

Abstract
Purpose:
Consensus molecular subtyping (CMS) of colorectal cancer (CRC) has potential to reshape the CRC landscape. We developed and validated an assay that is applicable on formalin fixed paraffin embedded (FFPE) samples of CRC and implemented the assay in a CLIA-certified laboratory.
Experimental design:
We performed an in silico experiment to build an optimal CMS classifier using a training set of 1329 samples from 12 studies and validation set of 1329 samples from 14 studies. We constructed assay based on Nanostring codesets for the top 472 genes, and performed analyses on paired flash frozen (FF)/FFPE samples from 175 CRCs to adapt the classifier to FFPE using a subset of genes found to be concordant between FF and FFPE, and tested the classifier`s reproducibility, repeatability, and validated in a CLIA-certified laboratory. We assessed prognostic significance of CMS in 345 patients pooled across 3 clinical trials.
Results:
The best classifier was Weighted Support Vector Machine with high accuracy across platforms and gene lists (>0.95), and the 472-gene model outperforming existing classifiers. We constructed subsets of 99 and 200 genes with high FF/FFPE concordance, and adapted FFPE-based classifier that had strong classification accuracy (>80%) relative to “gold standard” CMS. The classifier was reproducible to sample type, RNA quality, and demonstrated poor prognosis for CMS1-3 and good prognosis for CMS2 in metastatic CRC (p<0.001).
Conclusions:
We developed and validated a CRC CMS assay that is ready for use in clinical trials, to assess prognosis in standard-of-care settings and explore as predictor of therapy response.
Introduction:
Colorectal cancer (CRC) is the third most common cancer and a leading cause of cancer death worldwide. Several papers were published introducing CRC molecular subtyping systems; each partitioning colorectal cancer into three to six subtypes (1-7). The translational value of these works were limited by their relatively small sample sizes and lack of consensus regarding which of the six subtyping systems best captured the tumor heterogeneity and had superior utility as predictive and/or prognostic marker. In this context, Guinney, et al. (8) assembled the colorectal subtyping consortium (CRCSC) that sought to identify consensus molecular subtypes (CMS) by assembling a data base of gene expression measurements from 4,151 CRC patients from a collection of 18 international studies, having each of the six subtyping systems applied to each of these samples, and then using a network analysis to identify consensus clusters. The four consensus subtypes were identified primarily based on the biologic characteristics of colorectal cancer. However, findings by Guinney, et al. and subsequent other studies have demonstrated prognostic and predictive value of CMS in colorectal cancer (9-17).
In order to fully realize these potential benefits of CMS, it is necessary to have a robust, reliable single sample classifier to discern a CRC patient’s CMS from the tumor tissue. As part of an international consortium (8), we previously presented a Random Forest classifier that included 5973 genes and a “single sample” classifier based on nearest centroid predictor applied using 693 genes, built primarily using microarrays designed for use with flash frozen (FF) samples. Efforts are needed to build a more parsimonious single sample classifier using fewer genes that can be reliably run on RNA extracted from formalin-fixed paraffin embedded (FFPE) samples. In this paper, we introduce an FFPE-based CMS classifier using the NanoString platform that has strong accuracy for predicting the CMS in CRC samples including those from the CRCSC study. This gene classifier was discovered and validated in silico by using the CRCSC data sets and subsequently optimized based on degree of correlation across tissue types; FFPE vs. FF samples and platform type NanoString vs. Affymetrix. Subsequently, we validated this FFPE tissue based gene classifier in a CLIA-certified molecular diagnostic laboratory and demonstrate prognostic significance of CMS in CRC.
Material and Methods:
Development and Validation of CMS Classifier on CRCSC:
We performed in silico development and validation of the CMS classifier using the samples and datasets that were part of the “consensus” set in CRCSC, meaning that they had so-called “gold standard” CMS status, defined based on agreement among the six different subtyping systems, against which we could compare to assess classification accuracy of our CMS classifier. Details of the discovery and validation approach, data sets used, including tissue type, total number of samples, total number of consensus samples and preprocessing method are shown in figure-1A, supplementary table-1 and supplementary methods 1 and 2. Classification Modeling Strategies: The classification modelling strategies we considered included Linear Discriminant Analysis (18,19), Quadratic Discriminant Analysis (20,21), K-Nearest Neighbor (22,23), Random Forrest (8,24), Rotation Forrest (25,26), Weighted Support Vector Machine (wSVM)(27,28), Distance-Weighted Discrimination (DWD)(29,30), and Ensemble Methods comprised of voting schemes across these classifiers (supplementary methods 3). We split the training data set of V1 into subsets, containing 332, 332, 332, and 333 samples, respectively, for use in the four-fold cross validation model building strategy. For each modeling strategy, we applied the Quantile Normalization(30) and fit the model to each of the four ¾ subsets, optimizing tuning parameters using nested cross validation, and assessed accuracy for predicting the gold standard CMS on the left-out ¼ subset. Tuning parameters that showed the best accuracy were selected as optimal parameters for each subset. We summarized the predictive accuracy of each modeling strategy as a function of number of genes, allowing us to both assess which modeling strategy appears to be best and the minimum number of genes needed for accurate CMS classification. Choosing the best modeling strategy, we computed the classification accuracy in the validation data set V2 as well as the various subsets mentioned above, and summarized results again as a function of number of genes in the model. Gene Ranking Strategy: We designed a boosting procedure based on multi-class Adaboost (31) to order the genes (see supplementary methods 4), which amounts to a forward stage-wise additive selection in which samples were repeatedly re-weighted at each step so the next best gene focused more on samples misclassified on previous steps, resulting in a list of genes ranked in descending order of classification importance. By using the same reduced gene sets for each classification method, we were able to gain a straightforward comparison of which method appears to perform better, to find the minimum gene set size yielding good classification performance, and to fairly compare the various methods at any desired model size. wSVM classifier: Our results revealed that the best performing classifier was the wSVM. The user calls the wSVM function with an N by P matrix of expression values for P genes for each of N samples with the column names as Entrez IDs, and the function will quantile normalize the data and apply the wSVM to get class predictions. After pairwise coupling, for sample I, we obtain probabilities of each CMS, πij such that , with αi=maxj {πij} indicating the highest CMS class probability for that sample, which we consider a measure of CMS classification confidence. We have two possible rules to classify a sample into a CMS group based on these measures:
Figure-1:


Flowchart showing approach development and validation of CMS classifier on colorectal cancer subtyping consortium (CRCSC) (1A) and development of NanoString classifier on colorectal cancer samples.
Most Likely CMS: Classify sample i into the most likely CMS, {j: πij =max (πij)}, regardless of classification confidence αi.
Most Likely CMS with a Confidence Threshold: Classify sample i into the most likely CMS as long as the classification confidence αi is above some threshold λ (e.g. 0.50 or higher), and otherwise consider indeterminate {Choose CMS j: πij =max (πij) if πij >λ, otherwise CMS indeterminate}. Indeterminate samples are heterogenous tumors containing characteristics of multiple CMS, so could also be called “mixed CMS”, as done for 13% of total samples by Guinney et al. (8).
Generation of Gene Signature Classifier in CRC Samples:
Summary of the Approach for Development of NanoString Classifier:
We used a novel strategy to port the CMS classifier designed for Affymetrix platform on FF samples over to the NanoString/FFPE setting that efficiently utilizes the vast information available to us in the CRCSC data sets and overcomes inconsistencies in mRNA quality between FF and FFPE samples (Figure 1-B and supplementary Figure-1).
Affymetrix 133-2 Plus2.0 and NanoString CodeSets Based Gene Expression Assays:
Flash frozen and FFPE tumor samples from randomly selected 175 patients including 95 men and 80 women with stage I-IV colon cancer were included in the first phase of the study to build a FFPE classifier. Seventy-two out of 175 samples were included in the CRCSC, therefore, the “gold standard” CMS based on Affymetrix expression array was known and only Nanostring assay was run on RNA extracted from FF and FFPE tissue samples of these tumors. Additional FF and FFPE samples from 103 tumors were identified from our institutional biorepository. The “gold standard” CMS was not known for these samples, therefore, Affymetrix 133-2 Plus2.0 was run on FF samples to identify the CMS as the “gold standard”. Subsequently, Nanostring assay was run on RNA extracted from FF and FFPE samples from these tumors. All samples were derived from primary colon/rectum resection specimens without preoperative tumor targeted therapy. The clinico-pathologic features of the patient population are shown in Table-1 and supplementary methods 5. Briefly, tumor areas with higher than 60% tumor cellularity from the gland forming tumor and higher than 20% tumor cellularity from signet ring or mucinous tumor were manually macro-dissected from FF or FFPE tissue sections to enrich for the demarcated tumor area. In more than 2/3 samples, superficial and deep (invasive border) areas of the tumor were included for the macro dissection. RNA was extracted using Qiagen’s AllPrep DNA/RNA kits (QIAGEN, Netherlands) per manufacturer’s instructions. Each sample was quantitated using the qubit fluorometer, with yields ranging from .9 μg to 26 μg. Each sample was run on the Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA) to determine the RNA Integrity Number (RIN) for the FF samples and the DV200 value for the FFPE samples. The FFPE samples included in the development of single sample classifier had 18% to 79% of the RNA having greater than 200 intact nucleotides. Gene expression analysis with Affymetrix 133-2 Plus2.0 was performed as described previously (supplementary methods 6). We designed a custom set of NanoString CodeSets (NanoString Technologies, Seattle, WA) with 472 signature probes, selected from the top 500 genes from the boosting procedure and 28 reference probes. The NanoString CodeSets for each gene was chosen to be the genomic region that was most highly correlated with the fRMA level-expression summary of the gene by Affymetrix 133-2 Plus2.0 probe set (Affymetrix, Santa Clara, CA). The genes with at least 0.70 correlation between the CodeSets and gene level summary were included in the customized CodeSets. The 28 reference CodeSets were selected from the reference CodeSets on the NanoString PanCan array plusR, and selecting genes with evidence of no difference in CMS in our preliminary data (32-36). The assay was performed as per the NanoString guidelines (supplementary methods 7) with 10 patient samples, a positive and negative control on each cartridge. Raw data from the nsolver software was transferred to the bioinformatics group where the custom CMS classifier algorithm in the form of R script was used to determine which samples belonged to a particular CMS.
Table-1:
Clinicopathologic features of patients and samples included in the assay development and validation in the research molecular diagnostic laboratory
| Characteristics | n | |
|---|---|---|
| Age | <50 years | 39 |
| >50 years | 136 | |
| Gender | Male | 95 |
| Female | 80 | |
| Tumor location | Right colon | 75 |
| Left and sigmoid colon | 92 | |
| Rectum | 5 | |
| Multiple primary tumors | 3 | |
| pT stage | pT1 | 0 |
| pT2 | 16 | |
| pT3 | 133 | |
| pT4 | 18 | |
| pT4a | 3 | |
| pT4b | 1 | |
| pN stage | pN0 | 76 |
| pN1 | 55 | |
| pN1a | 2 | |
| pN1b | 5 | |
| pN1c | 0 | |
| pN2 | 27 | |
| pN2a | 4 | |
| pN2b | 2 | |
| pN3 | 0 | |
| pNX | 4 | |
| pM stage | pM0 | 165 |
| pM1 | 5 | |
| pMX | 5 | |
| Grade | Low (well or moderately differentiated) | 145 |
| High (poorly differentiated) | 30 | |
| Time between date of surgery and gene expression analysis | < 5years | 20 |
| 5-10 years | 95 | |
| >10 years | 60 | |
| Samples | Matched flash frozen and formalin fixed paraffin embedded | 149 |
| Only formalin fixed paraffin embedded | 12 | |
| Only flash frozen | 4 | |
Validation at the Research Molecular Diagnostic Laboratory:
The NanoString CodeSets were technically validated by running two samples with “gold standard” CMS known from the CRCSC data on three different lots of CodeSets. The old lot of CodeSets and new lot of CodeSets were run together in the same run and accuracy in identifying the CMS by these CodeSets was assessed by linear regression. We tested repeatability of CMS assay across 4 different runs by same technician in 12 samples, reproducibility with different technician in 12 samples, and reproducibility with different input RNA quantity (50-500ng) for 6 samples using the same CodeSets on the same nCounter used for prior experiments. We also tested reproducibility of CMS between colonoscopy biopsies and surgically resected primary CRC by running customized CodeSets on matched biopsies and resection samples, using same CodeSets and nCounter and laboratory personnel.
Assessing Performance of CMS Classifier Assay in A CLIA Certified Laboratory:
The NanoString assay with top 200 genes (CRC CMS-200) and top 99 genes (CRC CMS-100),was further validated at our CLIA-certified Molecular Diagnostic Laboratory (MDL) to apply this assay as an integral biomarker for a phase II clinical trial (NCT034365630) assessing safety and efficacy of dual TGF-β trap: anti-PD-L1 molecule M7824 (EMD-Serono) in CMS4 subtype CRC. Thirty-five tumor samples from stage II/III primary colon cancer, previously used for validation at the research molecular diagnostic laboratory, were used to validate the assay across 10 runs for a total of 120 reactions. All 35 samples were included in the CRCSC study and gold standard CMS was known for these samples, and the laboratory technician was blinded from the gold standard CMS for those samples. Input for the assay was 250 ng of total RNA extracted from FFPE tumor tissue with 20% or higher tumor cellularity. Accuracy, analytical sensitivity, and analytical specificity were assessed by comparing calls from the MDL CMS panel with “gold standard” CRCSC Affymetrix calls. Reproducibility was assessed across original run and at least 3 additional repeat runs without re-extraction of RNA. Repeat runs were also performed with re-extracted RNA and by 2 technicians.
Assessing Performance of CMS Classifier as a Prognostic Marker in Stage IV Colorectal Cancer:
Patients with a CMS determination from the NanoString based gene expression score were pooled from three separate sources, including two clinical trials; NCT03436563 (n=91) and a phase II trial assessing Trametinib and Durvalumab in microsatellite stable colorectal cancer (n=19, NCT03428126) and Assessment of Targeted Therapies Against Colorectal Cancer (ATTACC, NCT01196130) Screening Protocol (n=235) (37). The ATTACC samples and the samples from the Trametinib/Durvalumab study were characterized by CRC CMS-100 assay at the research molecular diagnostic laboratory, while samples from patients enrolled in M7824 clinical trial were characterized by CRC CMS-200 performed at the CLIA compliant molecular diagnostic laboratory. Median overall survival was calculated from date of stage IV diagnosis to death or date of last follow up, which was censored. Survival patterns were visualized with Kaplan Meier survival curves and compared using the log-rank test. Graphs were generated using IBM SPSS Statistics 24.
The study was approved by the institutional review board (IRB) with an informed consent from each subject or each subject`s guardian for the clinical trial samples. The work on samples from subjects not enrolled in the clinical trial was approved by the IRB with waiver of informed consent. The study has been conducted as per the ethical guidelines of U.S. common rule.
Results:
Performance of CRCSC Classifier on CRCSC Data Sets:
We selected the wSVM model as it had the best performance in the Training Data V1 using 4-fold cross-validation (Supplementary Figure 2, Supplementary Table 2 and supplementary material 8). The 4-group classification accuracy of the wSVM model on the validation data set V2 was 0.955 for the full model (5973 genes), and still outstanding for models involving smaller gene numbers, with 4-group classification accuracies of 0.959, 0.932, and 0.898 for models with 500, 75, and 20 genes, respectively. The performance of the wSVM classifier for the out-of-sample subset (V2o), the RNAseq subset (TCGA), and the Affymetrix subset (V2a) were comparable to the overall validation performance (V2), suggesting the classifier was robust to platform and has good out-of-sample performance, relatively evenly across CMS (Supplementary Tables 3-7, supplementary material 9). We chose a wSVM classifier with 472 genes to move forward with further validation. This classifier yielded an overall 96.3% classification accuracy in the Affymetrix subset V2a, with accuracies of 0.966, 0.967, 0.932, and 0.971 for CMS1, CMS2, CMS3, and CMS4, respectively. The CMS structure was remarkably persistent being highly consistent in training and validation datasets (heat map in Supplemental Figure-3). Further comparison on our classifier with classifiers described by Guinney et. al. is shown in the supplementary Table-8. Performance of 472 gene CRCSC classifier based on single Affymetrix probe gene set and the classifier performance by classification confidence are described in supplementary material 10 and 11 and supplementary Figure-4. Supplementary Table-9 shows all of the misclassified samples along with the corresponding wSVM class probabilities (πij) for each CMS, classification confidence (αi), and indication of whether this sample could be considered a “CMS mixture” (i.e. (πij >0.20 for multiple CMS) and if the “gold standard” was a part of that mixture. From this, we see that most of the “misclassified samples” had lower classification confidence αi, and many had evidence of being CMS mixtures, with the “gold standard” CMS being a component of the mixtures.
Nanostring CRCSC Classifier Optimization Based on Correlation between FF and FFPE Tumor Samples:
The sample-specific correlations of FF and FFPE measurements were very high for most samples (Figure 2a, Supplementary Table 10A and supplementary material 12), with a small number of samples with low correlations tending to have poorer RNA quality for their FF samples (p=0.0077) but not FFPE (p=0.28, Figures 2B and 2C). A histogram of the gene-specific correlation of FF and FFPE measurements for each of the 472 classifier genes is found in Figure 2D and summarized in supplementary Table 10B. This histogram demonstrates the high level of variability across genes in terms of concordance of paired FF/FFPE gene expression measurements, and the remarkable consistency of the gene-specific concordances across batches (Supplementary Figure 5) suggests that this concordance is a consistent characteristic of the gene/probe set and not a random technical factor. This motivated us to select a subset of genes showing high FF/FFPE concordance for use in our FFPE classifier, choosing the top 100 genes in terms of FF/FFPE correlation for the CMS-100 classifier and the top 200 genes for CMS-200.
Figure 2:

Sample-wise and Gene-wise correlation of paired FF/FFPE Samples: Figure- 2A: Histogram of sample-wise Spearman correlation of paired FF/FFPE values across all 472 CMS genes on Nanostring assay, with threshold of 0.75 marked with red vertical line. Figure-2B and 2C: Association of sample-wise FF/FFPE with RNA Quality: Scatterplot of gene-specific Spearman correlation of FF/FFPE vs. RNA quality of FF samples (2B, based on RIN) and FFPE samples (2C, based on % with 200nt). Figure-2D: Histogram of gene-wise Spearman correlation of paired FF/FFPE values based on samples with sample-wise correlation > 0.75, with thresholds to determine the top 100 and top 200 genes indicated by red and blue vertical lines, respectively.
NanoString FFPE Classifier Performance:
Figure 3 shows the classification accuracy of CMS-100 on FFPE and FF samples, CMS-472 on FF samples, and the Affy FF-100 and Affy FF-472 based on the Affymetrix validation data V2a, with accuracy split out by confidence threshold α and proportion of unclassified samples. The CRC CMS-100 model applied to FFPE samples had 4-group accuracy of 0.80 with 0.81 for CRCSC samples and 0.78 for non-CRCSC samples. For samples with high confidence (αi>0.80 or 0.90), the performance was better with 4-group accuracy of 0.86 and 0.89, respectively. For FF samples, the CMS-100 had 4-group classification accuracy of 0.80, 0.74 for the CRCSC samples and 0.88 for non-CRCSC samples, and 4-class accuracy of 0.87 and 0.92 for samples classified with high confidence αi =0.80 or 0.90, respectively (supplementary material 13). These performed comparably to the CMS-472, the 472 gene classifier on FF samples, and not much worse than CMS-100 in an idealized non-clinical setting based on batch-corrected Affymetrix data from the CRCSC studies.
Figure-3:

Bar chart and table showing 4-class accuracy of CMS classifiers, along with number (proportion) of samples classified to each CMS. We assess accuracy for classifier with top 100 genes in terms of FF/FFPE correlation for FFPE and FF, computed based on Nanostring measurements for FFPE and FF in current study (Nano FFPE-100, Nano FF-100) and based on Affymetrix measurements for FF in the Affy CRCSC validation data set (V2a, Affy FF-100), and for the full 472 gene classifier applied to FF samples run on Nanostring platform for current study (Nano FF-472) and FF samples run on Affymetrix in the Affymetrix CRCSC validation data set (Affy FF-472). Performance is summarized overall and for subsets of samples with high classification confidence (α>0.50, 0.80 or 0.90).
Supplementary figures 6A and 6B plot the 4-class accuracy vs. confidence level αi for FFPE and FF samples, demonstrating that samples classified with high confidence were more likely to be accurately classified. Supplementary figures 6C and 6D plot the 4-class accuracy vs. RNA quality, defined by %200nt (FFPE) or RIN (FF), demonstrating that there is little if any association of CMS accuracy with RNA quality, suggesting that the performance of classifier is robust to RNA quality in this study. One gene out of the 100 was mistakenly left off an order of the Nanostring codesets for some of the validation study, so the corresponding classifier CMS-100 that was validated has 99 genes. We confirmed the performance of the 100 and 99 gene classifier was concordant.
The CRC CMS-100 assay with 99 genes was 100% reproducible in predicting a CMS across different runs (12 samples= 48 runs), between two laboratory personnel (12 samples) and with different RNA input concentration (n=6). The reproducibility between biopsy and resection was 91% with 15 of 17 patients had same CMS between matched biopsy and resection specimens (Supplementary Table-11). All (12 from left colon and 5 from right colon) biopsy samples were procured from same tumor as surgically resected specimens. Tissue sections from RNA were derived from FFPE blocks that were generated for the clinical use. The two cases with discrepant CMS between biopsy and resection were sporadic CRC without any known predisposing condition or preoperative tumor targeted therapy. To determine impact of tumor location and histopathologic features on reproducibility of CMS, another pathologist reviewed Hematoxylin & Eosin stained sections of primary tumor from surgical resections of those included in assessment of inter-run reproducibility, InterTech reproducibility, reproducibility across different RNA concentration and reproducibility between biopsy and resection (n=30). In 19 samples both superficial and deep area of the tumor were macro dissected, 4 samples had only superficial and 7 had only deep area of the tumor macro dissected. Five tumors were poorly differentiated, one with mucinous histology and 25 tumors were moderately differentiated including one with mucinous histology. Due to high reproducibility across runs, technicians and RNA concentration, we did not observe any difference in CMS call among samples with different areas of macro dissection or histologic parameters. We also did not observe significant difference in probability of a CMS in the context of histologic parameters. However, two (of 17) samples that showed discrepancy for CMS between biopsy and surgical resection had only deep area of the tumor macro dissected from the resection specimens. We also did not find histologic features unique to 7 samples that were discrepant for the CMS between research laboratory and CLIA certified laboratory.
Performance of CRC CMS-200 in CLIA-certified Molecular Diagnostic Laboratory:
On initial run, 32/35 samples were accurately assigned the CMS as compared to the gold standard based on “most likely CMS”, i.e. with confidence threshold of αi>0.50. Three misclassified samples, with 0.50 and 0.57 “most likely CMS” probability on the initial run had “most likely CMS” probability in the borderline range (≥0.43 & <0.57) on one of the repeat runs. They were classified as mixed CMS as they had almost equal probability of two CMS (CMS2 and CMS4) with one of them matching the gold standard CMS. With 0.50 most likely CMS probability, these three samples are considered errors when forcing a single CMS call, resulting in 91% analytical sensitivity and specificity. However, if a confidence threshold of >0.57 is used, then in all three samples in all runs had CMS as per the gold standard, and the assay had 100% analytical sensitivity and analytical specificity. Inter run reproducibility was assessed from 3 separate extractions from 4 unique patient samples for a total of 12 cases. These 12 cases were run across 3 separate NanoString Runs and by 2 technologists. There was 100% concordance for the CMS classification among all 3 runs with an average standard deviation of ± 0.002 for the “most likely CMS” probability. The inter tech reproducibility was 100% for the CMS classification between both technicians with an average standard deviation of ± 0.002 for the CMS probability. Intra run reproducibility was assessed among 4 samples run in triplicate on a single NanoString run. There was 100% concordance for the CMS classification among all 3 runs with an average standard deviation of ± 0.012 for the CMS probability. Comparing CMS reproducibility with CMS-100 (99 genes) vs. 200 gene assay demonstrated 97% reproducibility with only 1 of 35 samples showing discordant CMS. List of CMS100 (99 genes) and CMS200 test and 16 housekeeping genes is shown in Supplementary Table-12. These reproducibility and repeatability findings were deemed up to the level of a CLIA certified assay to determine CMS 4 vs. other CMS for FFPE tumor samples from patients enrolled in the clinical trial targeting patients with CMS4 colorectal cancer, as described in the methods.
KRAS-BRAF Mutational Status and Prognostic Relevance of CMS by the NanoString CMS Classifier:
To confirm the expected biologic performance of the assay, we surveyed a set of mCRC patients enrolled in clinical trials and ATTACC Protocol (Table-2). Higher frequency of KRAS mutation was observed with CMS 3 (66%) and CMS 4 (50%) samples. BRAF mutations were identified only in CMS 1 (50%), CMS 4(8%), and mixed (12%) subtype samples (Figure-4A). We did not find significant difference in any of the clinicopathologic and molecular characteristics between samples that were classified as mixed vs. those that were classified in to one of CMS. Using the CMS-100 (99 genes) classifier, we were able to identify significant differences in overall survival by CMS, consistent with prior studies (9,14). Specifically, patients with a CMS2 tumor had the best survival with a median of 46.1 months from stage IV diagnosis (95% CI: 36.6, 58.1), patients with a CMS1 or CMS3 tumor had the poorest survival after a stage IV diagnosis with median survival times of 23.2 (95% CI: 19.3, 59.2) or 21.4 (95% CI: 15.8, 34.6) months, respectively. Patients with a CMS4 tumor had a survival pattern that was in between that of CMS2 and CMS1 or CMS3 with a median survival time of 35.3 months (95% CI: 32.2, 40.0) (Figure-4B).
Table-2:
Patient characteristics of the cohorts utilized to correlate CMS and overall survival
| N = 345 | ||
|---|---|---|
| Mean Age at Initial Diagnosis (SD) | 50.9 (11.5) | |
| Mean Age at Stage IV Diagnosis (SD) | 51.5 (11.5) | |
| Sex | ||
| Male | 187 (54.2) | |
| Female | 158 (45.8) | |
| Race/Ethnicity | ||
| Non-Hispanic (NH) White | 258 (74.8) | |
| NH African American | 32 (9.3) | |
| Hispanic | 27 (7.8) | |
| NH Asian | 22 (6.4) | |
| Other/Unknown | 6 (1.7) | |
| Stage at Initial Diagnosis | ||
| I | 5 (1.4) | |
| II | 24 (7.0) | |
| III | 110 (31.9) | |
| IV | 204 (59.1) | |
| NA | 2 (0.6) | |
| KRAS mutation status | ||
| wild type | 107 (31.0) | |
| canonical mutation | 147 (42.6) | |
| NA | 91 (26.4) | |
| NRAS mutation status | ||
| wild type | 236 (68.4) | |
| canonical mutation | 17 (4.9) | |
| non-canonical mutation | 1 (0.3) | |
| NA | 91 (26.4) | |
| BRAF mutation status | ||
| wild type | 229 (66.4) | |
| v600 | 20 (5.8) | |
| other mutation | 5 (1.4) | |
| NA | 91 (26.4) | |
| MSI status | ||
| MSS | 177 (51.3) | |
| NA | 168 (48.7) | |
| Consensus Molecular Subtype | ||
| 1, Immune | 12 (3.5) | |
| 2, Canonical | 117 (33.9) | |
| 3, Metabolic | 21 (6.1) | |
| 4, Mesenchymal | 161 (46.7) | |
| Mixed | 34 (9.9) | |
Figure-4:
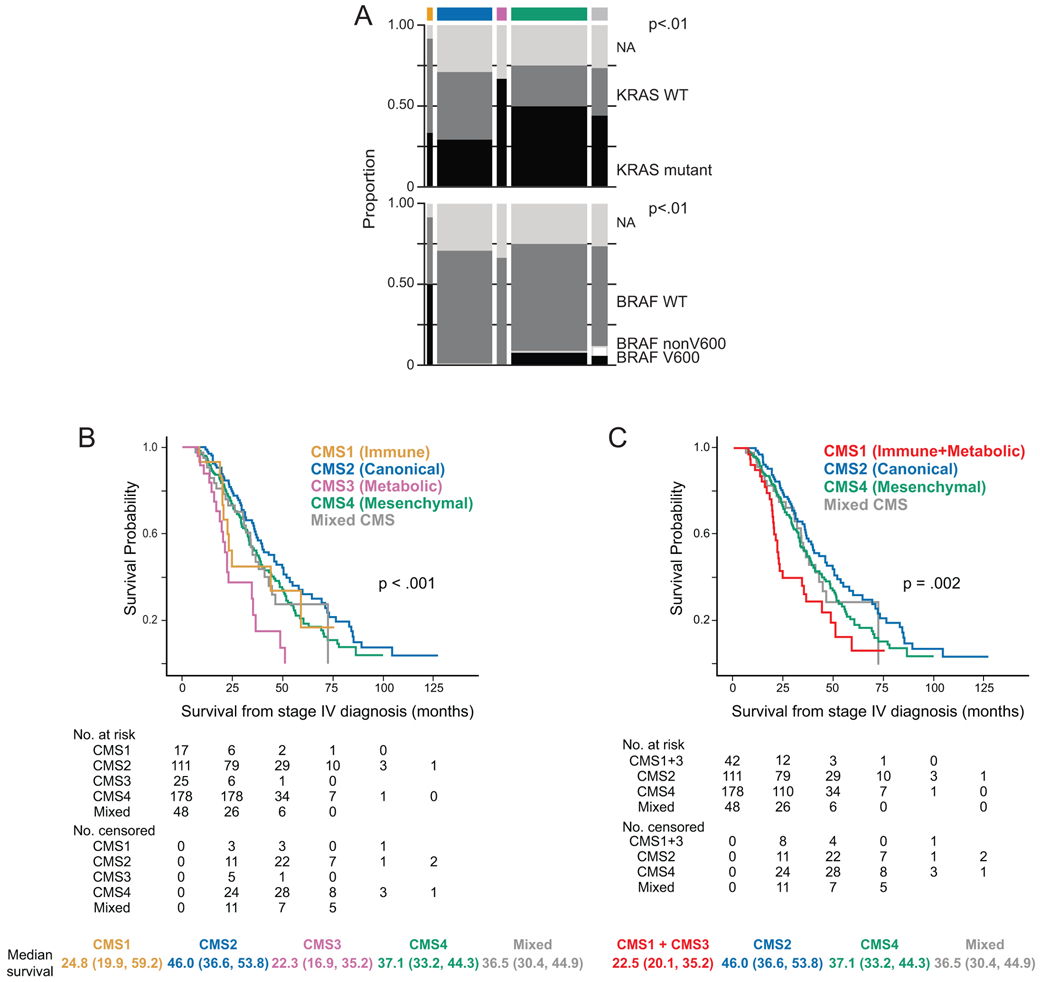
Distribution of KRAS and BRAF mutations across CMS (4A) and correlation of CMS with overall survival (4B) in stage IV colorectal cancer.
Discussion:
Consensus molecular subtyping has great potential to reshape the landscape of CRC treatment and contribute to the development of new precision therapeutic approaches. However, to realize this potential, it is necessary to transform the CMS based on network analysis of multiple gene expression datasets into a clinical test requires an assay that is reproducible across platforms and tissue types, has high classification accuracy, and is able to generate CMS in a single sample setting. We achieved this objective using a three step approach for building the classifier; in silico testing of various classification strategies considering various gene list sizes on CRCSC data generated on Affymetrix platform and determining that wSVM is the optimal system, a gene reduction exercise to select genes with best concordance across gene expression profiling platforms and tissue types to ensure optimal performance in FFPE samples, updating the wSVM using the CRCSC training set based on this reduced number of genes, and then using this classifier on measurements from the Nanostring assay on FFPE samples after transforming these values onto the scale of the Affymetrix data on FF samples that dominated the CRCSC training set.
Rather than just choosing a classification strategy in an ad hoc fashion, we used a systematic, rigorous strategy to rank the genes based on their classification value and to compare a large number of classification strategies for a wide range of model sizes. This allowed us to find out which strategy performed best, the wSVM, and to determine how parsimonious a classifier could be without sacrificing substantial classification accuracy. Given that the classification literature clearly demonstrates that no one classification strategy is optimal for all data sets, the consideration of multiple approaches is important when building classification signatures. Moreover, findings from the Microarray Quality Control (MAQC) project from FDA has shown that even slight differences in the statistical analysis led to discrepancies in biological interpretation (38). High accuracy in predicting CMS by nearly all statistical methods gives credence to the utility of our CMS assay in accurately classifying a colon cancer in one of the CMS.
The wSVM classifier we built using this strategy applied to training data, consisting largely of batch-corrected Affymetrix gene expression measurements from FF samples, performed exceptionally well in the CRCSC validation data. Our custom design of CodeSets best capitulating the signal in our training data, and our strategy of starting with more genes than necessary, then narrowing to a subset with evidence of high FF/FFPE correlation further mitigated the influence of FFPE on classification performance. Our quantile normalization strategy was sufficient to obtain reasonable performance for small (99 or 200) gene FFPE NanoString classifier. This strategy allowed us to efficiently utilize our data resources, using the enormous data on FF samples to train the classifier and collecting a smaller set of paired FF/FFPE samples to identify genes with high FF/FFPE concordance and map the FFPE NanoString expression values to the scale of the FF Affymetrix expression values, leading to our novel strategy for building the classifier. The consistency of gene-specific FF/FFPE concordance across batches provides strong support for this strategy.
The high concordance we observed in CMS for CRC samples between a research molecular testing laboratory and a CLIA-certified clinical molecular diagnostic laboratory indicate robust performance of the assay. High inter-laboratory reproducibility is likely due to similarities in the pre-analytical and analytical processes between two laboratories. Another reason for our high inter-laboratory reproducibility is the use of NanoString n-counter technology that utilizes non-amplified nucleic acids without any reverse transcription step and is applicable to multiple samples. Ragulan et. al.(39) demonstrated high classification accuracy and reproducibility of NanoString based subtyping-classification between FF and FFPE tissue samples of colorectal cancer. In spite of significant differences in the CRC classes and validation approach, this study and our study indicate that NanoString is a reliable platform to develop and validate gene expression based signature using FFPE samples of CRC.
Guinney et al. (8) found that approximately 87% of CRC tumors classified cleanly into a single CMS, but approximately 13% were “mixed CMS”, not outliers or a fifth subtype but heterogeneous samples containing characteristics of multiple CMS. We also found similar proportions of “mixed CMS” samples in our analyses. Clinically, patients with mixed CMS tumors could be treated multiple ways. One option would be to include any “mixed CMS” sample with a high enough probability of CMSx as a potential candidate for any targeted therapy that has been validated as a precision therapeutic for CMSx (x=1, 2, 3, or 4), which of course would require prospective validation before clinical application.
There is increasing evidence of the prognostic and predictive utility of CMS. Lenz et al. (9) using a NanoString based assay in a large cohort of patients with metastatic or advanced colorectal cancer enrolled in CALGB/SWOG 80305 phase III clinical trial, demonstrated that there is significant difference in overall survival by CMS with median survival of 40 months in CMS2 vs. median survival of 15 months in CMS1. The NanoString assay used for the CALGB/SWOG 80305 and our study differed significantly. Lenz et al. developed a customized NanoString based genes that were derived from some of the large data sets with published gold standard CMS labels, including, The Cancer Genome Atlas and other studies (5,13). Only genes that are common to these three data sets and those assessed in the CALGB/SWOG 80405 panel are used. While the genes included in our NanoString based assay were all derived from CRCSC database. Similar prognostic trends were observed by other groups including in patients enrolled in FIRE3 study comparing Cetuximab vs. Bevacizumab with FOLFIRI in metastatic colorectal cancer patients and by Mooi et. al.(14). As research only classifiers, these methodologies are not designed for application for individual patients or suitable for use in prospective patient assignment. In contrast, our classifier as deployed in a clinical lab is suitable for classifying individual patients with the rigor needed for guiding clinical management.
Our CLIA validated assay has potential of integral, integrated and exploratory marker. Hypotheses being explored include focused immunotherapy in CMS1, which represents a subgroup with evidence of higher immune infiltrates and activated T-cells. CMS2 represents a group with best overall survival from EGFR inhibition in retrospective assessment of the CALGB/SWOG 80405 trial, while CMS1 benefited from VEGF inhibition (PMID 31042420). CMS4 has active stromal signature and an immune modulating strategy has been proposed. For example, in the clinical trial (NCT034365630) assessing safety and efficacy of dual TGF-β trap: anti-PD-L1 molecule M7824 (EMD-Serono), CMS assay was used as an intergral biomarker to select patients with CMS4. Expanded efforts in this trial or other ongoing trials can be done looking to identify other CMS where efficacy of either M7824 or other drug can be assessed based on its mechanism of action and CMS biology. As an integrated assay, all patients can be prospectively tested to identify CMS. The interim analysis then looks at all comers, and if negative, then looks at CMS specific subgroups, with plan to continue the second half of the randomized study. Finally as an exploratory biomarker, a retrospective analysis can be done with the high quality CLIA assay to look for a CMS signal but also to minimize the risk of inconsistent assays when designing the follow up study. To support the goal of dissemination of a robust CMS classifier for retrospective or prospective utilization, the NanoString CodeSets and supporting bioinformatics information can be found at http://qcsrlshinypro.mdanderson.edu/CMSclia/.
Unavailability of matched samples prevented us from assessing CMS accuracy between primary and metastatic tissues in our study. Fontana E et.al (40) using publically available data from Khambata-Ford dataset (41), demonstrated no significant difference in CMS distribution between localized vs. metastatic disease. The impact of sample site on CMS classification is necessary to determine host organ influence and metastasis associated evolution of gene expression in CRC.
In summary, we have developed, validated and demonstrated prognostic utility of a CRC-CMS assay using FFPE samples. This CLIA validated assay provides a foundation to expand its utility to assess prognosis in a standard of care setting and explore the assay as a predictor of response to therapy in clinical trials.
Supplementary Material
Statement of translational relevance: In this manuscript, we have developed a gene expression assay for consensus molecular subtyping of colorectal cancer that shows prognostic relevance of CRC CMS. The assay is validated in CLIA-certified laboratory and is applicable for clinical trials in the current format.
Acknowledgments:
This work is funded by National Cancer Institute through Assay Validation For High Quality Markers For NCI-Supported Clinical Trials (UH2CA207101) and MD Anderson Cancer Center SPORE in Gastrointestinal Cancer (P50 CA221707). Part of this research was performed in MD Anderson’s Core facilities which is supported in part by the National Institutes of Health through Cancer Center Support Grant CA016672. Part of the validation work in the clinical lab was funded as part of a clinical trial (NCT03436563) funded by EMD-Serono. We thank Kim-Anh Vu in MD Anderson’s Department of Anatomic Pathology for helping with the figures.
References:
- 1.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Villamil B, Romera-Lopez A, Hernandez-Prieto S, Lopez-Campos G, Calles A, Lopez-Asenjo JA, et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behavior. BMC Cancer 2012;12:260 doi 10.1186/1471-2407-12-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlicker A, Beran G, Chresta CM, McWalter G, Pritchard A, Weston S, et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics 2012;5:66 doi 10.1186/1755-8794-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 2013;19(5):619–25 doi 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10(5):e1001453 doi 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Sousa EMF, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med 2013;19(5):614–8 doi 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 7.Budinska E, Popovici V, Tejpar S, D'Ario G, Lapique N, Sikora KO, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol 2013;231(1):63–76 doi 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21(11):1350–6 doi 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenz HJ, Ou FS, Venook AP, Hochster HS, Niedzwiecki D, Goldberg RM, et al. Impact of Consensus Molecular Subtype on Survival in Patients With Metastatic Colorectal Cancer: Results From CALGB/SWOG 80405 (Alliance). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2019;37(22):1876–85 doi 10.1200/JCO.18.02258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwon Y, Park M, Jang M, Yun S, Kim WK, Kim S, et al. Prognosis of stage III colorectal carcinomas with FOLFOX adjuvant chemotherapy can be predicted by molecular subtype. Oncotarget 2017;8(24):39367–81 doi 10.18632/oncotarget.17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song N, Pogue-Geile KL, Gavin PG, Yothers G, Kim SR, Johnson NL, et al. Clinical Outcome From Oxaliplatin Treatment in Stage II/III Colon Cancer According to Intrinsic Subtypes: Secondary Analysis of NSABP C-07/NRG Oncology Randomized Clinical Trial. JAMA oncology 2016;2(9):1162–9 doi 10.1001/jamaoncol.2016.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stintzing S WP, Lenz HJ et al. Consensus molecular subgroups (CMS)of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevasizumab in the FIRE3 (AIO KRK-0306) trial. . JCO 2017;35(Suppl 15):3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Annals of oncology : official journal of the European Society for Medical Oncology 2016;27(8):1386–422 doi 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 14.Mooi JK, Wirapati P, Asher R, Lee CK, Savas P, Price TJ, et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: molecular analysis of the AGITG MAX clinical trial. Annals of oncology : official journal of the European Society for Medical Oncology 2018;29(11):2240–6 doi 10.1093/annonc/mdy410. [DOI] [PubMed] [Google Scholar]
- 15.Becht E, de Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(16):4057–66 doi 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 16.Lal N, White BS, Goussous G, Pickles O, Mason MJ, Beggs AD, et al. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(1):224–33 doi 10.1158/1078-0432.CCR-17-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okita A, Takahashi S, Ouchi K, Inoue M, Watanabe M, Endo M, et al. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018;9(27):18698–711 doi 10.18632/oncotarget.24617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang D, Quan Y, He M, Zhou B. Comparison of linear discriminant analysis methods for the classification of cancer based on gene expression data. J Exp Clin Cancer Res 2009;28:149 doi 10.1186/1756-9966-28-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, Hastie T, Tibshirani R. Regularized linear discriminant analysis and its application in microarrays. Biostatistics 2007;8(1):86–100 doi 10.1093/biostatistics/kxj035. [DOI] [PubMed] [Google Scholar]
- 20.Arevalillo JM, Navarro H. A new method for identifying bivariate differential expression in high dimensional microarray data using quadratic discriminant analysis. BMC Bioinformatics 2011;12 Suppl 12:S6 doi 10.1186/1471-2105-12-S12-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hastie T, Tibshirani R. Efficient quadratic regularization for expression arrays. Biostatistics 2004;5(3):329–40 doi 10.1093/biostatistics/5.3.329. [DOI] [PubMed] [Google Scholar]
- 22.Ayyad SM, Saleh AI, Labib LM. Gene expression cancer classification using modified K-Nearest Neighbors technique. Biosystems 2019;176:41–51 doi 10.1016/j.biosystems.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Kumar MA, Ewoldt RH, Zukoski CF. Intrinsic nonlinearities in the mechanics of hard sphere suspensions. Soft Matter 2016;12(36):7655–62 doi 10.1039/c6sm01310d. [DOI] [PubMed] [Google Scholar]
- 24.Huynh-Thu VA, Geurts P. Unsupervised Gene Network Inference with Decision Trees and Random Forests. Methods Mol Biol 2019;1883:195–215 doi 10.1007/978-1-4939-8882-2_8. [DOI] [PubMed] [Google Scholar]
- 25.Stiglic G, Rodriguez JJ, Kokol P. Rotation of random forests for genomic and proteomic classification problems. Adv Exp Med Biol 2011;696:211–21 doi 10.1007/978-1-4419-7046-6_21. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez JJ, Kuncheva LI, Alonso CJ. Rotation forest: A new classifier ensemble method. IEEE Trans Pattern Anal Mach Intell 2006;28(10):1619–30 doi 10.1109/TPAMI.2006.211. [DOI] [PubMed] [Google Scholar]
- 27.Chan WH, Mohamad MS, Deris S, Zaki N, Kasim S, Omatu S, et al. Identification of informative genes and pathways using an improved penalized support vector machine with a weighting scheme. Comput Biol Med 2016;77:102–15 doi 10.1016/j.compbiomed.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Abdi MJ, Hosseini SM, Rezghi M. A novel weighted support vector machine based on particle swarm optimization for gene selection and tumor classification. Comput Math Methods Med 2012;2012:320698 doi 10.1155/2012/320698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H, Lu X, Liu Y, Haaland P, Marron JS. R/DWD: distance-weighted discrimination for classification, visualization and batch adjustment. Bioinformatics 2012;28(8):1182–3 doi 10.1093/bioinformatics/bts096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franks JM, Cai G, Whitfield ML. Feature specific quantile normalization enables cross-platform classification of molecular subtypes using gene expression data. Bioinformatics 2018;34(11):1868–74 doi 10.1093/bioinformatics/bty026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji Zhu HZ, Saharon Rosset and Trevor Hastie. Multi-class AdaBoost. 2009. 349–60 p. [Google Scholar]
- 32.Chen DT, Davis-Yadley AH, Huang PY, Husain K, Centeno BA, Permuth-Wey J, et al. Prognostic Fifteen-Gene Signature for Early Stage Pancreatic Ductal Adenocarcinoma. PLoS One 2015;10(8):e0133562 doi 10.1371/journal.pone.0133562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ligibel JA, Cirrincione CT, Liu M, Citron M, Ingle JN, Gradishar W, et al. Body Mass Index, PAM50 Subtype, and Outcomes in Node-Positive Breast Cancer: CALGB 9741 (Alliance). J Natl Cancer Inst 2015;107(9) doi 10.1093/jnci/djv179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prat A, Galvan P, Jimenez B, Buckingham W, Jeiranian HA, Schaper C, et al. Prediction of Response to Neoadjuvant Chemotherapy Using Core Needle Biopsy Samples with the Prosigna Assay. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(3):560–6 doi 10.1158/1078-0432.CCR-15-0630. [DOI] [PubMed] [Google Scholar]
- 35.Veldman-Jones MH, Lai Z, Wappett M, Harbron CG, Barrett JC, Harrington EA, et al. Reproducible, Quantitative, and Flexible Molecular Subtyping of Clinical DLBCL Samples Using the NanoString nCounter System. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(10):2367–78 doi 10.1158/1078-0432.CCR-14-0357. [DOI] [PubMed] [Google Scholar]
- 36.Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 2015;8:54 doi 10.1186/s12920-015-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overman MJ, Morris V, Kee B, Fogelman D, Xiao L, Eng C, et al. Utility of a molecular prescreening program in advanced colorectal cancer for enrollment on biomarker-selected clinical trials. Annals of oncology : official journal of the European Society for Medical Oncology 2016;27(6):1068–74 doi 10.1093/annonc/mdw073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodsaid FM, Amur S, Aubrecht J, Burczynski ME, Carl K, Catalano J, et al. Voluntary exploratory data submissions to the US FDA and the EMA: experience and impact. Nature reviews Drug discovery 2010;9(6):435–45 doi 10.1038/nrd3116. [DOI] [PubMed] [Google Scholar]
- 39.Ragulan C, Eason K, Fontana E, Nyamundanda G, Tarazona N, Patil Y, et al. Analytical Validation of Multiplex Biomarker Assay to Stratify Colorectal Cancer into Molecular Subtypes. Scientific reports 2019;9(1):7665 doi 10.1038/s41598-019-43492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fontana E, Eason K, Cervantes A, Salazar R, Sadanandam A. Context matters-consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Annals of oncology : official journal of the European Society for Medical Oncology 2019;30(4):520–7 doi 10.1093/annonc/mdz052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25(22):3230–7 doi 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.