

Abstract
Objective.
The objective of this exploratory study was to determine if perturbations in gut microbial composition and the gut metabolome could be linked to individuals with obesity and osteoarthritis (OA).
Methods.
Fecal samples were collected from obese individuals diagnosed with radiographic hand plus knee OA (n=59), defined as involvement of at least 3 joints across both hands, and a Kellgren-Lawrence (KL) grade 2–4 (or total knee replacement) in at least one knee. Controls (n=33) were without hand OA and with KL grade 0–1 knees. Fecal metabolomes were analyzed by a UHPLC/Q Exactive HFx mass spectrometer. Microbiome composition was determined in fecal samples by 16S ribosomal RNA amplicon sequencing (rRNA-seq). Stepwise logistic regression models were built to determine microbiome and/or metabolic characteristics of OA.
Results.
Untargeted metabolomics analysis indicated that OA cases had significantly higher levels of di- and tripeptides and significant perturbations in microbial metabolites including propionic acid, indoles, and other tryptophan metabolites. Pathway analysis revealed several significantly perturbed pathways associated with OA including leukotriene metabolism, amino acid metabolism and fatty acid utilization. Logistic regression models selected metabolites associated with the gut microbiota and leaky gut syndrome as significant predictors of OA status, particularly when combined with the rRNA-seq data.
Conclusions.
Adults with obesity and knee plus hand OA have distinct fecal metabolomes characterized by increased products of proteolysis, perturbations in leukotriene metabolism, and changes in microbial metabolites compared with controls. These metabolic perturbations indicate a possible role of dysregulated proteolysis in OA.
Keywords: obesity, osteoarthritis, microbiome, metabolomics, proteolysis
INTRODUCTION
Osteoarthritis (OA) is the most common form of arthritis and a leading cause of disability, affecting over 300 million people worldwide1. Major risk factors for OA include age, obesity, prior joint injuries, and genetics2,3. In an earlier study, we found that metabolomics could distinguish urine samples obtained from individuals with radiographic knee OA and obesity who progressed in severity of disease over an 18 month period, compared with individuals who were non-progressors, suggesting that metabolism plays a role in OA development4. That study showed that hippurate and trigonelline were important metabolites in differentiating progressors and non-progressors. These compounds are potential markers of gut microbiome activity5, indicating that the microbiome may be playing a role in the pathogenesis of OA. Emerging evidence shows that the gut microbiome may be an important factor in predicting and/or driving other forms of arthritis such rheumatoid arthritis (RA)6. Indeed, a dysbiosis of microbiome composition or activity has been proposed as a reason for the increased intestinal permeability seen in RA, explaining the characteristic rise in inflammation and joint destruction seen in this disease. If a similar mechanism exists for OA, this will provide an avenue to discover new mediators of OA and to identify new treatment options.
The investigation presented herein describes an untargeted metabolomics and microbiome analysis of fecal samples collected from individuals with obesity diagnosed with knee plus hand OA (OA cases) and controls with obesity but without hand or knee OA. This was a sub-study of a recently reported study (“parent study”) that compared the fecal microbiota, plasma cytokines, and serum lipopolysaccharide (LPS) levels between the OA cases and controls and examined the effect on the development of OA in germ-free mice transplanted with fecal samples from the cases and controls7. As noted in the parent study, we decided to enroll individuals with knee plus hand OA in attempt to enrich the study cohort with individuals more likely to have systemic metabolic factors related to the gut microbiome contributing to the development of multi-joint OA. Our goal here was to define fecal metabolic profiles that differentiated OA cases and controls to provide insight towards undiscovered mechanisms of OA and/or potential therapeutic targets. Furthermore, we used statistical modeling approaches to evaluate the ability of the combination of metabolomics and microbiome data to determine factors that influence OA status.
METHODS
Participants
This exploratory study used data and biospecimens collected from a subgroup of participants in the Johnston County Osteoarthritis Study (JoCo OA) who were enrolled in a study at the time of the most recent follow-up evaluation to determine the contribution of the gut microbiota to OA associated with obesity (specific details of the JoCo OA study can be found elsewhere8). The study was approved by the University of North Carolina (UNC) Institutional Review Board (IRB#15–1834). Details of subject recruitment, fecal sample collection and fecal microbiome analysis have been recently reported elsewhere7. Briefly, all participants (n=92) were older than 45 years of age and had obesity (BMI ≥ 30 kg/m2). OA cases had clinical and/or radiographic hand plus radiographic knee OA (defined as involvement of at least three joints across both hands, and a Kellgren-Lawrence (KL) grade 2–4 in at least one knee) while controls had no hand OA and KL grade 0–1 knees. When knee radiographs were evaluated at the time of recruitment, 50 participants were classified as cases and 42 as controls. The radiographs were subsequently evaluated by the study radiologist who determined 9 participants recruited as controls had KL grade of 2 rather than 1 in one knee. For the purposes of the present study, these individuals were reclassified as cases resulting in 59 cases and 33 controls. Descriptive statistics for participant characteristics were calculated using means and standard deviations for continuous variables and frequencies and percentages for categorical variables. Group differences were determined via t-tests or chi-square test or Fisher’s exact test as appropriate. Stool samples were collected by the participants using a collection kit and blood samples and clinical data were obtained following the JoCo OA study protocol8. Fecal microbiome analysis was performed by Illumina MiSeq 16S ribosomal RNA (rRNA) amplicon sequencing in the UNC Microbiome Core as described9–11. Taxa were summarized based at their taxonomic levels with QIIME 1.9.012.
Untargeted Fecal Metabolomics Analysis
Details of sample preparation, data acquisition and preprocessing are provided in the Supplementary Methods.
Multivariate and univariate statistical analysis
After filtering and pre-processing, picks were normalized and used for multivariate and univariate analyses13. Principal component analysis (PCA) was performed to assess data quality based on the tight clustering of the QC samples14. Partial least square discriminant analysis (PLS-DA) was used to identify differentiating peaks between OA cases and controls quantified by variable importance to projection (VIP) score for each peak15. VIP score is a measure of the variable’s importance in differentiating the phenotype in the PLS-DA model; higher values indicate that a variable makes more contribution differentiating the phenotype. PCA and PLS-DA were performed using SIMCA 15 (Sartorius Stedim Data Analytics AB, Umeå, Sweden) on the normalized, preprocessed untargeted data. For PLS-DA models, the automatic transformation function in SIMCA was used which log transforms variables with a high degree of skewness (sTest < −2 or > 2) or when 0 ≤ min/max < 0.1. Data were mean centered and pareto scaled as described16. Peaks with a VIP ≥ 1 were considered as important contributors to the model17. Models were evaluated using R2 which measures the percentage of the variance explained by the model, and Q2 which is a measure of predictive ability based on a 7-fold cross validation. R2X and R2Y represent the fraction of variance of the X and Y matrix. Two-sided Wilcoxon Rank Sum t-tests with the Satterthwaite correction for unequal variances were used to determine peaks that were statistically significant between OA cases and controls. P-values were not adjusted for multiple testing due to the exploratory, rather than confirmatory, nature of this study18. This discovery, hypothesis-generating approach has been utilized by our group in recent publications19,20. Fold changes were calculated using mean values for each peak. A positive fold change indicates a higher signal intensity in OA cases as compared to controls. Statistical analyses were conducted using SAS 9.4 (SAS Institute Inc, Cary, NC).
Identification and annotation of signals
Identification or annotation of peaks was performed through Progenesis QI (version 2.1, Waters Corporation) by matching to an in-house physical standards library (consisting of approximately 2000 compound standards run under the same instrument conditions as the study samples) as well as publicly available databases (HMDB, NIST, and METLIN)21,22. Assignments were made based on matches to exact mass (MS), MS/MS fragmentation pattern, isotopic ion pattern, or retention time (RT, for in-house library standards only). Evidence for identification or annotation is denoted by an ontology system developed by our lab as described previously19,20.
Pathway analysis
Pathway analysis was performed using the mummichog algorithm in Metaboanalyst’s MS Peaks to Pathways module23. All normalized features remaining after preprocessing were uploaded with mass-to-charge ratio (m/z), retention time, the p-value, and fold change between OA cases and controls for each peak. A p-value cutoff of 0.01 was used for the size of the permutation group that the algorithm used for selecting significant features for metabolite matching. A 3 ppm tolerance was used for mass accuracy for annotating peaks and identifying candidate pathways. All possible metabolites that were matched by m/z were searched in the Homo sapiens (human) [MFN] pathway library. The experimental list of metabolites was compared to a null distribution of randomly generated m/z features from the reference library to determine pathway significance24. Significance is reported as uncorrected p-values.
Logistic Regression modeling
Logistic regression models were built using SAS 9.4 to examine the odds of an OA diagnosis based on subject characteristics, metabolite peaks, and microbiome taxa. These variables were considered candidate predictors if there was an association with OA status at p < 0.10 based on either the t-test with Satterthwaite correction or chi-square test. Continuous variables were standardized prior to modeling. For each logistic regression model, a stepwise selection procedure starting with the candidate predictors used an entry criterion of p < 0.1 and a removal criterion of p > 0.05 to select the final models. Goodness of fit for each final model was assessed using the Hosmer-Lemeshow goodness-of-fit test. If the Hosmer-Lemeshow test was statistically significant (p < 0.05) then the distribution of the continuous variables was examined to determine if categorizing any of the variables at the median would improve the goodness of fit. The receiver operating characteristic curves (ROCs) were generated for each model, and the area under the curve (AUC) and the 95% confidence interval were calculated. Each model’s AUC was compared with the subject characteristics only model to determine whether the AUCs were significantly different using the “ROCCONTRAST” statement in SAS PROC LOGISTIC.
RESULTS
Participant demographics and clinical data
Characteristics of the 92 study participants are provided in Table 1. The two groups (OA cases vs controls) did not differ significantly in terms of age, gender, or race. BMI was significantly higher (p= 0.002) in OA cases (35.9 ± 4.2 kg/m2) than controls (33.3 ± 3.3 kg/m2). OA cases had a significantly (p<0.001) higher KL score compared to controls and 14 OA cases had undergone unilateral knee replacement surgery, consistent with our study design. There were no between group differences in use of NSAIDs, proton pump inhibitors, laxatives, or opioids (Supplementary Table S1).
Table 1:
Subject Characteristics
| Control (n=33) | Case (n=59) | p-value* | |
|---|---|---|---|
|
| |||
| Age years, mean (SD) | 71.0 (6.6) | 73.2 (6.8) | 0.139 |
| Female, N (%) | 21 (63.6%) | 48 (81.4%) | 0.060 |
| Race, N (%) | 0.274 | ||
| Black | 15 (45.5%) | 20 (33.9%) | |
| White | 18 (54.5%) | 39 (66.1%) | |
| BMI, mean (SD) | 33.3 (3.3) | 35.9 (4.2) | 0.002 |
| Maximum KL score, n (%) | <0.001 | ||
| 0 | 1 (3.0%) | 0 | |
| 1 | 32 (97.0%) | 0 | |
| 2 | 0 | 11 (18.6%) | |
| 3 | 0 | 13 (22.0%) | |
| 4 | 0 | 21 (35.6%) | |
| Replacement | 0 | 14 (23.7%) | |
BMI, body mass index; KL, Kellgren Lawrence
Continuous variables tested using t-test with Satterthwaite correction, categorical variables tested using chi-square test or Fisher’s Exact Test.
Metabolomics analysis and OA status
After filtering and pre-processing, 18,850 peaks were normalized and used for downstream analyses. The unsupervised PCA shows that the QC study pools clustered in the center of the study samples from which they were derived (Supplemental Figure S1). Supervised multivariate analysis by PLS-DA of the untargeted data was able to differentiate OA cases and controls with good model statistics (R2Y = 0.98, R2X= 0.325, Q2 = 0.64) (Figure 1). Table 2 shows the top 100 most statistically significant important metabolites by p-value based on t-test between OA cases and controls that could be identified or annotated using the in-house physical standards library or publicly available MS databases. The majority of annotated peaks were di- and tri-peptides, all of which were higher in OA cases than controls. Many of these top 100 metabolites were also differentiators of OA cases and controls in the PLS-DA model as indicated by a VIP > 1 (Table 2). Additional identifications and annotations are provided for all peaks with a p<0.05 between OA cases and controls (Supplementary Table S2). Although we use p-values to indicate peaks/metabolites of interest, those with a VIP ≥ 1 and/or large fold changes (absolute value ≥ 2) are particularly important for differentiating OA cases and controls. The MS Peaks to Pathways module in Metaboanalyst, which uses retention time and m/z values rather than identifications/annotations, identified several metabolic pathways that were significantly perturbed between OA cases and controls. The two most significantly perturbed pathways (Figure 2) included leukotriene metabolism (p < 0.0001) and tryptophan metabolism (p < 0.0001). Other significant (p < 0.05) pathway perturbations included those involved in fatty acid synthesis/metabolism (R Group Synthesis, Glycosylphosphatidylinositol (GPI)-anchor biosynthesis), amino acid metabolism (glycine, serine, alanine, and threonine metabolism) and pyruvate metabolism.
Figure 1:
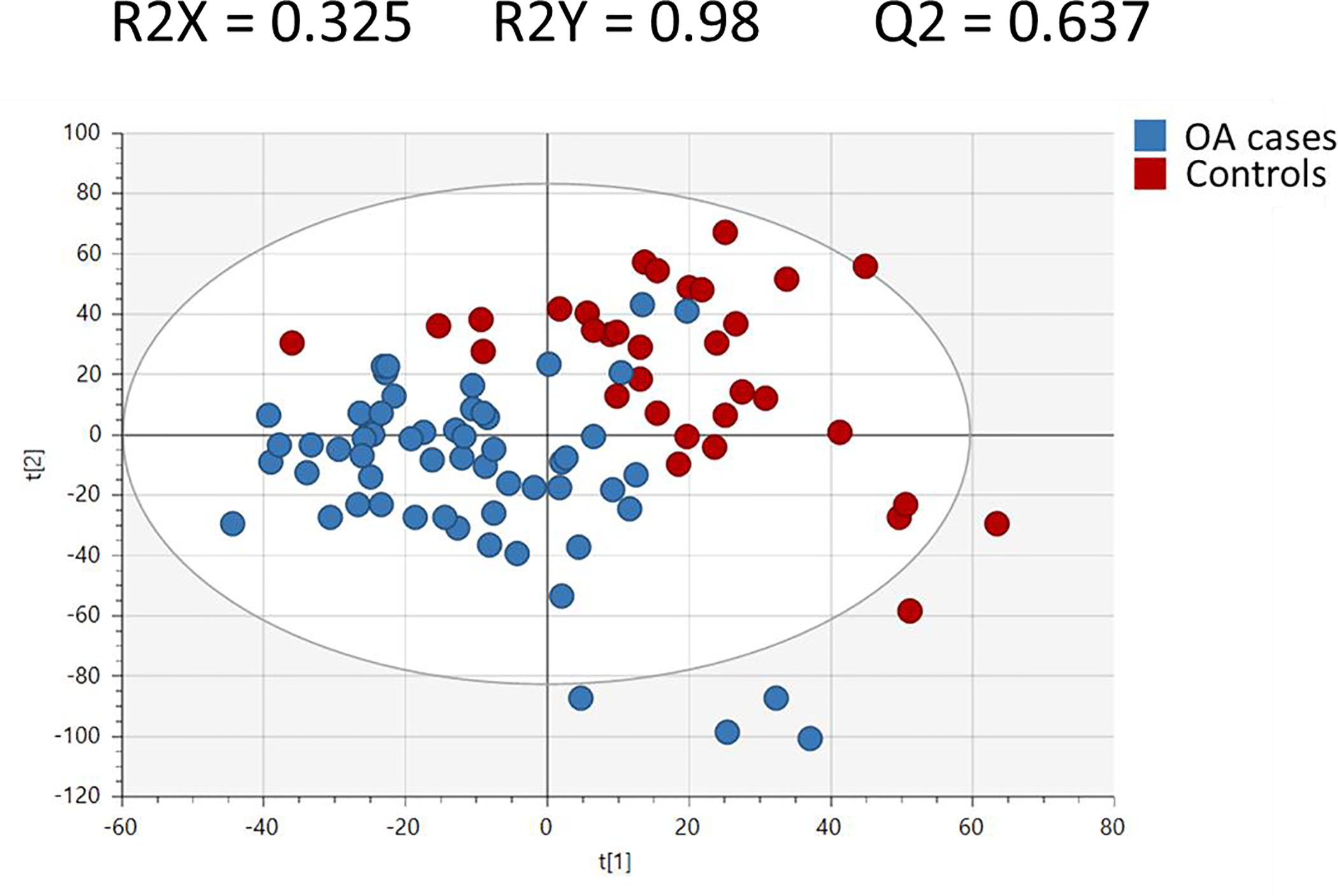
Partial Least Squares-Determinant Analysis (PLS-DA) plot demonstrating the separation of OA cases and controls using supervised multivariate analysis. t[1] and t[2] denote the first and second components respectively. R2 is the percentage of the variance explained by the model. Q2 is a measure of predictive ability based on a 7-fold cross validation. R2X and R2Y refer to the amount of variation of the X matrix (metabolites) and Y matrix (OA status) explained by the model.
Table 2:
Top 100 Assigned Metabolites by P-Value Between Cases and Controls
| Compound Name | Ontology Level | VIP | p-value | Fold Change |
|---|---|---|---|---|
| Sinapyl Alcohol | PDd | 1.5 | 5.4E-06 | 2.0 |
| Tsangane L 3-Glucoside | PDd | 1.2 | 1.2E-05 | 1.8 |
| Ser Thr Tyr | PDa | 1.3 | 1.8E-05 | 1.9 |
| Tyr-Glu-Lys | PDa | 1.1 | 7.4E-05 | 1.8 |
| Dynorphin B (10–13) | PDc | 1.2 | 7.4E-05 | 2.7 |
| DG(14:0/18:4(6Z,9Z,12Z,15Z)/0:0) | PDb | 1.1 | 9.6E-05 | 1.6 |
| Ala Val Leu | PDa | 1.1 | 1.1E-04 | 1.6 |
| N(6)-Methyllysine | PDc | 1.7 | 1.3E-04 | 1.8 |
| Ile-Glu-Arg | PDa | 1.0 | 1.5E-04 | 1.8 |
| Alanyl-Lysine | PDd | 1.4 | 1.5E-04 | 2.0 |
| Ala-Leu-Lys | PDa | 1.0 | 2.0E-04 | 1.5 |
| Val Thr Tyr | PDa | 0.9 | 2.1E-04 | 1.5 |
| Palmatine | PDd | 1.2 | 2.1E-04 | 1.7 |
| L-Tryptophan | OL2b | 1.4 | 2.2E-04 | 1.8 |
| His Val Tyr | PDa | 1.3 | 2.4E-04 | 1.8 |
| Phe-Gln-Lys | PDa | 1.2 | 2.6E-04 | 1.8 |
| Thr Leu Lys | PDa | 1.2 | 3.1E-04 | 1.6 |
| Ser-Leu-Lys | PDc | 1.5 | 3.8E-04 | 2.1 |
| Norzolmitripan | PDc | 2.3 | 4.0E-04 | 4.3 |
| Ser-Leu-Lys | PDc | 1.0 | 4.0E-04 | 1.4 |
| Valyl-Asparagine | PDc | 1.0 | 4.0E-04 | 1.5 |
| Lysopc(14:1(9Z)) | PDc | 1.3 | 4.7E-04 | 2.3 |
| Gln Phe Leu | PDa | 0.9 | 4.8E-04 | 1.4 |
| Ala Ile Leu Asp | PDc | 0.9 | 5.1E-04 | 1.4 |
| Leu-Thr-Arg | PDc | 1.5 | 5.8E-04 | 1.8 |
| Gly Ala Ile | PDa | 1.0 | 6.1E-04 | 1.6 |
| Met Tyr Leu | PDa | 1.2 | 6.1E-04 | 1.5 |
| Thr-Phe-Lys | PDc | 1.2 | 6.1E-04 | 1.9 |
| Glu Leu Trp | PDa | 0.7 | 6.1E-04 | 1.7 |
| Val Asn Lys | PDc | 1.1 | 6.2E-04 | 1.8 |
| Leucyl-Glycine | PDc | 1.5 | 6.7E-04 | 1.5 |
| Ser Val Lys | PDd | 1.0 | 7.7E-04 | 1.6 |
| Ile-Gln-Arg | PDc | 0.9 | 8.0E-04 | 1.5 |
| Met-Ile-Lys | PDa | 1.1 | 8.5E-04 | 1.7 |
| Phe-Glu-Lys | PDc | 1.2 | 8.7E-04 | 1.7 |
| Leu-Thr-Lys | PDc | 1.1 | 8.7E-04 | 1.5 |
| Gln Met Phe | PDa | 1.0 | 8.9E-04 | 1.5 |
| Leu-Asp-Arg | PDa | 1.0 | 9.0E-04 | 1.8 |
| 3-[(3-Methylbutyl)Nitrosoamino]-2-Butanone | PDc | 1.4 | 9.1E-04 | 1.6 |
| Dhelwangin | PDb | 1.5 | 9.7E-04 | 2.5 |
| Val-Pro-Lys | PDd | 1.2 | 9.8E-04 | 1.7 |
| Met-Ala-Lys | PDd | 1.4 | 9.9E-04 | 2.2 |
| Gly Tyr Phe | PDa | 0.9 | 1.0E-03 | 1.7 |
| Asn-Ile-Arg | PDa | 1.1 | 1.0E-03 | 2.0 |
| Dynorphin B (10–13) | PDd | 1.2 | 1.0E-03 | 2.8 |
| Phe-Ala-Lys | PDa | 1.2 | 1.1E-03 | 1.7 |
| Leu-Trp-Arg | PDa | 1.6 | 1.1E-03 | 2.9 |
| Val-Gly-Lys | PDd | 1.1 | 1.1E-03 | 1.7 |
| Trp Val Asp | PDa | 1.1 | 1.2E-03 | 1.8 |
| Thr Ile Ile | PDa | 0.9 | 1.2E-03 | 1.5 |
| Ile Thr Phe | PDa | 0.8 | 1.2E-03 | 1.5 |
| Phe-Met-Lys | PDa | 1.3 | 1.2E-03 | 2.6 |
| Tyrosyl-Valine | PDc | 0.8 | 1.3E-03 | 1.6 |
| Asn-Glu-Arg | PDd | 1.4 | 1.3E-03 | 2.0 |
| Cys-Val-Arg | PDd | 1.3 | 1.4E-03 | 3.1 |
| Tyr-Phe-Lys | PDa | 1.2 | 1.4E-03 | 1.7 |
| Leukotriene B4 3-Aminopropylamide | PDd | 0.9 | 1.4E-03 | 1.5 |
| Val Ala Gly Val Ala | PDc | 0.9 | 1.4E-03 | 1.4 |
| His Thr Leu | PDa | 1.0 | 1.4E-03 | 1.5 |
| Trp-Leu-Lys | PDa | 1.3 | 1.5E-03 | 2.1 |
| Gln Leu Lys | PDa | 1.0 | 1.6E-03 | 1.4 |
| N-Succinyl-L,L-2,6-Diaminopimelate | PDc | 1.4 | 1.6E-03 | −1.8 |
| Ile-Leu-Arg | PDa | 0.9 | 1.7E-03 | 1.7 |
| Gly Leu Tyr | PDa | 0.8 | 1.7E-03 | 1.4 |
| Trp Thr Phe | PDa | 2.9 | 1.7E-03 | 2.7 |
| His Phe Phe | PDa | 1.0 | 1.8E-03 | 1.7 |
| Ala Val Lys | PDc | 1.0 | 1.8E-03 | 1.8 |
| Gln-Asp-Arg | PDc | 1.1 | 1.8E-03 | 1.8 |
| His-Val-Arg | PDd | 1.0 | 1.9E-03 | 2.1 |
| Val Tyr | PDa | 0.8 | 1.9E-03 | 1.6 |
| Leu-Glu-Arg | PDa | 0.9 | 1.9E-03 | 1.8 |
| Penicilloic Acid | PDd | 1.1 | 1.9E-03 | 1.5 |
| Phe-Leu-Arg | PDa | 1.1 | 1.9E-03 | 1.9 |
| Asp-Ala-Arg | PDd | 1.1 | 2.0E-03 | 1.7 |
| Gln Leu Met | PDa | 0.9 | 2.1E-03 | 1.3 |
| Leu Ala Arg Glu Leu | PDd | 1.7 | 2.1E-03 | 3.1 |
| Serotonin | OL2b | 1.6 | 2.2E-03 | 1.7 |
| Trp-Leu-Lys | PDa | 1.6 | 2.2E-03 | 2.0 |
| Trp-Ile-Arg | PDd | 1.3 | 2.3E-03 | 2.2 |
| Ornithine | OL1 | 1.3 | 2.3E-03 | −1.6 |
| Phe-Thr-Arg | PDc | 1.4 | 2.3E-03 | 2.1 |
| Arginine | OL2b | 1.3 | 2.4E-03 | 1.6 |
| Tyr-Gln-Lys | PDd | 1.4 | 2.5E-03 | 2.5 |
| Tyr Thr Thr | PDa | 0.8 | 2.5E-03 | 1.5 |
| Phe Ser Gly | PDc | 1.0 | 2.5E-03 | 1.5 |
| Gly Leu Val | PDa | 0.9 | 2.6E-03 | 1.4 |
| Glycyl-Arginine | PDb | 1.0 | 2.6E-03 | 2.1 |
| Ile-Gln-Arg | PDd | 1.1 | 2.6E-03 | 1.8 |
| Thr Tyr Phe | PDa | 0.9 | 2.7E-03 | 1.6 |
| Leu-Asp-Arg | PDa | 1.0 | 2.7E-03 | 1.5 |
| Indole | PDa | 1.0 | 2.7E-03 | 1.5 |
| Dihydro-2,4-Dimethyl-6-(1-Methylpropyl)-4H-1,3,5-Dithiazine | PDd | 1.8 | 2.8E-03 | 3.8 |
| Phe-Glu-Arg | PDa | 1.3 | 2.8E-03 | 1.8 |
| Valyl-Lysine | PDc | 1.0 | 3.1E-03 | 1.7 |
| Gly Val Ile Gly Ile | PDd | 1.3 | 3.1E-03 | 1.9 |
| Asn Leu Lys | PDa | 0.8 | 3.1E-03 | 1.4 |
| Ile-Leu-Arg | PDa | 1.0 | 3.2E-03 | 2.0 |
| Val-Glu-Lys | PDc | 1.0 | 3.2E-03 | 1.6 |
| Pro-Leu-Lys | PDc | 1.7 | 3.2E-03 | 1.9 |
| Ile Thr Trp | PDa | 1.1 | 3.2E-03 | 1.7 |
VIP (variable importance to projection) scores were calculated using the PLS-DA model from Figure 1. P-values were calculated using a two-sided t-test with Satterthwaite correction. Positive fold changes indicate a higher mean value in subjects with OA compared to non-OA. OL1 refers to an in-house library match by MS, MS/MS, and RT; OL2a refers to an in-house library match by MS and RT; OL2b refers to an in-house library match by MS and MS/MS; PDa refers to a public database match by MS and experimental MS/MS (NIST or METLIN); PDb refers to a public database match by MS and theoretical MS/MS (HMDB); PDc refers to a public database match by MS and isotopic similarity; PDd refers to a public database match by MS only. Ontology levels OL1, OL2a, and OL2b correspond to level 1 of the Metabolomics Standards Initiative (MSI) standards (identification using two independent and orthogonal data), whereas levels PDa, PDb, PDc, and PDd correspond to level 2 of the MSI standards (putatively annotated compounds)59. Repeated metabolite names indicate multiple peaks matched to the same metabolite.
Figure 2.
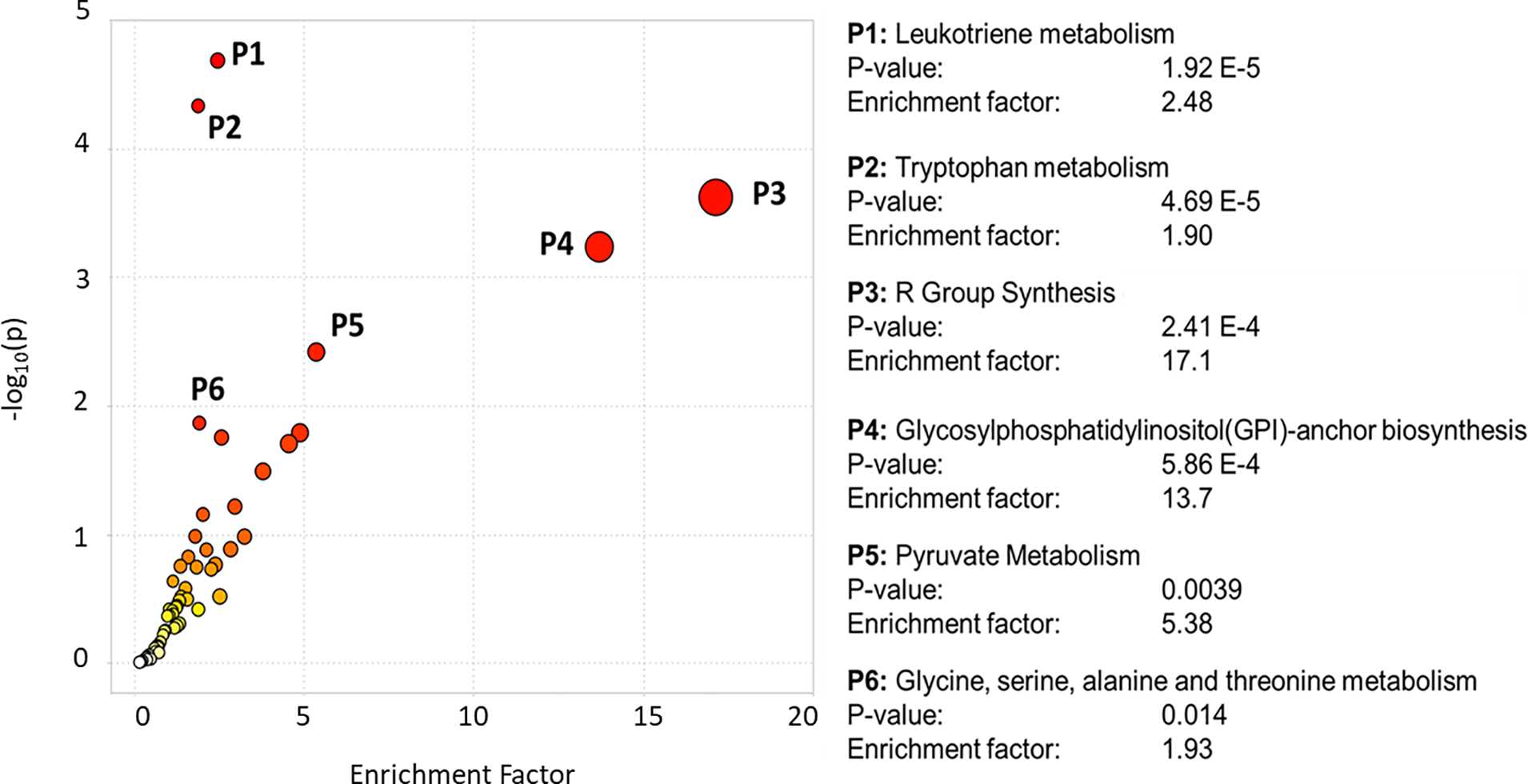
Metabolite pathway enrichment. P-values and fold changes were calculated for OA cases vs controls for over 18,850 peaks, and imported into the “MS Peaks to Pathways” module in Metaboanalyst. A 3 ppm tolerance was used for mass accuracy and a p-value cutoff of 0.01 was used for the mummichog algorithm.
Relationship between fecal metabolites and the fecal microbiome
A list of metabolites was curated from the literature25–27 that have been shown to be direct products of the microbiome, or whose levels are strongly influenced by microbiome activity (Supplementary Table S3). Each of the 18,850 peaks from the normalized untargeted data were searched for any potential matches to these microbiome-related metabolites. Table 3 shows a list of peaks with these identifications/annotations that had a p-value < 0.1. Because this is an exploratory study, a less stringent p-value was used to identify potential differences in these microbiome-related metabolites. Among these compounds are metabolites related to tryptophan metabolism, which agrees with the pathway analysis results. Most of these peaks also had a VIP > 1 (Table 3) in the PLS-DA model, indicating their importance is distinguishing OA cases from controls.
Table 3.
Assignments Related to Microbial Metabolites with p < 0.1 Between OA Cases vs Controls
| Compound Name | Ontology Level | VIP | p-value | Fold Change |
|---|---|---|---|---|
| Indole | PDa | 1.0 | 0.003 | 1.5 |
| Propionic Acid | PDc | 1.7 | 0.007 | −2.6 |
| Indole | PDa | 1.2 | 0.022 | 2.1 |
| Indole-3-Acetate | OL1 | 1.3 | 0.025 | 2.0 |
| Suberate | OL2b | 0.9 | 0.026 | −1.3 |
| Phenylacetate | PDc | 1.0 | 0.030 | 1.5 |
| Beta-L-Fucose | PDc | 0.9 | 0.031 | 2.6 |
| Tryptophanol | PDd | 1.0 | 0.032 | 1.9 |
| Phenylethanolamine | OL2a | 0.8 | 0.034 | 1.3 |
| Hippuric Acid | PDb | 1.5 | 0.034 | −2.4 |
| Pyruvic Acid | PDc | 1.0 | 0.056 | −1.4 |
| Epinephrine Sulfate | PDd | 1.4 | 0.060 | −1.8 |
| Shikimic Acid | OL2a | 0.7 | 0.061 | −1.3 |
| Aspartyl-Serine | PDc | 1.1 | 0.062 | −1.6 |
| Sarcosine | OL2a | 1.0 | 0.071 | 1.5 |
| Indole | PDa | 0.7 | 0.071 | 1.3 |
| P-Hydroxybenzylalcohol | PDc | 0.5 | 0.075 | −1.2 |
| Shikimic Acid | PDc | 1.1 | 0.079 | 1.4 |
| Hypoxanthine | OL1 | 0.7 | 0.080 | −1.3 |
| Hypoxanthine | OL2a | 0.6 | 0.084 | −1.3 |
| 4-Ethylphenol | PDd | 0.5 | 0.084 | −1.2 |
| N-Acetyl-D-Glucosamine | OL1 | 0.7 | 0.084 | −1.3 |
| Beta-D-Glucose | PDc | 1.1 | 0.087 | −1.3 |
| N-Acetyl-D-Glucosamine | OL2a | 0.8 | 0.093 | −1.5 |
| Indolepyruvate | PDb | 0.9 | 0.094 | 15.1 |
| Indoleacetaldehyde | OL2b | 1.6 | 0.094 | 1.2 |
| 3-Methylindole | PDc | 0.7 | 0.096 | −1.3 |
Displayed metabolites are peaks that matched to the microbial metabolite list in Table S3. VIP (variable importance to projection) scores were calculated using the PLS-DA model from Figure 1. P-values were calculated using a two-sided t-test with Satterwaite correction. Positive fold changes indicate a higher value in OA as compared to non-OA. OL1 refers to an in-house library match by MS, MS/MS, and RT; OL2a refers to an in-house library match by MS and RT; OL2b refers to an in-house library match by MS and MS/MS; PDa refers to a public database match by MS and experimental MS/MS (NIST orMETLIN); PDb refers to a public database match by MS and theoretical MS/MS (HMDB); PDc refers to a public database match by MS and isotopic similarity; PDd refers to a public database match by MS only. Ontology levels OL1, OL2a, and OL2b correspond to level 1 of the Metabolomics Standards Initiative (MSI) standards (identification using two independent and orthogonal data), whereas levels PDa, PDb, PDc, and PDd correspond to level 2 of the MSI standards (putatively annotated compounds)59. Repeated metabolite names indicate multiple peaks matched to the same metabolite.
Logistic regression was used to identify important predictors of OA status and to increase our understanding of the relationship between OA status and these predictors. The candidate predictors that had p-value < 0.1 based on the t-test or chi-square test were gender, BMI, 2,998 metabolite peaks, and 13 microbiome taxa. Table 4 summarizes the results for 5 logistic regression models. Model 1 included subject characteristics, and the final model included BMI with higher standardized BMI values increasing the odds of an OA diagnosis. Model 2 included only metabolite peaks as predictors, and the final model included two unknown peaks. One peak was dichotomized at the median to improve model fit. Higher standardized peak intensities and values greater than the median for the other peak were associated with increased odds of an OA diagnosis. Predictors for model 3 included only microbiome-related metabolites, and the final model included hippuric acid (PDb) and propionic acid (PDc), and higher standardized peak intensities were associated with decreased odds of an OA diagnosis. Model 4 included both the microbiome-related metabolites and microbiome taxa as predictors, and the final model included hippuric acid (PDb), propionic acid (PDc), and the taxon k_Bacteria;p_Firmicutes;c_Clostridia;o_Clostridiales;f_;g_ with higher standardized values of the 3 predictors being associated with increased odds of OA. Model 5 included the microbiome-related metabolites, microbiome taxa, and BMI as predictors, and the final model included N-acetyl-D-glucosamine (OL1), hippuric acid (PDb), propionic acid (PDc), k_Bacteria;p_Firmicutes;c_Clostridia;o_Clostridiales;f_;g_, and BMI with higher standardized values of the metabolites and microbiome taxa associated with decreased odds of OA and higher standardized BMI values associated with increased odds of OA. Figure S2 shows the ROC curves for the 5 models. The AUC for all models that included microbiome taxa and/or metabolites was higher than the AUC for the subject characteristics model. The AUC for the model with microbiome-related metabolites, microbiome genus, and BMI (model 5) was significantly higher compared to the subject characteristics model 1 (p=0.01).
Table 4.
Logistic Regression Models Used to Identify Predictors of OA Status.
| Model | Candidate Predictors | Final Predictors** Odds Ratios with 95% Confidence Interval | Final Model Goodness of fit p-value* | Area Under the Curve with 95% Confidence Interval |
|---|---|---|---|---|
| 1. Subject Characteristics Only | • Gender • BMI |
BMI • OR=2.37, 95%CI (1.30, 4.31) |
0.203 | 0.71 (0.60, 0.82) |
| 2. Metabolites Only | • 2,998 of the 18,850 peaks from the untargeted analysis associated with OA (p < 0.1) | Unknown Peak (3.74_231.6499 m/z) • OR=4.54, 95%CI (2.00, 10.29) High vs Low intensity for Unknown Peak (6.61_391.2138 m/z) † • OR=5.90, 95%CI (1.92, 18.13) |
0.253 | 0.82 (0.73, 0.90) |
| 3. Microbiome-Related Metabolites Only | • 27 identified/annotated metabolites related to the microbiome associated with OA (p < 0.1) | Hippuric Acid (PDb) • OR=0.53, 95%CI (0.32,0.90) Propionic Acid (PDc) • OR=0.44, 95%CI (0.25, 0.77) |
0.292 | 0.75 (0.64, 0.85) |
| 4. Micobiome-Related Metabolites, Microbiome genus†† | • 27 identified/annotated metabolites related to the microbiome associated with OA (p < 0.1) • 13 out of 236 microbiome taxa associated with OA (p < 0.1) |
Hippuric Acid (PDb) • OR=0.45, 95%CI (0.26, 0.80) Propionic Acid (PDc) • OR=0.43, 95%CI (0.25, 0.76) k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__;g_ • OR=0.31, 95%CI (0.13, 0.78) |
0.664 | 0.80 (0.70, 0.91) |
| 5. Micobiome-Related Metabolites, Microbiome genus††, and BMI | • 27 identified/annotated metabolites related to the microbiome and associated with OA (p < 0.1) • 13 of the 236 microbiome genus†† associated with OA (p < 0.1) • BMI |
N-Acetyl-D-Glucosamine (OL1) • OR=0.48, 95%CI (0.27, 0.85) Hippuric Acid (PDb) • OR=0.49, 95%CI (0.25, 0.94) Propionic Acid (PDc) • OR=0.40, 95%CI (0.20, 0.78) k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__;g__ • OR=0.28, 95%CI (0.11, 0.67) BMI • OR=4.35, 95%CI (1.73, 10.97) |
0.278 | 0.88 (0.81, 0.96) |
Hosmer-Lemeshow Goodness of Fit Test
Final predictors selected using stepwise logistic regression with entry criteria p-value < 0.1 and exit criteria p-value > 0.05
peak 6.61_391.2138m/z was dichotomized at the median to improve model fit.
If genus level information was not available, the lowest available taxon was used.
Unmatched peaks are displayed in the format retention time_mass.
AUC in bold is significantly higher than the AUC for the subject characteristics model.
BMI, body mass index; OR, odds ratio; CI, confidence interval
DISCUSSION
Several studies have used metabolomics to differentiate individuals with and without OA using samples from serum, urine, synovial fluid, and subchondral bone28–37. Studies investigating synovial fluid have found that OA subjects have significant alterations in fatty acid metabolism, glycerolipid metabolism, glycerophospholipid metabolism, sphingolipids, amino acid metabolism, leukotriene/arachidonic acid metabolism, glutathione metabolism and tricarboxylic acid cycle activity33–35. Metabolomics analysis of subchondral bone, performed by Swank et al., demonstrated that subjects with OA had altered sphingolipid metabolism, purine/pyrimidine metabolism, and metabolism of amino acids32. These investigations show that the immediate metabolic environment in the OA joint is characterized by alterations in energy utilization, oxidative stress, and inflammatory status. Analysis of serum, plasma and urine samples have shown that microbially-related metabolites (e.g., hippuric acid, trigonelline), amino acids (particularly arginine, ornithine, and branched chain amino acids), purines, and phosphatidylcholines are markers of OA status29–31,36,37. This shows that biospecimens that are systemic or local to the joints can capture many of the same metabolic changes in OA. Our analysis of fecal material from individuals with obesity and hand plus knee OA also showed changes in amino acids/peptides, inflammatory metabolites (leukotrienes), and microbial metabolites in accordance with these previous studies, indicating that fecal samples are able to capture many of the important metabolic changes associated with OA that are seen in other biospecimen types28–37.
We observed that the most prominent differentiators in the fecal metabolome between OA cases and controls were di- and tri-peptides which were overall elevated in the OA subjects (Table 2). This suggests that intestinal proteolysis was increased in those with OA. Dysregulated proteolysis in the GI tract has been recognized as an important component of inflammatory diseases affecting the GI tract, particularly inflammatory bowel disease (IBD)38–42. Sources of proteases in the GI tract come from the pancreas, intestinal epithelial cells, immune cells, and the microbiota. Additionally, the microbiota also produce protease inhibitors (e.g., serpins43) which aid in regulating proteolytic activity in the gut. An imbalance in this protease/protease inhibitor system has been shown to have multiple deleterious effects on intestinal integrity including degradation of tight junctions, matrix remodeling, processing of inflammatory mediators (e.g., cytokines), and apoptosis of intestinal epithelial cells42. This causes the degradation and increased permeability of the intestinal barrier, leading to a “leaky gut” phenomenon where components of the intestine can cross into the bloodstream. This includes components of the microbiome including LPS – a potent immunostimulatory molecule that leads to increased systemic inflammation when present in the circulation44 and which has been associated with knee OA severity45. Additionally, an increase in mucosal inflammation occurs due to exposure of mucosal immune cells to luminal antigens, further damaging the intestinal epithelium and increasing permeability46. Supporting this, our pathway analysis results showed that leukotriene metabolism was the most significantly perturbed metabolic pathway between OA cases and controls (Figure 2), indicating that changes in inflammatory signaling, in addition to changes in proteolysis, were characteristic of OA. This cascade of events connects dysregulated intestinal proteolysis with damage to the intestinal epithelium and a potential increase in systemic inflammation.
In the parent study7, serum LPS was found to be significantly higher in the OA cases than controls, suggesting that these individuals had increased intestinal permeability. No differences in microbiome diversity were observed, although specific taxa were nominally significant prior to adjusting p-values for false discovery rates7. In the current study, logistic regression using fecal metabolomics data in combination with BMI and the rRNA-seq data was superior to either factor alone in identifying individuals with OA. This suggests that all three of these areas play important roles in OA status.
The lack of differences in microbial diversity between OA cases and controls reported in the parent study indicate that metabolites are a main driver of our models. For this sub-study, we included models containing 1) BMI only, 2) metabolite peaks only, 3) microbial metabolites only, 4), microbial genus and microbial metabolites, and 5) BMI, microbial genus, and microbial metabolites. This process allowed us to identify the discriminatory power of metabolites, microbiome measurements, and BMI alone or in combination. While models 2–4 (metabolites and/or microbiome data without BMI) gave higher AUCs than model 1 (BMI only), the combination of all three provided the best discriminatory power, indicating that all three aspects are important in differentiating OA cases and controls.
Of particular interest was the finding that lower standardized peak intensities for the short chain fatty acid (SCFA), propionic acid, was significantly associated with an increase in the odds of having hand plus knee OA. Produced by the microbiome, changes in SCFAs have been shown to be indicative of gut dysbiosis and play a role in maintaining the integrity of the intestinal wall as well as influencing gut inflammatory status47,48. The observation that propionic acid was decreased in participants with OA indicates an imbalance of SCFA levels consistent with increased intestinal permeability. Additionally, our logistic regression models showed that the lower standardized levels of a Clostridia taxon (k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__;g__ ) was statistically associated with an increased odds in an OA diagnosis. Given that Clostridia are major butyrate producers in the gut49, this supports the finding that decreased SCFA production may be contributing to OA status. Furthermore, SCFAs are known to affect energy metabolism of the host50, potentially explaining the significant alterations in carbohydrate/amino acid metabolism that was found in the pathway analysis. Altogether, this data indicates that the inflammatory/proteolytic environment in the intestine of individuals with OA is associated with a decrease in SCFAs/SCFA-producing microbes, which may be contributing to increased intestinal permeability.
Interestingly, our analysis revealed that participants with OA had differences in tryptophan metabolic products, particularly the microbial product indole and its metabolites (indole-3-acetate, tryptophanol, indolepyruvate, indoleacetaldehyde) (Table 3). Produced by the microbiome from dietary tryptophan, indoles play a prominent role in maintaining intestinal barrier integrity through their actions on the aryl hydrocarbon receptor (AhR). As agonists, indoles activate AhR in the intestinal epithelium and aid in maintaining tight junction integrity and regulating intestinal immune status which aid in decreasing epithelial permeability51–54. Other metabolites of tryptophan that are produced by the host, such as kynurenine and serotonin, are also agonists for AhR and can contribute to maintaining intestinal health55,56. This is seemingly in contrast to the observation that cases had increased intestinal permeability and inflammation, as indicated by higher serum LPS, changes in SCFAs, and perturbations in leukotriene metabolism. Because tryptophan – an essential amino acid - comes from dietary protein, the increased proteolysis seen here in OA cases is likely producing higher amounts of free tryptophan in the intestine, leading to an overall increase in tryptophan and its metabolites. Even though these AhR agonists are increased in cases, their protective effects do not seem to be able to counteract the observed increase in intestinal permeability. Significantly increasing dietary tryptophan may be a potential nutritional intervention strategy in OA, as this could potentially take advantage of the enhanced proteolysis and increase the concentration of AhR agonists high enough to counteract the intestinal permeability. Interestingly, individuals with inflammatory GI diseases, such as IBD, have shown a decreased expression of AhR57. If this also occurs in OA, these individuals would be less responsive to the protective effects of tryptophan metabolites. Further investigation of intestinal expression of AhR would provide insight to the viability of this treatment strategy. Increasing SCFAs in the diet (particularly through increasing dietary fiber) has been proposed as a treatment option for gastrointestinal and inflammatory disorders by maintaining integrity of the intestinal epithelium, regulating immune cell activity, and modulating metabolism/energy status58. Given our findings with reduced SCFAs in OA, this may be another potential nutritional intervention strategy for this disease.
Hippuric acid, a mammalian-microbial co-metabolite derived from phenylalanine and dietary polyphenols, was decreased in OA case fecal samples and lower standardized levels were statistically associated with an increased odds ratio of an OA diagnosis. In a previous study by our group, hippuric acid was increased in the urine of OA progressors as compared to non-progressors4. The difference in hippuric acid levels seen between these two studies may be due to a difference in biospecimen type or may indicate an increased absorption of hippuric acid in OA. Additionally, differences in diet, particularly in polyphenol-rich foods, may also account for this observation.
Strengths and Limitations
One of the strengths of this study was the use of an unbiased approach to determine metabolic and/or microbial features that differentiate OA cases from controls. Using this method, several novel metabolic features of OA were observed. Additionally, the use of logistic regression models provided a method to combine metabolite and microbiome information to further identify features of OA. However, the methods used in this study were exploratory in nature and were not developed to specifically target microbial metabolites. Further verification will be needed to discern the role of theses metabolites in OA. Furthermore, the sample size for this study is comparatively small, so these findings should be re-investigated using a larger number of subjects and other cohorts. This exploratory study utilized a convenience sample, and thus no formal power/sample size calculations were performed, and no adjustments were made for multiple testing. The high cross-validation score of the PLS-DA model (Q2) indicates that this metabolomic differentiation is likely to be reproduced in other similar sample populations. Future replication of our results in other populations will be needed. Lastly, the OA cases had a significantly higher BMI than non-OA controls, therefore some of the metabolic differences may be related to differences in BMI rather than OA status. Further investigation will be needed to delineate metabolic changes due to BMI and OA status.
Conclusion
Our study presents a novel observation that metabolites related to intestinal proteolysis, intestinal permeability and the microbiome are altered in OA. These findings may lead to exciting new therapeutic options. Future investigations are needed to study the expression and activity of individual proteases in the intestine (both host and microbial). Furthermore, the assessment of intestinal AhR status and the effectiveness of increasing dietary tryptophan in OA should also be investigated. Altogether, these studies would provide more mechanistic information connecting intestinal health with OA status and its potential for therapeutic targeting.
Supplementary Material
Funding
Supported by the Arthritis Foundation, the National Center for Advancing Translational Sciences (NCATS) (UL1TR002489), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (P30AR072580). The Johnston County Osteoarthritis Project is funded in part by the Centers for Disease Control and Prevention (U01 DP006266). The UNC Microbiome and Gnotobiotic Cores are supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK034987 and P30 DK056350) and the Office of the Director, NIH (P40 OD010995). The metabolomics analysis was funded, in part, by the Nutrition Obesity Research Center (P30DK056350 - Zeisel, Mayer-Davis, MPI) and the NIH Common Fund Eastern Regional Comprehensive Metabolomics Resource Core Grant (#U24DK097193 -Sumner, PI).
Footnotes
Conflict of interest statement
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
This data is available at the NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Project ID PR001207 (ST001914). The data can be accessed directly via it’s Project DOI: http://dx.doi.org/10.21228/M8PQ6M
REFERENCES
- 1.Safiri S, Kolahi AA, Smith E, et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann Rheum Dis. Published online 2020:819–828. doi: 10.1136/annrheumdis-2019-216515 [DOI] [PubMed] [Google Scholar]
- 2.Lawrence RC, Felson DT, Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26–35. doi: 10.1002/art.23176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glyn-Jones S, Palmer AJR, Agricola R, et al. Osteoarthritis. Lancet. 2015;386(9991):376–387. doi: 10.1016/S0140-6736(14)60802-3 [DOI] [PubMed] [Google Scholar]
- 4.Loeser RF, Pathmasiri W, Sumner SJ, et al. Association of urinary metabolites with radiographic progression of knee osteoarthritis in overweight and obese adults: an exploratory study. Osteoarthritis Cartilage. 2016;24(8):1479–1486. doi: 10.1016/j.joca.2016.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palau-Rodriguez M, Tulipani S, Queipo-Ortuño MI, Urpi-Sarda M, Tinahones FJ, Andres-Lacueva C. Metabolomic insights into the intricate gut microbial-host interaction in the development of obesity and type 2 diabetes. Front Microbiol. 2015;6(OCT):1–12. doi: 10.3389/fmicb.2015.01151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taneja V Arthritis susceptibility and the gut microbiome. FEBS Lett. 2014;588(22):4244–4249. doi: 10.1016/j.febslet.2014.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loeser RF, Arbeeva L, Kelley K, et al. Association of increased serum lipopolysaccharide but not microbial dysbiosis with obesity-related osteoarthritis. Arthritis Rheumatol. 2021;doi: 10.10(Epub ahead of print). doi: 10.1002/art.41955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan JM, Linder GF, Renner JB, Fryer JG. The impact of arthritis in rural populations. Arthritis Rheum. 1995;8(4):242–250. doi: 10.1002/art.1790080407 [DOI] [PubMed] [Google Scholar]
- 9.Arnold JW, Roach J, Fabela S, et al. The pleiotropic effects of prebiotic galacto-oligosaccharides on the aging gut. Microbiome. 2021;9(1):1–19. doi: 10.1186/s40168-020-00980-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azcarate-Peril MA, Butz N, Cadenas B, et al. An attenuated Salmonella enterica serovar typhimurium strain and galacto-oligosaccharides accelerate clearance of Salmonella infections in poultry through modifications to the gut microbiome. Appl Environ Microbiol. 2018;84(5):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allali I, Arnold JW, Roach J, et al. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017;17(1):1–16. doi: 10.1186/s12866-017-1101-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high- throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth0510-335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonso A, Marsal S, Julià A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front Bioeng Biotechnol. 2015;3(MAR):1–20. doi: 10.3389/fbioe.2015.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broadhurst D, Goodacre R, Reinke SN, et al. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics. 2018;14(6). doi: 10.1007/s11306-018-1367-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galindo-Prieto B, Eriksson L, Trygg J. Variable influence on projection (VIP) for orthogonal projections to latent structures (OPLS). J Chemom. 2014;28(8):623–632. doi: 10.1002/cem.2627 [DOI] [Google Scholar]
- 16.van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics. 2006;7:1–15. doi: 10.1186/1471-2164-7-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bujak R, Daghir-Wojtkowiak E, Kaliszan R, Markuszewski MJ. PLS-based and regularization-based methods for the selection of relevant variables in non-targeted metabolomics data. Front Mol Biosci. 2016;3(JUL):1–10. doi: 10.3389/fmolb.2016.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bender R, Lange S. Adjusting for multiple testing - When and how? J Clin Epidemiol. 2001;54(4):343–349. doi: 10.1016/S0895-4356(00)00314-0 [DOI] [PubMed] [Google Scholar]
- 19.Li YY, Douillet C, Huang M, Beck R, Sumner SJ, Styblo M. Exposure to inorganic arsenic and its methylated metabolites alters metabolomics profiles in INS-1 832/13 insulinoma cells and isolated pancreatic islets. Arch Toxicol. 2020;94(6):1955–1972. doi: 10.1007/s00204-020-02729-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y-Y, Ghanbari R, Pathmasiri W, et al. Untargeted metabolomics: Biochemical perturbations in golestan cohort study opium users inform intervention strategies. Front Nutr. 2020;7:2020–2021. doi: 10.3389/fnut.2020.584585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colin SA, O’Maille G, Want EJ, et al. METLIN - A metabolite mass spectral database. Ther Drug Monit. 2005;27(6):747–751. http://www.ncbi.nlm.nih.gov/pubmed/16404815%5CnISI:000233812400016 [DOI] [PubMed] [Google Scholar]
- 22.Wishart DS, Feunang YD, Marcu A, et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D617. doi: 10.1093/nar/gkx1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong J, Soufan O, Li C, et al. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46(W1):W486–W494. doi: 10.1093/nar/gky310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S, Park Y, Duraisingham S, et al. Predicting network activity from high throughput metabolomics. PLoS Comput Biol. 2013;9(7). doi: 10.1371/journal.pcbi.1003123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–1267. [DOI] [PubMed] [Google Scholar]
- 26.Zheng X, Xie G, Zhao A, et al. The footprints of gut microbial-mammalian co-metabolism. J Proteome Res. 2011;10(12):5512–5522. doi: 10.1021/pr2007945 [DOI] [PubMed] [Google Scholar]
- 27.Li M, Wang B, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A. 2008;105(6):2117–2122. doi: 10.1073/pnas.0712038105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Likhodii S, Zhang Y, et al. Classification of osteoarthritis phenotypes by metabolomics analysis. BMJ Open. 2014;4(11):1–7. doi: 10.1136/bmjopen-2014-006286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q, Li H, Zhang Z, Yang F, Chen J. Serum metabolites as potential biomarkers for diagnosis of knee osteoarthritis. Dis Markers. 2015;2015. doi: 10.1155/2015/684794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Sun G, Likhodii S, et al. Metabolomic analysis of human plasma reveals that arginine is depleted in knee osteoarthritis patients. Osteoarthritis Cartilage. 2016;24(5):827–834. doi: 10.1016/j.joca.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 31.Senol O, Gundogdu G, Gundogdu K, Miloglu FD. Investigation of the relationships between knee osteoarthritis and obesity via untargeted metabolomics analysis. Clin Rheumatol. 2019;38(5):1351–1360. doi: 10.1007/s10067-019-04428-1 [DOI] [PubMed] [Google Scholar]
- 32.Yang G, Zhang H, Chen T, et al. Metabolic analysis of osteoarthritis subchondral bone based on UPLC/Q-TOF-MS. Anal Bioanal Chem. 2016;408(16):4275–4286. doi: 10.1007/s00216-016-9524-x [DOI] [PubMed] [Google Scholar]
- 33.Mickiewicz B, Kelly JJ, Ludwig TE, et al. Metabolic analysis of knee synovial fluid as a potential diagnostic approach for osteoarthritis. J Orthop Res. 2015;33(11):1631–1638. doi: 10.1002/jor.22949 [DOI] [PubMed] [Google Scholar]
- 34.Kim S, Hwang J, Kim J, Ahn JK, Cha HS, Kim KH. Metabolite profiles of synovial fluid change with the radiographic severity of knee osteoarthritis. J Bone Spine. 2017;84(5):605–610. doi: 10.1016/j.jbspin.2016.05.018 [DOI] [PubMed] [Google Scholar]
- 35.Carlson AK, Rawle RA, Wallace CW, et al. Characterization of synovial fluid metabolomic phenotypes of cartilage morphological changes associated with osteoarthritis. Osteoarthritis Cartilage. 2019;27(8):1174–1184. doi: 10.1016/j.joca.2019.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamers RJAN, van Nesselrooij JHJ, Kraus VB, et al. Identification of an urinary metabolite profile associated with osteoarthritis. Osteoarthritis Cartilage. 2005;13(9):762–768. doi: 10.1016/j.joca.2005.04.005 [DOI] [PubMed] [Google Scholar]
- 37.Zhai G, Wang-Sattler R, Hart DJ, et al. Serum branched-chain amino acid to histidine ratio: A novel metabolomic biomarker of knee osteoarthritis. Ann Rheum Dis. 2010;69(6):1227–1231. doi: 10.1136/ard.2009.120857 [DOI] [PubMed] [Google Scholar]
- 38.Van Spaendonk H, Ceuleers H, Witters L, et al. Regulation of intestinal permeability : The role of proteases. World J Gastroenterol. 2017;23(12):2106–2123. doi: 10.3748/wjg.v23.i12.2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carroll IM, Maharshak N. Enteric bacterial proteases in inflammatory bowel disease-pathophysiology and clinical implications. World J Gastroenterol. 2013;19(43):7531–7543. doi: 10.3748/wjg.v19.i43.7531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid-Schonbein GW. Inflammation and the autodigestion hypothesis. Microcirculation. 2009;16(4):289–306. doi: 10.1080/10739680902801949.Inflammation [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jablaoui A, Kriaa A, Mkaouar H, et al. Fecal serine protease profiling in inflammatory bowel diseases. Front Cell Infect Microbiol. 2020;10:21. doi: 10.3389/fcimb.2020.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vergnolle N Protease inhibition as new therapeutic strategy for GI diseases. Gut. 2016;65:1215–1224. doi: 10.1136/gutjnl-2015-309147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mkaouar H, Akermi N, Mariaule V, et al. Siropins, novel serine protease inhibitors from gut microbiota acting on human proteases involved in inflammatory bowel diseases. Microb Cell Fact. 2016;15(1):1–13. doi: 10.1186/s12934-016-0596-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh SS, Wang J, Yannie PJ, Ghosh S. Intestinal barrier dysfunction, LPS translocation, and disease development. J Endocr Soc. 2020;4(2):1–15. doi: 10.1210/jendso/bvz039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang ZY, Stabler T, Pei FX, Kraus VB. Both systemic and local lipopolysaccharide (LPS) burden are associated with knee OA severity and inflammation. Osteoarthritis Cartilage. 2016;24(10):1769–1775. doi: 10.1016/j.joca.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: Bad, good or context dependent. Mucosal Immunol. 2017;10(2):307–317. doi: 10.1038/mi.2016.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venegas DP, De Fuente MK, Landskron G, et al. Short chain fatty acids ( SCFAs ) - mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. doi: 10.3389/fimmu.2019.00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Cuesta-zuluaga J, Mueller NT, Álvarez-quintero R, et al. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients. 2018;11(1):51. doi: 10.3390/nu11010051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. Commensal clostridia: Leading players in the maintenance of gut homeostasis. Gut Pathog. 2013;5(1):1–8. doi: 10.1186/1757-4749-5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7(3):189–200. doi: 10.1080/19490976.2015.1134082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Xu K, Liu H, et al. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front Cell Infect Microbiol. 2018;8(FEB):1–22. doi: 10.3389/fcimb.2018.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. PNAS. 2020;117(32). doi: 10.1073/pnas.2000047117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Natividad JM, Agus A, Planchais J, et al. Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab. 2018;28(5):737–749.e4. doi: 10.1016/j.cmet.2018.07.001 [DOI] [PubMed] [Google Scholar]
- 54.Brandstätter O, Schanz O, Vorac J, et al. Balancing intestinal and systemic inflammation through cell type-specific expression of the aryl hydrocarbon receptor repressor. Sci Rep. 2016;6(May):1–17. doi: 10.1038/srep26091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–3198. doi: 10.4049/jimmunol.0903670.AN [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manzella C, Singhal M, Alrefai WA, Saksena S, Dudeja PK, Gill RK. Serotonin is an endogenous regulator of intestinal CYP1A1 via AhR. Sci Rep. 2018;8(1):1–13. doi: 10.1038/s41598-018-24213-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monteleone I, Rizzo A, Sarra M, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141(1):237–248.e1. doi: 10.1053/j.gastro.2011.04.007 [DOI] [PubMed] [Google Scholar]
- 58.Gill PA, van Zelm MC, Muir JG, Gibson PR. Review article: short chain fatty acids as potential therapeutic agents in human gastrointestinal and inflammatory disorders. Aliment Pharmacol Ther. 2018;48(1):15–34. doi: 10.1111/apt.14689 [DOI] [PubMed] [Google Scholar]
- 59.Sumner LW, Amberg A, Barrett D, et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics. 2007;3(3):211–221. doi: 10.1007/s11306-007-0082-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li YY, Stewart DA, Ye XM, et al. A metabolomics approach to investigate kukoamine B - A potent natural product with anti-diabetic properties. Front Pharmacol. 2019;9(JAN):1–16. doi: 10.3389/fphar.2018.01575 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This data is available at the NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Project ID PR001207 (ST001914). The data can be accessed directly via it’s Project DOI: http://dx.doi.org/10.21228/M8PQ6M