

ABSTRACT
Metabolic acidosis, defined as a plasma or serum bicarbonate concentration <22 mmol/L, is a frequent consequence of chronic kidney disease (CKD) and occurs in ~10–30% of patients with advanced stages of CKD. Likewise, in patients with a kidney transplant, prevalence rates of metabolic acidosis range from 20% to 50%. CKD has recently been associated with cognitive dysfunction, including mild cognitive impairment with memory and attention deficits, reduced executive functions and morphological damage detectable with imaging. Also, impaired motor functions and loss of muscle strength are often found in patients with advanced CKD, which in part may be attributed to altered central nervous system (CNS) functions. While the exact mechanisms of how CKD may cause cognitive dysfunction and reduced motor functions are still debated, recent data point towards the possibility that acidosis is one modifiable contributor to cognitive dysfunction. This review summarizes recent evidence for an association between acidosis and cognitive dysfunction in patients with CKD and discusses potential mechanisms by which acidosis may impact CNS functions. The review also identifies important open questions to be answered to improve prevention and therapy of cognitive dysfunction in the setting of metabolic acidosis in patients with CKD.
Keywords: acidosis, chronic kidney disease, cognitive dysfunction, klotho, motor function
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Chronic kidney disease (CKD) causes complex endocrine and metabolic disturbances, leading to bone disease and excessive cardiovascular morbidity and mortality. Among these endocrine disturbances are reduced levels of α-klotho and calcitriol and an increase in fibroblast growth factor 23 (FGF23), parathyroid hormone (PTH) and accumulation of uraemic toxins, including elevation of serum phosphate, as well as reduced erythropoietin levels and anaemia. Advanced stages of CKD also entail salt and water retention, along with hyperkalaemia and metabolic acidosis [1]. More recently, cognitive dysfunction and impaired motor functions have been associated with CKD and recognized as another complication that impacts on the quality of life of affected patients [2–6]. The term cognitive dysfunction is not very well defined but generally includes deficits in declarative learning, related memory formation and sensory processing. Also, sleep problems and mood disorders are linked to altered brain function in patients with CKD. Motor deficits are found in these patients that may encompass not only aberrations in central nervous system (CNS) functioning, but also remodelling of peripheral nerve and muscle configurations. This review focuses on the role of metabolic acidosis as a potential risk factor or contributor to the development of cognitive dysfunction and motor deficits in patients with CKD. We will review the evidence that associates metabolic acidosis with impaired brain functions, discuss potential mechanisms and raise questions that should be addressed both clinically as well as in model organisms to provide a better understanding of this problem.
Metabolic acidosis in patients with CKD
Metabolic acidosis is a common complication of patients with CKD and increases in prevalence with the progression of kidney disease [7]. Between 10% and 20% of patients with Stage G4 CKD have overt metabolic acidosis, which increases to 30–40% of patients with Stage G5 CKD (Figure 1) [1, 9–11]. Acidosis is defined here as a process causing a positive hydrogen (H+) balance in (extracellular) fluid compartments and encompasses overt acidosis when serum or plasma bicarbonate (HCO3−) [or total carbon dioxide (CO2)] falls to <22 mmol/L and/or a blood pH <7.36 as well as eubicarbonataemic acidosis when systemic HCO3− and pH are within normal limits but acid accumulation occurs in some organs (see also below). This definition is somewhat arbitrary, as only a few risk analyses have assessed the threshold for upper and lower HCO3− levels that associate with disease risks and serum pH might influence the association between serum HCO3−, renal failure and other disease risks [12]. As discussed below, such an analysis is also missing for the association between HCO3− levels and cognitive dysfunction. However, an association study in an elderly population for HCO3− level and all-cause mortality suggests that mortality is lowest in individuals with HCO3− values from 23 to 26 mmol/L [13]. In patients with CKD Stages G3 and G4, all-cause mortality is lowest between 23 and 32 mmol/L [14]. Overt metabolic acidosis is also frequently encountered in kidney transplant recipients and the prevalence reported in various studies ranges between 20% and 50% of patients [7, 15]. In these patients, metabolic acidosis may be caused by not only reduced kidney function, but also promoted by some immunosuppressants such as calcineurin inhibitors, immunological factors, the process of donation and donor characteristics and diet [15].
FIGURE 1.
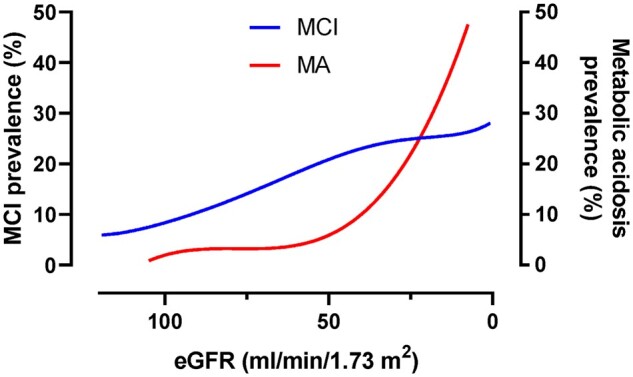
Prevalence of MCI and metabolic acidosis (MA) in patients with reduced kidney function. The prevalence of MCI and MA as a function of eGFR is estimated from several studies that reported the prevalence of MCI or MA in patients with reduced kidney function [1, 3, 5, 7, 8]. Cross-sectional studies analysing the prevalence of both clinical entities in the same cohort of patients have not been reported to date.
More recently, a novel concept of eubicarbonataemic acidosis has been introduced postulating accumulation of acid equivalents in a tissue under conditions of normal systemic acid–base balance [9, 10]. In kidney disease, acid retention in the kidney may drive kidney disease progression and possibly also some of the systemic alterations, such as changes in circulating hormones (i.e. endothelin or the renin–angiotensin–aldosterone system), while blood pH, HCO3− and partial pressure of arterial CO2 remain within normal limits. This concept is based mostly on observations in rat CKD models and lower urinary citrate excretion in patients with CKD [16, 17]. Unfortunately, no methods exist that allow measurement of local tissue pH with sufficient spatial and chemical resolution in vivo in humans to corroborate this model. An immediate consequence of this model would be the need to treat acidosis with alkali equivalents at early stages of kidney disease and before the occurrence of overt systemic acidosis to delay further progression of kidney disease. The implementation of a more holistic assessment of the metabolic acidosis of CKD may need to take into consideration multiple parameters, such as urinary citrate and ammonium, blood pH, serum HCO3− (or total CO2) and, in the future, other new early diagnostic markers or the direct measurement of tissue pH [18].
Multiple processes, conditions and diseases can lead to acidosis, but this review focuses on metabolic acidosis in the setting of CKD. Chronic metabolic acidosis is a condition in which the daily acid load exceeds the capacity of (remnant) kidneys to excrete H+ and regenerate HCO3− consumed by metabolism. In CKD, this is typically caused by the diminished capacity of kidneys to excrete acids and generate new HCO3− rather than an augmented daily acid load, although the latter can aggravate pre-existing acidosis, for example, in diabetic nephropathy with ketoacidosis. The main process that causes acidosis in CKD is the loss of ammoniagenesis, which acts as the main adaptive mechanism to excrete acid in the form of ammonium and to regenerate HCO3− [9, 19].
Cognitive dysfunction, motor dysfunction and acidosis in patients with CKD
Overt metabolic acidosis is a hallmark of advanced stages of CKD, while a higher risk of developing mild cognitive impairment (MCI) has been observed already in the early stages of CKD [3]. Only a few studies have examined the association between acidosis or serum/plasma HCO3− and brain functions. These will be discussed next.
Dobre et al. [2] examined memory and cognition in the cross-sectional SPRINT-MIND (Systolic Blood Pressure Intervention Trial Memory and Cognition in Decreased Hypertension) cohort, including 2853 hypertensive non-diabetic participants. The mean age of participants was 68 years, the mean estimated glomerular filtration rate (eGFR) was 71 mL/min/1.73 m2 and 30% had CKD. About 20% of participants had a HCO3− level <24 mmol/L. In this cohort, a 1 mmol/L lower HCO3− level associated with poorer performance in various tests on global cognitive or executive functions. This association persisted even after correction for eGFR and albuminuria. However, the positive association was attenuated when corrected for structural brain abnormalities typical for CKD detected by brain magnetic resonance imaging in a representative subset of patients. There was some specificity, as HCO3− did not associate with tests of memory, attention or language. Of note, the cognitive domains associated with low HCO3− in the SPRINT-MIND cohort are distinct from those found in patients with CKD, suggesting that distinct mechanisms may be responsible.
In a subset of participants in the Health, Aging and Body Composition (Health ABC) Study, a longitudinal study in older individuals (70–79 years), the association between serum HCO3− level and functional limitation was examined [4]. Functional limitation was scored based on the ability to walk a short distance and to climb stairs on two consecutive occasions 6 months apart with a follow-up of from 3 to 6 years. A total of 1544 participants were analysed with ~10% having HCO3− levels <23 mmol/L, while nearly 35% had levels >26 mmol/L. CKD was most prevalent in the low-HCO3− group. Participants with low HCO3− also had a lower blood pH and lower partial pressure of CO2, demonstrating that low HCO3− levels were unlikely to be due to respiratory problems. Those with the lowest HCO3− levels had the highest risk of developing functional limitations during the follow-up and this association persisted after multiple adjustments. Of note, both CKD and low HCO3− were independent risk factors for functional limitation. Some participants developed severe functional limitations, and this was associated with both a low HCO3− and a low blood pH, while blood pH showed no effect for milder functional limitations. Similar findings on the association of low HCO3− with lower gait speed and quadriceps strength had been reported previously in 2675 participants of the National Health and Nutrition Examination Survey 1999–2002 [20]. About 23% of participants had bicarbonate levels <23 mmol/L and were more likely to have CKD or diabetes. The association also persisted after multiple adjustments. However, this study was based on a single HCO3− measurement and the outcome measures probably reflected muscle function rather than central coordination.
Afsar and Elsurer [21] examined a small cohort of 65 patients on haemodialysis, measuring standard biochemical and clinical parameters as well as indicators of cognitive function, depression and sleep quality using well-established tests. In this cohort, lower venous blood HCO3− was associated with lower sleep quality, while no significant association was detected for cognitive functions and depression. However, this study lacked a control group, and as most of these patients had HCO3− levels <22 mmol/L, stratification could not be performed. The authors speculated that poor sleep quality was caused mainly by sleep apnoea. Another cross-sectional study with 190 CKD patients and 100 healthy patients in Nigeria found a negative association between serum HCO3− level and global cognitive impairment [22]. We identified only one study conducted in children with CKD that looked at the association between HCO3− and executive functions [23]. Acidosis was defined in this cohort as a serum HCO3− ≤20 mmol/L at baseline. Blood pressure variability and several cognitive tests as well as parental assessments of childrens’ cognitive function were analysed in children >6 years of age and with a median follow-up of 11.6 years. Most children were in CKD Stages G2–3. About 20% of children had HCO3− levels <20 mmol/L and this was associated with higher blood pressure, lower eGFR and higher proteinuria. However, neither HCO3− nor blood pressure was independently associated with executive functions. An interaction was found between blood pressure variability and HCO3− levels with executive functions, which showed that low HCO3− together with high blood pressure variability was associated with a worse score for executive functions. Thus one potential interpretation of this association is that HCO3− modifies the well-known effect of high blood pressure on the risk for reduced executive functions: as discussed below, pH alters the vascular reactivity of brain vessels and may modify this relationship.
Interventional studies using alkalinizing therapies in patients with CKD and acidosis have not examined their impact on cognitive function. One study reported the effect of oral sodium HCO3− supplements in a small study with 20 patients with an eGFR between 15 and 25 mL/min/1.73 m2 and serum HCO3− in the range of 20–24 mmol/L for 6 weeks. Alkali therapy improved the sit-to-stand time, while not changing hand grip strength [24]. While the sit-to-stand time might also include some aspects of central coordination, it may also be explained by increasing lower limb muscle strength. Likewise, de Brito-Ashurst et al. [25] reported that mid-arm muscle circumference increased in patients with CKD and acidosis when given alkali therapy. However, this improvement was linked to an overall improvement in nutritional status and again is unlikely to reflect motor control.
Metabolic acidosis or CKD as a cause of cognitive dysfunction?
The composition of the extracellular compartment is altered in patients with CKD, with imbalances in electrolytes and minerals, accumulation of uraemic toxins, volume expansion and accumulation of acids. Some of these disturbances are ameliorated in patients receiving peritoneal dialysis or haemodialysis, which also improves cognitive dysfunction and mood alterations. Other problems may be introduced by these treatment modalities, such as a reduction in brain blood flow or rapid alterations in acid–base parameters in haemodialysis. Unfortunately the association between acidosis or its amelioration by renal replacement therapy and cognitive dysfunction has not received much attention. While the associations between both cognitive dysfunctions and CKD as well as cognitive dysfunction and metabolic acidosis in CKD have become more apparent in recent years, it remains unclear how much can be directly attributed to metabolic acidosis. This is inherent to epidemiological associations, which usually do not establish causal links and direction of any dependencies. Moreover, there are other conditions with acidosis in which cognitive dysfunction and motor deficits are less common; in patients with tubulopathies such as inborn or acquired forms of distal renal tubular acidosis, cognitive and motor deficits are not part of the normal disease spectrum unless severe hypokalaemia develops, which can lead to muscle paralysis [26, 27]. Likewise, proximal renal tubular acidosis due to mutations in the SLC4A4 transporter does not cause cognitive dysfunction [28]. In rare cases of proximal tubular acidosis (type II RTA), intellectual disabilities may occur but are explained in part by the direct role of these genes in the CNS in addition to their role in regulating systemic acid–base homeostasis. Indeed, many genes that are expressed in the kidney are also expressed in the brain, often contributing to transport of ions as well as concentration control of electrolytes [29–31]. Examples include mutations in carbonic anhydrase II, which is expressed in kidney and in various CNS structures, as its mutations can lead to local calcifications in the brain [32], and the OCRL gene that leads to Lowe syndrome [33]. On the other hand, in patients with intact kidney function and chronic hypercapnia or patients with sleep apnoea and intermittent episodes of hypercapnia hypoxaemia, reduced vascular reactivity to CO2/pH and cognitive dysfunction have been reported. Clearly, better-powered and detailed clinical association studies are required to further dissect the possible contribution of acidosis as a risk factor for the development of cognitive dysfunction. Furthermore, intervention studies with correction of acid–base status could also provide evidence for a causal link between acidosis and cognitive dysfunction. Ideally these studies would be embedded into some of the larger trials aiming to reduce loss of kidney function with alkalizing therapies.
Mechanisms by which acidosis may impact on kidney disease progression
Acidosis has been shown to accelerate progression of CKD by multiple mechanisms. However, the relative contribution of each mechanism to reduced kidney function is unknown. We will briefly discuss these mechanisms here and examine later whether they might also be relevant for the brain:
Accumulation of ammonium in the kidney tissue leading to activation of the alternative complement pathway. This causes local inflammation, fibrosis and ultimately reduced kidney function [34]. Additionally, lower urine pH may lead to activation of the alternative complement pathway [35].
Renal tissue H+ retention has been proposed to stimulate the (local) production of hormones like endothelin, angiotensin II and aldosterone, which in turn cause renal inflammation and fibrosis [9, 36, 37].
α-Klotho is a protein required for FGF23 signalling that also has anti-inflammatory and renoprotective effects. Its levels fall early with a decrease in GFR and alkali therapy protects renal α-klotho levels in CKD patients [38].
Extrarenal inflammation. The effects of extracellular acidosis on immune cells have been covered by multiple studies [39], but except for indirect associations, a pH-dependent modulation of the immune response in CKD has not been shown. Oral HCO3− supplementation given to a hypertensive kidney disease rat model activated polarization of macrophages to the anti-inflammatory M2 type, suggesting that acidosis might regulate splenic immune responses in CKD [40].
Can acidosis contribute to cognitive dysfunction in CKD?
The pH-dependent mechanisms that may contribute to progression of renal disease should also be considered as possible modifiers of brain function. This poses three questions: Can acidosis of CKD contribute to cognitive dysfunction? Can the same or similar mechanisms contribute to kidney disease and brain dysfunction? Which other pH-dependent mechanisms may cause cognitive dysfunction?
The relationship between systemic pH and HCO3− levels and brain tissue and cerebrospinal fluid (CSF) pH is only partially understood. In 1969, repeated measurements in four humans showed that 5 days of acid or alkali load produced much narrower changes in the CSF pH compared with arterial pH [41]. Similar findings were obtained from patients with CKD compared with normal individuals [42]. Multiple studies have demonstrated that pH and HCO3− are lower in the CSF than in plasma in normal conditions and reductions in both parameters are highly attenuated in the CSF during metabolic acidosis [43]. In steady-state conditions, pH and HCO3− are expected to be the same in the CSF and brain extracellular fluid (ECF). However, in non-steady-state conditions, the values of these parameters tend to dissociate between these compartments [44]. Acidaemia causes smaller, but significant, changes in the same direction in brain ECF, even when CSF pH is unaltered [45, 46]. However, very small changes in the ECF pH during exercise, physical work and acute or chronic metabolic acidosis are capable of altering ventilatory responses and cerebral blood flow (CBF) [43]. Therefore, chronic low-grade metabolic acidosis may alter brain acid–base status and blood flow.
Even though chronic acidaemia may well translate into a more acidic environment in the brain, it is currently unclear to what extent eubicarbonataemic metabolic acidosis can affect the brain acid–base balance. Given that the acid–base changes in brain ECF are smaller than in arterial pH in individuals during overt chronic metabolic acidosis, one would need to account for very small changes (if any) in brain local acid–base status in cases of subclinical acidosis. Moreover, arguments that negative effects of subclinical acidosis on kidneys occur because of augmented ammoniagenesis cannot be applied to the brain. Also, while ammonia toxicity in the brain is well described [46], CKD is not a state of higher blood ammonia levels and therefore an accumulation of ammonium in the brain is unlikely [47]. Thus the accumulation of H+ and/or in brain tissue in eubicarbonataemic metabolic acidosis is unlikely to cause cognitive dysfunction. Recently, astrocytes have been shown to secrete HCO3− in response to purinergic signalling and consequently protect extracellular pH of the brain in response to a higher metabolic demand [48]. The in-tandem organization of the blood–brain barrier and astrocytes generates a ‘buffering wall’ in the brain, attenuating changes in extracellular pH and HCO3−. This involves a set of HCO3− importing or exporting transporters located in various brain cell types that, when absent in rodents, can affect diverse brain functions [49]. While hypoxia and hypercapnia impact the expression of several of these transporters [50, 51], the effect of CKD or metabolic acidosis has not been examined.
Another family of proteins involved in local and systemic control of pH homeostasis is carbonic anhydrase, which catalyses the hydration of CO2 to form carbonic acid (H2CO3) and subsequently H+ and HCO3−. At least nine isoforms of carbonic anhydrase are present in the human brain in different areas and cell types and their importance is highlighted by mutations in carbonic anhydrase II or IV, which causes intellectual disabilities or blindness. Pharmacological inhibition of carbonic anhydrase in young rats lowered intracellular pH in the cerebral cortex and cerebellum and increased CSF HCO3− levels without altering CSF pH [52]. In animal models, genetic ablation of some carbonic anhydrase isoforms or inhibition with acetazolamide reduced carbonic anhydrase activity in the brain and caused amnesia in object recognition tests [53, 54]. At least in animal models, carbonic anhydrase activators enhance memory formation [53]. Fear conditions and consolidation were also affected. In humans, carbonic anhydrase inhibitors such as topiramate are used to treat migraine and prevent epilepsy but impair processes involved in memory formation. Also, acetazolamide given for prevention of high-altitude sickness has similar effects on memory. In both cases, particularly emotional memory is affected. While the exact mechanisms by which carbonic anhydrases act on memory are mostly elusive, some evidence suggests that changes in local HCO3− concentrations affect neurotransmitter fluxes and excitability of GABAergic neurons [53], which in turn can affect motor learning [55].
α-Klotho is a protein required for FGF23 signalling and is mostly expressed in the kidney, parathyroid glands and choroid plexus. It exists as a membrane-bound form and after cleavage as soluble α-klotho circulating in the blood. The kidney is the main source of soluble α-klotho and the expression of α-klotho in the kidney decreases rapidly in the course of acute or chronic kidney disease [56]. In the brain, α-klotho deficiency impacts immune functions and complete absence of α-klotho is associated with Parkinson-like motor deficits that may be attributed in part to highly elevated calcitriol levels [57]. α-Klotho may also be protective of cognitive function [58, 59]. While calcitriol is typically rather low in patients with CKD, the local expression and concentration of α-klotho in the brain has not been characterized. Alkali therapy, by protecting kidney function, could indirectly influence brain function via klotho. A pilot study with CKD patients showed that HCO3− supplementation restored urinary but not serum α-klotho levels [38]. Additional studies are necessary to examine the role of α-klotho in the brain, its role in CKD and its sensitivity to systemic and/or local changes in acid–base status.
Metabolic acidosis of CKD could also affect the brain indirectly by altering the release of hormones into the bloodstream. Circulating renin–angiotensin–aldosterone system (RAAS) components have low permeability across the blood–brain barrier and are unlikely to be causes of cognitive dysfunction [60]. However, high levels of circulating endothelin-1 disturb the integrity of the blood–brain barrier, cause brain microvascular dysfunction and impair cognitive function [61, 62]. While blood endothelin levels are associated with both acidosis and CKD, and alkali therapy reduced plasma and urinary endothelin-1 levels in CKD patients [37], a recent clinical trial did not observe any reduction in urinary endothelin levels in acidotic CKD patients receiving alkali [63].
Although acidosis has been proposed as a direct immunomodulatory factor [39, 64], the effects of chronic metabolic acidosis on systemic inflammation are controversial and still poorly understood [65]. Short-term exposure to HCO3− in drinking water reduced the abundance of neutrophils and shifted macrophages towards an M2 phenotype in human blood [40]. If chronic metabolic acidosis can affect systemic inflammation, it could also affect brain functions in CKD patients. Indeed, inflammation per se can disrupt the blood–brain barrier, making it potentially more permeable to neurotoxic substances and metabolites, including uraemic toxins [66–68]. Also, inflammation increases the risk of vascular damage in the brain that again may alter cognitive function [69]. Pro-inflammatory cytokines such as tumour necrosis factor and interleukin-1β (IL-1β) can directly act on the brain or mediate effects via afferent nerves [70]. Locally, pH could also affect inflammation. In animals, chronic hypercapnia induces an inflammatory response in the midbrain, brainstem and cerebrum followed by changes in glutamatergic, serotonergic and catecholaminergic neurotransmission [71, 72]. Whether this response is triggered by local acidosis or other mediators is unclear, but it seems to involve IL-1β signalling, which is often enhanced in patients with CKD. As discussed above, α-klotho is highly expressed in the choroid plexus and if its expression at this site follows the same pattern of decline as in the kidney and parathyroid glands of patients and animal models of CKD, the reduced levels of α-klotho may increase brain inflammation [73]. The absence of brain α-klotho in a murine model stimulated the expression of pro-inflammatory cytokines and macrophage invasion into the brain. Also, microglial cells were activated. α-Klotho appears to act in part via suppression of the NLRP3 inflammasome in macrophages. Strikingly, α-klotho levels in the brain decline with age and similarities in cognitive dysfunction in patients with CKD and elderly patients with age-related decline in cognitive functions have been noted. Whether lower α-klotho levels in the brain are a common pathway in these different clinical entities remains to be explored.
Additionally, brain metabolism and CBF may be pH sensitive and contribute to cognitive dysfunction in patients with CKD. Brain metabolism is controlled by multiple factors including blood flow and pH-dependent mechanisms [48, 74]. Cerebral glutamine uptake was reduced in normal human subjects with ammonium chloride loading for 3 or 6 days and in CKD patients with severe acidosis [75]. Glutamine transport in the brain might be affected by the activity of HCO3− transporters. The astrocytic–neuronal lactate shuttle model proposes that astrocytes respond to a higher metabolic demand by stimulating glycolysis and exporting lactate to the extracellular space, which can fuel the energy metabolism of neurons [76]. Interestingly, a recent study demonstrated that astrocytes secrete HCO3− in concert with local neuronal energy demand [48]. As for glutamine, lactate transporters seem to be affected by HCO3− transport activity as well as by other H+-dependent mechanisms [77].
Acid–base status also directly impacts synaptic activity, which is the largest sink of energy equivalents in the brain [78]. As a rule of thumb, acidosis tends to reduce neuronal excitability and alkalosis to increase it [79]. While acidic pH is associated with lower synaptic activity, H+ has also been implicated in excitotoxicity [80]. Exposure of murine brain slices to an acidic perfusate overexcites murine pyramidal neurons and astrocytes, both impairing activity of GABAergic neurons [29–31, 81, 82]. The impaired activity of GABAergic neurons has been documented in experimental CKD models [68]. Therefore, given the close relationship between brain metabolism and acid–base status, it is tempting to speculate that a chronic low-grade metabolic acidosis might affect brain metabolism by regulating the transport of key substrates for energy metabolism and synaptic activity, besides other potential indirect effects via modulation of CBF and glycaemic control [83].
Both high and low CBF can affect brain function [84]. While decreased kidney function is associated with reduced CBF [85], other concurrent and common conditions in CKD, such as anaemia, can cause brain hyperperfusion [86]. Acid–base status also alters CBF, with metabolic and respiratory acidosis increasing CBF and alkalosis having the opposite effect [41, 87, 88]. However, in a CKD mouse model, the CBF induced by hypercapnia was initially increased but eventually attenuated, suggesting altered vascular reactivity and possible secondary consequences on brain metabolism [89]. Therefore it could be speculated that acidosis may harm the brain by elevating CBF and causing excitotoxicity. In the SPRINT study, serum HCO3− levels were associated with cognitive and executive performance, but not with CBF [2]. In another study with 2645 participants, high CBF in CKD patients was associated with a lower prevalence of stroke and dementia when compared with low-CBF patients [85]. Therefore, while theoretical aspects support that an elevation of CBF in response to chronic metabolic acidosis might drive cognitive dysfunction, high-quality data are still missing.
Chronic metabolic acidosis also results in renal magnesium wasting and hypomagnesaemia is often one of the features of acidosis. Even though there are few data linking magnesium and CKD progression, in the Atherosclerosis Risk in Communities study, higher dietary magnesium was associated with a lower risk of CKD [90]. Acidosis-associated hypomagnesaemia might contribute to cognitive dysfunction. Magnesium regulates neuronal transmission, protein synthesis and energy metabolism and magnesium deficiency mostly affects nervous and cardiovascular systems, resulting in weakness, tremors and even seizures. Magnesium is also implicated in memory function and neuronal plasticity, acting as an allosteric modulator of N-methyl-D-aspartate receptors involved in long-term potentiation. Another possible mechanism could be the association of low magnesium and neuroinflammation [91]. In a mouse model, a low-magnesium diet resulted in brain neuroinflammation, affecting the hippocampus and cortex, and impaired memory formation. Low magnesium is also associated with vascular damage that might affect CBF [92]. Last, low magnesium levels accelerate the loss of renal α-klotho expression [93].
SUMMARY AND OUTLOOK
A number of epidemiological studies demonstrate an association between low HCO3− and mild cognitive and motor impairment in the general population and identify an even more accentuated association in patients with CKD. Mechanistic studies demonstrating a causal link between acid–base status and brain function are not available yet and possible mechanisms can only be inferred (see hypothetical model in Figure 2). Thus a first step is to examine whether low HCO3− and/or acidosis are a cause of or only coexist with disturbed CNS function. A second step consists in examining which mechanisms modulated by low HCO3−/acidosis can affect CNS functions. This may allow identification of biomarkers to predict or monitor alterations of CNS function as well as to find pathways or molecular targets for treatment. A third step is to design clinical trials that test whether amelioration of acidosis is not only beneficial in preserving residual kidney function, but also positively impacts the loss of central brain functions or has the ability to reverse these changes. The growing recognition that kidney disease affects central and peripheral neuronal function is a critical step towards better preservation of brain organ functions in patients with reduced kidney function. A better understanding of the factors linking kidney disease and impaired brain function is necessary and acidosis/low HCO3− needs to be considered among other factors such as uraemic toxins, vascular changes or alterations in neurotransmitters and brain metabolism.
FIGURE 2.

Model summarizing possible links between CKD, metabolic acidosis and cognitive dysfunction and motor deficits. Reduced kidney function causes, among other consequences, accumulation of uraemic toxins and metabolic acidosis, and both may act on the brain to reduce central functions. In addition, immune functions are altered by both factors, leading to higher levels of pro-inflammatory factors that may also affect the brain. Reduced kidney function is also associated with reduced renal expression of α-klotho and reduced circulating levels of soluble α-klotho. Whether brain α-klotho expression is directly affected has not been examined. α-Klotho has neuroprotective functions.
ACKNOWLEDGEMENTS
The authors of this review have been supported by the HORIZON EU COST action CA19127-CONNECT (Cognitive Decline in Nephro-Neurology). C.I.D.Z. is supported by ERC advanced, ZonMw, NWO-ALW, Medical NeuroDelta, INTENSE (LSH-NWO), Vriendenfonds Albinisme and BIG EMC.
APPENDIX
CONNECT collaborators are
Giovambattista Capasso; Alexandre Andrade; Maie Bachmann; Inga Bumblyte; Adrian Constantin Covic; Pilar Delgado; Nicole Endlich; Andreas Engvig; Denis Fouque; Casper Franssen; Sebastian Frische; Liliana Garneata; Loreto Gesualdo; Konstantinos Giannakou; Dimitrios Goumenos; Ayşe Tugba Kartal; Laila-Yasmin Mani; Hans-Peter Marti; Christopher Mayer; Rikke Nielsen; Vesna Pešić; Merita Rroji (Molla); Giorgos Sakkas; Goce Spasovski; Kate I. Stevens; Evgueniy Vazelov; Davide Viggiano; Lefteris Zacharia; Ana Carina Ferreira; Jolanta Malyszko; Ewout Hoorn; Andreja Figurek; Robert Unwin; Carsten A. Wagner; Christoph Wanner; Annette Bruchfeld; Marion Pepin; Andrzej Więcek; Dorothea Nitsch; Ivo Fridolin; Gaye Hafez; Maria José Soler; Michelangela Barbieri; Bojan Batinić; Laura Carrasco; Sol Carriazo; Ron Gansevoort; Gianvito Martino; Francesco Mattace Raso; Ionut Nistor; Alberto Ortiz; Giuseppe Paolisso; Daiva Rastenytė; Gabriel Stefan; Gioacchino Tedeschi; Ziad A. Massy; Boris Bikbov; Karl Hans Endlich; Olivier Godefroy; Jean-Marc Chillon; Anastassia Kossioni; Justina Kurganaite; Norberto Perico; Giuseppe Remuzzi; Tomasz Grodzicki; Francesco Trepiccione; Carmine Zoccali; Mustafa Arici; Peter Blankestijn; Kai-Uwe Eckardt; Danilo Fliser; Eugenio Gutiérrez Jiménez; Maximilian König; Ivan Rychlik; Michela Deleidi; George Reusz.
Contributor Information
Pedro H Imenez Silva, Institute of Physiology, University of Zurich, Zürich, Switzerland; National Center of Competence in Research NCCR Kidney.CH, Zürich, Switzerland.
Robert Unwin, Department of Renal Medicine, Royal Free Hospital, University College London, London, UK.
Ewout J Hoorn, Department of Internal Medicine, Erasmus Medical Center, University Medical Center Rotterdam, Rotterdam, The Netherlands.
Alberto Ortiz, Department of Nephrology and Hypertension, IIS-Fundacion Jimenez Diaz, Universidad Autonoma de Madrid, Madrid, Spain.
Francesco Trepiccione, Biogem Institute of Molecular Biology and Genetics, Ariano Irpino, Italy; Department of Translational Medical Sciences, University of Campania “Luigi Vanvitelli”, Naples, Italy.
Rikke Nielsen, Department of Biomedicine–Anatomy, University of Aarhus, Aarhus, Denmark.
Vesna Pesic, Department of Physiology, Faculty of Pharmacy, University of Belgrade, Belgrade, Serbia.
Gaye Hafez, Department of Pharmacology, Faculty of Pharmacy, Altinbas University, Istanbul, Turkey.
Denis Fouque, CarMeN, INSERM 1060, Université Claude Bernard Lyon 1, Lyon, France; Service de Néphrologie, Lyon-Sud Hospital, Pierre-Bénite, France.
Ziad A Massy, Department of Nephrology, Ambroise Paré University Hospital, Assistance Publique Hôpitaux de Paris, Boulogne-Billancourt, France; Centre de Recherche en Epidémiologie et Santé des Populations, Institut National de la Santé et de la Recherche Médicale U1018-Team 5, Université de Versailles Saint-Quentin-en-Yvelines, University Paris Saclay, Villejuif, France.
Chris I De Zeeuw, Department of Neuroscience, Erasmus Medical Center, University Medical Center Rotterdam, Rotterdam, The Netherlands; Netherlands Institute for Neuroscience, Royal Dutch Academy of Art and Science, Amsterdam, The Netherlands.
Giovambattista Capasso, Biogem Institute of Molecular Biology and Genetics, Ariano Irpino, Italy; Department of Translational Medical Sciences, University of Campania “Luigi Vanvitelli”, Naples, Italy.
Carsten A Wagner, Institute of Physiology, University of Zurich, Zürich, Switzerland; National Center of Competence in Research NCCR Kidney.CH, Zürich, Switzerland.
CONNECT Action (Cognitive Decline in Nephro-Neurology European Cooperative Target):
Giovambattista Capasso, Alexandre Andrade, Maie Bachmann, Inga Bumblyte, Adrian Constantin Covic, Pilar Delgado, Nicole Endlich, Andreas Engvig, Denis Fouque, Casper Franssen, Sebastian Frische, Liliana Garneata, Loreto Gesualdo, Konstantinos Giannakou, Dimitrios Goumenos, Ayşe Tuğba Kartal, Laila-Yasmin Mani, Hans-Peter Marti, Christopher Mayer, Rikke Nielsen, Vesna Pešić, Merita Rroji, Giorgos Sakkas, Goce Spasovski, Kate I Stevens, Evgueniy Vazelov, Davide Viggiano, Lefteris Zacharia, Ana Carina Ferreira, Jolanta Malyszko, Ewout Hoorn, Andreja Figurek, Robert Unwin, Carsten Wagner, Christoph Wanner, Annette Bruchfeld, Marion Pepin, Andrzej Wiecek, Dorothea Nitsch, Ivo Fridolin, Gaye Hafez, Maria José Soler Romeo, Michelangela Barbieri, Bojan Batinić, Laura Carrasco, Sol Carriazo, Ron Gansevoort, Gianvito Martino, Francesco Mattace Raso, Ionut Nistor, Alberto Ortiz, Giuseppe Paolisso, Daiva Rastenytė, Gabriel Stefan, Gioacchino Tedeschi, Ziad Massy, Boris Bikbov, Karl Hans Endlich, Olivier Godefroy, Jean-Marc Chillon, Anastassia Kossioni, Justina Kurganaite, Norberto Perico, Giuseppe Remuzzi, Tomasz Grodzicki, Francesco Trepiccione, Carmine Zoccali, Mustafa Arici, Peter Blankestijn, Kai-Uwe Eckardt, Danilo Fliser, Eugenio Gutiérrez Jiménez, Maximilian Konig, Ivan Rychlik, Michela Deleidi, and George Reusz
FUNDING
This article is published as part of a supplement financially supported by the COST Action CA19127-Cognitive Decline in Nephro-Neurology: European Cooperative Target (CONNECT). P.H.I.S. and C.A.W. have been supported by the Swiss National Science Foundation–financed NCCR Kidney. CH. A.O. was supported by the FIS/Fondos FEDER [PI18/01366, PI19/00588, PI19/00815, DTS18/00032, ERA-PerMed-JTC2018 (KIDNEY ATTACK AC18/00064 and PERSTIGAN AC18/00071, ISCIII-RETIC REDinREN RD016/0009)], Sociedad Española de Nefrología, FRIAT and Comunidad de Madrid en Biomedicina B2017/BMD-3686 CIFRA2-CM.
AUTHORS’ CONTRIBUTIONS
P.H.I.S. and C.A.W. drafted the review. All co-authors read and commented on the review. All authors approved the manuscript.
CONFLICT OF INTEREST STATEMENT
C.A.W. has received honoraria or grant support from Medice, Salmon Pharma, Ardelyx, Chugai and Bayer. A.O. has received consultancy or speaker fees or travel support from Astellas, AstraZeneca, Amicus, Amgen, Fresenius Medical Care, Bayer, Sanofi-Genzyme, Menarini, Kyowa Kirin, Alexion, Otsuka and Vifor Fresenius Medical Care Renal Pharma and is the Director of the Catedra Mundipharma-UAM of diabetic kidney disease and the Catedra AstraZeneca-UAM of CKD and electrolytes. R.U. is currently employed by AstraZeneca BioPharmaceuticals R&D, Early CVRM (Cardiovascular, Renal and Metabolism), Cambridge UK and Gothenburg, Sweden.
REFERENCES
- 1. Moranne O, Froissart M, Rossert J. et al. Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol 2009; 20: 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dobre M, Gaussoin SA, Bates JT. et al. Serum bicarbonate concentration and cognitive function in hypertensive adults. Clin J Am Soc Nephrol 2018; 13: 596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Viggiano D, Wagner CA, Martino G. et al. Mechanisms of cognitive dysfunction in CKD. Nat Rev Nephrol 2020; 16: 452–469 [DOI] [PubMed] [Google Scholar]
- 4. Yenchek R, Ix JH, Rifkin DE. et al. Association of serum bicarbonate with incident functional limitation in older adults. Clin J Am Soc Nephrol 2014; 9: 2111–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Viggiano D, Wagner CA, Blankestijn PJ. et al. Mild cognitive impairment and kidney disease: clinical aspects. Nephrol Dial Transplant 2020; 35: 10–17 [DOI] [PubMed] [Google Scholar]
- 6. Pépin M, Ferreira AC, Arici M. et al. Cognitive disorders in patients with chronic kidney disease: specificities of clinical assessment. Nephrol Dial Transplant [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skiba K, Gojowy D, Szotowska M. et al. Metabolic acidosis in kidney transplant recipients. Pol Arch Intern Med 2018; 128: 587–593 [DOI] [PubMed] [Google Scholar]
- 8. Kim HJ, Kang E, Ryu H. et al. Metabolic Acidosis Is Associated with Pulse Wave Velocity in Chronic Kidney Disease: Results from the KNOW-CKD Study. Scientific Rep 2019; 9: 16139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wesson DE. The continuum of acid stress. Clin J Am Soc Nephrol 2021; doi: 10.2215/CJN.17541120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Madias NE. Metabolic acidosis and CKD progression. Clin J Am Soc Nephrol 2021; 16: 310–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Melamed ML, Raphael KL.. Metabolic acidosis in CKD: a review of recent findings. Kidney Med 2021; 3: 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kajimoto S, Sakaguchi Y, Asahina Y. et al. Modulation of the association of hypobicarbonatemia and incident kidney failure with replacement therapy by venous pH: a cohort study. Am J Kidney Dis 2021; 77: 35–43 [DOI] [PubMed] [Google Scholar]
- 13. Raphael KL, Murphy RA, Shlipak MG. et al. Bicarbonate concentration, acid–base status, and mortality in the health, aging, and body composition study. Clin J Am Soc Nephrol 2016; 11: 308–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Navaneethan SD, Schold JD, Arrigain S. et al. Serum bicarbonate and mortality in stage 3 and stage 4 chronic kidney disease. Clin J Am Soc Nephrol 2011; 6: 2395–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ritter A, Mohebbi N.. Causes and consequences of metabolic acidosis in patients after kidney transplantation. Kidney Blood Press Res 2020; 45: 792–801 [DOI] [PubMed] [Google Scholar]
- 16. Goraya N, Simoni J, Sager LN. et al. Urine citrate excretion as a marker of acid retention in patients with chronic kidney disease without overt metabolic acidosis. Kidney Int 2019; 95: 1190–1196 [DOI] [PubMed] [Google Scholar]
- 17. Gianella FG, Prado VE, Poindexter JR. et al. Spot urinary citrate-to-creatinine ratio is a marker for acid–base status in chronic kidney disease. Kidney Int 2021; 99: 208–217 [DOI] [PubMed] [Google Scholar]
- 18. Raphael KL, Kraut JA.. Assessing acid–base status in patients with CKD: does measurement of blood pH matter? Am J Kidney Dis 2021; 77: 9–11 [DOI] [PubMed] [Google Scholar]
- 19. Bürki R, Mohebbi N, Bettoni C. et al. Impaired expression of key molecules of ammoniagenesis underlies renal acidosis in a rat model of chronic kidney disease. Nephrol Dial Transplant 2015; 30: 770–781 [DOI] [PubMed] [Google Scholar]
- 20. Abramowitz MK, Hostetter TH, Melamed ML.. Association of serum bicarbonate levels with gait speed and quadriceps strength in older adults. Am J Kidney Dis 2011; 58: 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Afsar B, Elsurer R.. Association between serum bicarbonate and pH with depression, cognition and sleep quality in hemodialysis patients. Ren Fail 2015; 37: 957–960 [DOI] [PubMed] [Google Scholar]
- 22. Egbi OG, Ogunrin O, Oviasu E.. Prevalence and determinants of cognitive impairment in patients with chronic kidney disease: a cross-sectional study in Benin City, Nigeria. Ann Afr Med 2015; 14: 75–81 [DOI] [PubMed] [Google Scholar]
- 23. Harshman LA, Kogon AJ, Matheson MB. et al. Bicarbonate, blood pressure, and executive function in pediatric CKD—is there a link? Pediatr Nephrol 2020; 35: 1323–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abramowitz MK, Melamed ML, Bauer C. et al. Effects of oral sodium bicarbonate in patients with CKD. Clin J Am Soc Nephrol 2013; 8: 714–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Brito-Ashurst I, Varagunam M, Raftery MJ. et al. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009; 20: 2075–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopez-Garcia SC, Emma F, Walsh SB. et al. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant 2019; 34: 981–991 [DOI] [PubMed] [Google Scholar]
- 27. Viggiano D, Bruchfeld A, Carriazo S. et al. Brain dysfunction and tubulo-interstitial kidney diseases. Nephrol Dial Transplant [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Igarashi T, Inatomi J, Sekine T. et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet 1999; 23: 264–266 [DOI] [PubMed] [Google Scholar]
- 29. Rahmati N, Kunzelmann K, Xu J. et al. Slc26a11 is prominently expressed in the brain and functions as a chloride channel: expression in Purkinje cells and stimulation of V H+-ATPase. Pflugers Arch 2013; 465: 1583–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rahmati N, Vinueza Veloz MF, Xu J. et al. SLC26A11 (KBAT) in Purkinje cells is critical for inhibitory transmission and contributes to locomotor coordination. eNeuro 2016; 3: ENEURO.0028-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rahmati N, Hoebeek FE, Peter S. et al. Chloride homeostasis in neurons with special emphasis on the olivocerebellar system: Differential roles for transporters and channels. Front Cell Neurosci 2018; 12: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bosley TM, Salih MA, Alorainy IA. et al. The neurology of carbonic anhydrase type II deficiency syndrome. Brain 2011; 134: 3502–3515 [DOI] [PubMed] [Google Scholar]
- 33. Loi M. Lowe syndrome. Orphanet J Rare Dis 2006; 1: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nath KA, Hostetter MK, Hostetter TH.. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest 1985; 76: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peake PW, Pussell BA, Mackinnon B. et al. The effect of pH and nucleophiles on complement activation by human proximal tubular epithelial cells. Nephrol Dial Transplant 2002; 17: 745–752 [DOI] [PubMed] [Google Scholar]
- 36. Khanna A, Simoni J, Hacker C. et al. Increased endothelin activity mediates augmented distal nephron acidification induced by dietary protein. J Am Soc Nephrol 2004; 15: 2266–2275 [DOI] [PubMed] [Google Scholar]
- 37. Wesson DE, Simoni J, Broglio K. et al. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Physiol Renal Physiol 2011; 300: F830–F837 [DOI] [PubMed] [Google Scholar]
- 38. Hage V, Villain C, Pelletier S. et al. Bicarbonate supplement restores urinary klotho excretion in chronic kidney disease: a pilot study. J Ren Nutr 2019; 29: 285–288 [DOI] [PubMed] [Google Scholar]
- 39. Erra Díaz F, Dantas E, Geffner J.. Unravelling the interplay between extracellular acidosis and immune cells. Mediators Inflamm 2018; 2018: 1218297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ray SC, Baban B, Tucker MA. et al. Oral NaHCO3 activates a splenic anti-inflammatory pathway: evidence that cholinergic signals are transmitted via mesothelial cells. J Immunol 2018; 200: 3568–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fencl V, Vale JR, Broch JA.. Respiration and cerebral blood flow in metabolic acidosis and alkalosis in humans. J Appl Physiol 1969; 27: 67–76 [DOI] [PubMed] [Google Scholar]
- 42. Mitchell RA, Carman CT, Severinghaus JW. et al. Stability of cerebrospinal fluid pH in chronic acid–base disturbances in blood. J Appl Physiol 1965; 20: 443–452 [DOI] [PubMed] [Google Scholar]
- 43. Siesjö BK. The regulation of cerebrospinal fluid pH. Kidney Int 1972; 1: 360–374 [DOI] [PubMed] [Google Scholar]
- 44. Teppema LJ, Barts PWJA, Evers JAM.. Effects of metabolic arterial pH changes on medullary ECF pH, CSF pH and ventilation in peripherally chemodenervated cats with intact blood–brain barrier. Respir Physiol 1984; 58: 123–136 [DOI] [PubMed] [Google Scholar]
- 45. Javaheri S, De Hemptinne A, Vanheel B. et al. Changes in brain ECF pH during metabolic acidosis and alkalosis: a microelectrode study. J Appl Physiol Respir Environ Exerc Physiol 1983; 55: 1849–1853 [DOI] [PubMed] [Google Scholar]
- 46. Oja SS, Saransaari P, Korpi ER.. Neurotoxicity of ammonia. Neurochem Res 2017; 42: 713–720 [DOI] [PubMed] [Google Scholar]
- 47. Vaziri ND, Khazaeli M, Nunes ACF. et al. Effects of end-stage renal disease and dialysis modalities on blood ammonia level. Hemodial Int 2017; 21: 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Theparambil SM, Hosford PS, Ruminot I. et al. Astrocytes regulate brain extracellular pH via a neuronal activity-dependent bicarbonate shuttle. Nat Commun 2020; 11: 5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Choi I, Beedholm K, Dam VS. et al. Sodium bicarbonate cotransporter NBCn1/Slc4a7 affects locomotor activity and hearing in mice. Behav Brain Res 2021; 401: 113065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen L-M, Choi I, Haddad GG. et al. Chronic continuous hypoxia decreases the expression of SLC4A7 (NBCn1) and SLC4A10 (NCBE) in mouse brain. Am J Physiol Regul Integr Comp Physiol 2007; 293: R2412–R2420 [DOI] [PubMed] [Google Scholar]
- 51. Kanaan A, Douglas RM, Alper SL. et al. Effect of chronic elevated carbon dioxide on the expression of acid–base transporters in the neonatal and adult mouse. Am J Physiol Regul Integr Comp Physiol 2007; 293: R1294–R1302 [DOI] [PubMed] [Google Scholar]
- 52. Johanson CE, Parandoosh Z, Dyas ML.. Maturational differences in acetazolamide-altered pH and HCO3 of choroid plexus, cerebrospinal fluid, and brain. Am J Physiol 1992; 262: R909–R914 [DOI] [PubMed] [Google Scholar]
- 53. Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018; 28: 713–721 [DOI] [PubMed] [Google Scholar]
- 54. Blandina P, Provensi G, Passsani MB. et al. Carbonic anhydrase modulation of emotional memory. Implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem 2020; 35: 1206–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wulff P, Schonewille M, Renzi M. et al. Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci 2009; 12: 1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Drew DA, Katz R, Kritchevsky S. et al. Association between soluble Klotho and change in kidney function: the health aging and body composition study. J Am Soc Nephrol 2017; 28: 1859–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kosakai A, Ito D, Nihei Y. et al. Degeneration of mesencephalic dopaminergic neurons in klotho mouse related to vitamin D exposure. Brain Res 2011; 1382: 109–117 [DOI] [PubMed] [Google Scholar]
- 58. Dubal DB, Yokoyama JS, Zhu L. et al. Life extension factor klotho enhances cognition. Cell Rep 2014; 7: 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shardell M, Semba RD, Rosano C. et al. Plasma klotho and cognitive decline in older adults: findings from the InCHIANTI study. J Gerontol A Biol Sci Med Sci 2016; 71: 677–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jackson L, Eldahshan W, Fagan SC. et al. Within the brain: the renin–angiotensin system. Int J Mol Sci 2018; 19:876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fouda Abdelrahman Y, Fagan Susan C, Ergul A.. Brain vasculature and cognition. Arterioscler Thromb Vasc Biol 2019; 39: 593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Faraco G, Moraga A, Moore J. et al. Circulating endothelin-1 alters critical mechanisms regulating cerebral microcirculation. Hypertension 2013; 62: 759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bovée DM, Roksnoer LCW, Kooten C V. et al. Effect of sodium bicarbonate supplementation on the renin–angiotensin system in patients with chronic kidney disease and acidosis: a randomized clinical trial. J Nephrol 2020; doi: 10.1007/s40620-020-00944-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal 2013; 25: 2263–2271 [DOI] [PubMed] [Google Scholar]
- 65. Kellum JA, Song M, Li J.. Science review: Extracellular acidosis and the immune response: clinical and physiologic implications. Crit Care 2004; 8: 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shah B, Jagtap P, Sarmah D. et al. Cerebro-renal interaction and stroke. Eur J Neurosci 2021; 53: 1279–1299 [DOI] [PubMed] [Google Scholar]
- 67. Bobot M, Thomas L, Moyon A. et al. Uremic toxic blood–brain barrier disruption mediated by AhR activation leads to cognitive impairment during experimental renal dysfunction. J Am Soc Nephrol 2020; 31: 1509–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liabeuf S, Pepin M, Franssen CF et al. Chronic kidney disease and neurological disorders: are uremic toxins the missing piece of the puzzle? Nephrol Dial Transplant. doi: 10.1093/ndt/gfab223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tanaka S, Okusa MD.. Crosstalk between the nervous system and the kidney. Kidney Int 2020; 97: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miranda AS, Cordeiro TM, Dos Santos Lacerda Soares TM. et al. Kidney–brain axis inflammatory cross-talk: from bench to bedside. Clin Sci (Lond) 2017; 131: 1093–1105 [DOI] [PubMed] [Google Scholar]
- 71. Burgraff NJ, Neumueller SE, Buchholz KJ. et al. Brainstem serotonergic, catecholaminergic, and inflammatory adaptations during chronic hypercapnia in goats. FASEB J 2019; 33: 14491–14505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Burgraff NJ, Neumueller SE, Buchholz KJ. et al. Midbrain and cerebral inflammatory and glutamatergic adaptations during chronic hypercapnia in goats. Brain Res 2019; 1724: 146437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhu L, Stein LR, Kim D. et al. Klotho controls the brain–immune system interface in the choroid plexus. Proc Natl Acad Sci USA 2018; 115: E11388–E11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Deitmer JW, Theparambil SM, Ruminot I. et al. Energy dynamics in the brain: contributions of astrocytes to metabolism and pH homeostasis. Front Neurosci 2019; 13: 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tizianello A, De Ferrari G, Garibotto G. et al. Renal metabolism of amino acids and ammonia in subjects with normal renal function and in patients with chronic renal insufficiency. J Clin Invest 1980; 65: 1162–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Magistretti PJ, Pellerin L, Rothman DL. et al. Energy on demand. Science 1999; 283: 496–497 [DOI] [PubMed] [Google Scholar]
- 77. Noor SI, Pouyssegur J, Deitmer JW. et al. Integration of a ‘proton antenna’ facilitates transport activity of the monocarboxylate transporter MCT4. FEBS J 2017; 284: 149–162 [DOI] [PubMed] [Google Scholar]
- 78. Harris JJ, Jolivet R, Attwell D.. Synaptic energy use and supply. Neuron 2012; 75: 762–777 [DOI] [PubMed] [Google Scholar]
- 79. Sinning A, Hübner CA.. Minireview: pH and synaptic transmission. FEBS Lett 2013; 587: 1923–1928 [DOI] [PubMed] [Google Scholar]
- 80. Choi DW. Excitotoxicity: still hammering the ischemic brain in 2020. Front Neurosci 2020; 14:579953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhao H, Cai Y, Yang Z. et al. Acidosis leads to neurological disorders through overexciting cortical pyramidal neurons. Biochem Biophys Res Commun 2011; 415: 224–228 [DOI] [PubMed] [Google Scholar]
- 82. Huang L, Zhao S, Lu W. et al. Acidosis-induced dysfunction of cortical GABAergic neurons through astrocyte-related excitotoxicity. PLoS One 2015; 10: e0140324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mannon EC, O’Connor PM.. Alkali supplementation as a therapeutic in chronic kidney disease: what mediates protection? Am J Physiol Renal Physiol 2020; 319: F1090–F1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Karbowski J. Constancy and trade-offs in the neuroanatomical and metabolic design of the cerebral cortex. Front Neural Circuits 2014; 8: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sedaghat S, Vernooij MW, Loehrer E. et al. Kidney function and cerebral blood flow: the Rotterdam Study. J Am Soc Nephrol 2016; 27: 715–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sprick JD, Nocera JR, Hajjar I. et al. Cerebral blood flow regulation in end-stage kidney disease. Am J Physiol Renal Physiol 2020; 319: F782–F791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bucciarelli RL, Eitzman DV.. Cerebral blood flow during acute acidosis in perinatal goats. Pediatr Res 1979; 13: 178–180 [DOI] [PubMed] [Google Scholar]
- 88. Fencl V, Miller TB, Pappenheimer JR.. Studies on the respiratory response to disturbances of acid–base balance, with deductions concerning the ionic composition of cerebral interstitial fluid. Am J Physiol 1966; 210: 459–472 [DOI] [PubMed] [Google Scholar]
- 89. Choi B, Crouzet C, Lau WL. et al. Cerebral blood flow in chronic kidney disease. J Stroke Cerebrovasc Dis 2021; doi: 10.1093/ndt/gfaa134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tin A, Grams ME, Maruthur NM. et al. Results from the atherosclerosis risk in communities study suggest that low serum magnesium is associated with incident kidney disease. Kidney Int 2015; 87: 820–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tsuji R, Inoue H, Uehara M. et al. Dietary magnesium deficiency induces the expression of neuroinflammation-related genes in mouse brain. Neuropsychopharmacol Rep 2021; 41: 230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rodelo-Haad C, de Mier MV, Díaz-Tocados JM. et al. The role of disturbed Mg homeostasis in chronic kidney disease comorbidities. Front Cell Dev Biol 2020; 8:543099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sakaguchi Y, Hamano T, Matsui I. et al. Low magnesium diet aggravates phosphate-induced kidney injury. Nephrol Dial Transplant 2019; 34: 1310–1319 [DOI] [PubMed] [Google Scholar]