acute myeloid leukemia (AML) with dronabinol has a dual effect in overriding the differentiation blockage as a mean of treating leukemia. Dronabinol, a (−)-Δ9-Tetrahydrocannabinol (THC) isomer which induces leukemia cell apoptosis at high concentrations, also induces hypomethylation of Ogt transcription site promoting OGT mRNA and protein increase [139]. Maturation marker protein tyrosine phosphatase receptor type C (CD45) promotes cell maturation and needs to be glycosylated in order to be expressed on cell surface, thus increased O-GlcNAc promotes cell maturation which correlated with increased CD45 upon dronabinol treatment. Thus, maturation of leukemic blasts is dependent on O-GlcNAc, providing a novel druggable target for leukemia. Elevated O-GlcNAc in ALDH+ esophageal cancer stem cells (ECSCs) which are responsible for increased tumor initiation, metastases, resistance to therapy [140–142] promotes tumorigenesis in an immune escape dependent mechanism. Decreased O-GlcNAcylation through knockdown of OGT in ECSCs increased CD8+ T-cell granzymeB release to induce apoptosis of neighboring aldehyde dehydrogenase ALDH+ ECSCs while elevated OGT in ECSCs derived exosomes increased programmed cell death protein 1 (PD-1) expression on CD8+ T-cells, which interacts with PD-1 ligand (PDL-1) on tumor cells in an inhibitory anti-tumor immunity mechanism (Figure 2) [143]. Thus, increased O-GlcNAc protects ALDH+ ECSCs from CD8+ T-cell by increasing PD-1 expression [142]. Overall, these findings reinforce the protumorigenic role of increased O-GlcNAc and provide novel insights on regulation of tumor immunity and targets that may reduce tumor progression.
Targeting O-GlcNAc
Small molecule inhibitors of OGT and thus O-GlcNAcylation are limited and currently not highly effective or selective [144]. However, recent studies have identified novel pathways which involve indirect or synergistic inhibition of OGT which may help for future development of OGT targeted therapy. A recent study suggested that pharmacological inhibition of GFAT1 by using 6-diazo-5-oxonorleucine (DON), reduces O-GlcNAc levels and renders cancer cells more sensitive to apoptosis upon diamide-induced oxidative stress in breast cancer cells [145]. High dosage of DON lead to apoptosis in AML cells within 24 hours while low dose treatment of DON lead to AML cell differentiation and apoptosis following 4 to 7 days of treatment, stressing the importance of evaluating off target effects when inhibiting the HBP with DON [146]. Similarly, Deng et al. reported that silencing of GTPase RhoA effector, Rho-associated coiled-coil forming protein kinase 2 (ROCK2), seemingly exhibiting high expressions in osteosarcoma and correlating with poor prognosis, inhibits tumor proliferation in vivo and in vitro by inducing tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apoptosis, specifically via degradation of OGT, which in turn reduces O-GlcNAc levels and decrease tumor growth [147]. An interesting reciprocal regulation of OGT was elucidated in colon cancer, where X-linked inhibitor of apoptosis (XIAP) interacts with OGT and is O-GlcNAcylated on Ser406. However, upon binding with OGT, XIAP performs E3 ligase activity on OGT, promoting its ubiquitination and degradation and ultimately reducing cell proliferation, but this activity requires Ser406 O-GlcNAcylation on XIAP, demonstrating a novel inhibition route for OGT and O-GlcNAc levels [148].
A recent study proposes a synthetic lethality of OGT inhibition with cyclin dependent kinase 9 (CDK9) inhibitor treatment which blocks phosphorylation of C-terminal domain (CTD) of RNA Pol II, inhibiting transcription as identified in prostate cancer cells and organoids [149]. In addition, doxorubicin, a chemotherapeutic agent was shown to have synergistic effects with OGT inhibitor OSMI-1 the combination of which increased apoptotic cell death through the activation of both the p53 and mitochondrial Bcl2 pathways [150]. Importantly, it was recently shown that breast cancer cells that exhibit increased sensitivity to OSMI-1 correlate with tamoxifen resistance [151]. Upon treatment with OSMI-1, tamoxifen resistant cells induce an increase in transcription of tumor suppressor ERBB receptor feedback inhibitor 1 (ERRFI1), an inhibitor of ERBB2 signaling which drives tamoxifen resistance in estrogen receptor α (ERα)-positive breast cancers [152, 153],and whose expression is associated with positive outcome in patients. OGT regulation of ERFF1 is likely occurring through epigenetic methylation of the ERFF1 promoter speculated to be O-GlcNAc rich inducing decreased ERFF1 expression [151, 154]. Thus, OGT inhibition may increase the role of ERBB receptor tyrosine kinase (RTK) inhibitors to reduce cancer progression. These findings elucidate novel mechanisms of O-GlcNAc regulation of cancer progression as a downstream or upstream effector, and simultaneously suggest that inhibition of O-GlcNAc sensitizes cells to targeted therapies, which may open new endeavors for considering therapy that targets OGT in combination with other targeted therapies.
Future Directions
In this review, we discuss the prominent role of widely recognized increased O-GlcNAcylation in cancer; promoting growth through recently elucidated mechanisms of cellular signaling pathways, metabolic and transcriptional pathways, and changes in tumor microenvironment. New O-GlcNAc detection methods are improving [155] that are critically needed to increase our understanding the role of O-GlcNAc signaling in cancer biology and other disease states. Despite encouraging findings suggesting that reduction of O-GlcNAc levels via pharmacological inhibition of OGT can reduce cancer cell growth in vitro, the suitability and efficacy of these inhibitors in vivo is still in question and need to be further explored. A summary of all developed OGT inhibitors has previously been reviewed [156] and the main challenge remains finding alternative compounds with increased efficacy and thus test both anti-cancer effects but also toxicities of these compounds in vivo. In addition, given the wide and not-entirely known substrates of OGT, understanding the effects of its inhibition need to be further addressed, including the role of OGT in adult tissues. For example, OGT knockout (KO) in germline cells is embryonically lethal which suggests a critical role of O-GlcNAc in development [157]. More recently, cell specific induced OGT-KO models have developed to study the role of OGT in adult biology including in cardiomyocytes [158], pancreatic beta cells [159], T-cell [160], B-cell [161], neurons [162–164], adipocytes [165], and intestinal epithelial cells [166] in which deregulation of cellular pathways that generally led to block in normal functions implicating an important role of OGT in adult biology but also possible toxicities in reducing total O-GlcNAcylation. These findings further complicate the possibility of pharmacologically inhibiting OGT in cancer patients. However, given the aforementioned success with inhibiting O-GlcNAc levels to reduce tumor growth in vivo with genetic targeting such as shRNA, it remains a possibility that reducing O-GlcNAc can be beneficial in treating cancers. There is a need for developing more robust OGT inhibitors and in vivo testing to determine whether a therapeutic window exists to reduce O-GlcNAcylation in some tumors, especially as combination treatment to treat cancers.
Highlights.
The post-translational modification O-GlcNAcylation is found to be elevated in nearly all cancers.
This nutrient sensitive intracellular glycosylation can regulate a diverse set of cytosolic and nuclear proteins.
O-GlcNAcylation regulates signaling pathways including transcription, metabolic and immune regulation in cancer cells.
This review highlight most recent understanding of mechanisms by which O-GlcNAcylation regulates cancer cell signaling.
Acknowledgments
We want to thank Giang Le Ming for critical reading of manuscript. This work was supported by UO1CA244303 (MJR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chiaradonna F, Ricciardiello F, and Palorini R, The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells, 2018. 7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akella NM, Ciraku L, and Reginato MJ, Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol, 2019. 17(1): p. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darley-Usmar VM, Ball LE, and Chatham JC, Protein O-linked β-N-acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. Journal of molecular and cellular cardiology, 2012. 52(3): p. 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephen HM, Adams TM, and Wells L, Regulating the Regulators: Mechanisms of Substrate Selection of the O-GlcNAc Cycling Enzymes OGT and OGA. Glycobiology, 2021. 31(7): p. 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker MP, Peterson KR, and Slawson C, O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers (Basel), 2021. 13(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leney AC, et al. , Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc Natl Acad Sci U S A, 2017. 114(35): p. E7255–E7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butkinaree C, Park K, and Hart GW, O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta, 2010. 1800(2): p. 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hart GW, Nutrient regulation of signaling and transcription. Journal of Biological Chemistry, 2019. 294(7): p. 2211–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copeland RJ, Han G, and Hart GW, O-GlcNAcomics--Revealing roles of O-GlcNAcylation in disease mechanisms and development of potential diagnostics. Proteomics Clin Appl, 2013. 7(9–10): p. 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quik M, Hokke CH, and Everts B, The role of O-GlcNAcylation in immunity against infections. Immunology, 2020. 161(3): p. 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferron M, et al. , Protein O-GlcNAcylation in Cardiac Pathologies: Past, Present, Future. Front Endocrinol (Lausanne), 2018. 9: p. 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nie H and Yi W, O-GlcNAcylation, a sweet link to the pathology of diseases. J Zhejiang Univ Sci B, 2019. 20(5): p. 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma X, et al. , The emerging link between O-GlcNAcylation and neurological disorders. Cell Mol Life Sci, 2017. 74(20): p. 3667–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caldwell SA, et al. , Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene, 2010. 29(19): p. 2831–42. [DOI] [PubMed] [Google Scholar]
- 15.Phueaouan T, et al. , Aberrant O-GlcNAc-modified proteins expressed in primary colorectal cancer. Oncol Rep, 2013. 30(6): p. 2929–36. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Q, et al. , O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol, 2012. 29(2): p. 985–93. [DOI] [PubMed] [Google Scholar]
- 17.de Queiroz RM, et al. , Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the p53 Pathway in Ovarian Cancer Cells. J Biol Chem, 2016. 291(36): p. 18897–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang SZ, et al. , Regulation of pancreatic cancer TRAIL resistance by protein O-GlcNAcylation. Lab Invest, 2020. 100(5): p. 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itkonen HM, Loda M, and Mills IG, O-GlcNAc Transferase - An Auxiliary Factor or a Full-blown Oncogene? Mol Cancer Res, 2021. 19(4): p. 555–564. [DOI] [PubMed] [Google Scholar]
- 20.Ferrer CM, Sodi VL, and Reginato MJ, O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol, 2016. 428(16): p. 3282–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu D, et al. , Functional Analysis of O-GlcNAcylation in Cancer Metastasis. Front Oncol, 2020. 10: p. 585288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jozwiak P, et al. , O-GlcNAcylation and Metabolic Reprograming in Cancer. Front Endocrinol (Lausanne), 2014. 5: p. 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Justice RW, et al. , The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev, 1995. 9(5): p. 534–46. [DOI] [PubMed] [Google Scholar]
- 24.Xu T, et al. , Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development, 1995. 121(4): p. 1053–63. [DOI] [PubMed] [Google Scholar]
- 25.Grusche FA, Richardson HE, and Harvey KF, Upstream regulation of the hippo size control pathway. Curr Biol, 2010. 20(13): p. R574–82. [DOI] [PubMed] [Google Scholar]
- 26.Moroishi T, et al. , The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell, 2016. 167(6): p. 1525–1539 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao B, et al. , Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev, 2007. 21(21): p. 2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordenonsi M, et al. , The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell, 2011. 147(4): p. 759–72. [DOI] [PubMed] [Google Scholar]
- 29.Mohseni M, et al. , A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol, 2014. 16(1): p. 108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dupont S, et al. , Role of YAP/TAZ in mechanotransduction. Nature, 2011. 474(7350): p. 179–83. [DOI] [PubMed] [Google Scholar]
- 31.Aragona M, et al. , A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell, 2013. 154(5): p. 1047–1059. [DOI] [PubMed] [Google Scholar]
- 32.Benham-Pyle BW, Pruitt BL, and Nelson WJ, Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science, 2015. 348(6238): p. 1024–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim KM, et al. , Shear stress induced by an interstitial level of slow flow increases the osteogenic differentiation of mesenchymal stem cells through TAZ activation. PLoS One, 2014. 9(3): p. e92427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Codelia VA, Sun G, and Irvine KD, Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol, 2014. 24(17): p. 2012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu FX, et al. , Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell, 2012. 150(4): p. 780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X, et al. , Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest, 2015. 125(5): p. 2123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller E, et al. , Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol, 2012. 19(8): p. 955–62. [DOI] [PubMed] [Google Scholar]
- 38.Wennmann DO, et al. , The Hippo pathway is controlled by Angiotensin II signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis, 2014. 5: p. e1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu FX, et al. , Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev, 2013. 27(11): p. 1223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park HW, et al. , Alternative Wnt Signaling Activates YAP/TAZ. Cell, 2015. 162(4): p. 780–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mo JS, et al. , Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol, 2015. 17(4): p. 500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azzolin L, et al. , Role of TAZ as mediator of Wnt signaling. Cell, 2012. 151(7): p. 1443–56. [DOI] [PubMed] [Google Scholar]
- 43.Azzolin L, et al. , YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell, 2014. 158(1): p. 157–70. [DOI] [PubMed] [Google Scholar]
- 44.Fan R, Kim NG, and Gumbiner BM, Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A, 2013. 110(7): p. 2569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang W, et al. , The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem, 2012. 287(31): p. 26245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, et al. , AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol, 2015. 17(4): p. 490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan EH, et al. , The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene, 2005. 24(12): p. 2076–86. [DOI] [PubMed] [Google Scholar]
- 48.Zhao B, et al. , A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev, 2010. 24(1): p. 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao B, et al. , TEAD mediates YAP-dependent gene induction and growth control. Genes Dev, 2008. 22(14): p. 1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zanconato F, Cordenonsi M, and Piccolo S, YAP/TAZ at the Roots of Cancer. Cancer Cell, 2016. 29(6): p. 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moroishi T, Hansen CG, and Guan KL, The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer, 2015. 15(2): p. 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu FX, Zhao B, and Guan KL, Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell, 2015. 163(4): p. 811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng C, et al. , Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol Cell, 2017. 68(3): p. 591–604 e5. [DOI] [PubMed] [Google Scholar]
- 54.Li X, et al. , OGT regulated O-GlcNAcylation promotes papillary thyroid cancer malignancy via activating YAP. Oncogene, 2021. 40(30): p. 4859–4871. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, et al. , The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun, 2017. 8: p. 15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Itkonen HM, et al. , O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res, 2013. 73(16): p. 5277–87. [DOI] [PubMed] [Google Scholar]
- 57.Sodi VL, et al. , mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol Cancer Res, 2015. 13(5): p. 923–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Itkonen HM, et al. , High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics, 2019. 9(8): p. 2183–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deliot N and Constantin B, Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim Biophys Acta, 2015. 1848(10 Pt B): p. 2512–22. [DOI] [PubMed] [Google Scholar]
- 60.Hantute-Ghesquier A, et al. , TRPM Family Channels in Cancer. Pharmaceuticals (Basel), 2018. 11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fiorio Pla A and Gkika D, Emerging role of TRP channels in cell migration: from tumor vascularization to metastasis. Front Physiol, 2013. 4: p. 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu K, et al. , TRPM7 overexpression enhances the cancer stem cell-like and metastatic phenotypes of lung cancer through modulation of the Hsp90alpha/uPA/MMP2 signaling pathway. BMC Cancer, 2018. 18(1): p. 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luanpitpong S, et al. , A novel TRPM7/O-GlcNAc axis mediates tumour cell motility and metastasis by stabilising c-Myc and caveolin-1 in lung carcinoma. Br J Cancer, 2020. 123(8): p. 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das PK, Islam F, and Lam AK, The Roles of Cancer Stem Cells and Therapy Resistance in Colorectal Carcinoma. Cells, 2020. 9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma NS, et al. , O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability. Theranostics, 2019. 9(12): p. 3410–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akella NM, et al. , O-GlcNAc Transferase Regulates Cancer Stem-like Potential of Breast Cancer Cells. Mol Cancer Res, 2020. 18(4): p. 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deniaud E, et al. , Overexpression of Sp1 transcription factor induces apoptosis. Oncogene, 2006. 25(53): p. 7096–105. [DOI] [PubMed] [Google Scholar]
- 68.Sen N, Gui B, and Kumar R, Role of MTA1 in cancer progression and metastasis. Cancer Metastasis Rev, 2014. 33(4): p. 879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie X, et al. , O-GlcNAc modification regulates MTA1 transcriptional activity during breast cancer cell genotoxic adaptation. Biochim Biophys Acta Gen Subj, 2021. 1865(8): p. 129930. [DOI] [PubMed] [Google Scholar]
- 70.Li DQ, et al. , Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer Res, 2012. 72(2): p. 387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao J, et al. , Proteomic analysis of the OGT interactome: novel links to epithelial-mesenchymal transition and metastasis of cervical cancer. Carcinogenesis, 2018. 39(10): p. 1222–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.D’Errico I, et al. , Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC1-alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc Natl Acad Sci U S A, 2011. 108(16): p. 6603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Housley MP, et al. , A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem, 2009. 284(8): p. 514857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu X, et al. , RANBP2 Activates O-GlcNAcylation through Inducing CEBPalpha-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes. Cancers (Basel), 2021. 13(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruan HB, et al. , O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab, 2012. 16(2): p. 226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim YJ, et al. , O-GlcNAc stabilizes SMAD4 by inhibiting GSK-3beta-mediated proteasomal degradation. Sci Rep, 2020. 10(1): p. 19908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carvalho-Cruz P, et al. , Cellular glycosylation senses metabolic changes and modulates cell plasticity during epithelial to mesenchymal transition. Dev Dyn, 2018. 247(3): p. 481–491. [DOI] [PubMed] [Google Scholar]
- 78.Cork GK, Thompson J, and Slawson C, Real Talk: The Inter-play Between the mTOR, AMPK, and Hexosamine Biosynthetic Pathways in Cell Signaling. Front Endocrinol (Lausanne), 2018. 9: p. 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hart GW, Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front Endocrinol (Lausanne), 2014. 5: p. 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jozwiak P, et al. , Mitochondrial O-GlcNAc Transferase Interacts with and Modifies Many Proteins and Its Up-Regulation Affects Mitochondrial Function and Cellular Energy Homeostasis. Cancers (Basel), 2021. 13(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gallagher EJ and LeRoith D, Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol Rev, 2015. 95(3): p. 727–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Larsson SC, Mantzoros CS, and Wolk A, Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J Cancer, 2007. 121(4): p. 856–62. [DOI] [PubMed] [Google Scholar]
- 83.Peairs KS, et al. , Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol, 2011. 29(1): p. 40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alsheikh HAM, et al. , Normalizing glucose levels reconfigures the mammary tumor immune and metabolic microenvironment and decreases metastatic seeding. Cancer Lett, 2021. 517: p. 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Z, et al. , O-GlcNAcase Expression is Sensitive to Changes in O-GlcNAc Homeostasis. Front Endocrinol (Lausanne), 2014. 5: p. 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Decourcelle A, et al. , Evidence of a compensatory regulation of colonic O-GlcNAc transferase and O-GlcNAcase expression in response to disruption of O-GlcNAc homeostasis. Biochem Biophys Res Commun, 2020. 521(1): p. 125–130. [DOI] [PubMed] [Google Scholar]
- 87.Lin CH, et al. , Feedback Regulation of O-GlcNAc Transferase through Translation Control to Maintain Intracellular O-GlcNAc Homeostasis. Int J Mol Sci, 2021. 22(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Warburg O, Wind F, and Negelein E, The Metabolism of Tumors in the Body. J Gen Physiol, 1927. 8(6): p. 519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nie H, et al. , O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat Commun, 2020. 11(1): p. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lei Y, et al. , O-GlcNAcylation of PFKFB3 is required for tumor cell proliferation under hypoxia. Oncogenesis, 2020. 9(2): p. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou L, et al. , Glutamine-fructose-6-phosphate transaminase 2 (GFPT2) promotes the EMT of serous ovarian cancer by activating the hexosamine biosynthetic pathway to increase the nuclear location of beta-catenin. Pathol Res Pract, 2019. 215(12): p. 152681. [DOI] [PubMed] [Google Scholar]
- 92.Singh JP, et al. , O-GlcNAcase targets pyruvate kinase M2 to regulate tumor growth. Oncogene, 2020. 39(3): p. 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsieh TJ, et al. , Suppression of Glutamine:fructose-6-phosphate amidotransferase-1 inhibits adipogenesis in 3T3-L1 adipocytes. J Cell Physiol, 2012. 227(1): p. 108–15. [DOI] [PubMed] [Google Scholar]
- 94.Baldini SF, et al. , The Nutrient-Dependent O-GlcNAc Modification Controls the Expression of Liver Fatty Acid Synthase. J Mol Biol, 2016. 428(16): p. 3295–3304. [DOI] [PubMed] [Google Scholar]
- 95.Sodi VL, et al. , Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene, 2018. 37(7): p. 924–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raab S, et al. , Dual regulation of fatty acid synthase (FASN) expression by O-GlcNAc transferase (OGT) and mTOR pathway in proliferating liver cancer cells. Cell Mol Life Sci, 2021. 78(13): p. 5397–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Very N, et al. , Cross regulation between mTOR signaling and O-GlcNAcylation. J Bioenerg Biomembr, 2018. 50(3): p. 213–222. [DOI] [PubMed] [Google Scholar]
- 98.Tan W, et al. , Posttranscriptional regulation of de novo lipogenesis by glucose-induced O-GlcNAcylation. Mol Cell, 2021. 81(9): p. 1890–1904 e7. [DOI] [PubMed] [Google Scholar]
- 99.Wang J, et al. , The positive feedback between ACSL4 expression and O-GlcNAcylation contributes to the growth and survival of hepatocellular carcinoma. Aging (Albany NY), 2020. 12(9): p. 7786–7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen Y, et al. , O-GlcNAcylation Enhances NUSAP1 Stability and Promotes Bladder Cancer Aggressiveness. Onco Targets Ther, 2021. 14: p. 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu M, et al. , O-GlcNAcylation of ITGA5 facilitates the occurrence and development of colorectal cancer. Exp Cell Res, 2019. 382(2): p. 111464. [DOI] [PubMed] [Google Scholar]
- 102.Mason B, et al. , Fbxl17 is rearranged in breast cancer and loss of its activity leads to increased global O-GlcNAcylation. Cell Mol Life Sci, 2020. 77(13): p. 2605–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu W, et al. , O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J Hepatol, 2017. 67(2): p. 310–320. [DOI] [PubMed] [Google Scholar]
- 104.Zhao Z, et al. , [MicroRNA-424 inhibits autophagy and proliferation of hepatocellular carcinoma cells by targeting ATG14]. Nan Fang Yi Ke Da Xue Xue Bao, 2021. 41(7): p. 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu J, et al. , miR-424–5p represses the metastasis and invasion of intrahepatic cholangiocarcinoma by targeting ARK5. Int J Biol Sci, 2019. 15(8): p. 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ye Z, et al. , LINC00922 promotes the proliferation, migration, invasion and EMT process of liver cancer cells by regulating miR-424–5p/ARK5. Mol Cell Biochem, 2021. 476(10): p. 3757–3769. [DOI] [PubMed] [Google Scholar]
- 107.Vaiana CA, Kurcon T, and Mahal LK, MicroRNA-424 Predicts a Role for beta-1,4 Branched Glycosylation in Cell Cycle Progression. J Biol Chem, 2016. 291(3): p. 1529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feng D, et al. , O-GlcNAcylation of RAF1 increases its stabilization and induces the renal fibrosis. Biochim Biophys Acta Mol Basis Dis, 2020. 1866(3): p. 165556. [DOI] [PubMed] [Google Scholar]
- 109.Ning D, et al. , The crosstalk network of XIST/miR-424–5p/OGT mediates RAF1 glycosylation and participates in the progression of liver cancer. Liver Int, 2021. 41(8): p. 1933–1944. [DOI] [PubMed] [Google Scholar]
- 110.Qiang A, Slawson C, and Fields PE, The Role of O-GlcNAcylation in Immune Cell Activation. Front Endocrinol (Lausanne), 2021. 12: p. 596617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loos JA and Roos D, Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. 3. Stimulation by tuberculin and allogenic cells. Exp Cell Res, 1973. 79(1): p. 136–42. [PubMed] [Google Scholar]
- 112.Nakaya M, et al. , Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity, 2014. 40(5): p. 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jones RG and Thompson CB, Revving the engine: signal transduction fuels T cell activation. Immunity, 2007. 27(2): p. 173–8. [DOI] [PubMed] [Google Scholar]
- 114.DeBerardinis RJ and Cheng T, Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene, 2010. 29(3): p. 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Abramowitz LK, et al. , Blocked O-GlcNAc cycling disrupts mouse hematopoeitic stem cell maintenance and early T cell development. Sci Rep, 2019. 9(1): p. 12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Golks A, et al. , Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J, 2007. 26(20): p. 4368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lund PJ, Elias JE, and Davis MM, Global Analysis of O-GlcNAc Glycoproteins in Activated Human T Cells. J Immunol, 2016. 197(8): p. 3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Machacek M, et al. , Elevated O-GlcNAcylation enhances pro-inflammatory Th17 function by altering the intracellular lipid microenvironment. J Biol Chem, 2019. 294(22): p. 8973–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu B, et al. , The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat Commun, 2019. 10(1): p. 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang WH, et al. , NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U S A, 2008. 105(45): p. 17345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oh H and Ghosh S, NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev, 2013. 252(1): p. 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vaeth M and Feske S, NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Res, 2018. 7: p. 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li X, et al. , Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med, 2017. 214(4): p. 1093–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X, et al. , O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity, 2019. 50(4): p. 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hwang JS, et al. , Lipopolysaccharide (LPS)-stimulated iNOS Induction Is Increased by Glucosamine under Normal Glucose Conditions but Is Inhibited by Glucosamine under High Glucose Conditions in Macrophage Cells. J Biol Chem, 2017. 292(5): p. 1724–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jha AK, et al. , Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity, 2015. 42(3): p. 419–30. [DOI] [PubMed] [Google Scholar]
- 127.Mazzone M, Menga A, and Castegna A, Metabolism and TAM functions-it takes two to tango. FEBS J, 2018. 285(4): p. 700–716. [DOI] [PubMed] [Google Scholar]
- 128.Surdziel E, et al. , Multidimensional pooled shRNA screens in human THP-1 cells identify candidate modulators of macrophage polarization. PLoS One, 2017. 12(8): p. e0183679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rabold K, et al. , Cellular metabolism of tumor-associated macrophages - functional impact and consequences. FEBS Lett, 2017. 591(19): p. 3022–3041. [DOI] [PubMed] [Google Scholar]
- 130.Aras S and Zaidi MR, TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer, 2017. 117(11): p. 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goswami KK, et al. , Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol, 2017. 316: p. 1–10. [DOI] [PubMed] [Google Scholar]
- 132.Mantovani A, et al. , Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol, 2013. 229(2): p. 176–85. [DOI] [PubMed] [Google Scholar]
- 133.Poh AR and Ernst M, Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol, 2018. 8: p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moriwaki K and Asahi M, Augmented TME O-GlcNAcylation Promotes Tumor Proliferation through the Inhibition of p38 MAPK. Mol Cancer Res, 2017. 15(9): p. 1287–1298. [DOI] [PubMed] [Google Scholar]
- 135.Hinshaw DC, et al. , Hedgehog signaling regulates metabolism and polarization of mammary tumor-associated macrophages. Cancer Res, 2021. [DOI] [PMC free article] [PubMed]
- 136.Rodrigues Mantuano N, et al. , Hyperglycemia Enhances Cancer Immune Evasion by Inducing Alternative Macrophage Polarization through Increased O-GlcNAcylation. Cancer Immunol Res, 2020. 8(10): p. 1262–1272. [DOI] [PubMed] [Google Scholar]
- 137.Yang Y, et al. , OGT suppresses S6K1-mediated macrophage inflammation and metabolic disturbance. Proc Natl Acad Sci U S A, 2020. 117(28): p. 16616–16625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Malaker SA, et al. , Identification of Glycopeptides as Posttranslationally Modified Neoantigens in Leukemia. Cancer Immunol Res, 2017. 5(5): p. 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kampa-Schittenhelm KM, et al. , Epigenetic activation of O-linked beta-N-acetylglucosamine transferase overrides the differentiation blockage in acute leukemia. EBioMedicine, 2020. 54: p. 102678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang L, et al. , ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod Pathol, 2014. 27(5): p. 775–83. [DOI] [PubMed] [Google Scholar]
- 141.Zhang G, et al. , Esophageal cancer tumorspheres involve cancer stem-like populations with elevated aldehyde dehydrogenase enzymatic activity. Mol Med Rep, 2012. 6(3): p. 519–24. [DOI] [PubMed] [Google Scholar]
- 142.Yuan Y, et al. , Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8(+) T cells. Cancer Lett, 2021. 500: p. 98–106. [DOI] [PubMed] [Google Scholar]
- 143.Goodman A, Patel SP, and Kurzrock R, PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol, 2017. 14(4): p. 203–220. [DOI] [PubMed] [Google Scholar]
- 144.Trapannone R, Rafie K, and van Aalten DM, O-GlcNAc transferase inhibitors: current tools and future challenges. Biochem Soc Trans, 2016. 44(1): p. 88–93. [DOI] [PubMed] [Google Scholar]
- 145.Walter LA, et al. , Inhibiting the Hexosamine Biosynthetic Pathway Lowers O-GlcNAcylation Levels and Sensitizes Cancer to Environmental Stress. Biochemistry, 2020. 59(34): p. 3169–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Asthana A, et al. , Hexosamine biosynthetic pathway inhibition leads to AML cell differentiation and cell death. Molecular cancer therapeutics, 2018. 17(10): p. 2226–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Deng X, et al. , ROCK2 mediates osteosarcoma progression and TRAIL resistance by modulating O-GlcNAc transferase degradation. Am J Cancer Res, 2020. 10(3): p. 781–798. [PMC free article] [PubMed] [Google Scholar]
- 148.Seo HG, et al. , Mutual regulation between OGT and XIAP to control colon cancer cell growth and invasion. Cell Death Dis, 2020. 11(9): p. 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Itkonen HM, et al. , Inhibition of O-GlcNAc Transferase Renders Prostate Cancer Cells Dependent on CDK9. Mol Cancer Res, 2020. 18(10): p. 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee SJ and Kwon OS, O-GlcNAc Transferase Inhibitor Synergistically Enhances Doxorubicin-Induced Apoptosis in HepG2 Cells. Cancers (Basel), 2020. 12(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barkovskaya A, et al. , Inhibition of O-GlcNAc transferase activates tumor-suppressor gene expression in tamoxifen-resistant breast cancer cells. Sci Rep, 2020. 10(1): p. 16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shou J, et al. , Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst, 2004. 96(12): p. 926–35. [DOI] [PubMed] [Google Scholar]
- 153.Knowlden JM, et al. , Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology, 2003. 144(3): p. 1032–44. [DOI] [PubMed] [Google Scholar]
- 154.Yang X and Qian K, Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol, 2017. 18(7): p. 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Burt RA, et al. , Novel antibodies for the simple and efficient enrichment of native O-GlcNAc modified peptides. Mol Cell Proteomics, 2021: p. 100167. [DOI] [PMC free article] [PubMed]
- 156.Ju Kim E, O-GlcNAc Transferase: Structural Characteristics, Catalytic Mechanism and Small-Molecule Inhibitors. Chembiochem, 2020. 21(21): p. 3026–3035. [DOI] [PubMed] [Google Scholar]
- 157.O’Donnell N, et al. , Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol, 2004. 24(4): p. 1680–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Medford HM and Marsh SA, The role of O-GlcNAc transferase in regulating the gene transcription of developing and failing hearts. Future Cardiol, 2014. 10(6): p. 801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Alejandro EU, et al. , Disruption of O-linked N-Acetylglucosamine Signaling Induces ER Stress and beta Cell Failure. Cell Rep, 2015. 13(11): p. 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Swamy M, et al. , Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol, 2016. 17(6): p. 712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wu JL, et al. , O-GlcNAcylation is required for B cell homeostasis and antibody responses. Nat Commun, 2017. 8(1): p. 1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ruan HB, et al. , O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell, 2014. 159(2): p. 306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Su C and Schwarz TL, O-GlcNAc Transferase Is Essential for Sensory Neuron Survival and Maintenance. J Neurosci, 2017. 37(8): p. 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lagerlof O, et al. , The nutrient sensor OGT in PVN neurons regulates feeding. Science, 2016. 351(6279): p. 1293–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li M-D, et al. , Adipocyte OGT governs diet-induced hyperphagia and obesity. Nature Communications, 2018. 9(1): p. 5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhao M, et al. , Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol Med, 2018. 10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]