

Abstract
T cell activation, a key early event in the adaptive immune response, is subject to elaborate transcriptional control. Here, we examined how the activities of eight major transcription factor (TF) families are integrated to shape the epigenome of naïve and activated CD4 and CD8 T cells. By leveraging extensive polymorphisms in evolutionarily divergent mice, we identified the “heavy lifters” positively influencing chromatin accessibility. Members of Ets, Runx, and TCF/Lef TF families occupied the vast majority of accessible chromatin regions, acting as “housekeepers”, “universal amplifiers”, and “placeholders”, respectively, at sites that maintained or gained accessibility upon T cell activation. Additionally, a small subset of strongly induced immune response genes displayed a non-canonical TF recruitment pattern. Our study provides a key resource and foundation for the understanding of transcriptional and epigenetic regulation in T cells and offers a new perspective on the hierarchical interactions between critical TFs.
INTRODUCTION
T lymphocytes are principal cellular effectors and regulators of vertebrate immunity, critical for protection against infectious agents. Activated T cells undergo rapid proliferation and acquire specialized effector functions that allow migration to sites of infection, recruitment and stimulation of other immune cells, or direct killing of infected cells. Following pathogen clearance, the activated T cell pool contracts, leaving behind a clonally expanded population of long-lived memory T cells that retain specialized features acquired during their activation1.
Activated T cells can differentiate into distinct types of effector cells with divergent functional characteristics tailored for protection against different types of microbial and abiotic challenges1,2. Additionally, chronic antigen stimulation can result in a particular activation state known as dysfunction or exhaustion3, while local environments can invoke further distinctive features in tissue-resident T cells4,5. Thus, T cells can acquire a wide range of functional states that likely represent variations on a core program executed through elaborate transcriptional regulation.
The gene expression state of a cell is primarily determined by transcription factors (TFs) that bind to specific DNA sequence motifs and tune the transcriptional output of the associated genes by promoting or opposing the recruitment and activity of RNA polymerase. Human and mouse genomes encode ~1400 TFs, that can be subdivided into families based on structural similarity in their DNA binding domains6,7. TF families may contain many members capable of binding similar DNA sequence motifs8,9. TFs extensively interact with each other to combinatorially regulate transcription10. Their activity frequently relies on the recruitment of cofactors that lack sequence specific DNA-binding capacity, but can modify chromatin to influence its accessibility to other nuclear factors11,12.
The epigenetic and transcriptional changes underlying T cell activation are driven by the concerted activity of many TFs. Although dozens of non-redundant, functionally important TFs have been identified, the regulatory logic underlying their coordinated activity is poorly understood2,13,14. It is still to a large degree unclear which TFs act as activators vs. repressors, and which TFs have more general vs more specialized functions in defining T cell activation states. Moreover, while many pairs of TFs have been shown to bind to overlapping sets of targets, the significance of these interactions is largely unknown.
To gain new insights into the transcriptional regulation of T cell activation states, we have leveraged naturally occurring polymorphisms in TF binding motifs in mice15,16. The wild-derived inbred strain Cast/EiJ (Cast) has roughly 20 million genetic variants relative to the C57BL/6 (B6) strain, many of which overlap with cis-regulatory elements16. By linking these polymorphisms to allelic imbalances in transcription, chromatin accessibility, TF and cofactor binding in (B6/Cast) F1 mice we have dissected the mechanisms underlying transcriptional and epigenetic regulation in T cells undergoing activation in response to an acute viral infection. We found that positive regulation of chromatin accessibility and gene expression in naïve T cells is overwhelmingly dependent on just a few TF families, including Ets, Runx, and TCF/Lef. Representative members of these families occupy most accessible chromatin regions and interactions between them define distinct epigenetic responses to T cell activation. Ets1 binding defined regulatory elements overlapping with promoters of housekeeping genes, whose accessibility was largely unchanged upon T cell activation despite the recruitment of a variety of activation-dependent TFs and cofactors. Conversely, Runx1 dynamically bound to nearly all accessible chromatin regions in a manner that was strongly dependent on its own motif as well as motifs for other TFs induced or repressed upon activation. Runx motifs positively affected the accessibility of these elements suggesting that Runx TFs promiscuously amplify the activity of multiple TFs. Finally, TCF1 binding defined elements whose accessibility was typically lost upon T cell activation unless displaced by other activation-induced TFs. In addition to these canonical behaviors that defined most of the chromatin accessibility changes upon T cell activation, we found a small set of regulatory elements associated with immune response genes that showed a non-canonical pattern of Ets1 and TCF1 recruitment. This observation suggests that expression of immune response genes, which are strongly induced upon T cell activation, may be regulated in an atypical manner through recruitment of TFs whose function is normally dedicated to other cellular processes. Together, our study provides new insights into the relationships between TFs that regulate T cell activation.
RESULTS
TF motifs that regulate chromatin accessibility in T cells
While naïve and activated T cells express many functionally important TFs, it is unclear which of these factors act as determinants of chromatin accessibility. To address this question, we performed ATAC-seq on naïve CD4 and CD8 cells isolated from uninfected B6/Cast F1 mice and activated virus-specific T cells isolated on day 7 post-infection with LCMV Armstrong and analyzed the effects of TF binding motif polymorphisms on allele-specific chromatin accessibility (Fig. 1a, Extended data Fig. 1a, b, Supplementary Table 1). We identified a limited number of motifs that strongly affected chromatin accessibility (Fig. 1b, Extended data Fig. 1c). Ets, Runx, and specific bZIP and IRF family motifs showed a strong positive association with chromatin accessibility across cell types and activation states, with Ets motifs showing the strongest association. In contrast, other motifs showed cell state-specific effects: Sox and bHLH motif variants preferentially affected chromatin accessibility in naïve cells, whereas T-box and specific bZIP or bZIP-IRF4 composite (Batf) motifs had a greater effect in activated cells (Fig. 1b, Extended data Fig. 1c).
Fig. 1: Effect of TF-binding motif polymorphisms on allele-specific chromatin accessibility in naïve and activated T cells.
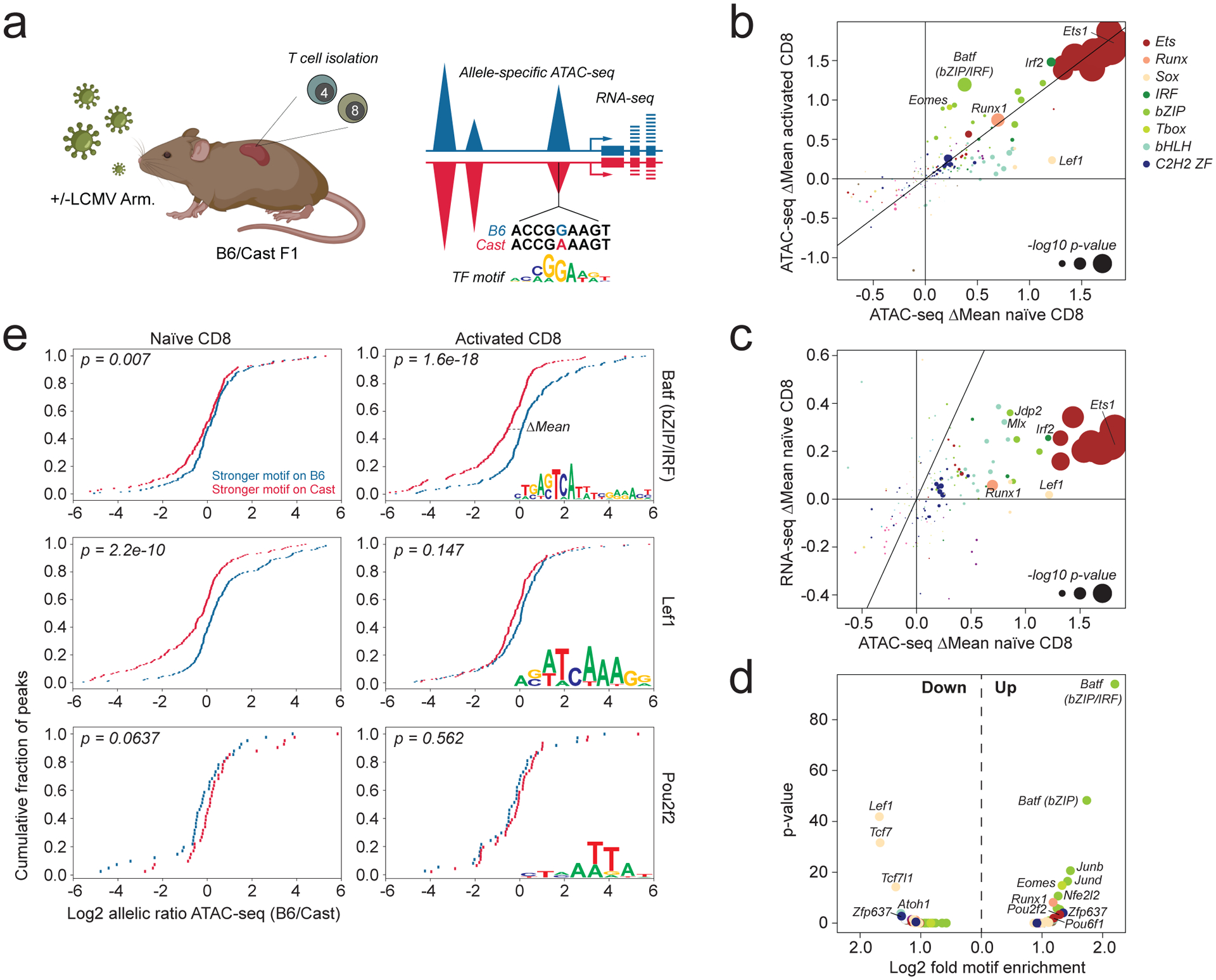
a: Experimental schematic. F1 (B6/Cast) mice were infected with 2×105 p.f.u. LCMV Armstrong i.p. injection or left uninfected. LCMV-specific CD4 and CD8 T cells were isolated on day 7 post-infection based on staining with I-Ab LCMV GP66–77 and H-2Db LCMV NP396–404 tetramers, respectively. Naïve CD44−CD62L+ TCRβ+ CD4 and CD8 T cells were isolated from uninfected mice. Sorted cell populations were subject to allele-specific ATAC-seq and RNA-seq analysis. Sequencing reads were aligned to a pseudo-diploid B6/Cast genome. Reads were counted as B6 or Cast-specific depending on the better genome alignment. After summing up all variant containing reads across the gene or peak region, the allelic ratio of RNA-seq or ATAC-seq signal was calculated as B6-specific reads/Cast-specific reads. Peaks containing variant-containing transcription factor binding motifs (TF motif) were divided into those containing stronger matches on the B6 allele and those containing stronger matches on the Cast allele. The mean allelic ratios of these two peak sets were compared using a t-test. The difference in mean allelic ratio between the two peak sets is denoted as ΔMean. This is further illustrated in panel e. Figure created with BioRender.com.
b: Effect of TF binding motif variants on chromatin accessibility measured as ΔMean in naïve and activated CD8 T cells. Data points are colored by TF family and scaled according to the −log10 p-value of a t-test comparing the mean allelic ratios of peak sets with stronger matches on the B6 vs Cast allele. The most significant p-value obtained across the two cell types (naïve and activated CD8) was used for scaling. Variant-containing motifs with <50 occurrences throughout the genome were excluded from the plot.
c: Effect of TF binding motif variants on chromatin accessibility and gene expression in naïve CD8 T cells. To determine the effect of motif variants on allelic bias in gene expression, peaks were linked to the nearest gene.
d: TF binding motif enrichment in peaks gaining or losing chromatin accessibility (fold change >2) compared to peaks whose accessibility was unchanged in activated vs naïve CD8 T cells. (Fisher’s exact test, one-sided p-values corrected by Benjamini-Hochberg Procedure).
e: Distribution of allelic ratios in ATAC-seq counts for peaks containing variants in select TF binding motifs. ΔMean describes the difference in mean allelic ratio between the blue peaks and the red peaks. (two-sided p-values were obtained from t-test for the null hypothesis that the independent allelic ratios of blue and red cis-elements have identical expected value μ).
A comparison between CD4 and CD8 T cells revealed that TF-binding motifs affecting chromatin accessibility naïve cells were very similar (Extended data Fig. 1d). In contrast, several motifs selectively affected chromatin accessibility in activated CD4 vs CD8 T cells (Extended data Fig. 1e). Variation in T-box motifs preferentially affected activated CD8 vs CD4 T cells, consistent with increased expression of the T-box family TFs T-bet and Eomes in the former (Extended data Fig. 1e,f). Conversely, TCF7/Lef1 motifs preferentially affected activated CD4 vs CD8 T cells, consistent with stronger activation-induced downregulation of Tcf7 in the latter (Extended data Fig. 1e,f). These results suggest that a relatively limited number of TF families is responsible for driving chromatin accessibility in T cells, with some showing graded cell type and cell state-specific activity.
While chromatin accessibility changes induced upon T cell activation were positively correlated with nearby gene expression, we also identified many individual peaks for which this association was not evident, raising the possibility that gene expression changes may be driven by TFs distinct from those affecting chromatin accessibility (Extended data Fig. 2a). However, we found that changes in aggregated ATAC-seq counts from multiple peaks linked to the same gene were almost always associated with gene expression changes, with the exception of a small subset of cell cycle related genes whose transient modulation was uncoupled from chromatin accessibility changes (Extended data Fig. 2b,c). Thus, most gene expression changes are strongly associated with cumulative accessibility changes across regulatory elements within a locus. Consistently, we found that TF binding motif polymorphisms affecting chromatin accessibility typically also affected nearby gene expression to varying degrees (Fig. 1c). Together, these observations suggest that, under our study conditions, most gene expression and chromatin accessibility changes are driven by the same core set of sequence-specific TFs.
While some motifs implicated in activation-induced chromatin remodeling were also identified through conventional motif enrichment analysis, others were only identified by allele-specific analysis, including the naïve T cell-specific activity of certain bHLH motifs (Fig. 1d). Moreover, conventional motif enrichment analysis was unable to distinguish between activating and repressive activity. For example, while both Batf and Pou2f2 motifs were enriched among peaks gaining accessibility upon T cell activation, allele-specific analysis suggested that the former acted as a strong activator selectively in activated cells, while the latter acted as a weak repressor in naïve cells (Fig. 1e). Thus, the effects of genetic variation provided new insights into chromatin regulation.
Ets1, Runx1, and TCF1 occupy most of the accessible genome
We next sought to characterize the interplay between TFs whose binding motifs were implicated in regulating chromatin accessibility. We first characterized binding of TFs whose motifs were most strongly linked to chromatin accessibility in naïve T cells. Analysis of Ets1, Runx1, and TCF1 binding by CUT&RUN revealed many TF-occupied sites covering ~93.6% of the accessible genome in naïve T cells (Fig. 2a–b, Extended data Fig. 3a–b). Ets1, Runx1, and TCF1-bound sites extensively overlapped, with ~51.4% of sites bound by all three factors. Strikingly, these “triple occupied” sites accounted for nearly all (86.8%) Ets1-bound elements, which were strongly enriched for promoters and 1st exons of genes (Fig. 2c). Smaller subsets of sites were bound by both Runx1 and TCF1 (22.9%), or Runx1 alone (8.1%) (Fig. 2b). Thus, most accessible chromatin regions in naïve T cells are occupied by some combination of Ets1, Runx1, and TCF1.
Fig. 2: Runx1, TCF1 and Ets1 occupy the majority of accessible chromatin regions.
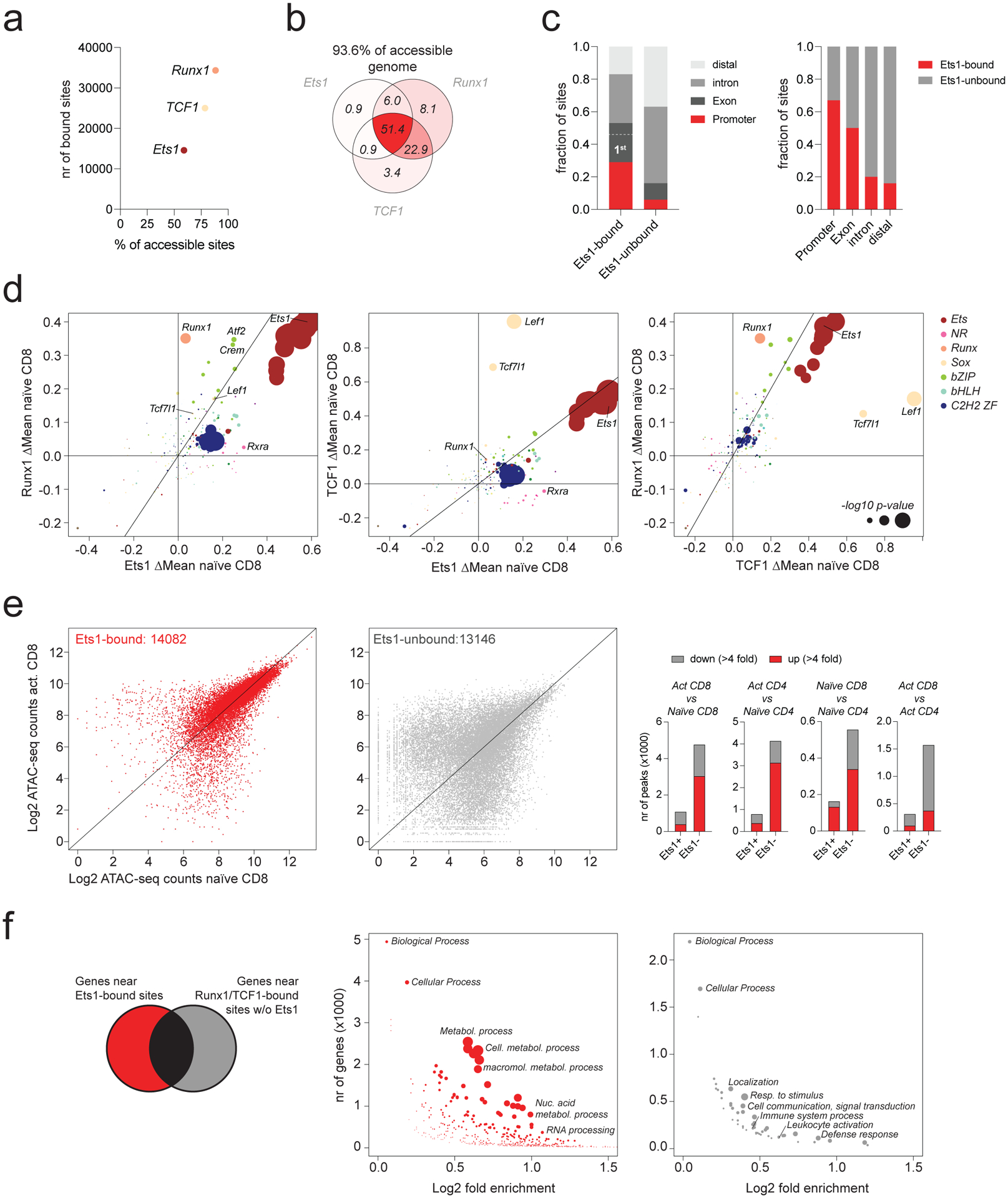
a: Runx1, TCF1, and Ets1 binding in naïve CD8 T cells measured by CUT&RUN. The number of ATAC-seq peaks bound by each TF is shown, together with the fraction of all accessible peaks (>100 ATAC-seq reads in naïve T cells) that is bound by each factor.
b: The fraction of all accessible peaks (>100 ATAC-seq reads in naïve T cells) that is bound by each combination of Runx1, TCF1, and Ets1.
c: Composition of Ets1-bound and –unbound regulatory elements (left). Fraction of ATAC-seq peaks overlapping with promoters, exons, introns, and distal elements bound by Ets1 (right).
d: Effect of genetic variation in TF-binding motifs on allelic bias in Runx1, Ets1, and TCF1 binding in naïve T cells. Data points are colored by TF family and scaled according to the −log10 p-value of a two-sided t-test comparing the allelic ratios between peaks with stronger matches on the B6 vs Cast allele.
e: Chromatin accessibility at ATAC-seq peaks bound by TCF1 or Runx1 with (Ets1-bound) or without Ets1 (Ets1-unbound) (left). Number of differentially accessible peaks (>100 reads in at least one cell type) across various comparisons (right).
f: GO term enrichment analysis of non-overlapping genes nearest to Ets1-bound vs. unbound peak sets. Data points are scaled according to the FDR.
The observation that Ets1-bound sites were typically co-occupied by all three TFs raised the possibility that these elements were bound non-specifically. To determine if TF binding to these regions was motif-dependent, we assessed the effects of motif polymorphisms on allele-specific TF occupancy at Ets1-bound sites. We found that binding of all three TFs was modulated by polymorphisms in their respective TF-binding motifs (Fig. 2d). Thus, Runx1, TCF1 and Ets1 binding were dependent on intact Runx, Sox, and Ets motifs, respectively. Additionally, binding of Runx1 and TCF1 were also strongly affected by variation in Ets motifs (Fig. 2d, Extended data Fig. 3c). Thus, TF binding to shared targets was motif-dependent and most likely specific. Moreover, Ets motifs, which act as the strongest determinants of chromatin accessibility, enhanced the binding of other transcription factors at shared targets.
Importantly, chromatin accessibility of Ets1-bound sites was to a large degree identical across naïve and activated CD4 and CD8 T cells (Fig. 2e). Conversely, sites bound by Runx1 or TCF1 in the absence of Ets1 underwent widespread activation-induced chromatin remodeling and showed frequent differential accessibility (Fig. 2e). Accordingly, genes near Ets1-bound chromatin regions were constitutively expressed at high levels and enriched for genes involved in metabolic processes, while genes associated with Runx1- or TCF1-bound regions not co-occupied by Ets1 were differentially expressed between cell types and enriched for genes involved in a variety of processes, including localization, cellular communication, and leukocyte activation (Fig. 2f, Extended data Fig. 3d–e). Thus, Ets1 binding defines a set of constitutively accessible genetic elements associated with housekeeping genes, which are typically co-occupied by Runx1 and TCF1 and account for more than half of the accessible genome in naïve T cells.
Ets1 binding defines constitutively accessible chromatin
We next sought to understand how the functions of activation-induced TFs were integrated with the pre-existing chromatin landscape. Because Ets1-bound elements did not undergo major accessibility changes in activated vs naïve T cells, it was possible that TFs whose expression or activity were modulated upon T cell activation were not recruited to these sites. To address this question, we determined the binding patterns for select representatives of bZIP, bHLH, IRF, Tbox, and Rel TF families in activated CD4 and CD8 T cells 7 days post infection with LCMV Armstrong. CUT&RUN of c-Jun, Bhlhe40, IRF4, T-bet, and NFATc1 revealed that each of these factors bound to ~20–50% of accessible regions in activated T cells (Fig. 3a–b). Interestingly, while most of the binding sites of these factors overlapped with Ets1 and underwent only minor changes in accessibility during T cell activation (Fig. 3c, d), only those that did not overlap with Ets1 binding underwent dramatic activation-induced chromatin remodeling (Fig. 3d). Thus, although activation-induced TFs are recruited to sites with and without Ets1 binding, their activity seems to be restricted selectively to the latter set of elements.
Fig. 3: Pre-accessible Ets1-bound chromatin regions are not responsive to activation-induced TFs.
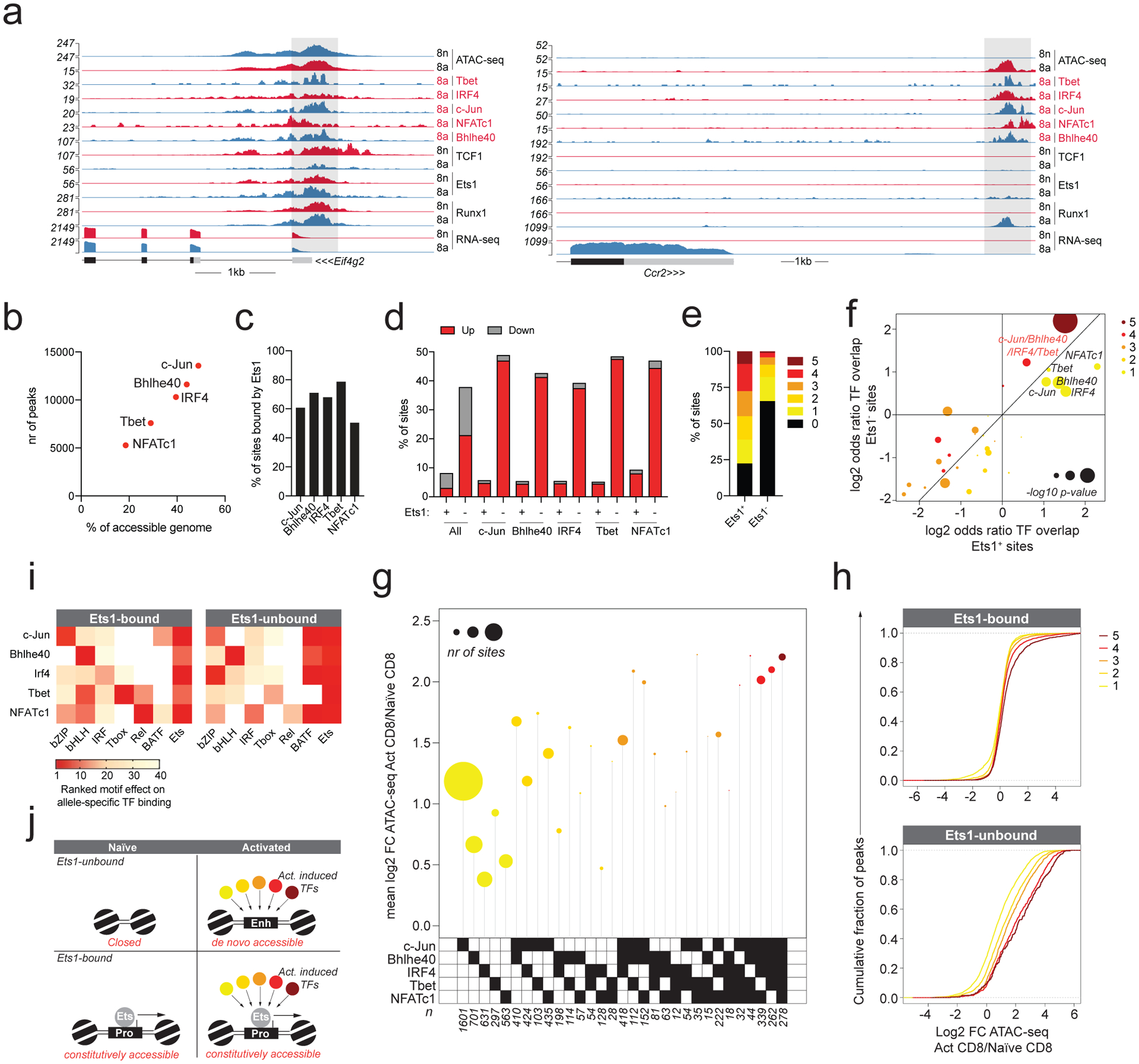
a: Gene tracks of RNA-seq, ATAC-seq and CUT&RUN read coverage at the Eif4g2 and Ccr2 loci in naïve and activated CD8 T cells.
b: c-Jun, Bhlhe40, IRF4, Tbet, and NFATc1 binding in activated CD8 T cells determined by CUT&RUN. The number of ATAC-seq peaks bound by each TF is shown together with the fraction of all accessible peaks (>100 ATAC-seq reads in activated T cells) that is bound by each factor.
c: Fraction of TF-bound ATAC-seq peaks co-bound by Ets1.
d: Fraction of sites bound by the indicated combination of TFs with >4 fold higher (up) or lower (down) chromatin accessibility in activated vs naïve CD8 T cells.
e: Fraction of Ets1-bound and -unbound ATAC-seq peaks that is bound by 0 to 5 activation-induced TFs.
f: Enrichment of TF co-localization at Ets1-bound and -unbound sites occupied by at least one activation-induced TF in activated CD8 T cells. p-values were calculated from one-sided Fisher’s exact test followed by Benjamini-Hochberg Procedure.
g: Fold change in chromatin accessibility in activated vs naïve CD8 T cells at Ets1-unbound ATAC-seq peaks occupied by distinct combinations of activation-induced TFs. Data points are scaled according to the number of peaks in each set.
h: Fold change in chromatin accessibility in activated vs naïve CD8 T cells at Ets1-bound and -unbound ATAC-seq peaks occupied by 1–5 activation-induced TFs.
i: Effect of TF-binding motif variation on allelic bias in TF occupancy at Ets1-bound vs –unbound ATAC-seq peaks. Each CUT&RUN experiment is shown on the Y-axis. X-axis shows the most statistically significant motif for each family.
j: Model of TF binding and chromatin accessibility changes at Ets1-bound and -unbound regulatory elements.
We considered the possibility that specific combinations of TFs present at sites not bound by Ets1 were responsible for driving accessibility changes. However, we found that binding sites of activation-induced TFs overlapped more extensively at Ets1-bound vs -unbound sites (Fig. 3e). At sites bound by at least one activation-induced TF, co-occupancy by all five factors was much more common than expected by chance and similarly enriched at both Ets1-bound and -unbound elements (Fig. 3f). Moreover, while certain combinations of activation-induced TFs occurred more frequently than others at Ets1-unbound sites, all combinations were associated with activation-induced increases in chromatin accessibility (Fig. 3g). In general, binding of a greater number of activation-induced TFs to Ets1-unbound elements was associated with greater changes in accessibility (Fig. 3h). Thus, differential changes in chromatin accessibility at Ets1-bound and -unbound sites induced upon T cell activation are unlikely to be accounted for by differential recruitment of activation-induced TFs.
While we considered the possibility that activation-induced TFs were recruited to constitutively accessible Ets1-bound sites in a manner independent of their respective binding motifs, analysis of allele-specific TF occupancy was inconsistent with this idea (Fig. 3i). To the contrary, we found that at Ets1-bound cis-regulatory elements, binding of each activation-induced TF was dependent on the presence of its respective TF-binding motif. Additionally, binding of these TFs at Ets1-unbound elements was modulated by polymorphisms in bZIP/IRF composite (Batf) motifs. Thus, recruitment of activation-induced TFs to both pre-accessible Ets1-bound, and de novo accessible Ets1-unbound elements was motif-dependent. Together, these observations suggest a simple model in which the functions of activation-induced TFs are overlaid onto the pre-existing chromatin landscape of naïve T cells (Fig. 3j).
Immune response genes recruit Ets1 upon T cell activation
While Ets1 binding was largely similar between activated and naïve T cells, our analysis revealed a small subset of sites (375 or 2.5% of Ets1 bound sites) that gained Ets1 binding de novo upon T cell activation (Fig. 4a, Extended data Fig. 4a). In contrast to other Ets1 targets, these elements were typically much less accessible in naïve T cells and were nearest to immune response related genes whose expression was strongly induced upon T cell activation (Fig. 4b, c). While the majority of constitutively bound Ets1 targets overlapped with promoters and 1st exons of genes, these upregulated Ets1 (Ets1up) sites typically overlapped with intronic and distal regulatory elements that were almost always co-occupied by multiple activation-induced TFs (Fig. 2c, 4d, e). In contrast to most Ets1-bound sites, Ets1up sites strongly gained accessibility upon T cell activation and underwent even more dramatic changes in accessibility than Ets1-unbound sites that recruited a similar number of activation-induced TFs (Fig. 4f). The observation that despite high Ets1 expression, Ets1up sites were not bound in naïve T cells raised the possibility that these sites lacked canonical Ets1 motifs. Indeed, Ets1up sites were relatively depleted of intact Ets motifs and typically matched the canonical Ets motifs to a lesser degree (Fig. 4g), but were enriched for stronger bZIP (Batf) and T-box motifs compared to other Ets1-bound sites.
Fig. 4: A small subset of immune response-related elements recruits Ets1 de novo upon T cell activation.
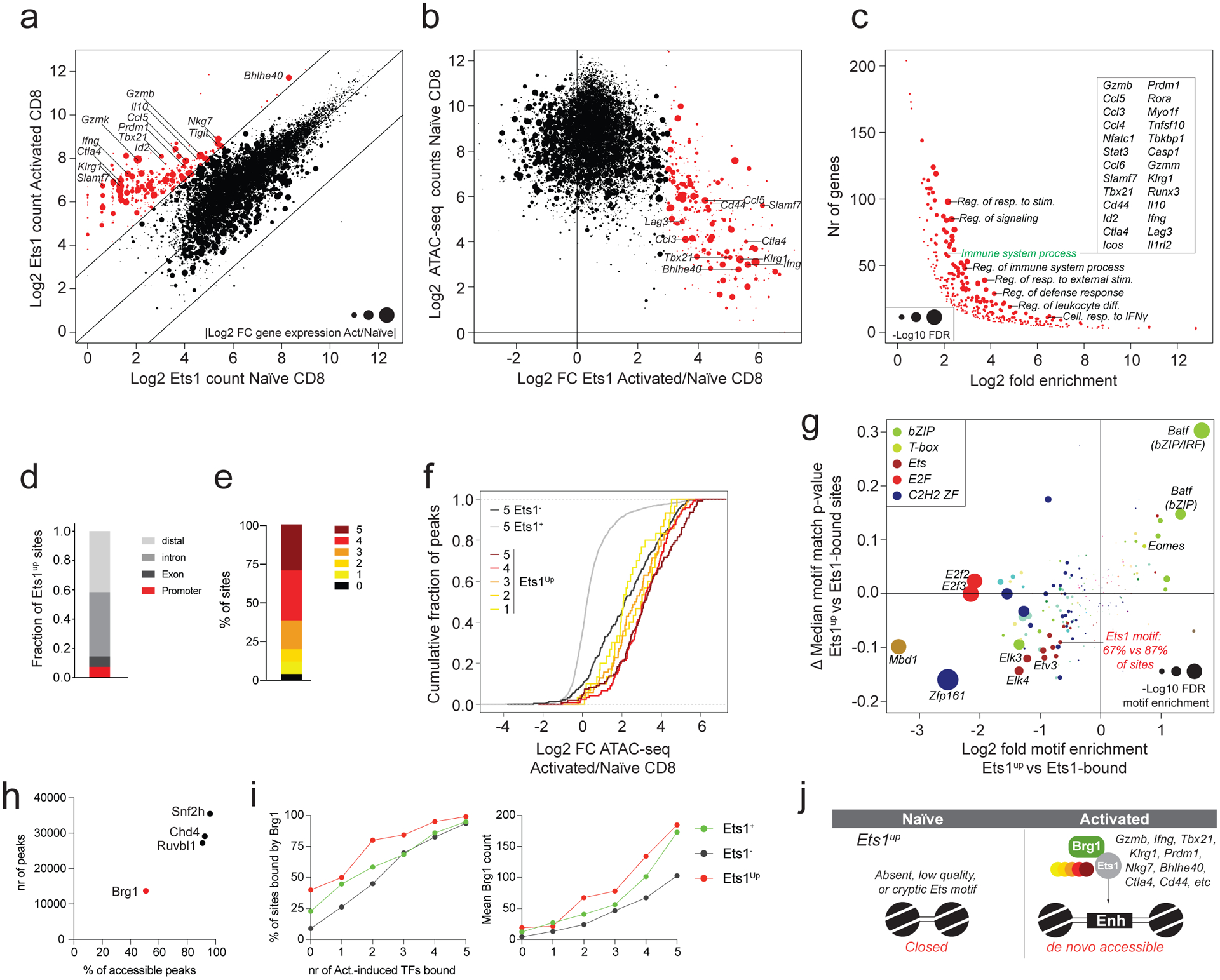
a: Ets1 CUT&RUN counts at ATAC-seq peak regions in naïve and activated CD8 T cells. Data points are scaled according to the absolute log2 fold change in expression of the nearest gene in activated vs naïve CD8 T cells. A subset of peaks with >8-fold more Ets1 binding in activated vs naïve CD8 T cells (Ets1up sites) is highlighted in red.
b: Chromatin accessibility in naïve CD8 T cells plotted against the log2 fold change in Ets1 binding in activated vs naïve CD8 T cells. Ets1up sites are highlighted in red.
c: Gene ontology term enrichment analysis for genes nearest to ATAC-seq peaks gaining Ets1 binding upon T cell activation (red dots in panel a-b).
d: Cis-regulatory element composition of Ets1up sites.
e: Fraction of Ets1up sites bound by 0 to 5 activation-induced TFs.
f: Changes in chromatin accessibility in activated vs naïve CD8 T cells at Ets1up sites bound by 1–5 activation-induced TFs. Ets1-bound (Ets1+) and -unbound (Ets1−) elements co-occupied by 5 activation-induced TFs are shown as a reference.
g: TF binding-motif enrichment and difference in median (ΔMedian) motif match p-value at Ets1up vs Ets1-bound ATAC-seq peaks.
h: Snf2h, Chd4, Ruvbl1, and Brg1 binding measured by CUT&RUN in activated CD8 T cells. The number of ATAC-seq peaks bound by each TF is shown, together with the fraction of all accessible peaks (>100 ATAC-seq reads in activated T cells) that is bound by each factor.
i: Fraction of Ets1-bound (Ets1+), Ets1-unbound (Ets1−) and Ets1up peaks co-bound by 0 to 5 activation-induced TFs that are bound by Brg1 in activated CD8 T cells (left). Mean Brg1 counts at the corresponding peak sets (right).
j: Model of TF activity at Ets1up elements.
Another factor that could contribute to the lack of Ets1 binding to Ets1up sites in naïve T cells is that these “non-canonical” sites require prior chromatin remodeling to render them accessible for Ets1 binding. A corollary to this model is that Ets1 alone would not be sufficient for the recruitment of the remodeling factors. To address this possibility, we analyzed the binding patterns of key subunits of four ATP-dependent chromatin remodelers: Snf2h, Chd4, Ruvbl1, and Brg1 in activated T cells17. While Snf2h, Chd4, and Ruvbl1 were almost universally present at all TF-bound regions, binding of Brg1, the ATPase subunit of SWI/SNF, was restricted to a smaller subset of sites (Fig. 4h, Extended data Fig. 4b). We found that at Ets1-bound, -unbound, and Ets1up sites alike, the binding of increasing numbers of activation-induced TFs was associated with recruitment of Brg1 (Fig. 4i). Analysis of the effects of TF-binding motif polymorphisms on allele-specific Brg1 recruitment revealed that in addition to Ets motifs, Tbox, bZIP, IRF, and bZIP/IRF composite motifs acted as major positive regulators of Brg1 recruitment (Extended data Fig. 4c). Thus, while intact Ets motifs might contribute to its binding or retention, Brg1 is typically not recruited to sites that are bound by Ets1 in the absence of activation-induced transcription factors.
Together, our results suggest that Ets motifs have a dominant effect on chromatin accessibility throughout the genome. While binding of Ets1 is typically restricted to constitutively accessible elements associated with promoters of housekeeping genes, a small subset of Ets1-bound enhancers associated with immune response related genes recruits Ets1 de novo upon T cell activation and undergoes dramatic chromatin remodeling. These Ets1up sites contain fewer and weaker Ets motifs and the binding of Ets1 to these sites may require assistance from other activation-induced TFs and the chromatin remodeler Brg1 (Fig. 4j).
Runx1 amplifies genome-wide chromatin accessibility changes
Our analysis suggested that similar to Ets motifs, Runx motifs also contributed to chromatin accessibility in a largely constitutive manner (Fig. 1b). However, in contrast to Ets1, we found that Runx1 binding was detectable at nearly all accessible sites across the genome, including those gaining or losing accessibility upon T cell activation (Fig. 5a–c). Activation induced widespread changes in Runx1 binding that correlated with chromatin accessibility changes (Fig. 5d). Thus, in contrast to Ets1, which binds mostly to constitutively accessible sites, Runx1 is redistributed upon T cell activation. This observation suggests that Runx1 is recruited to a very large fraction of its binding sites in a non-autonomous manner.
Fig. 5: Activation-dependent TF partners influence Runx1 localization.
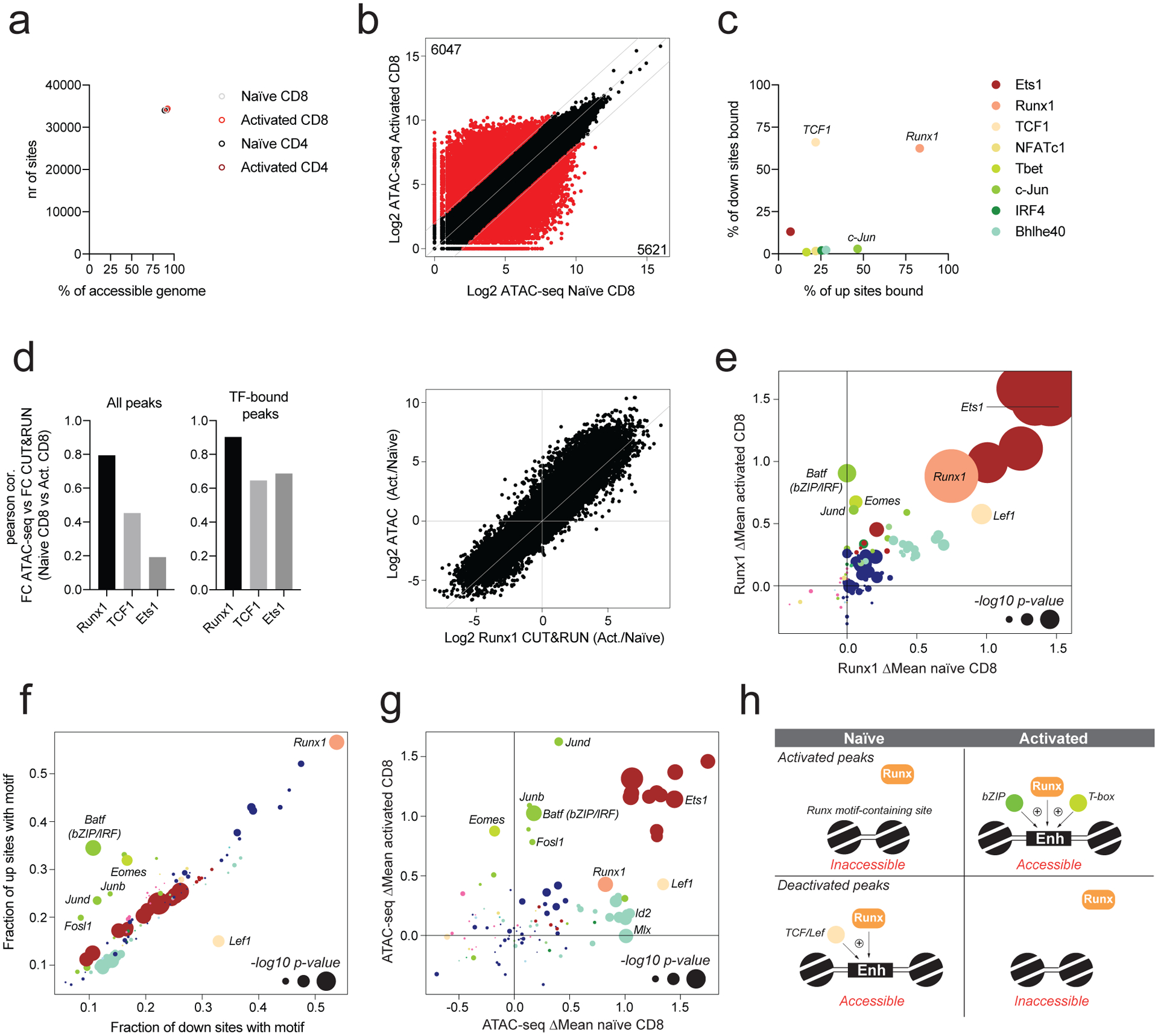
a: Runx1 binding was determined in naïve and activated CD4 and CD8 T cells using CUT&RUN. The number of ATAC-seq peaks bound by Runx1 is shown, together with the fraction of all accessible peaks (>100 ATAC-seq reads) that is bound by Runx1.
b: Chromatin accessibility in naïve and activated CD8 T cells across all ATAC-seq atlas peaks. Peaks with >4-fold differential accessibility are highlighted in red. Number of peaks with higher and lower chromatin accessibility in naïve and activated CD8 T cells is indicated.
c: Fraction of differentially accessible chromatin regions (highlighted in panel b) bound by each transcription factor.
d: Pearson correlation between changes in chromatin accessibility and changes in TF binding intensity for Runx1, TCF1, and Ets1 in activated vs naïve CD8 T cells. Analysis was done for all peaks or only the peaks bound by the respective TF. Scatter plot showing the fold change in Runx1 binding and fold change in chromatin accessibility in activated vs naïve CD8 T cells.
e: Effect of TF binding motif variation on Runx1 binding in naïve and activated CD8 T cells. Only peaks that were differentially accessible in naïve and activated CD8 T cells (panel b) were included in the analysis. Data points are colored by TF family and scaled according to the −log10 p-value of a two-sided t-test comparing allelic ratios between peaks with stronger motif matches on the B6 vs Cast allele. The most significant p-value obtained across the two cell types (naïve and activated CD8) was used for scaling.
f: TF-binding motif occurrence at peaks gaining (up sites) or losing accessibility (down sites) upon T cell activation. Motif significance of global allelic imbalance was calculated using two-sided t-test using the same approach as in Fig. 1e.
g: Effect of TF binding motif variation on chromatin accessibility in naïve and activated CD8 T cells. A two-sided t-test was used to calculate motif p-values as described in Fig. 1e. Only peaks that were differentially accessible in naïve and activated CD8 T cells (panel b) were included in the analysis.
h: Model of Runx1 activity at sites gaining or losing chromatin accessibility upon T cell activation.
To determine the mechanisms underlying Runx1 redistribution, we analyzed the effects of TF binding site polymorphisms on allele-specific Runx1 binding at sites strongly gaining or losing (>4-fold) chromatin accessibility in activated vs. naïve T cells. We found that polymorphisms in Runx motifs, which were the most abundant motifs at these sites, strongly affected Runx1 binding in both naïve and activated T cells (Fig. 5e, f). Lef1 (Sox) motifs, which were highly enriched at sites losing accessibility upon T cell activation, selectively affected Runx1 binding in naïve T cells. Conversely, bZIP and Tbox motifs, enriched at sites that gained accessibility, selectively affected Runx1 occupancy in activated T cells. These results suggest that distinct TFs contribute to Runx1 localization in naïve and activated T cells. Importantly, we found that polymorphisms in Runx motifs affected the accessibility of these activation-dependent Runx1-targets, suggesting that Runx1 is not just passively recruited to all accessible sites, but actively contributes to increasing chromatin accessibility (Fig. 5g). Based on these observations, we propose that Runx TFs may act as promiscuous “amplifiers” that can synergize with other activation state-dependent TFs (Fig. 5h).
Context dependent role of TCF1 during T cell activation
In contrast to Runx1 binding to sites either gaining or losing accessibility upon T cell activation, we found TCF1 bound preferentially to sites that lost accessibility (Fig. 5c). TCF1 bound sites that did not overlap with Ets1 became less accessible upon T cell activation, consistent with the downregulation of TCF1 and the related TF Lef1 (Fig. 6a, Extended data Fig. 1f, 5a). Thus, loss of chromatin accessibility upon T cell activation may, to a substantial degree, be explained by downregulation of TCF1 and Lef1, whose motifs show a positive association with chromatin accessibility in naïve T cells (Fig. 1b).
Fig. 6: Context-dependent functions of TCF1 in naïve and activated CD4 and CD8 T cells.
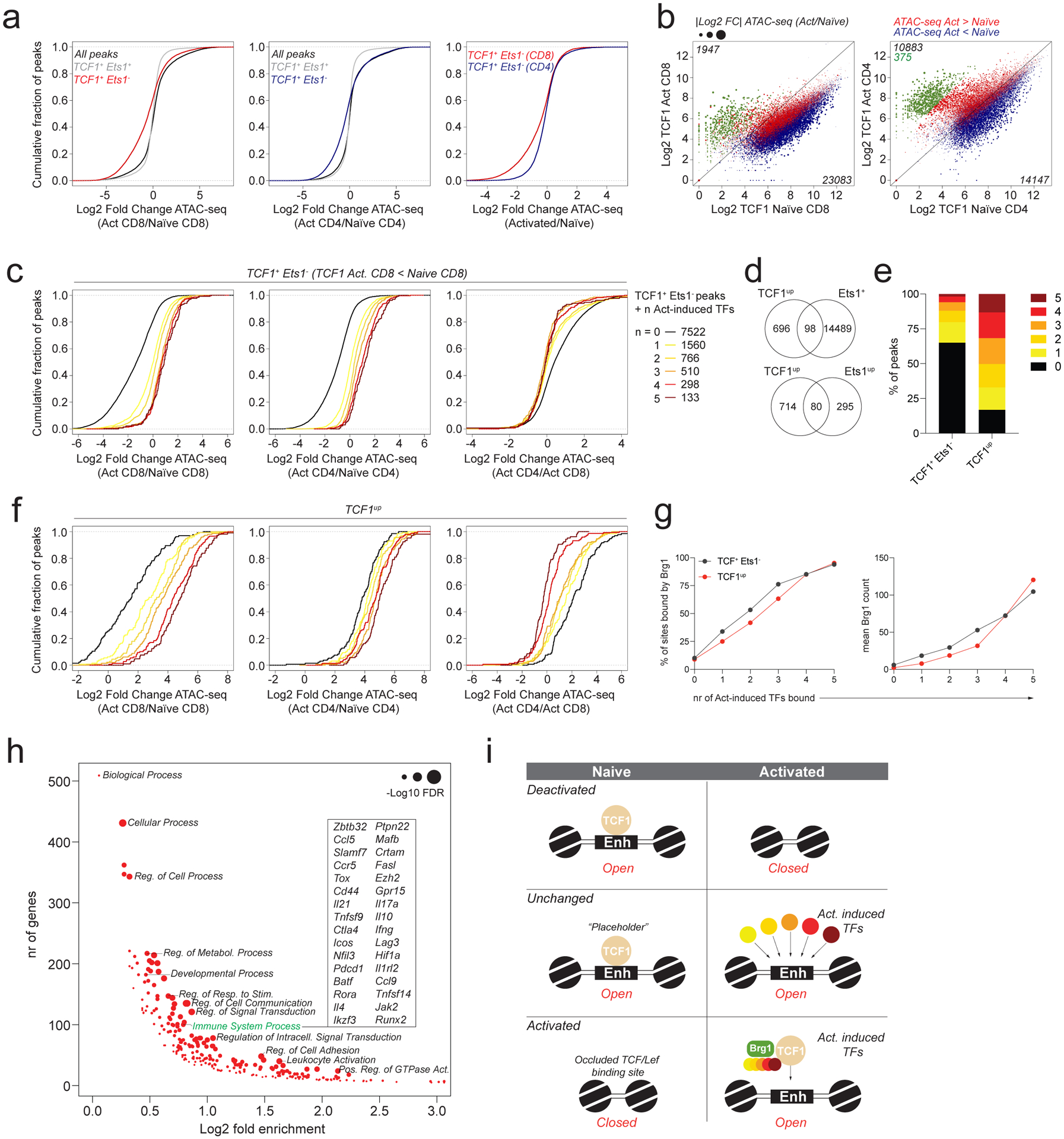
a: Chromatin accessibility changes between naïve and activated CD4 and CD8 T cells at ATAC-seq peaks bound by TCF1 with or without Ets1. All peaks denotes peaks with >100 reads in one or more cell types.
b: TCF1 CUT&RUN signal at ATAC-seq peak regions in naïve and activated CD4 and CD8 T cells. Data points are scaled according to the fold change in chromatin accessibility and colored blue or red according to the directionality of the accessibility change. A subset of peaks that has strong preferential binding of TCF1 in activated T cells was identified in CD4 T cells and highlighted in green.
c: Chromatin accessibility changes between naïve and activated CD4 and CD8 T cells at Ets1+TCF1− ATAC-seq peaks bound by 0–5 activation-induced TFs. Analysis was restricted to peaks with lower TCF1 binding in activated vs naïve CD8 T cells.
d: Overlap between TCF1up peaks (green peaks in panel b) and Ets1-bound ATAC-seq peaks (top) or Ets1up peaks (bottom).
e: Fraction of TCF1+Ets1− peaks and TCF1up peaks bound by 0–5 activation-induced TFs.
f: Chromatin accessibility changes in naïve and activated CD4 and CD8 T cells at TCF1up peaks bound by 0–5 activation-induced TFs.
g: Fraction of TCF1+Ets1− peaks and TCF1up peaks co-bound by 0–5 activation-induced TFs that are bound by Brg1 in activated CD8 T cells (left). Mean Brg1 count at the corresponding peak sets (right).
h: Gene ontology term enrichment analysis for genes nearest to TCF1up peaks (green dots in panel b).
i: Model of TF activity at TCF1-bound regulatory elements.
Importantly, activation-induced downregulation of TCF1 was much more pronounced in CD8 than in CD4 T cells, consistent with a stronger reduction in chromatin accessibility in the former (Extended data Fig. 1f, Fig. 6a). Accordingly, we observed a loss of TCF1 binding intensity by CUT&RUN in activated vs naïve CD8 T cells affecting nearly all TCF1 targets (Fig. 6b). Interestingly, while activation-induced changes in TCF1 binding were associated with corresponding changes in accessibility, we found that many sites that lost TCF1 binding by as much as 4-fold in activated vs naïve CD8 T cells still maintained or even increased their accessibility (Fig. 6b, Extended data Fig. 5b). This observation suggested that loss of TCF1 might be compensated for by the recruitment of other activating TFs. Consistent with this notion, the TCF1 occupied sites that did not recruit activation-induced TFs strongly lost chromatin accessibility upon T cell activation, while those recruiting more activation-induced TFs maintained or gained accessibility (Fig. 6c). Thus, in naïve CD8 T cells TCF1 may frequently act as a placeholder for activation-induced TFs. Furthermore, we observed that sites at which TCF1 was displaced by multiple activation-induced TFs were similarly accessible in activated CD4 vs CD8 T cells despite the selective maintenance of TCF1 levels in the former, suggesting that TCF1 became dispensable for the maintenance of chromatin accessibility at these sites after T cell activation (Fig. 6c). Conversely, sites at which TCF1 was not displaced by one or more activation-induced TFs became differentially accessible in activated CD4 and CD8 T cells, in accordance with varying amounts of TCF1 in these cells (Fig. 6c).
Our analysis also revealed a small subset of sites (794 sites or ~3% of all TCF1 targets) that acquired TCF1 binding only upon T cell activation (Fig. 6b, Extended data Fig. 5c). These sites were largely identical in CD4 and CD8 T cells, although they were less strongly bound in the latter. Thus, despite a global reduction in TCF1 binding intensity in activated CD8 T cells, a small subset of sites still recruited TCF1 de novo upon T cell activation. The vast majority of these TCF1up peaks were not bound by Ets1 but overlapped substantially with sites combinatorially bound by activation-induced TFs (Fig. 6d, e). TCF1up sites strongly gained accessibility upon T cell activation, particularly when bound by multiple activation-induced TFs (Fig. 6f). Depending on the number of activation-induced TFs that was bound, TCF1up sites responded differentially to activation in CD4 vs CD8 T cells. Specifically, when fewer activation-induced TFs were recruited, chromatin remodeling occurred in a CD4 T cell-specific manner. This observation further supports the notion than activation-induced TFs may compensate for the loss of TCF1 at a subset of targets following T cell activation.
The observation that despite high TCF1 expression, TCF1up sites were not bound in naïve T cells suggested that TCF1 alone was not sufficient to render these sites accessible. Thus, despite the previously described role of TCF1 as a pioneer factor capable of binding to inaccessible repressed chromatin regions, its activity was not sufficient to induce accessibility at a subset of its potential binding sites. Analysis of TCF1up sites revealed that the majority of these regions contained intact and high quality TCF1/Lef1 motifs, but were relatively depleted of Ets, Zinc Finger, and bHLH motifs, the latter of which had a positive effect on chromatin accessibility selectively in naïve T cells (Extended data Fig. 5d,e, 1b). Conversely, bZIP motifs were strongly enriched at TCF1up sites and accordingly we found that most of these sites were bound by c-Jun (Extended data Fig. 5f). Finally, we observed that sites bound by TCF1 in the absence of activation-induced TFs were rarely occupied by Brg1, whose recruitment was associated with the binding of activation-induced TFs (Fig. 6g). Together, these observations suggest that at a subset of targets, TCF1 cannot bind to its cognate motif in an autonomous manner but requires assistance from activation-induced TFs. Interestingly, we found that similar to Ets1up sites, TCF1up sites were associated with immune response-related genes, as well as genes involved in signal transduction, cell adhesion and cellular communication (Fig. 6h). Thus, regulatory elements associated with some of the most strongly induced and well-characterized immune response genes showed rather atypical patterns of Ets1 and TCF1 recruitment that were not representative of the prevalent function of these TFs. While TCF1 typically acted as a naïve T cell-specific activator, our results suggest that it plays additional previously unappreciated roles as a placeholder for activation-induced TFs and as an activation-dependent cofactor (Fig. 6i).
Graded activity of chromatin-modifying TFs in T cell subsets
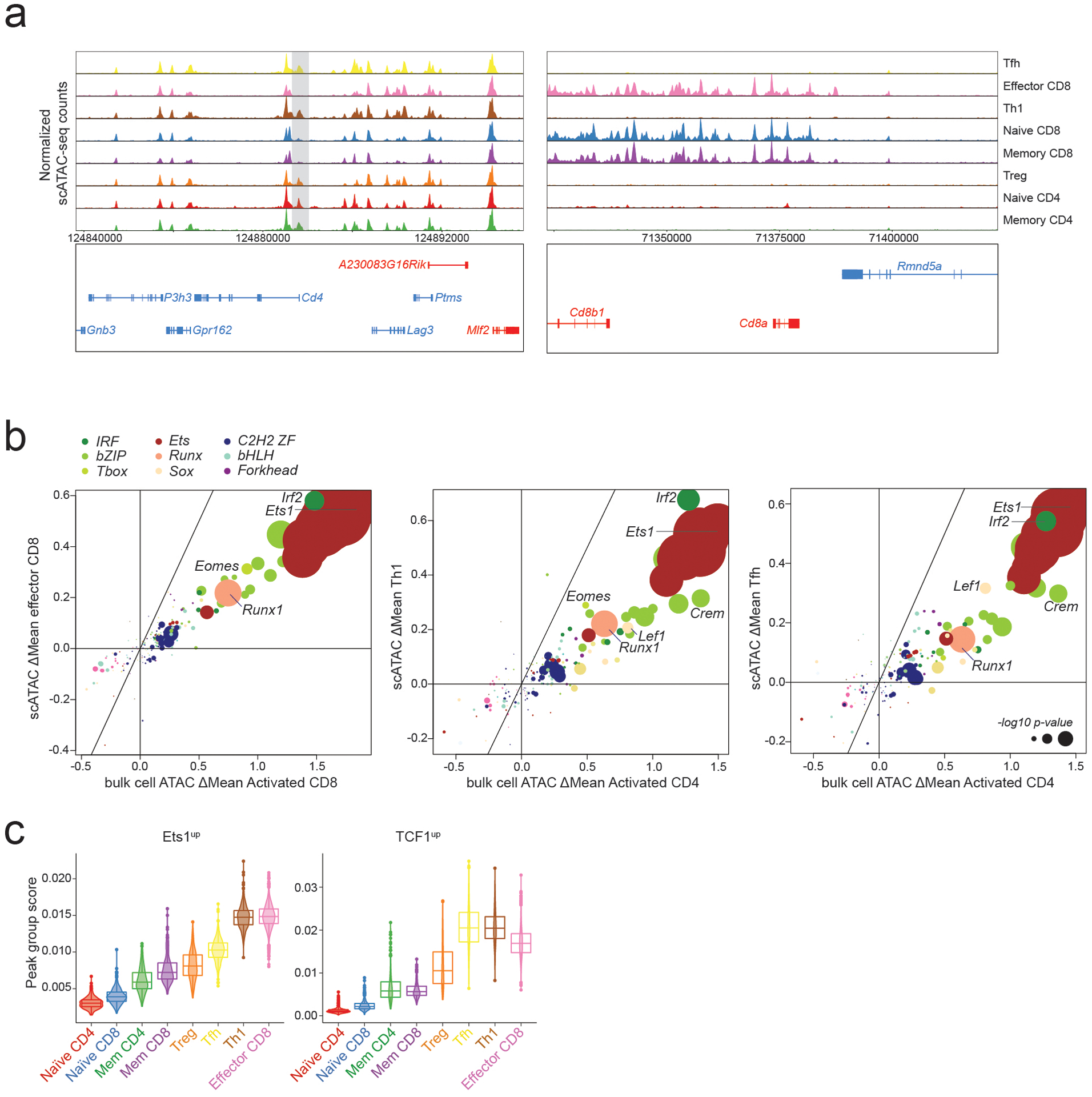
To assess the generalizability of our observations across distinct T cell subsets, we performed single cell (sc) ATAC-seq analysis on total splenic CD4 and CD8 T cells from LCMV-infected (B6/Cast) F1 animals on day 7 post-infection. This analysis revealed several distinct T cell subsets, including naïve and memory CD4 and CD8 T cells, regulatory T (Treg) cells, Th1 cells, Tfh cells, and a spectrum of effector CD8 T cells (Fig. 7a–b, Extended data Fig. 6a). To identify regulators of chromatin accessibility across these subsets, we analyzed the effects of TF binding motif polymorphisms on allele-specific pseudo-bulk scATAC-seq counts from these populations. Motifs affecting chromatin accessibility across T cell subpopulations were largely similar to those identified in bulk cell analyses (Extended data Fig. 6b). Consistently, genetic variation in Ets, Runx, IRF, and bZIP motifs strongly affected chromatin accessibility in all subsets. A comparison between the two main subpopulations of CD4 T cells arising in response to viral infection, Th1 cells and Tfh cells, revealed a stronger effect of Tbox motif variation on chromatin accessibility in the former, and a stronger effect of Sox motif variants in the latter (Fig. 7c). Accordingly, we found that chromatin regions bound by T-bet were more accessible in Th1 cells, while regions bound by TCF1 were more accessible in Tfh cells. Importantly, this effect was observed only at regions not bound by Ets1 (Fig. 7d). Consistent with our bulk cell analysis, Ets1-bound sites showed very high chromatin accessibility across all T cell subsets, while regions bound by activation-induced TFs in the absence of Ets1 were dynamically regulated (Fig. 7e). Similarly, Ets1up sites and TCF1up sites showed dynamic regulation with much higher accessibility across all activated T cell subsets compared to naïve T cells from the same mice (Extended data Fig. 6c). Together, our observations suggest that the major drivers of chromatin accessibility are similar across T cell subsets responding to acute LCMV infection, with graded activity of Sox and T-box family TFs at sites not bound by Ets1 accounting for many of the differences between Th1 and Tfh cells.
Fig. 7: Drivers of chromatin accessibility across T cell subpopulations.
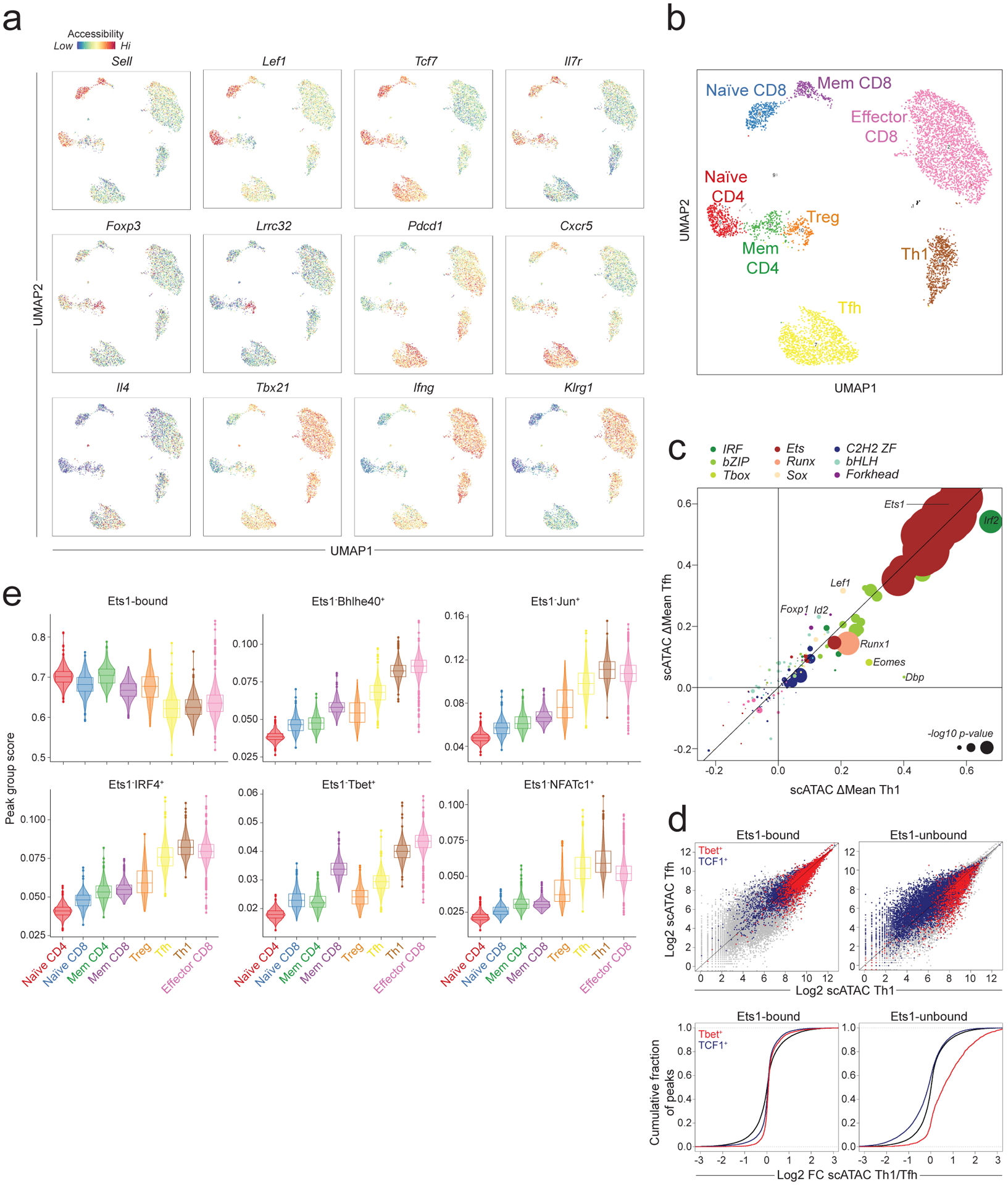
a: Chromatin accessibility at selected loci across T cell subpopulations in LCMV infected (B6/Cast) F1 mice measured by single cell (sc) ATAC-seq on total splenic CD4 and CD8 T cells (n=6043 cells).
b: Subset annotation.
c: Effect of genetic variation in TF binding motifs on allele-specific pseudo-bulk scATAC-seq counts in Th1 and Tfh cells. Data points are colored by TF family and scaled according to the −log10 p-value of a two-sided t-test comparing the allelic ratios between peaks with stronger matches on B6 vs Cast allele.
d: Pseudo-bulk scATAC-seq data showing accessibility in Th1 vs. Tfh cells at Ets1-bound or -unbound sites co-occupied by T-bet or by TCF1. Grey dots (top) and black lines (bottom) show all accessible peaks.
e: Violin plots and box plots showing accessibility of indicated peak groups across T cell subsets (n = 6043 cells). The center line in the box plots represents the median, the box limits represent the 25th and 75th percentiles, and the whiskers are the minimum/maximum values within 1.5 times the interquartile range. n = the number of cells in each cell annotation (420 Naïve CD4, 344 Naïve CD8, 234 Mem CD4, 231 Mem CD8, 198 Treg, 1321 Tfh, 652 Th1, 2583 Effector CD8).
DISCUSSION
T cell activation induces transcriptional and epigenetic changes that underlie the acquisition of specialized effector functions. Here we leveraged natural genetic variation between laboratory and wild-derived inbred mice to identify the TFs whose DNA-binding motifs shape chromatin accessibility and gene expression in naïve and activated T cells during acute LCMV infection. Most TF-binding motifs showed a positive effect on chromatin accessibility, with only few dedicated repressive motifs identified, including nuclear receptor and specific zinc-finger motifs. TF families whose motifs had either no, or only a limited effect included E2F, Forkhead, Gata, STAT and Smad. Some TFs belonging to these families likely carry out relatively specialized functions affecting only minor subsets of the loci to which they bind or acting transiently shortly after acute stimulation.
Considering the many TFs expressed in T cells, our studies unexpectedly identified just a few “heavy lifters” whose binding motifs most strongly affected chromatin accessibility. Three representative TFs belonging to these families, Ets1, Runx1, and TCF1 together occupied ~94% of all accessible chromatin regions in naïve T cells. Ets1 binding defined peaks enriched at promoters of housekeeping genes whose accessibility remained largely unchanged upon T cell activation. Interestingly, most of the binding sites for activation-induced TFs overlapped with Ets1-bound peaks, suggesting that these factors only affect chromatin accessibility at a small fraction of the regions that they occupy.
In addition to constitutively bound sites, a subset of elements acquired Ets1 binding de novo upon T cell activation and were preferentially associated with immune response genes. Although the exact mechanisms underlying the de novo recruitment of Ets1 remain unknown, we postulate that in addition to the presence of weaker Ets motifs, barriers to Ets1 binding may include tightly packed chromatin associated with repressive histone modifications and DNA methylation. In accordance with this model, it was reported that methylation of an intronic regulatory element in the Foxp3 locus could prevent the binding and activity of Ets1 at its consensus motifs18. Additionally, both activation-dependent and -independent partners, including Runx1 and bZIP/AP1 family TFs, could stabilize Ets1 binding at distinct sets of regulatory elements19–22. Finally, phosphorylation of Ets1 at multiple residues can destabilize DNA binding or promote cofactor recruitment suggesting additional mechanisms that could drive activation-state specific binding site selection23–26
T cells express multiple Ets family TFs that play non-redundant roles in their development or activation27–33. While all family members share a highly conserved Ets domain and recognize a consensus motif, their binding sites in vivo may either be shared or member-specific34–36. A comparison of binding sites for three Ets family TFs in a human T cell line revealed that redundant binding of multiple factors was common and enriched at transcription start sites of housekeeping genes, while non-redundant binding was rare and enriched at cell type-specific enhancers37,38. Based on these observations, we speculate that the majority of constitutively bound Ets1 targets identified in our study are co-occupied by multiple Ets family TFs, while Ets1up sites may be bound specifically by Ets1. Importantly, genetic variation in Ets motifs also affected chromatin accessibility and TF binding at sites not occupied by Ets1, suggesting that other family members may bind to these elements. Together, our results suggest that Ets family TFs play a uniquely dominant role in shaping the transcriptome and epigenome of T cells.
In contrast to Ets1, many Runx1 binding sites were activation state-dependent. Runx motifs were present at most differentially accessible ATAC-seq peaks and affected allele-specific chromatin accessibility. Thus, Runx family TFs may act as universal amplifiers of gene regulatory activity. Consistent with this model, Runx TFs have been reported to interact with a large variety of TFs, including Ets1, TCF1, Lef1, Gata3, Bcl11b, Tbet, Rorγt, Foxp1, and Foxp320,39–43. Interactions with some of these TFs can enhance or stabilize Runx1 binding and transactivation potential19,39. In addition to Runx1, T cells also express Runx2, and Runx3, which may bind dynamically to cell state-specific regulatory elements to carry out partially redundant functions during T cell development and activation44. A non-redundant role for Runx3 in CD8 lineage specification has been extensively characterized and could potentially be mediated by its association with a unique set of cofactors40,45,46. While our study suggests that intact Runx motifs have a net-positive effect on chromatin accessibility in mature T cells, Runx TFs have also been shown to engage in gene repression and associate with co-repressors, including TLE, and G9a40,47,48. Therefore, the division of labor between individual Runx family TFs and their roles as activators vs. repressors remains to be fully elucidated.
We found that TCF1-bound sites typically lost chromatin accessibility upon T cell activation. These changes occurred selectively at sites not co-occupied by Ets1 or activation-induced TFs and were more pronounced in CD8 vs CD4 T cells consistent with lower levels of TCF1 in the former. Together with the positive effect of intact TCF/Lef motifs on chromatin accessibility, these observations are consistent with the notion that TCF1 acts primarily as an activator49. Interestingly, we identified a substantial fraction of sites at which TCF1 was displaced by activation-induced TFs. Despite the loss of TCF1, these elements maintained their accessibility upon T cell activation. It is tempting to speculate that this placeholder activity of TCF1 may underlie its function in maintaining differentiation potential or “stem-ness”. Finally, we also identified a subset of TCF1 targets that was selectively bound only in activated T cells and frequently co-occupied by activation-induced TFs. It remains unclear what prevents TCF1 from binding to its activation-dependent sites in naïve T cells. While TCF1 is capable of binding and remodeling closed chromatin regions marked by repressive histone modifications, this capacity alone was apparently not sufficient to drive accessibility at these sites49.
Our findings offer a new perspective on the interplay between the activities of critical TFs that define T cell identity and improve our understanding of transcriptional and epigenetic regulation in resting and activated CD4 and CD8 T cells.
METHODS
Mice
Animals were housed at the Memorial Sloan Kettering Cancer Center (MSKCC) animal facility under specific pathogen free (SPF) conditions on a 12-hour light/dark cycle under ambient conditions with free access to food and water. All studies were performed under protocol 08-10-023 and approved by the MSKCC Institutional Animal Care and Use Committee. Mice used in this study had no previous history of experimentation or exposure to drugs. Male Cast/Eij mice were purchased from Jackson Laboratory and bred to female B6 mice to generate F1 offspring. Adult mice (> 6 weeks old) were used for experiments.
LCMV infection
LCMV Armstrong was grown in BHK cells and viral titers were determined using a plaque assay in Vero cells. Mice were infected with 2 × 105 plaque forming units (p.f.u.) by intraperitoneal (i.p.) injection.
Antibodies
The following antibodies were used for CUT&RUN: Runx1 (Thermo Fisher: PA5–19638), TCF1 (Cell Signaling Technology: 2203S), Ets1 (Cell Signaling Technology: 14069), T-bet (Santa Cruz Biotechnology: sc-21003 X), IRF4 (Santa Cruz Biotechnology: sc-6059), c-Jun (Abcam: ab31419), NFATc1 (Biolegend: 649607), Bhlhe40 (Novus Biologicals: NB100–1800SS), Brg1 (Abcam: ab110641), Chd4 (Abcam: ab72418), Ruvbl1 (Proteintech group: 10210–2-AP), Snf2h (Abcam: ab3749).
The following clones of fluorescently conjugated antibodies were obtained from BD Biosciences, Biolegend, Thermo Fisher, or Tonbo Bioscience and used for flow cytometry at a 1:400 dilution: CD45 (30-F11), TCRβ (H57–597), CD4 (RM4–5), CD8a (53–6.7), CD44 (IM7), CD62L (MEL-14).
Cell Staining and Flow Cytometry
Single cell suspensions of spleens were prepared in ice-cold cell isolation buffer (PBS with 2mM EDTA and 1% FCS) and subjected to red blood cell lysis using ACK buffer (150mM NH4Cl, 10mM KHCO3, 0.1mM Na2EDTA, pH7.3). T cells were enriched using Dynabeads FlowComp mouse CD4 and CD8 kits, according to manufacturer’s protocol. Cell surface antigens were stained at 4 degrees for 15 min using a mixture of fluorophore-conjugated antibodies. PE labeled H-2Db LCMV NP396–404 tetramers were generated by mixing biotinylated monomers (NIH tetramer core facility) with PE-labeled streptavidin. Class I tetramer staining was carried out for 30 min in ice-cold cell isolation buffer using a 1:200 dilution of tetramer. Staining with PE-labeled I-Ab LCMV GP66–77 tetramers (NIH tetramer core facility) was carried out for 30 minutes at 37 degrees in complete RPMI using a 1:200 dilution of tetramer. Cells were washed and passed through a 100 μm nylon mesh prior to sorting on a BD Aria II flow cytometer. Post-sort purity was routinely analyzed and typically higher than 95%. Naïve T cell populations were sorted as CD44−CD62L+ CD4 and CD8 T cells from uninfected mice. Activated cells for CUT&RUN were sorted as bulk CD44+ CD4 and CD8 T cells on day 7–8 post-infection. Activated and memory cells for RNA-seq and ATAC-seq were isolated as tetramer+ CD4 and CD8 T cells on day 7 or day 60 post-infection, respectively. Flow cytometry data was acquired using FACS Diva (BD) and analyzed using Flowjo.
RNA-Seq Library Preparation and Sequencing
Cell populations were double sorted straight into 1ml Trizol reagent (Thermo Fisher: 15596018). After addition of 200 μL chloroform, RNA was precipitated from the aqueous phase by addition of isopropanol and linear acrylamide. RNA was washed with 75% ethanol and resuspended in RNase-free water. After RiboGreen quantification and quality control by Agilent BioAnalyzer, 3.4ng total RNA underwent amplification using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech catalog #63488), with 12 cycles of amplification. Subsequently, 300ng of amplified cDNA was used to prepare libraries with the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using 8 cycles of PCR. Samples were barcoded and run on a HiSeq 4000 in a PE100 run, using the HiSeq 3000/4000 SBS Kit (Illumina). An average of 32M paired reads were generated per sample and the percent of mRNA bases per sample ranged from 51% to 73%.
ATAC-Seq Library Preparation and Sequencing
ATAC-seq libraries were prepared as described50. 5×104 cells were washed in ice cold PBS and lysed using ATAC-seq lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Following lysis, cells were incubated in 1x transposition buffer (Nextera TD buffer) containing 2.5 μL Nextera Tn5 transposase for 30 min at 37 degrees. Transposed DNA fragments were isolated using QIAGEN MinElute Reaction Cleanup kit and amplified using barcoded primers with Illumina adaptor sequences. The number of cycles used for library amplification was determined using a quantitative PCR side-reaction. Amplified libraries were purified and size selected using Ampure XP beads using consecutive purifications with bead-to-sample ratios of 0.5 and 1.8. After PicoGreen quantification and quality control by Agilent BioAnalyzer, libraries were pooled equimolar and run on a HiSeq 2500 in Rapid Mode in a PE100 run, using the HiSeq Rapid SBS Kit (Illumina). The loading concentration was 9.8pM and a 5% spike-in of PhiX was added to the run to increase diversity and for quality control purposes. The run yielded an average of 24M reads per sample.
CUT&RUN Library Preparation and Sequencing
CUT&RUN libraries were prepared as described by Skene et al51, with modifications15. All CUT&RUN experiments were performed on freshly sorted cell populations. For NFATc1 CUT&RUN sorted cells were re-stimulated in vitro for 1 hour at 37°C in the presence of 50 ng/mL phorbol-12-myristate-13-acetate (PMA, Sigma-Aldrich, P8139) and 500 ng/mL ionomycin (Sigma-Aldrich, I0634) with 1 μg/mL brefeldin A (Sigma-Aldrich, B6542). Cells were collected in a V-bottom 96 well plate by centrifugation and washed in antibody buffer (buffer 1 (1x permeabilization buffer from eBioscience Foxp3/Transcription Factor Staining Buffer Set diluted in nuclease free water, 1X EDTA-free protease inhibitors, 0.5mM spermidine) containing 2mM EDTA). Cells were incubated with antibodies (1:200 dilution) for 1h on ice. After 2 washes in buffer 1, cells were incubated with pA/G-MNase at 1:4000 dilution in buffer 1 for 1h at 4 degrees. Cells were washed twice in buffer 2 (0.05% (w/v) saponin, 1X EDTA-free protease inhibitors, 0.5mM spermidine in PBS) and resuspended in calcium buffer (buffer 2 containing 2mM CaCl2) to activate MNase. Following a 30-minute incubation on ice, 2x stop solution (20mM EDTA, 4mM EGTA in buffer 2) was added and cells were incubated for 10 min in a 37 degree incubator to release cleaved chromatin fragments. Supernatants were collected by centrifugation and DNA was extracted using a QIAGEN MinElute kit.
CUT&RUN libraries were prepared using the Kapa Hyper Prep Kit (Kapa Biosystems KK8504) and Kapa UDI Adaptor Kit (Kapa Biosystems KK8727) according to manufacturers protocol with the modifications described below. A-tailing temperature was reduced to 50 degrees to avoid melting of short DNA fragments and reaction time was increased to 1h to compensate for reduced enzyme activity as described by52. Following the adaptor ligation step, 3 consecutive rounds of Ampure purification were performed using a 1.4x bead to sample ratio to remove excess unligated adapters while retaining short adaptor-ligated fragments. Libraries were amplified for an average of 15 cycles using a 10 s 60°C annealing/extension step to enrich for shorter library fragments. Following amplification, libraries were purified using 3 consecutive rounds of Ampure purification with a 1.2x bead to sample ratio to remove amplified primer dimers while retaining short library fragments. A 0.5x Ampure purification step was included to remove large fragments prior to sequencing. After PicoGreen quantification and quality control by Agilent BioAnalyzer, libraries were pooled equimolar and run on a HiSeq 4000 in a PE50 run, using the HiSeq 3000/4000 SBS Kit (Illumina). The loading concentration was 2nM and a 5% spike-in of PhiX was added to the run to increase diversity and for quality control purposes.
Computational analysis of sequencing data
Alignment of F1 allele-specific reads
The F1 allele-specific reads were obtained from high throughput sequencing experiments, including ATAC-seq, RNA-seq and CUT&RUN. Raw sequencing reads were run through the previously described diploid genome alignment pipeline16. Briefly, to ensure unbiased mapping of sequencing reads from F1 mice, a pseudo-Cast genome was constructed by modifying the B6 reference genome (GRCm38) with genetic polymorphisms detected in the wild-derived inbred CAST/EiJ strain53. Next, the sequencing reads were aligned to the B6 and pseudo-Cast genome in parallel and the genomic coordinates of pseudo-Cast genome aligned reads were converted back to B6 coordinates. The allelic origin of each variant-containing read was determined based on the CIGAR string and alignment scores. For reads mapped to exactly the same location, the alignment with higher score was retained. For the non-variant-containing reads, the diploid genome alignment yielded identical scores on both genomes, therefore one of the alignments was selected randomly. With this strategy, the final alignment file contains allele-specific reads that are assigned to either B6 or Cast alleles, as well as invariant reads which do not carry allelic origin information.
RNA-seq data analysis
The pseudo-Cast genome was transformed from GRCm38 by incorporating high quality strain-specific variants (score ≥ 228). The RNA-seq data of naïve and activated T cells were aligned using STAR54, implemented in the diploid genome analysis pipeline with the following parameter settings: $STAR --runMode alignReads --readFilesCommand zcat --outSAMtype BAM --outFilterMultimapNmax 1 --outFilterMatchNmin 30 --alignIntronMin 20 --alignIntronMax 20000 --alignEndsType Local. Allelic ratio of gene expression was obtained by counting the allele-specific read pileup allocated to genes. The gene annotation GTF file was downloaded from ENSEMBL database at ftp.ensembl.org/ensembl/pub/release-83/gtf/. Plots were generated using Graphpad Prism or R.
ATAC-seq data analysis
Similar to RNA-seq, the ATAC-seq data of naïve and activated T cells were aligned using STAR54 with the splicing alignment feature switched off. The chromatin accessible peaks were called using MACS255: $macs2 call peak -t $inputFile -f BED -n $outputDir -g mm -p 1e-2 --nomodel --shift 75 --extsize 200 --keep-dup all --call-summits. IDR procedure was used to evaluate the peak reproducibility among replicates56. After removing the irreproducible peaks (IDR value < 0.05), the chromatin accessibility atlas was constructed by aggregating all the reproducible peaks from naïve and activated cell types. For each accessible peak, the nearest gene was assigned, and the peak was further categorized as promoter, intronic, exonic and intergenic cis-elements, respectively. Plots were generated using Graphpad Prism or R.
CUT&RUN data analysis
The paired-end CUT&RUN data of different transcription factors were mapped to the diploid genome using STAR54 with the splicing alignment feature switched off: $STAR --runMode alignReads --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 1 --outFilterMatchNmin 40 --outFilterMatchNminOverLread 0.4 --seedSearchStartLmax 15 --alignIntronMax 1 --alignEndsType Local. The aligned read pairs were retained if the fragment length was between 50 to 500 bp. Reads aligned to multiple loci were removed from further analysis. Peaks were called using MACS2 with its --nomodel setting55. IDR procedure was used to evaluate the peak reproducibility as described above56. A peak atlas was generated for individual TFs by retaining the reproducible peaks (IDR value < 0.2). CUT&RUN peaks that do not overlap ATAC-seq peaks were removed from the atlas. Plots were generated using Graphpad Prism or R.
Identification of active TF binding motifs
Experimentally determined Mus musculus motifs identified by ChIP-seq, PBM, SELEX, or HocoMoco were retrieved from CIS-BP database57. The motif position weight matrix was scanned using FIMO58 against the target peak sequences of both alleles for ATAC-seq and CUT&RUN data with default parameters. For motifs containing genetic variants, the degree of motif match on the B6 and Cast alleles was determined based on the significance of FIMO p value. For a given TF binding motif, the match strength was subsequently associated with the allelic imbalance of peaks containing the particular motif genome-wide. Peaks containing less than ten allele-specific reads were excluded from this analysis. A two-sided t-test was used to determine if stronger motif match on B6 or Cast alleles were associated with allelic imbalance of TF binding intensity or chromatin accessibility.
TF binding motif enrichment analysis
The same motifs used for identifying active TF binding motifs were identified within the peak sets of interest and background peaks using FIMO58 as described above. We defined a peak containing a motif if the motif resides within 300 bp distance from the peak summit. The non-parametric Fisher’s exact test was used to identify the motifs enriched in the target peak group.
Single cell (sc) ATAC-seq analysis
For the scATAC experiment, total CD4 and CD8 T cells were sorted from pooled splenocytes of 3 LCMV Armstrong infected (B6/Cast) F1 mice on day 7 post-infection. Sorted CD4 and CD8 T cells were mixed at equal ratios and used as input material for scATAC-seq analysis. Libraries were prepared according to the 10x single-cell ATAC–seq protocol from 10x Genomics Chromium (Single Cell ATAC Reagent Kits User Guide (CG000168, Rev A)). Briefly, cells were centrifuged (300g, 5 min, 4 °C) and permeabilized with 100 μl chilled lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% IGEPAL-CA630, 0.01% digitonin and 1% BSA). The sample was incubated on ice for 3–5 min and resuspended with 1 ml chilled wash buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20 and 1% BSA). After centrifugation (500g, 5 min, 4 °C), the pellet was resuspended in 100 μl chilled Nuclei buffer (2000153, 10x Genomics). Nuclei were counted using a haemocytometer, and finally the nucleus concentration was adjusted to 3,000 nuclei per μl. We used 15,360 nuclei as input for tagmentation. Nuclei were diluted to 5 μl with 1× nuclei buffer (10x Genomics) and mixed with ATAC buffer (10x Genomics) and ATAC enzyme (10x Genomics) for tagmentation (60 min, 37 °C). Single-cell ATAC–seq libraries were generated using the Chromium Chip E Single Cell ATAC kit (10x Genomics, 1000086) and indexes (Chromium i7 Multiplex Kit N, Set A, 10x Genomics, 1000084) following the manufacturer’s instructions. Final libraries were quantified using a Qubit fluorimeter (Life Technologies) and the nucleosomal pattern was verified using a BioAnalyzer (Agilent). Libraries were sequenced on a NovaqSeq6000 (Illumina) with the following read lengths: 50 + 8 + 16 + 50 (Read1 + Index1 + Index2 + Read2).
scATAC-seq data was aligned to the hybrid mouse genome using the following strategy. Files CAST_EiJ.mgp.v5.snps.dbSNP142.vcf and CAST_EiJ.mgp.v5.indels.dbSNP142.normed.vcf with SNP and indel genetic variants between B6 and Cast genomes were obtained from the Mouse Genome Project53. MMARGE v1.059 was used to create a Cast pseudogenome by introducing all variants to the B6 genome. Gene annotations were obtained from GENCODE vM2560 and mapped from B6 to Cast genomic coordinates using MMARGE. scATAC-seq read alignment and preprocessing were done using cellranger-atac v1.2.0. For this, cellranger-atac mkref was used to create custom B6 and Cast references, and cellranger-atac count was used for alignment. Cast alignments were then mapped to B6 coordinates using MMARGE. Custom script was used to identify each read in both B6 and Cast alignments and declare the read B6- or Cast-specific if the alignment score was higher for that allele, and declare the read ambiguous if the two scores were equal. In this procedure, reads were treated as single-end. cellranger-atac was also used with the default mm10 reference refdata-cellranger-atac-mm10–1.2.0 for allele-agnostic scATAC-seq alignment.
ArchR v1.0.161 was used for the allele-agnostic scATAC-seq analysis. The data was preprocessed by removing cells with counts smaller than 7500 and larger than 1e5, and filtering out doublets with a doublet enrichment score larger than 2.0. Then the count matrix was generated using a tile size of 500bp, without binarizing the counts. After preliminary analysis, 17 cells were classified as likely B cells, based on high accessibility at genes Cd79a, Cd79b and low accessibility at Cd3g, and removed from the analysis. The resulting count matrix for 6043 cells was used for subsequent analysis. LSI dimensionality reduction was performed with 4 iterations, max cluster size of 25 and resolution of 1.5, and the number of variable features equal to 52004 (matching the number of peaks obtained from bulk ATAC-seq analysis) and otherwise default parameters. Clustering was performed with resolution 2.0 and otherwise default parameters. UMAP was generated with parameters nNeighbors = 30, minDist = 0.5, and spread = 3. Clusters were annotated with cell states based on ArchR gene scores for a selection of marker genes. Scores for each peak group defined using bulk ATAC-seq and CUT&RUN analysis were generated using the scATAC-seq peak matrix generated with ArchR (allowing up to a read count of 30 per peak per cell). For each cell, peak counts were summed over all peaks in the peak group and then divided by the total number of counts over all peaks in the cell to generate a peak group score.
Allele-specific scATAC-seq counts were calculated for peaks obtained from bulk ATAC-seq analysis. For each peak in each cell, reads with the same start and end positions that aligned to that peak were collapsed to avoid PCR duplicates. For cell states annotated in the above analysis, allele-specific pseudo-bulk count matrices were generated by summing the allele-specific counts for each peak across all cells within each cell state.
Statistics & Reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Extended Data
Extended Data Fig. 1. RNA-seq and ATAC-seq of naïve and activated CD4 and CD8 T cells.
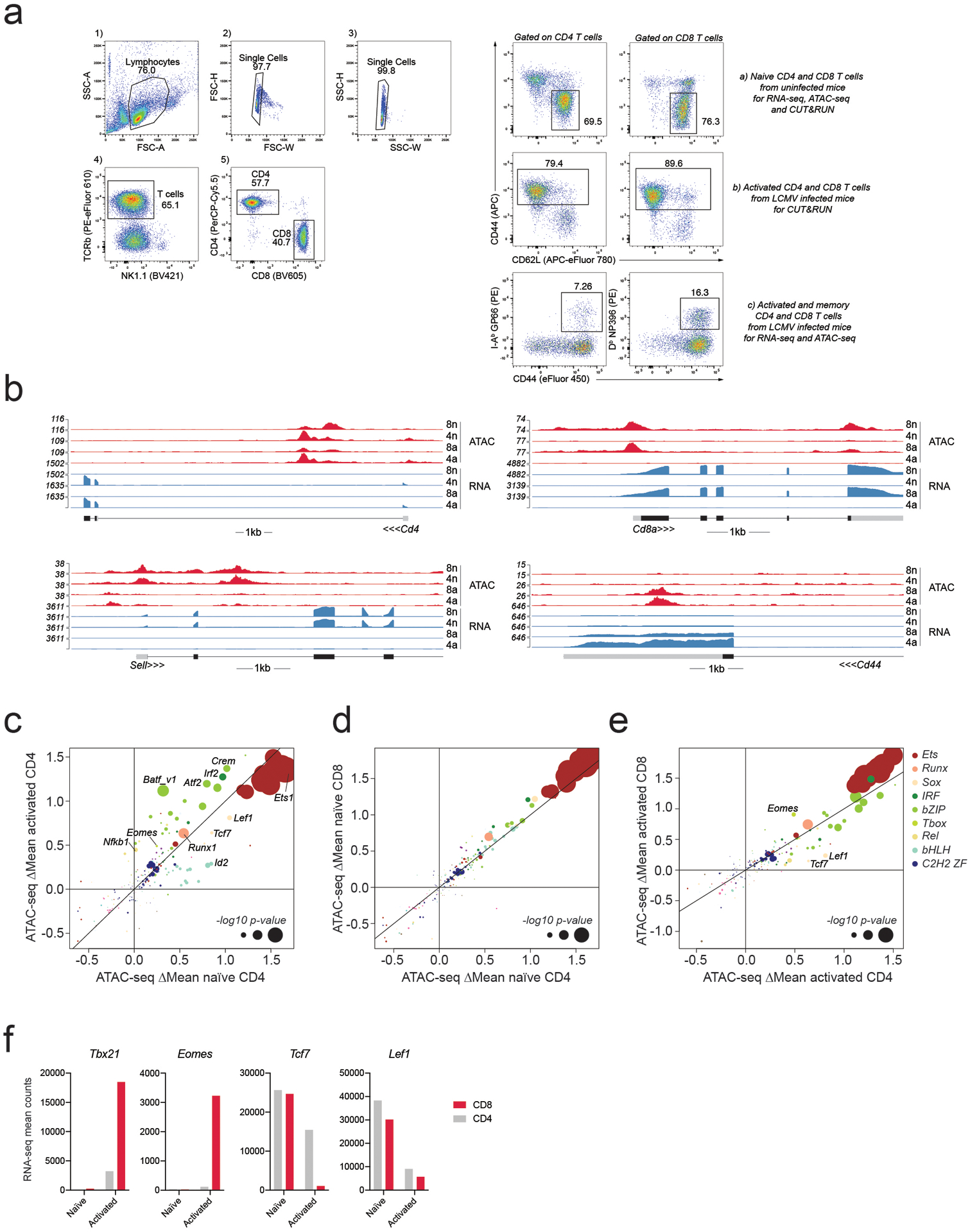
a: Example of gating strategy for cell isolation from the spleen. Live lymphocytes were identified based on FSC-A/SSC-A. Doublets were gated out and CD4 and CD8 T cells were identified as TCRβ+CD4+ and TCRβ+CD8+, respectively. Naïve CD4 and CD8 T cells used as input for RNA-seq, ATAC-seq, and CUT&RUN experiments were isolated from uninfected mice as CD44+CD62L− cells. Bulk activated CD4 and CD8 T cells used as input for CUT&RUN experiments were isolated as CD44+ cells from LCMV Armstrong infected mice on day 7–8 post-infection (p.i.). LCMV-specific activated and memory CD4 and CD8 T cells used as input for ATAC-seq and RNA-seq experiments were isolated based on tetramer staining on day 7 or day 60 p.i.
b: RNA-seq and ATAC-seq reads at the Cd4, Cd8a, Sell and Cd44 loci in naïve and activated CD4 and CD8 T cells.
c-e: Effect of TF binding motif variants on chromatin accessibility measured as ΔMean in naïve and activated CD4 T cells (A), naïve CD4 and CD8 T cells (B), activated CD4 and CD8 T cells (C). Data points are colored by TF family and scaled according to the −log10 p-value of a two-sided t-test comparing the mean allelic ratios between peaks with stronger matches on the B6 vs Cast allele. The most significant p-value obtained across the two cell types (naïve and activated CD8) was used for scaling. Motifs with <50 variant-containing motif occurrences were excluded from the plot.
f: Mean RNA-seq counts for Tbx21, Eomes, Tcf7 and Lef1 in naïve and activated CD4 and CD8 T cells.
Extended Data Fig. 2. Association between gene expression and chromatin accessibility changes.
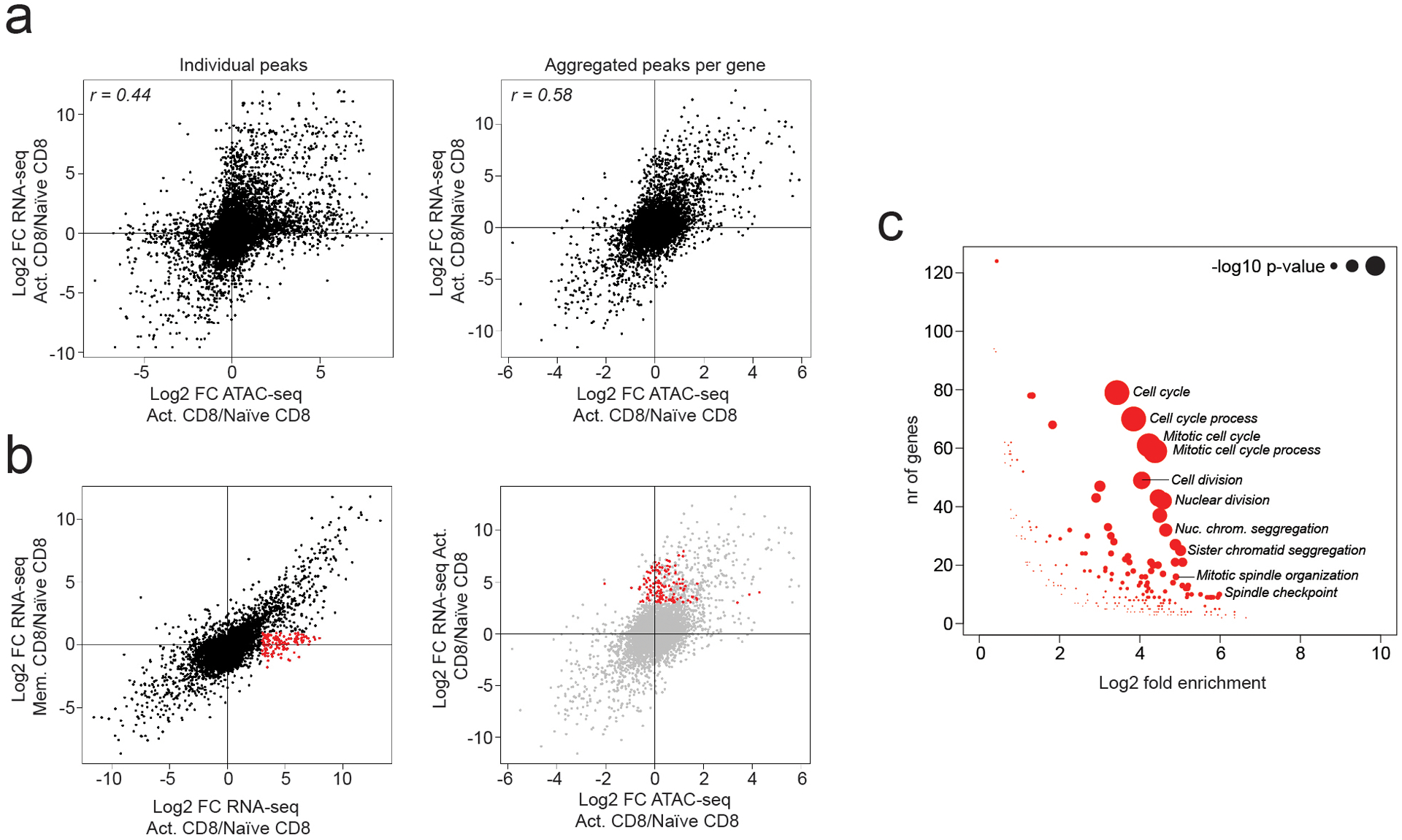
a: Correlation between changes in chromatin accessibility and gene expression in activated vs naïve CD8 T cells. Showing individual ATAC-seq peaks linked to the nearest gene (left) or the sum of counts across all ATAC-seq peaks linked to the same gene (right).
b: Fold change in gene expression in activated vs naïve CD8 T cells (x-axis) plotted against the fold change in gene expression in memory vs naïve CD8 T cells (y-axis). A subset of genes whose expression is transiently induced upon T cell activation is highlighted in red (left). Aggregated chromatin accessibility and gene expression changes for this gene set (right).
c: Gene ontology (GO) term enrichment analysis on genes highlighted in panel B
Extended Data Fig. 3. RNA-seq and ATAC-seq changes at Runx1, TCF1, and Ets1-bound genes.
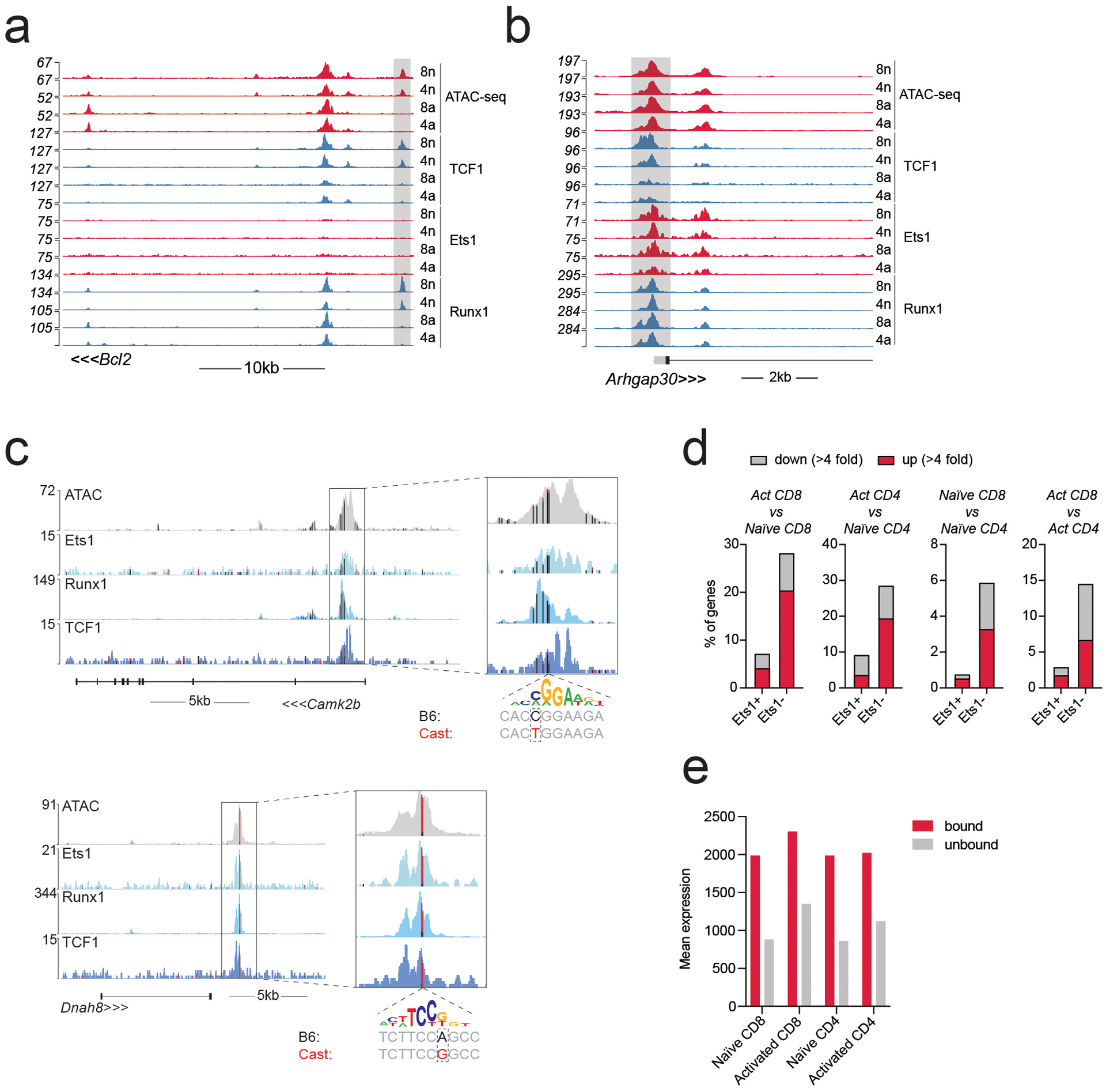
a-b: Gene tracks of RNA-seq, ATAC-seq and CUT&RUN coverage at the Bcl2 and Arhgap30 loci in naïve and activated CD8 T cells.
c: Track examples showing the effect of selected Ets1 motif variants on allelic bias in chromatin accessibility and TF binding in activated CD8 T cells from (B6/Cast) F1 mice. Vertical bars mark the position of genetic variants, with colors indicating the fraction of variant-containing reads coming from either the B6 (black) or Cast (red) allele. A genetic variant in the Camk2b locus results in a stronger Ets1 motif match on the B6 allele and a weaker match on the Cast allele and is associated with B6-specific chromatin accessibility and TF binding. The opposite pattern is observed at the Dnah8 locus.
d: Gene expression changes at genes (>100 reads in at least one cell type) nearest to Ets1-bound and -unbound ATAC-seq peaks.
e: Mean expression of genes nearest to Ets1-bound and -unbound ATAC-seq peaks.
Extended Data Fig. 4. TF and cofactor binding at Ets1-bound and –unbound chromatin regions.
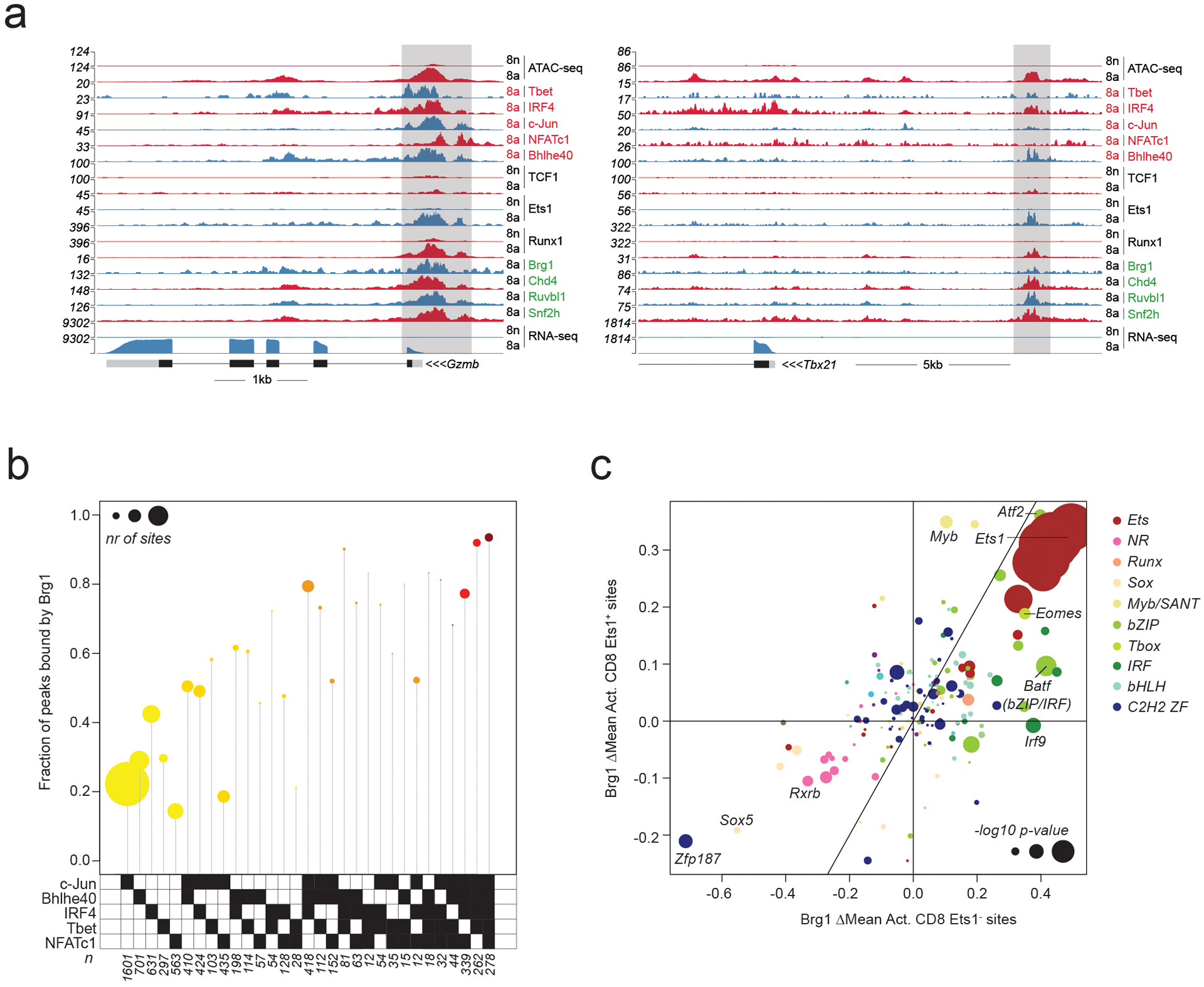
a: Gene tracks of RNA-seq, ATAC-seq and CUT&RUN coverage at the Gzmb and Tbx21 loci in naïve and activated CD8 T cells.
b: Fraction of peaks bound by Brg1 at Ets1-unbound ATAC-seq peaks occupied by distinct combinations of activation-induced TFs. Data points are scaled according to the number of peaks in each set.
c: Effect of TF binding motif variants on Brg1 occupancy at Ets1-bound (Ets1+) and Ets1-unbound (Ets1−) sites measured as ΔMean in activated CD8 T cells. Data points are colored by TF family and scaled according to the −log10 p-value of a t-test comparing the allelic ratios between peaks with stronger motif matches on the B6 vs Cast allele. The most significant p-value obtained across the two cell types (naïve and activated CD8) was used for scaling.
Extended Data Fig. 5. TF binding at TCF1-bound peak sets in naïve and activated CD4 and CD8 T cells.
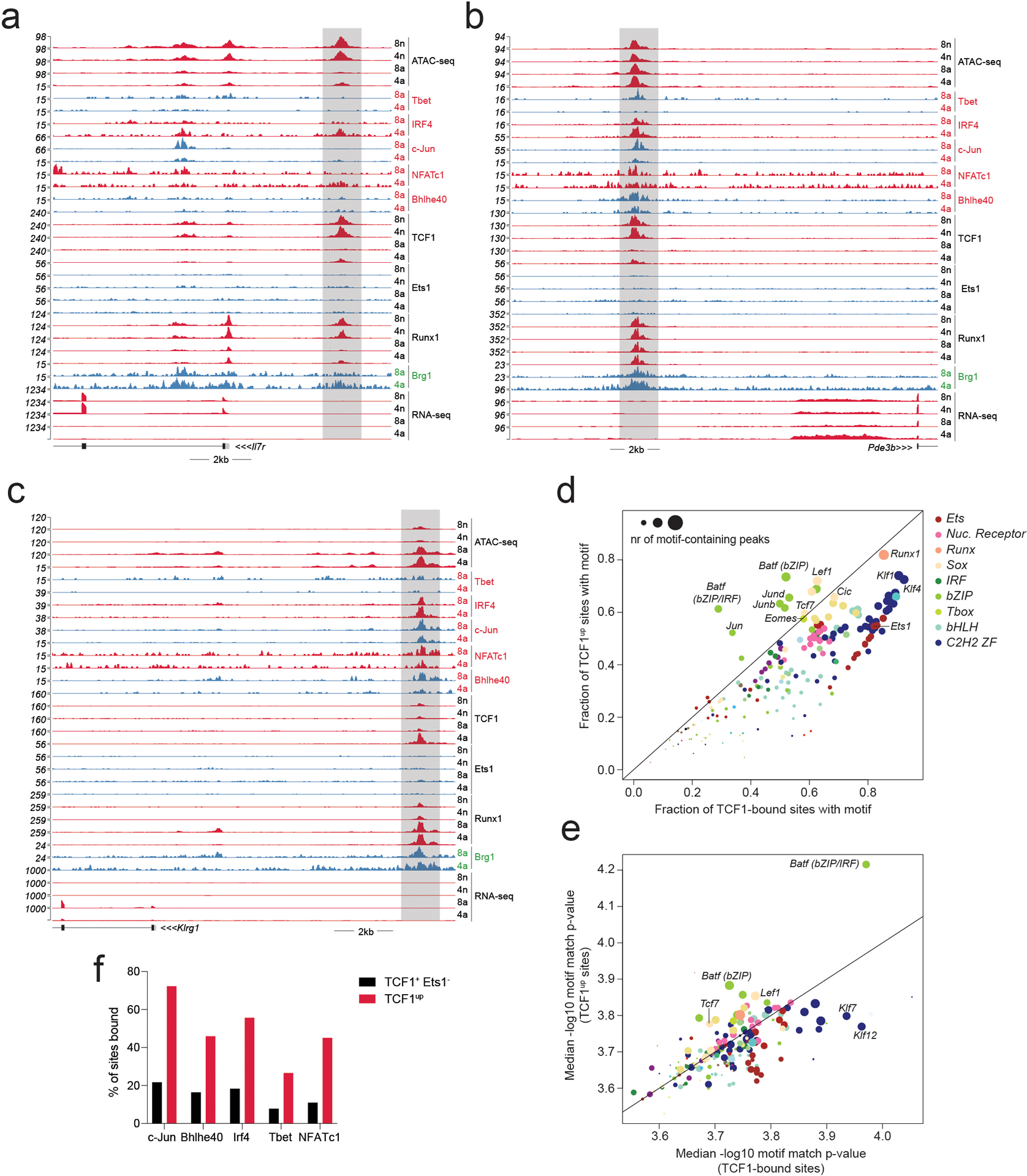
a-c: Gene tracks of RNA-seq, ATAC-seq and CUT&RUN read coverage at the Il7r, Pde3b and Klrg1 loci in naïve and activated CD4 and CD8 T cells.
d: Fraction of TCF1-bound sites and TCF1up sites containing specific TF-binding motifs.
e: Median motif match p-value at sites bound at TCF1-bound sites and TCF1up sites.
f: Percentage of TCF1+ Ets1− peaks and TCF1up peaks bound by each activation-induced TF.
Extended Data Fig. 6. Single cell ATAC-seq analysis of T cell subsets during acute LCMV infection.
a: Pseudo-bulk scATAC-seq tracks showing chromatin accessibility at CD4 and CD8 loci across splenic cell populations in LCMV infected (B6/Cast) F1 mice.
b: Effect of genetic variation in TF binding motifs on allele-specific ATAC-seq counts from bulk cell or single cell experiments. Data points are colored by TF family and scaled according to the −log10 p-value of a two-sided t-test comparing the allelic ratios between peaks with stronger matches on B6 vs Cast Allele.
c: Violin plots and box plots showing accessibility of indicated peak groups across T cell subsets. The center line in the box plots represents the median, the box limits represent the 25th and 75th percentiles, and the whiskers are the minimum/maximum values within 1.5 times the interquartile range. n = the number of cells in each cell annotation (420 Naïve CD4, 344 Naïve CD8, 234 Mem CD4, 231 Mem CD8, 198 Treg, 1321 Tfh, 652 Th1, 2583 Effector CD8)
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Rudensky lab for discussions and technical assistance. We thank the NIH tetramer core for providing critical reagents. This study was supported by NIH grants R01AI034206, U54 CA209975, NIH/NCI P30 CA008748, AACR-Bristol-Myers Squibb Immuno-oncology Research Fellowship 19-40-15-PRIT, National Natural Science Foundation of China (32170883 to Y.Z.), The Ludwig Center at Memorial Sloan Kettering Cancer Center, and the Parker Institute for Cancer Immunotherapy. A.Y.R. is an investigator with the Howard Hughes Medical Institute. We acknowledge the use of the MSKCC Single Cell Research Initiative (SCRI) and the Integrated Genomics Operation Core, funded by NIH/NCI Cancer Center Support Grant P30 CA008748, Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Footnotes
COMPETING INTERESTS
A.Y.R. is an SAB member, has equity in Sonoma Biotherapeutics and Vedanta Biosciences, and is a co-inventor or has IP licensed to Takeda that is unrelated to the content of this study. The remaining authors declare no competing interests.
CODE AVAILABILITY
The custom Shell and Python code used in this study are publicly available at Memorial Sloan Kettering Cancer Center website: http://cbio.mskcc.org/public/Leslie/zhongy/downloads/index8.html
DATA AVAILABILITY
All next generation sequencing data generated in this paper were deposited in the Gene Expression Omnibus (GEO) under SuperSeries accession numbers GSE166718 and GSE184006. Processed data are included with the manuscript as supplemental tables. The B6 reference genome (GRCm38) was downloaded from NCBI. Files CAST_EiJ.mgp.v5.snps.dbSNP142.vcf and CAST_EiJ.mgp.v5.indels.dbSNP142.normed.vcf with SNP and indel genetic variants between B6 and Cast genomes were obtained from the Mouse Genome Project53. Gene annotations were downloaded from ENSEMBL database at ftp.ensembl.org/ensembl/pub/release-83/gtf/ or obtained from GENCODE vM2560. Experimentally determined Mus musculus motifs identified by ChIP-seq, PBM, SELEX, or HocoMoco were retrieved from the CIS-BP database57.
REFERENCES
- 1.Kaech SM & Cui W Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12, 749–761, doi: 10.1038/nri3307 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu J, Yamane H & Paul WE Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28, 445–489, doi: 10.1146/annurev-immunol-030409-101212 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495, doi: 10.1146/annurev-immunol-041015-055318 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Mackay LK & Kallies A Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol 38, 94–103, doi: 10.1016/j.it.2016.11.004 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Milner JJ & Goldrath AW Transcriptional programming of tissue-resident memory CD8. Curr Opin Immunol 51, 162–169, doi: 10.1016/j.coi.2018.03.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaquerizas JM, Kummerfeld SK, Teichmann SA & Luscombe NM A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10, 252–263, doi: 10.1038/nrg2538 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Gray PA et al. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 306, 2255–2257, doi: 10.1126/science.1104935 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Badis G et al. Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723, doi: 10.1126/science.1162327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jolma A et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339, doi: 10.1016/j.cell.2012.12.009 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Gerstein MB et al. Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100, doi: 10.1038/nature11245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee TI & Young RA Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251, doi: 10.1016/j.cell.2013.02.014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spitz F & Furlong EE Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13, 613–626, doi: 10.1038/nrg3207 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Chang JT, Wherry EJ & Goldrath AW Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15, 1104–1115, doi: 10.1038/ni.3031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosokawa H & Rothenberg EV How transcription factors drive choice of the T cell fate. Nat Rev Immunol, doi: 10.1038/s41577-020-00426-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Veeken J et al. The Transcription Factor Foxp3 Shapes Regulatory T Cell Identity by Tuning the Activity of trans-Acting Intermediaries. Immunity 53, 971–984.e975, doi: 10.1016/j.immuni.2020.10.010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Veeken J et al. Natural Genetic Variation Reveals Key Features of Epigenetic and Transcriptional Memory in Virus-Specific CD8 T Cells. Immunity 50, 1202–1217.e1207, doi: 10.1016/j.immuni.2019.03.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hota SK & Bruneau BG ATP-dependent chromatin remodeling during mammalian development. Development 143, 2882–2897, doi: 10.1242/dev.128892 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polansky JK et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (Berl) 88, 1029–1040, doi: 10.1007/s00109-010-0642-1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wotton D, Ghysdael J, Wang S, Speck NA & Owen MJ Cooperative binding of Ets-1 and core binding factor to DNA. Mol Cell Biol 14, 840–850, doi: 10.1128/mcb.14.1.840 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim WY et al. Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J 18, 1609–1620, doi: 10.1093/emboj/18.6.1609 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang CY et al. Activation of the granulocyte-macrophage colony-stimulating factor promoter in T cells requires cooperative binding of Elf-1 and AP-1 transcription factors. Mol Cell Biol 14, 1153–1159, doi: 10.1128/mcb.14.2.1153 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bassuk AG & Leiden JM A direct physical association between ETS and AP-1 transcription factors in normal human T cells. Immunity 3, 223–237, doi: 10.1016/1074-7613(95)90092-6 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Pognonec P, Boulukos KE, Gesquière JC, Stéhelin D & Ghysdael J Mitogenic stimulation of thymocytes results in the calcium-dependent phosphorylation of c-ets-1 proteins. EMBO J 7, 977–983 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabault B & Ghysdael J Calcium-induced phosphorylation of ETS1 inhibits its specific DNA binding activity. J Biol Chem 269, 28143–28151 (1994). [PubMed] [Google Scholar]
- 25.Cowley DO & Graves BJ Phosphorylation represses Ets-1 DNA binding by reinforcing autoinhibition. Genes Dev 14, 366–376 (2000). [PMC free article] [PubMed] [Google Scholar]
- 26.Foulds CE, Nelson ML, Blaszczak AG & Graves BJ Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol Cell Biol 24, 10954–10964, doi: 10.1128/MCB.24.24.10954-10964.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bories JC et al. Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature 377, 635–638, doi: 10.1038/377635a0 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Muthusamy N, Barton K & Leiden JM Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 377, 639–642, doi: 10.1038/377639a0 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Mélet F, Motro B, Rossi DJ, Zhang L & Bernstein A Generation of a novel Fli-1 protein by gene targeting leads to a defect in thymus development and a delay in Friend virus-induced erythroleukemia. Mol Cell Biol 16, 2708–2718, doi: 10.1128/mcb.16.6.2708 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim CJ et al. The Transcription Factor Ets1 Suppresses T Follicular Helper Type 2 Cell Differentiation to Halt the Onset of Systemic Lupus Erythematosus. Immunity 49, 1034–1048.e1038, doi: 10.1016/j.immuni.2018.10.012 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Anderson MK, Hernandez-Hoyos G, Diamond RA & Rothenberg EV Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development 126, 3131–3148 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Yamada T, Park CS, Mamonkin M & Lacorazza HD Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Krüppel-like factors KLF4 and KLF2. Nat Immunol 10, 618–626, doi: 10.1038/ni.1730 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo CT et al. Ets transcription factor GABP controls T cell homeostasis and immunity. Nat Commun 8, 1062, doi: 10.1038/s41467-017-01020-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollenhorst PC, McIntosh LP & Graves BJ Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 80, 437–471, doi: 10.1146/annurev.biochem.79.081507.103945 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson CB et al. cis-acting sequences required for inducible interleukin-2 enhancer function bind a novel Ets-related protein, Elf-1. Mol Cell Biol 12, 1043–1053, doi: 10.1128/mcb.12.3.1043 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang CY, Petryniak B, Ho IC, Thompson CB & Leiden JM Evolutionarily conserved Ets family members display distinct DNA binding specificities. J Exp Med 175, 1391–1399, doi: 10.1084/jem.175.5.1391 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollenhorst PC, Shah AA, Hopkins C & Graves BJ Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev 21, 1882–1894, doi: 10.1101/gad.1561707 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollenhorst PC et al. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet 5, e1000778, doi: 10.1371/journal.pgen.1000778 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giese K, Kingsley C, Kirshner JR & Grosschedl R Assembly and function of a TCR alpha enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Genes Dev 9, 995–1008, doi: 10.1101/gad.9.8.995 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Verbaro DJ, Sakurai N, Kim B, Shinkai Y & Egawa T Cutting Edge: The Histone Methyltransferase G9a Is Required for Silencing of Helper T Lineage-Associated Genes in Proliferating CD8 T Cells. J Immunol 200, 3891–3896, doi: 10.4049/jimmunol.1701700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lazarevic V et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat Immunol 12, 96–104, doi: 10.1038/ni.1969 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang F, Meng G & Strober W Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol 9, 1297–1306, doi: 10.1038/ni.1663 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812, doi: 10.1038/nature08750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin B et al. Runx1 and Runx3 drive progenitor to T-lineage transcriptome conversion in mouse T cell commitment via dynamic genomic site switching. Proc Natl Acad Sci U S A 118, doi: 10.1073/pnas.2019655118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taniuchi I et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 111, 621–633, doi: 10.1016/s0092-8674(02)01111-x (2002). [DOI] [PubMed] [Google Scholar]
- 46.Egawa T, Tillman RE, Naoe Y, Taniuchi I & Littman DR The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med 204, 1945–1957, doi: 10.1084/jem.20070133 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levanon D et al. Transcriptional repression by AML1 and LEF-1 is mediated by the TLE/Groucho corepressors. Proc Natl Acad Sci U S A 95, 11590–11595, doi: 10.1073/pnas.95.20.11590 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Setoguchi R et al. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science 319, 822–825, doi: 10.1126/science.1151844 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Johnson JL et al. Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48, 243–257.e210, doi: 10.1016/j.immuni.2018.01.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218, doi: 10.1038/nmeth.2688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, doi: 10.7554/eLife.21856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu N et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e417, doi: 10.1016/j.cell.2018.03.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keane TM et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294, doi: 10.1038/nature10413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137, doi: 10.1186/gb-2008-9-9-r137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Q, Brown JB, Huang H & Bickel PJ Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779 (2011). [Google Scholar]
- 57.Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443, doi: 10.1016/j.cell.2014.08.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grant CE, Bailey TL & Noble WS FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018, doi: 10.1093/bioinformatics/btr064 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Link VM, Romanoski CE, Metzler D & Glass CK MMARGE: Motif Mutation Analysis for Regulatory Genomic Elements. Nucleic Acids Res 46, 7006–7021, doi: 10.1093/nar/gky491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773, doi: 10.1093/nar/gky955 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Granja JM et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411, doi: 10.1038/s41588-021-00790-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All next generation sequencing data generated in this paper were deposited in the Gene Expression Omnibus (GEO) under SuperSeries accession numbers GSE166718 and GSE184006. Processed data are included with the manuscript as supplemental tables. The B6 reference genome (GRCm38) was downloaded from NCBI. Files CAST_EiJ.mgp.v5.snps.dbSNP142.vcf and CAST_EiJ.mgp.v5.indels.dbSNP142.normed.vcf with SNP and indel genetic variants between B6 and Cast genomes were obtained from the Mouse Genome Project53. Gene annotations were downloaded from ENSEMBL database at ftp.ensembl.org/ensembl/pub/release-83/gtf/ or obtained from GENCODE vM2560. Experimentally determined Mus musculus motifs identified by ChIP-seq, PBM, SELEX, or HocoMoco were retrieved from the CIS-BP database57.