

Abstract
The Ces-2/E2A-HLF binding element (CBE) is recognized by Caenorhabditis elegans death specification gene product Ces-2 and human acute lymphocytic leukemia oncoprotein E2A-HLF. In an attempt to identify a cellular CBE-binding protein(s) that may be involved in apoptosis regulation in mammals, multiple nuclear binding complexes of CBE were identified in various mammalian cell lines and tissues by electrophoretic mobility shift assay. Cyclic AMP (cAMP)-responsive element (CRE)-binding protein (CREB) was present in one major CBE complex of Ba/F3 and TF-1 cells, and both in vitro-translated and Escherichia coli-synthesized CREB bound to CBE. Activation of CREB by cAMP-elevating chemicals or the catalytic subunit of protein kinase A (PKAc) resulted in induction of the CBE-driven reporter gene. Stimulation of Ba/F3 cells with interleukin-3 (IL-3) promptly induced phosphorylation of CREB at serine133 partially via a PKA-dependent pathway. Consistently, Ba/F3 cell survival in the absence of IL-3 was prolonged by activation of PKA. Conversely, treatment of cells with a PKA inhibitor or expression of the dominant negative forms of the regulatory subunit type I of PKA and CREB overrode the survival activity of IL-3. Last, the bcl-2 gene was demonstrated to be one candidate cellular target of the CREB-containing CBE complex, as mutations in the CRE and CBE sites significantly reduced the IL-3 inducibility of the bcl-2 promoter. Together, our results suggest that CREB is one cellular counterpart of Ces-2/E2A-HLF and is part of IL-3 dependent apoptosis regulation in hematopoietic cells.
During the development of the small nematode Caenorhabditis elegans, 131 of the 1,090 cells in the worm embryo undergo programmed cell death (46, 47). Genetic studies have identified mutations in 14 genes that specifically affect this process. Recent findings suggest that these genes define an evolutionarily conserved genetic pathway for programmed cell death from C. elegans to mammals. These mutations divide the process of programmed cell death into four distinct steps: determination of whether individual cells will undergo programmed cell death or adopt another fate (8, 9, 48); execution of the cell (8, 16); engulfment of the dying cell by a neighboring cell (10, 15); and degradation of the dead, engulfed cell (15, 45). Mutation in genes involved in the last three steps affects all dying cells, whereas mutation in genes in the cell death specification step affects only a small number of specific cell types.
Two genes that affect the death of only a small number of cells have been identified. The sister cells of the neurosecretory motor neurons (NMN) in the pharynx undergo programmed cell death depending on the expression of two cell death specification genes, ces-1 and ces-2 (8, 9, 48). Dominant gain-of-function mutations in the ces-1 gene and recessive loss-of-function mutations in the ces-2 gene allow these two neuron cells to survive ces-1 loss-of-function mutation adopts the wild-type phenotype; however, it suppresses the phenotype of ces-2 loss-of-function mutation in NSM sister neurons. Therefore, the ces-2 gene was suggested to encode a pro-apoptotic protein that in turn represses the function of the antiapoptotic ces-1 gene. Recently, the ces-2 gene was molecularly cloned, and the predicted protein possessed the characteristic functional domains of basic region-leucine zipper (bZIP) proteins (34). Not only did the amino acid sequence of Ces-2 share significant homology with E4BP4 and PAR family proteins, including HLF and TEF, in the basic DNA-binding domains, but also the DNA sequence recognized by the Ces-2 protein was identical to that of the PAR family and E4BP4.
On the other hand, B-cell-specific expression of a chimeric protein that fused HLF to the transcription factor E2A resulted in formation of human acute B-lineage leukemia (19, 21). This fusion protein, E2A-HLF, retains the DNA binding specificity of its parental HLF protein but has greater trans-activation and transformation potential (22, 58). Recently, anti-apoptotic activity was suggested to underlie the mechanism of transformation of the E2A-HLF gene (23). The close homology of the basic DNA-binding domain of HLF to that of the Ces-2 protein of C. elegans further suggests the involvement of E2A-HLF in the conserved cell death pathway. Since overexpression of E2A-HLF in a factor-dependent cell line resulted in prolonged survival, it may be that the targets of the E2A-HLF protein are involved in antiapoptotic functions induced by cytokines. These target genes may be normally under the control of a set of cellular regulators that recognize similar DNA sequences on promoters as E2A-HLF.
CREB is a 43-kDa bZIP transcription factor composed of a C-terminal basic DNA-binding domain, an adjacent leucine zipper dimerization domain, and a kinase-inducible transcriptional activation domain (36, 55). CREB binds to cyclic AMP (cAMP)-responsive element (CRE) (TGACGTCA) both as a homodimer and as a heterodimer in association with other members of the CREB/ATF family. Phosphorylation of Ser133 within the kinase-inducible transcriptional activation domain of CREB is required to induce the transcriptional activity of the protein. Phosphorylation of Ser133 activates CREB, at least in part, by facilitating its binding to the 256-kDa CREB-binding protein (5, 13). The CREB/CREB-binding protein complex can, in turn, interact with and activate the basal transcriptional machinery.
The initial aim of this study was to identify cellular proteins that bind to the Ces-2/E2A-HLF binding element (CBE) and are involved in apoptosis regulation in hematopoietic cells. One protein that we identified turned out to be a known bZIP protein, CREB. In this report, we demonstrated that CREB is one major binding component of the CBE-binding complexes in several different cell types. With interleukin-3 (IL-3) dependent Ba/F3 cells as a model system, we demonstrated that activation of the PKA/CREB pathway plays a role in the survival activity of IL-3 and that part of this PKA/CREB pathway is likely mediated through activation of some CBE-controlled antiapoptotic genes.
MATERIALS AND METHODS
Chemicals, cell lines and culture conditions.
Forskolin, IBMX (3-isobutyl-1-methylxanthine), and Myr-PKI (myristoylated protein kinase A [PKA] inhibitor 14–22 amide; catalog no. 476485) were purchased from Calbiochem-Novabiochem Co. (San Diego, Calif.). IPTG (isopropyl-β-d-thiogalactopyranoside), dimethyl sulfoxide, tetracycline and CdCl2 were purchased from Sigma (St. Louis, Mo.). IL-3 and granulocyte-macrophage colony-stimulating factor (GM-CSF) were purchased from R&D Systems (Minneapolis, Minn.). Ba/F3 and TF-1 are cytokine-dependent cell lines and were cultured in media containing murine IL-3 and human GM-CSF, respectively, as previously described (31, 57). 293T was cultured in the standard Dulbecco modified Eagle medium containing 10% fetal calf serum. PC12 cells were cultured in Dulbecco modified Eagle medium containing 5% fetal calf serum plus 10% horse serum and incubated in 10% CO2.
Screening of expression cDNA library with oligonucleotide probes.
Expression cDNA libraries of human leukocytes, mouse embryonic stem cells, and mouse 15-day embryos were purchased from Clontech (Palo Alto, Calif.). Screening of expression cDNA libraries with oligonucleotide probes was performed as described by Vinson et al. (49), with slight modifications. The cDNA libraries were plated and incubated at 42°C for 4 h. The first nitrocellulose filters (Millipore, Bedford, Mass.) which had been presoaked in 10 mM IPTG were overlaid on the plates and incubated for 4 h at 37°C, and the duplicate filters were incubated for an additional 4 h at 37°C. After removal from culture plates, the nitrocellulose filters were allowed to air dry for 15 min at room temperature and then subjected to the refolding and binding procedures as described in the vendor's instructions. Labeling of catenated CBE oligonucleotides with [32P]dCTP was performed as previously described (49). These positive plaques were further purified by rescreening three to four times, and the insert was amplified by PCR and then sequenced.
Nuclear extract preparation and EMSA.
Cells were treated under various conditions, and nuclear extracts were prepared as described by Dignam et al. (7). Binding reactions were performed as described by Chadosh (3). In brief, 5 μg of nuclear extract was incubated at 30°C for 15 min with a 32P-end-labeled double-stranded oligonucleotide probe (2 × 104 cpm) containing the consensus CBE sequence (CBE probe, 5′-GCTACATATTACGTAACAAGCGTT-3′; underlined nucleotides are the core sequence of CBE) or a 4-bp mismatched CBE sequence (M4 probe, 5′-GCTACATAacACGTgtCAAGCGTT-3′; mutilated nucleotides are shown in lowercase). In competitive electrophoretic mobility shift assays (EMSAs), reactions were performed in a total volume of 20 μl, and extracts were incubated with 10-, 100-, or 1,000-fold molar excess of competitive unlabeled oligonucleotides added 5 min prior to addition of the labeled probe. Sequences of the competitors are shown in Table 1. Supershift experiments were performed by incubating 1 μl of antiserum with nuclear extracts at 4°C for 30 min prior to the DNA binding reaction. Nondenaturing polyacrylamide gels containing 5% polyacrylamide were run at 4°C in 0.5× Tris-borate-EDTA. The gel was then dried under vacuum and analyzed by autoradiography. UV cross-linking of CBE-binding complexes was performed as described by Chadosh (3). Sequences of the CBE-like element-containing oligonucleotides used in the competitive EMSA are shown in Table 1.
TABLE 1.
CBE-like elements in the promoter of apoptosis-related genes
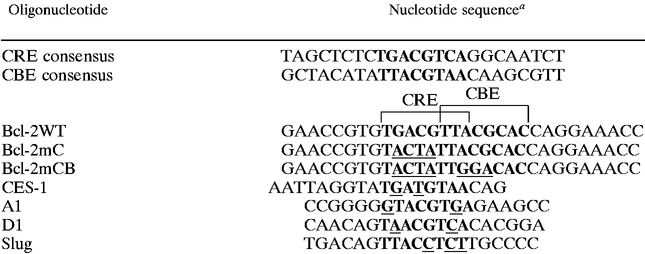 |
Boldface type highlights the CRE and CBE consensus sequences. Underlined nucleotides represent deviations from the consensus sequence.
≥90% competition of binding activity by 10-fold (+++), 100-fold (++), or 1,000-fold (+) concentration of the competitor oligonucleotides. − indicates ≤30% competition by a 1,000-fold concentration of the competitor.
ND, not determined.
Plasmid construction.
The reporter plasmids p3xCBE-Luc, p3xCRE-Luc, and p3xM4-Luc were constructed by inserting three copies of the CBE, M4 (see below), or CRE consensus sequence into the BgIII site of the pGL2-promoter vector (Promega). The sense-strand sequences of these oligonucleotides are as follows: CBE, 5′GATCTGCTACATATTACGTAACAAGCGTTG3′ (22); M4, 5′GATCTGCTACATAacACGTgtCAAGCGTTG3′ (22); and CRE, 5′GATCCAGAGATTGCCTGACGTCAGAGAGCTAGA3′ (43a). The Myr-AKT expression plasmid was kindly provided by Anke Klippel (28). The expression plasmid for the catalytic subunit of PKA (PKAc) was constructed by inserting the PKAc coding sequence into the pCMV vector. The plasmid expressing the dominant negative mutant of the PKA regulatory subunit (PKArDN), MT-REVAB-neo, which is under the control of the mouse metallothionein 1 promoter, was a gift from G. Stanley McKnight (6). The bcl-2 promoter-driven luciferase reporters containing wild-type (Bcl-2WT) or mutant (Bcl-2mC) CRE sites were kindly provided by Linda Boxer (54). The luciferase gene in Bcl-2WT was driven by a promoter located in the bcl-2 genomic locus from nucleotides −1640 to −1286 (relative to the translation initiation codon ATG; the CRE and CBE sites are located at positions −1559 and −1554, respectively). Plasmid Bcl-2mCB was derived from Bcl-2mC by introducing point mutations as shown in Table 1 into the putative CBE next to the CRE site in the bcl-2 promoter. A G-to-T point mutation at nucleotide 976 of CREB cDNA was introduced by PCR-assisted mutagenesis to generate the pcDNA3-CREBR287L vector. The dominant negative effect of CREBR287L on the wild-type CREB protein (52) was verified by EMSA. Plasmid pTRE-HA-CREBR287L is a construct in which a hemagglutinin epitope (HA)-tagged CREBR287L protein can be synthesized under a Tet-off inducible system (Clontech). This expression vector was constructed by first generating an intermediate vector (pJ3HA-CREBR287L) in which a DNA sequence encoding the HA tag was fused in frame to the 5′ end of the CREBR287L cDNA. The pJ3H vector (42), kindly provided by J. Chernoff, was used for this intermediate step. The DNA fragment spanning the HA-CREBR287L cDNA sequence was then released from pJ3HA-CREBR287L by digestion with HindIII and EcoRI, rendered blunt ended, and ligated into the flushed EcoRI site of the pTRE vector (Clontech) to yield the final construct, pTRE-HA-CREBR287L.
Establishment of Ba/F3 cell lines conditionally expressing CREBR287L.
To generate Ba/F3 derivatives that express HA-CREBR287L under the Tet-off inducible system (Clontech), Ba/F3 cells were cotransfected with plasmids pTRE-HA-CREBR287L and pTet-off (Clontech) by electroporation as previously described (4). After transfection, cells were selected in medium supplemented with G418 (800 μg/ml) and tetracycline (2 μg/ml) for 2 weeks. G418-resistant subclones were cultured in medium with or without tetracycline, and cell lysates were subjected to Western blot analysis for the expression and inducibility of HA-tagged CREBR287L. Two independent subclones, 4 and 8, expressing the highest levels of HA-CREBR287L under induced conditions, were selected for further analysis. The control cell line C3 was generated in the same way except that the pTRE-HA-CREBR287L vector was replaced with the empty expression vector pTRE.
Antibodies and Western blot analysis.
Antibody specific to CREB was previously described (18). Antibody specific for CREB phosphorylated at Ser133 was purchased from Upstate Biotechnology Inc. (Lake Placid, N.Y.). Monoclonal antibody to the HA epitope (12CA5) was purchased from Boehringer Mannheim. Antibodies for PKAr and PKAc were purchased from Transduction Laboratories (Lexington, Ky.). Cell lysates were prepared and fractionated by standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred onto a polyvinylidene difluoride nylon membrane by Western blotting. The membrane was probed with antibodies and visualized by an enhanced chemiluminescence system (Amersham, Buckinghamshire, England) as previously described (31).
Transient transfection and annexin V staining.
Ba/F3 cells cultured in IL-3-containing medium were washed with phosphate-buffered saline (PBS) and resuspended in RF buffer (60) (2 mM HEPES, 15 mM K2HPO4, 250 mM d-mannitol, 1 mM MgCl2 [pH 7.2]) at the density of 5 × 106 cells/ml. For each electroporation, 200 μl of cell suspension was mixed with 7.5 μg of expression plasmid and 2.5 μg of cotransfection marker pEFCD16/CD7/STOP. Cells were then electroporated using the RF modular of Gene Pulser II (Bio-Rad, Hercules, Calif.) at the following settings: 300 V, 15 bursts, 1 s between bursts, 2 ms of burst width, and frequency of 40 KHz. After electroporation, cells were recovered in IL-3-containing medium for 12 h prior to cytokine deprivation (for PKAc-expressing cells) or cadmium treatment (for PKArDN-expressing cells). PKAc-expressing cells were grown in 0.5% fetal bovine serum-containing, cytokine-free medium for 12 h; PKArDN- and CREBR287L-expressing cells were grown in medium containing 0.5% fetal bovine serum and 5 U of IL-3 per ml with or without 2 μM CdCl2 for 20 h. Cells were then washed with PBS, stained with biotin-conjugated annexin V (Catalog no. 1828690;) (Boehringer GmbH, Mannheim, Germany) and anti-CD16 antibody (MCA1193; Serotec), stained with R-phycoerythrin-conjugated streptavidin (Jackson ImmunoResearch Laboratories) and fluorescein isothiocyanate-conjugated secondary antibody (Jackson ImmunoResearch Laboratories), and analyzed by flow cytometry (4). In flow cytometry analysis, CD16-positive cells were gated to analyze their annexin V staining pattern.
Lipofection and luciferase assay.
PC12 (4 × 105) or 293 (2 × 105) cells were transiently transfected with 1 μg of the reporter plasmid DNA and various amounts of the expression plasmid (PKAc [0.1 μg] or pcDNA-3cREBR287L [0.5 μg]) by using Lipofectamine (Life Science, Gaithersburg, Md.) as instructed by the vendor. For IBMX and forskolin stimulation experiments, 1 μM IBMX and 100 μM forskolin were added to the culture medium. After 20 h, cell were washed with cold PBS and lysed in 40 μl of reporter lysis buffer (Promega) for 10 min on ice. Lysates were centrifuged at 15,000 rpm for 10 min. Twenty microliters of the supernatant was added to 100 μl of luciferase assay buffer (Promega) and applied to a luminometer (TD20/20; Turner Designs Instruments, Promega) to detect luciferase activity. Transfection of plasmid into Ba/F3 cells for luciferase assays was done as described above for annexin V staining analysis.
RESULTS
Multiple CBE-binding complexes in mammalian cells.
To identify the evolutionarily conserved Ces-2/E2A-HLF homologues in mammalian cells, we used a sensitive EMSA to explore various nuclear extracts and determine whether any of the nuclear factors could form a complex with CBE. The nuclear extract from Ba/F3 bound to the CBE oligonucleotide probe and formed multiple binding complexes in the native polyacrylamide gel (Fig. 1, lane 1). A complete inhibition was observed with an unlabeled CBE probe (Fig. 1, lane 2) but not with a 4-bp mismatched M4 probe (see Materials and Methods) (Fig. 1, lane 3), thus verifying the sequence specificity of these complexes. The fast-migrating group of complexes was constantly present in all extracts tested, including extracts from human GM-CSF-dependent TF-1 (lane 4), murine IL-2-dependent HT-2 (lane 5), human cervical carcinoma HeLa (lane 6), human hepatocarcinoma HepG2 (lane 7), murine fibroblast NIH 3T3 (lane 8), and Chinese hamster ovary carcinoma CHOP (lane 9) cell lines, and in extracts from thymocytes and splenocytes of BALB/c mice (lanes 10 and 11) and thymocytes of porcine origin (lane 12). The slow-migrating group of complexes was detectable in some extracts and was barely detectable in others, including HeLa, NIH 3T3, and mouse spleen cells (lanes 6, 8, and 11).
FIG. 1.

Multiple CBE-binding complexes exist in mammalian cells. Nuclear extracts were prepared from various cell lines and tissues (as indicated at the top) and subjected to EMSA with a 32P-labeled CBE probe. A 100-fold excess of unlabeled CBE probe (lane 2) or mutant CBE probe M4 (lane 3) was used to compete (Comp.) for the formation of specific complexes. Multiple binding complexes are categorized as slow-migrating (S) and fast-migrating (F′) groups. Each group is composed of at least two to three complexes. Thy, thymocytes; Spl., splenocytes.
Very little binding activity was detected in ion-free buffer. A drastic increase of binding activity was observed in buffers containing KCl from 50 to 200 μM, with a peak at around 133 μM (data not shown). All of these complexes seemed to require an optimal ionic concentration similar to that recommended for the binding of E2A-HLF to CBE, i.e., 133 mM (19). None of these binding complexes were heat resistant, and they rapidly diminished when the temperature exceeded 55°C (data not shown).
Identification of CREB in CBE-binding complexes.
To explore the molecular identities of these CBE-binding proteins, we applied UV to cross-link them to 32P-labeled CBE and then fractionated the proteins by SDS-PAGE to determine their molecular weights. As shown by EMSA, UV cross-linking did not interrupt the pattern of CBE-binding complexes (Fig. 2A, lanes 1 and 2) and binding specificity (lane 3). SDS-PAGE showed at least two major bands of 46 and 50 kDa that bound with the 32P-labeled CBE probe (Fig. 2A, lane 4). The 32P labeling of these two components by UV cross-linking was drastically inhibited by a 100-fold excess of unlabeled CBE (Fig. 2A, lane 5) but only slightly affected by the M4 probe (lane 6). From the sizes of these CBE-binding components and the sequence homology between CBE and the CRE, with only two nucleotide alterations, at the −3 and +3 positions of the core dyad symmetry, we postulated that one of the 46-kDa proteins was CREB.
FIG. 2.

CREB exists in CBE-binding complexes. (A) Molecular weight determination of CBE-binding proteins by UV cross-linking. The nuclear extract of Ba/F3 cells was mixed with a labeled CBE probe and irradiated with (+) or without (−) short-wavelength UV for 10 min (see Materials and Methods) in the presence (lanes 3 and 5) or absence (lanes 2 and 4) of competitor (Comp.) CBE or M4 (lane 6). One half of the sample was analyzed by EMSA (lanes 1 to 3), and the other half was analyzed by SDS-PAGE (lanes 4 to 6). Positions of molecular weight markers are marked at the left of lane 4. (B) Nuclear extracts prepared from Ba/F3 (lanes 1 to 3) and TF-1 (lanes 4 to 6) cells or in vitro-synthesized CREB (lanes 7 to 9) were subjected to EMSA. Rabbit preimmune serum (Pre.) or polyclonal anti-CREB antibody (Ab) (CREB) was used to perform supershift experiments. The position of the supershifted binding complex is indicated by a thick arrow. In the preimmune serum, there is an undetermined CBE-binding activity that did not supershift the CREB-containing complex but comigrated with the supershifted CREB-CBE complex. The positions for slow- and fast-migrating groups (S and F) are indicated as described in the legend to Fig. 1.
To verify our hypothesis, we performed three independent experiments. First, we demonstrated the presence of CREB in the fast-migrating group of CBE-binding complexes of hematopoietic cells by supershifting the specific complexes with anti-CREB antibody. Nuclear extracts from both Ba/F3 and TF-1 cells formed multiple complexes with the 32P-labeled CBE oligonucleotide (Fig. 2B), and complexes in the fast-migrating group were specifically supershifted to an upper position (Fig. 2B, lanes 3 and 6) with anti-CREB antibody. Incubation with the preimmune serum gave an extra binding complex that comigrated with the supershifted band but did not affect the CBE complexes (Fig. 2B, lanes 2 and 5; see also lane 8). Second, the complexes in the fast-migrating group were suggested to be the complexes of CREB homo- or heterodimers. The in vitro-translated CREB formed a complex with the CBE probe that comigrated with one of the complexes of the Ba/F3 nuclear extract (Fig. 2B, lane 7). The complex of the in vitro-translated CREB with CBE was supershifted in the presence of anti-CREB antibody but not by preimmune serum (Fig. 2B, lanes 8 and 9). However, CREB did not distinguish between the canonical CRE (underlined in the sequence that follows) (5′-AGAGATTGCCTGACGTCAGAGAGCTAG-3′) and CBE (5′-TTACGTAA-3′) and bound to both sequences equally well (data not shown). The binding complex of CREB with CBE was efficiently competed by the CRE oligonucleotide and vice versa (data not shown), suggesting that CREB binds to both elements with similar affinities.
Finally, we demonstrated that the recombinant CREB protein bound to CBE by expression screening (Table 2). We used a 32P-labeled CBE oligonucleotide to screen three expression libraries in Escherichia coli and demonstrated the binding of CBE by CREB in a mouse embryo library (Table 2). HLF and TEF (the PAR family members), ATF-2, E4BP4 and C/EBPβ were also identified during these screening experiments (Table 2). These findings suggested that there are at least four protein families that are able to bind CBE and that all may be involved in mediating CBE-dependent transactivation.
TABLE 2.
Representative CBE-binding cDNAs identified by expression screening
| Source of library | Identity of cDNA | No. of independent clones | No. of plaques screened (106) |
|---|---|---|---|
| Human leukocyte | E4BP4 | 5 | 1.44 |
| C/EBPβ | 6 | ||
| Mouse embryonic stem cell | TEF | 2 | 1.44 |
| ATF2 | 1 | ||
| Mouse 15-day embryo | HLF | 1 | 1.44 |
| TEF | 3 | ||
| C/EBPβ | 2 | ||
| CREB | 2 | ||
| ATF2 | 1 |
Activation of CBE reporter genes by cAMP and PKA signals.
Although CREB binds effectively to CBE, it is not clear whether CBE can indeed mediate CREB-dependent gene transactivation. To assess this possibility, we first examined whether elevation of cellular cAMP levels, one way to activate CREB, could stimulate the transcription of a CBE-driven luciferase reporter gene. As shown in Fig. 3A, treatment of transfected cells with forskolin, a cAMP-elevating agent, plus IBMX, an inhibitor of phosphodiesterase, increased the transcription activity of the CBE reporter by 2-fold (in 293T cells) or 50-fold (in PC12 cells). We next examined whether a similar effect could be achieved in cells overexpressing the constitutively active form of PKA, an upstream activator of CREB. The overexpressed PKAc was presumably constitutively active, due to the shortage of PKAr in the transfected cells. Figure 3B shows that in PC12 cells, PKAc can significantly activate the CBE-driven reporter compared to the reporter of the inactive CBE mutation M4 or the vector alone; however, the effect is not as dramatic as that of the CRE-driven reporter. A similar transactivation effect by PKAc on the CBE reporters was observed in experiments using IL-3-dependent Ba/F3 cells (Fig. 3C). Inclusion of the dominant negative mutant of CREB, CREBR287L, in the transactivation assays antagonized the stimulation effect of PKAc (Fig. 3B, lanes 3 and 6; Fig. 3C, lanes 2 to 4). Thus, these data suggested that the PKA/CREB pathway is able to activate the CBE-driven transcription in hematopoietic and nonhematopoietic cells.
FIG. 3.

Transcriptional activation of CBE reporter genes by the cAMP/PKA/CREB signaling pathway. (A) Activation of CBE reporter genes by cAMP-elevating agents. 293T and PC12 cells were transfected with p3xCBE-Luc (1 μg) and treated with or without forskolin plus IBMX (as indicated at the bottom). (B) CREB-dependent activation of the CBE reporter gene by PKA in PC12 cells. PC12 cells were transfected with the PKAc expression plasmid (0.1 μg) and various luciferase reporter plasmids (1 μg of each), together with dominant negative mutant CREBR287L (1 μg). (C) CREB-dependent activation of CBE reporter genes by PKA in Ba/F3 cells. Ba/F3 cells were electroporated with the PKAc expression plasmid (0.45 μg) and CBE luciferase reporter plasmid (3 μg), together with CREBR287L (0, 1.5 and 4 μg; shown as a triangle). Twenty hours after transfection, cell lysates were prepared and analyzed by luciferase assays. Data shown are representative results from three independent experiments performed in duplicate (in PC12 cells) or quadruplicate (in Ba/F3 cells). Luciferase activities are plotted in arbitrary units per microgram of total protein.
PKA-dependent CREB phosphorylation and cell survival in hematopoietic cells.
Phosphorylation of CREB at Ser133 is essential for the transcriptional activity of CREB. In this study, CREB was shown to be one of the major CBE-binding proteins in hematopoietic cells and was suggested to be required for activation of CBE-driven genes. We next wished to determine whether CREB could be phosphorylated at Ser133 by cytokines in cytokine-dependent cell lines. Rapid induction of phosphorylation of CREB by cytokine was demonstrated in TF-1, HT-2, Ba/F3, and C2GM cells by their own essential cytokines (data not shown). All cytokines tested induced a rapid phosphorylation of CREB at Ser133, within 5 to 10 min. Treatment of Ba/F3 cells with PKI (Fig. 4A, lanes 2, 3, and 4) partially decreased the phosphorylation level of CREB after IL-3 stimulation. A minor signal, representing the phosphorylated ATF-2 protein (Fig. 4A), was also induced by IL-3 and suppressed by PKI. These data were consistent with the notion that the phosphorylation of CREB at Ser133 induced by IL-3 was, at least partly, through the PKA pathway.
FIG. 4.

IL-3 induces CREB phosphorylation and survival function partly via the PKA pathway. (A) IL-3 induces PKA-dependent CREB Ser133 phosphorylation. Cytokine-starved Ba/F3 cells were pretreated with dimethyl sulfoxide (lanes 1 and 2) or PKI (lanes 3 and 4) for 30 min before being restimulated with IL-3 (IL3). After 5 min of stimulation, cells were harvested and analyzed by Western blot analysis with antibodies specific to phosphorylated CREB at Ser133 (-CREB) and to all forms of CREB (CREB). The star indicates the position of the phosphorylated ATF-2. (B) Increase of survival of Ba/F3 cells by cAMP-elevating agents. Viable cell number was measured by trypan blue staining after 18 h of incubation in cytokine-free medium without (−) or with (+) forskolin (FsK 100 μM) plus IBMX. Numbers of viable cells are presented as percentages of the number of cells initially seeded. Results shown are means ± standard deviations from two independent experiments performed in duplicate. (C) Decrease of cell viability by PKI. Ba/F3 cells were cultured in IL-3-containing medium supplemented with various doses of Myr-PKI (lanes 2 and 3, 10 and 20 μM). Cells cultured in cytokine-free medium were included as a negative control (lane 4). Numbers of viable cells under various treatments were determined and are presented as a percentage of the number of cells initially seeded. Shown are representative results from three independent experiments performed in duplicate.
We next investigated the potential role of the PKA/CREB pathway in apoptosis regulation in hematopoietic cells. Ba/F3 cells were treated with various doses of forskolin in the absence of murine IL-3, and only at levels higher than 100 μM was forskolin able to suppress cell death induced by cytokine deprivation (data not shown). IBMX further potentiated the death prevention effect of forskolin (Fig. 4B). The cAMP analogues dibutyryl- and 8-bromo-cAMP also showed an ability to prolong cell survival comparably to forskolin plus IBMX (data not shown). On the other hand, treatment of Ba/F3 cells with PKI in the presence of IL-3 significantly suppressed cell viability and increased apoptosis in a dose-dependent manner (Fig. 4C). These data suggested that activation of the cAMP/PKA/CREB pathway could possibly antagonize apoptotic signals in hematopoietic cells.
The PKA/CREB pathway plays a role in IL-3's survival activity.
To prove that the PKA/CREB pathway is involved in the IL-3-dependent survival signal, we next explored the effects of the dominant negative forms of CREB and PKA in Ba/F3 cells. Since CREBR287L forms a dimer with CREB/ATF family members and cannot bind to DNA, experiments using this dominant negative construct provided a stringent test for whether CREB is indeed part of the survival signal of IL-3. CREBR287L and PKArDN were transiently transfected into Ba/F3 cells along with a green fluorescence protein (GFP) or CD16 marker plasmid. In the regular culture media, both CREBR287L (Fig. 5A) and PKArDN (Fig. 5C, upper right) were expressed at a significant level in the GFP-positive population. In the presence of cadmium ion, the PKArDN levels were further increased due to increased transcription of the metallothionein promoter (Fig. 5C, lower right). An annexin V staining assay revealed that CREBR287L and PKArDN both effectively enhanced the percentage of apoptotic Ba/F3 cells in IL-3-containing culture medium. Apoptosis increased from 21 to 35% for cells transfected with CREBR287L (Fig. 5B) and from 8 to 29% for PKArDN-transfected cell (Fig. 5D, top). In the presence of cadmium ion, apoptosis increased from 14 to 48% (Fig. 5D, bottom). The promoter of the metallothionein gene, which drives PKArDN expression, was leaky, and both PKArDN protein expression and apoptosis enhancement were observed in the absence of cadmium ion (Fig. 5C and D).
FIG. 5.

Dominant negative forms of CREB and PKA reverse the antiapoptotic effect of IL-3 in Ba/F3 cells. (A and C) Expression of CREBR287L (A) and PKArDN (C) in Ba/F3 cells. Vectors pcDNA-3, pcDNA-3/CREBR287L, and MT-REVAB-neo (indicated as PKArDN) (7.5 μg of each construct) were transfected into Ba/F3 cells along with 2.5 μg of pCMV-GFP. Cells were allowed to express these proteins for 20 h in IL-3-containing medium without (A; C, top) or with (C, bottom) 2 μM cadmium chloride. Solid peaks indicate cells stained with control immunoglobulin G (IgG). Open peaks indicate cells stained with anti-CREB antibody (A) or anti-PKAr antibody (C). Open peaks in dotted lines are cells not expressing GFP (GFP −), and solid lines are cells expressing GFP (GFP +). Transfection efficiency was 10 to 12%. (B and D) Dominant negative forms of CREB (B) and PKAr (D) enhance apoptosis of Ba/F3 cells in the presence of IL-3. After transfection with the marker pCD16/7/Stop (2.5 μg) and the pcDNA-3 vector (7.5 or 18 μg), pcDNA-3/CREBR287L (18 μg), or MT-REVAB-neo (7.5 μg), cells were cultured in IL-3 containing medium with or without 2 μM cadmium chloride for 20 h. The percentages of apoptotic (annexin V-positive) cells in the transfected populations (CD16 positive) were analyzed and quantified as described in Materials and Methods. One set of representative data from three independent experiments is shown.
The role of CREB in IL-3's survival signal was also examined in Ba/F3 derivatives where the CREBR287L mutant was conditionally induced. For this experiment, two stable lines expressing CREBR287L, under the Tet-off inducible promoter (Clontech), were established. When tetracycline was removed, the expression level of CREBR287L was increased dramatically in both stable cell lines 4 and 8, but not in the control cell line C3 (Fig. 6A, top). Of note, in both cell lines 4 and 8, we also observed an increased expression of a protein band that comigrated with the endogenous CREB and was recognized by CREB but not by the HA antibody (Fig. 6A). EMSA with nuclear extracts made from these stable lines revealed that expression of CREBR287L by removing tetracycline from the culture medium dramatically decreased the level of fast-migrating complexes of CBE in both cell lines 4 and 8 and had no significant effect on the level of the slow-migrating complexes (Fig. 6B, top). The fast-migrating CBE complex in cell line C3 was not significantly altered by tetracycline deprivation. Serving as an internal control, the level of GATA-1 binding complex did not change in any cell lines tested regardless the presence or absence of tetracycline (Fig. 6B, bottom). These data indicated that we have successfully established two stable cell lines wherein the CREB-containing CBE-binding complexes can be specifically reduced by expression of the CREBR287L mutant.
FIG. 6.

Inducible expression of CREBR287L reduces CBE binding and viability of Ba/F3 cells. (A) Two Ba/F3 derivatives (clones 4 and 8) stably overexpressing CREBR287L under a Tet-off inducible system were established. The expression level of HA-CREBR287L (indicated by an arrowhead) in clones 4 and 8 in medium with (+) or without (−) tetracycline (Tet) was analyzed by immunoblotting with anti-HA and anti-CREB antibodies. C3, a Ba/F3 derivative expressing pTRE vector alone, is included as a negative control. (B) Decrease of CBE-binding activity in cells overexpressing CREBR287L. Nuclear extracts were prepared from each cell clone under conditions (with or without tetracycline) as indicated, and EMSA was then performed as described for Fig. 1 with a CBE (top) or GATA-1 (as an internal control; bottom) probe. The slow- and fast-migrating complexes are indicated as S and F, respectively. The asterisk indicates the position of the GATA-1 complex. (C) Suppression of cell viability by CREBR287L in stable lines. Cells cultured in the presence (+) or absence (−) of tetracycline for 72 h were washed and seeded in medium without IL-3. Sixteen hours after IL-3 depletion numbers of viable cell in each culture group were determined and are presented as percentages of the number of cells initially seeded. The data presented are averages from three independent experiments done in duplicate. ∗, 0.001 < P < 0.01 compared to lane 3; ∗∗, 0.001 < P < 0.01 compared to lane 5.
Consistent with results obtained from the transient assay (Fig. 5A and B), Ba/F3 cells with induced expression of CREBR287L were more sensitive to cytokine withdrawal-induced apoptosis (CWIA) (Fig. 6C). However, unexpectedly, with such a prominent induction of CREBR287L in both clones 4 and 8 (Fig. 6A), its effect on CWIA was no better than that observed in the transient assay. This suggests that activation of CREB plays only a partial role in IL-3's survival signal. Alternatively, the induced expression of the CREB or a CREB-like molecule through an unknown mechanism when the CREBR287L mutant was induced (Fig. 6A) may have compensated for the dominant negative effect of CREBR287L in these stable clones. Taken together, these data suggest that both PKA and CREB play a role in the antiapoptotic activity of IL-3 in Ba/F3 cells.
Next, we examined whether constitutive activation of PKA would antagonize IL-3 removal-induced apoptosis. To address this issue, PKAc was transfected into Ba/F3 cells together with the marker gene, either the GFP or CD16/7 chimeric protein expression plasmid. The level of PKAc increased dramatically in the GFP-positive population compared to that in the GFP-negative population (Fig. 7A, right). The annexin V staining assay revealed that IL-3 depletion caused death of about 50% of control Ba/F3 cells via apoptosis (Fig. 7B, Vector). However, the expression of PKAc significantly reduced apoptosis to 17% (Fig. 7B, PKAc). To examine whether CREB was a downstream effector of PKA in this assay system, we expressed PKAc together with the dominant negative mutant CREBR287L into Ba/F3 cells, and apoptosis percentage was monitored with annexin V staining. CREBR287L partly diminished the protection effect of PKAc, and the percentage of apoptotic cells increased from 17 to 40 (Fig. 7B). Taken together, our results indicated that CREB indeed functions as a part of the survival signals activated by PKA.
FIG. 7.

Expression of PKAc suppresses CWIA of Ba/F3 cells. (A) Expression of PKAc in transiently transfected Ba/F3 cells. Ba/F3 cells were transfected with pCMV vector or PKAc (7.5 μg of each), as indicated together with pCMV-GFP (2.5 μg). At 20 h after transfection, cells were analyzed for expression of PKAc by flow cytometry. The solid peaks indicate cells stained with rabbit normal serum, and the open peaks indicate cells stained with rabbit anti-PKAc antibody. The open peaks in dotted lines indicate the GFP-negative populations (GFP −), and solid lines show the GFP-positive populations (GFP +). Transfection efficiency was 10 to 12%. IgG, immunoglobulin G. (B) Suppression of CWIA by expression of PKAc. After transfection with the marker pCD16/7/Stop (2.5 μg) and pCMV vector or PKAc expression plasmid (2 μg of each) with or without CREBR287L (18 μg), cells were cultured in the absence of IL-3 for 20 h prior to being stained with anti-CD16 antibody and annexin V and analyzed by flow cytometry. Apoptotic percentages are as indicated at the upper right. Transfection efficiency was around 15%. One set of representative data from three independent experiments is shown.
Identification of bcl-2 as a CBE containing survival gene.
To investigate whether the CREB-dependent antiapoptotic effect involves activation of any CBE-containing survival gene(s), we first searched the DNA database for an apoptosis-related gene(s) whose promoter contains the consensus CBE or a CBE-like element. We thus identified a few candidate genes, including bcl-2 (54), A1 (14), D1 cyclin (17), and slug (44) (Table 1). Next, we examined whether the CBE-like elements in these candidate genes bound to known CBE-binding proteins (e.g., HLF) in vitro. To address this issue, we examined by competitive EMSA whether any of these CBE-like elements competed with the 32P-labeled consensus CBE (sequence is shown in Table 1) for binding to the ΔHLF protein, a truncated form of HLF containing only the DNA-binding domain. The CBE of the C. elegans ces-1 gene that is known to be recognized by the Ces-2 protein (i.e., one of two authentic CBE-binding proteins) (35) was included here as a positive control. As shown in Fig. 8A and summarized in Table 1, ΔHLF bound to the consensus CBE, and its binding was completely abolished by the presence of a 10-fold molar excess of the same unlabeled competitor (Fig. 8A, CBE). Under the same conditions, the CBE of the ces-1 gene did not manifest any significant competition until 1,000-fold excesses of oligonucleotides were present in the assay, suggesting that ΔHLF bound to the CBE of the ces-1 gene with low affinity (Fig. 8A, CES-1). On the other hand, among the mammalian genes tested, only the CBE-like element of bcl-2 showed a competition ability similar to that of the ces-1 CBE site (Fig. 8A, Bcl-2mC). The CBE-like elements from the D1 cyclin, slug, and A1 genes (Fig. 8A) were all incapable of competing with the consensus CBE site for binding to ΔHLF even at a 1,000-fold excess. Interestingly, mutation in the CBE sequence of the bcl-2 gene abrogated the ability to compete for binding to ΔHLF (Fig. 8A, Bcl-2mCB). These results suggest that the bcl-2 promoter contains a low-affinity CBE site whose affinity is similar to that of ces-1 CBE site. Next, using the same approach described above, we tested whether CREB can also bind to the CBE site of the bcl-2 gene. As shown in Fig. 8B, although CREB bound to the consensus CBE sequence with moderate affinity, CREB, like ΔHLF, bound to the CBEs of the ces-1 and bcl-2 (i.e., Bcl-2mC) genes with low affinity (Fig. 8B). The same CBE mutation (Bcl-2mCB) also abolished the binding ability of the CREB protein (Fig. 8B). The CBE-like element of the slug gene served as a negative control which did not bind to CREB at all (Fig. 8B, Slug). In conclusion, the human bcl-2 gene contains a low-affinity CBE site in its promoter, and this CBE site can also be recognized by CREB in vitro.
FIG. 8.

The CBE site in the bcl-2 promoter is important for IL-3 inducibility in hematopoietic cells. (A and B) Competition for ΔHLF (A) or CREB (B) binding to the CBE consensus by various CBE-like sequences. The consensus CBE sequence was labeled with 32P and incubated with either ΔHLF (A) or CREB (B) in the presence of a 10-, 100-, or 1,000-fold molar excess (indicated as a triangle) of competitive (Comp.) CBE-like sequences. The genes from which the competitive CBE-like sites were are indicated above the triangles, and their sequences are shown in Table 1. The competitive EMSAs were repeated three times, and a representative set of data is shown. (C and D) Both CRE and CBE contribute to PKA (C) or IL-3 (D) stimulation of bcl-2 reporter activity. (C) Ba/F3 cells were electroporated with various bcl-2 reporters as indicated together with a PKAc (+PKA) or control (−PKA) expression vector. Twenty hours after transfection, cell lysates were prepared and analyzed by luciferase assays. (D) Same as panel C except that the PKA expression vector was omitted, and the transfected cells were left in medium with or without IL-3 prior to being analyzed for luciferase activity. WT, mC, mCB denote reporter plasmids Bcl-2WT, Bcl-2mC, and Bcl-2mCB, respectively (see Materials and Methods). The data presented are averages from three independent experiments performed in duplicate.
We next investigated whether the bcl-2 CBE responded to PKA and/or IL-3 signaling in Ba/F3 cells. For this experiment, Bcl-2WT, Bcl-2mC, or Bcl-2mCB was cotransfected with the PKAc or control vector into Ba/F3 cells. As shown in Fig. 8C, cotransfection with the PKAc expression plasmid stimulated Bcl-2WT activity about threefold (Fig. 8C, WT). Clustered point mutations that abolished the CRE site (Bcl-2mC [Table 1]) resulted a reporter that was still responsive to PKAc stimulation, albeit to a lesser degree (∼2-fold) (Fig. 8C, mC). In contrast, Bcl-2mCB (Table 1) showed almost no response to PKAc (Fig. 8C, mCB). These results suggest that both CRE and CBE sites play a role in PKA stimulation of bcl-2 promoter activity. Next, using a similar reporter assay, we examined the role of these two DNA elements in IL-3's effect on bcl-2 promoter activity. Figure 8D shows that a similar effect is observed in this assay. That is, the luciferase activity of Bcl-2WT was stimulated ∼6-fold by IL-3; IL-3's stimulatory effect was diminished to ∼4-fold for Bcl-2mC, whereas Bcl-2mCB was nearly refractory to IL-3 stimulation. Taken together, these data suggest that both CRE and CBE contribute to IL-3 activation of the bcl-2 promoter and that these two sites are likely regulated by the PKA/CREB pathway.
DISCUSSION
The CBE is a DNA sequence recognized by death specification gene product Ces-2, leukemic oncoprotein E2A-HLF, adenovirus E4 gene transcription factor E4BP4, and PAR family transcription factors. Due to the functional and structural conservation of Ces-2 and E2A-HLF in the regulation of apoptosis, their downstream targets, the CBE-driven genes, are also suggested to be involved in death control. The initial purpose of this study was to look for cellular CBE-binding proteins that may regulate the same downstream targets as those of E2A-HLF and may be involved in positive or negative regulation of CWIA in hematopoietic cells. In this study, we have identified multiple CBE-binding complexes in nuclear extracts from various mammalian cells and tissues. A complex containing CREB was one of the major complexes in most of the cell lines and tissues tested. Consistent with this finding, by screening the E. coli expression cDNA libraries with radiolabeled CBE probes, we identified CREB as one of the CBE-binding proteins. Of note, among the CBE-binding proteins (Table 1), only E4BP4 and CREB have been demonstrated to be involved in apoptosis regulation of hematopoietic cells (reference 20 and this study). The role of C/EBPβ and HLF/TEF families in death control remains to be investigated.
We also demonstrated that the CBE-driven reporter gene is able to respond to PKA activation by either forskolin treatment or gene transfer and that this transactivation effect is CREB -dependent. Although the binding affinities of CREB to CBE and to CRE were indistinguishable in vitro (data not shown), PKA induced a better activation of the CRE-driven reporter than of the CBE-driven reporter. While the exact nature of this difference is not clear, it may suggest that additional factors are involved in PKA regulation of CBE- or CRE-mediated gene transcription. Of note, although a similar sequence motif named CRE-2 (TGCGTCA) was shown to be recognized by CREB and this binding motif alone conferred IL-3 inducibility on a heterologous promoter (53), neither the consensus CBE nor the CRE motif configured in the same way harbored this property (data not shown). A similar result was also observed with a putative CRE site located in the egr-1 promoter. That is, deletion of this element abolished the IL-3 inducibility of the egr-1 reporters, whereas a construct containing this DNA element alone upstream of the thymidine kinase promoter and chloramphenicol acetyltransferase gene failed to respond to IL-3 stimulation (29, 40). Together, these results indicated that sequences flanking the CRE or CBE site play a modulatory role in the transcriptional responses of these two DNA elements.
Like IL-3, other hematopoietic growth factors such as GM-CSF and IL-2 also induce Ser133 phosphorylation of CREB (reference 29 and our unpublished results). Although multiple signaling pathways triggered by a diverse array of stimuli were reported to lead to Ser133 phosphorylation and activation of CREB (see reference 43 for a review), the mechanism by which hematopoietic growth factors stimulate serine phosphorylation of this transcription factor is still unclear. In this report, we provide evidence that IL-3-stimulated CREB phosphorylation at Ser133 involves activation of the PKA pathway (Fig. 4A). Furthermore, we observed that the pharmacological inhibitors of phosphatidylinositol 3-kinase and SAPK2/p38 both diminished CREB phosphorylation induced by IL-3 stimulation (our unpublished data), suggesting that a combination of pathways regulate CREB activity in IL-3-dependent cells.
Elevation of intracellular cAMP contents by pharmacological agents induces apoptosis of some cell systems (26, 32) but triggers the opposite response, i.e., protects against cell death, in many other cell types, including MC/9 myeloid cells (41), neutrophilic polymorphonuclear leukocytes (37, 38, 51), U937 promonocytic leukemia cells (12), primary hepatocytes (11), T-cell hybridomas (24, 30), and RAW 264.7 macrophages (50). Consistent with the latter case, we observed that activation of PKA, a downstream effector of cAMP, by treatment of cells with chemical inducers or by ectopic expression of the catalytic subunit of PKA prolonged survival of Ba/F3 cells in the absence of IL-3. Our results together with others thus suggest that PKA is one cellular target frequently modulated by a variety of stimuli and, depending on the cellular context, activation of PKA leads to either cell survival or cell death. On the other hand, CREB is one important downstream factor activated by PKA. Constitutive activation of CREB protects human melanoma cells from UV-induced apoptosis (56) and contributes to the acquisition of the malignant phenotype (25). Recent studies with transgenic and knockout mice further demonstrated that CREB and its paralog, CREM, are important for cell survival. CREM-deficient mice, for example, exhibit a spermatogenesis defect secondary to enhanced apoptosis of germ cells (2). Overexpression of a dominant negative CREB transgene, moreover, induces apoptosis in T cells following growth factor stimulation (1). With IL-3-dependent Ba/F3 cells as a model system, we demonstrated that activation of the PKA/CREB pathway plays a role in the survival activity of IL-3.
The bcl-2 gene was recently demonstrated to be the target of the antiapoptotic effect of CREB in neuronal tissues (39) and in murine T helper cells (59), through a conserved CRE in the bcl-2 promoter. Intriguingly, our search for the CBE-containing apoptosis-related genes discovered that there is a low-affinity CBE site partially overlapping with the CRE located in the downstream regulatory element of the bcl-2 promoter (Table 1) (54). We further demonstrated that CREB binds to this CBE site with an affinity similar to that of its binding to the ces-1 CBE element, and that both CRE and CBE elements contribute to PKA or IL-3 activation of the bcl-2 reporter expression. Together, these results suggest that the bcl-2 gene is likely one cellular target of the CREB-containing CBE complex. The in vivo role of this CBE complex on bcl-2 gene transcription and its possible regulation by the PKA pathway remain to be determined. The Bcl-2 family proteins including Bcl-2 itself, Bcl-XL, and Mcl-1 were reported to be involved in the survival activity of IL-3 (4, 27, 33). While the detailed survival pathway triggered by IL-3 stimulation remains to be determined, our results suggest that the PKA/CREB pathway plays a role in this process and that part of this PKA/CREB pathway is likely mediated through activation of some CBE-controlled antiapoptotic genes.
ACKNOWLEDGMENTS
We thank Linda M. Boxer for providing bcl-2 promoter luciferase reporter genes, Jonathan Chernoff for providing pJ3H expression vector, Yueh-Liang Tsai for technical assistance, and Hsiu-ming Shih for critical comments on the manuscript.
This work was supported in part by an intramural fund from Academia Sinica, Taiwan (J.J.-Y.Y.) and grants NSC87-2314-B-001-016 and NSC88-2314-B-001-018 from the National Science Council of Taiwan (J.J.-Y.Y.).
W. Chen and Y.-L. Yu contributed equally to this work.
REFERENCES
- 1.Barton K, Muthusamy N, Chanyangam M, Fischer C, Clendenin C, Leiden J M. Defective thymocyte proliferation and IL-2 production in transgenic mice expressing a dominant-negative form of CREB. Nature. 1996;379:81–85. doi: 10.1038/379081a0. [DOI] [PubMed] [Google Scholar]
- 2.Blendy J A, Kaestner K H, Weinbauer G F, Nieschlag E, Schutz G. Severe impairment of spermatogenesis in mice lacking the CREM gene. Nature. 1996;380:162–165. doi: 10.1038/380162a0. [DOI] [PubMed] [Google Scholar]
- 3.Chadosh L A. Mobility shift DNA-binding assay using gel electrophoresis. In: Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Wiley Publishers; 1988. p. 12.2. [Google Scholar]
- 4.Chao J R, Wang J M, Lee S F, Peng H W, Lin Y H, Chou C H, Li J C, Huang H M, Chou C K, Kuo M L, Yen J J, Yang-Yen H F. mcl-1 is an immediate-early gene activated by the granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling pathway and is one component of the GM-CSF viability response. Mol Cell Biol. 1998;18:4883–4898. doi: 10.1128/mcb.18.8.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chrivia J C, Kwok R P, Lamb N, Hagiwara M, Montminy M R, Goodman R H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 6.Clegg C H, Correll L A, Cadd G G, McKnight G S. Inhibition of intracellular cAMP-dependent protein kinase using mutant genes of the regulatory type I subunit. J Biol Chem. 1987;262:13111–13119. [PubMed] [Google Scholar]
- 7.Dignam J, Lebovitz R, Roeder R. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis H M, Horvitz H R. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 9.Ellis R E, Horvitz H R. Two C. elegans genes control the programmed deaths of specific cells in the pharynx. Development. 1991;112:591–603. doi: 10.1242/dev.112.2.591. [DOI] [PubMed] [Google Scholar]
- 10.Ellis R E, Jacobson D M, Horvitz H R. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fladmark K E, Gjertsen B T, Doskeland S O, Vintermyr O K. Fas/APO-1(CD95)-induced apoptosis of primary hepatocytes is inhibited by cAMP. Biochem Biophys Res Commun. 1997;232:20–25. doi: 10.1006/bbrc.1997.6214. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Bermejo L, Perez C, Vilaboa N E, de-Blas E, Aller P. cAMP increasing agents attenuate the generation of apoptosis by etoposide in promonocytic leukemia cells. J Cell Sci. 1998;111:637–644. doi: 10.1242/jcs.111.5.637. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez G A, Montminy M R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 14.Grumont R J, Rourke I J, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 1999;13:400–411. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedgecock E M, Sulston J E, Thomson J N. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- 16.Hengartner M O, Ellis R E, Horvitz H R. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 17.Herber B, Truss M, Beato M, Muller R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene. 1994;9:1295–1304. [PubMed] [Google Scholar]
- 18.Hsueh Y P, Liang H E, Ng S Y, Lai M Z. CD28-costimulation activates cyclic AMP-responsive element-binding protein in T lymphocytes. J Immunol. 1997;158:85–93. [PubMed] [Google Scholar]
- 19.Hunger S P, Ohyashiki K, Toyama K, Cleary M L. Hlf, a novel hepatic bZIP protein, shows altered DNA-binding properties following fusion to E2A in t(17;19) acute lymphoblastic leukemia. Genes Dev. 1992;6:1608–1620. doi: 10.1101/gad.6.9.1608. [DOI] [PubMed] [Google Scholar]
- 20.Ikushima S, Inukai T, Inaba T, Nimer S D, Cleveland J L, Look A T. Pivotal role for the NFIL3/E4BP4 transcription factor in interleukin 3-mediated survival of pro-B lymphocytes. Proc Natl Acad Sci USA. 1997;94:2609–2614. doi: 10.1073/pnas.94.6.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inaba T, Roberts W M, Shapiro L H, Jolly K W, Raimondi S C, Smith S D, Look A T. Fusion of the leucine zipper gene HLF to the E2A gene in human acute B-lineage leukemia. Science. 1992;257:531–534. doi: 10.1126/science.1386162. [DOI] [PubMed] [Google Scholar]
- 22.Inaba T, Shapiro L H, Funabiki T, Sinclair A E, Jones B G, Ashmun R A, Look A T. DNA-binding specificity and trans-activating potential of the leukemia-associated E2A-hepatic leukemia factor fusion protein. Mol Cell Biol. 1994;14:3403–3413. doi: 10.1128/mcb.14.5.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inaba T, Inukai T, Yoshihara T, Seyschab H, Ashmun R A, Canman C E, Laken S J, Kastan M B, Look A T. Reversal of apoptosis by the leukaemia-associated E2A-HLF chimaeric transcription factor. Nature. 1996;382:541–544. doi: 10.1038/382541a0. [DOI] [PubMed] [Google Scholar]
- 24.Ivanov V N, Lee R K, Podack E R, Malek T R. Regulation of Fas-dependent activation-induced T cell apoptosis by cAMP signaling: a potential role for transcription factor NF-kappa B. Oncogene. 1997;14:2455–2464. doi: 10.1038/sj.onc.1201088. [DOI] [PubMed] [Google Scholar]
- 25.Jean D, Harbison M, McConkey D J, Ronai Z, Bar-Eli M. CREB and its associated proteins act as survival factors for human melanoma cells. J Biol Chem. 1998;273:24884–24890. doi: 10.1074/jbc.273.38.24884. [DOI] [PubMed] [Google Scholar]
- 26.Jondal M, Xue Y, McConkey D J, Okret S. Thymocyte apoptosis by glucocorticoids and cAMP. Curr Top Microbiol Immunol. 1995;200:67–79. doi: 10.1007/978-3-642-79437-7_5. [DOI] [PubMed] [Google Scholar]
- 27.Kinoshita T, Yokota T, Arai K, Miyajima A. Regulation of Bc1–2 expression by oncogenic Ras protein in hematopoietic cells. Oncogene. 1995;10:2207–2212. [PubMed] [Google Scholar]
- 28.Kulike G, Klippel A, Weber M J. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H J, Mignacca R C, Sakamoto K M. Transcriptional activation of egr-1 by granulocyte-macrophage colony-stimulating factor but not interleukin 3 requires phosphorylation of cAMP response element-binding protein (CREB) on serine 133. J Biol Chem. 1995;270:15979–15983. doi: 10.1074/jbc.270.27.15979. [DOI] [PubMed] [Google Scholar]
- 30.Lee M R, Liou M L, Liou M L, Yang Y F, Lai M Z. cAMP analogs prevent activation-induced apoptosis of T cell hybridomas. J Immunol. 1993;151:5208–5217. [PubMed] [Google Scholar]
- 31.Lee S F, Huang H M, Chao J R, Lin S, Yang-Yen H F, Yen J J. Cytokine receptor common beta chain as a potential activator of cytokine withdrawal-induced apoptosis. Mol Cell Biol. 1999;19:7399–7409. doi: 10.1128/mcb.19.11.7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lerner A, Kim D H, Lee R. The cAMP signaling pathway as a therapeutic target in lymphoid malignancies. Leuk Lymphoma. 2000;37:39–51. doi: 10.3109/10428190009057627. [DOI] [PubMed] [Google Scholar]
- 33.Leverrier Y, Thomas J, Perkins G R, Mangeney M, Collins M K, Marvel J. In bone marrow derived Baf-3 cells, inhibition of apoptosis by IL-3 is mediated by two independent pathways. Oncogene. 1997;14:425–430. doi: 10.1038/sj.onc.1200845. [DOI] [PubMed] [Google Scholar]
- 34.Metzstein M M, Hengartner M O, Tsung N, Ellis R E, Horvitz H R. Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans gene ces-2. Nature. 1996;382:545–547. doi: 10.1038/382545a0. [DOI] [PubMed] [Google Scholar]
- 35.Metzstein M M, Horvitz H R. The C. elegans cell death specification gene ces-1 encodes a snail family zinc finger protein. Mol Cell. 1999;4:309–319. doi: 10.1016/s1097-2765(00)80333-0. [DOI] [PubMed] [Google Scholar]
- 36.Montminy M R, Bilezikjian L M. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 37.Ottonello L, Gonella R, Dapino P, Sacchetti C, Dallegri F. Prostaglandin E2 inhibits apoptosis in human neutrophilic polymorphonuclear leukocytes: role of intracellular cyclic AMP levels. Exp Hematol. 1998;26:895–902. [PubMed] [Google Scholar]
- 38.Parvathenani L K, Buescher E S, Chacon-Cruz E, Beebe S J. Type I cAMP-dependent protein kinase delays apoptosis in human neutrophils at a site upstream of caspase-3. J Biol Chem. 1998;273:6736–6743. doi: 10.1074/jbc.273.12.6736. [DOI] [PubMed] [Google Scholar]
- 39.Riccio A, Ahn S, Davenport C M, Blendy J A, Ginty D D. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto K M, Fraser J K, Lee H J, Lehman E, Gasson J C. Granulocyte-macrophage colony-stimulating factor and interleukin-3 signaling pathways converge on the CREB-binding site in the human egr-1 promoter. Mol Cell Biol. 1994;14:5975–5985. doi: 10.1128/mcb.14.9.5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheid M P, Foltz I N, Young P R, Schrader J W, Duronio V. Ceramide and cyclic adenosine monophosphate (cAMP) induce cAMP response element binding protein phosphorylation via distinct signaling pathways while having opposite effects on myeloid cell survival. Blood. 1999;93:217–225. [PubMed] [Google Scholar]
- 42.Sells M A, Chernoff J. Epitope-tag vectors for eukaryotic protein production. Gene. 1995;152:187–189. doi: 10.1016/0378-1119(94)00685-l. [DOI] [PubMed] [Google Scholar]
- 43.Shaywitz A J, Greenberg M E. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 43a.Shepard A R, Zhang W, Eberhardt N L. Two CGTCA motifs and a GHF/Pit1 binding site mediate cAMP-dependent protein kinase A regulation of human growth hormone gene expression in rat anterior pituitary GC cells. J Biol Chem. 1994;269:1804–1814. [PubMed] [Google Scholar]
- 44.Stegmann K, Boecker J, Kosan C, Ermert A, Kunz J, Koch M C. Human transcription factor SLUG: mutation analysis in patients with neural tube defects and identification of a missense mutation (D119E) in the Slug subfamily-defining region. Mutat Res. 1999;406:63–69. doi: 10.1016/s1383-5726(99)00002-3. [DOI] [PubMed] [Google Scholar]
- 45.Sulston J E. Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos Trans R Soc Lond. 1976;275:287–297. doi: 10.1098/rstb.1976.0084. [DOI] [PubMed] [Google Scholar]
- 46.Sulston J E, Horvitz H R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 47.Sulston J E, Schierenberg E, White J G, Thomson J N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 48.Trent C, Tsung N, Horvitz H R. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vinson C R, LaMarco K L, Johnson P F, Landschulz W H, McKnight S L. In situ detection of sequence-specific DNA binding activity specified by a recombinant bacteriophage. Genes Dev. 1988;2:801–806. doi: 10.1101/gad.2.7.801. [DOI] [PubMed] [Google Scholar]
- 50.von-Knethen A, Lotero A, Brune B. Etoposide and cisplatin induced apoptosis in activated RAW 264.7 macrophages is attenuated by cAMP-induced gene expression. Oncogene. 1998;17:387–394. doi: 10.1038/sj.onc.1201926. [DOI] [PubMed] [Google Scholar]
- 51.Walker B A, Rocchini C, Boone R H, Ip S, Jacobson M A. Adenosine A2a receptor activation delays apoptosis in human neutrophils. J Immunol. 1997;158:2926–2931. [PubMed] [Google Scholar]
- 52.Walton K M, Rehfuss R P, Chrivia J C, Lochner J E, Goodman R H. A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol Endocrinol. 1992;6:647–655. doi: 10.1210/mend.6.4.1350057. [DOI] [PubMed] [Google Scholar]
- 53.Wang J M, Chao J R, Chen W, Kuo M L, Yen J J, Yang-Yen H F. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson B E, Mochon E, Boxer L M. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamamoto K K, Gonzalez G A, Biggs III W H, Montminy M R. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334:494–498. doi: 10.1038/334494a0. [DOI] [PubMed] [Google Scholar]
- 56.Yang Y M, Dolan L R, Ronai Z. Expression of dominant negative CREB reduces resistance to radiation of human melanoma cells. Oncogene. 1996;12:2223–2233. [PubMed] [Google Scholar]
- 57.Yen J J, Hsieh Y C, Yen C L, Chang C C, Lin S, Yang-Yen H F. Restoring the apoptosis suppression response to IL-5 confers on erythroleukemic cells a phenotype of IL-5-dependent growth. J Immunol. 1995;154:2144–2152. [PubMed] [Google Scholar]
- 58.Yoshihara T, Inaba T, Shapiro L H, Kato J Y, Look A T. E2A-HLF-mediated cell transformation requires both the trans-activation domains of E2A and the leucine zipper dimerization domain of HLF. Mol Cell Biol. 1995;15:3247–3255. doi: 10.1128/mcb.15.6.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F, Rincon M, Flavell R A, Aune T M. Defective Th function induced by a dominant-negative cAMP response element binding protein mutation is reversed by Bcl-2. J Immunol. 2000;165:1762–1770. doi: 10.4049/jimmunol.165.4.1762. [DOI] [PubMed] [Google Scholar]
- 60.Zheng Q, Chang D C. High-efficiency gene transfection by in situ electroporation of cultured cells. Biochem Biophy Acta. 1991;1088:104–110. doi: 10.1016/0167-4781(91)90158-i. [DOI] [PubMed] [Google Scholar]