

Abstract
Molecular excitons play a foundational role in chromophore aggregates found in light-harvesting systems and offer potential applications in engineered excitonic systems. Controlled aggregation of chromophores to promote exciton delocalization has been achieved by covalently tethering chromophores to deoxyribonucleic acid (DNA) scaffolds. Although many studies have documented changes in the optical properties of chromophores upon aggregation using DNA scaffolds, more limited work has investigated how structural modifications of DNA via bridged nucleotides and chromophore covalent attachment impact scaffold stability as well as the configuration and optical behavior of attached aggregates. Here we investigated the impact of two types of bridged nucleotides, LNA and BNA, as a structural modification of duplex DNA-templated cyanine (Cy5) aggregates. The bridged nucleotides were incorporated in the domain of one to four Cy5 chromophores attached between adjacent bases of a DNA duplex. We found that bridged nucleotides increase the stability of DNA scaffolds carrying Cy5 aggregates in comparison with natural nucleotides in analogous constructs. Exciton coupling strength and delocalization in Cy5 aggregates were evaluated via steady-state absorption, circular dichroism, and theoretical modeling. Replacing natural nucleotides with bridged nucleotides resulted in a noticeable increase in the coupling strength (≥10 meV) between chromophores and increased H-like stacking behavior (i.e., more face-to-face stacking). Our results suggest that bridged nucleotides may be useful for increasing scaffold stability and coupling between DNA templated chromophores.
Introduction
Molecular (Frenkel) excitons arise from the collective interaction among coupled chromophores that allows creation of excited states that are shared across participating chromophores. The shared excitation energy extends in a wavelike manner over an aggregate (i.e., excitons are delocalized).1−7 Excitons have garnered considerable research interest arising from their function in natural light-harvesting systems8−11 and their potential applications in artificial light harvesting, organic optoelectronics,12,13 and nanoscale computing.14,15 It follows that the realization of devices using excitonic systems requires controlled delocalization of excitons along a specific path (e.g., exciton wire). A variety of chromophores have been shown to spontaneously aggregate in solution,16−19 with reported delocalization over 60+ chromophores in self-assembled aggregates (i.e., -mers).19 In the absence of additional structural support or scaffolding, however, further control over the relative placement and orientations of individual dyes to influence how aggregation occurs remains elusive. Natural photosynthetic systems use proteins as a scaffold for chromophores to facilitate controlled delocalization for efficient energy transfer, but protein scaffolds present design challenges due to complex folding mechanisms and large number of possible amino acid combinations.20−23
Deoxyribonucleic acid (DNA)-based nanotechnology has emerged as an effective and accessible method to design scaffolded chromophore aggregate systems at subnanometer scales because of the simple design rules, sequence selectivity, small number of components, and commercial availability of custom-sequenced chromophore-conjugated DNA oligomers. DNA has been used successfully to induce chromophore aggregation by covalently attaching chromophores to specific bases and allowing strands to self-assemble to facilitate aggregation.24,25,34−43,26,44−53,27−33
Electronic interactions between aggregated chromophores are well approximated by an augmented Frenkel Hamiltonian54−58 of the following form:
| 1 |
where εme is the monomer transition energy, Jm,n is the exchange energy associated with transition dipole coupling between chromophores from a single exciton, Km,n is associated with the average energy of a pair of singly excited chromophores at sites m and n, Δm is associated with the energy of a single chromophore with two simultaneous excitations, and the operators B̂i and B̂i are exciton creation and annihilation operators, respectively. Note that this Hamiltonian does not contain the vibrational aspect of chromophores or impact of the bath (environment) on the individual chromophores or aggregates.
Of particular interest for this study is the magnitude of Jm,n, as this parameter quantifies the interaction that allows the single exciton to transfer, or “hop”, from the chromophore, m, in an aggregate to the adjacent chromophore, n. Because of this inherent transfer of excitation energy associated with Jm,n, we refer to this parameter as the excitonic hopping parameter. Note that Mass et al. recently provided an extensive discussion of previous terminology regarding Jm,n.28 The coupling arising from the Jm,n term in the Hamiltonian is responsible for exciton delocalization54,56−61 and is therefore of interest for designing an exciton wire.
Transfer of energy predominately occurs through a dipole–dipole interaction between the induced transition dipole moments (TDMs) of participating chromophores, and thus the magnitude of Jm,n is sensitive to interchromophore separation. Weak coupling has been observed for chromophores with base-pair (bp) separations of 2 bp or more,14,32,62,63 whereas strong coupling—resulting in exciton delocalization—has been reported with chromophore separations of 1 bp or less.24,26,32,37,64 Although attempts have been made to influence aggregation by changing separation distances between chromophores on a single strand33 and with chromophores positioned on opposing strands in a duplex,26,33 little information is available about how changes to the scaffold strands themselves affect exciton delocalization (Jm,n) and duplex stability in DNA-templated chromophore networks.
One significant challenge in designing the DNA scaffold or templating of chromophore networks is the instability in chromophore proximity and orientations due to the dynamic nature of the DNA scaffold itself (so-called DNA “breathing”). Breathing is a general term that encompasses several modes of thermally activated conformational changes to the DNA duplex.65 Fluctuations in the geometry of the DNA helix are thought to arise from multiple processes. These include low activation energy events such as temporary dissociation of the hydrogen bonds between complementary bases as well as higher activation energy events such as unstacking of bases along the helical axis.65 The latter typically occur at temperatures near the melting temperature of the duplex, but the former are thought to have an activation energy on the order of kBT at room temperature.65 Given that localized dissociation events, such as temporary base pair dissociation, can occur even at temperatures well below the melting temperature, one possible avenue to mitigate breathing is to use alternative nucleic acids that can increase hybridization affinity between complementary strands.
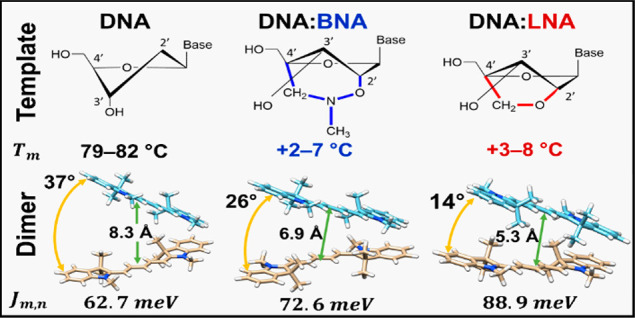
Bridged nucleotides are modified nucleotides of DNA or RNA in which the pentose sugar ring is “locked” into the 3′-endo conformation66 (N-type; Figure 1a). The N-type sugar pucker is typical of an A-form DNA duplex,67 which is associated with higher hybridization stability.68,69 The sugar is forced into this conformation by a bridge spanning the 2′ oxygen and the 4′ carbon of the deoxyribose sugar ring. The bridge restricts the sugar from adopting the 2′-endo conformation (S-type) found in a typical B-form DNA duplex and thus eliminates a flexural degree of freedom in the oligomer backbone. It follows that the resulting perturbation of the scaffold helix would influence the relative geometry between dyes that are covalently bonded to the scaffold strand in the vicinity of a modified nucleotide; however, to the authors’ knowledge, no studies to date have reported the influence of bridged nucleotides on exciton delocalization in covalently bonded DNA-templated chromophore aggregates.
Figure 1.

(a) Chemical structure of the deoxyribose sugar in DNA (left), LNA (center), and BNA (right) nucleotides. DNA shows the C2′-endo conformation found in the B-form DNA helix. LNA and BNA each show the ribose sugar locked into the C3′-endo conformation (found in the A-form helix) by a bridge connecting the 2′ oxygen to the 4′ carbon. (b) Sequences and chromophore locations for DNA-templated chromophore aggregates from zero (unlabeled) to four (tetramer) chromophores. Each design was reproduced with each of three scaffold variations: (1) DNA-only (DNA:DNA) (black), (2) seven LNA bases (DNA:LNA) (red), and (3) seven BNA bases (DNA:BNA) (blue). Inset (top right) shows the structure of the Cy5 chromophore and the phosphoramidite linkers to the DNA backbone.
Another important consideration in designing DNA-templated chromophore networks is the choice of conjugation. There have been successful attempts to use DNA to template chromophores to form aggregates by noncovalent binding to DNA;35 however, these types of chromophore aggregation are difficult to control in regard to the precise location and number of chromophores. Covalent bonding to the DNA scaffold offers a higher degree of control over chromophore placement and the number of chromophores in an aggregate or network. Chromophores can be attached covalently to DNA during the solid-state synthesis of oligonucleotides via a phosphoramidite method or as a postmodification of a synthesized oligonucleotide carrying an anchor group for attachment (e.g., NHS-ester method). The former method is more beneficial for creating excitonic wires on DNA, as it ensures that all dyes in the wire are present because of the “capping” step in the synthesis cycle. In contrast, the postmodification of an oligonucleotide with several chromophores is more likely to result in “missing” chromophores in a wire. To ensure the desired number of chromophores in the excitonic wire, we chose Cy5 attachment via a synthetically available dual-phosphoramidite method in which both ends of the dye are attached to the DNA via short linkers. As such, a chromophore is attached more rigidly to the DNA, thus restricting the orientations available to the attached chromophores. In comparison, however, with a single linker attachment to an internal nucleobase,2−4 which is often done via a postmodification, the dual-phosphoramidite method requires an interruption of the continuous DNA backbone, thus disrupting the base-stacking interactions that help stabilize the duplex.
In this study, we investigate exciton delocalization in cyanine (Cy5) chromophore aggregates templated sequentially along modified DNA duplex scaffolds to form linear aggregates (i.e., dimer, trimer, and tetramer) that serve as an exciton wire. We chose to conjugate the Cy5 chromophores to the DNA template with a rigid two-linker attachment between adjacent phosphate groups (see Figure 1). We report on the influence on exciton delocalization and thermodynamic stability arising from inclusion of either of two types of bridged nucleotides in DNA scaffold strands in the domain of attached chromophores. The first type is locked nucleic acid (LNA), which is a first-generation bridged nucleotide that contains a simple carbon linker across the deoxyribose sugar to restrict the sugar conformation68,70 (Figure 1a). We also examine a third-generation bridged nucleotide, 2′,4′-BNANC[N-Me] (hereafter BNA), which includes an additional nitrogen atom and methyl group in the carbon linker bridge.71 The restricted sugar conformation is thought to lead to a more rigid duplex70 compared with the DNA-only, i.e., DNA containing only natural bases because of less conformational flexibility in the phosphate backbone72 and more efficient base stacking,73 which, we hypothesize, may mitigate the occurrence of low activation energy breathing events. Conversely, the perturbation of the helix near the chromophores may instigate nucleation of breathing events,65 which, in principle, could lead to changes in relative position and orientation of chromophores within the templated aggregate, as evidenced by changes in optical properties in aggregates ranging from monomers (one chromophore) up to tetramers (four chromophores). As a result, we want to know if the inclusion of bridged nucleotides near attached chromophores can be used to alter the relative orientations of the chromophores within the aggregates to enhance exciton delocalization (i.e., increase Jm,n) to facilitate use of the aggregate as an exciton wire as well as to evaluate the ability of bridged nucleotides to counteract the destabilizing effect of inserting chromophores into the phosphate–sugar backbone of a DNA duplex.
Materials and Methods
Sample Preparation
Nucleic acid strands in lyophilized form were obtained from two different vendors. All initial dye-labeled, strands with only DNA, and strands containing LNA were obtained from Integrated DNA Technologies (Coralville, IA). Strands containing BNA and additional (duplicate) dye-labeled strands containing three and four Cy5 chromophores were obtained from Bio-Synthesis Inc. (Lewisville, TX). All Cy5-labeled strands, and unlabeled strands with LNA or BNA modifications, were purified by dual high-performance liquid chromatography (HPLC). Unlabeled strands containing only DNA were purified by standard desalting methods.
All strands were rehydrated with ultrapure water (UPW) (Barnstead Nanopure, Thermo Scientific) to a stock concentration of 100 μM. We verified concentrations by measuring the absorbance via a NanoDrop One microvolume ultraviolet–visible (UV–vis) spectrometer (Thermo Scientific) using the theoretical extinction coefficients provided by the manufacturers. Forty-two base-pair (bp) duplexes were prepared by combining equimolar amounts of complementary strands with UPW and a concentrated buffer (10× Tris-borate-EDTA [TBE], 150 mM MgCl2) to obtain 150 μL samples in 1× TBE 15 mM MgCl2. Except where noted, samples were prepared at a nominal 5 μM concentration (verified from absorbance values at 260 nm and the DNA scaffold extinction coefficient at 260 nm). All duplexes were annealed by using a Mastercycler Nexus polymerase chain reaction (PCR) cycler (Eppendorf). Samples were held at 98 °C for 15 min and then slowly cooled at a rate of 0.4 °C/min until they reached 25 °C. Samples were then allowed to equilibrate to room temperature (∼20 °C).
Melting Curves
Melting curves were obtained by using a Cary-5000 UV–vis–near-infrared (NIR) spectrophotometer (Agilent Technologies). Unlabeled 42 bp duplexes were prepared at 2 μM, whereas chromophore-labeled strands were prepared at 5 μM. All samples were prepared in 1× TBE with 15 mM MgCl2 and transferred to a 1 cm path length low-headspace quartz cuvette. The absorbance at 260 nm was monitored as temperature was increased from 25 up to 98 °C at a rate of 1 °C/min. Melting data were analyzed by using the first derivative of absorbance with respect to temperature. Experimental results were deconvolved into one or more Gaussian curves. Melting temperatures (Tms) were identified as the maximum value of each Gaussian curve. Melting temperatures reported in Figure 2 were derived from data collected while temperature was increasing. See section S1 of the Supporting Information for additional melting data, including hysteresis.
Figure 2.

(a) Example of an experimental melting curve (middle) and the derivative with respect to temperature (right) with Gaussian curves identifying the main transition (red) and the minor transition (green) for the dimer on DNA:DNA. (b) Melting temperatures for scaffolds containing zero to four covalently attached chromophores (2–5 μM samples in 1× TBE 15 mM MgCl2; absorbance at 260 nm monitored as temperature increased from 25 to 98 °C at 1 °C/min). Melting transition temperatures were determined to be inflections points in the melting curves. The solid data points indicate the main melting point, whereas the open data points refer to the minor “premelting” point. “0” chromophores denote duplex DNA without chromophores. The lines were added to highlight trends.
Nondenaturing Polyacrylamide Gel Electrophoresis (PAGE)
Samples (5 μM, 25 μL) were combined with 5 μL loading buffer [20% v/v Ficoll (Sigma-Aldrich) and 20% v/v bromophenol blue (Sigma-Aldrich)] and loaded into wells of 10% native PAGE gel (1.5 mm) casted with 1× TBE buffer. Samples were allowed to migrate through the gel for 90 min with 150 V of applied voltage at 15 °C in 1× TBE, 15 mM MgCl2 running buffer. The resultant gel was imaged in a FluoroChem Q imager (Alpha Innotech, San Leandro, CA) under epi-UV illumination.
Steady-State Absorption Spectroscopy
Steady-state, ensemble absorption spectroscopy was used to characterize the electronic structure of chromophore aggregates. Absorption spectra were recorded by using the Cary-5000 (Agilent) in dual-beam mode at 22 °C. Samples (50 μL, 5 μM, 1× TBE, 15 mM MgCl2) were transferred into a 1 cm path length quartz cuvette (50 μL capacity, Starna Cells). Absorption was monitored from 230 to 800 nm in 1 nm steps.
Circular Dichroism
The CD spectra were recorded by using a JASCO J-810 CD spectrophotometer (JASCO). Samples (100 μL, 5 μM, 1× TBE 15 mM MgCl2) were measured in a 1 cm path length quartz cuvette (100 μL capacity, JASCO) at room temperature (∼22 °C). The CD signal was monitored from 230 to 800 nm for three consecutive scans at 200 nm/min and averaged.
KRM Model Simulation Tool
Theoretical modeling was performed, based on the work of Kühn, Renger, and May (KRM),74 to estimate the relative positions and orientations of chromophores within the templated aggregates. Our in-house developed software26−28,31,75 was used to extract coupling information (Jm,n) and relative chromophore positions and orientations by simultaneously fitting experimental absorbance and CD data. The KRM Model Simulation Tool evaluates the energy eigenstates and eigenvalues of a Holstein-like Hamiltonian. An extended dipole approximation is employed to estimate the excitonic hopping parameter(s), Jm,n. Eigenstates and eigenvalues are used to compute theoretical absorbance and CD spectra which are then compared with experimental data. A stochastic gradient search method is employed to iteratively generate theoretical spectra to minimize the deviation from experimental results (see section S3 of the Supporting Information).
Results
To address our research questions and compare the effects of replacing natural nucleotides with LNA or BNA near chromophore aggregates on exciton delocalization, we obtained DNA oligomers that included from zero up to four chromophores on one DNA-only strand of a duplex to be hybridized with three different complementary strands resulting in a total of 15 samples. We chose to use commercially available cyanine-5 (Cy5) as the study chromophore given its high extinction coefficient and its already extensive characterization of spectral properties.
Construct Design
Each nucleic acid sample is a 42 bp duplex with identical base pair sequences. The Cy5 chromophores were internally bound in the DNA backbone by using a double-link attachment to phosphate groups on adjacent nucleotides (nt) near the center of a 42 nt DNA strand. Complementary strands are 42 nt with a sequence designed to hybridize with the dye-labeled strands by using Watson–Crick base pairing rules to form a duplex. Sequences were determined by using in-house developed sequence optimization software, and final designs were checked for the probability of forming unwanted secondary structures by using the Nucleic Acid Package (NUPACK; California Institute of Technology).76 Three types of complementary strands were prepared containing (1) DNA only, (2) seven LNA nucleotides, or (3) seven BNA nucleotides spanning the domain of the chromophores on the labeled strand (Figure 1b).
Characterization of the Scaffold
To determine the influence of bridged nucleotides on the stability and conformation of a DNA duplex scaffold, we used two approaches. We used melting curves to evaluate the duplex stability of each sample and a series of polyacrylamide gel electrophoresis (PAGE) experiments to investigate conformational changes of the DNA helix when substituting DNA bases with LNA or BNA.
Thermal Denaturation
We applied DNA thermal denaturation to quantify the change in thermodynamic stability upon substitution of DNA with BNA or LNA. Melting curves quantify thermodynamic stability in nucleic acid scaffolds as a function of melting temperature (Tm). The melting temperature in this context is defined as the temperature at which half of the base pairs in a DNA duplex are dissociated. In our analysis, the Tm for the 42 bp DNA-only duplex was 82.4 °C (Figure 2). Replacing seven consecutive nucleotides on one strand of the duplex with LNA increased Tm to 90.9 °C. Conversely, replacing the same seven nucleotides with BNA increased Tm to 89.5 °C. The higher melting temperatures suggest greater hybridization affinity between complementary strands and greater thermodynamic stability of the scaffold. In addition, we observed that constructs containing chromophores show multiple melting transitions, evident in the inflection points in the melting curves. By fitting Gaussian curves to the first derivative of our experimental melting curves, we can determine one or more melting points to compare Tm of scaffolds with only DNA to those with LNA or BNA substituted into the DNA duplex. In addition, melting curves also can reveal how chromophore interactions with the preceding may alter the results.
Inclusion of chromophores led to more complicated melting behaviors. In all cases, Gaussian peak fitting suggested that the chromophore-labeled scaffolds had a minor “premelting” transition at a lower temperature than the main melting transition (Figure 3 and section S1).
Figure 3.

PAGE images (10% gel; 1× TBE running buffer; 150 V; T = 15 °C; 90 min; epi-UV illumination). (a) PAGE image by lanes for a control strand with four added negatively charged phosphoryl (PO3–) groups to add extra charges to a 42 bp DNA duplex (1) versus an unmodified 42 bp DNA duplex (2). (b) PAGE image by lanes for unmodified 42 bp DNA duplex (2) versus 42 bp DNA duplex with seven LNA bases (3), 42 bp DNA duplex with seven BNA bases (4), and 42 bp DNA duplex with four attached Cy5 chromophores (5). (c) PAGE image for constructs containing four Cy5 chromophores. Lanes 5 and 6 contained DNA-only duplexes with pairs of Cy5 chromophores at each end (5) compared with four Cy5 chromophores in the center of the strand (6). Lanes 7 and 8 contained Cy5 tetramers with substituted LNA (red) and BNA (blue), respectively. (d) Sequences and design schematics corresponding to the gel images in (a–c). See section S2 of the Supporting Information for full gel images.
The theoretical Tm of 82.2 °C for the unlabeled, unmodified DNA sequence (Table 1, entry 1) strongly agreed with our experimental melting temperature of 82.4 °C. While insertion of one chromophore to the DNA:DNA duplex did not affect the temperature of the main transition, an additional transition at 79.5 °C was observed. The addition of a second and third chromophore to the unmodified duplex resulted in further decrease of both main and minor melting transitions to 80.2 and 76.7 °C, respectively, for two chromophores and 79.2 and 74.7 °C, respectively, for three chromophores. Adding a fourth chromophore further reduced the temperature of the main melting transition to 77.9 ± 0.3 °C for all samples. Therefore, for this last attachment scheme, we observed no appreciable difference in melting temperature between natural DNA samples and those modified with LNA or BNA. To estimate if the major and minor transitions were due to independent melting of DNA duplex arms on each side of the chromophore(s), we calculated the theoretical melting points of the corresponding duplex fragments (Table 1, entries 2–7). Observed minor melting temperatures are considerably higher than temperatures of duplex arms, suggesting that the major and minor transitions were not due to independent melting of the DNA duplex arms on either side of the chromophore(s).
Table 1. Theoretical Melting Temperatures for the 42 bp DNA:DNA Duplex Used in This Study and Several Partial Duplex Fragments.

Entry 1 contains the full 42 bp sequence used in this study. Bases at the midpoint of the sequence are shown in violet for reference. Entries 2–7 are partial fragments of the sequence in entry 1.
Theoretical melting temperatures were obtained by using an online software program provided by European Molecular Biology Laboratory77 (Cambridgeshire, UK) using the nearest-neighbor method “Allawi et al. 1997”, 15 mM MgCl2, and 89 mM TRIS. Salt correction was included by using the “SantaLucia et al. 1998” option.
Polyacrylamide Gel Electrophoresis (PAGE)
To further investigate changes to the DNA scaffold induced by inclusion of LNA/BNA, or resulting from chromophore addition and aggregation behavior, we used the relative migration rates of samples through the polyacrylamide gels to evaluate changes to the tertiary structure of the DNA scaffolds. We observed that in general the migration rate of each sample was unaffected by the inclusion of LNA or BNA, indicating that LNA and BNA did not significantly affect the tertiary structure of the duplexes (Figure 3b,c). When chromophores were added, however, each added chromophore resulted in slower gel migration. The slower migration rate was attributed to the increased bulk of dye–DNA constructs upon attaching chromophores to DNA.
To determine if additional factors, such as chromophore position or interchromophore attractive forces, contributed to the slower migration rate caused by the addition of chromophores, we used a control sample composed of an unlabeled duplex equipped with terminal charged phosphoryl (PO3–) groups. This design helped to determine if the change in migration rate could be attributed to the change in net charge introduced by adding positively charged Cy5 molecules. The control sample migrated at a slightly slower rate than the unmodified duplexes, which indicated that the additional negative charge on the chromophores did not alter the speed of gel migration (Figure 3a). To test the possible role of chromophore position, we designed another experiment using a total of four chromophores in which the gel migration rate with two chromophores that were located at each end of the duplex was contrasted with samples containing four chromophores at the center of the duplex. Specifically, we wanted to know if interchromophore attractive forces due to aggregation distorted the DNA scaffold and caused a significant change in the linearity of the DNA helix which would result in slower gel migration. We again observed no difference in migration rates (Figure 3c). This result suggested that the slower observed migration rate from inclusion of Cy5 chromophores was largely independent of chromophore position and interchromophore attractive forces.
Characterization of Aggregate Behavior
We used a combination of steady-state spectroscopy and theoretical modeling to investigate the influence of LNA and BNA on the optical behavior and geometry of Cy5 aggregates templated via modified duplex scaffolds.
Absorption Spectroscopy
Each scaffold type was characterized first with one attached Cy5 molecule (monomer). The Cy5 monomer sample on DNA:DNA exhibited an absorbance maximum at 647 nm. The maximum is assigned to the 0–0 energy transition, that is, the excitation of the monomer from the ground state to the lowest energy excited state (Figure 4a). A second absorbance band around 600 nm is assigned to the 0–1 vibronic transition, that is, the excitation from the ground state to the first electronic excited state including one quanta of vibration. Monomers on DNA:LNA and DNA:BNA scaffolds had similar and slightly higher energy absorbance maxima at 644 nm (0–0) and a similarly shifted 0–1 transition near 600 nm. All three monomer structures had an extinction coefficient of about 230000 M–1 cm–1 at the absorbance maximum, which is generally in agreement with published78 and manufacturer-stated values.
Figure 4.

Experimental absorbance and circular dichroism spectra for aggregates templated on DNA:DNA (black), DNA:LNA (red), and DNA:BNA (blue) duplex scaffolds with Cy5 monomers (a), dimers (b), trimers (c), and tetramers (d). Scaffolds with LNA or BNA contained seven bridged nucleotides spanning the domain of the chromophores on the complementary strand. All samples were 5 μM in 1× TBE 15 mM MgCl2; T = 22 °C for absorbance and room temperature (∼21 °C) for circular dichroism.
The Cy5 dimer sample on the DNA:DNA scaffold showed a hypsochromic (blue) shift in the absorbance maximum: that is, the redistribution of the oscillator strength was toward shorter wavelengths, relative to the DNA:DNA monomer (compare Figure 4b to Figure 4a). The shift in absorbance to higher energy transitions was consistent with an H-like (i.e., face-to-face stacking) molecular packing arrangement.79 The absorbance spectrum showed two main bands, with the most intense feature now at 601 nm. The other major band, which was slightly broader and of lower intensity, was centered around 643 nm. Dimers on DNA:LNA and DNA:BNA both showed a similar hypsochromic shift, with the maximum absorbance feature centered at 599 and 600 nm, respectively, and the broader, lower intensity bands centered at 645 and 644 nm, respectively. For all three scaffold types, the intensities of the two main absorbance features were comparable, and each contained an enhanced minor feature centered near 550 nm.
The trimer sample on DNA:DNA had two main absorbance bands with similar hypsochromic shifts relative to the corresponding dimer, with the most intense peak at 603 nm and a second feature around 645 nm (Figure 4c). The intensities of the two most prominent features were similar, as with the DNA:DNA dimer. Trimers on DNA:LNA and DNA:BNA showed absorption spectra that were nearly identical with each other, with the most intense peak blue-shifted (i.e., greater redistribution of oscillator strength) by 11 nm relative to the DNA:DNA trimer. In both cases, the shorter wavelength peak was significantly more intense than the lower energy band at 647 nm (Figure 4c). The comparable shift in peak wavelength intensity and the similar increase in peak intensity ratio between higher energy and lower energy absorbance bands suggested that the addition of either LNA or BNA into the DNA duplex scaffold near attached chromophores had a similar impact (when compared with each other) on the electronic structure of the attached chromophore aggregates.
Tetramers had absorbance spectra similar to the trimer series (Figures 4c and 4d). The DNA:DNA-scaffolded tetramer had a maximum absorbance band at 598 nm, with the second most intense band around 645 nm. The DNA:LNA and DNA:BNA-scaffolded tetramers again showed nearly identical absorbance spectra compared with each other, and both were distinct from the DNA:DNA version. Tetramers on both scaffold types had an absorbance maximum at 592 nm, which is blue-shifted by about 6 nm from the DNA:DNA tetramer maximum. The lower energy band was centered at 647 nm. As with the trimer series, the intensity of the lower energy feature was significantly lower than the higher energy feature (Figure 4c).
The greater blue-shift in absorbance maximum of the DNA:LNA and DNA:BNA templated aggregates than the DNA:DNA templated aggregates was consistent with the addition of LNA or BNA in the scaffold altering the orientations of the attached chromophores toward a more H-like configuration, suggesting the chromophores may have been more strongly coupled (i.e., greater exciton delocalization).
Circular Dichroism Spectroscopy
Circular dichroism (CD) spectroscopy measures the differential absorption of right- versus left-hand circularly polarized light. The CD rotational strength for a pair of coupled chromophores is proportional to a quadruple product80 with this form:
| 2 |
where 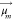 and
and 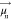 are the TDM
vectors for chromophore m and n,
respectively, and
are the TDM
vectors for chromophore m and n,
respectively, and 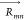 is the separation vector between the TDMs.
From this expression it is clear that the CD signal depends not only
on the coupling strength (Jm,n) but also on the proximity (|
is the separation vector between the TDMs.
From this expression it is clear that the CD signal depends not only
on the coupling strength (Jm,n) but also on the proximity (| |) and mutual
orientation of the participating
TDMs. For Cy5, a single chromophore is achiral (i.e., little to no
CD signal; see Figure 4b); therefore, any CD signal obtained from Cy5 aggregates in this
study is a signature of exciton delocalization.
|) and mutual
orientation of the participating
TDMs. For Cy5, a single chromophore is achiral (i.e., little to no
CD signal; see Figure 4b); therefore, any CD signal obtained from Cy5 aggregates in this
study is a signature of exciton delocalization.
The CD spectrum from the DNA:DNA dimer showed a well-defined positive feature at 660 nm (Figure 4b). Below 650 nm, the signal was negative, but weak (low intensity) and noisy (complex line shapes), which we attributed to the presence of multiple configurations of dimer orientations within the sample solution (aggregate heterogeneity). The DNA:LNA and DNA:BNA dimers showed similar spectral features, with both structures having a positive feature at 679 nm that was red-shifted by 19 nm from the DNA:DNA dimer and a negative feature at 601 nm that was not present in the DNA:DNA spectrum.
The Cy5 trimer on DNA:DNA showed the strongest and most well-defined CD signal (Figure 4c). A significant negative feature occurred at 684 nm and a strong positive feature at 655 nm that were both lower in energy than the 0–0 monomer transition. The negative band at 599 nm corresponded to the aggregate absorbance maximum. Trimers on DNA:LNA and DNA:BNA scaffolds showed CD spectra that were nearly identical when compared with each other and distinct from the DNA:DNA trimer. A negative CD feature corresponded to the absorbance maxima at 591 nm, and a positive feature appeared around 667 nm in both spectra, which was significantly red-shifted from the 0–0 monomer peak.
The DNA:DNA tetramer showed a negative feature at 686 nm and positive feature at around 655 nm, similar to the DNA:DNA trimer CD spectrum (Figure 4d). We observed a negative feature at 599 nm corresponding to the absorbance maximum of the aggregate. Although quite noisy, DNA:LNA and DNA:BNA versions both showed similar features when compared with each other, with a negative signal at 576 nm and two positive features at 620 and ∼665 nm.
Theoretical Modeling
We used our in-house KRM Model Simulation Tool27,28,31 to extract relative orientations of chromophores within each aggregate. Our model employs the Hamiltonian in eq 1 which is truncated to consider the case for single excitations (K and Δm terms are ignored) and augmented with terms to capture vibrational effects (see section S3). Each system Hamiltonian is diagonalized, and the allowed vibronic transitions and corresponding transition amplitudes (eigenvalues of the Hamiltonian) are convolved with Gaussian line shapes and summed to form a theoretical spectrum. This modeled spectrum enables comparison with experimental data to extract information related to coupling strength and chromophore spatial proximity and orientation relative to neighboring chromophores.
In Table 2, Jm,n indicates the electronic coupling strength between each pair of chromophores. In general, for H-like aggregates, smaller center-to-center distances (Rm,n in Table 2) between chromophores, short of introducing charge-transfer effects, will lead to larger values of Jm,n. The deviation from parallel between each pair of TDMs is given by the oblique angle—which we will call α—that describes the absolute angle between TDMs in three dimensions (see Figure 5). A smaller oblique angle implies that the packing is closer to parallel, which we expect to increase Jm,n for H-like packing arrangements. An example of simulated spectra and associated molecular geometry leading to the results stated in Table 2 are shown in Figure 6. For complete modeling results, see section S3.
Table 2. KRM Model Simulation Tool Fitting Results for Oblique Angle α (deg), Center-to-Center Distance R (Å), and Exciton Hopping Parameter J (meV) for Each Pair of TDMs within Each Aggregate for (a) Dimers, (b) Trimers, and (c) Tetramersa.

See section S3 for expanded modeling results, including diagrams that show the spatial arrangement of each modeled aggregate.
Figure 5.
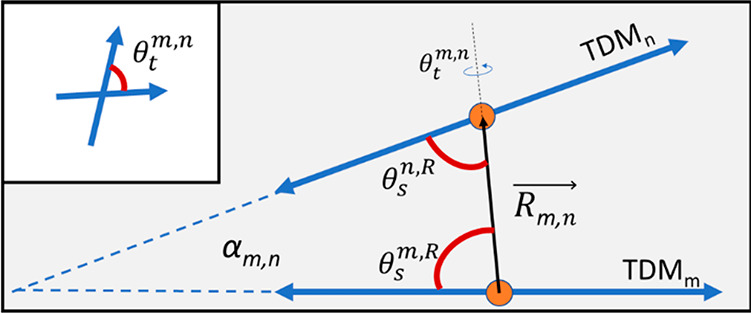
Diagram depicting relative orientation parameters for a pair of TDMs in an H-like stacking arrangement, including oblique angle, α, the center-to-center separation vector, R, the relative slip angles, θs, and the twist angle, θt (see section S3). Note that, here, α is shown in two dimensions for the special case of zero twist angle, but in general the angle spans three dimensions. The inset shows the twist angle looking along the separation vector from m to n. The slip angle for nonparallel TDMs will depend on which TDM is chosen as the reference.
Figure 6.

Example of KRM Model Simulation Tool outputs (dimer on DNA:DNA). (a) Experimental (black traces) absorbance (bottom) and circular dichroism (top) spectra shown with scaled theoretical spectra (dotted red traces). The vertical bars denote modeled absorbance transitions divided into proportions of purely electronic transitions (black) and transitions involving at least one quanta of vibration (red). (b) Example of the dimer configuration leading to the theoretical spectra shown in panel a. Molecular images were created by using Avogadro and visualized by using Chimera81 by assuming that the transition dipole moment is oriented along the long axis of the molecule. For complete modeling results for this study, see section S3.
Inclusion of LNA and BNA generally led to higher modeled values of Jm,n, regardless of the number of chromophores within an aggregate (Table 2). Dimers on all three scaffold types adopted an H-like packing arrangement. For the dimer on the DNA:DNA scaffold, the oblique angle α between the long axes of the chromophores was 37°, whereas dimers with LNA and BNA showed a more parallel arrangement with oblique angles of 14.1° and 26.3°, respectively. The excitonic hopping parameter Jm,n was 62.7 meV for the DNA:DNA dimer. The Jm,n parameter for LNA and BNA increased to 88.9 and 72.6 meV, respectively, which reflected stronger coupling due to enhanced H-packing (i.e., closer to parallel and face-to-face).
Trimers showed consistently smaller oblique angles between chromophores in the constructs containing LNA and BNA compared to DNA-only. The smaller angles led to more parallel H-packing arrangements and higher Jm,n values. Modeling results for all trimer scaffold types indicated that all three chromophores interacted with each other and suggested that exciton delocalization occurred over all three chromophores.
Results for tetramers on all three scaffolds indicated that three of the chromophores may have been in close proximity and significantly interacting, whereas the fourth was farther away. Results suggested that coupling between adjacent chromophores as indicated by larger Jm,n values was significantly stronger for constructs containing LNA or BNA (see section S3 for expanded KRM Model Simulation Tool results).
Discussion
First, we discuss scaffold behavior, in particular, how attachment of chromophores affected the stability and structure of the DNA:DNA, DNA:LNA, and DNA:BNA scaffolds. Incorporation of LNA and BNA was shown to be accompanied by increased duplex stability and changes in its tertiary structure. The increased thermal stability of LNA and BNA is believed to arise from the restricted 3′-endo sugar conformation that is associated with the A-form of the DNA helix rather than the B-form helix that is typically associated with natural DNA.73 Molecular dynamics simulations suggest that the perturbation of the helix toward the A-form is localized near the substituted nucleotides.70 The thermodynamic factors contributing to the stability provided by bridged nucleotides such as LNA remains an open question. Stability has previously been attributed to improved base stacking associated with the A-form (enthalpic)73,82,83 and to decreased entropy loss upon aggregation due to preorganization of the phosphate–sugar backbone (entropic).82,84 Our experimental CD results (see section S5) suggest that the B-form DNA helix acquired some features of the A-form in the region where seven bridged LNA or BNA nucleotides were incorporated. In particular, we noted a small blue-shift in the CD feature associated with the DNA (UV region) in constructs containing bridged nucleotides relative to otherwise identical constructs containing only natural DNA.
Previous work suggests that the contribution to Tm per substituted LNA and BNA nucleotide differs significantly depending on sequence and experiment design.82,85 In our experimental melting results for unlabeled duplex scaffold, we also observed that the inclusion of LNA or BNA led to stabilization of duplex scaffold manifested in a higher Tm compared to DNA-only scaffold.
Experimental melting results for chromophore-labeled samples confirmed that the attached chromophores destabilized the DNA duplex. Analysis of the derivative of the melting data revealed a more complex dissociation process compared to unlabeled duplexes. In all of the chromophore-labeled samples, we observed a minor “premelting” transition occurring at a lower temperature than the main melting transition. We suggest that the instability introduced by inserting chromophores into the backbone of one component strand in the duplex without accommodating for the size of the chromophore on the complementary strand disturbed the integrity of the duplex. We suggest that this disruption resulted in the dissociation of the chromophore domain at a lower temperature than the full duplex possibly by allowing nucleation of breathing events. Breathing is known to be a minor effect in a well-formed duplex, with only a tiny fraction of base pairs (∼10–5 or less)65,86 dissociated at any one time. We note, however, that disturbances to the helix are thought to increase the probability of local fluctuations (i.e., breathing).65 We propose that the destabilization from chromophore insertion can promote breathing at a lower temperature than the main melting transition. Although thermal stability in each construct was generally increased by the inclusion of LNA and BNA, as evidenced by a higher temperature for the main melting transitions (Figure 2) for up to three chromophores, the instability from chromophore insertion appeared to counteract the stabilizing effect of the bridged nucleotides. With the addition of the fourth chromophore, thermal stability gains from the bridged nucleotides were no longer observed.
Theoretical melting temperatures for DNA fragments (Table 1) suggest that although the templates showed some disruption, likely due to dye insertion, the duplexes appeared to melt as a single unit rather than as two separate fragments. The fragments representing the unmodified duplex sections on either side of the aggregates were found to have theoretical melting temperatures well below the observed premelting transitions, suggesting that base hybridization was occurring to a certain extent between the base pairs within the domain containing the chromophores.
Incorporation of the chromophores, indeed, might influence DNA tertiary structure (DNA conformations). For example, Mass et al. showed that interchromophore attractive forces are strong enough to influence scaffold conformation and serve to stabilize 4-arm DNA Holliday junctions with attached squaraine aggregates.28 To access the changes in the tertiary structure of the DNA scaffolds upon insertion of chromophores, we performed PAGE experiments. The results of the PAGE experiments indicated that each additional chromophore inserted into the scaffold caused a noticeable incremental decrease in the migration rate (see section S2), which we attribute to the added bulk of each chromophore. In contrast, the migration rate of the duplex scaffolds through the gel was insensitive to the position of the attached chromophores along the duplex (Figure 3c), suggesting that interchromophore interactions did not significantly contribute to the tertiary structure of the scaffolds. Additionally, the change in the construct charge due to the attachment of positively charged chromophores did not appear to be responsible for a slower migration rate of the duplex. Finally, it appears that the replacement of the natural nucleotides with the LNA or BNA did not appear to disturb the tertiary structure of the DNA compared with DNA-only samples.
Next, we proceed with a discussion of aggregate behavior. Absorbance results from all dimer, trimer, and tetramer aggregates all exhibited exciton delocalization behavior in a similar manner—they all showed a redistribution of oscillator strength toward shorter wavelengths compared with the monomer spectrum. The hypsochromic shift in absorbance, along with substantially reduced fluorescence (see section S6), was indicative of H-like packing geometries within the aggregates. Inclusion of either LNA or BNA induced greater hypsochromic shifts in the absorbance spectrum, suggesting that the presence of LNA/BNA in the scaffold favorably influenced relative chromophore orientation within each aggregate toward an enhanced H-like packing arrangement. Furthermore, inclusion of LNA or BNA had comparable impacts on the spectra relative to DNA-only constructs. We suggest that the observed spectral shifts arose predominantly from the locked conformation of the sugar ring in the backbone of LNA and BNA.
In general, the CD spectra of the aggregates showed complex line shapes with relatively low intensity, with the notable exception of the trimer on a purely DNA duplex. The complexity of the CD spectra may arise because (1) aggregates were not adopting stable configurations—due to local scaffold instability—or (2) a sample could contain a mix of aggregate orientations (heterogeneity) with competing CD signals. The low intensity in the CD signals could arise either from the reasons mentioned above or from geometrical factors. In eq 2, the rotational strength depends on the cross-product of the TDM vectors and the subsequent dot product with the separation vector, Rm,n. If one considers the case of coplanar TDM vectors, the cross-product will yield a vector that is normal to the plane of the TDMs. The separation vector in this case would necessarily lie in the same plane as the TDM vectors; therefore, the dot product would be zero and no CD signal is produced. As a result, one can expect the CD signal to go to zero as the twist angle (θt; see Figure 5) decreases to zero (chromophores become coplanar), even if the chromophores are strongly coupled (large Jm,n). To add further complexity, Jm,n also depends on the relative TDM orientations (see section S3). The CD signal is further convoluted by coupling to vibrational modes that lead to a mixing of electronic and vibronic states (see section S4). Given all these considerations, one must use caution when interpreting CD data. Although one might expect that strongly coupled chromophores (large Jm,n) would exhibit an intense CD signal, the geometric terms prevent direct interpretation. The intensity of the CD signal alone is not an exclusive indicator of the magnitude of Jm,n or the extent of exciton delocalization.
Consistent with the absorbance results, aggregates with either LNA or BNA showed similar CD features to each other that were distinct from those in DNA-only duplexes. Additionally, all aggregates with LNA or BNA showed a larger split between positive and negative features compared with corresponding aggregates on DNA-only scaffolds. All aggregates exhibited CD features at longer wavelengths (lower energies) than the lowest energy electronic monomer transition (0–0) that is consistent with the idea that additional energy states are accessed upon chromophore aggregation—a signature of molecular exciton delocalization.79,87
Although the (suspected) presence of multiple aggregate configurations in sample solutions prevented the KRM Model Simulation Tool from completely describing the observed spectra, theoretical modeling results supported the finding that LNA and BNA influence the packing arrangement of chromophores toward a more H-like arrangement and strengthened coupling. Modeling constructs with LNA and BNA returned smaller oblique angles (α) between transition dipole moments and enhanced Jm,n values compared with the same constructs on DNA-only scaffolds. The inability to completely describe aggregate spectra may be a result of the instability introduced in each scaffold duplex by inserting chromophores without accommodating for their physical size on the complementary strand, which contributed to variations in aggregate configurations. Alternatively, the difficulty in fitting the absorbance spectra of the higher order aggregates may suggest that our model should be refined to include additional coupling-related terms to account for charge transfer54,58 that may become significant at short interchromophore distances where wave function overlap is significant.58
Although our modeling supported the general finding of increased coupling strength with the inclusion of LNA/BNA, we acknowledge that the model cannot account for multiple potential orientations still present in a sample solution. The inability to fully reproduce the data with a single aggregate orientation, combined with the overall complexity of the aggregate CD signals, indicated that the presence of multiple aggregate orientations is likely. As a result, the modeling outputs represented a possible average orientation within each aggregate. The suspected variation in chromophore orientations may arise from the scaffold instability introduced from chromophore insertion that counteracted the stabilizing effect of LNA/BNA inclusion in the scaffold.
Conclusions
Steady-state optical characterization results suggested that our DNA/LNA/BNA-templated Cy5 chromophore aggregates supported exciton delocalization over as many as four chromophores. Furthermore, inclusion of bridged nucleotides (LNA or BNA) in the domain of the chromophores led to enhancements in coupling strength (Jm,n) as evidenced by additional shifts in absorbance and CD spectra and extracted from theoretical modeling (Table 2). Stronger coupling and a more rigid scaffold may lead to applications as an exciton wire; however, melting data suggested that the inclusion of chromophores in the DNA backbone had a destabilizing effect that partially counteracted the additional stability gained from inclusion of the synthetic nucleotides.
Realizing rational control of aggregate packing geometry and exciton delocalization for use as an exciton wire and gaining the full benefits of including LNA or BNA in the scaffold template will require a reduction in heterogeneity in aggregate orientations. Possible paths to reduced heterogeneity include decreasing the induced strain in the scaffold that resulted from the manner in which chromophores are inserted in the scaffold backbone or by altering the attachment method to include a shorter linker from the scaffold to the chromophore. The former may provide a more stable scaffold on which to template an aggregate network with reduced scaffold fluctuations, and the latter may, in principle, restrict the degrees of freedom and possible orientations available to the chromophores within the aggregate, leading to fewer possible aggregate configurations.
Acknowledgments
This research was supported wholly by the U.S. Department of Energy, Idaho National Laboratory, Laboratory Directed Research and Development project through blanket master contract 154754 between Battelle Energy Alliance and Boise State University, Release 15, except for the following: the circular dichroism spectrometer was made available through the Biomolecular Research Center (BRC) supported by NIH awards P20GM103408 and P20GM109095, the MJ Murdock Charitable Trust, and the Idaho State Board of Education. Molecular graphics images in Figure 6 were produced by using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) All authors have given approval to the final version of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.1c07602.
Melting study, polyacrylamide gel electrophoresis, KRM Model Simulation Tool description and results, vibrational influence in steady-state optical spectra, ultraviolet circular dichroism, steady-state fluorescence, and supplemental references (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Scholes G. D.; Smyth C. Perspective: Detecting and Measuring Exciton Delocalization in Photosynthetic Light Harvesting. J. Chem. Phys. 2014, 140, 110901. 10.1063/1.4869329. [DOI] [PubMed] [Google Scholar]
- Blancafort L.; Voityuk A. A. Exciton Delocalization, Charge Transfer, and Electronic Coupling for Singlet Excitation Energy Transfer between Stacked Nucleobases in DNA: An MS-CASPT2 Study. J. Chem. Phys. 2014, 140, 095102. 10.1063/1.4867118. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; Rumbles G.. Excitons in Nanoscale Systems. In Mater. Sustain. Energy A Collect. Peer-Reviewed Res. Rev. Artic. from Nat. Publ. Gr.; World Scientific Publishing Co.: 2010; pp 12–25. [Google Scholar]
- Monshouwer R.; Abrahamsson M.; Van Mourik F.; Van Grondelle R. Superradiance and Exciton Delocalization in Bacterial Photosynthetic Light-Harvesting Systems. J. Phys. Chem. B 1997, 101, 7241–7248. 10.1021/jp963377t. [DOI] [Google Scholar]
- Lim J. M.; Kim P.; Yoon M. C.; Sung J.; Dehm V.; Chen Z.; Würthner F.; Kim D. Exciton Delocalization and Dynamics in Helical π-Stacks of Self-Assembled Perylene Bisimides. Chem. Sci. 2013, 4, 388–397. 10.1039/C2SC21178E. [DOI] [Google Scholar]
- Dahlbom M.; Pullerits T.; Mukamel S.; Sundström V. Exciton Delocalization in the B850 Light-Harvesting Complex: Comparison of Different Measures. J. Phys. Chem. B 2001, 105, 5515–5524. 10.1021/jp004496i. [DOI] [Google Scholar]
- Eisfeld A.; Briggs J. S. The J-Band of Organic Dyes: Lineshape and Coherence Length. Chem. Phys. 2002, 281, 61–70. 10.1016/S0301-0104(02)00594-3. [DOI] [Google Scholar]
- Ishizaki A.; Fleming G. R. Quantum Coherence in Photosynthetic Light Harvesting. Annu. Rev. Condens. Matter Phys. 2012, 3, 333–361. 10.1146/annurev-conmatphys-020911-125126. [DOI] [PubMed] [Google Scholar]
- Engel G. S.; Calhoun T. R.; Read E. L.; Ahn T. K.; Mančal T.; Cheng Y. C.; Blankenship R. E.; Fleming G. R. Evidence for Wavelike Energy Transfer through Quantum Coherence in Photosynthetic Systems. Nature 2007, 446, 782–786. 10.1038/nature05678. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; Fleming G. R.; Olaya-Castro A.; Van Grondelle R. Lessons from Nature about Solar Light Harvesting. Nat. Chem. 2011, 3, 763–774. 10.1038/nchem.1145. [DOI] [PubMed] [Google Scholar]
- Mirkovic T.; Ostroumov E. E.; Anna J. M.; van Grondelle R.; Govindjee; Scholes G. D. Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms. Chem. Rev. 2017, 117, 249–293. 10.1021/acs.chemrev.6b00002. [DOI] [PubMed] [Google Scholar]
- Ostroverkhova O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. 10.1021/acs.chemrev.6b00127. [DOI] [PubMed] [Google Scholar]
- Brixner T.; Hildner R.; Köhler J.; Lambert C.; Würthner F. Exciton Transport in Molecular Aggregates - From Natural Antennas to Synthetic Chromophore Systems. Adv. Energy Mater. 2017, 7, 1700236. 10.1002/aenm.201700236. [DOI] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Davis P. H.; Lee J.; Kuang W.; Hughes W. L.; Graugnard E.; Yurke B.; Knowlton W. B. Excitonic and Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. 10.1021/ph500444d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaciłowski K. Digital Information Processing in Molecular Systems. Chem. Rev. 2008, 108, 3481–3548. 10.1021/cr068403q. [DOI] [PubMed] [Google Scholar]
- Jelley E. E. Spectral Absorption and Fluorescence of Dyes in the Molecular State [1]. Nature 1936, 138, 1009–1010. 10.1038/1381009a0. [DOI] [Google Scholar]
- Jelley E. E. Molecular, Nematic and Crystal States of 1:1′-Diethyl-ψ-Cyanine Chloride [12]. Nature 1937, 139, 631–632. 10.1038/139631b0. [DOI] [Google Scholar]
- Scheibe G. Über Die Veränderlichkeit Der Absorptionsspektren in Lösungen Und Die Nebenvalenzen Als Ihre Ursache. Angew. Chem. 1937, 50, 212–219. 10.1002/ange.19370501103. [DOI] [Google Scholar]
- Würthner F.; Kaiser T. E.; Saha-Möller C. R. J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials. Angew. Chem., Int. Ed. 2011, 50, 3376–3410. 10.1002/anie.201002307. [DOI] [PubMed] [Google Scholar]
- Snow C. D.; Nguyen H.; Pande V. S.; Gruebele M. Absolute Comparison of Simulated and Experimental Protein-Folding Dynamics. Nature 2002, 420, 102–106. 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- Baker D.; Sali A. Protein Structure Prediction and Structural Genomics. Science (Washington, DC, U. S.) 2001, 294, 93–96. 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Dill K. A.; MacCallum J. L. The Protein-Folding Problem, 50 Years On. Science (Washington, DC, U. S.) 2012, 338, 1042–1046. 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- Yeates T. O. Geometric Principles for Designing Highly Symmetric Self-Assembling Protein Nanomaterials. Annu. Rev. Biophys. 2017, 46, 23–42. 10.1146/annurev-biophys-070816-033928. [DOI] [PubMed] [Google Scholar]
- Asanuma H.; Fujii T.; Kato T.; Kashida H. Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol., C 2012, 13, 124–135. 10.1016/j.jphotochemrev.2012.04.002. [DOI] [Google Scholar]
- Asanuma H.; Shirasuka K.; Takarada T.; Kashida H.; Komiyama M. DNA-Dye Conjugates for Controllable H* Aggregation. J. Am. Chem. Soc. 2003, 125, 2217–2223. 10.1021/ja021153k. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Patten L. K.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. 10.1021/acs.jpca.7b04344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass O. A.; Wilson C. K.; Roy S. K.; Barclay M. S.; Patten L. K.; Terpetschnig E. A.; Lee J.; Pensack R. D.; Yurke B.; Knowlton W. B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. 10.1021/acs.jpcb.0c06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J. S.; Davis P. H.; Christy A.; Kellis D. L.; Kandadai N.; Toa Z. S. D.; Scholes G. D.; Yurke B.; Knowlton W. B.; Pensack R. D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10, 2386–2392. 10.1021/acs.jpclett.9b00404. [DOI] [PubMed] [Google Scholar]
- Hart S. M.; Chen W. J.; Banal J. L.; Bricker W. P.; Dodin A.; Markova L.; Vyborna Y.; Willard A. P.; Häner R.; Bathe M. Engineering Couplings for Exciton Transport Using Synthetic DNA Scaffolds. Chem. 2021, 7, 752. 10.1016/j.chempr.2020.12.020. [DOI] [Google Scholar]
- Barclay M. S.; Roy S. K.; Huff J. S.; Mass O. A.; Turner D. B.; Wilson C. K.; Kellis D. L.; Terpetschnig E. A.; Lee J.; Davis P. H.; et al. Rotaxane Rings Promote Oblique Packing and Extended Lifetimes in DNA-Templated Molecular Dye Aggregates. Commun. Chem. 2021, 4, 19. 10.1038/s42004-021-00456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham P. D.; Khachatrian A.; Buckhout-White S.; Deschamps J. R.; Goldman E. R.; Medintz I. L.; Melinger J. S. Resonance Energy Transfer in DNA Duplexes Labeled with Localized Dyes. J. Phys. Chem. B 2014, 118, 14555–14565. 10.1021/jp5065006. [DOI] [PubMed] [Google Scholar]
- Nicoli F.; Roos M. K.; Hemmig E. A.; Antonio M.; Di De R. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA- Duplexes. 2019, 120, 9941–9947. 10.1021/acs.jpca.6b10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heussman D.; Kittell J.; Kringle L.; Tamimi A.; Von Hippel P. H.; Marcus A. H. Measuring Local Conformations and Conformational Disorder of (Cy3)2 Dimer Labeled DNA Fork Junctions Using Absorbance, Circular Dichroism and Two-Dimensional Fluorescence Spectroscopy. Faraday Discuss. 2019, 216, 211–235. 10.1039/C8FD00245B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banal J. L.; Kondo T.; Veneziano R.; Bathe M.; Schlau-Cohen G. S. Photophysics of J-Aggregate-Mediated Energy Transfer on DNA. J. Phys. Chem. Lett. 2017, 8, 5827–5833. 10.1021/acs.jpclett.7b01898. [DOI] [PubMed] [Google Scholar]
- Ikeda S.; Okamoto A. Hybridization-Sensitive On-Off DNA Probe: Application of the Exciton Coupling Effect to Effective Fluorescence Quenching. Chem. - Asian J. 2008, 3, 958–968. 10.1002/asia.200800014. [DOI] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. J- vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a near-IR Chiroptical Reporter. Chem. Commun. 2013, 49, 5298–5300. 10.1039/c3cc42103a. [DOI] [PubMed] [Google Scholar]
- Teo Y. N.; Kool E. T. DNA-Multichromophore Systems. Chem. Rev. 2012, 112, 4221–4245. 10.1021/cr100351g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringle L.; Sawaya N. P. D.; Widom J.; Adams C.; Raymer M. G.; Aspuru-Guzik A.; Marcus A. H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148, 085101. 10.1063/1.5020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashida H.; Asanuma H.; Komiyama M. Alternating Hetero H Aggregation of Different Dyes by Interstrand Stacking from Two DNA-Dye Conjugates. Angew. Chem., Int. Ed. 2004, 43, 6522–6525. 10.1002/anie.200460870. [DOI] [PubMed] [Google Scholar]
- Kashida H.; Tanaka M.; Baba S.; Sakamoto T.; Kawai G.; Asanuma H.; Komiyama M. Covalent Incorporation of Methyl Red Dyes into Double-Stranded DNA for Their Ordered Clustering. Chem. - Eur. J. 2006, 12, 777–784. 10.1002/chem.200500111. [DOI] [PubMed] [Google Scholar]
- Häner R.; Samain F.; Malinovskii V. L. DNA-Assisted Self-Assembly of Pyrene Foldamers. Chem. - Eur. J. 2009, 15, 5701–5708. 10.1002/chem.200900369. [DOI] [PubMed] [Google Scholar]
- Fujii T.; Kashida H.; Asanuma H. Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory. Chem. - Eur. J. 2009, 15, 10092–10102. 10.1002/chem.200900962. [DOI] [PubMed] [Google Scholar]
- Hara Y.; Fujii T.; Kashida H.; Sekiguchi K.; Liang X.; Niwa K.; Takase T.; Yoshida Y.; Asanuma H. Coherent Quenching of a Fluorophore for the Design of a Highly Sensitive In-Stem Molecular Beacon. Angew. Chem., Int. Ed. 2010, 49, 5502–5506. 10.1002/anie.201001459. [DOI] [PubMed] [Google Scholar]
- Malinovskii V. L.; Wenger D.; Häner R. Nucleic Acid-Guided Assembly of Aromatic Chromophores. Chem. Soc. Rev. 2010, 39, 410–422. 10.1039/B910030J. [DOI] [PubMed] [Google Scholar]
- Garo F.; Häner R. A DNA-Based Light-Harvesting Antenna. Angew. Chem., Int. Ed. 2012, 51, 916–919. 10.1002/anie.201103295. [DOI] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. Synthesis and Properties of Squaraine -Modified DNA. Org. Biomol. Chem. 2012, 10, 8944–8947. 10.1039/c2ob26787j. [DOI] [PubMed] [Google Scholar]
- Probst M.; Wenger D.; Biner S. M.; Häner R. The DNA Three-Way Junction as a Mould for Tripartite Chromophore Assembly. Org. Biomol. Chem. 2012, 10, 755–759. 10.1039/C1OB06400B. [DOI] [PubMed] [Google Scholar]
- Probst M.; Langenegger S. M.; Häner R. A Modular LHC Built on the DNA Three-Way Junction. Chem. Commun. 2014, 50, 159–161. 10.1039/C3CC47490A. [DOI] [PubMed] [Google Scholar]
- Li S.; Langenegger S. M.; Häner R. Control of Aggregation-Induced Emission by DNA Hybridization. Chem. Commun. 2013, 49, 5835–5837. 10.1039/c3cc42706d. [DOI] [PubMed] [Google Scholar]
- Cunningham P. D.; Díaz S. A.; Yurke B.; Medintz I. L.; Melinger J. S. Delocalized Two-Exciton States in DNA Scaffolded Cyanine Dimers. J. Phys. Chem. B 2020, 124, 8042–8049. 10.1021/acs.jpcb.0c06732. [DOI] [PubMed] [Google Scholar]
- Sohail S. H.; Otto J. P.; Cunningham P. D.; Kim Y. C.; Wood R. E.; Allodi M. A.; Higgins J. S.; Melinger J. S.; Engel G. S. DNA Scaffold Supports Long-Lived Vibronic Coherence in an Indodicarbocyanine (Cy5) Dimer. Chem. Sci. 2020, 11, 8546–8557. 10.1039/D0SC01127D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Díaz S. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Hestand N. J.; Spano F. C. Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev. 2018, 118, 7069–7163. 10.1021/acs.chemrev.7b00581. [DOI] [PubMed] [Google Scholar]
- Spano F. C.; Agranovich V.; Mukamel S. Biexciton States and Two-Photon Absorption in Molecular Monolayers. J. Chem. Phys. 1991, 95, 1400–1409. 10.1063/1.461121. [DOI] [Google Scholar]
- Abramavicius D.; Palmieri B.; Mukamel S. Extracting Single and Two-Exciton Couplings in Photosynthetic Complexes by Coherent Two-Dimensional Electronic Spectra. Chem. Phys. 2009, 357 (1–3), 79–84. 10.1016/j.chemphys.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramavicius D.; Palmieri B.; Voronine D. V.; Šanda F.; Mukamel S. Coherent Multidimensional Optical Spectroscopy of Excitons in Molecular Aggregates; Quasiparticle versus Supermolecule Perspectives. Chem. Rev. 2009, 109, 2350–2408. 10.1021/cr800268n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestand N. J.; Spano F. C. Molecular Aggregate Photophysics beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res. 2017, 50, 341–350. 10.1021/acs.accounts.6b00576. [DOI] [PubMed] [Google Scholar]
- Eisfeld A.; Kniprath R.; Briggs J. S. Theory of the Absorption and Circular Dichroism Spectra of Helical Molecular Aggregates. J. Chem. Phys. 2007, 126, 104904. 10.1063/1.2464097. [DOI] [PubMed] [Google Scholar]
- Fidder H.; Knoester J.; Wiersma D. A. Optical Properties of Disordered Molecular Aggregates: A Numerical Study. J. Chem. Phys. 1991, 95, 7880–7890. 10.1063/1.461317. [DOI] [Google Scholar]
- Hestand N. J.; Spano F. C. Determining the Spatial Coherence of Excitons from the Photoluminescence Spectrum in Charge-Transfer J-Aggregates. Chem. Phys. 2016, 481, 262–271. 10.1016/j.chemphys.2016.06.005. [DOI] [Google Scholar]
- Graugnard E.; Kellis D. L.; Bui H.; Barnes S.; Kuang W.; Lee J.; Hughes W. L.; Knowlton W. B.; Yurke B. DNA-Controlled Excitonic Switches. Nano Lett. 2012, 12, 2117–2122. 10.1021/nl3004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melinger J. S.; Khachatrian A.; Ancona M. G.; Buckhout-White S.; Goldman E. R.; Spillmann C. M.; Medintz I. L.; Cunningham P. D. FRET from Multiple Pathways in Fluorophore-Labeled DNA. ACS Photonics 2016, 3, 659–669. 10.1021/acsphotonics.6b00006. [DOI] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Díaz S. A. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Von Hippel P. H.; Johnson N. P.; Marcus A. H. Fifty Years of DNA “Breathing”: Reflections on Old and New Approaches. Biopolymers 2013, 99, 923–954. 10.1002/bip.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengel J.; Petersen M.; Frieden M.; Koch T. Chemistry of Locked Nucleic Acids (LNA): Design, Synthesis, and Bio-Physical Properties. Lett. Pept. Sci. 2003, 10, 237–253. 10.1007/BF02484561. [DOI] [Google Scholar]
- Wengel J. Synthesis of 3′-C- and 4’-C-Branched Oligodeoxynucleotides and the Development of Locked Nucleic Acid (LNA). Acc. Chem. Res. 1999, 32, 301–310. 10.1021/ar980051p. [DOI] [Google Scholar]
- Koch T. Locked Nucleic Acids: A Family of High Affinity Nucleic Acid Probes. J. Phys.: Condens. Matter 2003, 15, S1861–S1871. 10.1088/0953-8984/15/18/317. [DOI] [Google Scholar]
- Kim S.-K.; Retes P.; Linse K. D.; Castro P.; Castro M. Bridged Nucleic Acids (BNAs) as Molecular Tools. J. Biochem. Mol. Biol. Res. 2015, 1, 67–71. 10.17554/j.issn.2313-7177.2015.01.13. [DOI] [Google Scholar]
- Ivanova A.; Rösch N. The Structure of LNA:DNA Hybrids from Molecular Dynamics Simulations: The Effect of Locked Nucleotides. J. Phys. Chem. A 2007, 111, 9307–9319. 10.1021/jp073198j. [DOI] [PubMed] [Google Scholar]
- Rahman S. M. A.; Seki S.; Obika S.; Yoshikawa H.; Miyashita K.; Imanishi T. Design, Synthesis, and Properties of 2′,4′-BNANC: A Bridged Nucleic Acid Analogue. J. Am. Chem. Soc. 2008, 130, 4886–4896. 10.1021/ja710342q. [DOI] [PubMed] [Google Scholar]
- Wenge U.; Wengel J.; Wagenknecht H.-A. Photoinduced Reductive Electron Transfer in LNA:DNA Hybrids: A Compromise between Conformation and Base Stacking. Angew. Chem., Int. Ed. 2012, 51, 10026–10029. 10.1002/anie.201204901. [DOI] [PubMed] [Google Scholar]
- Petersen M.; Nielsen C. B.; Nielsen K. E.; Jensen G. A.; Bondensgaard K.; Singh S. K.; Rajwanshi V. K.; Koshkin A. A.; Dahl B. M.; Wengel J.; et al. The Conformations of Locked Nucleic Acids (LNA). J. Mol. Recognit. 2000, 13, 44–53. . [DOI] [PubMed] [Google Scholar]
- Kühn O.; Renger T.; May V. Theory of Exciton-Vibrational Dynamics in Molecular Dimers. Chem. Phys. 1996, 204, 99–114. 10.1016/0301-0104(95)00448-3. [DOI] [Google Scholar]
- Huff J. S.; Turner D. B.; Mass O. A.; Patten L. K.; Wilson C. K.; Roy S. K.; Barclay M. S.; Yurke B.; Knowlton W. B.; Davis P. H.; et al. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. 10.1021/acs.jpcb.1c04517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. NUPACK: Analysis and Design of Nucleic Acid Systems. J. Comput. Chem. 2011, 32, 170–173. 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- MELTING Online; https://www.ebi.ac.uk/biomodels-static/tools/melting/melt.php (accessed 2021-06-10).
- Kang J.; Kaczmarek O.; Liebscher J.; Dähne L. Prevention of H-Aggregates Formation in Cy5 Labeled Macromolecules. Int. J. Polym. Sci. 2010, 2010, 1. 10.1155/2010/264781. [DOI] [Google Scholar]
- Kasha M.; Rawls H. R.; El-Bayoumi M. A. The Exciton Model In Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. 10.1351/pac196511030371. [DOI] [Google Scholar]
- Berova N.; Di Bari L.; Pescitelli G. Application of Electronic Circular Dichroism in Configurational and Conformational Analysis of Organic Compounds. Chem. Soc. Rev. 2007, 36, 914–931. 10.1039/b515476f. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- McTigue P. M.; Peterson R. J.; Kahn J. D. Sequence-Dependent Thermodynamic Parameters for Locked Nucleic Acid (LNA)-DNA Duplex Formation. Biochemistry 2004, 43, 5388–5405. 10.1021/bi035976d. [DOI] [PubMed] [Google Scholar]
- Nielsen K. E.; Singh S. K.; Wengel J.; Jacobsen J. P. Solution Structure of an LNA Hybridized to DNA: NMR Study of the d(CT(L)GCT(L)T(L)CT(L)GC):D(GCAGAAGCAG) Duplex Containing Four Locked Nucleotides. Bioconjugate Chem. 2000, 11, 228–238. 10.1021/bc990121s. [DOI] [PubMed] [Google Scholar]
- Jensen G. A.; Singh S. K.; Kumar R.; Wengel J.; Jacobsen J. P. A Comparison of the Solution Structures of an LNA:DNA Duplex and the Unmodified DNA:DNA Duplex. J. Chem. Soc. Perkin Trans. 2 2001, (7), 1224–1232. 10.1039/b008431j. [DOI] [Google Scholar]
- Tolstrup N.; Nielsen P. S.; Kolberg J. G.; Frankel A. M.; Vissing H.; Kauppinen S. OligoDesign: Optimal Design of LNA (Locked Nucleic Acid) Oligonucleotide Capture Probes for Gene Expression Profiling. Nucleic Acids Res. 2003, 31, 3758–3762. 10.1093/nar/gkg580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe J. R.; Nikolova E. N.; Eichhorn C. D.; Chugh J.; Hansen A. L.; Al-Hashimi H. M. Characterizing RNA Dynamics at Atomic Resolution Using Solution-State NMR Spectroscopy. Nat. Methods 2011, 8, 919–931. 10.1038/nmeth.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasha M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55. 10.2307/3571331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.