

Abstract
Aims
Identifying early right ventricular (RV) dysfunction and impaired vasodilator reserve is challenging in heart failure with preserved ejection fraction (HFpEF). We hypothesized that cardiac magnetic resonance (CMR)‐based exercise imaging and serial cyclic guanosine monophosphate (cGMP) measurements can identify dynamic RV‐arterial uncoupling and responsiveness to pulmonary vasodilators at early stages of the HFpEF syndrome.
Methods and results
Patients with HFpEF (n = 16), impaired left ventricular relaxation due to concentric remodelling (LVCR, n = 7), and healthy controls (n = 8) underwent CMR at rest and during supine bicycle exercise with simultaneous measurements of central haemodynamics and circulating cGMP levels, before and after oral administration of 50 mg sildenafil. At rest, mean pulmonary artery pressures (mPAP) were higher in HFpEF, compared with LVCR and controls (27 ± 2, 18 ± 1, and 11 ± 1, respectively; P = 0.01), whereas biventricular volumes, heart rate, and stroke volume were similar. During exercise, LVCR and HFpEF had a greater increase in the ratio of mPAP over cardiac output than controls (5.50 ± 0.77 and 6.34 ± 0.86 vs. 2.24 ± 0.55 in controls, P = 0.005). The ratio of peak exercise to rest RV end‐systolic pressure‐volume, a surrogate of RV contractility, was significantly reduced in LVCR and HFpEF (2.32 ± 0.17 and 1.56 ± 0.08 vs. 3.49 ± 0.35 in controls, P < 0.001) and correlated with peak exercise VO2 (R 2 = 0.648, P < 0.001). cGMP levels increased with exercise across the HFpEF spectrum (P < 0.05 vs. baseline), except when postcapillary pulmonary hypertension was present at rest (P = 0.73 vs. baseline). A single sildenafil administration failed to increase circulating cGMP levels and did not improve RV performance.
Conclusion
Exercise CMR identifies impaired RV‐arterial coupling at an early stage of HFpEF. Circulating cGMP levels phenocopy the haemodynamic spectrum in HFpEF but fail to increase after phosphodiesterase type 5 inhibition, endorsing the need for alternative interventions to increase cGMP signalling in HFpEF.
Keywords: Heart failure, Exercise, Haemodynamics, Cardiac magnetic resonance imaging, Phosphodiesterase type 5 inhibitor
Introduction
Right ventricular dysfunction (RVD) and pulmonary hypertension (PH) are common in heart failure with preserved ejection fraction (HFpEF). Depending on the imaging modality, parameters and cut‐off values to report RVD, its prevalence in HFpEF ranges from 18% to 50%. 1 In addition, commonly used echocardiographic criteria for RVD including tricuspid annular plane systolic excursion (TAPSE), fractional area change, right ventricular (RV) strain, and TAPSE over pulmonary artery systolic pressure ratio (TAPSE:PASP ratio), were shown to predict all‐cause mortality and heart failure hospitalizations. 1 , 2 , 3 , 4 , 5 The mechanisms contributing to RVD are multiple and include unfavourable left to RV interdependence, increased RV afterload with ventricular‐arterial uncoupling and reduced pulmonary vasodilator function. 6 , 7 , 8
Two major obstacles limit the development of effective strategies that can improve clinical outcome in HFpEF patients with RVD and PH. First, early diagnosis of RVD and PH is difficult because measurements of RV function and pulmonary haemodynamics are mostly made at rest, while HFpEF patients typically develop symptoms upon physical exercise. Echocardiographic evaluation of RV function is challenging during exercise, given the complex geometry of the RV, limited acoustic windows and high prevalence of COPD and obesity in HFpEF. 9 In contrast, real‐time cardiac magnetic resonance (CMR) imaging can circumvent these limitations and accurately evaluate biventricular volumes during strenuous exercise and free breathing. 10 , 11 Second, depressed vasodilator reserve is a key pathogenic mechanism of PH in heart failure, 7 caused in part by endothelial dysfunction and impaired cyclic guanosine monophosphate (cGMP) signalling. 12 Despite a strong preclinical rationale, therapeutic interventions with phosphodiesterase type 5 (PDE5) inhibition did not provide clinical benefit in the general HFpEF population. 13 , 14 , 15 , 16 The latter may be related to the large heterogeneity of the HFpEF spectrum, and the failure to identify the subset of patients who may benefit. A common limitation in most of these studies is the notorious absence of cGMP measurements as surrogate marker of pulmonary vasodilator reserve during exercise and/or responsiveness to vasodilator challenge.
We hypothesized that exercise RV performance can better identify dysfunction at earlier stages of the disease and that cGMP measurements can signal pulmonary vasodilator responsiveness in HFpEF. Therefore, the first aim of this study was to evaluate RV performance at rest and during exercise using CMR in carefully selected symptomatic HFpEF patients with and without PH (i.e. Stage C heart failure) and in asymptomatic patients with subclinical left ventricular (LV) diastolic dysfunction (i.e. Stage B heart failure). 17 The second aim was to simultaneously evaluate invasive haemodynamics during exercise CMR and measure circulating cGMP levels before and after loading dose of sildenafil, a type 5 phosphodiesterase inhibitor, and to test its potential to improve RVD.
Methods
Subjects
Heart failure with preserved ejection fraction patients were screened at the ambulatory heart failure clinic at our institution and 16 patients agreed to participate. HFpEF was defined by clinical symptoms of chronic heart failure (dyspnoea and fatigue), normal LV ejection fraction (LVEF ≥ 50%), and elevated left heart filling pressures [estimated by pulmonary artery occlusion pressure (PAOP)] at rest (>15 mmHg) and/or with exercise (≥25 mmHg). 18 , 19 No signs of congestion were present at clinical examination. Seven subjects with impaired LV relaxation pattern (E/A < 1) and at least moderate LV concentric remodelling due to long lasting primary arterial hypertension (further denoted as LVCR) were included at our ambulatory hypertension clinic. Patients with significant valvular heart disease (> mild stenosis, > moderate regurgitation), unstable coronary artery disease or history of myocardial infarction, hypertrophic or infiltrative cardiomyopathy, primary renal or hepatic disease, and significant ventilatory disease (FEV1 < 50%, TLC or VC < 70%) were excluded. Eight healthy control subjects voluntarily consented to participate in the study. All had a normal electrocardiogram, transthoracic echocardiogram, spirometry and normal rest and exercise haemodynamics on right heart catheterization. 20 All participants had to be able to perform at least 50 W on an upright bicycle stress test. The study protocol conformed to the Declaration of Helsinki and was approved by the local Ethics Committee. All participants provided written informed consent prior to inclusion.
Study protocol
All subjects were studied without interruption of their chronic medications. First, cardiopulmonary exercise testing with continuous monitoring of expiratory gases was performed on an upright cycle ergometer (ER900 and Oxycon Alpha, Jaeger, Germany), using a continuous ramp protocol until exhaustion. Outcome measures included peak oxygen consumption (VO2 peak), maximal power output (Pmax), peak heart rate and ventilatory efficiency, defined as the relationship between minute ventilation and carbon dioxide production (VE/VCO2 slope). Within 24 h, all subjects underwent exercise cardiac magnetic resonance (exCMR) imaging with simultaneous invasive pressure measurements. Immediately prior to exCMR, all subjects underwent a right heart catheterization. A 7‐Fr MRI‐compatible pulmonary artery catheter (Edwards Lifesciences, CA, USA) was inserted through the right internal jugular vein and guided to the proximal right main pulmonary artery under fluoroscopy, while a 20‐gauge arterial catheter was placed in the radial artery of the non‐dominant hand. In the exCMR suite, these catheters were attached to MRI‐compatible transducers that were connected to a Powerlab recording system (AD Instruments, Oxford, UK). Right atrial pressure and pulmonary and systemic artery pressures were continuously monitored during the protocol and analysed offline using LabChart V7.3.7 (AD Instruments). As per discussions with the Ethics Committee, PAOP was not measured at rest, nor during exercise in controls. All pressure measurements were obtained during unrestricted respiration and measured at end‐expiration (mean of three consecutive cardiac cycles), in accordance with previous invasive exercise haemodynamic studies. 21 , 22 Right atrial pressure and PAOP were measured at mid a wave. 23 Exercise workloads for the exCMR protocol were determined as 25%, 50%, and 66% of peak power (Pmax) achieved during previous cardiopulmonary exercise testing. Given the fact that supine exercise at 66% of peak power closely corresponds to maximal sustainable exercise intensity in an upright position, this workload will hereinafter be referred to as peak intensity. 11 Subjects were then given a single oral dose of 50 mg sildenafil and 30–60 min later CMR measurements were repeated at rest and at peak‐intensity exercise using the same wattage as during baseline evaluation.
Cardiac magnetic resonance equipment, image acquisition, and analysis
Biventricular volumes were measured using a free‐breathing real‐time CMR method that we previously validated against invasive standards. 11 In brief, subjects performed supine bicycle exercise within the CMR bore using a cycle ergometer with adjustable electronic resistance (Lode, Groningen, The Netherlands). Images were acquired on a Philips Achieva 1.5 T MRI system with a five‐element phased‐array coil (Philips Medical Systems, Best, The Netherlands). Using in‐house‐developed software (RightVol, Leuven, Belgium), LV and RV contours were manually traced on short‐axis images with compensation for respiratory phase and with simultaneous reference on horizontal long‐axis slices, enabling a clear identification of the atrioventricular plane. End‐diastolic and end‐systolic ventricular volumes were calculated through a summation of disk method and indexed for body surface area. From these measurements, stroke volume index (SVi), cardiax index (CI), and ejection fraction (EF) were inferred. In accordance with previous studies, SVi was determined from LV rather than RV volumes because patients with PH frequently develop tricuspid regurgitation, particularly during exercise. Because control subjects did not have PAOP measurements, total pulmonary vascular resistance (TPR) was defined as the ratio of mean pulmonary artery pressure (mPAP) to cardiac output (CO) to allow comparison with control group. Similarly, total systemic vascular resistance was calculated as the ratio of mean systemic artery pressure to CO. When comparing patient groups with available PAOP measurements, pulmonary vascular resistance (PVR) was calculated as (mPAP‐PAOP)/CO. Exercise reserve was defined as the difference between rest and peak exercise volume or haemodynamic measures. 24 Pulmonary vascular load was evaluated through the relationship between mean pulmonary artery pressure and cardiac output (mPAP/CO slope). Abnormal pulmonary vascular load during exercise was defined as a mPAP/CO slope > 3 mmHg/L/min. 25 Pulmonary artery pulse pressure was calculated as the difference between systolic and diastolic pulmonary artery pressure and pulmonary arterial compliance (CPA) as the ratio of RV stroke volume to pulmonary artery pulse pressure. The RV end‐systolic pressure–volume relationship (RVESPVR), a surrogate of RV contractility, was calculated as mPAP divided by RVESV. 26 Abnormal RV contractile reserve was defined as the ratio of peak exercise to resting RVESPVR (subsequently referred to as ‘RVESPVR ratio’) of <2. 27 Finally, we calculated the RV stroke volume to RV end‐systolic volume ratio (RV SV/ESV) as a simplified quantification of RV‐arterial coupling. 28 , 29
Blood samples
N‐terminal pro‐brain natriuretic peptide was analysed from standard blood samples. cGMP was determined pre‐sildenafil and post‐sildenafil, both at rest and at peak exercise, on blood samples collected from the radial artery catheter during the exCMR protocol. Measurements were carried out using a commercially available cGMP immunoassay (Cyclic GMP Elisa kit, Sanbio, Uden, The Netherlands).
Statistics
Data were analysed using SPSS Statistics version 26 (IBM Corporation, Amonk, NY, USA). Normality was ensured using the Shapiro–Wilk test, and variables are presented as means (±standard deviation) or as medians (with 25% and 75% interquartile range) accordingly. Baseline characteristics of the different groups were compared using a χ 2 test for categorical data and either a Kruskal–Wallis H test or a one‐way analysis of variance (ANOVA) with the Bonferroni post‐hoc correction for continuous variables. A mixed linear model with compound symmetric covariance matrix and the Bonferroni post‐hoc test for multiple comparisons were performed to evaluate the cardiac volume response to exercise within and between groups. For the comparison between pre‐sildenafil and post‐sildenafil measurements, we used a repeated measures ANOVA with exercise intensity as within‐subject effect and subject group (controls vs. LVCR vs. HFpEF) as between‐subject effect. Individual pressure‐flow relationships (mPAP/CO and PAOP/CO slopes) were calculated through linear regression of the serial measurements of mPAP (or PAOP) and CO. Differences in pressure‐flow slope coefficients between groups were compared using one‐way ANOVA. A P value of <0.05 was considered statistically significant. Pearson correlation coefficients were used to assess the univariate relationships between RVESPVR ratio, RV SV/ESV and VO2 peak during exercise. A two‐sided P value below 0.05 was considered as statistically significant.
Results
Clinical characteristics
Seven patients with LVCR and 16 patients with HFpEF were enrolled in the study. Both groups were older than the eight control subjects who consented with participation (Table 1 ). Except for arterial hypertension, comorbidities were most frequent in the HFpEF group (Table 1 ). In particular, a history of atrial fibrillation was highly frequent in HFpEF patients. Fourteen out of 16 HFpEF patients and five out of seven LVCR were on beta blocking agents, which were not discontinued before the examinations. HFpEF patients had a markedly higher NT‐proBNP level than LVCR and controls (both P < 0.001). As expected, exercise capacity (measured as VO2 peak) was markedly reduced in HFpEF and in LVCR (both P < 0.001) compared with controls. However, when expressed as percentage of expected peak VO2, the exercise capacity was only mildly reduced.
Table 1.
Clinical and biochemical characteristics and medication use
| Characteristic | Controls (n = 8) | LVCR (n = 7) | HFpEF (n = 16) | P value | |
|---|---|---|---|---|---|
| Age (years) | 56 ± 3 | 66 ± 3 | 72 ± 2 | <0.001 | |
| Male sex, n (%) | 5 (63) | 5 (71) | 7 (44) | 0.424 | |
| BMI (kg/m2) | 27.0 ± 1.8 | 29.6 ± 0.9 | 31.0 ± 1.3 | 0.194 | |
| AHT, n (%) | 2 (25) | 7 (100) | 15 (96) | <0.001 | |
| AF, n (%) | 0 (0) | 0 (0) | 13 (81)† | <0.001 | |
| Type 2 DM, n (%) | 0 (0) | 2 (29) | 8 (50)† | 0.035 | |
| CAD, n (%) | 0 (0) | 1 (14) | 2 (13) | 0.556 | |
| OSAS, n (%) | 0 (0) | 2 (29) | 6 (38) | 0.139 | |
| NYHA | I | N/A | 7 | 1 | — |
| II | N/A | 0 | 9 | ||
| III | N/A | 0 | 6 | ||
| IV | N/A | 0 | 0 | ||
| Medications | |||||
| ACE or ARB, n (%) | 2 (25) | 5 (71) | 10 (63) | 0.133 | |
| β‐blocker, n (%) | 1 (13) | 5 (71) | 14 (88) | 0.001 | |
| Loop diuretic, n (%) | 0 (0) | 1 (14) | 13 (81) | <0.001 | |
| Thiazide, n (%) | 2 (25) | 4 (57) | 4 (25) | 0.278 | |
| Spironolactone, n (%) | 0 (0) | 0 (0) | 6 (38) | 0.031 | |
| CCB, n (%) | 0 (0) | 5 (71) | 7 (44) | 0.015 | |
| Laboratory results | |||||
| Haemoglobin (g/dL) | 13.1 ± 0.3 | 13.4 ± 0.6 | 12.6 ± 0.3 | 0.379 | |
| Creatinine (mg/dL) | 0.82 (0.70–0.87) | 1.04 (0.80–1.18) | 1.04 (0.88–1.55)* | 0.025 | |
| eGFR (mL/min/1.73m2) | 98 (91–105) | 78 (63–91)* | 66 (35–78)* | 0.001 | |
| NT‐proBNP (pg/mL) | 44 (38–64) | 199 (100–241)* | 803 (344–1236) *, † | <0.001 | |
| CPET parameters | |||||
| Peak power (W) | 181 ± 30 | 130 ± 11 | 87 ± 8* | 0.001 | |
| Peak HR (bpm) | 162 ± 8 | 128 ± 7 | 115 ± 8* | 0.003 | |
| VO2 peak (mL/kg/min) | 28.2 ± 3.7 | 18.2 ± 1.9* | 14.7 ± 1.1* | <0.001 | |
| VO2 peak (% predicted) | 103 ± 7 | 85 ± 8 | 84 ± 5 | 0.103 | |
| VE/VCO2 (ratio) | 26.6 (25.4–28.8) | 32.2 (27.8–38.3)* | 35.6 (32.2–39.5)* | 0.015 | |
Data presented as mean± SE or median (25% and 75% percentile); P values for between‐group difference (one‐way ANOVA test, Kruskall–Wallis test or Pearson χ 2 test where appropriate).
P < 0.05 Bonferroni post‐hoc compared with controls.
P < 0.05 Bonferroni post‐hoc compared with LVCR.
AF, atrial fibrillation; AHT, arterial hypertension; BMI, body mass index; CCB, calcium channel blocker; DM, diabetes mellitus; eGFR, estimated glomerular filtration rate (calculated with CKD‐EPI formula); FEV1, forced expiratory volume in 1 s; HFpEF, heart failure with preserved ejection fraction; HR, heart rate; LVCR, left ventricular relaxation due to concentric remodelling; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; NYHA, New York Heart Association; OSAS, obstructive sleep apnoea syndrome; VE/VCO2, ratio of minute ventilation/carbon dioxide production;VO2 peak, peak oxygen uptake.
Echocardiography and baseline cardiac magnetic resonance data
At baseline, LV and RV dimensions and ejection fraction were comparable in the three groups (Table 2 ). HFpEF and LVCR patients showed significant LV concentric remodelling (relative wall thickening > 0.42). Left atrial volume and tricuspid regurgitation peak velocity were highest in the HFpEF group, whereas E/E' did not differ between the three groups. The high prevalence of atrial fibrillation in HFpEF complicated the comparison of the E/A ratio between groups.
Table 2.
Baseline echocardiographic and CMR parameters
| Parameter | Controls (n = 8) | LVCR (n = 7) | HFpEF (n = 16) | P value |
|---|---|---|---|---|
| Echocardiographic parameters of diastolic function | ||||
| LAVI (mL/m2) | 35 ± 3 | 45 ± 3 * | 58 ± 4 *, † | 0.043 |
| E/A (ratio) | 1.12 ± 0.11 | 0.74 ± 0.07 | 1.50 ± 0.37 | 0.078 |
| E/E (ratio) | 8.01 ± 0.94 | 8.04 ± 0.56 | 9.76 ± 0.91 | 0.316 |
| TR peak velocity (m/s) | N/A | 2.29 (2.09–2.61) | 2.71 (2.51–3.10)† | 0.042 |
| Estimated sPAP (mmHg) | N/A | 25.98 (22.49–31.08) | 38.10 (32.15–46.21) † | 0.002 |
| CMR parameters of left and right ventricular structure and function | ||||
| LVEDD (mm) | 48 ± 2 | 43 ± 4 | 47 ± 6 | 0.192 |
| LVEDVI, (mL/m2) | 75 ± 8 | 69 ± 6 | 66 ± 3 | 0.448 |
| LVMI (gm/m2) | 71 ± 11 | 86 ± 14 | 82 ± 18 | 0.146 |
| RWT (ratio) | 0.36 ± 0.05 | 0.56 ± 0.07 * | 0.48 ± 0.09 * | <0.001 |
| LVEF (%) | 63.1 ± 2.4 | 61.4 ± 1.9 | 61.7 ± 1.7 | 0.852 |
| RVEDVI (mL/m2) | 74 ± 9 | 65 ± 3 | 69 ± 4 | 0.564 |
| RVEF (%) | 61 ± 2 | 59 ± 1 | 56 ± 2 | 0.455 |
Data presented as mean ± SE or median (25% and 75% percentile); P values for between‐group difference (one‐way ANOVA test, Kruskall–Wallis test or Pearson χ 2 test where appropriate).
P < 0.05 Bonferroni post‐hoc compared with controls.
P < 0.05 Bonferroni post‐hoc compared with LVCR.
CMR, cardiac magnetic resonance; E/A, ratio of early (E) to late (A) mitral valve flow velocity; E/E, ratio of E over averaged medial and lateral tissue Doppler lengthening velocity; HFpEF, heart failure with preserved ejection fraction; HR, heart rate; LAVI, left atrial volume index; LVEDD, left ventricular end‐diastolic diameter; LVEDVI, LV end‐diastolic volume index; LVEF, left ventricular ejection fraction; LVCR, left ventricular relaxation due to concentric remodelling; LVMI, left ventricular mass index; RVEF, right ventricular ejection fraction; RWT, relative wall thickening; sPAP, systolic pulmonary artery pressure; TR, tricuspid regurgitation.
Biventricular function at rest and during exercise measured using cardiac magnetic resonance
Left ventricular end‐diastolic volume index and RV end‐diastolic volume index did not change from rest to peak‐intensity exercise in control subjects, while LV end‐systolic volume index and RV end‐systolic volume index decreased (P = 0.045 and P = 0.036, respectively, Supporting Information, Figure S1 ), which resulted in an increase of LVEF and RV ejection fraction (RVEF) (P < 0.01 for both, Figure 1 ). In patients with HFpEF, LVEF increased with incremental exercise intensity (+4.2%, P = 0.008), while RV end‐diastolic volume index and RV end‐systolic volume index both increased (P = 0.01 and P < 0.01, respectively), resulting in a small decline of RVEF (−3.3%, P = 0.012). Patients with impaired LV relaxation (LVCR) showed an intermediate response to exercise, with no change in LVEF or RVEF.
Figure 1.
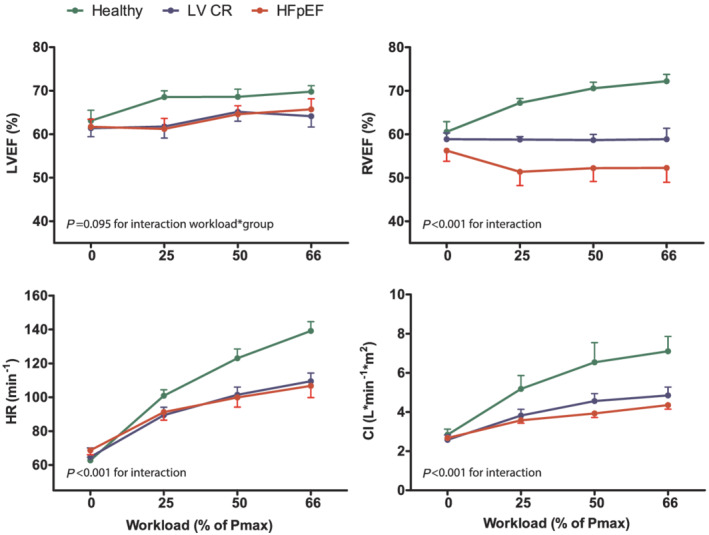
Comparison of change in LV and RV ejection fraction (LVEF and RVEF, respectively), heart rate and cardiac index (CI) during incremental exercise in patients with HFpEF, patients with impaired relaxation (LVCR) and healthy controls. Workloads are presented as a percentage of maximum power output (Pmax) determined during previous exercise testing. P values are shown for the interaction between group and exercise intensity using linear mixed models. Data are presented as means and SEM at each time point.
Compared with healthy controls, heart rate reserve was significantly reduced in HFpEF and LVCR (76 ± 5, 34 ± 4, and 45 ± 3 bpm, respectively, P < 0.01 for interaction exercise*group), in part attributable to the intake of beta blocking agents. Consequently, the increase in CI is smaller in the latter two groups (P < 0.01 for interaction exercise*group).
Pulmonary vascular function and right ventricular functional/contractile reserve
Resting pulmonary artery pressures and TPR were higher in HFpEF patients than in healthy controls and LVCR (P < 0.001 for both, Table S1 and Figure 2 A ), and these differences persisted after adjusting for age (P < 0.01 for both). While TPR did not change in control subjects with exercise, it increased significantly in LVCR and HFpEF (P = 0.04 and P = 0.02, respectively). PVR did not change significantly with exercise in either HFpEF or LVCR (P = 0.396 and P = 0.526, respectively, Table S1 ) due to the concomitant rise in PAOP, which was similar in those groups (Figure 3 ). However, the ratio of peak PAOP to peak workload normalized to body weight was higher in HFpEF (53 ± 6 mmHg/W/kg vs. 31 ± 3 mmHg/W/kg, respectively, P = 0.03). The mPAP/CO slopes in HFpEF and LVCR were significantly higher than in controls (both P < 0.01, Figure 2 B ). In contrast, systemic vascular resistance decreased in all three groups during exercise (P = 0.01, Table S1 ), albeit to a greater extent in controls. Resting CPA was higher in controls and LVCR than in HFpEF (P < 0.01, adjusted for age, Table S1 ). Because of the high basal CPA values, the absolute exercise‐induced reduction in CPA is also greatest in controls, but peak exercise CPA still remains significantly higher in controls than in HFpEF or LVCR (Figure 2 C ).
Figure 2.
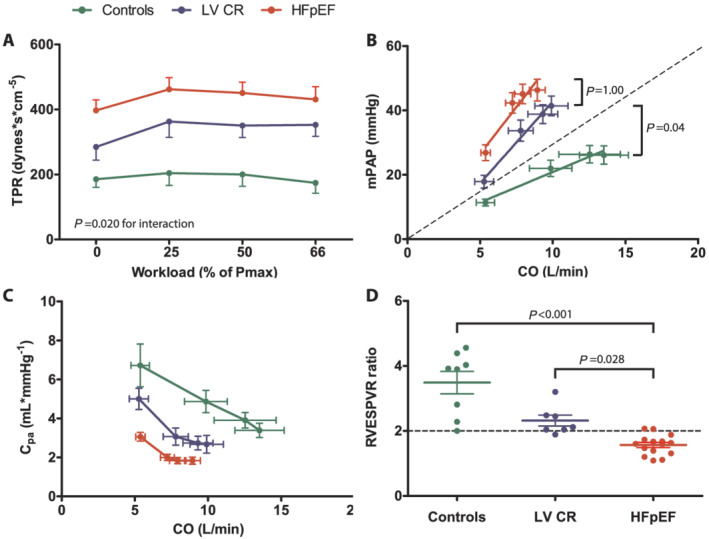
Right ventricular afterload and contractile reserve in healthy controls, patients with impaired relaxation (LVCR) and patients with heart failure and preserved ejection fraction (HFpEF). (A) Evolution of TPR (total pulmonary vascular resistance) from rest to peak‐intensity exercise. Workloads are presented as a percentage of maximum power output (Pmax) determined at previous cardiopulmonary exercise testing. P value indicates interaction between group and exercise intensity using linear mixed models. (B) Linear mean pulmonary artery pressure (mPAP)‐cardiac output (CO) relationships based on averages of serial measurements of mPAP and CO during exercise. P values are given for between‐group differences in mPAP/CO slope using one‐way ANOVA. The dashed line indicates the upper limit of normal (slope = 3). (C) Evolution of pulmonary artery compliance (CPA) with increasing cardiac output at four‐stage exercise test. (D) Individual data points of the ratio of peak exercise RV end‐systolic pressure volume ratio (RVESPVR) to rest RVESPVR. The horizontal dashed line indicates the normal value (=2). P values denote the between‐group differences using one‐way ANOVA.
Figure 3.
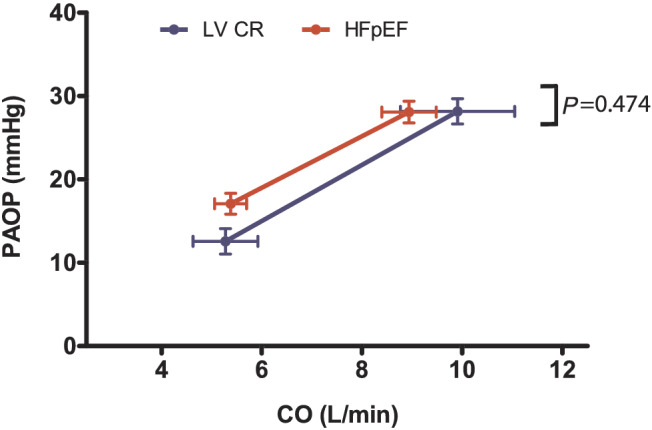
Evolution of PAOP over CO from rest to peak exercise in HFpEF patients and patients with impaired relaxation (LVCR). Data are presented as mean ± SEM. No difference is seen between the PAOP/CO slopes of the two patient groups, using an independent‐samples t‐test (P = 0.474).
When subdividing our HFpEF cohort into patients with or without PH at rest (mPAP > 25 mmHg, n = 8 for both), it appeared that the observed changes in RV afterload were mainly driven by the presence of PH at rest (Supporting Information, Figure S2 A, B, & C ). Importantly, as none of the patients showed an elevated resting PVR > 3 and/or a resting diastolic pressure gradient ≥ 7, all eight patients with elevated mPAP at rest were diagnosed with isolated postcapillary pulmonary hypertension (IpcPH). 30 The haemodynamic responses of HFpEF patients without IpcPH were analogous to LVCR.
Consistent with the observed gradients in RV afterload, HFpEF and LVCR had significantly reduced RV contractile reserve, as estimated by the RVESPVR ratio, compared with healthy controls (P < 0.01, Figure 2 D ). Consequently, RV‐arterial coupling was less effective in both patient groups, illustrated by the lack of RV SV/ESV increase during exercise (P < 0.01 compared with controls, Table S1 ). Both the dynamic change in RV SV/ESV and RV contractile reserve correlated strongly with CI reserve (R 2 = 0.471 and 0.666, respectively; both P < 0.001) and VO2 peak (R 2 = 0.316 and 0.648, respectively; both P < 0.001). In contrast, no associations were found between resting haemodynamic parameters (e.g. RV or LV volumes, LVEF, & RVEF) and VO2 peak.
Response to pulmonary vasodilator challenge during graded exercise and circulating cyclic guanosine monophosphate levels
Four HFpEF patients (including two patients with IpcPH at rest) were not able to perform a second, adequate exercise test after sildenafil intake because of poor exercise tolerance. After sildenafil administration, resting systemic blood pressure tended to decrease in all groups (Table S2 ) with concomitantly lower PAOP at rest in LVCR and HFpEF. Sildenafil also lowered resting mPAP and TPR in all groups, but only significantly in controls (P = 0.027 and P = 0.037, respectively). At peak exercise, mPAP, but not TPR, was still significantly lower in controls after sildenafil (P = 0.049). In patients with LVCR or HFpEF, sildenafil failed to significantly reduce PAOP, mPAP, or PVR at peak exercise. Furthermore, no statistically significant differences were observed in biventricular volume changes (data not shown) or CI rise (Figure 4 A ) during exercise following sildenafil administration. The mPAP/CO curves started at a lower mPAP, but their slopes were equal before and after sildenafil administration in all three groups (Figure 4 B ). In a subgroup analysis of HFpEF patients with IpcPH at rest, sildenafil seemed to reduce PVR (although not statistically significantly) but failed to improve ventriculo‐vascular coupling, as evidenced by the inadequate increase in RVESPV and a decline in RVEF and RV SV/ESV (Table S3 ). Consequently, no increase in SVi or CI at peak exercise was observed (P = 0.91 and P = 0.31, respectively).
Figure 4.
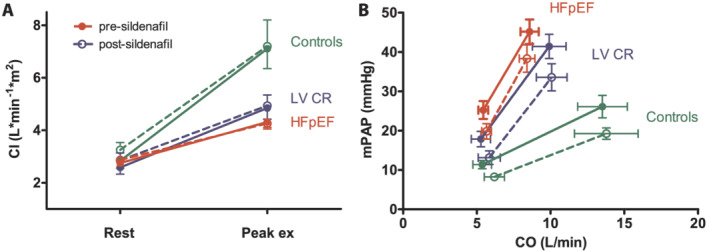
Cardiac index (CI) and pulmonary vascular reserve in patients with heart failure with preserved ejection fraction (HFpEF), patients with impaired relaxation (LVCR) and healthy controls before (solid lines) and after (dashed lines) sildenafil administration. (A) CI response from rest to peak‐intensity exercise. No significant difference is seen between before and after sildenafil administration, using repeated measures ANOVA. (B) Linear mean pulmonary artery pressure (mPAP)‐cardiac output (CO) relationships based on averages of serial measurements of mPAP and CO during exercise. Again, no significant difference in slopes is seen between before and after sildenafil administration, using repeated measures ANOVA (RMANOVA).
To investigate whether the failure of sildenafil to improve pulmonary vasoreactivity or RV contractile function was caused by impaired cGMP signal transduction, we serially measured arterial cGMP plasma levels in a subset of patients (Figure 5 ). Baseline arterial cGMP levels at rest did not differ between the four groups (P = 0.129). However, there is a tendency towards higher levels in the LVCR and HFpEF groups. In healthy controls, as well as in LVCR and HFpEF without IpcPH, circulating cGMP levels significantly increased upon exercise (P < 0.05 for all, Figure 5 A ). This response was absent in HFpEF with IpcPH (P = 0.73). After sildenafil, arterial cGMP levels modestly and selectively increased in HFpEF without IpcPH (P = 0.019 vs. pre‐sildenafil). In none of the four groups, sildenafil was associated with further increase in cGMP during exercise (P = 0.09, P = 0.43, P = 0.93, and P = 0.17 respectively, Figure 5 B ).
Figure 5.
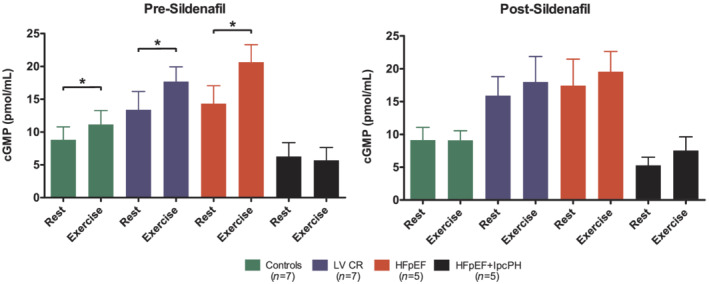
Effect of exercise and sildenafil intake on circulating cyclic guanosine monophosphate levels. Data are presented as mean with SEM error bars. * = P < 0.05 for difference between resting and peak‐exercise levels, using paired‐samples t‐tests.
Discussion
Right ventricular dysfunction carries an impaired prognosis in HFpEF, 1 , 9 , 31 but is often not recognized during evaluation at rest. In this study, we used for the first time simultaneous gold‐standard invasive pressure and CMR‐derived ventricular volume measurements during incremental exercise to comprehensively study RV performance in patients with a varying degree of LV diastolic dysfunction. We observed a significant and progressive reduction of RV contractile reserve (estimated by RVESPVR ratio) across the spectrum from asymptomatic LV diastolic dysfunction to HFpEF. Exercise CMR evaluation also revealed ineffective RV‐arterial coupling in patients with impaired relaxation, and to a greater extent in HFpEF, which was associated with a marked failure to increase circulating cGMP levels with exercise in the sickest patients with PH and which proved refractory to pharmacological intervention with the PDE5 inhibitor, sildenafil. Our data highlight the presence of impaired RV‐arterial coupling at an early stage in the HFpEF syndrome and call for early therapeutic interventions to effectively unload the right heart and improve its function.
Increased right ventricular afterload in heart failure with preserved ejection fraction
Elevation in LV filling pressures during exercise despite preserved LVEF is pathognomonic for HFpEF. 19 , 32 PH in HFpEF is primarily attributable to passive backward transmission of these filling pressures but can be further elevated by reduced left atrial compliance and exercise‐induced mitral regurgitation. 7 , 33 The subsequent chronic or repetitive pulmonary venous congestion induces pulmonary vasoconstriction and vascular remodelling, which further increases PVR. 34 , 35 In our patient cohort, PVR at rest was significantly higher in HFpEF patients, compared with patients with asymptomatic diastolic dysfunction. Importantly, the net vascular load that opposes RV ejection not only incorporates a resistive, non‐pulsatile component, reflected by PVR, but also a pulsatile component reflected by the pulmonary artery compliance (CPA). 36 The inverse relationship between PVR and CPA implies that a small rise in PVR in patients with low baseline resistance is accompanied by a bigger decrease in CPA, as observed in early pulmonary vascular disease. 37 This relation is even further accentuated during exercise, since elevated PAOP shifts the CPA/PVR curve leftwards and downwards, indicating that elevation of PAOP further lowers CPA for any given PVR. 38 Given the similar increase in PAOP during exercise in HFpEF and LVCR patients, the mPAP/CO slopes and CPA decrease were similar in both patient groups (Figure 2 B and C ). The relative reduction of CPA from rest to peak exercise is much more pronounced, compared with the relative increase in PVR, indicating that dynamic RV‐arterial uncoupling in HFpEF is mainly driven by an increase in pulsatile afterload.
Exercise‐induced right ventricular‐arterial uncoupling in heart failure with preserved ejection fraction
Optimal RV‐arterial coupling implies that RV contractility matches RV afterload so that flow output occurs at minimal energy cost. In our HFpEF cohort, the increase in RV contractility during exercise was insufficient as RV‐arterial coupling, approximated by the RV SV/ESV ratio, deteriorated from rest to peak exercise (Table S2 ). Given the close relation between RVEF and RV SV/ESV, it is not surprising that this was accompanied by a dynamic decrease in RVEF in this patient group (Figure 1 ). Furthermore, the exercise CMR technique enabled us to clearly demonstrate an impaired increase in ventricular contractility during exercise in patients without any signs or symptoms of heart failure, but with early diastolic dysfunction (i.e. Stage B heart failure) (Figure 2 ). These patients, however, preserve RV‐arterial coupling at peak exercise, as evidenced by an unchanged RV SV/ESV ratio and represent an intermediate haemodynamic phenotype between healthy controls and HFpEF patients.
The reduced RV contractile reserve in both the HFpEF and LV CR group may suggest intrinsic myocardial disease. However, the pathophysiology of RV dysfunction in HFpEF is still largely unclear, and possible mechanisms include microvascular dysfunction, atrial fibrillation, and impaired RV contractile response due to beta‐adrenergic receptor desensitization. 39 , 40 In addition, the marked rise in RA pressure during exercise in both the LVCR and HFpEF group imply the presence of RV diastolic dysfunction, which is likely to be caused by the same pathophysiological process affecting the LV. Indeed, in a study by Borlaug et al., both the diastolic and systolic reserve of the LV and RV were intimately related and contributed to increased RA pressure and PAOP, impaired CO reserve, and limitation in exercise capacity. 21 Interestingly, CMR and RV biopsy studies demonstrated diffuse RV myocardial fibrosis in HFpEF patients, similar to the earlier described LV fibrosis in these patients. 41 , 42
Overall, both RV contractile reserve and the dynamic change in RV SV/ESV correlated strongly with CI reserve and peak VO2. In contrast, no associations were found between resting haemodynamic parameters (e.g. RV or LV volumes, LVEF, & RVEF) and VO2 peak, emphasizing even more the importance of exercise testing in unmasking ventriculo‐vascular alterations in HFpEF. These data also highlight the importance of the right heart as an important determinant of functional capacity among HFpEF patients.
Cyclic guanosine monophosphate signalling and response to phosphodiesterase type 5 inhibition in heart failure with preserved ejection fraction spectrum
Multiple lines of evidence identified impaired NO‐cGMP signalling as a central player in the pathophysiology of both LV diastolic dysfunction and pulmonary vascular dysfunction. 43 , 44 While preliminary studies in PH due to left heart disease suggested potential benefit of enhanced NO‐cGMP signalling using PDE5 inhibitors, data from small randomized controlled trials have yielded conflicting results. 13 , 14 , 15 , 16 More recently, two retrospective studies provided some evidence for the use of PDE5 inhibition in patients with combined precapillary and postcapillary PH. 45 , 46 In our study, single administration of sildenafil did not improve RV or pulmonary vascular function, neither in patients with more advanced stage of the disease (i.e. with increased pulmonary artery pressures at rest) nor in patients with early stage LV diastolic dysfunction (impaired relaxation). This may not be a surprising finding, as our HFpEF cohort only included patients with IpcPH. However, we did observe an abnormal exercise‐induced increase in PVR in both the LVCR and the HFpEF group, which has been previously reported by others. 47 In five HFpEF patients with IpcPH, peak exercise PVR exceeded the value of 2.1 WU, which is considered the upper limit or normal for patients > 50 years old. 48 However, this exertional increase in PVR is more likely attributable to hypoxaemia‐induced pulmonary vasoconstriction and the upstream transmission of a high left atrial pressure, rather than the result of effective pulmonary vascular remodelling. 49 The absence of advanced pulmonary vascular disease in our HFpEF group is further illustrated by the similar rise in TPG in both the LVCR group and the HFpEF group (Figure S3 ).
Sildenafil primarily acts on NO‐mediated, cGMP‐dependent pulmonary vasodilation and has not been proven to change diastolic properties of the LV or left atrium. Our data show that RV afterload in early stages of type 2 PH is predominantly determined by pulsatile afterload, rather than the load imposed by a steady flow, and is therefore unlikely to respond to sildenafil. Another, more biological explanation is that inadequate production rather than excessive breakdown causes limited cGMP signalling. This can be related to both increased nitrosative/oxidative stress in cardiomyocytes and down‐regulation of atrial natriuretic peptide receptors in the pulmonary vascular beds. 50 , 51 Alternatively, insufficient spill over of the intracellular NO‐dependent cGMP signalling into the blood stream has been suggested. 13 Our data are consistent with these hypotheses, because circulating cGMP levels were significantly lower in the sickest group of patients (HFpEF with pulmonary hypertension) and did not respond to exercise or sildenafil administration. Soluble guanylate cyclase (sGC) activators were not commercially available at the time of our initial study design, but the neutral findings with PDE5 inhibition could favour the use of a sGC activator in future studies. On the other hand, recent clinical trials with direct sGC stimulators in HFpEF have failed. 52 , 53
One major explanation for the negative trial results in HFpEF is the large heterogeneity in pathophysiology across the patient cohorts enrolled. To identify those patients with combined precapillary and postcapillary PH who might benefit from targeted PH therapy, accurate haemodynamic assessment is required. 45 , 46 In this context, more detailed phenotyping by means of exercise imaging might help identify other patient subgroups (e.g. with a prominent increase in PVR and/or RV dysfunction during exercise) who could benefit from therapies targeting RV‐arterial coupling, but this remains to be elucidated. 54 , 55 Second, therapy might be initiated too late at a stage of the disease where irreversible microvascular and myocardial remodelling preclude NO‐cGMP‐based interventions. Studying both functional (LV and RV exercise haemodynamics) and structural parameters (extracellular volume measurement) using CMR might identify earlier stages of the disease (i.e. in asymptomatic patients), 19 , 56 which are better responsive to targeted interventions. In this respect, endurance training programmes have already been shown to improve diastolic function and exercise capacity in asymptomatic patients with diastolic dysfunction. 57
Limitations
First, the small sample size may have increased the probability of type II statistical errors. However, the unparalleled accuracy of exercise CMR in HFpEF enabled robust and statistically significant measurements. Second, PAOP measurements were not obtained in healthy controls for safety reasons. Third, for the estimation of the contractile function, several assumptions had to be made. The calculation of the end‐systolic pressure–volume point was simplified by assuming the volume at zero pressure (V0) to be zero at all times while in reality it may vary with EDV. 58 Furthermore, end‐systolic pressure was approximated as mPAP in all groups, although in case of pulmonary hypertension, it might be better estimated as systolic pulmonary artery pressure. 59 Nevertheless, because RV contractile reserve was derived as the ratio from rest to peak exercise and given the constant relationship between systolic and mPAP during exercise, the effect of this modification is expected to be minimal. Likewise, by using the RV SV/ESV ratio to quantify RV‐arterial coupling, the effect of PCWP in pulmonary arterial load calculations is disregarded. RV SV/ESV is related to outcome in a PH cohort with different PH aetiology, 29 but its prognostic value in an exclusive type 2 PH population still needs to be explored. Finally, pulse pressure measurement using a fluid‐filled catheter can be challenging due to catheter ringing. 60 However, usage of high‐fidelity micromanometer‐tipped catheters is not possible in a CMR environment.
Conclusion
Identifying early RV dysfunction and impaired vasodilator reserve is challenging in HFpEF, but it is a prerequisite for better risk stratification and earlier therapeutic intervention. Exercise CMR identifies RV dysfunction and impaired RV‐arterial coupling at an early stage of HFpEF. Circulating cGMP levels, a surrogate marker for residual pulmonary vasodilator reserve, phenocopy the haemodynamic spectrum in HFpEF but fail to increase after PDE5 inhibition, which endorses the need for alternative interventions to increase cGMP signalling in HFpEF.
Conflict of interest
None declared.
Funding
This work was supported by a PhD Fellowship scholarship (Grant 11Z2515N to TP), a Research Project grant (Grant G0A2514N) from the Research Foundation ‐ Flanders (FWO), Belgium, and the AstraZeneca chair in cardiology at KU Leuven (to SJ).
Supporting information
Table S1. Baseline haemodynamics from rest to peak exercise.
Table S2. Biventricular function and haemodynamics from rest to peak exercise before and after sildenafil administration.
Table S3. Biventricular function and haemodynamics from rest to peak exercise before and after sildenafil administration in HFpEF with and without isolated postcapillary pulmonary hypertension (IpcPH).
Figure S1. Comparison of biventricular volume changes during incremental exercise in patients with HFpEF, patients with impaired relaxation (LVCR) and healthy controls.
Figure S2. Right ventricular afterload and contractile reserve in healthy controls, patients with impaired relaxation (LVCR) and patients with heart failure and preserved ejection fraction with (HPpEF+IpcPH) or without isolated postcapillary pulmonary hypertension (HFpEF).
Figure S3. Evolution of transpulmonary gradient (TPG) over CO from rest to peak exercise in HFpEF patients and patients with impaired relaxation (LVCR).
Acknowledgements
The authors gratefully acknowledge Kris Byloos and Guido Putzeys for their assistance with the CMR examinations, and Hilde Gillijns for her assistance with laboratory analyses.
Petit, T. , Claessen, G. , Claeys, M. , La Gerche, A. , Claus, P. , Ghysels, S. , Delcroix, M. , Ciarka, A. , Droogne, W. , Van Cleemput, J. , Willems, R. , Voigt, J.‐U. , Bogaert, J. , and Janssens, S. (2021) Right ventricular and cyclic guanosine monophosphate signalling abnormalities in stages B and C of heart failure with preserved ejection fraction. ESC Heart Failure, 8: 4661–4673. 10.1002/ehf2.13514.
References
- 1. Gorter TM, Hoendermis ES, van Veldhuisen DJ, Voors AA, Lam CS, Geelhoed B, Willems TP, van Melle JP. Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta‐analysis. Eur J Heart Fail 2016; 18: 1472–1487. [DOI] [PubMed] [Google Scholar]
- 2. Guazzi M, Dixon D, Labate V, Beussink‐Nelson L, Bandera F, Cuttica MJ, Shah SJ. RV contractile function and its coupling to pulmonary circulation in heart failure with preserved ejection fraction: stratification of clinical phenotypes and outcomes. J Am Coll Cardiol Img 2017; 10: 1211–1221. [DOI] [PubMed] [Google Scholar]
- 3. Lam CS, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction: a community‐based study. J Am Coll Cardiol 2009; 53: 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mohammed SF, Hussain I, AbouEzzeddine OF, Takahama H, Kwon SH, Forfia P, Roger VL, Redfield MM. Right ventricular function in heart failure with preserved ejection fraction: a community‐based study. Circulation 2014; 130: 2310–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Santas E, Palau P, Guazzi M, de la Espriella R, Miñana G, Sanchis J, Bayes‐Genís A, Lupón J, Chorro FJ, Núñez J. Usefulness of right ventricular to pulmonary circulation coupling as an indicator of risk for recurrent admissions in heart failure with preserved ejection fraction. Am J Cardiol 2019; 124: 567–572. [DOI] [PubMed] [Google Scholar]
- 6. Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA. Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J 2014; 35: 3452–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segers VF, Brutsaert DL, De Keulenaer GW. Pulmonary hypertension and right heart failure in heart failure with preserved left ventricular ejection fraction: pathophysiology and natural history. Curr Opin Cardiol 2012; 27: 273–280. [DOI] [PubMed] [Google Scholar]
- 8. Guazzi M, Naeije R. Pulmonary hypertension in heart failure. J Am Coll Cardiol 2017; 69: 1718–1734. [DOI] [PubMed] [Google Scholar]
- 9. Gorter TM, van Veldhuisen DJ, Bauersachs J, Borlaug BA, Celutkiene J, Coats AJS, Crespo‐Leiro MG, Guazzi M, Harjola VP, Heymans S, Hill L, Lainscak M, Lam CSP, Lund LH, Lyon AR, Mebazaa A, Mueller C, Paulus WJ, Pieske B, Piepoli MF, Ruschitzka F, Rutten FH, Seferovic PM, Solomon SD, Shah SJ, Triposkiadis F, Wachter R, Tschope C, de Boer RA. Right heart dysfunction and failure in heart failure with preserved ejection fraction: mechanisms and management. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2018; 20: 16–37. [DOI] [PubMed] [Google Scholar]
- 10. Lurz P, Muthurangu V, Schievano S, Nordmeyer J, Bonhoeffer P, Taylor AM, Hansen MS. Feasibility and reproducibility of biventricular volumetric assessment of cardiac function during exercise using real‐time radial k‐t SENSE magnetic resonance imaging. J Magn Reson Imaging: JMRI 2009; 29: 1062–1070. [DOI] [PubMed] [Google Scholar]
- 11. La Gerche A, Claessen G, Van de Bruaene A, Pattyn N, Van Cleemput J, Gewillig M, Bogaert J, Dymarkowski S, Claus P, Heidbuchel H. Cardiac MRI: a new gold standard for ventricular volume quantification during high‐intensity exercise. Circ Cardiovasc Imaging 2013; 6: 329–338. [DOI] [PubMed] [Google Scholar]
- 12. MüNzel T, Feil R, MüLsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by cyclic guanosine 3′,5′‐cyclic monophosphate–dependent protein kinase. Circulation 2003; 108: 2172–2183. [DOI] [PubMed] [Google Scholar]
- 13. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, Lesinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, Trial R. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309: 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, van Veldhuisen DJ, Voors AA. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J 2015; 36: 2565–2573. [DOI] [PubMed] [Google Scholar]
- 15. Andersen MJ, Ersboll M, Axelsson A, Gustafsson F, Hassager C, Kober L, Borlaug BA, Boesgaard S, Skovgaard LT, Moller JE. Sildenafil and diastolic dysfunction after acute myocardial infarction in patients with preserved ejection fraction: the Sildenafil and Diastolic Dysfunction After Acute Myocardial Infarction (SIDAMI) trial. Circulation 2013; 127: 1200–1208. [DOI] [PubMed] [Google Scholar]
- 16. Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase‐5 inhibition in a 1‐year study. Circulation 2011; 124: 164–174. [DOI] [PubMed] [Google Scholar]
- 17. Writing Committee M , Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128: e240–e319. [DOI] [PubMed] [Google Scholar]
- 18. Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De Keulenaer G, Leite‐Moreira AF, Borbely A, Edes I, Handoko ML, Heymans S, Pezzali N, Pieske B, Dickstein K, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 2007; 28: 2539–2550. [DOI] [PubMed] [Google Scholar]
- 19. Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail 2010; 3: 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009; 34: 888–894. [DOI] [PubMed] [Google Scholar]
- 21. Borlaug BA, Kane GC, Melenovsky V, Olson TP. Abnormal right ventricular‐pulmonary artery coupling with exercise in heart failure with preserved ejection fraction. Eur Heart J 2016; 37: 3293–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maron BA, Cockrill BA, Waxman AB, Systrom DM. The invasive cardiopulmonary exercise test. Circulation 2013; 127: 1157–1164. [DOI] [PubMed] [Google Scholar]
- 23. Houston BA, Tedford RJ. What we talk about when we talk about the wedge pressure. Circ Heart Fail 2017; 10. [DOI] [PubMed] [Google Scholar]
- 24. Haddad F, Vrtovec B, Ashley EA, Deschamps A, Haddad H, Denault AY. The concept of ventricular reserve in heart failure and pulmonary hypertension: an old metric that brings us one step closer in our quest for prediction. Curr Opin Cardiol 2011; 26: 123–131. [DOI] [PubMed] [Google Scholar]
- 25. Herve P, Lau EM, Sitbon O, Savale L, Montani D, Godinas L, Lador F, Jais X, Parent F, Gunther S, Humbert M, Simonneau G, Chemla D. Criteria for diagnosis of exercise pulmonary hypertension. Eur Respir J 2015; 46: 728–737. [DOI] [PubMed] [Google Scholar]
- 26. Sanz J, Sánchez‐Quintana D, Bossone E, Bogaard HJ, Naeije R. Anatomy, function, and dysfunction of the right ventricle. J Am Coll Cardiol 2019; 73: 1463–1482. [DOI] [PubMed] [Google Scholar]
- 27. Claessen G, La Gerche A, Voigt JU, Dymarkowski S, Schnell F, Petit T, Willems R, Claus P, Delcroix M, Heidbuchel H. Accuracy of echocardiography to evaluate pulmonary vascular and RV function during exercise. J Am Coll Cardiol Img 2016; 9: 532–543. [DOI] [PubMed] [Google Scholar]
- 28. Sanz J, García‐Alvarez A, Fernández‐Friera L, Nair A, Mirelis JG, Sawit ST, Pinney S, Fuster V. Right ventriculo‐arterial coupling in pulmonary hypertension: a magnetic resonance study. Heart 2012; 98: 238–243. [DOI] [PubMed] [Google Scholar]
- 29. Vanderpool RR, Pinsky MR, Naeije R, Deible C, Kosaraju V, Bunner C, Mathier MA, Lacomis J, Champion HC, Simon MA. RV‐pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart 2015; 101: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galiè N, Humbert M, Vachiery J‐L, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37: 67–119. [DOI] [PubMed] [Google Scholar]
- 31. Aschauer S, Kammerlander AA, Zotter‐Tufaro C, Ristl R, Pfaffenberger S, Bachmann A, Duca F, Marzluf BA, Bonderman D, Mascherbauer J. The right heart in heart failure with preserved ejection fraction: insights from cardiac magnetic resonance imaging and invasive haemodynamics. Eur J Heart Fail 2016; 18: 71–80. [DOI] [PubMed] [Google Scholar]
- 32. Phan TT, Shivu GN, Abozguia K, Sanderson JE, Frenneaux M. The pathophysiology of heart failure with preserved ejection fraction: from molecular mechanisms to exercise haemodynamics. Int J Cardiol 2012; 158: 337–343. [DOI] [PubMed] [Google Scholar]
- 33. Naeije R, Gerges M, Vachiery JL, Caravita S, Gerges C, Lang IM. Hemodynamic phenotyping of pulmonary hypertension in left heart failure. Circ Heart Fail 2017; 10. [DOI] [PubMed] [Google Scholar]
- 34. Moraes DL, Colucci WS, Givertz MM. Secondary pulmonary hypertension in chronic heart failure: the role of the endothelium in pathophysiology and management. Circulation 2000; 102: 1718–1723. [DOI] [PubMed] [Google Scholar]
- 35. Delgado JF, Conde E, Sanchez V, Lopez‐Rios F, Gomez‐Sanchez MA, Escribano P, Sotelo T, Gomez de la Camara A, Cortina J, de la Calzada CS. Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur J Heart Fail 2005; 7: 1011–1016. [DOI] [PubMed] [Google Scholar]
- 36. Vonk Noordegraaf A, Westerhof BE, Westerhof N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69: 236–243. [DOI] [PubMed] [Google Scholar]
- 37. Lankhaar JW, Westerhof N, Faes TJ, Marques KM, Marcus JT, Postmus PE, Vonk‐Noordegraaf A. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol 2006; 291: H1731–H1737. [DOI] [PubMed] [Google Scholar]
- 38. Tedford RJ, Hassoun PM, Mathai SC, Girgis RE, Russell SD, Thiemann DR, Cingolani OH, Mudd JO, Borlaug BA, Redfield MM, Lederer DJ, Kass DA. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation 2012; 125: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle–pulmonary circulation unit. Circulation 2009; 120: 992–1007. [DOI] [PubMed] [Google Scholar]
- 40. Lohse MJ, Engelhardt S, Danner S, Böhm M. Mechanisms of beta‐adrenergic receptor desensitization: from molecular biology to heart failure. Basic Res Cardiol 1996; 91: 29–34. [DOI] [PubMed] [Google Scholar]
- 41. Patel RB, Li E, Benefield BC, Swat SA, Polsinelli VB, Carr JC, Shah SJ, Markl M, Collins JD, Freed BH. Diffuse right ventricular fibrosis in heart failure with preserved ejection fraction and pulmonary hypertension. ESC Heart Fail 2020; 7: 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, Riley SJ, Subramanya V, Brown EE, Hopkins CD, Ononogbu S, Mandell KP, Halushka MK, Steenbergen C, Rosenberg AZ, Tedford RJ, Judge DP, Shah SJ, Russell SD, Kass DA, Sharma K. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC: Heart Fail 2020; 8: 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013; 62: 263–271. [DOI] [PubMed] [Google Scholar]
- 44. Greene SJ, Gheorghiade M, Borlaug BA, Pieske B, Vaduganathan M, Burnett JC Jr, Roessig L, Stasch JP, Solomon SD, Paulus WJ, Butler J. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc 2013; 2: e000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kramer T, Dumitrescu D, Gerhardt F, Orlova K, Ten Freyhaus H, Hellmich M, Baldus S, Rosenkranz S. Therapeutic potential of phosphodiesterase type 5 inhibitors in heart failure with preserved ejection fraction and combined post‐ and pre‐capillary pulmonary hypertension. Int J Cardiol 2019; 283: 152–158. [DOI] [PubMed] [Google Scholar]
- 46. Opitz CF, Hoeper MM, Gibbs JSR, Kaemmerer H, Pepke‐Zaba J, Coghlan JG, Scelsi L, D'Alto M, Olsson KM, Ulrich S, Scholtz W, Schulz U, Grünig E, Vizza CD, Staehler G, Bruch L, Huscher D, Pittrow D, Rosenkranz S. Pre‐capillary, combined, and post‐capillary pulmonary hypertension. J Am Coll Cardiol 2016; 68: 368–378. [DOI] [PubMed] [Google Scholar]
- 47. Singh I, Rahaghi FN, Naeije R, Oliveira RKF, Systrom DM, Waxman AB. Right ventricular‐arterial uncoupling during exercise in heart failure with preserved ejection fraction. Chest 2019; 156: 933–943. [DOI] [PubMed] [Google Scholar]
- 48. Oliveira RKF, Agarwal M, Tracy JA, Karin AL, Opotowsky AR, Waxman AB, Systrom DM. Age‐related upper limits of normal for maximum upright exercise pulmonary haemodynamics. Eur Respir J 2016; 47: 1179–1188. [DOI] [PubMed] [Google Scholar]
- 49. Lewis GD, Bossone E, Naeije R, Grünig E, Saggar R, Lancellotti P, Ghio S, Varga J, Rajagopalan S, Oudiz R, Rubenfire M. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation 2013; 128: 1470–1479. [DOI] [PubMed] [Google Scholar]
- 50. van Heerebeek L, Hamdani N, Falcao‐Pires I, Leite‐Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012; 126: 830–839. [DOI] [PubMed] [Google Scholar]
- 51. Tsutamoto T, Kanamori T, Wada A, Kinoshita M. Uncoupling of atrial natriuretic peptide extraction and cyclic guanosine monophosphate production in the pulmonary circulation in patients with severe heart failure. J Am Coll Cardiol 1992; 20: 541–546. [DOI] [PubMed] [Google Scholar]
- 52. Armstrong PW, Lam CSP, Anstrom KJ, Ezekowitz J, Hernandez AF, O'Connor CM, Pieske B, Ponikowski P, Shah SJ, Solomon SD, Voors AA, She L, Vlajnic V, Carvalho F, Bamber L, Blaustein RO, Roessig L, Butler J. Effect of vericiguat vs placebo on quality of life in patients with heart failure and preserved ejection fraction. JAMA 2020; 324: 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Udelson JE, Lewis GD, Shah SJ, Zile MR, Redfield MM, Burnett J, Parker J, Seferovic JP, Wilson P, Mittleman RS, Profy AT, Konstam MA. Effect of praliciguat on peak rate of oxygen consumption in patients with heart failure with preserved ejection fraction. JAMA 2020; 324: 1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Senni M, Paulus WJ, Gavazzi A, Fraser AG, Diez J, Solomon SD, Smiseth OA, Guazzi M, Lam CS, Maggioni AP, Tschope C, Metra M, Hummel SL, Edelmann F, Ambrosio G, Stewart Coats AJ, Filippatos GS, Gheorghiade M, Anker SD, Levy D, Pfeffer MA, Stough WG, Pieske BM. New strategies for heart failure with preserved ejection fraction: the importance of targeted therapies for heart failure phenotypes. Eur Heart J 2014; 35: 2797–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoeper MM, Lam CSP, Vachiery JL, Bauersachs J, Gerges C, Lang IM, Bonderman D, Olsson KM, Gibbs JSR, Dorfmuller P, Guazzi M, Galie N, Manes A, Handoko ML, Vonk‐Noordegraaf A, Lankeit M, Konstantinides S, Wachter R, Opitz C, Rosenkranz S. Pulmonary hypertension in heart failure with preserved ejection fraction: a plea for proper phenotyping and further research. Eur Heart J 2017; 38: 2869–2873. [DOI] [PubMed] [Google Scholar]
- 56. Mordi IR, Singh S, Rudd A, Srinivasan J, Frenneaux M, Tzemos N, Dawson DK. Comprehensive echocardiographic and cardiac magnetic resonance evaluation differentiates among heart failure with preserved ejection fraction patients, hypertensive patients, and healthy control subjects. J Am Coll Cardiol Img 2018; 11: 577–585. [DOI] [PubMed] [Google Scholar]
- 57. Nolte K, Schwarz S, Gelbrich G, Mensching S, Siegmund F, Wachter R, Hasenfuss G, Dungen HD, Herrmann‐Lingen C, Halle M, Pieske B, Edelmann F. Effects of long‐term endurance and resistance training on diastolic function, exercise capacity, and quality of life in asymptomatic diastolic dysfunction vs. heart failure with preserved ejection fraction. ESC Heart Fail 2014; 1: 59–74. [DOI] [PubMed] [Google Scholar]
- 58. Trip P, Kind T, van de Veerdonk MC, Marcus JT, de Man FS, Westerhof N, Vonk‐Noordegraaf A. Accurate assessment of load‐independent right ventricular systolic function in patients with pulmonary hypertension. J Heart Lung Transplant 2013; 32: 50–55. [DOI] [PubMed] [Google Scholar]
- 59. Redington AN, Rigby ML, Shinebourne EA, Oldershaw PJ. Changes in the pressure‐volume relation of the right ventricle when its loading conditions are modified. Br Heart J 1990; 63: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tedford RJ. Determinants of right ventricular afterload (2013 Grover Conference series). Pulm Circ 2014; 4: 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Baseline haemodynamics from rest to peak exercise.
Table S2. Biventricular function and haemodynamics from rest to peak exercise before and after sildenafil administration.
Table S3. Biventricular function and haemodynamics from rest to peak exercise before and after sildenafil administration in HFpEF with and without isolated postcapillary pulmonary hypertension (IpcPH).
Figure S1. Comparison of biventricular volume changes during incremental exercise in patients with HFpEF, patients with impaired relaxation (LVCR) and healthy controls.
Figure S2. Right ventricular afterload and contractile reserve in healthy controls, patients with impaired relaxation (LVCR) and patients with heart failure and preserved ejection fraction with (HPpEF+IpcPH) or without isolated postcapillary pulmonary hypertension (HFpEF).
Figure S3. Evolution of transpulmonary gradient (TPG) over CO from rest to peak exercise in HFpEF patients and patients with impaired relaxation (LVCR).