

Abstract
BACKGROUND AND AIM:
Genetic alterations in intrahepatic cholangiocarcinoma (iCCA) are increasingly well characterized, but their impact on outcome and prognosis remains unknown.
APPROACH AND RESULTS:
This bi-institutional study of patients with confirmed iCCA (n = 412) used targeted next-generation sequencing of primary tumors to define associations among genetic alterations, clinicopathological variables, and outcome. The most common oncogenic alterations were isocitrate dehydrogenase 1 (IDH1; 20%), AT-rich interactive domain–containing protein 1A (20%), tumor protein P53 (TP53; 17%), cyclin-dependent kinase inhibitor 2A (CDKN2A; 15%), breast cancer 1–associated protein 1 (15%), FGFR2 (15%), polybromo 1 (12%), and KRAS (10%). IDH1/2 mutations (mut) were mutually exclusive with FGFR2 fusions, but neither was associated with outcome. For all patients, TP53 (P < 0.0001), KRAS (P = 0.0001), and CDKN2A (P < 0.0001) alterations predicted worse overall survival (OS). These high-risk alterations were enriched in advanced disease but adversely impacted survival across all stages, even when controlling for known correlates of outcome (multifocal disease, lymph node involvement, bile duct type, periductal infiltration). In resected patients (n = 209), TP53mut (HR, 1.82; 95% CI, 1.08–3.06; P = 0.03) and CDKN2A deletions (del; HR, 3.40; 95% CI, 1.95–5.94; P < 0.001) independently predicted shorter OS, as did high-risk clinical variables (multifocal liver disease [P < 0.001]; regional lymph node metastases [P < 0.001]), whereas KRASmut (HR, 1.69; 95% CI, 0.97–2.93; P = 0.06) trended toward statistical significance. The presence of both or neither high-risk clinical or genetic factors represented outcome extremes (median OS, 18.3 vs. 74.2 months; P < 0.001), with high-risk genetic alterations alone (median OS, 38.6 months; 95% CI, 28.8–73.5) or high-risk clinical variables alone (median OS, 37.0 months; 95% CI, 27.6-not available) associated with intermediate outcome. TP53mut, KRASmut, and CDKN2Adel similarly predicted worse outcome in patients with unresectable iCCA. CDKN2Adel tumors with high-risk clinical features were notable for limited survival and no benefit of resection over chemotherapy.
CONCLUSIONS:
TP53, KRAS, and CDKN2A alterations were independent prognostic factors in iCCA when controlling for clinical and pathologic variables, disease stage, and treatment. Because genetic profiling can be integrated into pretreatment therapeutic decision-making, combining clinical variables with targeted tumor sequencing may identify patient subgroups with poor outcome irrespective of treatment strategy.
Intrahepatic cholangiocarcinoma (iCCA) is an aggressive malignancy with rising incidence and mortality,(1,2) marked genetic heterogeneity,(3) and limited treatment options.(4) Complete resection of localized iCCA remains the only potentially curative treatment, but recurrence rates are high.(4) Furthermore, resection is not an option for most patients who present with advanced unresectable disease, for whom systemic chemotherapy remains the primary treatment strategy but has limited benefit. Recent clinical trials have suggested promising results for treatments targeting tumors with isocitrate dehydrogenase 1/2 (IDH1/2) and FGFR2 alterations(5,6); however, the impact of these therapies in a minority of patients with these alterations is unclear.
Lymph node metastasis and multifocal liver disease are powerful predictors of poor outcome after resection in iCCA(7,8) and are often used for prognostication and treatment allocation. However, some patients with unfavorable clinical criteria experience prolonged survival, whereas others with favorable clinical factors (e.g., solitary lesions, lymph node–negative) recur quickly and die shortly after surgery. Similarly, reliable outcome predictors for patients with advanced disease are lacking.
Recent studies have made inroads in characterizing mutational patterns and identifying genetic alterations with potential prognostic and therapeutic significance.(9,10) In most studies, however, such alterations have been noted in a minority of patients. Given the rarity and marked genomic heterogeneity of iCCA,(3) it has been difficult to characterize the complex interplay among alterations in individual somatic alterations and related pathways and to determine those alterations with prognostic value.(5,6,11) Thus, the primary aim of the present study was to characterize the mutational landscape of primary iCCA in a large cohort of patients, covering the broad spectrum of disease extent, with the goal of defining broadly applicable prognostic genomic alterations juxtaposed with known clinicopathologic predictors of outcome.
Patients and Methods
STUDY DESIGN AND PATIENTS
This study included patients with histologically confirmed primary iCCA treated at Memorial Sloan Kettering Cancer Center (MSKCC; USA) or Erasmus Medical Center (the Netherlands). The cohort included patients who underwent curative-intent resection at MSKCC (1993–2018) or Erasmus (2005–2015) (postoperative mortalities excluded) and patients treated nonoperatively at MSKCC (2008–2019). Systemic chemotherapy regimens were determined by the treating oncologist; some patients were treated with hepatic arterial infusion chemotherapy (HAIC), as described.(12,13) HAIC consisted of continuous infusion of floxuridine into the liver circulation through a surgically implanted hepatic pump in combination with concurrent systemic chemotherapy (primarily gemcitabine+platinum). Data were collected from prospectively maintained databases and supplemented with medical record review. Patients with previous clinical tumor genetic profiling or with available banked tumor tissue/pathological slides for retrospective genomic analysis were included. A pathologist blinded to tumor genotype reviewed slides to confirm the diagnosis and assess liver parenchymal disease (i.e., cirrhosis, steatosis), coded as present or absent. In the unresected cohort, cirrhosis was based on clinical grounds as cirrhotic liver morphology could not be assessed pathologically due to small tissue samples (e.g., liver biopsy). The diagnosis of chronic viral hepatitis was based on positive serum markers for chronic hepatitis B and C. Of note, data from 109 (31 resected, 78 unresected) patients have been reported.(11) The study was approved by the institutional review boards at MSKCC and Erasmus. All patients provided written informed consent for targeted-sequencing and in accordance with the ethical guidelines of the Declaration of Helsinki.
Resected tumors were staged according to American Joint Committee on Cancer’s (AJCC’s) eighth edition classification.(14) Early in the study time frame, hepatoduodenal ligament lymphadenectomy was performed selectively if clinically warranted but was subsequently routine. Patients with clinically node-negative disease had comparable survival to patients with histologically node-negative disease (P = 0.20; Supporting Fig. S1); both were categorized as “lymph node–negative.” Patients with lymph node metastasis and/or multifocal disease were considered “clinical high risk” for recurrence and death; those without these features were deemed “clinical low risk.” In unresected patients, staging was based on imaging and available pathology. Suspicious lymph node metastasis on imaging was categorized as “lymph node–positive.”(14) The reasons for unresectability included locally advanced disease (multifocal liver disease, lymph node metastasis, or extensive vascular and/or biliary involvement) or distant metastatic disease (M1).
GENOMIC PROFILING
All specimens were sequenced in the clinical laboratories of the Molecular Diagnostics Service at MSKCC using the MSK-Integrated Mutation Profiling of Actionable Cancer Targets (IMPACT) assay, a clinically validated hybridization capture-based targeted next-generation sequencing array that can detect mutations, copy-number alterations (CNAs), and select rearrangements.(15) The assay was expanded during the study period, and of the 412 cases, 40 were sequenced using the initial 341-gene assay, 147 using the 410-gene assay, and the remaining 225 using the 468-gene assay.(15) Sequencing and analysis were performed as described.(15,16) Briefly, all slides were re-reviewed by experienced attending pathologists (E.V. or C.S.) to identify tumor and normal liver tissue for DNA extraction. All samples had >60% tumor content, and DNA isolated from the primary tumor and matched normal liver tissue or blood was sequenced. All classes of genomic alterations were determined and called against the patient’s matched normal sample. Genomic data are available at cBioPortal (www.cbioportal.org/study/summa ry?id=ihch_msk_2021).
Genes sequenced in at least 70% of samples and recurrent oncogenic alterations occurring in ≥5% of all patients were identified. Genetic alterations were filtered for oncogenic variants using OncoKB (http://oncokb.org), (17) a precision oncology knowledge base that tracks cancer gene alterations. Only oncogenic alterations (oncogenic or likely oncogenic by OncoKB) were included in statistical analyses. We evaluated 12 canonical signaling pathways from The Cancer Genome Atlas (TCGA) PanCancer Atlas, including cell cycle, Hippo, Myc, Notch, nuclear factor erythroid 2–related factor 2 (NRF2), phosphoinositide 3-kinase (PI3K), receptor tyrosine kinase (RTK)/RAS, TGF-β, tumor protein P53 (TP53), Wnt, epigenetic, and DNA damage repair (DDR).(18,19)
Tumor mutation burden (TMB) was calculated as the total number of somatic non–silent protein–coding mutations divided by the coding region captured in each MSK-IMPACT panel (341 genes, 0.98 Mb; 410 genes, 1.06 Mb; 468 genes, 1.22 Mb), as we have validated.(20)
Variation in genomic alterations stratified by disease extent was analyzed for the entire cohort. Because complete staging information was not available for all patients who did not undergo resection or exploration, patients were divided into three groups (solitary liver tumor, multifocal liver tumor, and metastatic disease [lymph node with or without distant sites]) that correlated with increasing AJCC stage.
STATISTICAL ANALYSIS
Recurrence-free survival (RFS) after resection was calculated from surgery date until documented recurrence or death. Patients alive and recurrence-free at last follow-up were censored. Patients were followed every 3–6 months; physical examination, carbohydrate antigen 19–9 (CA19–9) level, and cross-sectional imaging were performed at each visit. Time of recurrence was defined as the time of the first imaging that reported definitive or suspicious new tumors or, for patients with biopsy-proven recurrence, the date of positive cytological or histological results.
Overall survival (OS) was calculated from the time of liver resection until death. Patients alive at last follow-up were censored. Five-year outcome estimates with two-sided 95% CIs are reported. For unresected patients, OS was calculated from date of diagnosis until death or last follow-up. For the entire cohort and for resected versus unresected comparisons, OS was calculated from the date of diagnosis.
Individual genes and pathways were evaluated for association with RFS/OS using univariable Cox proportional hazards regression. Associations between mutational status and tumor histological characteristics were assessed with Fisher’s exact test. Kaplan-Meier estimates were calculated for 5-year survival and median survival. Histopathologic variables were evaluated for associations with RFS/OS using univariable Cox proportional hazards regression, and HAIC was treated as a time-dependent variable. When testing multiple genes/characteristics, P values were adjusted for multiple testing using the false discovery rate approach within outcome; Q values (adjusted P values) < 0.05 were considered significant.
Clinicopathological multivariable models were constructed by including all clinical/pathological factors significant in univariable analysis and retaining factors with P < 0.05. For the unresected cohort, suspicious lymphadenopathy was included due to clinical importance. The final multivariable models were constructed by adding genomic factors significant in univariable analysis to the final clinicopathological model. All tests were two-sided, and all analyses were performed using R, version 4.0.0.
Results
CLINICAL AND PATHOLOGIC FEATURES OF THE COHORT
Overall, 412 patients with iCCA from the two institutions were included, of whom 390 were treated at MSKCC and 22 at Erasmus (Table 1). The median age was 64 years, and the male:female ratio was approximately 1:1. Sixty-four percent (264/412) of the patients had multifocal or nodal with or without distant metastatic disease, and 51% underwent resection as a primary treatment.
TABLE 1.
Demographic and Clinicopathological Characteristics
| Characteristic | Overall (N = 412) | Solitary liver tumor (n = 148) | Multifocal liver disease (n = 86) | Metastatic disease (n = 178) | Q | |
|---|---|---|---|---|---|---|
| All patients | Age (years) at diagnosis, median (range) | 64 (19–89) | 66 (19–89) | 63 (29–78) | 61 (31–87) | 0.01 |
| Sex | 0.50 | |||||
| Female | 222/412 (54%) | 85/148 (57%) | 49/86 (57%) | 88/178 (49%) | ||
| Male | 190/412 (46%) | 63/148 (43%) | 37/86 (43%) | 90/178 (51%) | ||
| BMI, median (range) | 27.5 (17.6–59.8) | 27.7 (19.6–59.8) | 27.7 (17.6–54.7) | 27.1 (17.6–52.1) | 0.60 | |
| Diabetes | 84/411 (20%) | 35/147 (24%) | 18/86 (21%) | 31/178 (17%) | 0.50 | |
| Chronic hepatitis | 33/412 (8.0%) | 15/148(10%) | 4/86 (4.7%) | 14/178 (7.9%) | 0.50 | |
| Hepatitis B | 19/412 (4.6%) | 8/148 (5.4%) | 1/86 (1.2%) | 10/178 (5.6%) | 0.40 | |
| Hepatitis C | 16/412 (3.9%) | 7/148 (4.7%) | 3/86 (3.5%) | 6/178 (3.4%) | 0.80 | |
| Cirrhosis | 29/403* (7.2%) | 10/144 (6.9%) | 8/85 (9.4%) | 11/174 (6.3%) | 0.80 | |
| PSC | 6/412 (1.5%) | 2/148 (1.4%) | 2/86 (2.3%) | 2/178 (1.1%) | 0.80 | |
| Smoking | 0.30 | |||||
| Current smoker | 41/409 (10%) | 20/146 (14%) | 9/85 (11%) | 12/178 (50%) | ||
| Former smoker | 166/409 (41%) | 50/146 (34%) | 39/85 (46%) | 77/178 (6.7%) | ||
| Never smoked | 202/409 (49%) | 76/146 (52%) | 37/85 (44%) | 89/178 (43%) | ||
| HAIC | 65/412 (16%) | 10/148 (6.8%) | 32/86 (37%) | 23/178 (12%) | <0.001 | |
| Tumor grade | 0.02 | |||||
| Well differentiated | 15/392 (3.8%) | 8/144 (5.6%) | 4/81 (4.9%) | 3/167 (1.8%) | ||
| Moderately differentiated | 231/392 (59%) | 92/144 (64%) | 53/81 (65%) | 86/167 (51%) | ||
| Poorly differentiated | 146/392 (37%) | 44/144 (31%) | 24/81 (30%) | 78/167 (47%) | ||
| Treatment group | <0.001 | |||||
| Resected | 209/412 (51%) | 132/148 (89%) | 33/86 (38%) | 44/178 (25%) | ||
| Unresected | 203/412 (49%) | 16/148 (11%) | 53/86 (62%) | 134/178 (75%) | ||
| ECOG PS | 0.01 | |||||
| 0 | 168/390 (43%) | 75/139 (54%) | 33/82 (40%) | 60/169 (36%) | ||
| 1 | 208/390 (53%) | 60/139 (43%) | 49/82 (60%) | 99/169 (59%) | ||
| 2–3 | 14/390 (3.6%) | 4/139 (2.9%) | 0/82 (0%) | 10/169 (5.9%) | ||
| CA19–9 elevated (>40 U/mL) | 190/307 (62%) | 46/93 (49%) | 38/63 (60%) | 106/151 (70%) | 0.01 | |
| Resected patients only | Characteristic | Overall (N = 209) | Solitary liver tumor (n = 132) | Multifocal liver disease (n = 33) | Metastatic disease (n = 44) | Q |
| Systemic chemotherapy | 94/207 (45%) | 45/132 (34%) | 13/32 (41%) | 36/43 (84%) | <0.001 | |
| Neoadjuvant | 29/209 (14%) | 6/132 (4.5%) | 8/33 (24%) | 15/44 (34%) | <0.001 | |
| Adjuvant | 76/205 (37%) | 39/131 (30%) | 9/32 (28%) | 28/42 (67%) | <0.001 | |
| Pathological tumor size (cm), median (range) | 6.0 (1.1–24.0) | 5.6 (1.1–18.5) | 6.1 (2.0–15.0) | 7.2 (2.7–24.0) | 0.04 | |
| LVI | 108/208 (52%) | 58/131 (44%) | 16/33 (48%) | 34/44 (77%) | 0.002 | |
| Perineural invasion | 57/186 (31%) | 33/120 (28%) | 6/27 (22%) | 18/39 (46%) | 0.07 | |
| Positive margin status | 25/209 (12%) | 14/132 (11%) | 6/33 (18%) | 5/44 (11%) | 0.50 | |
| Liver steatosis | 67/199 (34%) | 51/127 (40%) | 9/32 (28%) | 7/40 (18%) | 0.04 | |
| Periductal infiltration | 23/163 (14%) | 13/107 (12%) | 3/23 (13%) | 7/33 (21%) | 0.50 | |
| BD type | 0.80 | |||||
| Small | 176/205 (86%) | 114/132 (86%) | 27/31 (87%) | 35/42 (83%) | ||
| Large | 23/205 (11%) | 13/132 (9.8%) | 4/31 (13%) | 6/42 (14%) | ||
| Indeterminate | 6/205 (2.9%) | 5/132 (3.9%) | 0/31 (0%) | 1/42 (2.4%) | ||
| Positive lymph node status | 42/209 (20%) | 0/132 (0%) | 0/33 (0%) | 42/44 (95%) | <0.001 |
All data are n (%) unless noted. Denominators < 412 (for entire cohort) and <209 (for resected cohort) in overall column reflect missing data. Bold indicates significance.
In a small subset of patients (n = 9), an adequate assessment of cirrhosis was not possible.
Abbreviations: BMI, body mass index; ECOG, Eastern Cooperative Oncology Group; PS, performance status.
Patients were stratified according to disease extent (Table 1). Older patients tended to present with solitary liver tumors, and patients with more advanced disease tended to be younger. Higher-grade tumors were more likely to be metastatic at the time of presentation. No other clinicopathologic variables were associated with disease extent. Of 148 patients with solitary liver tumors, 89% (132/148) underwent resection, with the remainder frequently deemed unresectable owing to significant major vascular invasion or extensive biliary involvement. Patients with multifocal disease infrequently underwent operative intervention as multiple tumors are a relative contraindication to surgical resection(21); in the majority of such cases, multifocality was not appreciated preoperatively. Hepatic arterial infusion chemotherapy was administered largely to patients with unresectable, multifocal liver disease, with or without regional nodal disease.
The cohort was then stratified and analyzed by treatment. A total of 209 patients underwent resection, 94/207 (45%) of whom received additional systemic treatment (the use of any systemic chemotherapy could not be confirmed in two patients), including adjuvant HAIC with 5-fluoro-2-deoxyuridine with or without systemic chemotherapy (n = 6) and adjuvant systemic therapy only (n = 70), predominantly gemcitabine with or without platinum or capecitabine (n = 51) (no perioperative targeted therapies). Of the remaining 203 patients who did not undergo resection, 107 (53%) had unresectable locally advanced disease (i.e., bilateral multifocal disease or major vascular invasion), and 96 (47%) had metastatic disease. Suspicious lymph node involvement based on imaging was present in 31% (63/203) at locoregional sites only (hepatoduodenal ligament) and 30% (60/203) at distant nodal basins. Of patients who did not undergo resection, 197/203 (97%) received palliative chemotherapy, predominantly gemcitabine+platinum (145/197, 74%). Only 9/203 (4%) patients received a mitogen-activated protein kinase kinase inhibitor; no other first-line targeted agents were used. HAIC with concurrent systemic chemotherapy (primarily gemcitabine+oxaliplatin) was used in 54/203 (27%) unresected patients.
MUTATIONAL LANDSCAPE OF iCCA
Targeted DNA sequencing on 412 samples identified a total of 1,551 genetic alterations. The most common inactivating mutations were found in IDH1 (20%), AT-rich interactive domain–containing protein 1A (ARID1A; 20%), TP53 (17%), breast cancer 1–associated protein 1 (BAP1; 15%), and polybromo 1 (PBRM1; 12%) (Fig. 1A; Supporting Table S1). Activating mutations were found in KRAS (10%) and RAS/RAF kinases, including BRAF (5%), NRAS (3%), RASA1 (3%), and neurofibromin 1 (NF1; 1.7%). Ninety-two oncogenic fusions were identified in 78/412 patients (19%); 53 fusions in 47/412 (11%) patients involved FGFR2, and 38 were known in-frame FGFR2 fusions. Focal CNAs were noted in multiple genes, with cyclin-dependent kinase inhibitor 2A (CDKN2A) homozygous deletions (del) being the most common (13%; Fig. 1A). Rare amplifications in RTKs, such as Erb-B2 receptor tyrosine kinase 2 (ERBB2) (n = 7), EGFR (n = 6), and MET (n = 3), were identified. The median TMB per sample was 2.6 mutations/MB (interquartile range, 1.8–3.9); only 2 patients had TMB > 40 (consistent with microsatellite instability). Overall, most genes belonged to four key pathways, with alterations in epigenetic regulators being most frequent (246/412, 60%), followed by RTK/RAS signaling (196/412, 48%), TP53 (97/412, 24%), and cell cycle (85/412, 21%) pathways.
FIG. 1.
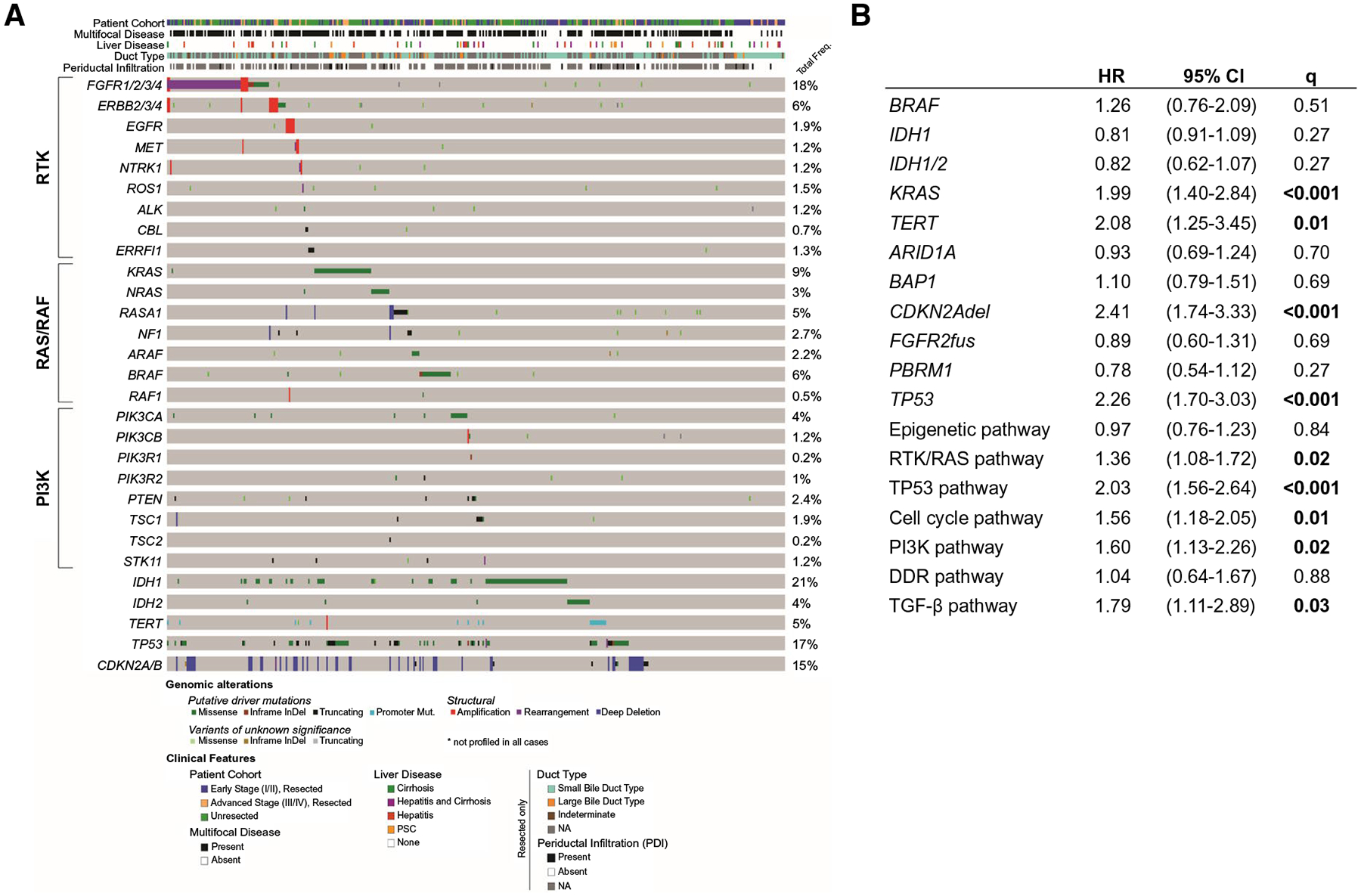
(A) OncoPrint representation of the genetic alterations occurring in the entire cohort (N = 412), including detailed breakdown of the RTK, RAS/RAF, and PI3K pathway genes, IDH1/2, and survival-associated genes. (B) Cox regression for overall survival by genetic features in all iCCA (N = 412, 287 events). Abbreviations: ARAF, A-Raf protooncogene; CBL, Cbl proto-oncogene; ERFFI1, ERBB receptor feedback inhibitor 1; NTRK, neurotrophic receptor tyrosine kinase; PTEN, phosphatase and tensin homolog; ROS1, ROS protooncogene 1; STK11, serine/threonine kinase 11; TSC1/2, TSC complex subunit 1/2.
Next, we examined mutual exclusivity and co-occurrence of enriched pathways and mutated genes (mut) across the cohort. IDH1/2mut tumors were significantly mutually exclusive from FGFR2 fusions (fus) (τ = −0.19, Q = 0.002), CDKN2Adel (τ = −0.17, Q = 0.008), TP53mut (τ = −0.15, Q = 0.02), and telomerase reverse transcriptase (TERT) mut (τ = −0.13, Q = 0.04). In addition, there was significant co-occurrence of TERT and TP53 aberrations (τ = 0.19, Q = 0.002) (Supporting Fig. S2). PI3K (τ = 0.21, Q = 0.001), TGF-β (τ = 0.19, Q = 0.002), cell cycle (τ = 0.18, Q = 0.004), and RTK/RAS (τ = 0.16, Q = 0.01) pathways showed co-occurrence with TP53mut (Supporting Table S2). In contrast, IDH1/2mut were mutually exclusive with most pathways (RTK/RAS [τ = −0.24, Q < 0.001], cell cycle [τ = −0.15, Q = 0.02], TP53 [τ = −0.15, Q = 0.02], TGF-β [τ = −0.13, Q = 0.04]) and not associated with any other pathway defined by the TCGA PanCancer Atlas.
RELATIONSHIP BETWEEN MUTATIONAL PROFILE AND CLINICAL OUTCOMES
We leveraged the size and clinical annotation of this cohort to determine the clinical significance of recurrent genetic alterations. Patients with alterations in the TP53, RTK/RAS, cell cycle, PI3K, or TGF-β pathways had shorter survival than those without (Fig. 1B). Specifically, for all patients, TP53mut, KRASmut, TERTmut, and CDKN2Adel were significantly associated with shorter OS (Figs. 1B and 2). Notably, only deep deletions in CDKN2A (n = 52) were associated with significantly shorter OS (P < 0.001), while presumed monoallelic CDKN2A alterations (n = 9) appeared to have little impact (Fig. 2C). Significant survival differences were not observed in patients harboring IDH1/2mut or FGRF2fus compared to wild type (wt) (Fig. 2E), although IDH1/2mut tumors showed a trend toward an improved OS (P = 0.08). In addition, the frequency of FGFR2fus (P = 0.34) and IDH1/2mut (P = 0.26) did not differ between resected and unresected patients, and there was no significant difference in the predictive power of either mutation in the resected group. Of note, IDH1mut and IDH2mut were not associated with outcome when analyzed together or separately (data not shown). IDH1/2mut tumors were less often associated with elevated CA19–9 levels (44% vs. 67%; P = 0.002), whereas peers with TP53mut (76% vs. 58% wt; Q = 0.04) and CDKN2Adel showed higher CA19–9 levels (83% vs. 58% wt; Q = 0.01) (Supporting Table S3). Of note, in the 29 patients with cirrhosis, 6 (21%) had TERT promoter alterations versus 14 (4%) of the 374 cirrhosis-free cases (Q = 0.008), and in the 6 patients with primary sclerosing cholangitis (PSC), 5 (83%) had a TP53mut (Q = 0.007) (Supporting Table S4).
FIG. 2.
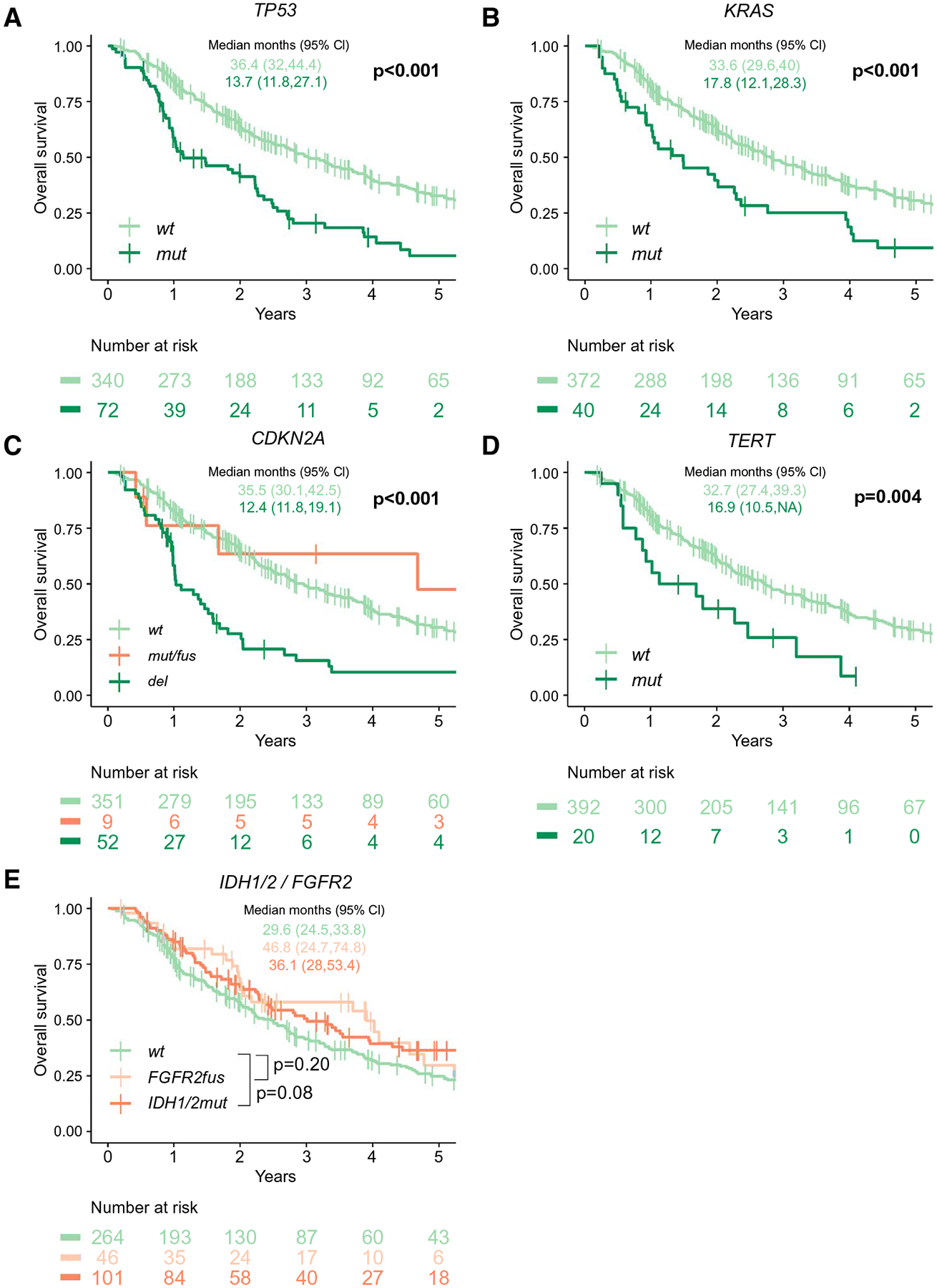
Effect of TP53 (A), KRAS (B), CDKN2A (C), TERT (D), IDH1/2 (E), and FGFR2 (E) mutation status on overall survival in the whole cohort (N = 412, 287 events).
RELATIONSHIP BETWEEN GENETIC ALTERATIONS AND CLINICAL STAGING AND TREATMENT
We next analyzed genomic alterations stratified by treatment group—resected versus unresected—to determine if mutational profiles were associated with primary treatment allocation (Fig. 3; Supporting Table S1). Across the landscape of genetic alterations, tumors in both groups had similar genetic signatures, and neither group was dominated by specific alterations in individual genes or canonical signaling pathways. However, there was an association between high-risk genetic alterations (i.e., those associated with shorter OS in the entire cohort [TP53mut, KRASmut, CDKN2Adel]) and disease extent (Tables 2–4). (TERTmut omitted due to its strong correlation with TP53mut.) The incidence of both TP53mut and CDKN2Adel increased progressively from early (i.e., solitary tumors) to later stage disease (i.e., multifocal liver or nodal with or without distant metastatic disease) (Table 2). KRASmut was more common in advanced iCCA but had similar prevalence in solitary and multifocal tumors. Indeed, in the resected cohort, 65% of TP53mut (n = 24/37), 53% of KRASmut (n = 10/19), 64% of CDKN2Adel (n = 14/22), and 64% (40/63) of all genetic high-risk patients had either multifocal disease or metastatic disease (nodal or distant). In contrast, of the resected patients without any high-risk genetic alterations, only 25% (37/146) had multifocal or metastatic disease (P < 0.001). The genetic high-risk cohort also comprised >50% (17/33) of the resected multifocal disease group but only 21% (11/53) of the unresected multifocal disease group (P = 0.004). In a multivariable model, all three high-risk genetic alterations were significant predictors of poor outcome, independent of disease extent (Table 3), with CDKN2Adel having the strongest impact (HR, 2.5; 95% CI, 1.7–3.8).
FIG. 3.
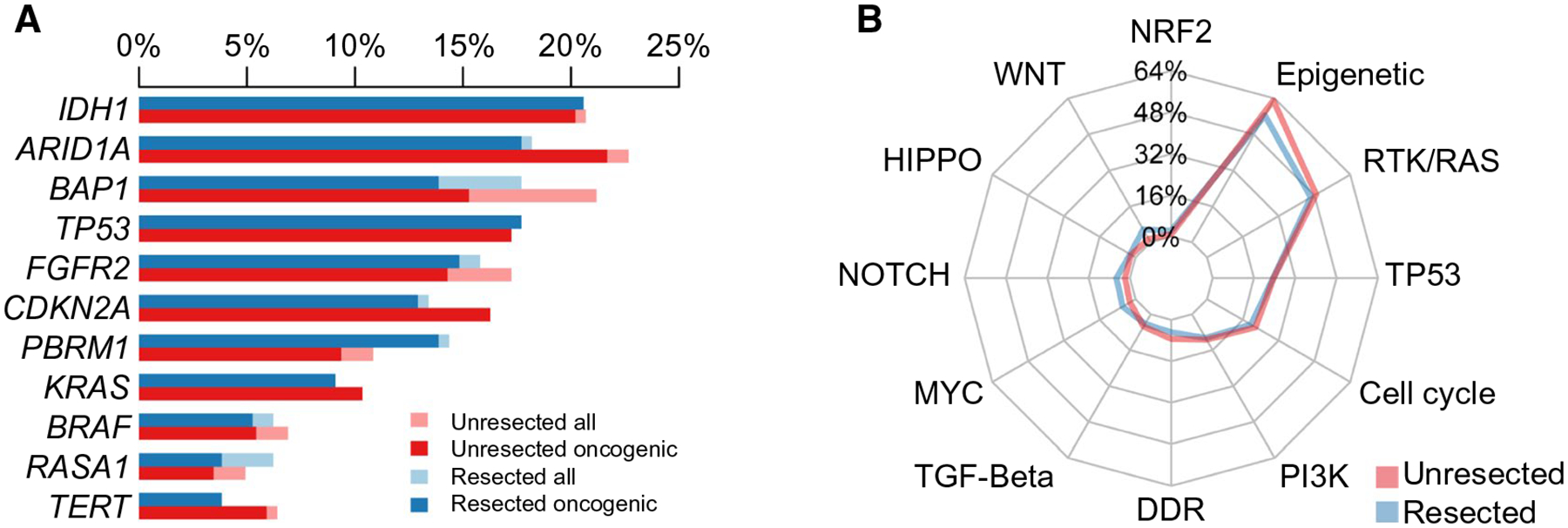
(A) Distribution of driver mutations or structural genetic alterations in iCCA occurring with a frequency of >5% in the entire cohort (N = 412), stratified by treatment group (resected and unresected). (B) Spider plot illustrating frequencies of alterations across 12 canonical signaling pathways in the entire cohort (N = 412), stratified by treatment group.
TABLE 2.
Association Between Disease Extent and Genetic Alterations
| Characteristic | Solitary liver tumor (n = 148), n (%) | Multifocal liver disease (n = 86), n (%) | Nodal ± distant metastases (n = 178), n (%) | Q |
|---|---|---|---|---|
| TP53mut | 16 (11%) | 15 (17%) | 41 (23%) | 0.02 |
| KRASmut | 11 (7%) | 6 (7%) | 23 (13%) | 0.20 |
| CDKN2Adel | 8 (5%) | 10 (12%) | 34 (19%) | 0.003 |
Bold indicates significance.
TABLE 4.
OS Estimates Stratified by Disease Extent and Genetic Alteration
| Solitary liver tumor | Multifocal liver disease | Nodal with or without distant metastases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Events | Median OS, months (95% CI) | n | Events | Median OS, months (95% CI) | n | Events | Median OS, months (95% CI) | |
| All | 148 | 85 | 63.6 (55.6–84.9) | 86 | 56 | 30.0 (26.1–44.4) | 178 | 146 | 15.8 (13.3–19.0) |
| TP53 | |||||||||
| wt | 132 | 72 | 69.4 (58.6–87.1) | 71 | 44 | 40.6 (26.1–51.5) | 137 | 111 | 17.3 (14.2–21.4) |
| mut | 16 | 13 | 28.3 (12.3–NA) | 15 | 12 | 26.6 (21.8–NA) | 41 | 35 | 11.8 (9.9–26.7) |
| KRAS | |||||||||
| wt | 137 | 74 | 72.5 (58.6–87.9) | 80 | 50 | 30.7 (26.1–45) | 155 | 128 | 17.3 (13.7–20.6) |
| mut | 11 | 11 | 33.1 (17.8–NA) | 6 | 6 | 25.5 (17.9–NA) | 23 | 18 | 12.1 (5.9–24.1) |
| CDKN2A | |||||||||
| wt | 140 | 80 | 63.6 (55.6–84.9) | 76 | 47 | 32.8 (26.7–47.2) | 144 | 116 | 17.7 (14.2–23.9) |
| del | 8 | 5 | 74.8 (19.1–NA) | 10 | 9 | 19.3 (11.8–NA) | 34 | 30 | 11.9 (9.9–15.5) |
| Any genetic high risk | |||||||||
| wt | 121 | 64 | 72.5 (61–88.1) | 58 | 32 | 44.4 (26.8–67.7) | 100 | 81 | 18.7 (15.7–25.6) |
| alt | 27 | 21 | 47.6 (19.1–74.8) | 28 | 24 | 24.4 (17.9–32) | 78 | 65 | 12.1 (10.1–17.5) |
P values were adjusted for multiple comparisons within outcome using the false discovery rate correction. Abbreviations: alt, altered; ECOG, Eastern Cooperative Oncology Group; PS, performance status.
TABLE 3.
Influence of Clinicopathological and Genetic Alterations on OS (N = 412, 287 events)
| Characteristic | HR | 95% CI | P |
|---|---|---|---|
| Disease extent | |||
| Solitary liver tumor | Ref | ||
| Multifocal liver disease | 1.16 | 0.71–1.88 | 0.60 |
| Metastatic disease | 2.13 | 1.39–3.24 | <0.001 |
| Resected | 2.60 | 1.81–3.73 | <0.001 |
| ECOG PS | |||
| 0 | Ref | ||
| 1 | 1.33 | 0.98–1.79 | 0.07 |
| 2–3 | 3.28 | 1.65–6.53 | <0.001 |
| Log CA19-9 | 1.06 | 1.00–1.12 | 0.05 |
| CDKN2Adel | 2.50 | 1.65–3.78 | <0.001 |
| TP53mut | 1.87 | 1.31–2.68 | <0.001 |
| KRASmut | 1.94 | 1.25–3.01 | 0.003 |
Bold indicates significance.
The relative impact of individual genomic alterations was then analyzed within each disease extent category (Table 4). TP53mut and KRASmut appeared to have a greater impact on OS in patients with solitary liver tumors, while CDKN2Adel was a more potent predictor in patients with multifocal liver and nodal or distant metastases. When analyzed as a group, the presence of any one or more high-risk genetic alteration corresponded to shorter median OS across all disease extent categories (Table 4).
MUTATIONAL STATUS AND CLINICAL PREDICTORS OF OUTCOME IN RESECTED iCCA
Next, we integrated clinical variables with genetic predictors specifically for patients who underwent resection. Median RFS and OS for the resected cohort (n = 209) were 18.4 (95% CI, 13.4–24.2) and 46.4 (95% CI, 41.2–61.3) months, respectively; 5-year RFS and OS were 22% (95% CI, 17–29) and 42% (95% CI, 35–50), respectively. Among resected patients, the presence of any one or more high-risk genetic alterations (TP53mut, KRASmut, and/or CDKN2Adel) was associated with shorter RFS and OS compared to wt tumors (Fig. 4A). The impact of each individual alteration on median survival was TP53mut = 25.8 months (95% CI, 16.1–35.7) versus TP53wt = 59.9 months (95% CI, 46.4–74.2), KRASmut = 27.6 months (95% CI, 12.1–52.6) versus KRASwt = 52.3 months (95% CI, 43.0–68.0), and CDKN2Adel = 14.8 months (95% CI, 10.0–37.0) versus CDKN2Awt = 52.6 months (95% CI, 45.2–67.2). PI3K pathway alterations (n = 22) were also associated with a shorter OS compared to wt tumors: 33.0 (95% CI, 20.4-not available [NA]) versus 52.6 (95% CI, 44.9–68.0) months (Fig. 4A).
FIG. 4.
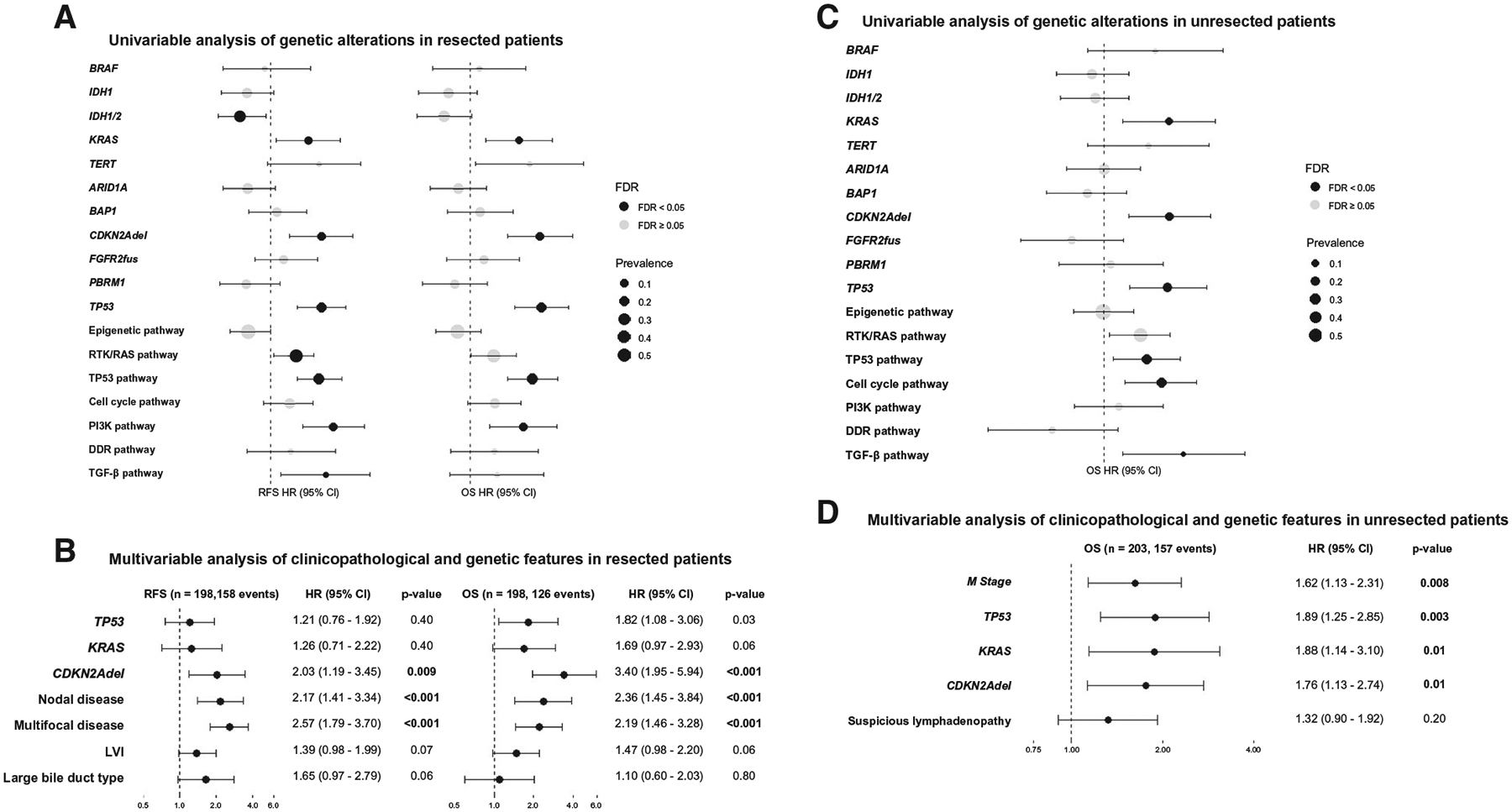
Univariable and multivariable analysis of genetic and clinicopathological features with RFS and OS in (A,B) resected and (C,D) unresected patients. (A) Univariable analysis of genetic features in resected patients (n = 209, 166 RFS events, 130 OS events). (B) Multivariable analysis of clinicopathological and genetic features in resected patients (n = 198). (C) Univariable analysis of genetic features in unresectable patients (n = 203, 157 events). (D) Multivariable analysis of clinicopathological and genetic features in unresectable patients (n = 203). P values were adjusted for multiple comparisons within outcome using the false discovery rate correction. Abbreviations: FDR, false discovery rate; M stage=metastatic disease.
Several clinical variables were associated with a significant risk of recurrence and death, including large tumor size (>5 cm), multifocal liver disease, regional lymph node metastases, lymphovascular invasion (LVI), perineural invasion, periductal infiltration (PDI), and large bile duct (BD) type by univariable analysis (Supporting Table S5). On multivariable analysis, nodal disease, multifocal liver disease, and LVI remained independently associated with RFS and OS (Supporting Table S5), whereas large BD type remained an independent predictor for shorter time to recurrence but not to death. In the clinicopathological multivariable model, TP53mut and CDKN2Adel remained independent predictors of shorter OS, in addition to multifocal liver disease and nodal disease (Fig. 4B), whereas KRASmut and LVI trended toward statistical significance (P = 0.06). Due to overfitting, we did not formally evaluate the independent prognostic value of PDI in the multivariable analysis. However, even after inclusion of BD type and PDI, the results did not change (data not shown). TP53mut was associated with LVI (76% vs. 47% wt; Q = 0.02), nodal disease (43% vs. 15% wt; Q < 0.001), and large BD type (33% vs. 7% wt; Q = 0.02); KRASmut was associated with perineural invasion (65% vs. 27% wt; Q = 0.04) and large BD type (37% vs. 9% wt; Q = 0.02), and CDKN2Adel was associated with multifocal disease (55% vs. 25% wt; Q = 0.04) (Supporting Tables S6 and S7).
To assess risk stratification by genetic profile in resected patients, the low-risk (solitary, node-negative) and high-risk (multifocal and/or nodal disease) clinical groups were stratified by the presence (“genetic high risk”) or absence (“genetic low risk”) of at least one alteration in TP53, KRAS, or CDKN2A (Fig. 5A,B). Patients with both or without any high-risk clinical or genetic factors represented the extremes of outcome (median OS, 18.3 and 95% CI, 14.8–29.7 vs. OS, 74.2 and 95% CI, 61.3–95.1 months, respectively, P < 0.001). Within this wide range, however, the interaction between clinical and genetic variables further stratified patients into outcome groups. Specifically, RFS and OS in clinical low-risk patients were significantly reduced in the presence of at least one high-risk genetic alteration (genetic high risk) (Fig. 5A,B); similarly, outcomes in the high-risk clinical group were improved if no high-risk genetic alterations were present (genetic low-risk).
FIG. 5.
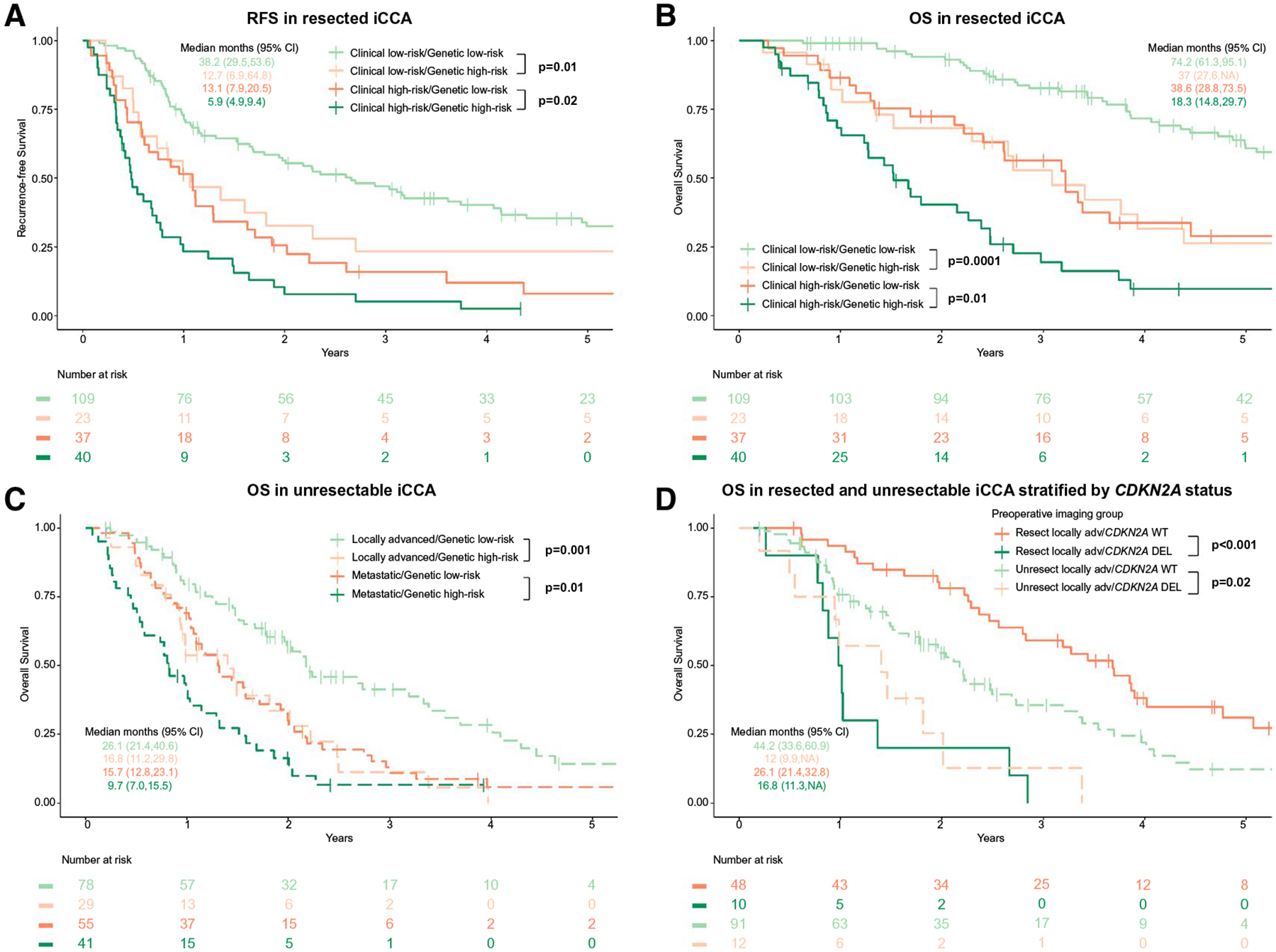
Effect of clinical and genetic high-risk group (TP53/KRAS/CDKN2A alterations) on (A) RFS (n = 209, 166 events) and (B) OS (n = 209, 130 events) after resection. (C) OS stratified by genetic risk groups and disease extent (locally advanced vs. metastatic, n = 203, 157 events) in patients with unresectable disease. (D) OS by CDKN2Adel status in resected/unresected patients of similar disease extent: resected clinical high-risk (n = 77) and unresected locally advanced (n = 103).
RISK STRATIFICATION IN UNRESECTABLE DISEASE
Next, we examined the utility of mutational profiling to stratify outcome in patients who did not undergo resection. In patients with unresectable disease (n = 203), median OS was 24.1 months (95% CI, 18.7–29.9) in those with locally advanced disease compared to 13.1 months (95% CI, 11.7–15.8) in those with distant metastases. On univariate analysis, TP53mut, KRASmut, and CDKN2Adel, as well as alterations in the RTK/RAS and TGF-β pathways, were associated with shorter OS (Fig. 4C). On multivariate analysis, only metastatic disease and TP53mut, KRASmut, and CDKN2Adel remained independent predictors of worse survival (Fig. 4D), recapitulating the findings observed in resected patients.
Alterations in TP53, KRAS, and CDKN2A stratified OS in unresected patients (locally advanced, P = 0.001; metastatic, P = 0.01; Fig. 5C). Median OS was 26.1 months (95% CI, 21.4–40.6) in patients with locally advanced, genetic low-risk tumors versus 16.8 months (95% CI, 11.1–29.8) in those with genetic high-risk tumors. In patients with metastatic disease, median OS was 15.7 months (95% CI, 12.8–23.1) for genetic low-risk tumors versus 9.7 months (95% CI, 7.0–15.5) with high-risk features, such that specific genetic alterations were universal predictors of poorer outcome, irrespective of disease extent or primary treatment modality.
In unresected patients with locally advanced iCCA treated with any chemotherapy (n = 103), survival was improved in genetic low-risk (n = 75; OS, 26.1 months, 95% CI, 23.5–40.6) compared to genetic high-risk tumors (n = 28; OS, 17.5 months, 95% CI, 11.2–29.8; P = 0.001). Within this group, HAIC was a strong predictor of improved survival (n = 50; HR, 0.45, 95% CI, 0.27–0.77; P = 0.003), independent of genomic profile. In fact, when compared with resected patients with similar disease extent (i.e., clinical high-risk, n = 77), the median OS for unresected, locally advanced patients was shorter (24.1, 95% CI, 18.7–29.9, vs. 32.0, 95% CI, 26.7–41.3, months), but the reduced HR of the HAIC treatment resulted in outcomes more comparable to resected, clinical high-risk patients. By contrast, patients in the locally advanced subgroup with genetic high-risk tumors generally had poor outcomes across all treatments. In particular, clinical high-risk CDKN2Adel patients (n = 14) submitted to resection had a median OS of 12.2 months (95% CI, 10.5-NA) versus 16.8 months (95% CI, 11.3-NA) for locally advanced patients with CDKN2Adel tumors who did not undergo resection (n = 12) (Fig. 5D). Alterations in TP53 or KRAS, on the other hand, did not negate apparent treatment outcome differences between these two groups (clinical high-risk resected: TP53mut 26.7 months [n = 24; 95% CI, 17.7–33.6] and KRASmut 27.1 months [n = 10; 95% CI, 13.3-NA] versus locally advanced unresected: TP53mut 11.8 months [n = 16; 95% CI, 10.1-NA and KRASmut 9.4 months [n = 8; 95% CI, 6.6-NA]). CDKN2Adel was therefore the strongest negative predictor of OS among the high-risk genetic variables across treatment groups.
Discussion
Several recent studies have highlighted the genetic heterogeneity of iCCA.(22) In the present analysis, we describe targeted genomic sequencing results from 412 iCCA tumor samples, representing one of the largest series combining genomics with detailed clinical annotation of treatment and outcomes. The data demonstrate the clinical utility of routine genomic analysis in patients with iCCA both for prognostication and for treatment recommendations.
As has been described, we observed that no single genetic alteration occurs in >25% of patients; however, we identified a high incidence of somatic alterations in the epigenetic (60%), RTK/RAS (48%), TP53 (24%), and cell cycle (21%) pathways, consistent with previous reports from other Western centers.(22,23) FGFR2fus were present in 11% of patients, a similar incidence as described recently (13.6%).(24) In our study, TP53 and KRAS alteration prevalence was higher than that reported from other Western centers (6%−10%).(23,25,26) In the present series, we identify an independent predictive value of mutations in TP53 and KRAS and deletions in CDKN2A for oncological outcome in patients treated for iCCA, even after adjustment for clinicopathological confounders. Specifically, the presence of any one of these high-risk genetic alterations independently predicted shorter survival, regardless of disease stage or treatment, reflecting their prognostic significance when combined with known prognostic clinical variables.(4,8,27) While the genetic and clinical high-risk features tended to coexist, there was a large proportion of patients with high-risk genetic alterations in the setting of clinically more favorable tumors.
The association of somatic mutations in TP53 with shorter survival in resected iCCA has been reported,(22,28) suggesting an association with more aggressive disease and poor outcome.(28) In the present study, TP53mut was also associated with LVI, nodal disease, and large BD type, consistent with an aggressive phenotype, as shown in other liver cancers.(29) Associations between KRASmut, perineural invasion, large BD type, and worse outcome after iCCA resection have also been reported.(22,30,31) The prognostic implications of TP53 and KRAS alterations in univariate analyses were described in a prior analysis of 321 biliary tract cancer samples, including 224 iCCAs.(32) In contrast to that study, here we demonstrate that each of these genetic high-risk mutations associates with outcomes independently for resected and unresectable disease, even when stratified by disease extent and known pathologic variables. Our study also uniquely identifies an association between alterations in CDKN2A and survival outcomes in patients with iCCA irrespective of disease extent or treatment, building on our findings in advanced disease.(11) We also found that a subgroup with CDKN2Adel and high-risk clinical features had universally poor survival regardless of treatment, suggesting that these patients do not benefit from resection.
Divergent from previous studies that have reported predominantly large BD type iCCA (41%−59%),(33–37) the present study mainly consisted of small BD tumors. Potential reasons for this finding include a small number of patients with PSC or liver fluke infection,(35,38,39) two etiologies associated with large BD type iCCA.(36) Consistent with previous reports,(34,35,40–43) patients with iCCA submitted to resection with large BD type had worse prognosis in univariate analysis. However, histopathological subtype was not an independent predictor of postoperative survival in multivariate analysis, perhaps due to a significant association between large BD type and KRASmut and TP53mut.
There are conflicting data regarding the association between other commonly identified genetic alterations and outcomes. The most commonly reported actionable alterations in iCCA are IDH1/2mut and FGFR2fus, with significant mutual exclusivity between IDH1/2mut and CDKN2Adel, TP53mut, TERTmut, and FGFR2fus, consistent with previous work.(44) IDH1/2 alterations may represent a distinct disease mechanism as oncogenic IDH1/2 mutations are known to acquire neomorphic activity to produce the oncometabolite 2-hydroxyglutarate, with subsequently an inhibitory effect on α-ketoglutarate-dependent enzymes.(45,46) While IDH1/2mut and FGFR2fus remain predictive biomarkers for patients with these mutations receiving targeted therapies, particularly in advanced disease,(47,48) our data do not support a major prognostic role for all patients. For IDH1/2mut, this finding contrasts with a number of studies that have shown that IDH1/2mut may be associated with improved survival.(49,50) Other efforts to define the relationship of IDH1/2mut with survival have shown no impact(51,52) or even poorer survival.(23) We suspect that the heterogeneity in the literature related to inconclusive association of IDH1/2mut with outcome may reflect different proportions of resected and unresected patients in each study; of note, we observed an improvement in RFS in resected patients with IDH1/2mut and a trend toward increased OS, consistent with a prior evaluation in patients treated surgically.(49)
Multifocal disease and lymph node metastasis constituted clinical stratification, given their predictive power and availability prior to resection (versus histological features). The addition of genetic information, however, greatly improved risk stratification, identifying additional patient subgroups that may obtain either significant or marginal benefit from treatment. For instance, we demonstrate that patients with multifocal disease or lymph node involvement without high-risk genetic alterations have superior outcomes after resection. Despite relative contraindications to surgical resection in clinical high-risk patients, there appears to be a significant survival improvement in the absence of high-risk genetic features. On the other hand, patients with CDKN2A deletions are more likely to have multifocal disease and/or lymph node involvement, as well as universally poor outcomes irrespective of whether they underwent resection. Patients with this genetic profile combined with high-risk clinical features did not appear to benefit from resection, and they may be best treated initially with systemic therapy alone or in combination with HAIC, which resulted in survival rates better than systemic chemotherapy, consistent with our experience.(53) Surgical resection should be reserved in these instances as there appears to be quite limited benefit.
As recent data supporting the use of targeted therapies for tumors with IDH1 mutations(6) and FGFR fusions(5) now highlight the critical need for tumor mutational profiling in iCCA, we propose that deleterious mutations and alterations in KRAS, TP53, and/or CDKN2A are also key variables that can guide therapeutic interventions in these patients. In our large cohort, the ability to stratify by mutation, as well as treatment or disease extent, uncovers specific genetic aberrations that identify a population more likely to have a more aggressive disease course after resection. For this patient subgroup, the risk–benefit equation would seem to favor nonoperative management, given the potential morbidity associated with hepatic resection and limited benefit of intervention. Validation of these results in larger populations remains necessary, but the findings uncover a genetically defined population worthy of further investigation.
Limitations of this study include inherent selection bias associated with retrospective analysis of patients from two tertiary institutions; the findings may not fully account for geographic/demographic variation of iCCA.(54) The present cohort certainly reflects a non–liver fluke, non–hepatitis B endemic population in Western countries distinct from previous studies with mainly Asian populations. Additionally, our study used annotations in OncoKB, in which several variants of unknown significance (VUS) are often poorly characterized. However, large-scale efforts to characterize most VUS eventually demonstrate no clinical significance.(41) Finally, our effort focused primarily on genetic events in iCCA and does not offer additional insight into the epigenetic, transcriptional, and cell-extrinsic mechanisms that contribute to heterogeneous disease biology and thus outcome. Our observation that extensive clinical and genetic annotation allows prediction of outcomes in patients with iCCA highlights, however, the importance of driver mutations in tumor biological behavior.
Study patients also received various adjuvant and locoregional treatments which were not controlled for in statistical analyses. Certain pathologic characteristics were unavailable for some specimens, but this occurred in <10% of samples. Additionally, while some patients were treated prior to the introduction of contemporary chemotherapy, this group accounted for only 1% of all patients. Finally, resected tumors were staged according to AJCC guidelines, whereas staging was based on imaging and available pathology for unresected patients. We have taken great care to avoid direct statistical comparisons between the two treatment groups, and the concordance of the high-risk clinical stage based on preoperative imaging versus postsurgical histopathology was otherwise excellent (86% [180/209] in the resected cohort). Reanalysis of the risk stratification by CDKN2Adel status in resected and unresected patients of similar disease extent based on preoperative imaging only did not change the findings of the study (data not shown).
In conclusion, we identified high-risk mutations that stratify outcome in patients with iCCA and enhance prognostic modeling based on clinical variables alone. Pretreatment genetic profiling adds critical information to known clinicopathologic factors to inform therapeutic decision-making in patients with iCCA and to identify potential benefit from currently available treatment options.
Supplementary Material
Acknowledgment:
We acknowledge Erin Patterson, Ph.D. (Memorial Sloan Kettering Cancer Center), for editorial assistance.
Supported in part by the National Institutes of Health/National Cancer Institute (P30 CA008748 Cancer Center Support Grant, Cycle for Survival), and the Clinical and Translational Science Center at Weill Cornell Medical Center and Memorial Sloan Kettering Cancer Center (UL1TR00457, to L.M.P.).
Potential conflict of interest:
Dr. Harding consults for and received grants from Bristol-Myers Squibb. He consults for Eisai, Exelexis, Merck, Cytomax, QED, and Zymeworks. Dr. Cercek advises Bayer and Array. She received grants from GlaxoSmithKline, RGenix, and Seattle Genetics. Dr. Solit advises Pfizer and BridgeBio. He advises and owns stock in Loxo and Lilly Oncology. Dr. Kingham is on the speakers’ bureau for Olympus Surgical. Dr. Lowery advises Agios and Roche. She is on the speakers’ bureau for Novartis.
Abbreviations:
- AJCC
American Joint Committee on Cancer
- ARID1A
AT-rich interactive domain–containing protein 1A
- BAP1
breast cancer 1–associated protein 1
- BD
bile duct
- CA19–9
carbohydrate antigen 19–9
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- DDR
DNA damage repair
- del
deletion
- ERBB2
Erb-B2 receptor tyrosine kinase 2
- fus
fusion
- HAIC
hepatic arterial infusion chemotherapy
- iCCA
intrahepatic cholangiocarcinoma
- IDH1/2
isocitrate dehydrogenase 1/2
- LVI
lymphovascular invasion
- MSKCC
Memorial Sloan Kettering Cancer Center
- mut
mutant
- NA
not available
- OS
overall survival
- PBRM1
polybromo 1
- PDI
periductal infiltration
- PI3K
phosphoinositide 3-kinase
- PSC
primary sclerosing cholangitis
- RFS
recurrence-free survival
- RTK
receptor tyrosine kinase
- TERT
telomerase reverse transcriptase
- TMB
tumor mutation burden
- TP53
tumor protein P53
- WT
wild type
Footnotes
These data were presented in part at the 2017 American Society of Clinical Oncology Annual Meeting.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.31829/suppinfo.
REFERENCES
- 1).Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001;33:1353–1357. [DOI] [PubMed] [Google Scholar]
- 3).Brandi G, Farioli A, Astolfi A, Biasco G, Tavolari SJO. Genetic heterogeneity in cholangiocarcinoma: a major challenge for targeted therapies. Oncotarget 2015;6:14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Doussot A, Groot-Koerkamp B, Wiggers JK, Chou J, Gonen M, DeMatteo RP, et al. Outcomes after resection of intrahepatic cholangiocarcinoma: external validation and comparison of prognostic models. J Am Coll Surg 2015;221:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Abou-Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]; Abou-Alfa GK and Zhu AX (1st author and senior author) contributed equally.
- 7).de Jong MC, Nathan H, Sotiropoulos GC, Paul A, Alexandrescu S, Marques H, et al. Intrahepatic cholangiocarcinoma: an international multi-institutional analysis of prognostic factors and lymph node assessment. J Clin Oncol 2011;29:3140–3145. [DOI] [PubMed] [Google Scholar]
- 8).Endo I, Gonen M, Yopp AC, Dalal KM, Zhou Q, Klimstra D, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96. [DOI] [PubMed] [Google Scholar]
- 9).Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–1010. [DOI] [PubMed] [Google Scholar]
- 10).Zhu AX, Borger DR, Kim Y, Cosgrove D, Ejaz A, Alexandrescu S, et al. Genomic profiling of intrahepatic cholangiocarcinoma: refining prognosis and identifying therapeutic targets. Ann Surg Oncol 2014;21:3827–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 2018;24:4154–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Kemeny NE, Schwartz L, Gönen M, Yopp A, Gultekin D, D’Angelica MI, et al. Treating primary liver cancer with hepatic arterial infusion of floxuridine and dexamethasone: does the addition of systemic bevacizumab improve results. Oncology 2011;80:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Jarnagin WR, Schwartz LH, Gultekin DH, Gönen M, Haviland D, Shia J, et al. Regional chemotherapy for unresectable primary liver cancer: results of a phase II clinical trial and assessment of DCE-MRI as a biomarker of survival. Ann Oncol 2009;20:1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, et al. The eighth edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin 2017;67:93–99. [DOI] [PubMed] [Google Scholar]
- 15).Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015;17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017;2017:PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 2018;173:321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Ding L, Bailey MH, Porta-Pardo E, Thorsson V, Colaprico A, Bertrand D, et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell 2018;173:305–320.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 2018;36:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Weber SM, Ribero D, O’Reilly EM, Kokudo N, Miyazaki M, Pawlik TM. Intrahepatic cholangiocarcinoma: expert consensus statement. HPB (Oxford) 2015;17:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JSC, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun 2014;5:5696. [DOI] [PubMed] [Google Scholar]
- 23).Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427–1434. [DOI] [PubMed] [Google Scholar]
- 25).Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol 2018;68:959–969. [DOI] [PubMed] [Google Scholar]
- 26).Chan-on W, Nairismägi M-L, Ong CK, Lim WK, Dima S, Pairojkul C, et al. Exome sequencing identifies distinct mutational patterns in liver fluke–related and non-infection-related bile duct cancers. Nat Genet 2013;45:1474–1478. [DOI] [PubMed] [Google Scholar]
- 27).Wang Y, Li J, Xia Y, Gong R, Wang K, Yan Z, et al. Prognostic nomogram for intrahepatic cholangiocarcinoma after partial hepatectomy. J Clin Oncol 2013;31:1188–1195. [DOI] [PubMed] [Google Scholar]
- 28).Simbolo M, Vicentini C, Ruzzenente A, Brunelli M, Conci S, Fassan M, et al. Genetic alterations analysis in prognostic stratified groups identified TP53 and ARID1A as poor clinical performance markers in intrahepatic cholangiocarcinoma. Sci Rep 2018;8:7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouzé E, Blanc J-F, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol 2017;67:727–738. [DOI] [PubMed] [Google Scholar]
- 30).Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One 2014;9:e115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Chen TC, Jan YY, Yeh TS. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann Surg Oncol 2012;19(Suppl. 3):S675–S681. [DOI] [PubMed] [Google Scholar]
- 32).Javle M, Bekaii-Saab T, Jain A, Wang Y, Kelley RK, Wang K, et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 2016;122:3838–3847. [DOI] [PubMed] [Google Scholar]
- 33).Aishima S, Kuroda Y, Nishihara Y, Iguchi T, Taguchi K, Taketomi A, et al. Proposal of progression model for intrahepatic cholangiocarcinoma: clinicopathologic differences between hilar type and peripheral type. Am J Surg Pathol 2007;31:1059–1067. [DOI] [PubMed] [Google Scholar]
- 34).Hayashi A, Misumi K, Shibahara J, Arita J, Sakamoto Y, Hasegawa K, et al. Distinct clinicopathologic and genetic features of 2 histologic subtypes of intrahepatic cholangiocarcinoma. Am J Surg Pathol 2016;40:1021–1030. [DOI] [PubMed] [Google Scholar]
- 35).Komuta M, Govaere O, Vandecaveye V, Akiba J, Van Steenbergen W, Verslype C, et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology 2012;55:1876–1888. [DOI] [PubMed] [Google Scholar]
- 36).Liau J-Y, Tsai J-H, Yuan R-H, Chang C-N, Lee H-J, Jeng Y-M. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features. Mod Pathol 2014;27:1163–1173. [DOI] [PubMed] [Google Scholar]
- 37).Akita M, Fujikura K, Ajiki T, Fukumoto T, Otani K, Azuma T, et al. Dichotomy in intrahepatic cholangiocarcinomas based on histologic similarities to hilar cholangiocarcinomas. Mod Pathol 2017;30:986–997. [DOI] [PubMed] [Google Scholar]
- 38).Carpino G, Cardinale V, Folseraas T, Overi D, Grzyb K, Costantini D, et al. Neoplastic transformation of the peribiliary stem cell niche in cholangiocarcinoma arisen in primary sclerosing cholangitis. Hepatology 2019;69:622–638. [DOI] [PubMed] [Google Scholar]
- 39).Komuta M, Spee B, Borght SV, De Vos R, Verslype C, Aerts R, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology 2008;47:1544–1556. [DOI] [PubMed] [Google Scholar]
- 40).Chung T, Rhee H, Nahm JH, Jeon Y, Yoo JE, Kim Y-J, et al. Clinicopathological characteristics of intrahepatic cholangiocarcinoma according to gross morphologic type: cholangiolocellular differentiation traits and inflammation- and proliferation-phenotypes. HPB (Oxford) 2020;22:864–873. [DOI] [PubMed] [Google Scholar]
- 41).Hoffman-Andrews L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci 2018;4:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Kozaka K, Sasaki M, Fujii T, Harada K, Zen Y, Sato Y, et al. A subgroup of intrahepatic cholangiocarcinoma with an infiltrating replacement growth pattern and a resemblance to reactive proliferating bile ductules: “bile ductular carcinoma.” Histopathology 2007;51:390–400. [DOI] [PubMed] [Google Scholar]
- 43).Nakanuma Y, Sato Y, Harada K, Sasaki M, Xu J, Ikeda H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J Hepatol 2010;2:419. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nakanuma Y contributed to half of this review and the remaining four authors equally contributed to the other half.
- 44).Javle MM, Murugesan K, Shroff RT, Borad MJ, Abdel-Wahab R, Schrock AB, et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J Clin Oncol 2019;37:4087. [Google Scholar]
- 45).Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2018;36:276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Lowery MA, Abou-Alfa GK, Burris HA, Janku F, Shroff RT, Cleary JM, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol 2017;35:4015. [Google Scholar]; Co-Senior authors: Zhu AX, Abou-Alfa GK.
- 49).Wang P, Dong Q, Zhang C, Kuan P-F, Liu Y, Jeck WR, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Wang J, Zhang Z-G, Ding Z-Y, Dong W, Liang H-F, Chu L, et al. IDH1 mutation correlates with a beneficial prognosis and suppresses tumor growth in IHCC. J Surg Res 2018;231:116–125. [DOI] [PubMed] [Google Scholar]
- 51).Kipp BR, Voss JS, Kerr SE, Barr Fritcher EG, Graham RP, Zhang L, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol 2012;43:1552–1558. [DOI] [PubMed] [Google Scholar]
- 52).Goyal L, Govindan A, Sheth RA, Nardi V, Blaszkowsky LS, Faris JE, et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015;20:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Cercek A, Boerner T, Tan BR, Chou JF, Gönen M, Boucher TM, et al. Assessment of hepatic arterial infusion of floxuridine in combination with systemic gemcitabine and oxaliplatin in patients with unresectable intrahepatic cholangiocarcinoma: a phase 2 clinical trial. JAMA Oncol 2019;6:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet 2012;44:690–693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.