

SUMMARY
CARMILs are large, multidomain, membrane-associated proteins that regulate actin assembly and Rho-family GTPases, but their role in inflammatory signaling is not defined. Tandem mass tag mass spectrometry indicated that, in fibroblasts, CARMIL1 associates with interleukin (IL)-1 signaling molecules. Immunoprecipitation of cells transfected with CARMIL1 mutants showed that the leucine-rich repeat (LRR) region of CARMIL1 associates with IL-1 receptor type 1 (IL-1R1) and IL-1 receptor-associated kinase (IRAK). Knockout of CARMIL1 by CRISPR-Cas9 reduced IL-1-induced ERK activation by 72% and MMP3 expression by 40%. Compared with CARMIL1 wild-type (WT), cells expressing mutant CARMIL1 lacking its LRR domain exhibited 45% lower ERK activation and 40% lower MMP3 expression. In fibroblasts transduced with a cell-permeable, TAT CARMIL1 peptide that competed with IL-1R1 and IRAK binding to the LRR of CARMIL1, collagen degradation was reduced by 43%. As the LRR of CARMIL1 evidently regulates IL-1 signaling, CARMIL1 could become a target for anti-inflammatory drug development.
In Brief
CARMILs regulate actin assembly, but their role in inflammation is unknown. Wang et al. show that fibroblasts require CARMIL1 for IL-1 signaling. Knockout of CARMIL1 reduces IL-1 signaling and collagen degradation. Cell-permeable, CARMIL1-binding peptides that block its interaction with IL-1 signaling molecules inhibit collagen degradation. CARMIL1 is a promising target for anti-inflammatory drug development.
Graphical Abstract
INTRODUCTION
Interleukin-1 (IL-1) is an important inflammatory mediator that promotes the degradation and remodeling of extracellular matrices (ECMs) by increasing the expression of matrix metalloproteinases (MMPs); these processes are centrally involved in several inflammatory diseases, including rheumatoid arthritis (Dinarello, 2019), acute lung injury, Still’s disease, and chronic periodontitis (Hönig et al., 1989). Binding of IL-1 to its signaling receptor (IL-1RI) leads to the recruitment of IL-1R accessory protein (IL-1RAcP; Wesche et al., 1997b). The activated IL-1R signaling complex subsequently recruits myeloid differentiation factor 88 (MyD88), an adaptor protein that binds IL-1R-associated kinases 1 and 2 (IRAK1 and IRAK2) (Wesche et al., 1997a; Muzio et al., 1997). IRAK is rapidly phosphorylated, then disengages from MyD88 (Wesche et al., 1997a) and activates signal transduction cascades, including the mitogen-activated protein kinase (MAPK) family members ERK (MacGillivray et al., 2000), JNK (Burns et al., 1998), and p38 (McDermott and O’Neill, 2002).
Anchorage-dependent cells such as gingival fibroblasts and chondrocytes are directly involved in the matrix degradation seen, respectively, in the high-prevalence inflammatory diseases periodontitis and rheumatoid arthritis. For IL-1 signal transduction in fibroblasts and chondrocytes, IL-1 receptor type 1 (IL-1R1) must be recruited to focal adhesions (FAs), which is followed by activation of the FA kinase (Arora et al., 1995), Ca2+ release from the endoplasmic reticulum (Lo et al., 1998; Luo et al., 1997), and activation of ERK (MacGillivray et al., 2000; Wang et al., 2003). All of these processes are involved in the signaling system that leads to increased expression of MMPs (Wang et al., 2010; Reunanen et al., 1998). Despite extensive investigation, the regulation of IL-1 signaling in anchorage-dependent stromal cells is not well defined.
FAs form at sites of integrin receptor binding and clustering and comprise complex aggregates of cytoskeletal, scaffolding, adhesion, and signaling proteins (Wehrle-Haller and Imhof, 2002; Burridge and Chrzanowska-Wodnicka, 1996). The spatial co-localization of these interacting molecules provides a pivotal regulatory locus for signal transduction. In IL-1 signaling, the organization of actin filament networks in FAs is critical for activation of ERK (MacGillivray et al., 2000). In this phenomenon, high-density actin filament networks and actin-binding proteins in FAs provide a poorly defined scaffold (Murphy-Ullrich, 2001) that enables the assembly of IL-1R-associated signaling molecules into a functional complex (McCulloch et al., 2006). There is also functional interdependence of IL-1 signaling on the organization of the actin cytoskeleton, because IL-1 induces transient cell contraction of the actin filament network (Zhu et al., 1998). Currently, little is known about the processes by which the IL-1 signaling machinery is recruited to FAs.
The assembly of FAs is currently being investigated with several technologies, including imaging (Morimatsu et al., 2015), proteomics (Manninen and Varjosalo, 2017), and bioinformatics (Horton et al., 2015), which are providing new insights into the composition, organization, and regulation of these complexes. In previous tandem mass tag mass spectrometry experiments, we analyzed FA proteins isolated from human fibroblasts that were involved in IL-1 signaling. We found that actin capping protein (CP) and its associated proteins regulate the recruitment of IL-1 signaling proteins to FAs, IL-1-induced ERK activation, and MMP expression (Wang et al., 2018). Accordingly, CP and/or its associated proteins may be critical for regulating tissue destruction in IL-1-mediated inflammatory diseases. CP is a highly conserved, 64-kDa heterodimeric protein that binds to the fast-growing, barbed ends of actin filaments and blocks the exchange of actin monomers to regulate filament growth (Maruyama et al., 1990; Wear et al., 2003). However, CP does not exhibit structural features (Zwolak et al., 2010) that would indicate its ability to interact directly with IL-1R1 or other IL-1 signaling proteins (i.e., IRAK and MyD88). Accordingly, we searched for potential CP-associated proteins that would interact directly with IL-1 signaling proteins.
A diverse group of proteins interact with CP through a conserved, CP-interaction (CPI) motif; notably, Capping Protein Arp2/3 myosin I linker (CARMIL) (Zwolak et al., 2013), which binds CP with sub-nanomolar affinity (Edwards et al., 2014), recruits CP to specific subcellular locations, and modulates its actin capping activity via allosteric mechanisms (Edwards et al., 2014). CARMIL-family proteins are large, highly conserved, multidomain homodimers. The biochemical, structural, and cellular properties of CARMIL proteins suggest that they may interact with signaling networks to regulate the dynamics of the actin cytoskeleton at membranes. Indeed, various domains of these CARMIL isoforms associate with plasma membranes, vimentin filaments, SH3-containing class I myosin, the dual-GEF Trio, and other adaptors and signaling molecules. Their structural features suggest that CARMILs play a variety of membrane-associated functions related to actin assembly and signaling.
The three vertebrate isoforms—CARMIL1, −2, and −3—are defined by conserved differences in their amino-acid sequences. CARMIL1 is strongly implicated in aberrant inflammatory processes and is expressed in abundance by fibroblasts (Stark et al., 2017). CARMIL1 (Zwolak et al., 2013) is an ~1,370-aa, multidomain protein that comprises a non-canonical pleckstrin homology (PH) domain (involved in plasma membrane targeting), 16 leucine-rich repeats (LRRs), a large helical domain (HD), a conserved ~30-aa CPI motif, a ~14-aa CARMIL-specific interaction (CSI) motif, and a proline-rich domain (PRD) that contains six canonical SH3-binding sites. CARMIL1 regulates CP through the CPI and CSI motifs (Zwolak et al., 2013). In this study, we examined the role of CARMIL1 and its LRR region and CPI-CSI motifs (CP-binding domain) in regulating the generation of IL-1 signaling in spreading fibroblasts.
RESULTS
CARMIL1 Is Required for IL-1-Induced ERK Activation and MMP Expression
As CARMIL localizes to the plasma membrane in an CP-independent manner (Liang et al., 2009; Yang et al., 2005), we examined whether CARMIL1 is enriched in FAs after IL-1 stimulation. Based on comparisons with BSA-coated bead preparations, which model non-specific adhesive interactions, we found that CARMIL1 was only present in collagen-coated, bead-associated protein preparations (Figure 1A). As a negative control, collagen-coated bead-associated preparations were immunoblotted for GAPDH, a cytosolic protein expressed at low levels in FA preparations. As a positive control, collagen-coated bead-associated preparations were immunoblotted for the FA-associated protein, paxillin.
Figure 1. CARMIL1 Associates with IL-1 Signaling Proteins to Mediate ERK Phosphorylation.

(A) Whole-cell lysates and BSA- and collagen-coated bead-associated proteins were prepared from human gingival fibroblasts (HGFs) that had been treated without or with IL-1 (20 ng/mL) for indicated time periods. Lysates were immunoblotted for CARMIL1, paxillin, and GAPDH. Densitometry data are expressed as means ± SE. Differences between indicated groups were assessed by ANOVA.
(B) CARMIL1 and IgG (top) immunoprecipitates (IPs) were prepared from whole-cell lysates of HGFs that had been treated without or with IL-1 (20 ng/mL) for 45 min and immunoblotted for IL-1R1, IRAK, CARMIL1, and IgG, respectively. Densitometry data of the ratios of IL-1R1/CARMIL1 and IRAK/CARMIL1 are expressed as means ± SE. IL-1R1 and IgG (bottom left) and IRAK and IgG (bottom right) IPs were prepared from whole-cell lysates of HGFs that had been treated without or with IL-1 (20 ng/mL) for 45 min and immunoblotted for CARMIL1, IL-1R1, IRAK, and IgG, respectively. Densitometry data of ratio of CARMIL1/IL-1R1 and CARMIL1/IRAK are expressed as means ± SE.
(C) HGFs were transfected with siRNA control or siRNA CARMIL1 for 48 h and then treated with either PBS or IL-1 (20 ng/mL) for 45 min. Lysates were immunoblotted for p(T202/Y204)-ERK and total ERK. Lysates of siRNA control and siRNA CARMIL1-transfected cells were immunoblotted for CARMIL1 to confirm CARMIL1 KD. β-actin was used for loading control. Ratios of p(T202/Y204)-ERK to ERK were quantified by densitometry, which were assessed by ANOVA and are expressed as means ± SE.
We conducted immunoprecipitation experiments to examine whether IL-1 treatment enhanced the association between CARMIL1 and the IL-1 signaling protein IL-1R1 or IRAK (Figure 1B). These data showed that IL-1 strongly increased the association between IL-1R1 or IRAK and CARMIL1, whereas immunoprecipitations conducted with control immunoglobulin G (IgG) showed faint signals with no specific changes of blot density between the different proteins (Figure 1B).
IL-1-induced signaling can promote the expression of proteins that degrade the ECM, such as MMPs; the expression of these proteins is dependent on MAPK signaling (Mengshol et al., 2000). Because ERK activation stimulated by IL-1 is dependent on FAs (Lo et al., 1998), we examined the role of CARMIL1 in IL-1-induced ERK activation and MMP1/MMP3/MMP9/MMP14 expression in human gingival fibroblasts (HGFs) transfected with small interfering RNA (siRNA) to CARMIL1. Compared with control siRNA, knockdown (KD) of CARMIL1 by siRNA substantially diminished IL-1-induced ERK phosphorylation (by ~68%; Figure 1C, *p < 0.01) and also reduced MMP3 mRNA expression (~50% at 24 h after IL-1 stimulation; Figure 2, *p < 0.01). In cells with siRNA KD of CARMIL1, there were no statistically significant reductions of MMP1, −9, and −14 that were attributable to CARMIL1 KD after treatment with IL-1 (p > 0.2). Arising from the analyses of mRNA for the MMPs, we also examined MMP3 protein in separate experiments. Cells were subjected to the same treatment regimen that was used for those cells in which mRNA was analyzed. Cell lysates were immunoblotted for MMP3, and a separate immunoblot was conducted for GAPDH in cell lysates. Consistent with the mRNA data, KD of CARMIL1 blocked the increased of IL-1-induced MMP3 protein expression at 6 and 24 h (Figure 2, top right).
Figure 2. CARMIL1 Regulates IL-1-Induced MMP Expression.

HGFs were transfected with siRNA control (scrambled sequence) or siRNA CARMIL1 for 48 h and treated without or with IL-1 (20 ng/ml) for 6 h and 24 h. Total RNA was extracted; and MMP1, MMP3, MMP9, and MMP14 mRNA levels were quantified by qRT-PCR analysis. Values are means ± SE from n = 3 independent experiments. In cells with siRNA KD of CARMIL1, there were no statistically significant reductions of MMP1, −9, and −14 that were attributable to CARMIL1 KD after treatment with IL-1 (p > 0.2). There was increased mRNA (p < 0.05) for MMP9 after IL-1 treatment in cells that had been pre-treated with siRNA for CARMIL1. Top right: cell lysates were immunoblotted for MMP3, and a separate immunoblot was conducted for GAPDH in cell lysates. These cells were subjected to the same treatment regimen that was used for the other cells in which mRNA was analyzed.
Arising from these experiments, a CARMIL1 knockout (KO) cell line was generated from immortalized HGFs-hTERT (CARMIL1 WT [wild-type]; Applied Biological Materials, Richmond, BC, Canada) using CRISPR-Cas9 genome editing technology (STAR Methods). Immunoblot analysis of the CARMIL1 KO cells showed deletion of CARMIL1 expression compared with that in CARMIL1 WT cells (Figure S1A). Similar to cells treated with siRNA, CARMIL1 KO cells created by CRISPR-Cas9 exhibited markedly diminished IL-1-induced ERK phosphorylation (~72%; Figure S1A, *p < 0.01) and reduced MMP3 expression (~39%; Figure S1B, *p < 0.01). Further, and as expected because of the actin-filament-modifying role of CARMIL, KD of CARMIL1 markedly affected the formation and size of FAs (Figure S2) and the ability of cells to form actin-filament-enriched extensions that are involved in degradation of the ECM (Figures S3 and S4). These data indicate that CARMIL1 is involved in the regulation of IL-1-induced MAPK signaling that lead to MMP expression in gingival fibroblasts and the development of the adhesions and extensions that are associated with collagen remodeling.
CARMIL1-LRR Domain Interacts with the IL-1 Signaling Proteins IL-1R1 and IRAK to Enable IL-1-Induced ERK Phosphorylation and MMP3 Expression
CARMIL contains several domains, including a PH domain, a LRR domain, a central HD, a CPI-CSI motif, and a PRD (Zwolak et al., 2013) (Figure 3A). As LRR domains in many different proteins mediate protein-protein interactions (Kobe and Kajava, 2001), we examined the associations of the LRR domain or the CP-binding domain of CARMIL1 with IL-1R1 and IRAK. CARMIL1 KO cells were transfected with glutathione S-transferase (GST)-tagged, full-length CARMIL1 (Carmil1), deleted LRR domain (designated as Carmil1 δLRR), or deleted CP-binding domain (designated as Carmil1 δCP). Immunoblotting showed that full-length and deleted-domain CARMIL1 proteins were, indeed, expressed by CARMIL1 KO cells (Figure 3A, bottom left). Immunoprecipitation experiments showed that both IL-R1 and IRAK associated with the full-length CARMIL1 and the deleted CP-binding domain mutant, while there was minimal association with the deleted LRR domain (Figure 3A, bottom middle and right). These data indicate that the LRR of CARMIL1 mediates association with the IL-1 signaling molecules, IL-1R1 and IRAK.
Figure 3. LRR Domain of CARMIL1 Is Required for IL-1-Induced ERK Activation and MMP3 Expression.

(A) Top: domain architecture of CARMIL: PH, pleckstrin homology; HD, central helical domain; LRR, leucine-rich repeat; CPI-CSI, CPI-CARMIL-specific interaction motif (CP-binding domain); and PRD, carboxy-terminal. CARMIL1 KO cells were transfected with GST-tagged full-length CARMIL1 (Carmil1), deleted LRR domain (Carmil1 δLRR), and deleted CP-binding domain (Carmil1 δCP). Whole-cell lysates were immunoblotted for GST antibody to confirm transfection (bottom left). GST immunoprecipitation was conducted with CARMIL1 KO cells transfected with GST-tagged full-length CARMIL1 (Carmil1), deleted LRR domain (Carmil1 δLRR), and deleted CP-binding domain (Carmil1 δCP), then immunoblotting was conducted with IL-1R1 and GST (bottom middle) and with IRAK and GST (bottom right).
(B) Whole-cell lysates from CARMIL1 KO and CARMIL1 KO cells transfected with GST-tagged CARMIL1 plasmids in (A), plated on fibronectin (FN) and treated without or with IL-1 (20 ng/mL) were immunoblotted for p(T202/Y204)-ERK and total ERK. Lysates of CARMIL1 WT and CARMIL1 KO cells were immunoblotted for CARMIL1 to confirm CARMIL1 KO. β-actin was used for loading control. Ratios of p(T202/Y204)-ERK to ERK were quantified by densitometry and are indicated as the percent mean ratios of densities ± SE of 4 replicates from 3 separate experiments.
(C) MMP3 mRNA levels were quantified in the above transfected cells that were treated without or with IL-1 (20 ng/mL) for 6 h and 24 h by qRT-PCR analysis, respectively. Values are means ± SE from n = 3 independent experiments.
We assessed the impact of the LRR domain and the CP-binding domain on IL-1-induced ERK phosphorylation. In CARMIL KO cells transfected with full-length CARMIL1 (Carmil1) or deleted CP-binding domain (Carmil1 δCP), IL-1-induced ERK phosphorylation was much higher than in CARMIL KO cells or cells transfected with the deleted LRR domain (Carmil1 δLRR) (Figure 3B). Similarly, we examined the role of LRR and CP-binding domains on MMP3 expression in response to IL-1. In CARMIL KO cells or CARMIL1 KO cells transfected with the deleted LRR domain, IL-1-induced MMP3 expression was much lower compared with that in cells transfected with either WT CARMIL1 (Carmil1) or the deleted CP-binding domain (Carmil1 δCP) (Figure 3C). In cells expressing the LRR domain deletion mutant, MMP3 expression in response to IL-1 was comparable to that in CARMIL1 KO cells. These results suggest that the LRR domain, but not the CP-binding domain, of CARMIL1 is important for IL-1-induced MMP3 expression.
Cell-Permeable Peptides Blocking CARMIL1-Dependent Interactions with IL-1RI and IRAK Inhibit IL-1 Signaling
The C terminus of CARMIL1 comprises a CP binding and uncapping motif that includes the CARMIL homology domain 3 (CAH3, residues 965–1,038) (Jung et al., 2001; Yang et al., 2005; Uruno et al., 2006; Fujiwara et al., 2010). These previous studies showed that peptides with sequences corresponding to the sequences of the CPI motifs can inhibit CP and uncap CP-bound actin filaments. We designed and synthesized competitive peptides (LRR and CP) that compete with the LRR domain and CPI-CSI motifs of CARMIL1; these peptides contained cell-penetrating TAT sequences and were labeled with fluorescein isothiocyanate (FITC) (see STAR Methods for peptide sequence). The rationale for the design of the peptides was based on choosing sequences within one of the repeated motifs in the LRR domain in particular that would likely be exposed in aqueous environments, with enhanced solubility with minimal numbers of hydrophobic residues. We also synthesized a control peptide that contained the same amino acids as the LRR domain peptide, but the amino acid sequence was scrambled.
WT HGFs were incubated with the peptides (10 μM for 8 h); flow cytometry and microscopy showed 100% transduction of these cells (Figure 4A). We examined whether these peptides affect the association of CARMIL1 with IL-1 signaling proteins by immunoprecipitation. After treating peptide-transduced cells with vehicle or IL-1, CARMIL1 was immunoprecipitated from whole-cell lysates. The CARMIL1 immunoprecipitates were immunoblotted for CARMIL1, IL-1R1, and IRAK (Figure 4B). Cells that were transduced with the LRR peptide exhibited reductions of association of CARMIL1 with IL-1R1 (55%) or IRAK (39%) compared with the control peptide. There was no significant change of the association of CARMIL1 with IL-1R1 or IRAK in cells transduced with the CP peptide. These data indicate that the LRR domain of CARMIL1 mediates the association of CARMIL1 with the IL-1 signaling proteins IL-1R1 and IRAK. Accordingly, we examined the impact of these peptides on ERK activation and MMP3 expression in response to IL-1. There was reduced IL-1-induced ERK phosphorylation (56%, Figure 4C) and MMP3 expression (45% at 6 h; 40% at 24 h; Figure 4D) in cells transduced with the LRR peptide compared with the control peptide after IL-1 stimulation. In contrast, cells transduced with the CP peptide did not show changes of ERK phosphorylation or MMP3 expression in response to IL-1. Collectively, these data indicate that the LRR domain of CARMIL1 impacts IL-1-induced ERK activation and MMP3 expression by providing an adaptor that physically focuses IL-1 signaling proteins such as IL-1R1 and IRAK.
Figure 4. Effect of Cell-Permeable CARMIL1 Peptides on IL-1-Induced ERK Activation and MMP3 Expression.

(A) HGFs were incubated with FITC-labeled, cell-penetrating TAT LRR peptide (1 μM/mL) and CP peptide (1 μM/mL) overnight and analyzed by flow cytometry and viewed by fluorescence microscopy.
(B) HGFs transduced with FITC-labeled, cell-penetrating TAT control peptide, LRR peptide, and CP peptide were treated without or with IL-1 (20 ng/mL) for 45 min. CARMIL1 IPs were prepared from whole-cell lysates of the cells above and immunoblotted for IL-1R1 and CARMIL1 (left), and for IRAK and CARMIL1 (right). The histograms show the percent mean ratios of densities ± SE of IL-1R1 to CARMIL1 or of IRAK to CARMIL1, respectively. Data from three independent experiments were averaged, and the mean ratios ± SE (n = 3 replicates) are indicated.
(C) Whole-cell lysates from transduced cells in (B) treated without or with IL-1 (20 ng/mL) for 45 min were immunoblotted for p(T202/Y204)-ERK and total ERK. Ratios of p(T202/Y204)-ERK to ERK were quantified by densitometry and indicated as the percent mean ratios of densities ± SE of 3 replicates from 3 separate experiments.
(D) MMP3 mRNA levels were quantified in the aforementioned transduced cells that were treated without or with IL-1 (20 ng/mL) for 6 h and 24 h by qRT-PCR analysis, respectively. Values are means ± SE from n = 3 independent experiments.
Cell-Permeable Peptides Blocking CARMIL1-Dependent Interactions with IL-1RI and IRAK Inhibit IL-1-Induced Collagen Degradation
We assessed the contribution of the LRR domain or the CP-binding domain of CARMIL1 in IL-1-induced collagen degradation. HGFs were transduced with control peptide, LRR peptide, or the CP peptide. Cells were plated on fibrillar collagen gels for 24 h and fixed, but not permeabilized, to ensure that the collagen degradation assessed by immunostaining (antibody recognition of a collagenase-generated, 3/4-length degradation neo-epitope in collagen fibrils) was only extracellular and not intracellular (Figure 5). Images of degraded collagen were projected on the z axis, and each cell-associated 3/4 collagen particle was counted in the region of interest containing the cell only (i.e., pericellular collagenolysis). LRR-peptide-transduced cells showed 43% fewer numbers of labeled particles exhibiting pericellular collagen degradation compared with the control-peptide-transduced cells (p < 0.05, Figure 5B). The CP-peptide-transduced cells showed no difference in collagen degradation compared with the control-peptide-transduced cells (p > 0.2; Figure 5B). Collectively, these data indicate that the LRR of CARMIL1 regulates collagen degradation in response to IL-1.
Figure 5. LRR Domain of CARMIL1 Is Associated with Inhibition of Collagen Degradation.

(A) Staining for 3/4 collagen fragments (red), actin filaments (phalloidin; blue), FITC-labeled peptide (green), and intact collagen fibrils (gray) by confocal reflectance (CR) in HGFs and HGF cells transduced with control peptide, LRR peptide, and CP-peptide plated on 3D, type I collagen gels and treated without or with IL-1 (20 ng/mL).
(B) Quantification of collagen degradation stained with a 3/4 collagen neoepitope antibody in the different treatments and conditions mentioned above. 3/4 collagen intensity levels were normalized to the overall average of all intensity values derived with ImageJ using 3/4 collagen signals. Values are means ± SE from n = 3 independent experiments.
DISCUSSION
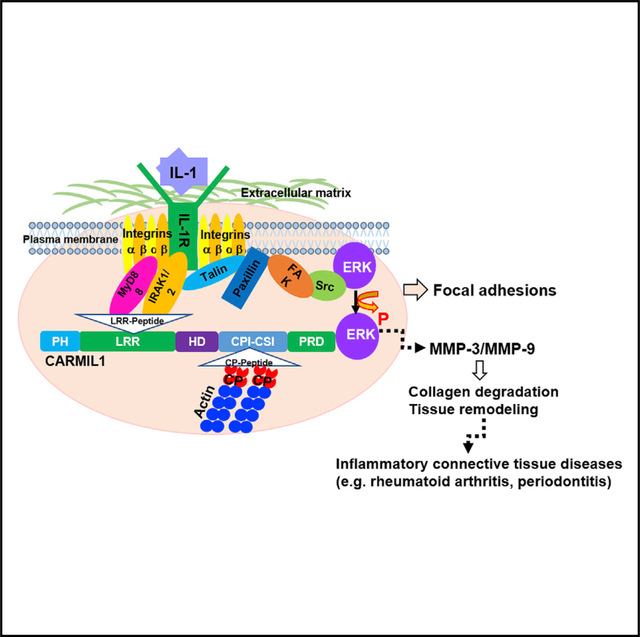
IL-1 signaling mediates ECM degradation in several different inflammatory diseases, including periodontitis (Boch et al., 2001). Gingival fibroblasts express very high levels of IL-1RIs (Qwarnstrom et al., 1988) that are spatially restricted to matrix adhesions (MacGillivray et al., 2000; Qwarnström et al., 1991, 1988). We and others have shown that, in anchorage-dependent cells, IL-1R1 must be sequestered in FAs to enable IL-1 signaling (Arora et al., 1995; Lo et al., 1998; Qwarnström et al., 1991). FAs, which are enriched with integrins (Geiger et al., 2009), also contain the IL-1 receptor-associated protein and IRAK (MacGillivray et al., 2000), which are essential for IL-1 signaling. The novelty of the present report is that CARMIL1 may act as an adaptor protein in which its LRR region interacts with IL-1R1 and IRAK, thereby concentrating IL-1 signaling complexes at the edges of spreading cells. Notably, CARMIL is recruited to the plasma membrane and only at cell edges undergoing active protrusion (Fujiwara et al., 2014). Thus, in addition to its well-described role in controlling actin assembly, CARMIL1 may be important in the regulation of FA-dependent pathophysiological processes such as IL-1-induced inflammation and matrix degradation, which occurs in cells that are spreading and exploring the extracellular matrix (Figure 6).
Figure 6. Proposed Model for Function of CARMIL1 in IL-1 Signaling.

In spreading cells, FAs are formed under the extracellular matrix. In the FA complexes, the leucine-rich repeat region of CARMIL1 regulates the recruitment of effector molecules IL-1R1 and IRAK to facilitate the regulatory processes involved in IL-1-mediated ERK to MMP pathway.
The approximation of IL-1 receptors with IL-1 receptor-associated proteins in FAs (MacGillivray et al., 2000; Qwarnstrom et al., 1988) restricts the generation of downstream signals in anchorage-dependent cells that lead to increased expression of MMP3 and MMP9 and degradation of the ECM (McCulloch et al., 2006; Wang et al., 2009). The central findings in this report are that CARMIL1 is enriched in FAs, is recruited to the IL-1 signaling complex, and is required for IL-1 signal transduction. We found that, in HGFs, the LRR of CARMIL1 associates with the IL-1 signaling molecules IL-1R1 and IRAK and regulates IL-1-mediated ERK activation, MMP3 expression, and collagen degradation. These findings highlight CARMIL1 as a critical protein that is required for the assembly and signal transduction function of IL-1RI complexes in anchorage-dependent cells.
We found that, in the CRISPR-generated cell line in which CARMIL1 has been deleted, there was marked IL-1-induced ERK activation in spite of CARMIL deletion using CRISPR. We note, however, that this level of ERK activation in response to IL-1 is ~40% less than control cells (Figure 3B) and that the cells in which CARMIL1 with the deletion of the LRR domain exhibits IL-1-induced ERK activation that is very similar to the CARMIL1-depleted cells. These findings indicate that while IL-1 can stimulate ERK phosphorylation to some extent that is independent of CARMIL, there is also a large component of IL-1-induced ERK activation that requires CARMIL1 and that, in particular, this requirement is centered around the leucine-rich domain. The rationale for this argument is further bolstered by the finding that when CARMIL1 is expressed in cells without the actin CP-binding domain, then ERK activation is similar to that in control cells. Further, we note that, in contrast to the CRISPR-generated CARMIL1 KO cells, short-term siRNA reduction of CARMIL1 in HGFs induced a much larger reduction of IL-1-induced ERK activation. This difference could reflect the adaptation of cells in culture and the development of compensatory mechanisms in which relatively less IL-1 signaling is restricted to the FAs of these cells.
CARMIL1 is a multifunctional actin regulator consisting of an N-terminal domain that includes LRRs that enable extensive interactions with other regulatory proteins (Stark et al., 2017) and a 51-residue region near the C terminus that comprises a CP binding and uncapping motif that includes the CARMIL homology domain 3 (CAH3, residues 965–1,038) (Fujiwara et al., 2010; Jung et al., 2001; Yang et al., 2005; Uruno et al., 2006). Our data showing that the leucine-rich domains of CARMIL1 mediate binding to IL-1RI and IRAK indicate that CARMIL1 may act as an adaptor protein in which its LRR region interacts with IL-1 signaling proteins to facilitate IL-1-mediated matrix degradation.
Degradation of the extracellular matrix is fundamental to IL-1-driven inflammatory diseases. For complex signaling systems such as IL-1, anchorage of signaling proteins to specific organelles such as FAs is required to promote effective signal transduction (Aplin and Juliano, 1999). Based on the structure of CARMIL1 and its LRR and CPI-CSI domain (Edwards et al., 2014; Stark et al., 2017; Zwolak et al., 2013), we synthesized TAT-fusion peptides labeled with FITC and assessed their cellular uptake by fluorimetry. Our results showed that a peptide competing with CP binding to CARMIL1 did not inhibit IL-1-induced collagen degradation, whereas a peptide that competed with IL-1R1 and IRAK binding to the LRR of CARMIL1 strongly inhibited collagen degradation. We conclude that the LRR of CARMIL1 could regulate IL-1-mediated collagen degradation in HGFs and could be a target for anti-inflammatory drug development.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, C. A. McCulloch: christopher.mcculloch@utoronto.ca.
Materials Availability
Investigators who are interested in obtaining cells or peptides described herein must be willing to complete a Materials Transfer Agreement with the University of Toronto.
Data and Code Availability
There are no unpublished custom code, software, or algorithms in this manuscript.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human gingival fibroblasts (HGFs) were isolated from explants obtained during periodontal surgeries from adult (ages 35–50 years) male and female patients. Biopsies of gingiva were obtained with informed patient consent under a University of Toronto human ethics experimental protocol. Cells were cultured in α-minimal essential medium and mass cultures were grown up, mixed from different donor biopsies and used at passages 5–10. Media were supplemented with 10% fetal bovine serum (FBS) and 146 units/ml penicillin G, 50 μg/ml gentamycin sulfate, and 0.25 μg/ml amphotericin B (Fungizone). HGFs were used between passages 5 and 10.
Immortalized human gingival fibroblasts (hTERT cells) were purchased from Applied Biological Materials Inc. (Richmond, BC) and maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS and antibiotics. Cells were cultured at 37 °C in a humidified incubator supplied with 5% CO2 and passaged with 0.25% trypsin containing 1 mM EDTA.
METHOD DETAILS
Reagents
Mouse monoclonal antibody to Carmil, goat anti-mouse IgG antibodies and rabbit antibodies to IL-1R1 and IRAK-1 were obtained from Abcam (Cambridge, United Kingdom). Rabbit monoclonal antibodies to phospho-p44/42 MAPK and polyclonal antibodies to p44/42 MAPK were obtained from Cell Signaling Technology (Danvers, MA, USA). On-targetplus Smartpool small interfering RNA (siRNA) targeted at CARMIL1 and Dharmafect 1 transfection reagent were purchased from GE Dharmacon (Lafayette, CO, USA). Rabbit antibodies to a fragment of collagenase-cleaved type-1 collagen was purchased from Immunoglobe (Himmelstadt, Germany). Mouse monoclonal antibodies to β-actin and bovine plasma fibronectin were obtained from Sigma-Aldrich (St. Louis, MO). Type-1 bovine collagen (5010) was from Advanced BioMatrix (San Diego, CA). Alexa Fluor® 488 goat anti-mouse, Alexa Fluor® 568 goat anti-rabbit IgG antibodies, and rhodamine phalloidin were from Life Technologies (Burlington, ON). IL-1β was purchased from R&D Systems (Minneapolis, MN). Cell-penetrating TAT- Control peptide (GRKKRRQRRRPQNGAGYEMGVCVENRKVLCRSKPCNRA-Lys-FITC), LRR peptide (GRKKRRQRRRPQTLVHLDLSGNVLRGDDLSHMYNFLAQPNK-Lys- FITC) and CP peptide (GRKKRRQRRRPQGVSFDLPAQADTLHSANKSRVKMRGKRRPQTRA-Lys- FITC) were synthesized by Biomatik (Cambridge, ON)
CARMIL1 constructs
For GST-tagged deleted domain CARMIL1 constructs, PCR products containing WT CARMIL1, lack of LRR domain or CP-binding domain fragments were ligated into pcDNA3.1 and transformed into DH5α-competent Escherichia coli cells (Invitrogen; Burlington, ON). The construct was sequenced (ACGT Corp., Toronto, ON) and transformed into BL21 (DE3) Escherichia coli. (Agilent, Mississauga, ON) for GST-tagged-protein expression and purification. Transfections of the CARMIL1 KO cell line with constructs were performed with FuGENE® HD (Promega, Madison, WI).
siRNA knockdown
HGFs were seeded at 30% confluence 24 h prior to transfection and were incubated with 10 nM scrambled siRNA (negative control; QIAGEN, Hilden, Germany) or On-Targetplus (LRRC16A isoform) CARMIL! siRNA (Dharmacon) and DharmaFECT 1 transfection reagent 1 for 48 h following the manufacturer’s instructions. Cells were trypsinized and replated on FN (10 mg/ml) for 3 h in medium containing 1% serum before stimulating with 20 ng/ml IL-1 or vehicle control in serum-free medium for various time points. Whole cell lysates were collected, and protein concentrations were determined by BCA assay. Equal amounts of total proteins from each treatment condition were separated on 10% acrylamide gels and immunoblotted to estimate the effectiveness of CARMIL1 knockdown (KD) and IL-1-induced ERK activation.
CARMIL1 deletion by CRISPR/Cas9
An CARMIL1 KO cell line was generated from Immortalized HGFs-hTERT by Applied Biological Materials Inc. (Richmond, BC) using CRISPR/Cas9 genome editing technology. Sequencing of the target regions was done via Sanger sequencing. sgRNA were designed CARMIL1 gene (Homo sapiens, NM_001173977.1) RNAs (sgRNAs) 5ʹ-GGGAGGTGTTTGTAATTTGG-3ʹ and 5ʹ-TTCTTGGGATGCCTTTACAC-3ʹ targeting human CARMIL1 exon were chosen. The selected sgRNAs were cloned into a sgRNA/Cas9 expression vector and introduced to Immortalized HGFs-hTERT cells.
Immunoblotting
Cells were lysed in RIPA buffer (Sigma) containing phenylmethylsulfonyl fluoride, NaVO4 and a protease inhibitor cocktail. The lysates were centrifuged at 10,000 rpm for 5 min and the supernatant separated for analysis. BCA analysis was conducted to ensure that equal amounts of protein were separated on 8 or 10% acrylamide gels, immunoblotted with appropriate primary and secondary antibodies as indicated, and imaged with an Odyssey Fc infrared detection system (LI-COR Biotechnology; Lincoln, NE).
Immunoprecipitation
Cells were lysed in 1% Tris-NaCl-Triton immunoprecipitation buffer (20 mM Tris at pH 7.5, 1% Triton X-100, 0.1% SDS, 150 mM NaCl) containing 1 mM PMSF, 1 mM NaVO3, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Equal amounts of protein from cleared extracts were immunoprecipitated with the Dynabeads immunoprecipitation protocol (Invitrogen, Carlsbad).
Immunostaining and confocal microscopy
Cells were seeded on to glass, or fibronectin-coated glass, or fibrillar collagen gels and incubated for various times, fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton-X-100 for 10 min, then blocked for 20 min with 1% bovine serum albumin (BSA) in PBS, incubated with primary antibody overnight at 4°C, incubated with labeled secondary antibody for 1 hr. at room temperature, and counterstained with fluorescence-conjugated phalloidin and 4ʹ,6-diamidino-2-phenylindole. Immunostained cells were visualized by confocal microscopy ( × 40 objective, TCS SP8; Leica, Wetzlar, Germany).
Collagen degradation assays
Collagen gels were prepared as described (Coelho et al., 2017) using bovine type I collagen (1 mg/mL; Advanced BioMatrix) on glass coverslips. Cells were incubated overnight on gels, washed, fixed with 4% PFA, and stained with rhodamine phalloidin and a neoepitope antibody (Immunoglobe) that recognizes degraded type I collagen (3/4 fragment) to visualize pericellular collagen degradation. To ensure that only pericellular collagen was immunostained, cells were not permeabilized. Only the optical sections that showed cells directly in contact with the collagen gel were used for analysis of collagen degradation. For imaging, at least 30 cells for each experimental condition were analyzed.
Real-time quantitative PCR
After IL-1 stimulation in low-serum (1% fetal bovine serum) minimal essential medium, total RNA was extracted from Cells using a RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. Total RNA samples (1 μg as determined by a Thermo Fisher Scientific NanoDrop ND-1000 spectrophotometer) were reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. Real-time quantitative PCR was performed on a CFX96 Touch real-time PCR detection system according to the product protocol using SsoFast EvaGreen Supermix (Bio-Rad) and validated Human MMP-1, MMP-3, MMP-9, MMP14 and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) primers. PCR reactions were performed twice, and all assays were performed in triplicate. The relative expression of the MMP1, MMP3, MMP9 and MMP14 genes to the housekeeping gene GAPDH was calculated using the 2−ΔΔCt method.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were conducted in quadruplicate and were repeated ≥ 3 times (n) for each indicated experiment. For all continuous variables, mean and standard errors were computed. A Student’s t test was used for 2-sample comparisons, and statistical significance was set at a type I error rate of p < 0.05. For multiple comparisons, ANOVA followed by the post hoc Tukey’s test were used. Data were analyzed with Prism and Excel software.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-CARMILI | Abcam | Cat. # 52681 |
| Rabbit polyclonal anti-IL-1R1 | Abcam | Cat. #106278 |
| Rabbit monoclonal anti-IRAK1 | Abcam | Cat. #180747. RRID EPR18630 |
| Rabbit monoclonal anti- phospho-p44/42 MAPK | Cell Signaling | Cat. # 4376S |
| Rabbit polyclonal anti-p44/42 MAPK | Cell Signaling | Cat. #9102 |
| Rabbit polyclonal anti-3/4 collagen fragment | Immunoglobe | Cat. #0217–050 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat. #A5441 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Recombinant Human IL-1β | R&D | Cat. #201-LB |
| Bovine plasma fibronectin | Sigma-Aldrich | Cat. # F4759 |
| Type I bovine collagen | Advanced Biomatrix | Cat. #5010 |
| Cell-penetrating TAT- Control peptide (GRKKRRQRRRPQNGAGYEMGVCVENRKVLCRSKPCNRA-Lys-FITC | Biomatik and this paper | Control peptide |
| LRR peptide (GRKKRRQRRRPQTL. VHLDLSGNVLRGDDLSHMYNFLAQPNK-Lys- FITC | Biomatik and this paper | LRR peptide |
| CP peptide (GRKKRRQRRRPQGVSFDLPAQADTLHSANKSRVKMRGKRRPQTRA-Lys- FITC | Biomatik and this paper | CP peptide |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| CARMIL1 KO human gingival fibroblasts | Applied Biological Materials and this paper | N/A |
| Wild type TERT-immortalized human gingival fibroblasts. (hTERT Cells) | Applied Biological Materials | T0026 |
| Human gingival fibroblasts, passages 5–10 | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| siRNA targeting sequence CARMIL1 55604 | Dharmacon | On-targetplus Smartpool |
| Primers for MMP-1, MMP-3, MMP-9, MMP14 and GAPDH | Acgt | N/A |
| Recombinant DNA | ||
|
| ||
| GST-tagged deleted domain CARMIL1 constructs | Custom DNA constructs and this paper | LRR and CP deletions |
Highlights.
CARMIL1 associates with IL-1 signaling molecules
CARMIL1 knockout reduces IL-1-induced ERK activation and MMP3 expression
The leucine-rich repeat region of CARMIL1 associates with IL-1 signaling proteins
Cell-permeable, CARMIL1-binding peptides block IL-1-induced collagen degradation
ACKNOWLEDGMENTS
C.A.M. is supported by a Government of Canada, Canada Research Chair award in Matrix Dynamics (Tier 1). The research was supported by a Canadian Institutes of Health research grant (MOP-503020) to C.A.M. G.P.D. and C.A.M. are also supported by an R01 grant (R01HL132950) from the NIH.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107781.
REFERENCES
- Aplin AE, and Juliano RL (1999). Integrin and cytoskeletal regulation of growth factor signaling to the MAP kinase pathway. J. Cell Sci. 112, 695–706. [DOI] [PubMed] [Google Scholar]
- Arora PD, Ma J, Min W, Cruz T, and McCulloch CA (1995). Interleukin-1-induced calcium flux in human fibroblasts is mediated through focal adhesions. J. Biol. Chem. 270, 6042–6049. [DOI] [PubMed] [Google Scholar]
- Boch JA, Wara-aswapati N, and Auron PE (2001). Interleukin 1 signal transduction–current concepts and relevance to periodontitis. J. Dent. Res. 80, 400–407. [DOI] [PubMed] [Google Scholar]
- Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, and Tschopp J (1998). MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 273, 12203–12209. [DOI] [PubMed] [Google Scholar]
- Burridge K, and Chrzanowska-Wodnicka M (1996). Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12, 463–518. [DOI] [PubMed] [Google Scholar]
- Coelho NM, Arora PD, van Putten S, Boo S, Petrovic P, Lin AX, Hinz B, and McCulloch CA (2017). Discoidin Domain Receptor 1 Mediates Myosin-Dependent Collagen Contraction. Cell Rep. 18, 1774–1790. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2019). The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 15, 612–632. [DOI] [PubMed] [Google Scholar]
- Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, and Cooper JA (2014). Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 15, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, and Hammer JA 3rd. (2010). Direct observation of the uncapping of capping protein-capped actin filaments by CARMIL homology domain 3. J. Biol. Chem. 285, 2707–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, Piszczek G, and Hammer JA (2014). Capping protein regulatory cycle driven by CARMIL and V-1 may promote actin network assembly at protruding edges. Proc. Natl. Acad. Sci. USA 111, E1970–E1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, and Bershadsky AD (2009). Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 10, 21–33. [DOI] [PubMed] [Google Scholar]
- Hönig J, Rordorf-Adam C, Siegmund C, Wiedemann W, and Erard F (1989). Increased interleukin-1 beta (IL-1 beta) concentration in gingival tissue from periodontitis patients. J. Periodontal Res. 24, 362–367. [DOI] [PubMed] [Google Scholar]
- Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Frémillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D, et al. (2015). Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 17, 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Remmert K, Wu X, Volosky JM, and Hammer JA 3rd. (2001). The Dictyostelium CARMIL protein links capping protein and the Arp2/3 complex to type I myosins through their SH3 domains. J. Cell Biol. 153, 1479–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B, and Kajava AV (2001). The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11, 725–732. [DOI] [PubMed] [Google Scholar]
- Liang Y, Niederstrasser H, Edwards M, Jackson CE, and Cooper JA (2009). Distinct roles for CARMIL isoforms in cell migration. Mol. Biol. Cell 20, 5290–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YY, Luo L, McCulloch CA, and Cruz TF (1998). Requirements of focal adhesions and calcium fluxes for interleukin-1-induced ERK kinase activation and c-fos expression in fibroblasts. J. Biol. Chem. 273, 7059–7065. [DOI] [PubMed] [Google Scholar]
- Luo L, Cruz T, and McCulloch C (1997). Interleukin 1-induced calcium signalling in chondrocytes requires focal adhesions. Biochem. J. 324, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillivray MK, Cruz TF, and McCulloch CA (2000). The recruitment of the interleukin-1 (IL-1) receptor-associated kinase (IRAK) into focal adhesion complexes is required for IL-1beta -induced ERK activation. J. Biol. Chem. 275, 23509–23515. [DOI] [PubMed] [Google Scholar]
- Manninen A, and Varjosalo M (2017). A proteomics view on integrin-mediated adhesions. Proteomics 17 (3–4), 1600022. 10.1002//pmic201600022. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Kurokawa H, Oosawa M, Shimaoka S, Yamamoto H, Ito M, and Maruyama K (1990). Beta-actinin is equivalent to Cap Z protein. J. Biol. Chem. 265, 8712–8715. [PubMed] [Google Scholar]
- McCulloch CA, Downey GP, and El-Gabalawy H (2006). Signalling platforms that modulate the inflammatory response: new targets for drug development. Nat. Rev. Drug Discov. 5, 864–876. [DOI] [PubMed] [Google Scholar]
- McDermott EP, and O’Neill LA (2002). Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1. J. Biol. Chem. 277, 7808–7815. [DOI] [PubMed] [Google Scholar]
- Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, and Brinckerhoff CE (2000). Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 43, 801–811. [DOI] [PubMed] [Google Scholar]
- Morimatsu M, Mekhdjian AH, Chang AC, Tan SJ, and Dunn AR (2015). Visualizing the interior architecture of focal adhesions with high-resolution traction maps. Nano Lett. 15, 2220–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy-Ullrich JE (2001). The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J. Clin. Invest. 107, 785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Ni J, Feng P, and Dixit VM (1997). IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615. [DOI] [PubMed] [Google Scholar]
- Qwarnstrom EE, Page RC, Gillis S, and Dower SK (1988). Binding, internalization, and intracellular localization of interleukin-1 beta in human diploid fibroblasts. J. Biol. Chem. 263, 8261–8269. [PubMed] [Google Scholar]
- Qwarnström EE, MacFarlane SA, Page RC, and Dower SK (1991). Interleukin 1 beta induces rapid phosphorylation and redistribution of talin: a possible mechanism for modulation of fibroblast focal adhesion. Proc. Natl. Acad. Sci. USA 88, 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reunanen N, Westermarck J, Häkkinen L, Holmström TH, Elo I, Eriksson JE, and Kähäri VM (1998). Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathways. J. Biol. Chem. 273, 5137–5145. [DOI] [PubMed] [Google Scholar]
- Stark BC, Lanier MH, and Cooper JA (2017). CARMIL family proteins as multidomain regulators of actin-based motility. Mol. Biol. Cell 28, 1713–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruno T, Remmert K, and Hammer JA 3rd. (2006). CARMIL is a potent capping protein antagonist: identification of a conserved CARMIL domain that inhibits the activity of capping protein and uncaps capped actin filaments. J. Biol. Chem. 281, 10635–10650. [DOI] [PubMed] [Google Scholar]
- Wang Q, Downey GP, Choi C, Kapus A, and McCulloch CA (2003). IL-1 induced release of Ca2+ from internal stores is dependent on cell-matrix interactions and regulates ERK activation. FASEB J. 17, 1898–1900. [DOI] [PubMed] [Google Scholar]
- Wang Q, Rajshankar D, Branch DR, Siminovitch KA, Herrera Abreu MT, Downey GP, and McCulloch CA (2009). Protein-tyrosine phosphatase-alpha and Src functionally link focal adhesions to the endoplasmic reticulum to mediate interleukin-1-induced Ca2+ signaling. J. Biol. Chem. 284, 20763–20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rajshankar D, Laschinger C, Talior-Volodarsky I, Wang Y, Downey GP, and McCulloch CA (2010). Importance of protein-tyrosine phosphatase-alpha catalytic domains for interactions with SHP-2 and inter-leukin-1-induced matrix metalloproteinase-3 expression. J. Biol. Chem. 285, 22308–22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Delcorde J, Tang T, Downey GP, and McCulloch CA (2018). Regulation of IL-1 signaling through control of focal adhesion assembly. FASEB J. 32, 3119–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wear MA, Yamashita A, Kim K, Maéda Y, and Cooper JA (2003). How capping protein binds the barbed end of the actin filament. Curr. Biol. 13, 1531–1537. [DOI] [PubMed] [Google Scholar]
- Wehrle-Haller B, and Imhof B (2002). The inner lives of focal adhesions. Trends Cell Biol. 12, 382–389. [DOI] [PubMed] [Google Scholar]
- Wesche H, Henzel WJ, Shillinglaw W, Li S, and Cao Z (1997a). MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7, 837–847. [DOI] [PubMed] [Google Scholar]
- Wesche H, Korherr C, Kracht M, Falk W, Resch K, and Martin MU (1997b). The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases). J. Biol. Chem. 272, 7727–7731. [DOI] [PubMed] [Google Scholar]
- Yang C, Pring M, Wear MA, Huang M, Cooper JA, Svitkina TM, and Zigmond SH (2005). Mammalian CARMIL inhibits actin filament capping by capping protein. Dev. Cell 9, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Xiong W, Rodgers G, and Qwarnstrom EE (1998). Regulation of interleukin 1 signalling through integrin binding and actin reorganization: disparate effects on NF-kappaB and stress kinase pathways. Biochem. J. 330, 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Uruno T, Piszczek G, Hammer JA 3rd, and Tjandra N (2010). Molecular basis for barbed end uncapping by CARMIL homology domain 3 of mouse CARMIL-1. J. Biol. Chem. 285, 29014–29026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Yang C, Feeser EA, Ostap EM, Svitkina T, and Dominguez R (2013). CARMIL leading edge localization depends on a non-canonical PH domain and dimerization. Nat. Commun. 4, 2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are no unpublished custom code, software, or algorithms in this manuscript.