

Abstract
The cytokines of the transforming growth factor–β (TGF-β) family promote the growth and differentiation of multiple tissues but the role of only the founding member, TGF-β, in regulating the immune responses has been extensively studied. TGF-β is critical to prevent the spontaneous activation of self-reactive T cells and sustain immune homeostasis. In contrast, in the presence of proinflammatory cytokines, TGF-β promotes the differentiation of effector T helper 17 (TH17) cells. Abrogating TGF-β receptor signaling prevents the development of interleukin-17 (IL-17)–secreting cells and protects mice from TH17 cell–mediated autoimmunity. Here, we found that the receptor of another member of TGF-β family, bone morphogenetic protein receptor 1α (BMPR1α), regulates T helper cell activation. We found that the differentiation of TH17 cells from naïve CD4+ T cells was inhibited in the presence of BMPs. Abrogation of BMPR1α signaling during CD4+ T cell activation induced a developmental program that led to the generation of inflammatory effector cells expressing large amounts of IL-17, IFN-γ, TNF family cytokines, and transcription factors defining the TH17 cell lineage. We found that TGF-β and BMPs co-operated to establish effector cell functions and the cytokine profile of activated CD4+ T cells. Together, our data provide insight into the immunoregulatory function of BMPs.
Introduction
The transforming growth factor-β (TGF-β) family of cytokines are important for promoting the homeostasis of various tissues and the differentiation of select immune cell subsets (1, 2). Family members, including TGFβ and bone morphogenetic proteins (BMPs), are produced by both stromal and immune cells (3, 4). Different TGF-β family cytokines may have overlapping or opposing functions, although these effects are often dependent on cellular and cytokine context. A founding member of the family, TGF-β, is an essential regulator of T cell development and constrains self-reactivity of peripheral T cells and their responses to antigenic stimulation (5, 6). Mice deficient in TGF-β1 gene succumb to uncontrolled inflammation and systemic, lethal autoimmune disease, which is mediated by exaggerated activation of T helper (Th) effector cells, which produce IFN-γ (Th1) or interleukin-4 (Th2) (7, 8). These effects are due, in part, to the requirement of TGF-β for the development of Foxp3+ regulatory T (Treg) cells, an immunoregulatory T cell subset (9, 10). Thus, TGF-β exerts cell-intrinsic and cell-extrinsic effects that prevent the activation of self-reactive peripheral T cells (11). Conversely, in an inflammatory environment TGF-β supports the generation of pathogenic Th17 cells, which secrete IL-17 (12). Loss of TGF-β signaling in T cells prevents IL-17 secretion and protects mice from autoimmune encephalomyelitis after immunization with self-antigen (13). In promoting Th17 cell generation, TGF-β cooperates with the pro-inflammatory cytokines IL-1, IL-6 or IL-21 and IL-23 (14-17). Activated T cells, including Th17 and Treg cells are the main source of TGF-β, which induces and sustains Th17 cell differentiation (18). These findings underscore the importance of context-sensitive signaling and autocrine or paracrine TGF-β production for local immunoregulation.
Despite our increased understanding of how TGF-β regulates T cell functions, the immunoregulatory roles of many other members of the TGF-β cytokine family, especially bone morphogenetic proteins (BMPs) remain largely unknown. BMPs represent the largest subgroup of TGF-β cytokine family (19). They control a wide range of biological activities in various cell types and play critical roles in morphogenesis of various tissues and organs. BMPs bind heteromeric complexes of type I (BMP receptor 1α, BMPR1α and receptor 1β, BMPR1β) and type II (BMPR2) receptors to activate signal transduction pathways involving Mothers Against Decapentaplegic homologs (or Smad)1/5/8. BMPs can also regulate myeloid, B, Natural killer, and peripheral T cells during infection, inflammation, and cancer (4, 20, 21). BMP2/4 or activin A increase the ability of TGF-β to promote generation of adaptive Treg cells (aTreg), which arise during immune responses to limit inflammation (20, 22). Furthermore, BMPs increase phosphorylation of Runt-related transcription factor 1 (Runx1), which promotes IL-2 gene expression in conventional T cells and, in concert with Foxp3, inhibits in Treg cells (23). BMPs are also involved in restraining inflammation, but their exact role remains controversial (3).
To test the role of BMPs in peripheral T cells we generated mice lacking BMPR1α in T cells and found that BMPR1α-deficient T cells preferentially differentiated into Th17 cells after activation. Transcriptome analysis suggested that loss of BMPR1α enhanced expression of transcripts involved in Th17 lineage commitment and inflammatory effector T cells. We showed that immunization with Complete Freund’s Adjuvant (CFA) stimulated stronger pro-inflammatory responses in BMPR1α-deficient mice than in wild-type mice. Similarly, adoptive transfer of BMPR1α-deficient CD4+ cells into lymphopenic hosts induced more severe colitis than cells isolated from wild-type mice. These findings indicate that BMPs may oppose TGF-β signaling in CD4+ T cells and describe a role for BMPR1α during CD4+ T cell lineage commitment and effector responses. Our data underscore the importance of interactions between TGF-β family members in immunoregulation.
Results
BMPs inhibit Th17 differentiation
To understand whether BMPs may directly affect T cell responses, we examined the expression of BMP receptors in murine T cell subsets. We found that activated cells expressed more BMPR1α transcript and protein than naive and Treg cells (Fig. 1A and B). Similarly, after in vitro stimulation we observed that BMPR1α transcription was quickly increased in CD4+ T cells (Fig. 1C). In contrast, BMPR2 transcripts were highly expressed in Treg cells and weren’t affected by T cell activation (fig. S1A). Thus, our data suggest that activated CD4+ T cells may be preferentially sensitive to BMPs because of activation-induced expression of BMPR1α.
Fig. 1. BMP signaling regulates T cell activation and lineage commitment.
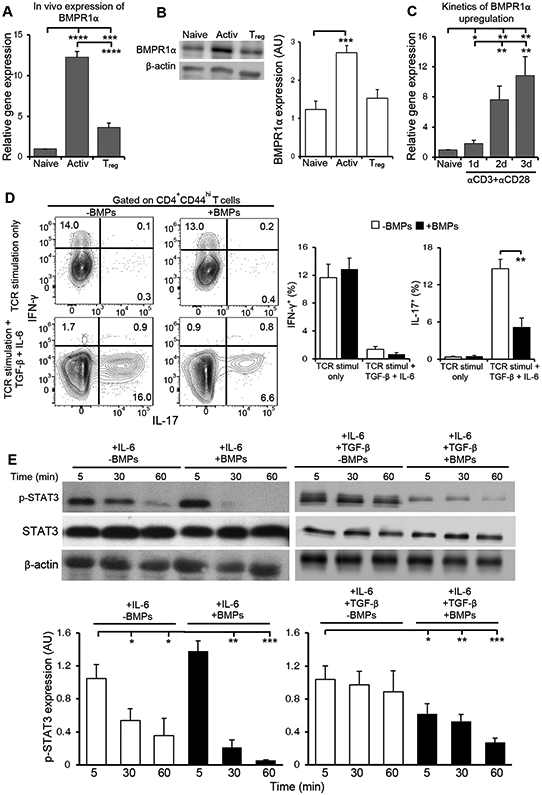
(A) qRT-PCR analysis of BMPR1α mRNA expression in sorted CD4+ naive, activated and Treg cells. Data are means ± SD pooled from 3 independent sorts. (B) Western blot analysis of BMPR1α abundance in lysates from sorted CD4+ naive, activated and Treg cells. Blots (left) are representative of three independent experiments. Normalized band intensity data (right) are means ± SD pooled from all experiments. (C) qRT-PCR analysis of BMPR1α in sorted, naive CD4+ T cells stimulated with antibodies against CD3 and CD28 for the indicated times. Results are means ± SD pooled from three independent experiments. (D) Flow cytometry analysis of IFN-γ and IL-17 in CD4+ T cells stimulated with antibodies against CD3 and CD28 for 4 days and BMP2/4/7 or TGF-β and IL-6, as indicated. Dot plots (left) representative of four independent stimulations. The frequency of cytokine producing cells (right) are means ± SD pooled from all experiments. (E) Western blot analysis of p-STAT3 and STAT3 in CD4+ T cells stimulated for the indicated times with IL-6 or IL-6 and TGF-β in the absence or presence of BMP2/4/7. Blots (top) are representative of four independent experiments. Normalized band intensity data (bottom) are means ± SD pooled from all experiments. *P<0.05, **P< 0.01, ***P< 0.001, ****p< 0.0001 as determined by Student’s t- test.
To determine the effects of BMPs on activated T cells, naive CD4+ T cells were stimulated with plate-bound antibodies against CD3 and CD28 in the absence or presence of BMP 2, 4 and 7. When cytokines were included to skew differentiation into Th1, Th2 and Th17 cells, we found that BMPs did not inhibit expression of T-bet and IFN-γ, which are essential for the generation of Th1 effector cells (fig. S1B). Similarly, BMPs did not inhibit expression of GATA3 and IL-4, which promote development of Th2 effector cells. Remarkably, BMP exposure inhibited the expression of IL-17 (fig. S1B). Flow cytometry analysis confirmed these results, indicating that whereas BMPs did not affect IFN-γ expression, BMP exposure greatly reduced the secretion of IL-17 (Fig. 1D). Together, our results indicate that direct BMPs stimulation restricts the differentiation of Th17 cells.
To understand how BMP signaling inhibits Th17 cell differentiation, we tested the effects of BMPs on STAT phosphorylation downstream of TGF-β and IL-6 receptor stimulation. We found that BMPs treatment reduced IL-6-mediated STAT3 activation. Although the initial phosphorylation of STAT3 in CD4+ T cells activated in the presence of IL-6 was comparable with and without BMPs, it was sustained only in the absence of BMPs (Fig. 1E). In CD4+ T cells activated in the presence of both IL-6 and TGF-β, STAT3 phosphorylation was much stronger and stable over time in the absence of BMPs (Fig. 1E). Given that the transcription of Rorc and IL-17 are STAT3-dependent, these data suggest that BMPs reduce the activity of signaling pathways necessary for the development of Th17 cells (24).
BMPR1α signaling controls generation of Th17 CD4+ T cells in vivo
To further determine the effects of BMP signaling, we used previously characterized mice lacking the BMPR1α gene in T cells (BMPR1αT−) (25, 26). Loss of BMPR1α does not alter the proportions or absolute T cell numbers in the thymus and peripheral organs, with the exception that fewer Treg cells developed than in wild-type mice (26). Even with a decreased proportion of Treg cells, BMPR1α-deficiency in T cells did not precipitate autoimmune disease, even in mice up to 6 months old. Deletion of the BMPR1α gene abrogated BMP signaling as indicated by the loss of Smad5phosphorylation in activated CD4+ T cells (Fig. 2A). However, we found no change in the phosphorylation of Smad2, which is downstream of the TGF-β receptor (Fig. 2A). Thus, these data validate efficient specific inactivation of the BMP receptor (1). When BMPR1α-sufficient and -deficient CD4+ T cells were activated in vitro, without exogenous cytokines, both cell types increased activation markers CD44, CD25 and decreased CD62L, but a larger fraction of BMPR1α-deficient CD4+ T cells produced IFN-γ (Fig. 2, B and C). Consistent with our earlier observation that BMPs stimulation reduces Th17 development (Fig. 1D), we found that in the presence of TGF-β and IL-6 activation of BMPR1α-deficient CD4+ T cells generated more IL-17 producing cells than activation of wild-type cells (Fig. 2D). This effect was dependent upon the combined effects of both the inflammatory cytokine IL-6 and TGF-β (fig. S2). In contrast to studies using the small molecule dorsomorphin, an AMPK inhibitor that blocks BMP receptor signaling and impedes Th17 cell differentiation, these data demonstrated that under Th17 polarizing conditions BMPR1α-deficient CD4+ T cells preferentially differentiated into Th17 cells in vitro (27).
Fig. 2. BMPR1α restricts CD4+ T cells IL-17 production.
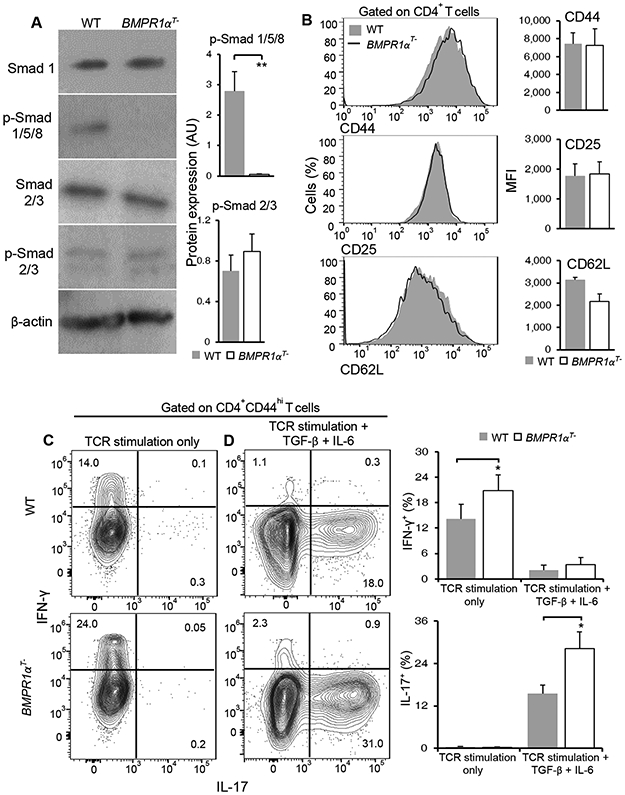
(A) Western blot analysis of total and phosphorylated Smad1/5/8 and Smad 2/3 in activated wild-type (WT) and BMPR1αT− CD4+ T cells. Blots (left) are representative of three independent experiments. Normalized band intensity data (right) are means ± SD pooled from all experiments. (B) Flow cytometry analysis of the indicated cell surface markers on activated WT and BMPR1αT− CD4+ T cells. Histograms (left) are representative of four independent analyses. Mean florescence intensity (MFI) data (right) are means ± SD from all experiments. (C and D) Flow cytometry analysis of cytokine production by CD4+ T cells from WT and BMPR1αT− mice stimulated with antibodies against CD3 and CD28 in medium alone (C), or in the presence of TGFβ and IL-6 (D), as indicated. Contour plots are representative of four independent stimulations. The frequency of cytokine producing cells are means ± SD pooled from all experiments. *P<0.05, **P< 0.01, as determined by Student’s t-test.
To understand the effects of BMP signaling in vivo, we characterized the response of wild-type and BMPR1αT− mice immunized with CFA. Flow cytometry analysis of the draining lymph nodes indicated that a greater proportion of BMPR1α-deficient CD4+ T cells than wild-type CD4+ T cells expressed markers of activation, which include decreased CD62L and increased CD44, CD25, 4-1BB, and ICOS. These data suggested that absence of BMPR1α is not associated with impaired activation (Fig. 3A). Similar to our in vitro results, we found that CD4+ T cells activated in BMPR1αT− mice produced more IFN-γ and IL-17 secreting effector cells upon antigenic re-stimulation than CD4+ T cells activated in wild-type mice (Fig. 3B). The stronger inflammatory responses of mice lacking BMPR1α in T cells resulted in increased footpads swelling when compared to wild-type mice (Fig. 3C). Consistent with the known role of TGF-β in Th17 cell differentiation in the context of inflammation, these data indicated that abrogation of BMPR1 α signaling further increased proportion of CD4+ T cells producing IL-17 (12). Thus, our analysis suggests that BMPR1α signaling in CD4+ T cells opposes TGFβ-mediated Th17 differentiation.
Fig. 3. BMPR1α reduces the generation of pro-inflammatory CD4+ T cells in vivo.

(A) Flow cytometry analysis of the indicated activation markers on CD4+ T cells isolated from the popliteal lymph nodes of WT and BMPR1αT− mice injected with CFA in the footpad and analyzed after 5 days. Contour plots are representative of three independent experiments. The frequency of naive, activated, and CD25+ cells and activation marker MFI are means ± SD from all experiments. (B) ELISPOT analysis of cytokine production by T cells isolated from WT and BMPR1αT− mice 7 days after booster immunization with class II MHC restricted LCMV gp61-80 peptide in CFA. Images (left) were quantified spot forming units (SFU). Quantified data (right) are means ± SD pooled from three independent experiments. (C) Analysis of footpad thickness in WT and BMPR1αT− mice injected with CFA in the footpad and analyzed after 5 days. Images (top) are representative of three independent experiments. Quantified data (bottom) are means ± SD pooled from all experiments. *P< 0.05, **P< 0.01, ***P< 0.001, as determined by Student’s t-test.
Cell-intrinsic BMPR1α activity controls Th17 lineage specification
The effects of BMPR1α-deficiency on Th17 lineage bias may be due to their increased cytokine responses, because of an effect of BMPR1α during T cell development, or due to a cell-intrinsic effect of BMPR1α. To begin to address this question in T cells with a fixed antigen specificity and affinity, we crossed BMPR1αT− mice to mice expressing a transgenic TCR specific for an analog of pigeon cytochrome C (PCC) (28). Similar to our earlier results (Fig. 2C), when purified transgenic CD4+ T cells from BMPR1αT− mice were activated by peptide antigen, these cells produced more IFN-γ secreting cells than cells isolated from wild-type mice (fig. S3A). To mimic an infection-like environment we added a low concentration of LPS to cultured cells. Antigen specific cells activated in the presence of LPS, BMPR1αT− CD4+ T cells were biased to differentiate into Th17 cells (fig. S3B), similar to stimulation in the presence of exogenous IL-6 and TGF-β (fig. S3C). These data indicate that the preference for Th17 development is not due to gross changes in the TCRs that are selected for during T cell development. Furthermore, when PCC-specific BMPR1α-deficient and -sufficient CD4+ T cells were mixed together and co-cultured in the presence of either IL-6 and TGF-β, or IL-21 and TGF-β, a larger proportion of BMPR1α-deficient cells secreted IL-17 than wild-type cells (Fig. 4A). In this experiment, the abundance of CD25 was similar on both populations of CD4+ T cells treated with TGF-β and IL-6 (Fig. 4B), indicating there was no defect in activation in this condition. Furthermore, the abundance of CCR6 was increased in BMPR1α-deficient cells and the abundance of CCR5 was the same in both populations (Fig. 4B). Together, these data indicate that cell-intrinsic BMPR1α signaling biases CD4+ T cell development towards the Th17 lineage.
Fig. 4. Cell-intrinsic BMPR1α expression suppresses the generation of Th17 CD4+ T cells.
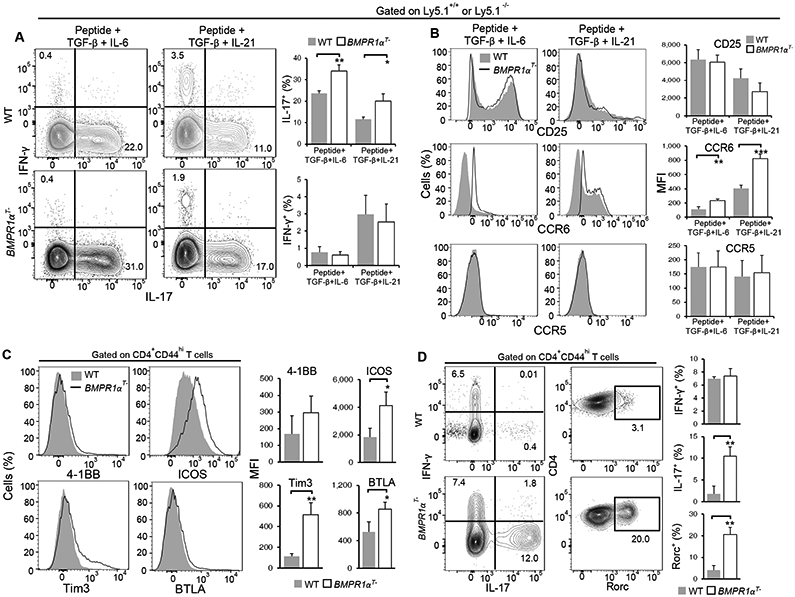
(A and B) Flow cytometry analysis of IFN-γ and IL-17 production (A) and surface marker expression (B) on co-cultured TCR transgenic WT and BMPR1αT− CD4+ T cells stimulated with antigenic peptide and the indicated cytokines for 6 days. Contour plots and histograms (left) are representative of three independent experiments. The frequency of cytokine producing cells and marker mean fluorescence intensity (MFI) (right) are means ± SD from all experiments. (C and D) Flow cytometry analysis of surface marker expression (C), IFN-γ and IL-17 production (D) and Rorc abundance (D) in lymph node T cells isolated 5 days after TCR transgenic WT and BMPR1αT− mice were immunized with antigenic peptide and CFA. Histograms and contour plots (left) are representative of three independent experiments. The frequency of cytokine producing cells and marker MFI (right) are means ± SD from all experiments. *P< 0.05, **P< 0.01, ***P< 0.001, as determined by Student’s t-test.
To compare activation of antigen specific cells in vivo, we immunized wild-type and BMPR1αT− mice expressing transgenic TCR with PCC peptide and CFA. Analysis of T cell populations in popliteal lymph nodes demonstrated increased activation of BMPR1α-deficient CD4+ T cells, as indicated by increased amounts of the co-stimulatory molecules 4-1BB, ICOS, and Tim3, and inhibitory molecule BTLA (Fig. 4C). Whereas IFN-γ was produced by activated CD4+ T cells both in wild-type and BMPR1αT− mice, substantial numbers of IL-17 producing cells were only present in mice lacking BMPR1α in T cells (Fig. 4D). Consistent with increased cytokine production in BMPR1αT− mice, the proportion of cells expressing Rorc was also increased in BMPR1αT− mice when compared to wild-type mice (Fig. 4D). These results suggest that under inflammatory conditions in vivo, BMPR1α-deficient cells differentiated primarily into IL-17 producing CD4+ T cells.
RNAseq identifies BMPR1α controls Th17 cell effector functions
To determine how BMPR1α signaling affects molecular circuits controlling CD4+ T cell lineage commitment, we examined the global gene expression profiles of naive and activated wild-type and BMPR1αT− CD4+ T cells using RNA sequencing (RNAseq). Overall, when gene expression profiles were compared, 4183 and 4746 genes were found differentially expressed between naive and activated BMPR1α-sufficient and -deficient CD4+ T cells, respectively (fig. S4, A and B). A subset of 678 of these genes were differentially expressed between activated BMPR1α-sufficient and -deficient CD4+ T cells. Principal component analysis performed on both activated cells subsets demonstrated different gene expression profiles. (Fig. 5, A and B and Table S1). When we interrogated the expression of genes assembled from published reports that define Th1, Th2, Th17 and aTreg cell subsets, we found that T-bet, IFN-γ, IL-12rb1, IL-12rb2, IL-17a, IL- 17f, Rorc, Rora, IL-21, Stat1 and Stat4 were highly expressed in both BMPR1α-deficient and - sufficient CD4+ T cells (fig. S4C) (29-33). All of these genes are essential to establish and sustain Th1 and in particular, Th17 cell differentiation (24, 34). We found that BMPR1α-deficient cells expressed more transcripts for pivotal genes regulating Th17 cell specification and effector functions including Rorc, IL-23R, IL-17a and IL-17f. This pattern of gene expression is remarkably different from activated CD4+ T cells developing in TGF-β- or TGF-βRII-deficient mice, which do not express genes associated with Th17 cell differentiation and remain protected from Th17 cell mediated autoimmunity (7, 8, 11, 35). Activated BMPR1α-deficient cells also did not express or expressed only low amounts of Th2 cell signature genes like Gata3, IL-4, IL-4ra, IL-5 and Areg but expressed higher amounts of IL-13 (31). Similarly, BMPR1α-deficient cells did not increase transcription of genes associated with aTreg generation, such as Foxp3, Nr4a1, Nr4a3 and Gpr83 (36-38).
Fig. 5. BMPR1α alters Th1/Th17 gene expression.

(A) Principal component analysis of genes expressed by activated BMPR1α-sufficient and -deficient CD4+ T cells. Analysis included gene expression data from three samples of BMPR1α-sufficient and -deficient cells. (B) Genes differentially expressed in activated CD4+ T cells from WT and BMPR1αT− mice. Analysis included genes differentially expressed between three samples of BMPR1α-sufficient and - deficient cells. Colors show log2 fold change of the expression difference. (C) Metascape GO analysis of genes differentially expressed by activated WT and activated BMPR1α-deficient CD4+ T cells. (D) Transcript levels, fold change expression and p-values of genes differentially expressed in activated CD4+ T cells from WT and BMPR1αT− mice, related to inflammatory, immune and metabolic responses. Vertical bars represent average values of gene expression levels (fpkm) ± SD. All data are representative of three biological replicates.
When we performed gene set enrichment analysis using Metascape web suite to associate gene sets with cellular processes described by Gene Ontology (GO) terms, we found that the network topology indicated extensive connections between clusters and substantial overlap of gene sets (Fig. 5C and Table S2) (33). All major clusters of GO terms were broadly related to inflammatory and immune response, control of cytokine production, signaling and cell adhesion. Separate network clusters related to TNF, IL-17, IL-12, IL-13 and Jak-STAT signaling, indicated molecular changes in these pathways differentially affected activated wild-type and BMPR1α-deficient CD4+ T cells. By focusing on biological processes primarily related to inflammatory and immune responses we found that activated BMPR1α-deficient CD4+ T cells overexpressed transcripts for the cytokines Tnf, lymphotoxin (LT-α), Csf2 (GM-CSF), RANKL (TNFSF11), and IL-10 (Fig. 5D). This cytokine pattern largely overlaps with the transcriptional signatures of in vitro generated Th17 cells and is consistent with pathological Th17 cells found in autoimmune diseases (17, 24, 34). Furthermore, BMPR1α-deficient cells expressed higher amounts of transcripts encoding receptors for IL-1 (IL-1R1), IL-17c (IL-17RE) and IL-23 (IL-23R). All three cytokines are either essential to promote induction and terminal maturation of Th17 cells or are required to sustain their effector functions (12, 16, 39-42). BMPR1α-deficient cells had elevated expression of signaling molecules that facilitate Th17 generation and increase production of pro-inflammatory cytokines, such as Aif1, Cish, and Dusp14 (43-45). BMPR1α-deficient cells also displayed elevated expression of proenkephalin (Penk), Tlr6, IRAK1BP1, and coronin2a (Coro2a), which promote production of pro-inflammatory cytokines and Th1/Th17 responses, and Msh5 and Nqo1, which protects cells against hypoxia and oxygen radicals generated during an inflammatory response (46-51). In summary, the gene expression pattern of Th lineage canonical markers substantiated functional and flow cytometry studies and strongly supports our conclusion that deficient BMPR1α signaling impacts Th lineage commitment by promoting generation of Th17 effector cells.
Loss of BMPR1α alters cytokine and TCR-dependent signaling in CD4+ T cells
Th17 lineage bias could be caused by modulation of BMP and/or TGF-β signaling pathways resulting from absence of functional BMPR1α. Downregulation of Smad4, a shared component of BMP and TGF-β signaling pathways, leads to Th17 cell mediated gastrointestinal inflammation (52). However, our analysis showed equal quantities of Smad4 were present in naive and activated, BMPR1α-deficient and sufficient CD4+ cells (Fig. 6A). We also established that phosphorylation of Smad2/3 in response to different concentrations of TGF-β was the same in BMPR1α-sufficient and -deficient CD4+ T cells (Fig. 6B). Thus, the lack of BMPR1 α did not affect T cell sensitivity to TGF-β signaling. In fact, stimulation with TGF-β does not change BMP2/4 induced Smad1 phosphorylation, further suggesting that membrane proximal signals mediated by either TGF-β or BMPR1α are independent in T cells (20).
Fig. 6. BMPR1α mediates TCR and cytokine-dependent signaling.
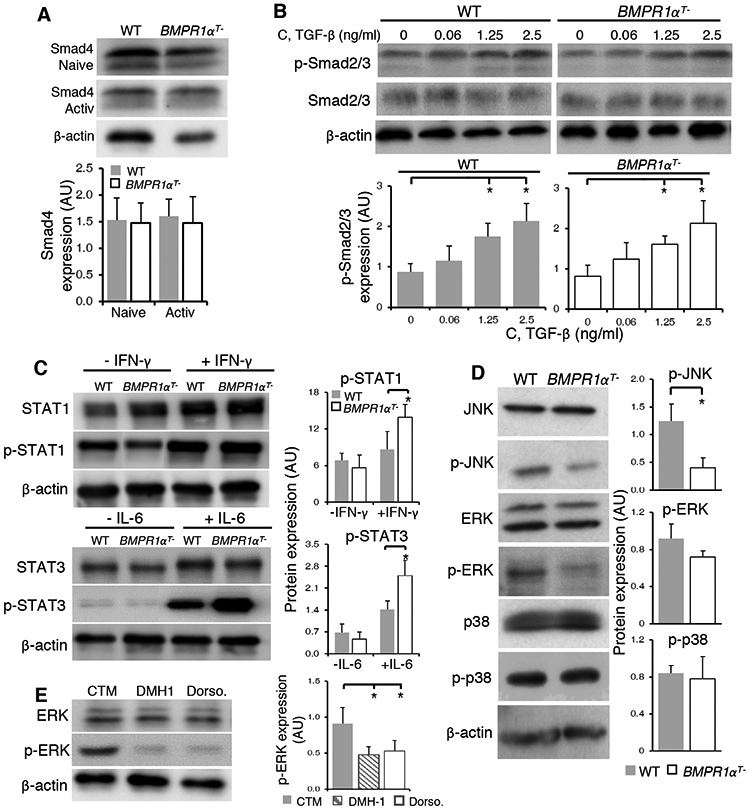
(A) Western blot analysis of Smad4 in naive and activated CD4+ T cells. Blots (top) are representative of four independent experiments. Normalized band intensity data (bottom) are means ± SD from all experiments. (B) Western blot analysis of total and phosphorylated Smad 2/3 in CD4+ T cells from WT and BMPR1αT− mice stimulated for 1 hour with plate-bound antibodies against CD3 and CD28 in the presence of indicated concentrations of TGF-β. Blots (top) are representative of four independent stimulations. Normalized band intensity data (bottom) are means ± SD from all experiments. (C) Western blot analysis of STAT1 and STAT3 phosphorylation in lysates from WT or BMPR1αT− CD4+ T cells directly isolated and lysed (−IFN-γ and −IL-6) or activated for 5 min with plate-bound antibodies against CD3 and CD28 in the presence of IFN-γ or IL-6. Blots (left) are representative of four independent experiments. Normalized band intensity data (right) are means ± SD from all experiments. (D) Western blot analysis of Jnk, Erk and p38 phosphorylation in lysates from WT or BMPR1αT− CD4+ T cells activated with Con A for 2 days. Blots (left) are representative of four independent experiments. Normalized band intensity data (right) are means ± SD from all experiments. (E) Western blot analysis of Erk phosphorylation in WT CD4+ T cells activated with plate-bound antibodies against CD3 and CD28 in the presence of inhibitors DMH1 or dorsomorphin for 24 hours. Blots (left) are representative of three independent experiments. Normalized band intensity data (right) are means ± SD from all experiments. *P< 0.05, as determined by Student’s t-test.
The Th17 lineage bias that we observed could result from BMPR1α signaling affecting events downstream of cytokine receptors. To examine this possibility, we probed the phosphorylation of STAT3 and STAT1, after stimulation of wild-type and BMPR1α-deficient CD4+ T cells with IL-6 and IFN-γ. We found that STAT3 and STAT1 are hyper-phosphorylated in activated BMPR1α-deficient cells after cytokine stimulation (Fig. 6C). Increased STAT3 and STAT1 phosphorylation in BMPR1α-deficient cells is associated with decreased expression of inhibitory molecules SOCS3 and SIGIRR (IL-1R8) known to inhibit IL-17 and IFN-γ production (Fig. 5D) (53-56). Another signaling molecule, Cish, elevated in BMPR1α-deficient cells, decreases TCR signaling but also inhibits IL-2 signaling, mediated by STAT5, which increases Th17 cell differentiation (44, 57, 58). In summary, gene expression changes resulting from impaired BMP signaling support the intrinsic bias of BMPR1α-deficient cells to differentiate into Th17 effector cells and are consistent with the BMP inhibition of Th17 generation in wild-type cells.
Sensitivity to TGF-β signaling in T cells is set in response to TCR stimulation by MAP activation of kinase pathways, which promote T cell proliferation and differentiation into effector cells (59). To investigate signaling events downstream of the TCR we examined phosphorylation of JNK, ERK and p38 kinases in wild-type and BMPR1α-deficient CD4+ T cells after stimulation with antibodies against CD3 and CD28 (Fig. 6D). MAP kinases are known to phosphorylate the linker region of R-Smads regulating not only their nuclear translocation and degradation but also induction of Rorc and IL-17 transcription (60, 61). Whereas p38 and Erk phosphorylation were similar in wild-type and BMPR1α-deficient cells, JNK activity was reduced in activated BMPR1α-deficient cells. This is consistent with a report of JNK pathway inhibiting Th17 cell generation (62). JNK activity may be further reduced in BMPR1α-deficient cells by upregulation of DUSP16 phosphatase known to preferentially dephosphorylate JNK (Fig. 5D) (45, 63). In contrast, ERK, but not p38 or JNK phosphorylation was decreased in CD4+ T cells activated in the presence of small molecule inhibitors of BMPR1α signaling and other kinases, which also decrease Th17 cell differentiation (Fig. 6E and fig. S5) (64, 65). Collectively, these results underscore the importance of crosstalk between signaling pathways in regulating Th17 lineage specification but also point to differences between targeted genetic ablation of BMPR1α and use of BMP signaling inhibitors.
BMPR1α-deficient CD4+ T cells are highly pathogenic in vivo
To understand the consequence of BMPR1α-mediated suppression of IL-17 effector cell development, we investigated immunopathology in a mouse model of inflammatory bowel disease (66, 67). Naive CD4+CD45RBhigh cells sorted from either wild-type and BMPR1αT− mice were transferred into TCRα chain knockout mice (TCRα−), and recipients were monitored and scored for weight loss and hallmark symptoms of colitis. We found that weight loss was comparable (Fig. 7A), however other colitis symptoms were more severe in recipients of BMPR1α-deficient cells than recipients of wild-type cells (Fig. 7, B and C). Necropsy analysis revealed recipients of BMPR1α-deficient cells had shorter colon length than recipients of wild-type cells, indicative of more severe inflammation and colitis (Fig. 7D). We also found by histological analysis that recipients of wild-type and BMPR1α-deficient cells had prominent lymphocytic infiltrate involving mucosa and submucosa (Fig. 7E). When lamina propria cells were isolated and stained for cytokines and Rorc expression, recipients of BMPR1α-deficient cells expressed much higher levels of inflammatory cytokines, IFN-γ and IL-17, and transcription factor Rorc than recipients of wild-type cells (Fig. 7F). Sorted BMPR1α-deficient donor CD4+ T cells from lamina propria expressed more IL-6, IL-1β and IL-17 pro-inflammatory cytokine transcripts than wild-type donor cells (Fig. 7G). Similar results were obtained after feeding mice dextran sodium sulfate to chemically induce colitis (fig. S6). Collectively, these results demonstrated that BMPR1α-deficient effector CD4+ T cells mediate an exaggerated inflammatory response and can contribute to immune pathology.
Fig. 7. Adoptive transfer of BMPR1α-deficient CD4+CD45RBhigh cells induces more severe colitis.

(A to F) TCRα− recipient mice received a transfer of either WT or BMPR1αT− CD4+CD45RBhigh cells. (A to C) Weight loss, bleeding and diarrhea were scored over time. (D) Total colon length was assessed on day 21. (E) Histological analysis of colonic cross-sections by hematoxylin and eosin staining was performed on the indicated mice on day 21. Scale bars equal 100 μM. (F) Flow cytometry analysis of cytokines and Rorc in CD4+ T cells isolated from the colons of recipient mice. (G) qRT-PCR analysis of IL-6, IL-1β, IL-17, Rorc and IFN-γ transcripts in CD4+ T cells isolated by flow cytometry sorting from colons of recipient mice. All results are representative of four mice per group analyzed in three independent experiments. Quantified data are means ± SD from all experiments. *P< 0.05, **P< 0.01, ***P< 0.001, ****p< 0.0001, as determined by Student’s t-test.
Discussion
While the many functions of TGF-β in regulating immune system homeostasis and generation of T cell subsets have been extensively studied, the role of BMPs and their receptors in controlling initiation, maintenance and resolution of immune responses, is much less appreciated (4). Due to the number of BMP ligands far exceeding the number of available receptors, it is more practical to study the role of BMPs by eliminating receptors. This strategy also avoids confusion associated with diverse effects of BMPs on multiple lineages of hematopoietic cells and possible indirect effects on T cells. Additionally, this approach has previously been established where deleting transforming growth factor beta receptor II (TGF-βRII) was used to assess the role of TGF-β in T cells (7, 8). We show that BMPR1α is weakly expressed in naive cells and expression is strongly enhanced upon activation. Thus, studying the effects of deleting BMPR1α gene provided insight on the immunoregulatory role of BMPs in activated T cells.
Deletion of BMPR1α gene did not substantially impact survival and expansion of peripheral CD4+ T cells. Although wild-type and BMPR1αT− mice have similar proportions and absolute cell numbers in the thymus and peripheral organs, in the absence of BMPR1α, Treg cells are reduced (26). In contrast to T cell specific deletion of TGF-βRII, which causes lymphoproliferation, multi-organ leukocyte infiltration and early lethality in mice, loss of BMPR1α did not result in similar spontaneous autoimmune phenotype (7, 8). This lack of prominent autoimmunity in BMPR1αT− mice may be explained by our data indicating that there is a low amount of BMPR1α in naive CD4+ T cells, making these cells less sensitive to the lack of BMPs. Only after activation did BMPR1α-deficient cells produced higher proportions of IFN-γ secreting cells than wild-type cells, which correlated with the increased abundance of BMPR1α after activation.
We showed that in wild-type CD4+ T cells, BMPs inhibit induction of Rorc and IL-17 expression. This effect of BMPs on the transcriptional program of T cell activation is associated with impaired phosphorylation of STAT3, indicating crosstalk between signaling pathways downstream of IL-6 receptor and BMPR1α. However, BMPR1α-deficient cells also generated more IFN-γ secreting cells after in vitro stimulation or after stimulation in vivo. This suggested that BMPR1α may also suppress Th1 development in conditions which do not support Th17 cell differentiation. In contrast, when BMPs were directly added to T cell culture medium, which may activate multiple receptors, we found no effect on Th1 cells differentiation. Increased Th1 differentiation of BMPR1α-deficient cells resembled CD4+ T cells with impaired TGF-β signaling, which do not differentiate into Th17 cells, but instead produce more Th1 cells when activated in conditions inducing Th17 cells (13, 39). At the same time, activation of CD4+ T cells in the presence of LPS, Mycobacterium tuberculosis or zymosan or selective neutralization of IL-6 or TGF-β changed proportions of Th1 and Th17 cells (12). Additionally, abrogation of BMPR1α signaling may also dysregulate BMP-dependent control of receptors for IL-1, IL-23 and IL-17 and downstream signaling molecules. We also observed that expression of metabolic regulators such as Msh5, NqoI and Egln3, and molecules controlling response to pro-inflammatory cytokines such as SOCS3, Sigirr and Cish was changed in activated BMPR1α-deficient cells to increase cell response and persistence in inflammatory environment. It is plausible that lack of BMPR1 α tonic signaling may program CD4+ T cells in unmanipulated BMPR1αT− mice to respond robustly to inflammation. Collectively, these findings demonstrate that the outcome of T cell activation may be determined by both intrinsic signaling framework and is context sensitive, depending on the spectrum of inflammatory cytokines present in the activating environment.
Our data suggested that the effect of BMPR1α did not rely on blocking TGF-β signaling, as shown by studies of Smad2/3 phosphorylation and Smad4 abundance, but instead involved modulation of STAT3 and MAP kinase responses. The extent of signaling crosstalk, and molecular regulation of heterogeneity and stability of Th lineages, especially Th17 cells, are not well understood (68-70). CD4+ lineage defining transcription factors have permissive or bivalent histone modification in activated Th subsets, which contribute to T cell plasticity (77). Transitions between Th1 and Th17 cells or effector cells co-producing IL-17 and IFN-γ are found in cell lineage tracing studies in experimental autoimmune encephalomyelitis or inflammatory bowel disease, but the role of individual transcription factors and cytokine receptors in the differentiation and function of these effector cells remains controversial (72-74). Our findings revealed an important role for BMPR1α in regulating Th cell developmental programs and uncovered new immunoregulatory functions of the TGF-β family members.
Analysis of gene expression also identified a number of signal transduction molecules, known from previous studies to regulate production of inflammatory cytokines, which transcription is controlled by BMPR1α. Further analysis of these molecules will provide new insight into not only how BMPR1α signaling affects CD4+ T cell polarization but may help identify molecules that sustain function of inflammatory cells. Transcriptional profiling was also instrumental to understand the broad impact of BMPR1α signaling and identify biological processes and metabolic regulators which supply BMPR1α-deficient cells with effector functions. Gene set enrichment analysis established that BMPR1α signaling is primarily responsible for regulation of various aspects of inflammation and immune responses of T cells. This scope of cellular functions regulated by BMPR1 α is consistent with BMPs role in controlling chronic inflammation, organ fibrosis and homeostasis (75). Altogether, we validated the gene expression pattern using functional and flow cytometry studies, strongly supporting our conclusion that deficient BMPR1α signaling promotes generation of Th17 effector cells and enhances inflammatory responses.
Members of the TGF-β cytokine family may have opposing functions with regards to Th17 subset specification and regulation of inflammation. Transcription profiling offered molecular evidence that in contrast to TGF-βRII-deficient helper cells, BMPR1α-deficient cells expressed transcription factors and a cytokine profile associated with Th17 cell lineage (5, 13). In particular, BMP7 reverses the pro-inflammatory activity of TGF-β and protects from chronic organ injury (76, 77). Similarly, BMPs are produced in multiple tissues in response to infection or various stimuli causing cell damage, block inflammation, and restore tissue homeostasis (3, 4). In contrast, blockade of BMP signaling in rheumatoid arthritis patients with specific inhibitors augments inflammation induced by IL-17 (78). Consistent with our results, BMPs ameliorate intestinal inflammation and protect from mucosal damage (79, 80). This role of BMPs contrasts with recent reports that dorsomorphin and DMH1, which are small molecule inhibitors of BMP receptors and other kinases, impede proliferation of human and mouse T cells and block Th17 differentiation (27, 81, 82). Dorsomorphin inhibits BMPR1α (Alk3), BMPR1β (Alk6) and activin receptor-like kinase-2 (Alk2), which all share BMP ligands, as well as TGF-βRII, VEGFR2 (Flk1/KDR) and AMP-activated protein kinase (AMPK), an energy sensor in T cells which is necessary for accumulation of Th17 cells (64, 65, 83, 84). DMH1 inhibitor is more specific but in addition to BMPR1α, inhibits activin receptor-like kinase 1 (Alk1) and Alk2 (64, 65, 85). Activin receptors are competitively bound by activins and BMPs which changes their signaling from Smad2/3 to Smad1/5/8 and makes inhibition studies difficult to interpret (83). In our studies we used dorsomorphin and DMH1 at concentrations which did not inhibit T cell proliferation, and were lower than in previous studies, but this treatment still inhibited MAP kinase phosphorylation downstream of TCR stimulation. While our studies showed that BMPs signaling through the BMPR1α inhibited generation of Th17 cells, studies targeting other receptors are necessary to dissect complexity of immune regulation by cytokines of TGF-β family.
Multiple factors impact immunoregulation mediated by BMPs and TGF-β in tissues including their cellular source and bioavailability. Paracrine or autocrine production of TGF-β by T cells is essential for both regulation of tolerance and promotion of Th17 cells (5). Localized production and tissue availability controls the response to BMPs, but how their production is regulated in healthy tissues and during immune responses remains largely unknown (3, 75). One possibility is that BMPs are predominantly produced by stromal or epithelial cells and represent signaling cues exchanged between local tissues and the immune system. Once secreted, TGF-β and BMPs precursors associate with soluble or matrix molecules present in extracellular space (4, 86). These interactions affect proteolytic cleavage and maturation, degradation and diffusion, regulate binding to membrane receptors, and represent an important level of regulation of TGF-β and BMPs signaling. Proteolytic processing of TGF-β by furin convertase is essential for regulating peripheral T cell activation, but how furin or other proteases regulate maturation of BMP precursors is not known (87). Identifying cellular sources and uncovering how production, secretion and processing of BMPs are regulated will reveal new mechanisms of local immunoregulation and provide rationale for their therapeutic application. Thus, our findings provide a framework for broader understanding of how TGF-β and BMPs regulate T cells in immune tolerance and autoimmunity.
Materials and Methods
Mice
TCRα− mice were purchased from Jackson Laboratory (88). BMPR1αT− mice were generated by crossing BMPR1α conditional knockout mice with mice expressing CD4cre and Foxp3GFP reporter (25, 89, 90). Foxp3GFP and BMPR1αT− mice were crossed with mice expressing TCR specific for analog of peptide derived from pigeon cytochrome C (PCC) (91). All mice were on the C57BL6 genetic background. Mice were bred and housed in specific pathogen-free conditions in the animal facility of Old Dominion University. All experiments were approved by IACUC. Both female and male mice were used in experiments and we have not observed any difference in T cell development and activation between sexes. Mice were 6- to 12-week-old for all experiments.
T cell activation and polarization
For in vitro activation, CD4+ cells were purified by flow cytometry sorting or with magnetic beads and stimulated with plate-bound antibodies against CD3 (10 μg/ml, 2C11) and CD28 (1 μg/ml, 37.51) (both from BD Biosciences), Con A (2 μg/ml; Sigma) or analog of PCC peptide (PCC50V54A) (5 μM) in the presence of antigen presenting cells in αMEM media (HyClone) supplemented with 10% Fetal Bovine Serum (Atlanta Biologicals), 2 mM L-glutamine, dextrose, essential and non-essential amino acids, sodium pyruvate, sodium bicarbonate, antibiotics, and 2-β-mercaptoethanol.
To produce polarized effector cells, CD4+ T cells were purified by flow cytometry sorting or with magnetic beads and stimulated with plate-bound antibodies against CD3 and CD28. For Th1 differentiation, cells were stimulated in the presence of IL-12 (10 ng/ml; PeproTech) and antibody against IL-4 (10 μg/ml; BD Bioscience). For Th2 differentiation, cells were stimulated in the presence of IL-4 (10 ng/ml; PeproTech), and antibody against IFN-γ (10 μg/ml; BD Bioscience) and IL-12 (10 μg/ml; BD Bioscience). For Th17 differentiation, cells were stimulated in the presence of IL-6 (20 ng/ml; PeproTech) and TGF-β (3 ng/ml; PeproTech). Cells were cultured and analyzed after 4 days. To examine BMP signaling, T cells were cultured with BMP2, BMP4 and BMP7 (3 ng/ml each; PeproTech). To inhibit TGF-β signaling, SB525334 (10 nM; Selleckchem) was added to cell culture. To inhibit BMP signaling, T cells were incubated in the presence of dorsomorphin (1 μM; Tocris Bioscience) or DMH1 (10 μM; Tocris Bioscience).
For co-culture, total lymph nodes and spleen from PCC transgenic mice were combined. The cells from wild-type and BMPR1αT− mice were then mixed 1:1 and stimulated with PCC peptide and either IL-6 (20 ng/ml; PeproTech) and TGF-β (3 ng/ml; PeproTech) or IL-21 (100 ng/ml; PeproTech) and TGF-β (3 ng/ml; PeproTech). Cell proportions in the co-culture were determined to be 60% BMPR1α-sufficient and 40% BMPR1α-deficient using CD45.1 and CD45.2 staining. Cells were cultured and analyzed after 6 days.
In vivo activation and immunization
For in vivo activation, mice were immunized in the footpad with 100 μg antigenic peptide PCC50V54A (Anaspec) emulsified in CFA. After 5 days, animals were sacrificed and popliteal draining lymph nodes were isolated.
Adoptive transfer of CD4+ CD45RBhigh T cells into lymphopenic mice
Naive CD4+CD45RBhigh T cells were flow cytometry sorted from spleens of wild-type and BMPR1αT− mice and 5x105 cells per mouse was transferred intravenously into TCRα knockout C57BL6 mice. Mice were monitored every 2-3 days for 3 weeks.
DSS-induced colitis
Experimental colitis was induced by administration of 2.5% (weight/volume) DSS (molecular weight, 40,000 kDa; MP Biomedicals) dissolved in sterile drinking water ad libitum for 6 days to mice. Mice were monitored daily and euthanized after 7 days for histological assessment, flow cytometry and gene expression analysis.
Assessment of intestinal inflammation
Body weight, occult or gross blood lost per rectum and stool consistency were assessed as briefly described (92). The baseline clinical score was determined on day 0. Scoring went as follows, no weight loss was scored as 0, weight loss of 1% - 5% as 1; 5% - 10% as 2; 10% - 20% as 3; and more than 20% as 4. For stool consistency, a score of 0 was assigned for well-formed pellets, 2 points for semi-formed stools, and 4 points for liquid stools. For bleeding, a score of 0 points was assigned for no blood, 2 points for positive occult blood test (Sure-Vue™ Fecal Occult Blood Test; Fisher HealthCare) and 4 points for gross bleeding. For CD4+CD45RBhigh T cell transfer weight loss, bleeding and diarrhea scores were shown. For experimental colitis induced with DSS all scores were added together and divided by three, resulting in a total clinical score.
Histological analysis
A small section of the distal colon was removed and fixed in 4% formaldehyde. Paraffin-embedded tissues were sectioned and stained with hematoxylin and eosin by Eastern Virginia Medical School. Imaging and pathological analysis was performed at the Department of Pathology at the University of Pittsburgh School of Medicine.
Isolation of lamina propria lymphocytes
Colons were excised from the mice, opened longitudinally and washed in ice cold PBS to remove all fecal matter. The colon was then cut into approximately 1.5 cm pieces and incubated twice in 15 ml of HBSS supplemented with 2 mM EDTA and 5% FBS for 15 min at 37°C with shaking. After each incubation, the epithelial layer was removed by vortexing and passing through a 100 μm strainer and new solution was added. After the second EDTA wash, the pieces were incubated in 15 ml HBSS supplemented with 10 mM HEPES for 15 min at 37°C with shaking. The pieces were strained, cut into fine pieces and placed in 10 ml digestion solution containing 5% FBS, 1.0 mg/ml each of Collagenase I, II and IV (Worthington), and 0.1 mg/ml DNase I (Sigma). Digestion was performed by incubating the pieces for 20 min at 37°C with shaking. After digestion, the solution was vortexed and passed through a 70 μm strainer. The supernatants were washed once in cold HBSS and again in PBS with 2% FBS. The cells were immediately used for experiments.
Flow cytometry
Single cell suspensions were prepared from lymph nodes, spleen or cells were isolated from intestinal wall and stained with antibodies labeled with FITC, PE, PE-Cy5, PE-Cy7, APC, APC-Cy7, Alexa Fluor 680, Alexa Fluor 780, BV421, BV450 or biotin. Antibodies were purchased from eBioscience, BD Biosciences or BioLegend. Following antibodies were used: CD4 (GK1.5), CD44 (IM7), CD25 (PC61), CD62L (MEL-14), 4-1BB (17B5), ICOS (7E.17G9), Tim3 (8B.2C12), BTLA (8F4), Klrg-1 (2F1), CCR6 (29-2L17), CCR5 (HM-CCR5), CD45RB (16A), CD45.1 (A20) and CD45.2 (104). For intracellular cytokine staining cells were isolated from lymphoid organs or activated in vitro. Before staining, cells were incubated for 3 hours with 10 μg/ml Brefeldin A (BD Biosciences), 50 ng/ml PMA (Sigma) and 1 μg/ml Ionomycin (MP Biomedical) in T cell culture medium. After the 3 hours incubation period, cells were stained for surface markers first and then for cytokines using Cytofix/Cytoperm kit (BD Biosciences) and antibodies specific for IFN-γ (XMG1.2), IL-17 (TC11-18H10) labeled with fluorochromes. For intracellular staining of transcription factors, cells were stained first for surface markers and then for Rorc (Q31-378) using Transcription Factor Staining Buffer kit (eBioscience). All flow cytometry samples were ran on a BD FACSCanto II or LSRII (BD Bioscience) and analyzed using FlowJo software (Tree Star Inc.). Cell sorting was done on MoFlo (Cytomation) or Insight (BD Biosciences) cell sorters.
Enzyme-linked immunosorbent spot assay
Wild-type and BMPR1αT− mice were immunized with 100 μg of LCMV peptide gp61-80 (Anaspec) in CFA in the footpads. After one month, a booster dose was applied and mice were sacrificed 7 days later. Draining (popliteal) lymph nodes and spleen cells (2x105 cells per well) were isolated and cytokine secreting cells were quantified using ELISPOT assay and ELISPOT Ready-SET-Go kit reagents (eBioscience). Cytokines were detected using antibodies specific for IFN-γ (capture: R4-6A2; detection: XMG1.2), IL-4 (capture: 11B11; detection: BVD6-24G2), IL-17 (capture: eBio17CK15A5; detection: eBio17B7), IL-2 (capture: JES6-1A12; detection: JES6-5H4) and IL-10 (capture: JES5-16E3; detection: JES5-2A5) and spots were quantified with ImmunoSpot software (CTL) on an S6 Macro reader.
Gene expression analysis
RNA was prepared according to manufacturer’s instructions (PureLink® RNA kit, Life Technologies) and reverse-transcribed with SuperScript™ III (Life Technologies) per manufacturer’s instructions. Equal amounts of cDNA were used in triplicates to detect transcripts of BMPR1α, BMPR2, IL-6, IL-1β, IL-17, IFN-γ and Rorc using TaqMan® Universal Master Mix II (Life Technologies) in the StepOne™ Real-Time PCR System (Applied Biosystems). The transcript abundance of each gene was normalized to β-actin. For end-point PCR, transcription factors: IFN-γ, T-bet, IL-4, GATA3, IL-17, Rorc, BMPR1α and β-actin transcripts were amplified with GoTaq polymerase (Promega).
RNAseq and transcriptome analysis
Global analysis of gene expression was performed using Illumina Sequencing platform in Georgia Cancer Center Core Facility, Augusta University. Naive CD4+CD44−CD62L+Foxp3GFP− cells were flow sorted from lymph node and spleens of unmanipulated wild-type mice. Lymph node and spleen cells isolated from wild-type or BMPR1αT− mice were activated with Con A (2 μg/ml, Sigma) and LPS (10 ng/ml, Sigma) for 4 days and activated CD4+CD44+CD62L−Foxp3GFP− cells were flow cytometry sorted. At least three different samples were processed for each cell type. Total RNA was prepared using commercial kit (Qiagen). DNA for sequencing was produced with Illumina kit. RNAseq data analysis was performed using Tuxedo protocol as described in (93). Briefly, sequencing reads were aligned to reference genome using Tophat2, followed by estimation of RNA using Cufflinks 2.11. Differential gene expression analysis was performed using Cuffdiff application of Cufflinks. Genes were considered differentially expressed if fold expression was 1.5 or more and the difference was statistically significant. To visualize differences between gene expression profiles of activated wild-type and BMPR1α-deficient CD4+ T cells we performed principal component analysis (PCA). The gene lists subject to PCA analysis included all genes with expression levels above the threshold allowing for differential expression analysis in Cufflinks suite. Expression profiles of genes differentially expressed between activated BMPR1α-sufficient and deficient CD4+ T cells were visualized as a heat map. Expression profiles of genes differentially expressed between naive wild-type and activated BMPR1α-sufficient and deficient CD4+ T cells were visualized as volcano plots. All analyzes were done in R (version 3.4.3). Bioinformatics transcriptome analysis was performed in College of Public Health of Ohio State University. Gene Ontology and gene enrichment analyses were performed using Metascape (http://metascape.org) and network graphs were edited using Cytoscape (94, 95).
Western blot
Lymph node cells were isolated and CD4+ T cells were purified by magnetic beads. Purified CD4+ T cells (5x105) were lysed directly in Laemmli buffer with protease and phosphatase inhibitors (Halt™ Protease and Phosphatase Inhibitor Cocktail, Thermo Fisher) or stimulated with plate-bound antibodies against CD3 and CD28 for 5 min in the presence of cytokines IFN-γ (50 ng/ml, PeproTech) or IL-6 (20 ng/ml, PeproTech) and lysed. Proteins were separated by SDS-PAGE, blotted on PVDF membranes and probed with primary antibodies specific for STAT1 (D1K9Y), p-STAT1 (58D6), STAT3 (79D7) and p-STAT3 (D3A7) (all from Cell Signaling Technology, diluted 1:1000 – 1:2000) following incubation with secondary anti-mouse or anti-rabbit IgG antibody conjugated with horseradish peroxidase (Bio-Rad). Proteins were developed with Clarity Western ECL (Bio-Rad) substrate and filter images were acquired on ChemiDoc Touch Imaging system and analyzed using ImageLab software (Bio-Rad). To examine kinetics of STAT3 phosphorylation purified CD4+ T cells were stimulated with plate-bound antibodies against CD3 and CD28 for 5, 30 and 60 min. in the presence of IL-6 (20 ng/ml, PeproTech) and BMP2/4/7 (5ng/ml each, PeproTech) and in the absence or presence of TGF-β (3 ng/ml; PeproTech). For the detection of Smad 1 (D59D7), p-Smad 1/5/8 (D5B10), Smad 2/3 (L16D3), p-Smad 2/3 (138D4), Smad4 (D3R4N), JNK (56G8), p-JNK (81E11), ERK (137F5), p-ERK (D13.14.48), p38 (D13E1) and p-p38 (D3F9) (all antibodies from Cell Signaling Technology, diluted 1:1000 – 1:2000) purified CD4+ T cells (5x105) from wild-type or BMPR1αT− mice were stimulated in vitro for 2 days with Con A (2 μg/ml). To inhibit BMP signaling, wild-type T cells were activated with plate-bound antibodies against CD3 and CD28 for 24 hours in the presence of dorsomorphin (1 μM; Tocris Bioscience) or DMH1 (10 μM; Tocris Bioscience). After stimulation, cells (5x105) were lysed and analyzed as above. For Smad2/3 phosphorylation in different concentrations of TGF-β, purified CD4+ T cells from wild-type or BMPR1αT− mice were stimulated for 1 hour with plate-bound antibodies against CD3 and CD28 in the presence of different concentrations of TGF-β (Peprotech). For BMPR1α protein analysis, naive (CD4+CD44lowCD62Lhigh), activated (CD4+CD44highCD62Llow) and Treg (CD4+Foxp3GFP+) cells were flow sorted from Foxp3GFP mice and cells were lysed in Laemmli buffer. Blots were incubated with anti-BMPR1α antibody (MA5-17036, Thermo Fisher Scientific). For quantitative analysis Western blots samples were normalized for protein loading using β-actin (13E5, Sigma), and next normalized for abundance of a specific protein. Once total protein levels were normalized, relative levels of phosphorylated protein in different samples were compared. Relative intensities of phosphorylated proteins from different experiments were used to calculate mean and standard deviations.
Statistical analysis
p values were calculated with the two-tailed Student’s test for two-group comparison, as applicable, with Microsoft Excel Software and SigmaPlot. To calculate standard deviation of gene expression ratios we used bootstrap method (96).
Supplementary Material
Fig. S1. Signaling through BMPR1α alters T cell lineage commitment.
Fig. S2. Cytokine production in WT and BMPR1α-deficient cells activated in the presence of TGF-β or IL-6.
Fig. S3. Antigen specific BMPR1α-deficient CD4+ T cells generate increased proportion of proinflammatory cells.
Fig. S4. Transcriptional profiles of naive versus activated CD4+ T cells.
Fig. S5. Membrane proximal signaling in BMPR1α-deficient and -sufficient CD4+ T cells.
Fig. S6. DSS-induced colitis is more severe in BMPR1αT− mice.
Table S2. GO analysis between WT and BMPR1αT− mice using RNAseq.
Table S1. Differentially expressed genes between WT and BMPR1αT− mice using RNAseq.
Acknowledgments:
We thank EVMS Histology Services Lab for preparing tissue sections and ODU Core laboratory for flow cell sorting.
Funding:
This work was supported by funding from NIH (R21 AI097600 to P.K.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The RNAseq data reported in this paper are freely available at the National Center for Biotechnology Information Gene Expression Omnibus database (accession #GSE103124). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Massague J, TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A, Bone morphogenetic proteins: a critical review. Cell. Signal 23, 609–620 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Grgurevic L, Christensen GL, Schulz TJ, Vukicevic S, Bone morphogenetic proteins in inflammation, glucose homeostasis and adipose tissue energy metabolism. Cytokine Growth Factor Rev. 27, 105–118 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Chen W, Ten Dijke P, Immunoregulation by members of the TGFbeta superfamily. Nat. Rev. Immunol 16, 723–740 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Li MO, Flavell RA, TGF-beta: a master of all T cell trades. Cell 134, 392–404 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubtsov YP, Rudensky AY, TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol 7, 443–453 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Marie JC, Liggitt D, Rudensky AY, Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of Transforming Growth Factor-beta receptor. Immunity. 25, 441–454 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Li MO, Sanjabi S, Flavell RA, Transforming Growth Factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 25, 455–471 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Marie JC, Letterio JJ, Gavin M, Rudensky AY, TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med 201, 1061–1067 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM, Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25− regulatory T cells TGF-beta induction of trasnscription factor Foxp3. J. Exp. Med 198, 1875–1886 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li MO, Wan YY, Flavell RA, T cell-produced Transforming Growth Factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 26, 579–591 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B, TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17- producing T cells. Immunity. 24, 179–189 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B, Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol 7, 1151–1156 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK, Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441,235–238 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK, IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature 448, 484–487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O'Shea JJ, Cua DJ, The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol 10, 314–324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK, Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol 13, 991–999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO, Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 34, 396–408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyazono K, Kamiya Y, Morikawa M, Bone morphogenetic protein receptors and signal transduction. J. Biochem 147, 35–51 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Lu L, Ma J, Wang X, Wang J, Zhang F, Yu J, He G, Xu B, Brand DD, Horwitz DA, Shi W, Zheng SG, Synergistic effect of TGF-beta superfamily members on the induction of Foxp3+ Treg. Eur. J. Immunol 40, 142–152 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuczma M, Kraj P, Bone Morphogenetic Protein Signaling Regulates Development and Activation of CD4(+) T Cells. Vitam. Horm 99, 171–193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber S, Stahl FR, Schrader J, Luth S, Presser K, Carambia A, Flavell RA, Werner S, Blessing M, Herkel J, Schramm C, Activin A promotes the TGF-beta-induced conversion of CD4+CD25− T cells into Foxp3+ induced regulatory T cells. J. Immunol 182, 4633–4640 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S, Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446, 685–689 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, Littman DR, A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR, Generation of Bmpr/Alk3 conditional knockout mice. Genesis 32, 69–72 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Kuczma M, Kurczewska A, Kraj P, Modulation of bone morphogenic protein signaling in T-cells for cancer immunotherapy. J. Immunotoxicol 11, 319–327 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshioka Y, Ono M, Osaki M, Konishi I, Sakaguchi S, Differential effects of inhibition of bone morphogenic protein (BMP) signalling on T-cell activation and differentiation. Eur. J. Immunol 42, 749–759 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Kraj P, Pacholczyk R, Ignatowicz L, alpha/betaTCRs differ in the degree of their specificity for the positively selecting MHC/peptide ligand. J. Immunol 166, 2251–2259 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR, The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C, Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O'Shea JJ, Zhao K, Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, Gazit R, Adoro S, Glimcher L, Chan S, Kastner P, Rossi D, Collins JJ, Mathis D, Benoist C, A multiply redundant genetic switch 'locks in' the transcriptional signature of regulatory T cells. Nat. Immunol 13, 972–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tripathi SK, Lahesmaa R, Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol. Rev 261, 62–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A, Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limon P, Nakayama M, Leonard WJ, Flavell RA, Excessive Th1 responses due to the absence of TGF-beta signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc. Natl. Acad. Sci. U.S.A 110, 6961–6966 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY, Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Lu LF, Gavin MA, Rasmussen JP, Rudensky AY, G protein-coupled receptor 83 is dispensable for the development and function of regulatory T cells. Mol. Cell. Biol 27, 8065–8072 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, Ichinose H, Metzger D, Chambon P, Yoshimura A, Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol 14, 230–237 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ, Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467, 967–971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C, Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lubberts E, The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol 11,415–429 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR, IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol 8, 967–974 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Chinnasamy P, Lutz SE, Riascos-Bernal DF, Jeganathan V, Casimiro I, Brosnan CF, Sibinga NE, Loss of Allograft Inflammatory Factor-1 Ameliorates Experimental Autoimmune Encephalomyelitis by Limiting Encephalitogenic CD4 T-Cell Expansion. Mol. Med 21, 233–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer DC, Guittard GC, Franco Z, Crompton JG, Eil RL, Patel SJ, Ji Y, Van Panhuys N, Klebanoff CA, Sukumar M, Clever D, Chichura A, Roychoudhuri R, Varma R, Wang E, Gattinoni L, Marincola FM, Balagopalan L, Samelson LE, Restifo NP, Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med 212, 2095–2113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang CY, Li JP, Chiu LL, Lan JL, Chen DY, Chuang HC, Huang CY, Tan TH, Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J. Immunol 192, 1547–1557 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Shan F, Xia Y, Wang N, Meng J, Lu C, Meng Y, Plotnikoff NP, Functional modulation of the pathway between dendritic cells (DCs) and CD4+T cells by the neuropeptide: methionine enkephalin (MENK). Peptides 32, 929–937 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Morgan ME, Koelink PJ, Zheng B, den Brok MH, van de Kant HJ, Verspaget HW, Folkerts G, Adema GJ, Kraneveld AD, Toll-like receptor 6 stimulation promotes T-helper 1 and 17 responses in gastrointestinal-associated lymphoid tissue and modulates murine experimental colitis. Mucosal Immunol. 7, 1266–1277 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conner JR, Smirnova II, Moseman AP, Poltorak A, IRAK1BP1 inhibits inflammation by promoting nuclear translocation of NF-kappaB p50. Proc. Natl. Acad. Sci. U.S.A 107, 11477–11482 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang W, Ghisletti S, Saijo K, Gandhi M, Aouadi M, Tesz GJ, Zhang DX, Yao J, Czech MP, Goode BL, Rosenfeld MG, Glass CK, Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature 470, 414–418 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sekine H, Ferreira RC, Pan-Hammarstrom Q, Graham RR, Ziemba B, de Vries SS, Liu J, Hippen K, Koeuth T, Ortmann W, Iwahori A, Elliott MK, Offer S, Skon C, Du L, Novitzke J, Lee AT, Zhao N, Tompkins JD, Altshuler D, Gregersen PK, Cunningham-Rundles C, Harris RS, Her C, Nelson DL, Hammarstrom L, Gilkeson GS, Behrens TW, Role for Msh5 in the regulation of Ig class switch recombination. Proc. Natl. Acad. Sci. U.S.A 104, 7193–7198 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wildner G, Kaufmann U, What causes relapses of autoimmune diseases? The etiological role of autoreactive T cells. Autoimmun. Rev 12, 1070–1075 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Hahn JN, Falck VG, Jirik FR, Smad4 deficiency in T cells leads to the Th17- associated development of premalignant gastroduodenal lesions in mice. J. Clin. Invest 121, 4030–4042 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS, SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol 4, 540–545 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ, SOCS3 regulates the plasticity of gp130 signaling. Nat. Immunol 4, 546–550 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, Altuntas CZ, Sass Bak-Jensen K, McGeachy MJ, Do JS, Xiao H, Delgoffe GM, Min B, Powell JD, Tuohy VK, Cua DJ, Li X, The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity. 32, 54–66 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garlanda C, Dinarello CA, Mantovani A, The interleukin-1 family: back to the future. Immunity 39, 1003–1018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea JJ, Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26, 371–381 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Landsman T, Waxman DJ, Role of the cytokine-induced SH2 domain-containing protein CIS in growth hormone receptor internalization. J. Biol. Chem 280, 37471–37480 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Chang X, Liu F, Wang X, Lin A, Zhao H, Su B, The kinases MEKK2 and MeKK3 regulate transforming growth factor-beta-mediated helper T cell differentiation. Immunity 34, 201–212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo X, Wang XF, Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 19, 71–88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoon JH, Sudo K, Kuroda M, Kato M, Lee IK, Han JS, Nakae S, Imamura T, Kim J, Ju JH, Kim DK, Matsuzaki K, Weinstein M, Matsumoto I, Sumida T, Mamura M, Phosphorylation status determines the opposing functions of Smad2/Smad3 as STAT3 cofactors in TH17 differentiation. Nat. Commun 6, 7600 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coquet JM, Middendorp S, van der Horst G, Kind J, Veraar EA, Xiao Y, Jacobs H, Borst J, The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity 38, 53–65 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Musikacharoen T, Bandow K, Kakimoto K, Kusuyama J, Onishi T, Yoshikai Y, Matsuguchi T, Functional involvement of dual specificity phosphatase 16 (DUSP16), a c-Jun N-terminal kinase-specific phosphatase, in the regulation of T helper cell differentiation. J. Biol. Chem 286, 24896–24905 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, Hopkins CR, Lindsley CW, Hong CC, In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem. Biol 5, 245–253 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cross EE, Thomason RT, Martinez M, Hopkins CR, Hong CC, Bader DM, Application of small organic molecules reveals cooperative TGFbeta and BMP regulation of mesothelial cell behaviors. ACS Chem. Biol 6, 952–961 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F, Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 33, 279–288 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, Li X, Cua DJ, Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 43, 727–738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vahedi G, A CP, Hand TW, Laurence A, Kanno Y, O'Shea JJ, Hirahara K, Helper T-cell identity and evolution of differential transcriptomes and epigenomes. Immunol. Rev 252, 24–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burkett PR, Meyer zu Horste G, Kuchroo VK, Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J. Clin. Invest 125, 2211–2219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang C, Collins M, Kuchroo VK, Effector T cell differentiation: are master regulators of effector T cells still the masters? Curr. Opin. Immunol 37, 6–10 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O'Shea JJ, Zhao K, Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 30, 155–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B, Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol 12, 255–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E, Cutting edge: the pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J. Immunol 190, 4478–4482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krausgruber T, Schiering C, Adelmann K, Harrison OJ, Chomka A, Pearson C, Ahern PP, Shale M, Oukka M, Powrie F, T-bet is a key modulator of IL-23-driven pathogenic CD4(+) T cell responses in the intestine. Nat. Commun 7, 11627 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weiskirchen R, Meurer SK, BMP-7 counteracting TGF-beta1 activities in organ fibrosis. Front. Biosci 18, 1407–1434 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R, BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med 9, 964–968 (2003). [DOI] [PubMed] [Google Scholar]
- 77.Sugimoto H, LeBleu VS, Bosukonda D, Keck P, Taduri G, Bechtel W, Okada H, Carlson W Jr., Bey P, Rusckowski M, Tampe B, Tampe D, Kanasaki K, Zeisberg M, Kalluri R, Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med 18, 396–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varas A, Valencia J, Lavocat F, Martinez VG, Thiam NN, Hidalgo L, Fernandez-Sevilla LM, Sacedon R, Vicente A, Miossec P, Blockade of bone morphogenetic protein signaling potentiates the pro-inflammatory phenotype induced by interleukin-17 and tumor necrosis factor-alpha combination in rheumatoid synoviocytes. Arthritis Res. Ther 17, 192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takabayashi H, Shinohara M, Mao M, Phaosawasdi P, El-Zaatari M, Zhang M, Ji T, Eaton KA, Dang D, Kao J, Todisco A, Anti-inflammatory activity of bone morphogenetic protein signaling pathways in stomachs of mice. Gastroenterology 147, 396–406 e397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maric I, Wensveen TT, Smoljan I, Orlic ZC, Bobinac D, Bone Morphogenetic Proteins and signaling pathway in Inflammatory Bowel Disease. Advances in Pathogenesis and Management, Karoui Sami (Ed.), InTech, , (2012). [Google Scholar]
- 81.Martinez VG, Sacedon R, Hidalgo L, Valencia J, Fernandez-Sevilla LM, Hernandez-Lopez C, Vicente A, Varas A, The BMP pathway participates in human naive CD4+ T cell activation and homeostasis. PLoS One 10, e0131453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vogt J, Traynor R, Sapkota GP, The specificities of small molecule inhibitors of the TGFb and BMP pathways. Cell Signal. 23, 1831–1842 (2011). [DOI] [PubMed] [Google Scholar]
- 83.Mueller TD, Nickel J, Promiscuity and specificity in BMP receptor activation. FEBS Lett. 586, 1846–1859 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, Pelletier J, Piccirillo CA, Krawczyk CM, Divangahi M, Jones RG, The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Kirmizitas A, Meiklejohn S, Ciau-Uitz A, Stephenson R, Patient R, Dissecting BMP signaling input into the gene regulatory networks driving specification of the blood stem cell lineage. Proc. Natl. Acad. Sci. U.S.A 114, 5814–5821 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Umulis D, O'Connor MB, Blair SS, The extracellular regulation of bone morphogenetic protein signaling. Development 136, 3715–3728 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, Roebroek A, Belkaid Y, Creemers J, O'Shea JJ, T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature 455, 246–250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, et al. , Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature 360, 225–231 (1992). [DOI] [PubMed] [Google Scholar]
- 89.Kuczma M, Podolsky R, Garge N, Daniely D, Pacholczyk R, Ignatowicz L, Kraj P, Foxp3-deficient regulatory T cells do not revert into conventional effector CD4+ T cells but constitute a unique cell subset. J. Immunol 183, 3731–3741 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB, A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15, 763–774 (2001). [DOI] [PubMed] [Google Scholar]
- 91.Kraj P, Pacholczyk R, Ignatowicz H, Kisielow P, Jensen P, Ignatowicz L, Positive selection of CD4(+) T cells is induced in vivo by agonist and inhibited by antagonist peptides. J. Exp. Med 194, 407–416 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Araki A, Kanai T, Ishikura T, Makita S, Uraushihara K, Iiyama R, Totsuka T, Takeda K, Akira S, Watanabe M, MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J. Gastroenterol 40, 16–23 (2005). [DOI] [PubMed] [Google Scholar]
- 93.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, Yanguez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, Konig R, Stertz S, Garcia-Sastre A, Chanda SK, Meta- and Orthogonal Integration of Influenza "OMICs" Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe. 18, 723–735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Severiano A, Carrico JA, Robinson DA, Ramirez M, Pinto FR, Evaluation of jackknife and bootstrap for defining confidence intervals for pairwise agreement measures. PLoS One 6, e19539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Signaling through BMPR1α alters T cell lineage commitment.
Fig. S2. Cytokine production in WT and BMPR1α-deficient cells activated in the presence of TGF-β or IL-6.
Fig. S3. Antigen specific BMPR1α-deficient CD4+ T cells generate increased proportion of proinflammatory cells.
Fig. S4. Transcriptional profiles of naive versus activated CD4+ T cells.
Fig. S5. Membrane proximal signaling in BMPR1α-deficient and -sufficient CD4+ T cells.
Fig. S6. DSS-induced colitis is more severe in BMPR1αT− mice.
Table S2. GO analysis between WT and BMPR1αT− mice using RNAseq.
Table S1. Differentially expressed genes between WT and BMPR1αT− mice using RNAseq.