

Abstract
Aims
Children presenting with hypertrophic cardiomyopathy (HCM) in infancy are reported to have a poor prognosis, but this heterogeneous group has not been systematically characterized. This study aimed to describe the aetiology, phenotype, and outcomes of infantile HCM in a well‐characterized multicentre European cohort.
Methods and results
Of 301 children diagnosed with infantile HCM between 1987 and 2019 presenting to 17 European centres [male n = 187 (62.1%)], underlying aetiology was non‐syndromic (n = 138, 45.6%), RASopathy (n = 101, 33.6%), or inborn error of metabolism (IEM) (n = 49, 16.3%). The most common reasons for presentation were symptoms (n = 77, 29.3%), which were more prevalent in those with syndromic disease (n = 62, 61.4%, P < 0.001), and an isolated murmur (n = 75, 28.5%). One hundred and sixty‐one (53.5%) had one or more co‐morbidities. Genetic testing was performed in 163 (54.2%) patients, with a disease‐causing variant identified in 115 (70.6%). Over median follow‐up of 4.1 years, 50 (16.6%) underwent one or more surgical interventions; 15 (5.0%) had an arrhythmic event (6 in the first year of life); and 48 (15.9%) died, with an overall 5 year survival of 85%. Predictors of all‐cause mortality were an underlying diagnosis of IEM [hazard ratio (HR) 4.4, P = 0.070], cardiac symptoms (HR 3.2, P = 0.005), and impaired left ventricular systolic function (HR 3.0, P = 0.028).
Conclusions
This large, multicentre study of infantile HCM describes a complex cohort of patients with a diverse phenotypic spectrum and clinical course. Although overall outcomes were poor, this was largely related to underlying aetiology emphasizing the importance of comprehensive aetiological investigations, including genetic testing, in infantile HCM.
Keywords: Infant‐onset, Hypertrophic, Cardiomyopathy, Prognosis
Introduction
Hypertrophic cardiomyopathy (HCM) presenting in childhood has an estimated annual incidence of 0.24–0.47 per 100 000. 1 , 2 , 3 Nearly one quarter of paediatric HCM cases present in the first year of life, 4 where the reported annual incidence is 0.51–3.2 per 100 000. 1 , 2 , 3 The underlying aetiology in infant‐onset disease is more heterogeneous than that seen in later childhood, with a higher proportion of syndromic (e.g. RASopathy syndrome) or metabolic disease reported. 4 , 5 , 6 Historically, sarcomeric disease has been considered to be very rare infancy due to variable and age‐related incomplete penetrance. However, more recent publications have challenged this assumption and suggested that sarcomeric disease can present in the very young. 7 , 8 Patients with infant‐onset HCM are recognized to have a particularly poor long‐term prognosis, 4 , 6 , 9 , 10 , 11 , 12 , 13 , 14 with mortality largely attributed to heart failure. 4 , 6 , 9 However, no study to date has systematically described the aetiology, clinical features, and long‐term outcomes of infantile HCM. Understanding the spectrum and progression of disease along with an improved ability to predict long‐term outcomes would allow a more individualized approach to patient care, with important implications for clinical management. The aim of this study was to describe the aetiology, disease phenotype, and outcomes of infantile HCM in a well‐characterized multicentre European cohort.
Methods
Patient cohort
A retrospective, multicentre European cohort of children diagnosed with HCM in infancy between 1987 and 2019 was formed. Data were collected in 301 children from 17 European paediatric cardiac centres and included 159 patients included in previous reports. 4 , 15 The diagnosis of HCM was made if maximal left ventricular wall thickness (MLVWT) in any single segment was greater than two standard deviations above the body surface area‐corrected mean (Z score ≥ 2) and not explained by abnormal loading conditions. 16 The diagnosis of an underlying RASopathy syndrome, inborn error of metabolism (IEM), or neuromuscular disease (HCM phenocopies) was made by the local principal investigator following systematic assessment of phenotype, biochemical, and genetic testing. In the absence of an underlying diagnosis, the aetiology was classified as non‐syndromic, in line with previous publications. 4 Patients with confirmed disease‐causing sarcomeric variants were classified as non‐syndromic, but a separate analysis of their baseline characteristics and long‐term outcomes was performed. Eligible patients were identified by the principal investigator at each collaborating site.
Data collection
Anonymized, non‐invasive clinical data were collected from baseline evaluation, during follow‐up, and at last clinical review, including demographics; aetiology; co‐morbidities (cardiac or extra‐cardiac); symptoms; medication; family history; genetic testing; resting and ambulatory 12 lead electrocardiograms (ECG); 2D echocardiography; and surgical or catheter interventions. Heart failure symptoms were assessed using the Ross criteria (for symptom assessment below 5 years of age) 17 or the New York Heart Association (NYHA) functional classification (for those over 5 years of age). 17 Echocardiographic measurements were made according to current guidelines. 16 , 18 Left ventricular outflow tract (LVOT) obstruction was defined as a peak instantaneous gradient ≥ 30 mmHg. 16 Right ventricular outflow tract (RVOT) obstruction was defined as a peak instantaneous gradient ≥ 36 mmHg. 18 Impaired left ventricular (LV) systolic function was defined as a fractional shortening (FS) ≤ 28% or ejection fraction ≤ 55%. 18 Data were collected and verified by the principal investigator at each collaborating site.
Outcomes
The primary patient outcomes, taken from the last clinical encounter, were all‐cause mortality [congestive cardiac failure (CCF), sudden cardiac death (SCD), other cardiovascular (CV) death, and non‐CV death] or cardiac transplantation. Secondary outcomes included major arrhythmic cardiac events (MACE), defined as SCD or an equivalent event [appropriate implantable cardioverter defibrillator (ICD) therapy, aborted cardiac arrest, or sustained ventricular tachycardia (VT) with haemodynamic compromise] 16 ; atrial arrhythmias; and surgical/catheter‐based interventions. Outcomes were determined by the treating cardiologist at each site. Patients were classified as lost to follow‐up if last clinical review was more than 3 years from the end of study period (December 2019).
Genetics
Genetic testing was performed at the treating clinician's discretion. Data were collected from patients in whom genetic testing had been performed, including date of testing; size of gene panel; and variants identified (gene and protein change). Genetic testing use across different eras was investigated: pre‐2000; 2000–2004; 2005–2009; 2010–2014; and 2015 onwards. The pathogenicity of reported variants was reclassified by the authors according to the American College of Medical Genetics and Genomics (ACMG) classification. 19
Statistics
Body surface area was calculated from weight. 20 MLVWT and left atrial (LA) diameter measurements are expressed in millimetres and as body surface area‐corrected z scores. 21 , 22 Continuous variables are described as mean (± standard deviation) or median [interquartile range (IQR)], with three group comparisons conducted using analysis of variance (ANOVA) or Kruskal–Wallis tests, respectively. The distribution of categorical variables was compared using the χ 2 test or Fisher's exact test. A significance level of 0.05 was used for all comparisons.
Estimates of survival were obtained using the Kaplan–Meier product limit method. The association of clinical variables with the outcome of interest was assessed in a univariate Cox proportional hazard model. A P value of <0.1 was used to select variables for inclusion in a multivariable Cox proportional hazards regression model. Backwards selection techniques were used to identify variables that remained significant at 0.05 level. All statistical analyses were performed with STATA (Stata statistical software release 14; StataCorp LP, College Station, TX).
Ethics
This study complies with the Declaration of Helsinki. Local ethical approval was obtained at each participating site with waver of informed consent for retrospective, anonymized data. The data underlying this article cannot be shared publically as consent for dissemination of patient data was not obtained.
Results
Demographics and presentation
Three hundred and one patients with infant‐onset HCM were identified, of whom 187 (62.1%) were male. One hundred and thirty‐eight (45.8%) had non‐syndromic HCM, 101 (33.6%) had a RASopathy, and 49 (16.3%) had an IEM. Data on aetiology and reason for presentation are shown in Table 1 and Supporting Information, Table S1 . One hundred and sixty‐one patients (53.5%) had one or more co‐morbidities, which were more common in patients with a RASopathy (n = 76, 75.3%) or IEM (n = 35, 71.4%) (Supporting Information, Figure S1 ). The most common co‐morbidities varied by underlying diagnosis (Supporting Information, Table S2 ). Seventy‐seven (27.9%) had an additional cardiac lesion, most commonly an atrial or ventricular septal defect (n = 35) or valvar pulmonary stenosis (n = 22). Additional cardiac lesions were more common in those with a RASopathy syndrome (n = 42/101, 41.6%).
Table 1.
Clinical characteristic and long‐term outcome by aetiology
| Whole cohort (n = 306) | Non‐syndromic (138) | RASopathy (101) | IEM (49) | P value | ||||
|---|---|---|---|---|---|---|---|---|
| Non‐syndromic (138) | Non‐syndromic with genetic testing a (67) | Comparison by aetiology (n = 288) | Comparison by aetiology using reduced cohort (n = 217) a | |||||
| Male gender | 187 (62.1) | 88 (63.8) | 43 (65.4) | 66 (65.4) | 28 (57.1) | 0.609 | 0.607 | |
| FHx HCM (n = 294) | 69 (23.5) | 48 (37.5) | 33 (49.3) | 8 (8.1) | 12 (24.5) | <0.001 | <0.001 | |
| FHx SCD | 23 (7.6) | 14 (10.1) | 7 (10.9) | 2 (2) | 6 (12.2) | 0.026 | 0.048 | |
| Reason for referral (n = 263) | Symptomatic | 77 (29.3) | 18 (17.1) | 12 (19.4) | 35 (40.7) | 21 (43.8) | <0.001 | <0.001 |
| Antenatal diagnoses | 9 (3.4) | 1 (1) | 1 (1.6) | 5 (5.8) | 2 (6.3) | |||
| Murmur | 75 (28.5) | 45 (42.9) | 26 (41.9) | 23 (26.7) | 4 (8.3) | |||
| Family screening | 24 (8.0) | 21 (20) | 14 (22.6) | 0 | 2 (4.2) | |||
| Screening for associated condition | 17 (6.5) | 0 | 0 | 3 (3.4) | 11 (22.9) | |||
| Other | 50 (19.0) | 20 (19.1) | 9 (14.5) | 20 (23.3) | 7 (14.6) | |||
| Co‐morbidities | Any | 161 (53.5) | 50 (36.2) | 22 (32.8) | 76 (75.3) | 35 (71.4) | <0.001 | <0.001 |
| Cardiac (n = 276) | 77 (27.9) | 30 (21.7) | 13 (19.4) | 42 (41.6) | 4 (8.2) | <0.001 | <0.001 | |
| Initial clinical assessment | ||||||||
| Ross class > 1 (n = 271) | 99 (36.5) | 24 (21.4) | 11 (17.2) | 47 (49.5) | 24 (49.0) | <0.001 | <0.001 | |
| Any cardiac symptoms | 129 (42.9%) | 47 (34.1) | 62 (61.4) | 31 (63.3) | <0.001 | <0.001 | ||
| Pattern of hypertrophy (n = 267) | ASH | 118 (44.2) | 80 (65.6) | 53 (82.8) | 26 (28.6) | 6 (12.2) | <0.001 | <0.001 |
| Concentric | 64 (23.9) | 16 (13.1) | 4 (6.3) | 22 (24.2) | 24 (45.0) | |||
| Biventricular | 85 (31.8) | 22 (18.0) | 7 (10.9) | 40 (44.0) | 18 (36.7) | |||
| MWT (mm) | Median, IQR | 9 (7,12) | 9.3 (7.5,13.0) | 10.5 (8, 13) | 9 (7,11.8) | 9 (7,11) | 0.403 | 0.024 |
| MWT z score | Mean (±) | 10.8 (5.8) | 10.3 (4.0) | 11.6 (±5.3) | 9.3 (3.1) | 9.9 (3.9) | 0.823 | 0.946 |
| Impaired systolic function (n = 160) | 12 (7.5) | 3 (2.2) | 1 (1) | 1 (1) | 7 (14.3) | 0.001 | 0.001 | |
| LVOT obstruction (n = 265) | 160 (60.6) | 42 (43.8) | 22 (36.1) | 49 (62.8) | 6 (17.1) | <0.001 | <0.001 | |
| RVOT obstruction (>16 mmHg) (n = 155) | 66 (42.6) | 12 (20.3) | 6 (26.1) | 48 (72.7) | 5 (20) | <0.001 | <0.001 | |
| Outcome | ||||||||
| Died | 48 (15.9) | 11 (8.0) | 3 (4.5) | 16 (15.8) | 20 (40.8) | <0.001 | <0.001 | |
| SCD | 8 (2.7) | 3 | 2 | 2 | 3 | |||
| CCF | 14 (4.7) | 6 | 1 | 4 | 4 | |||
| Other CV | 4 (1.3) | 0 | 0 | 3 | 0 | |||
| Non‐CV | 20 (6.6) | 2 | 0 | 6 | 12 | |||
| Unknown | 2 (0.7) | 0 | 0 | 1 | 1 | |||
| Transplant | 6 | 3 | 1 | 1 | 2 | |||
| Mortality or transplant incidence rate/100 patient years | 3.1 (95% CI 2.41–4.11) | 1.33 (95% CI 0.73–2.39) | 0.87 (95% CI 0.33–2.32) | 2.39 (95% CI 1.47–3.91) | 13.22 (95% CI 8.62–20.29) | <0.001 | <0.001 | |
| Survival | 1 year | 86.2 (95% CI 81.7–89.7%) | 94.7 (95C% CI 89.2–97.4) | 96.0 (95% CI 88.3–99.2) | 88.0 (95% CI 79.8–93.0) | 65.1 (95%CI 50.0–76.7) | ||
| 5 years | 83.1 (95% CI 78.2% ‐ 87.0) | 93.0 (95% CI 86.9–96.3) | 96.0 (95% CI 88.3–99.2) | 85.7 (95%CI 77.0–91.3) | 57.9 (95% CI 42.5–70.6) | |||
| 10 years | 80.0 (95% CI 73.8–84.9) | 90.0 (95% CI 79.8–95.2) | 96.0 (95% CI 88.3–99.2) | 81.1 (95%CI 69.7–88.6) | 57.9 (95% CI 42.5–70.6) | |||
ASH, asymmetric septal hypertrophy; CCF, congestive cardiac failure; CI, confidence interval; CV, cardiovascular; FHx, family history; HCM, hypertrophic cardiomyopathy; IEM, inborn error of metabolism; IQR, interquartile range; LVOT, left ventricular outflow tract; MACE, major arrhythmic cardiac event; MWT, maximal wall thickness; NYHA, New York Heart Association; RVOT, right ventricular outflow tract; SCD, sudden cardiac death.
Patients in whom genetic testing has been performed.
Initial clinical phenotype
Data on initial clinical assessment are shown in Table 1 . Cardiac medication was started in 146 (54.7%): beta‐blockers (n = 137, 51.3%); diuretics (n = 39, 14.6%); heart failure therapy including angiotensin‐converting enzyme inhibitors (ACE‐I) and diuretics (n = 9, 3.4%); anti‐arrhythmics (n = 3, 1.1%); disopyramide (n = 2, 0.7%); and calcium channel blockers (n = 1, 0.4%). Five patients required cardioactive inotropic support at presentation, of which four had an IEM.
Genetics
Genetic testing strategy by era and aetiology is summarized in Figure 1 and Supporting Information, Table S3 . Genetic testing was performed in 163 (54.2%) patients, with a disease‐causing variant identified in 115 (70.6%) (Supporting Information, Table S4 ). Patients with a RASopathy were more likely to undergo genetic testing (RASopathy n = 72, 71.3% vs. IEM n = 24, 49.0% vs. non‐syndromic n = 67, 48.6%, P value 0.001), but there was no difference in the overall prevalence of genetic testing over time (Figure 1 , Supporting Information, Table S3 , P value 0.244).
Figure 1.
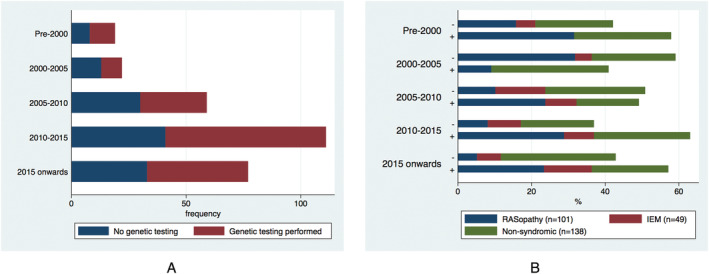
Use of genetic testing in infantile hypertrophic cardiomyopathy. (A) By era (P value 0.244). (B) By underlying aetiology and result. (+) represents identification of a centre‐reported variant of unknown significance or disease‐causing variant. (−) represents a negative genetic test. IEM, inborn error of metabolism.
Of patients classified as non‐syndromic, those who underwent genetic testing were more likely to have a family history of HCM (n = 33, 49.3% vs. n = 15, 24.6%, P value 0.004) and an asymmetric pattern of hypertrophy (n = 53, 82.8% vs. n = 27, 46.6%, P value < 0.001), but did not differ by the prevalence of co‐morbidities (n = 22, 32.8% vs. n = 28, 39.4%, P value 0.420). Of variants previously classified as pathogenic (P)/likely pathogenic (LP), after ACMG reclassification (n = 30), 20 variants remained pathogenic/likely pathogenic (MYBPC3 n = 7, MYH7 n = 9, KRAS n = 1, LZRT1 n = 1, MYBPC3 + MYBPC3 = 1) and 10 were reclassified as a VUS (MYBPC3 n = 1, MYH7 n = 2, RAF1 n = 1, TPM1 n = 1, ACTN n = 2, MYOM1 n = 1, PRKAG2 n = 1, PKP2 n = 1). Three patients had more than one variant identified [MYBPC3 (P) + MYBPC3 (LP); MYH7 (LP) + PRKAG2 (LP); MYH7 (LP) + PKP2 (VUS)]. Baseline demographics for those with and without a disease‐causing sarcomeric variant identified on genetic testing is described in Supporting Information, Table S5 .
For patients with a RASopathy following ACMG reclassification (n = 43), 33 patients had a single pathogenic/likely pathogenic variant (RAF1 n = 5, PTPN11 n = 17, RIT1 n = 6, HRAS n = 4, BRAF n = 1) and 5 had more than one variant [PTPNP11 (P) + MYH7 (VUS), LZRT1 (VUS) + MYH7 (VUS); PTPN11 (P) + MYH7 (LP); LZRT1 (LP) + HRAS (VUS); DSC2 (VUS) + SCN5A (VUS)].
Clinical follow‐up
Median length of follow‐up was 4.1 years (IQR 1.3–8.1, range 0–30.9 years). Eighty‐nine patients were followed up for 10 years or longer [non‐syndromic HCM n = 43 (48.3%); RASopathy n = 41 (46.1%); IEM n = 5 (5.6%)].
Interventions
Table 2 describes surgical and interventional procedures. An LV septal myectomy was performed in 28 patients (9.3%) (non‐syndromic n = 10, RASopathy n = 17, IEM n = 1) at median age 4.8 years (IQR 1.0–6.5, range 0–13.2), with concomitant mitral valve repair in 6. Twenty‐two patients (7.3%) (RASopathy n = 20, IEM n = 2) underwent RVOT obstruction relief (balloon pulmonary valvulopasty n = 9, surgical relief n = 18) at median age 7.3 months (IQR 4.1–10.8, range 0.8–112.3). Eight patients required more than one surgical procedure. Ten patients (3.3%) had an ICD inserted for primary (n = 6) or secondary (n = 3) prevention, at a median (range) age of 13.4 years (6.5–12.1) and 8.4 years (2.5–11.3), respectively, of which two had appropriate ICD therapies (Supporting Information, Table S6 ).
Table 2.
Interventions (catheter and surgical) during follow‐up
| Intervention | N | ||
|---|---|---|---|
| ICD | Primary | 6 | |
| Secondary | 3 | ||
| Unknown indication | 1 | ||
| Pacemaker | Sinoatrial disease | 2 | |
| AV block | 3 | ||
| Surgery | RVOT relief | 18 | |
| RVOT Patch | 11 | ||
| Valvotomy | 4 | ||
| Suprapulmonary PS relief (patch) | 1 | ||
| RVOT conduit | 1 | ||
| Unknown | 1 | ||
| Myectomy | 28 | ||
| With MV repair | 6 | ||
| Aortic valvotomy | 3 | ||
| Other cardiac surgery | ASD closure | 3 | |
| PDA ligation | 4 | ||
| Coarctation repair | 2 | ||
| LVAD | 2 | ||
| Sympathectomy | 1 | ||
| Multiple surgical procedures | Myectomy | 2 (n = 3), 3 (n = 1) | |
| Mitral valve replacement | 2 (previous MV plasty + myectomy; previous MV repair + RVOT relief) | ||
| RVOT relief followed by myectomy | 2 | ||
| Catheter interventions | Balloon pulmonary valvuloplasty | 9 (7 of whom required subsequent surgical intervention) | |
| PDA closure | 1 | ||
| EPS | Ablation | 5 (symptomatic SVT n = 4, WPW n = 1) | |
ASD, atrial septal defect; AV, atrioventricular; EPS, electrophysiology study; ICD, implantable cardioverter defibrillator; LVAD, left ventricular assist device; MV, myectomy; PDA, patent ductus arteriosus; PS, pulmonary stenosis; RVOT, right ventricular outflow tract; SVT, supraventricular tachycardia; WPW, Wolff–Parkinson–White syndrome.
Arrhythmias
Fifteen patients (4.9%) [non‐syndromic (n = 7, 5.1%); IEM (n = 5, 10.2%); RASopathy (n = 3, 3%)] had one or more MACE (sustained VT with haemodynamic compromise in eight; SCD in six; resuscitated cardiac arrest in four; and appropriate ICD therapy in two) during follow‐up (Supporting Information, Table S6 ), with an overall annual incidence rate of 0.88 [95% confidence interval (CI) 0.527–1.451]. Six MACE (43%) occurred during infancy; the remaining occurred at a mean age of 9.2 years (range 1.4–14.9). Eleven (3.7%) had supraventricular arrhythmias detected during follow‐up.
Mortality
A total of 253 patients (84.1%) were alive at last clinical follow‐up, including 6 (2.0%) who had undergone cardiac transplantation. Forty‐eight patients (15.9%) died: non‐CV 20 (6.6%) (infection n = 8, respiratory n = 3, metabolic acidosis n = 3, neurological insult n = 2, gastrointestinal bleed n = 1, not described n = 3); CCF 14 (4.7%); SCD 8 (2.7%); other CV 4 (1.3%); and unknown 2 (0.7%). Six patients (2%) were lost to clinical follow‐up. Figure 2 shows the cause and age at time of death by aetiology. Thirty‐two (66.7%) deaths occurred in infancy (IEM n = 15/23, 75.0%; RASopathy n = 10/16, 62.5%; non‐syndromic n = 7/11, 63.6%) most commonly secondary to non‐CV causes (IEM n = 9, 60%; RASopathy n = 4, 10%; non‐syndromic n = 1, 14.3%) or CCF (IEM n = 4, 26.7%; RASopathy n = 3, 30.0%; non‐syndromic n = 5, 71.4%). Of deaths occurring after early childhood (>5 years), four (80%) were sudden and one (20%) was heart failure related. Overall survival free from all‐cause mortality or transplant was 86.2% (81.7–89.7%) at 1 year and 83.1% (95% CI 78.2–87.0) at 5 years. Survival varied by aetiology (Table 1 , Figure 2 C ). Predictors of all‐cause mortality at baseline on multivariable analysis were an underlying diagnosis of an IEM [hazard ratio (HR) 4.40 (1.95–9.66), P = 0.070], cardiac symptoms [HR 3.26 (95% CI 1.42–7.48), P = 0.005], and impaired LV systolic function [HR 2.97 (95% CI 1.12–7.87), P = 0.028]. Of those with impaired systolic function at baseline, 6 (50%) died; cause of death was SCD (n = 1), CCF (n = 1), and non‐CV death (sepsis n = 3, metabolic acidosis n = 1). Predictors of CV mortality or transplantation on multivariable analysis were cardiac symptoms at diagnosis [HR 19.1 (95% CI 4.22–86.70), P < 0.001] and higher MLVWT [HR 1.19 per mm increase (95% CI 1.07–1.34), P = 0.002] (Table 3 ).
Figure 2.
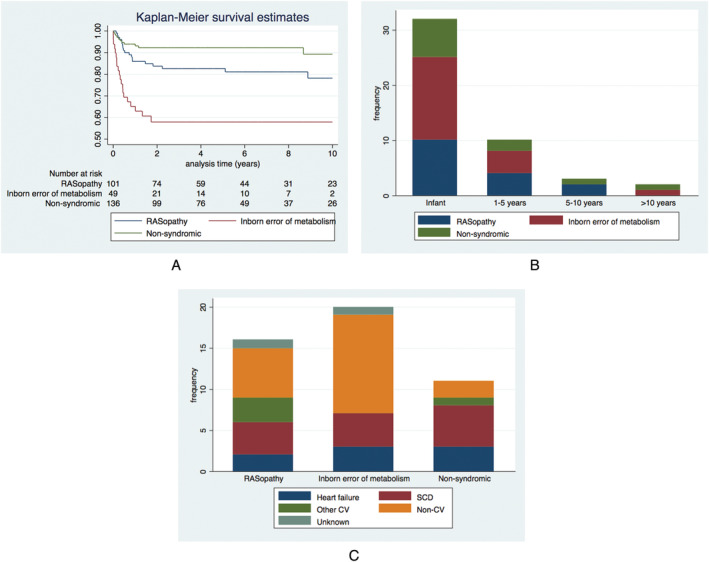
Long‐term survival of infantile hypertrophic cardiomyopathy. (A) Kaplan–Meier curves of transplant free survival by underlying aetiology. (B) Age at death by aetiology. (C) Cause of death by aetiology. CV, cardiovascular; SCD, sudden cardiac death.
Table 3.
Univariable and multivariable Cox regression analysis for predictors of outcome
| All‐cause mortality | Cardiovascular mortality or transplant | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Univariable analysis | Multivariable analysis | Univariable analysis | Multivariable analysis | ||||||
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | ||
| Female | 0.94 (0.527–1.695) | 0.849 | 0.85 (0.409–1.784) | 0.675 | |||||
| Aetiology | Non‐syndromic | Baseline | Baseline | ||||||
| RASopathy | 1.87 (0.867–4.029) | 0.110 | 1.10 (0.534–2.272) | 0.793 | |||||
| IEM | 6.40 (3.054–13.484) | <0.001 | 4.40 (1.950–9.659) | <0.001 | 2.45 (1.088–5.521) | 0.029 | 2.19 (0.938–5.125) | 0.070 | |
| Co‐morbidities | Cardiac | 0.82 (0.406–1.645) | 0.571 | 0.81 (0.328 0 1.983) | 0.640 | ||||
| Any | 0.63 (0.345–1.140) | 0.126 | 1.29 (0.631–2.653) | 0.482 | |||||
| Family history of HCM | 0.26 (0.095–0.735) | 0.011 | 0.44 (0.155–1.280) | 0.133 | |||||
| Family history of SCD | 1.11 (0.399–3.094) | 0.840 | 1.32 (0.401–4.340) | 0.649 | |||||
| Any symptoms at baseline | 3.96 (2.011–7.805) | <0.001 | 3.26 (1.419–7.483) | 0.005 | 7.84 (2.727–22.557) | <0.001 | 19.1 (4.222–86.704) | <0.001 | |
| LVOTO | 1.49 (0.775–2.852) | 0.233 | 0.66 (0.314–1.385) | 0.272 | |||||
| RVOTO | 1.37 (0.634–2.957) | 0.424 | 2.07 (0.736–5.844) | 0.167 | |||||
| Increasing maximal wall thickness | 1.01 (0.934–1.100) | 0.742 | 0.0437 | 1.19 (1.067–1.344) | 0.002 | ||||
| Impaired systolic function | 5.43 (2.117–13.248) | <0.001 | 2.97 (1.124–7.865) | 0.028 | 1.46 (0.190–11.217) | 0.717 | |||
CI, confidence interval; HCM, hypertrophic cardiomyopathy; HR, hazard ratio; IEM, inborn error of metabolism; LVOTO, left ventricular outflow tract obstruction; RVOTO, right ventricular outflow tract obstruction; SCD, sudden cardiac death.
Discussion
This multicentre study of infantile HCM is, to our knowledge, the first systematic description of infant‐onset HCM and describes a complex and varied cohort of patients with a diverse phenotypic spectrum, clinical course, and outcome. Overall prognosis was poor, but was largely dependent on underlying aetiology, which emphasizes the importance of making an accurate diagnosis in these patients. Genetic testing identified a disease‐causing variant in up to 70% and should be considered for all patients with infant‐onset HCM.
Aetiology of infantile hypertrophic cardiomyopathy
The aetiology of childhood HCM is more varied than that seen in adulthood, driven by a higher proportion of IEMs and RASopathies in patients presenting in infancy. 4 , 5 , 6 , 9 This is highlighted by the findings in this study, in which over 50% of patients had metabolic or syndromic disease. The proportion of patients with syndromic disease in particular was higher than previously reported in 328 patients with infantile HCM in the Pediatric Cardiomyopathy Registry (PCMR) 6 ; this may reflect differences in systematic screening of children with syndromes known to be associated with HCM, or more comprehensive aetiological investigations in the expert centres recruiting to this study.
A major strength of this study is the high frequency of genetic testing and diagnostic yield, with an identifiable underlying aetiology identified in the majority of the patients, emphasizing the importance of systematic and comprehensive aetiological investigations, including genetics, in infantile HCM. Almost half the patients were classified as having non‐syndromic disease, and a disease‐causing sarcomeric variant was identified in 60% of those in whom genetic testing was performed most commonly in MYBPC3 (n = 15) or MYH7 (n = 14), supporting the notion that sarcomeric disease can manifest at any age. 23 , 24 The lack of significant differences between those with a sarcomeric variant and those non‐syndromic patients in whom genetic testing was not performed suggests that a substantial proportion of these are also likely to have variants in one or more cardiac sarcomere protein genes. 25 , 26 Variants in the RAS‐MAPK pathway genes were identified in three non‐syndromic patients, in keeping with previous reports that non‐sarcomeric variants may contribute to the disease phenotype in children with apparently non‐syndromic disease. 8 Additionally, one patient with metabolic disease had a co‐existing pathogenic sarcomeric variant, which emphasizes the importance of considering dual pathology. Although case reports and small case series have described severe early‐onset disease associated with compound or heterozygote sarcomeric gene variants, 27 , 28 only one patient had a compound heterozygote variant in MYBPC3 and two further patients had an additional VUS in a cardiomyopathy‐associated gene. It is possible that compound heterozygosity or double homozygosity results in foetal demise in most cases, but these findings suggest that the majority of disease seen in infancy is caused by single pathogenic sarcomeric variants. The presence of variants in non‐HCM genes, although unlikely to explain the HCM phenotype alone, could act as modifiers for other, as yet unidentified, HCM disease‐causing variants. Previous studies have suggested that patients with de novo sarcomeric variants are at higher risk of adverse outcomes, including mortality. Data on inheritance pattern were not collected in this study, but future studies systematically investigating the prevalence of de novo variants and role of genotype in long‐term outcomes for infantile HCM would be useful.
A family history of HCM was present in a higher proportion of non‐syndromic patients than expected (38%), one quarter of patients with an IEM, and nearly 1 in 10 patients with a RASopathy syndrome, highlighting the need for performing family screening regardless of the aetiology. 23 , 24
Phenotypic characteristics
A major finding of this study is the high frequency of associated complex medical needs, with over half having an additional co‐morbidity. The most frequent co‐morbidities were cardiac or neurological, but this differed by underlying aetiology. As expected, co‐morbidities were most common in those with syndromic disease, but were also present in one‐third of those classified with non‐syndromic disease. It is possible that a proportion of the so‐called ‘non‐syndromic’ patients had undiagnosed syndromic disease as genetic testing was not universally performed. However, there was no difference in the prevalence of co‐morbidities between those undergoing genetic testing or those identified to have a disease‐causing sarcomere variant suggesting that co‐morbidities are also common in non‐syndromic infantile HCM patients. It is noteworthy that one‐third of patients with an IEM had no additional co‐morbidities; this could reflect under‐reporting of non‐cardiac features by cardiologists, a predominant cardiac phenotype in some IEM, or age‐related penetrance of non‐cardiac manifestations.
Consistent with previous reports, the HCM phenotype varied according to the underlying aetiology. 5 , 6 , 10 , 13 Concentric LV hypertrophy and biventricular hypertrophy were more common in those with syndromic disease; co‐existing RVOT obstruction was seen predominantly in RASopathy patients, and LVOT obstruction was rare in those with IEM. One‐third of patients had heart failure symptoms at presentation, and 12 (8%) had impaired LV systolic function, the majority of which (n = 7) had an IEM. Outlook was poor for this subgroup of patients, with 50% dying during follow‐up, although the majority from non‐cardiac causes. The finding of impaired systolic function should therefore prompt clinicians to look for an underlying syndromic or metabolic aetiology.
Long‐term outcomes
Diagnosis of HCM in infancy has been shown repeatedly to be associated with poor short‐term outcomes, 10 and in keeping with this, in our cohort, 1 year mortality was 14%. However, survival rates varied significantly according to the underlying aetiology, with non‐syndromic patients having a much better prognosis compared with IEM. The cause of death also differed by aetiology, with non‐CV causes accounting for the majority of deaths for IEM, whilst cardiac causes were more common for non‐syndromic HCM or RASopathies. One strength of this study is the long‐term follow‐up of patients (30% had over 10 years' follow‐up), allowing long‐term trends in survival to be investigated. For all aetiology groups, most deaths occurred during infancy, with survival curves plateauing in later follow‐up, suggesting that, for those infants who survive beyond 2 years, the prognosis is substantially better. Previous long‐term population studies have described differences in the mode of death during follow‐up, 9 with early deaths caused by heart failure and later deaths resulting from ventricular arrhythmias. Whilst similar trends were seen in this study, the majority of early deaths were non‐cardiac and 3 out of 11 arrhythmic deaths occurred during infancy. This highlights both the importance of risk stratification for SCD at all ages and the burden of non‐CV disease in infantile HCM.
The overall MACE rate (0.82/100 patient years) seen in this cohort was lower than that reported in children presenting above 1 year (approximately 1.2–1.4/100 patient years). 4 , 6 , 9 , 29 Importantly, over half of MACE were in patients with syndromic disease, including IEM (e.g. Pompe's and mitochondrial) and RASopathies, the majority of which occurred in infancy, challenging the concept that malignant arrhythmias are rare in syndromic and metabolic HCM. It is beyond the scope of this study to investigate risk stratification for SCD in infant‐onset disease, and further work to identify risk factors specific to the infantile HCM population is required.
Heart failure symptoms, 10 , 11 , 13 low birth weight, 10 degree of hypertrophy, 5 , 9 , 10 , 11 impaired systolic function, 9 , 11 ‘mixed’ phenotypes, 10 and underlying aetiology 9 , 11 have been described as important clinical predictors of worse outcomes. However, most studies to date have been limited by small patient numbers and a lack of detailed aetiological information, with infantile disease treated as a homogenous group for the purpose of analysis. In our cohort, all‐cause mortality and CV mortality or transplantation were associated not only with the presence of symptoms and echocardiographic phenotypic parameters but also with a diagnosis of IEM. This suggests that underlying aetiology as well as phenotype is important for prognosis and emphasizes the importance of a systematic approach to making an accurate diagnosis in this group of patients.
Limitations
This study is limited by inherent problems of retrospective studies, in particular missing or incomplete data, particularly in relation to genetic variant data. Variations in clinical assessment and patient management are inevitable as patients were recruited from multiple centres in different geographical locations. As genetic testing was performed at the participating clinicians' discretion and across different eras with different sized panels and gene sequencing techniques, it is beyond the scope of this study to discuss the clinical yield of genetic testing in infantile HCM. Although a high proportion of patients with a RASopathy syndrome or IEM had a disease‐causing variant identified on genetic testing, it is not known whether genetic testing results altered the final diagnosis or confirmed previous clinical suspicions. Further work to explore the age‐related and gene‐related penetrance of sarcomeric disease in infant HCM is needed. Although the mortality rate is unlikely to be affected by these missing data due to nationally recorded death data in participating countries, other outcomes, such as arrhythmic events or surgical interventions, could have been underestimated.
Conclusions
This large multicentre study of infantile HCM describes a complex and varied cohort of patients with a diverse phenotypic spectrum, genetic substrate, clinical course, and outcome. Prognosis depends on clinical presentation, disease phenotype, and the underlying aetiology, which emphasizes the importance of making an accurate diagnosis in these patients. Genetic testing identified a disease‐causing variant in up to 70% and should be considered for all patients with infant‐onset HCM. Arrhythmic events were rare but occurred during infancy and in patients with syndromic disease, highlighting the importance of a systematic approach to diagnosis, screening, and risk stratification even in very young patients with HCM.
Conflict of interest
None to declare.
Funding
This work was supported by the British Heart Foundation (grant number FS/16/72/32270) to G.N. and J.P.K. and the Association for European Paediatric and Congenital Cardiology (AEPC junior grant) to G.N. J.P.K. and E.F. are supported by Max's Foundation, Great Ormond Street Hospital Charity, and Great Ormond Street Hospital for Children. J.P.K. is the recipient of a Medical Research Council (MRC) Clinical Academic Research Partnership (CARP) award. This work is (partly) funded by the NIHR GOSH BRC. The views expressed are those of the author(s) and not necessarily those of the NHS, NIHR, or the Department of Health. This work was (partly) supported by Children's Memorial Health Institute (statutory grant number S176/2018) to L.Z.
Supporting information
Table S1. Aetiology of patients with infantile disease.
Table S2. Prevalence of comorbidities by underlying diagnosis.
Table S3. Use of genetic testing over time by underlying aaetiology.
Table S4. Results of genetic testing in non‐syndromic and RASopathy patients.
Table S5. Comparison of baseline demographics in Non‐syndromic patients according to genetic testing status.
Table S6. Description of clinical characteristics and outcomes of patients with major arrhythmic cardiac events.
Figure S1. Comorbidities according to aetiology in infantile HCM
Figure S2. Genetic testing strategy in infantile HCM
Norrish, G. , Kolt, G. , Cervi, E. , Field, E. , Dady, K. , Ziółkowska, L. , Olivotto, I. , Favilli, S. , Passantino, S. , Limongelli, G. , Caiazza, M. , Rubino, M. , Baban, A. , Drago, F. , Mcleod, K. , Ilina, M. , McGowan, R. , Stuart, G. , Bhole, V. , Uzun, O. , Wong, A. , Lazarou, L. , Brown, E. , Daubeney, P. E. F. , Lota, A. , Delle Donne, G. , Linter, K. , Mathur, S. , Bharucha, T. , Adwani, S. , Searle, J. , Popoiu, A. , Jones, C. B. , Reinhardt, Z. , and Kaski, J. P. (2021) Clinical presentation and long‐term outcomes of infantile hypertrophic cardiomyopathy: a European multicentre study. ESC Heart Failure, 8: 5057–5067. 10.1002/ehf2.13573.
References
- 1. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003; 348: 1639–1646. [DOI] [PubMed] [Google Scholar]
- 2. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, Tikanoja T, Paavilainen T, Simell O. Epidemiology of idiopathic cardiomyopathies in children and adolescents. a nationwide study in Finland. Am J Epidemiol 1997; 146: 385–393. [DOI] [PubMed] [Google Scholar]
- 3. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003; 348: 1647–1655. [DOI] [PubMed] [Google Scholar]
- 4. Norrish G, Field E, McLeod K, Ilina M, Stuart G, Bhole V, Uzun O, Brown E, Daubeney PEF, Lota A, Linter K, Mathur S, Bharucha T, Kok KL, Adwani S, Jones CB, Reinhardt Z, Kaski JP. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: a retrospective study in United Kingdom. Eur Heart J 2019; 40: 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population‐based study. Circulation 2005; 112: 1332–1338. [DOI] [PubMed] [Google Scholar]
- 6. Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA. Epidemiology and cause‐specific outcome of hypertrophic cardiomyopathy in children: findings from the pediatric cardiomyopathy registry. Circulation 2007; 115: 773–781. [DOI] [PubMed] [Google Scholar]
- 7. Marston NA, Han L, Olivotto I, Day SM, Ashley EA, Michels M, Pereira AC, Ingles J, Semsarian C, Jacoby D, Colan SD, Rossano JW, Wittekind SG, Ware JS, Saberi S, Helms AS, Ho CY. Clinical characteristics and outcomes in childhood‐onset hypertrophic cardiomyopathy. Eur Heart J 2021; 42: 1988–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaski JP, Syrris P, Shaw A, Alapi KZ, Cordeddu V, Esteban MT, Jenkins S, Ashworth M, Hammond P, Tartaglia M, McKenna WJ, Elliott PM. Prevalence of sequence variants in the RAS‐mitogen activated protein kinase signaling pathway in pre‐adolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2012; 5: 317–326. [DOI] [PubMed] [Google Scholar]
- 9. Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, Turner C, Davis AM, Semsarian C, Colan SD, Robertson T, Ramsay J, Justo R, Sholler GF, King I, Weintraub RG. Long‐term outcomes of hypertrophic cardiomyopathy diagnosed during childhood: results from a national population‐based study. Circulation 2018; 138: 29–36. [DOI] [PubMed] [Google Scholar]
- 10. Lipshultz SE, Orav EJ, Wilkinson JD, Towbin JA, Messere JE, Lowe AM, Sleeper LA, Cox GF, Hsu DT, Canter CE, Hunter JA, Colan SD. Risk stratification at diagnosis for children with hypertrophic cardiomyopathy: an analysis of data from the pediatric cardiomyopathy registry. Lancet 2013; 382: 1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suda K, Kohl T, Kovalchin JP, Silverman NH. Echocardiographic predictors of poor outcome in infants with hypertrophic cardiomyopathy. Am J Cardiol 1997; 80: 595–600. [DOI] [PubMed] [Google Scholar]
- 12. Maron BJ, Tajik AJ, Ruttenberg HD, Graham TP, Atwood GF, Victorica BE, Lie JT, Roberts WC. Hypertrophic cardiomyopathy in infants: clinical features and natural history. Circulation 1982; 65: 7–17. [DOI] [PubMed] [Google Scholar]
- 13. Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, Cox GF, Towbin JA, Connuck DM, Messere JE, Lipshultz SE. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the pediatric cardiomyopathy registry. Am Heart J 2012; 164: 442–448. [DOI] [PubMed] [Google Scholar]
- 14. Shaffer KM, Denfield SW, Schowengerdt KO, Towbin JA, Radovancevic B, Frazier OH, Price JK, Gajarski RJ. Cardiac transplantation for pediatric patients. with inoperable congenital heart disease. Tex Heart Inst J 1998; 25: 57–63. [PMC free article] [PubMed] [Google Scholar]
- 15. Ziolkowska L, Turska‐Kmiec A, Petryka J, Kawalec W. Predictors of long‐term outcome in children with hypertrophic cardiomyopathy. Pediatr Cardiol 2016; 37: 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and Management of Hypertrophic Cardiomyopathy of the european Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733–2779. [DOI] [PubMed] [Google Scholar]
- 17. Ross RD. The Ross classification for heart failure in children after 25 years: a review and an age‐stratified revision. Pediatr Cardiol 2012; 33: 1295–1300. [DOI] [PubMed] [Google Scholar]
- 18. Wyman Lai LM, Cohen M, and Geva T. Echocardiography in Pediatric and Congenital Heart Disease: From Fetus to Adult: Wiley Blackwell; 2009. [Google Scholar]
- 19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bailey BJ, Briars GL. Estimating the surface area of the human body. Stat Med 1996; 15: 1325–1332. [DOI] [PubMed] [Google Scholar]
- 21. Lopez L, Colan S, Stylianou M, Granger S, Trachtenberg F, Frommelt P, Pearson G, Camarda J, Cnota J, Cohen M, Dragulescu A, Frommelt M, Garuba O, Johnson T, Lai W, Mahgerefteh J, Pignatelli R, Prakash A, Sachdeva R, Soriano B, Soslow J, Spurney C, Srivastava S, Taylor C, Thankavel P, van der Velde M, Minich L. Relationship of echocardiographic Z scores adjusted for body surface area to age, sex, race, and ethnicity: the pediatric heart network Normal echocardiogram database. Circ Cardiovasc Imaging 2017; 10: e006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neilan TG, Pradhan AD, King ME, Weyman AE. Derivation of a size‐independent variable for scaling of cardiac dimensions in a normal paediatric population. Eur J Echocardiogr 2009; 10: 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lafreniere‐Roula M, Bolkier Y, Zahavich L, Mathew J, George K, Wilson J, Stephenson EA, Benson LN, Manlhiot C, Mital S. Family screening for hypertrophic cardiomyopathy: is it time to change practice guidelines? Eur Heart J 2019; 40: 3672–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Norrish G, Jager J, Field E, Quinn E, Fell H, Lord E, Cicerchia MN, Ochoa JP, Cervi E, Elliott PM, Kaski JP. Yield of clinical screening for hypertrophic cardiomyopathy in child first‐degree relatives. Circulation 2019; 140: 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2: 436–441. [DOI] [PubMed] [Google Scholar]
- 26. Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 2008; 358: 1899–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation 2000; 102: 1950–1955. [DOI] [PubMed] [Google Scholar]
- 28. Marziliano N, Merlini PA, Vignati G, Orsini F, Motta V, Bandiera L, Intrieri M, Veronese S. A case of compound mutations in the MYBPC3 gene associated with biventricular hypertrophy and neonatal death. Neonatology 2012; 102: 254–258. [DOI] [PubMed] [Google Scholar]
- 29. Bharucha T, Lee KJ, Daubeney PE, Nugent AW, Turner C, Sholler GF, Robertson T, Justo R, Ramsay J, Carlin JB, Colan SD, King I, Weintraub RG, Davis AM. Sudden death in childhood cardiomyopathy: results from a long‐term national population‐based study. J Am Coll Cardiol 2015; 65: 2302–2310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Aetiology of patients with infantile disease.
Table S2. Prevalence of comorbidities by underlying diagnosis.
Table S3. Use of genetic testing over time by underlying aaetiology.
Table S4. Results of genetic testing in non‐syndromic and RASopathy patients.
Table S5. Comparison of baseline demographics in Non‐syndromic patients according to genetic testing status.
Table S6. Description of clinical characteristics and outcomes of patients with major arrhythmic cardiac events.
Figure S1. Comorbidities according to aetiology in infantile HCM
Figure S2. Genetic testing strategy in infantile HCM