

Abstract
The heterogeneous expression of tumor-associated antigens limits the efficacy of chimeric antigen receptor (CAR)–redirected T cells (CAR-Ts) for the treatment of glioblastoma (GBM). We have found that chondroitin sulfate proteoglycan 4 (CSPG4) is highly expressed in 67% of the GBM specimens with limited heterogeneity. CSPG4 is also expressed on primary GBM-derived cells, grown in vitro as neurospheres (GBM-NS), which recapitulate the histopathology and molecular characteristics of primary GBM. CSPG4.CAR-Ts efficiently controlled the growth of GBM-NS in vitro and in vivo upon intracranial tumor inoculation. Moreover, CSPG4.CAR-Ts were also effective against GBM-NS with moderate to low expression of CSPG4. This effect was mediated by the in vivo up-regulation of CSPG4 on tumor cells, induced by tumor necrosis factor–α (TNFα) released by the microglia surrounding the tumor. Overall, the constitutive and TNFα-inducible expression of CSPG4 in GBM may greatly reduce the risk of tumor cell escape observed when targeted antigens are heterogeneously expressed on tumor cells.
INTRODUCTION
Glioblastoma (GBM) is the most lethal primary brain cancer, with standard treatment based on surgery, radiotherapy, and chemotherapy promoting an overall survival of about 15 months (1, 2). Immunotherapy may complement such treatments, and considerable optimism is presently given to T cells with redirected specificity via expression of chimeric antigen receptors (CAR-Ts). CARs are fusion proteins in which the binding moiety derived from a monoclonal antibody (mAb) is fused with a signaling molecule of the CD3/T cell receptor complex and costimulatory endodomains (3). Upon insertion in T cells, CARs confer human lymphocyte antigen (HLA)–independent cytotoxic activity to T cells and promote T cell proliferation and survival (4).
Crucial to the successful application of CAR-Ts in malignancies including GBM are the restricted expression pattern and the amount of expression of the targeted tumor-associated antigens (TAAs). In this regard, epidermal growth factor receptor variant III (EGFRvIII) is an attractive target antigen for GBM, because its expression is restricted to tumor cells (5–7). In contrast, typical GBM-associated antigens for CAR targeting, such as interleukin-13 receptor subunit α−2 (IL-13Rα2), HER2, and EphA2, are not only generally overexpressed by GBM tumor cells but also detectable in some normal tissues (8–11). Another common feature of the GBM-associated antigens so far targeted with CAR-Ts is the marked intratumoral heterogeneity of expression, which promotes tumor immune escape due to antigen loss (12–15). Finally, although they are frequently used to assess the efficacy of new therapies, established GBM cell lines do not mirror the heterogeneity of the histopathology and molecular characteristics of primary GBM (16, 17).
To overcome these limitations, we have selected to target chondroitin sulfate proteoglycan 4 (CSPG4) in GBM and to use primary glioma-derived cells grown as neurospheres (GBM-NS) as a tumor model. CSPG4 is a cell surface type I transmembrane protein critical for tumor progression and metastasis (18–20). CSPG4 is overexpressed, with limited intra- and intertumoral heterogeneity, by many types of solid tumors (21–25). Notably, the CSPG4 protein is not or barely detectable in normal tissues (18, 22, 25). We have now found that CSPG4 is expressed in considerable amounts not only in GBM specimens but also in GBM-NS, which are particularly relevant for assessing the efficacy of potential therapies because they recapitulate the molecular properties of the primary GBM when expanded in vitro or engrafted in immunodeficient mice (16, 17).
RESULTS
CSPG4 is expressed in GBM specimens and characteristic of more aggressive tumors
The immunohistochemistry of a cohort of 46 GBM specimens showed that CSPG4 was highly expressed in 67% (31 of 46) of the cases and in lower amounts in 33% (15 of 46) of the samples. Among specimens with low expression, CSPG4 was undetectable in only one specimen (Table 1). CSPG4 was expressed in GBM across different molecular subtypes (26). Among GBM with high CSPG4 expression, 36% were assigned to the proliferative/classical subtype, 39% to the mesenchymal subtype, and 25% to the proneural subtype (table S1). The pattern of expression in GBM specimens with high amounts of CSPG4 was diffuse and intense in tumor cells located in the central mass of the tumor and in tumor-infiltrating cells (Fig. 1, A to C). CSPG4 can be expressed by vessels during wound healing and by tumor-associated vessels (26). We found that CSPG4 was coexpressed with α-SMA (smooth muscle actin) in pericytes localized in the vessel wall of GBM (Fig. 1D) (27). In contrast, vessels in normal brain are smaller than those in GBM and do not express CSPG4 (fig. S1). Furthermore, CSPG4 expression in tumor cells was associated with that of nestin, a marker defining dedifferentiated and more aggressive GBM (Fig. 1E) (28). We obtained GBM specimens from patients treated with conventional treatment (neurosurgery, radiotherapy, and chemotherapy with temozolomide) (2) at a single institution and analyzed them by immunohistochemistry. We thus correlated the CSPG4 expression with patients’ overall survival and found that individuals with a high percentage of tumor cells expressing CSPG4 had poorer overall survival (P = 0.002) (Fig. 1F). Patients affected by GBM highly expressing CSPG4 had shorter median survival than those with GBM expressing low amounts of CSPG4 (6.5 months versus 16 months) [ratio, 2.462; 95% confidence interval (CI) of ratio, 1.922 to 3.001]. Twenty-three percent (7 of 30) of GBM with high expression of CSPG4 contained methylated O6-methylguanine-DNA methyltransferase (MGMT) promoter compared to 26.7% (4 of 15) of GBM expressing low amounts of CSPG4 (Fisher’s exact test, P = 0.7) (Table 1). In a multivariate analysis and a Cox proportional hazard regression model analysis considering MGMT methylation and age as variables, CSPG4 expression remained the only significant predictor of overall survival [P = 0.0003, exp(b), 0.27; 95% CI, 0.1331 to 0.5488].
Table 1. Characteristics of patients, GBM specimens, and corresponding neurospheres.
BT, brain tumor; MES, mesenchymal; PN, proneural; CLAS, classical; ND, not determined; Unmet, unmethylated; Met, methylated; OS, overall survival; F, female; M, male.
| GBM | CSPG4 expression | % CSPG4 GBM-NS | OS | Subtype | MGMT methylation | Age | Gender | |
|---|---|---|---|---|---|---|---|---|
| Specimens | Vessels | |||||||
| BT168 | +++ | ++ | 49.0 | 6.0 | MES | Unmet | 78 | F |
| BT205 | + | + | 85.0 | 2.0 | PN | Unmet | 73 | M |
| BT423 | ++ | ++ | 98.7 | 6.5 | CLAS | Met | 43 | F |
| BT273 | + | +/− | 85.9 | 14.0 | CLAS | Met | 59 | M |
| BT275 | + | ++ | 97.4 | 15.0 | CLAS | Met | 67 | F |
| BT299 | ++ | + | 99.0 | 6.5 | PN | Unmet | 50 | M |
| BT308 | + | ++ | 96.0 | 3.0 | PN | Unmet | 76 | M |
| BT326 | + | ++ | 63.0 | 6.5 | PN | Unmet | 28 | M |
| BT328 | + | +/− | 83.2 | 6.0 | MES | Unmet | 56 | M |
| BT358 | ++ | ++ | 95.6 | 22.0 | MES | Unmet | 64 | F |
| BT373 | ++ | ++ | 55.4 | 3.0 | PN | Unmet | 65 | M |
| BT417 | + | ++ | 90.0 | 11.0 | CLAS | Met | 53 | M |
| BT462 | + | ++ | 42.5 | 10.0 | CLAS | Unmet | 71 | M |
| BT482 | ++ | + | 82.6 | 8.0 | MES | Unmet | 38 | M |
| BT517 | ++ | ++ | 91.9 | 8.0 | CLAS | Unmet | 48 | M |
| BT137 | + | ++ | 6.0 | PN | Unmet | 37 | F | |
| BT140 | + | ++ | 10.0 | MES | Unmet | 47 | F | |
| BT150 | + | ++ | 10.0 | CLAS | Unmet | 61 | M | |
| BT157 | + | +/− | 9.0 | MES | Unmet | 54 | M | |
| BT202 | + | ++ | 4.0 | MES | Unmet | 70 | M | |
| BT206 | + | ++ | 4.5 | CLAS | Unmet | 69 | M | |
| BT211 | + | ++ | 4.5 | MES | ND | 61 | M | |
| BT209 | + | ++ | 4.5 | PN | Unmet | 56 | M | |
| BT219 | + | + | 5.0 | MES | Unmet | 55 | M | |
| BT235 | ++ | ++ | 24.5 | CLAS | Met | 70 | M | |
| BT261 | ++ | ++ | 3.0 | CLAS | Unmet | 65 | M | |
| BT334 | ++ | ++ | 9.0 | CLAS | Unmet | 63 | M | |
| BT513 | ++ | + | 13.0 | MES | Unmet | 58 | M | |
| BT241 | + | ++ | 13.0 | ND | Unmet | 70 | M | |
| BT248 | + | ++ | 6.0 | ND | Met | 64 | M | |
| BT347 | ++ | ++ | 29.0 | ND | Met | 57 | F | |
| BT302 | +/− | ++ | 16.0 | 60.0 | MES | Unmet | 19 | F |
| BT337 | +/− | ++ | 69.0 | 17.5 | PN | Met | 65 | M |
| BT379 | +/− | ++ | 97.0 | 16.0 | CLAS | Unmet | 73 | M |
| BT422 | +/− | +/− | 59.1 | 36.0 | CLAS | Unmet | 70 | M |
| BT480 | +/− | ++ | 59.0 | 13.0 | MES | Unmet | 69 | F |
| BT483 | +/− | ++ | 97.7 | 15.0 | PN | Met | 54 | M |
| BT487 | +/− | ++ | 70.6 | 24.0 | MES | Unmet | 72 | M |
| BT500 | +/− | ++ | 84.0 | 9.5 | CLAS | Unmet | 67 | F |
| BT155 | +/− | ++ | 13.0 | CLAS | Unmet | 46 | F | |
| BT245 | +/− | ++ | 46.0 | PN | Met | 57 | F | |
| BT274 | − | + | 18.0 | PN | Unmet | 50 | M | |
| BT279 | +/− | + | 15.0 | CLAS | Unmet | 60 | F | |
| BT283 | +/− | + | 9.5 | CLAS | Met | 46 | F | |
| BT490 | +/− | ++ | 16.0 | PN | Unmet | 76 | F | |
| BT175 | +/− | + | 12.5 | ND | Unmet | 63 | M | |
Fig. 1. CSPG4 is expressed in GBM specimens and GBM-NS and associated with more aggressive disease.
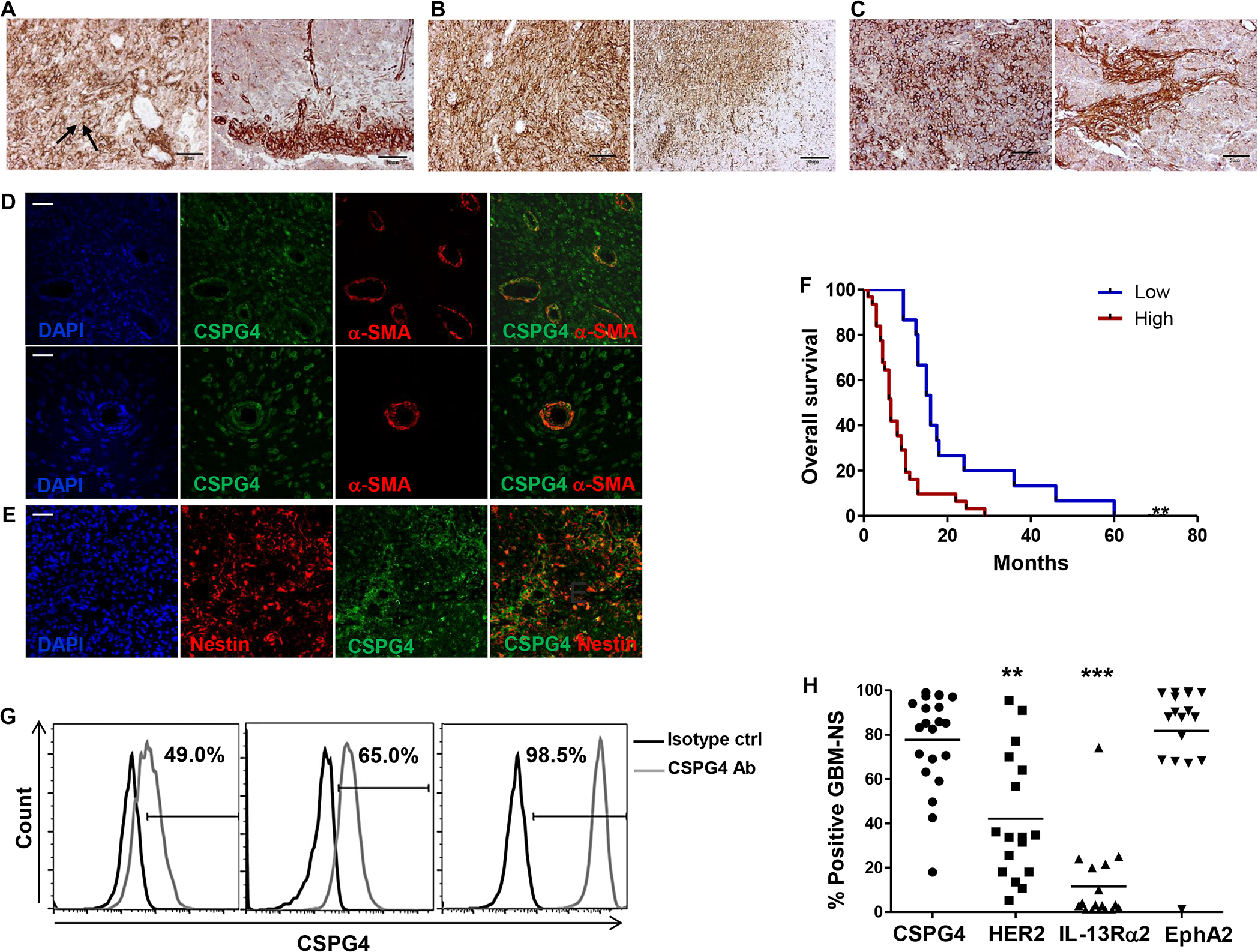
(A to C) Immunohistochemistry of GBM specimens. (A and B) Representative GBM specimens with high CSPG4 expression [BT168 (A) and BT299 (B)]. CSPG4 is mainly located in the plasma membrane of the tumor cells (arrows), with lower signal within the cytoplasm. Tumor cells within the tumor mass and tumor cells infiltrating the parenchyma both express CSPG4. (C) Representative GBM specimen with moderate-low CSPG4 expression (BT308). Tumor clusters containing both tumor cells and tumor-associated pericytes expressing CSPG4 are identified as CSPG4–α-SMA double positive within the normal parenchyma (scale bars, 50 μm). (D) Representative GBM specimen (BT168) evaluated by immunofluorescence show the presence of blood vessels within the tumor mass. Tumor-associated pericytes are identified as CSPG4 (green) and α-SMA (red) double positive (merge) (scale bars, 50 μm). (E) Representative GBM specimen evaluated by immunofluorescence (BT168). Nuclei that have been visualized by 4′,6-diamidino-2-phenylindole (DAPI) staining (blue), nestin (red), and CSPG4 (green) are highly coexpressed (merge) by tumor cells (scale bar, 50 μm). (F) Kaplan-Meier survival curves for patients with low-moderate and high CSPG4 expression in tumor cells (**P = 0.002). (G) Flow cytometry histograms showing the expression of CSPG4 in GBM-NS. Moderate-low expression (CSPG4ML) (left panel), moderate-high expression (CSPG4MH) (middle panel), and high expression (CSPG4H) (right panel) are represented. (H) Scatter plots showing the expression of CSPG4 (n = 23), HER-2 (n = 17), IL-13Rα2 (n = 17), and EphA2 (n = 17) in GBM-NS [**P < 0.001 and ***P < 0.0001, analysis of variance (ANOVA)].
GBM-NS express CSPG4
To establish GBM tumor models that recapitulate the main features of the originating GBM both in vitro and in vivo upon engraftment in immunodeficient mice (16), we established in vitro GBM-NS from primary tumors (Table 1). GBM-NS were then characterized for the expression of CSPG4 and other surface molecules currently targeted with CAR-Ts, such as HER2, IL-13Rα2, and EphA2 (10, 11, 29). Seventy percent of GBM-NS had high CSPG4 expression (percentage of positive cells ranging from 71 to 99%) (CSPG4H), 17% had moderate-high expression (percentage of positive cells ranging from 51 to 70%) (CSPG4MH), and 13% had moderate-low expression (percentage of positive cells, <50%) (CSPG4ML) (Fig. 1G and Table 1). The expression of CSPG4 in GBM-NS was comparable to that of EphA2 but significantly higher than that of HER2 and IL-13Rα2 (P < 0.0001) (Fig. 1H).
CSPG4.CAR-Ts target GBM-NS in vitro
To evaluate the susceptibility of GBM-NS to CSPG4-mediated targeting, T lymphocytes obtained from six healthy donors were transduced to express the CSPG4.CAR, encoding the 4–1BB (30) costimulatory endodomain, via gammaretroviral vector gene transfer. CSPG4.CAR was detectable in 75 ± 9.1% of CD3+ T cells using flow cytometry by day 7 of culture (fig. S2A), including both CD8+ and CD4+ T cells (69.9 ± 2.5% versus 25.8 ± 0.4%, respectively) (fig. S2B). We then tested the cytolytic activity of CSPG4.CAR-Ts against 19 GBM-NS in coculture experiments. CSPG4.CAR-Ts cocultured with GBM-NS at effector to target (E:T) ratios ranging from 1:5 to 2:5 efficiently eliminated both CSPG4H and CSPG4ML GBM-NS by day 3 of coculture (1.5 ± 4.8 and 1.8 ± 5.2 residual GBM-NS, respectively) (Fig. 2, A and B). In contrast, GBM-NS continued to grow in the presence of control T cells (cTs) (74.6 ± 20.8% residual GBM-NS) (Fig. 2, A and B). CSPG4.CAR-Ts showed a T helper 1 (TH1)–type cytokine profile in response to GBM-NS, as seen by the release of interferon-γ (IFN-γ) and IL-2 (Fig. 2, C and D). CSPG4.CAR-Ts, but not cTs, proliferated in response to GBM-NS, as evaluated using a carboxyfluorescein diacetate succinimidyl ester (CFSE)–based dilution assay on day 7 of coculture (66 ± 7.4% versus 9 ± 1.5%, respectively; n = 23; P < 0.01) (Fig. 2, E and F). In a standard 51Cr release assay, CSPG4.CAR-Ts lysed GBM-NS (Fig. 2G).
Fig. 2. CSPG4.CAR-Ts target GBM-NS in vitro.
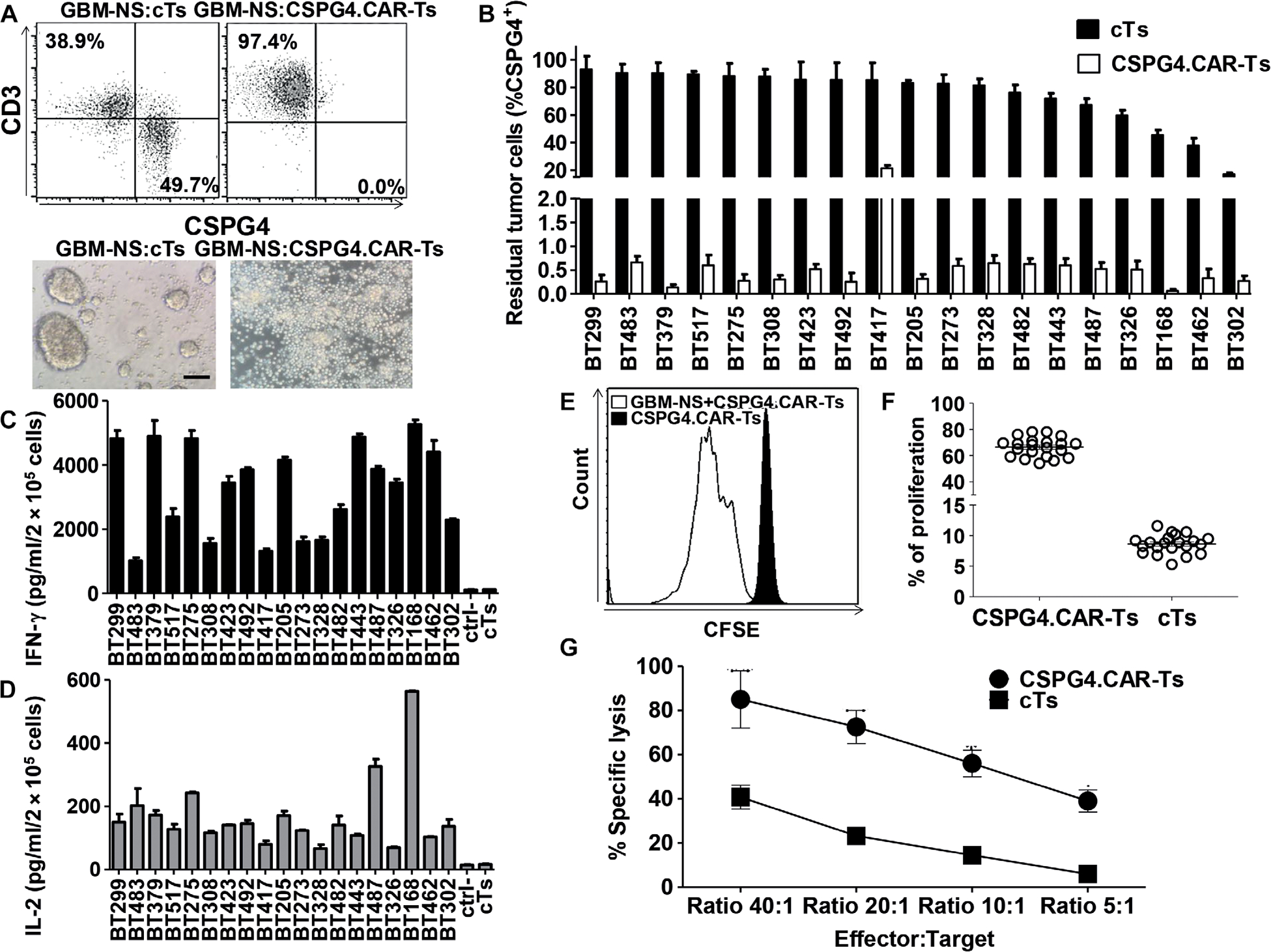
GBM-NS were cocultured in vitro with either cTs or CSPG4.CAR-Ts in GBM-NS serum-free medium [Dulbecco’s modified Eagle’s medium (DMEM)/F12] and B27 supplement. (A) Representative dot plots (upper panel) and microscopic images (lower panel) showing the cytotoxic activity of CSPG4.CAR-Ts compared to cTs. Flow cytometry plots show GBM-NS and T cells on day 3 of coculture as assessed by flow cytometry using CD3 and CSPG4 as markers for T cells and GBM-NS, respectively (scale bar, 200 μm). (B) Bar graph showing the percentage of residual tumor cells in cocultures of CSPG4.CAR-Ts or cTs against 19 GBM-NS at an E:T ratio of 1:5. GBM-NS are ordered by CSPG4 expression (highest to lowest CSPG4-expressing cell lines). Data are means ± SD of T cell preparations obtained from six different healthy donors. (C and D) IFN-γ (C) and IL-2 (D) released by either cTs or CSPG4.CAR-Ts into the supernatant of coculture with GBM-NS within 24 hours. The established cell line T98G was used as a CSPG4-negative target (ctrl). (E) Representative flow cytometry histograms showing the proliferation of CSPG4.CAR-Ts in response to GBM-NS compared to CSPG4.CAR-Ts alone, as evaluated by the CFSE dilution assay on day 7 of coculture in one representative of six donors. (F) Scatter plot showing the proliferation of CSPG4.CAR-Ts in response to 23 GBM-NS compared to cTs. (G) Cytotoxic activity of cTs and CSPG4.CAR-Ts evaluated in a 6-hour 51Cr release assay. Data are means ± SD of T cells generated from two donors against a total of four GBM-NS (BT168-NS, BT308, BT462-NS, and BT275-NS).
Tumor growth is effectively controlled by intracranial administration of CSPG4.CAR-Ts in GBM-NS xenograft models
In the first model, nude mice were implanted intracranially with GBM-NS CSPG4H (96%, BT308-NS) modified to express green fluorescent protein (GFP)/firefly luciferase (FFluc). CSPG4.CAR-Ts and cTs were injected intratumorally 2 weeks later (Fig. 3A), and tumor growth was monitored by in vivo imaging (Fig. 3, B and C). Mice treated with cTs developed neurological signs of tumor growth and required euthanasia by day 60. In contrast, CSPG4.CAR-Ts effectively controlled tumor growth (Fig. 3, B and C) and improved survival (Fig. 3D) (P < 0.0001), with 6 of 10 mice tumor-free up to day 180, whereas 4 of 10 mice developed neurological signs of tumor growth and required euthanasia. None of the mice inoculated with either cTs or CSPG4.CAR-Ts developed signs of graft-versus-mouse disease. In the second model, nude mice were implanted intracranially with CSPG4ML (49%, BT168-NS) modified to express GFP/FFluc. CSPG4.CAR-Ts and cTs were injected intratumorally 1 week later (Fig. 3E). Mice treated with cTs developed neurological signs of tumor growth and required euthanasia by day 80. In this model, CSPG4.CAR-Ts also controlled tumor growth (Fig. 3, F and G) and improved survival (Fig. 3H) (P < 0.0001). Administration of CSPG4. CAR-Ts also consistently prolonged the survival of mice implanted with two other GBM-NS, CSPG4H (97%, BT275-NS) and CSPG4ML (42%, BT462-NS), respectively (P < 0.0001) (fig. S3, A and B), and of mice implanted with the established GBM tumor cell line U87-MG (fig. S4, A to D).
Fig. 3. CSPG4.CAR-Ts control tumor growth in GBM-NS xenograft models.
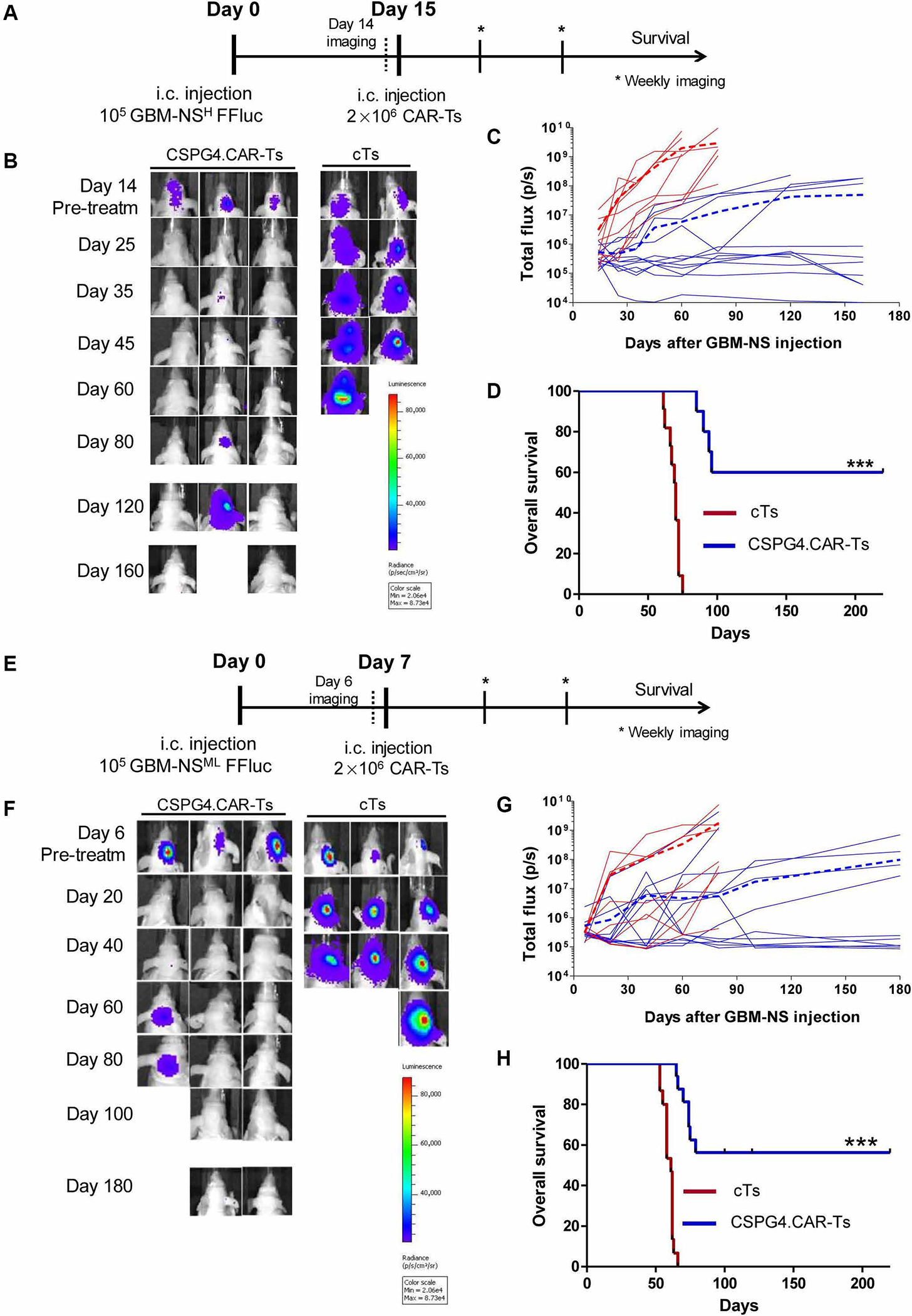
(A) Nude mice implanted intracranially (i.c.) with GBM-NS CSPG4H (96%, BT308-NS). GBM-NS were labeled with GFP/FFluc. Mice were injected intratumorally with either CSPG4. CAR-Ts or cTs at day 15. (B) Representative in vivo imaging illustrating the growth of GFP/FFluc BT308-NS in treated mice. (C) Line graphs of tumor flux (photons per second) versus time of all BT308-NS–bearing mice treated with cTs (red lines) or CSPG4.CAR-Ts (blue lines). The dotted lines represent the mean photon flux for cTs (dotted red line) and CSPG4.CAR-Ts (dotted blue line). (D) Kaplan-Meier survival curves of BT308-NS–bearing mice injected intratumorally with either CSPG4.CAR-Ts or cTs (***P < 0.0001). (E) Nude mice implanted intracranially with GBM-NS CSPG4ML (49%, BT168-NS). GBM-NS were labeled with GFP/FFluc. Mice were injected intratumorally with either CSPG4. CAR-Ts or cTs at day 7 because BT168-NS showed faster growth in vivo as compared to BT308-NS. (F) Representative in vivo imaging illustrating the growth of GFP/FFluc BT168-NS in treated mice. (G) Line graphs of tumor flux (photons per second) versus time of all BT168-NS–bearing mice treated with cTs (red lines) or CSPG4.CAR-Ts (blue lines). The dotted lines represent the mean photon flux for cTs (dotted red line) and CSPG4.CAR-Ts (dotted blue line). (H) Kaplan-Meier survival curves of BT168-NS–bearing mice injected intratumorally with either CSPG4.CAR-Ts or cTs (***P < 0.0001).
In parallel experiments using GFP/FFluc-labeled T cells (31), we observed that CSPG4.CAR-Ts persisted for 9 to 10 days upon intratumoral inoculation in mice, whereas cTs were almost undetectable by day 3 (Fig. 4, A and B). We confirmed, by flow cytometry, the presence of CSPG4.CAR-Ts within the tumor 1 week after infusion (60 ± 5% of hCD45+CD3+ cells), whereas T cells were undetectable in mice receiving cTs (Fig. 4C). Notably, in mice treated with CSPG4.CAR-Ts and developing tumors, immunofluorescence of the explanted tumors showed CSPG4 expression comparable to that of tumors explanted from mice treated with cTs (Fig. 4, D and E). These tumors also coexpressed nestin, recapitulating the features of the parental GBM specimen (BT168) (Fig. 4, D and E). CSPG4.CARs encoding either CD28 or the combination of CD28 and 4–1BB endodomains were also tested, but they only partially eliminated GBM-NS in vitro (fig. S5). Similarly, in vivo, they generally showed limited antitumor activity (fig. S6) and short persistence (fig. S7). As observed in mice treated with CSPG4.CAR-Ts encoding 4–1BB, tumor recurrence in mice treated with CSPG4.CAR encoding CD28 or CD28/4–1BB occurred without evidence of antigen loss by tumor cells (fig. S8).
Fig. 4. CSPG4.CAR-Ts do not cause tumor escape due to antigen loss.
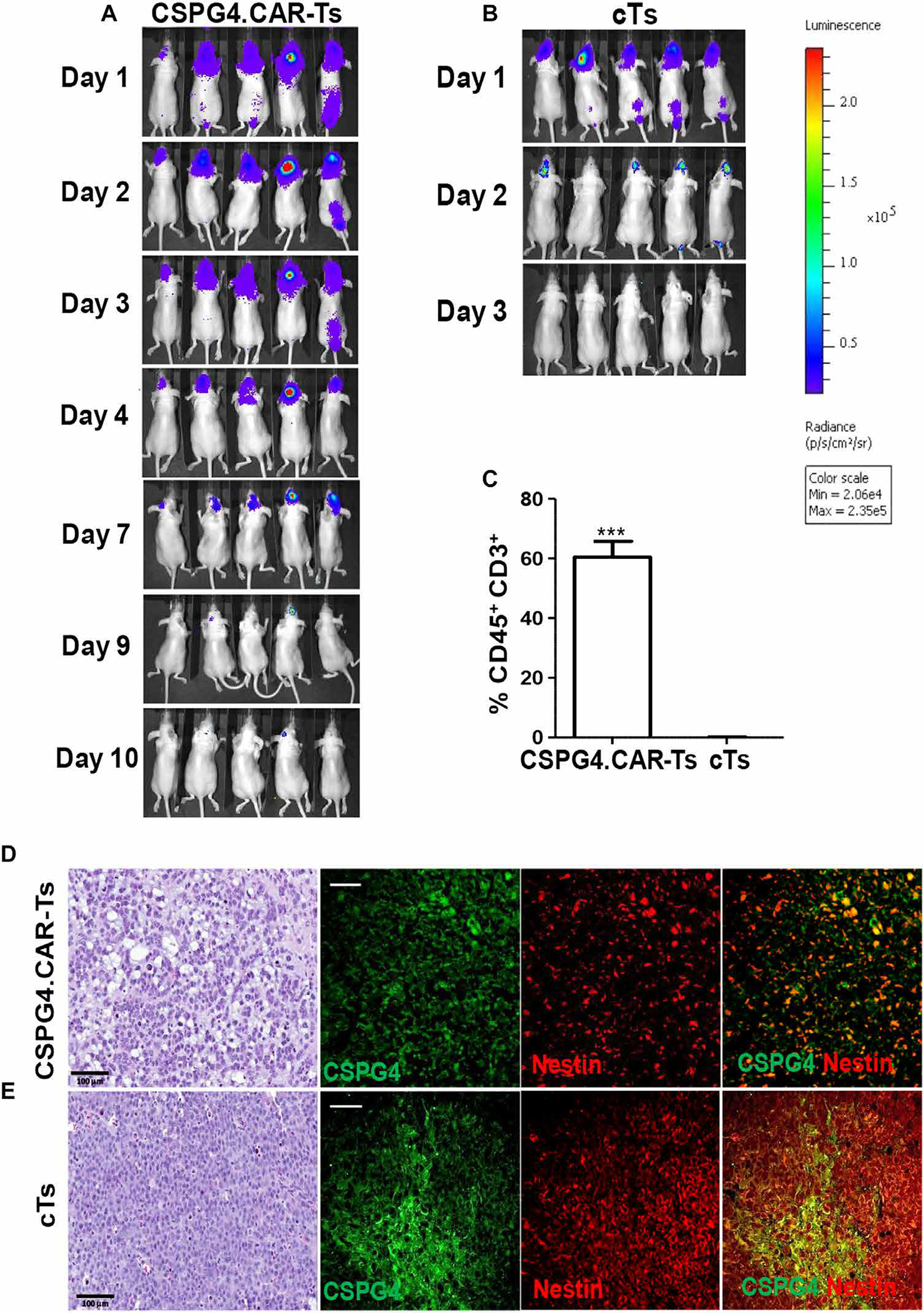
(A and B) Nude mice implanted intracranially with GBM-NS CSPG4H (96%, BT308-NS) were infused intratumorally with either CSPG4.CAR-Ts (A) or cTs (B) labeled with GFP/FFluc. Bioluminescence imaging shows the persistence of CSPG4.CAR-Ts (A) and cTs (B). (C) Bar graph showing human T cells detected intratumorally in xenograft gliomas removed from mice treated with CSPG4.CAR-Ts or cTs. Human CD45+CD3+ T cells were detected by flow cytometry. (D and E). Immunohistochemistry and immunofluorescence of GBM-NS CSPG4ML (49%, BT168-NS) glioma removed from mice previously treated with either CSPG4.CAR-Ts (D) or cTs (E). Hematoxylin and eosin staining of gliomas from mice treated with CSPG4.CAR-Ts shows a disruption in the architecture as compared to glioma from mice treated with cTs. Immunofluorescence of gliomas shows that CSPG4 and nestin expression is highly preserved in gliomas treated with either cTs or CSPG4.CAR-Ts (scale bars, 50 μm).
GBM-NS up-regulates PD-L1 upon exposure to IFN-γ
CSPG4.CAR-Ts encoding the 4–1BB endodomain were detectable for 8 to 10 days after intratumor inoculation, which is likely insufficient to consistently eliminate all tumor cells. We observed that CSPG4.CAR-Ts released different amounts of IFN-γ in response to GBM-NS in vitro, which was independent from the amount of CSPG4 expression. To explore this phenomenon, we analyzed expression of PD-L1 (programmed cell death ligand 1) in GBM-NS. As shown in fig. S9, GBM-NS express low amounts of PD-L1, but PD-L1 is up-regulated upon exposure to IFN-γ in vitro. Notably, the expression of PD-L1 by GBM-NS inversely correlated with the amount of IFN-γ released by CSPG4. CAR-Ts in response to GBM-NS in vitro (fig. S9), and PD-L1 blockade enhanced IFN-γ released by CSPG4.CAR-Ts in response to GBM-NS. PD-L1 up-regulation by GBM-NS was also observed in vivo in response to CSPG4.CAR-Ts (fig. S9), suggesting that the PD-1/PD-L1 axis contributes to limiting the efficacy of CAR-Ts in GBM.
CSPG4 is up-regulated on GBM-NS upon engraftment in xenograft models
Because CSPG4.CAR-Ts promoted excellent antitumor activity in mice engrafted with CSPG4ML GBM-NS, without evidence of immune escape of CSPG4-negative tumor cells, we investigated whether the expression of CSPG4 was up-regulated in vivo. Tumor cells from BT168-NS xenografts explanted 1 week after intracranial inoculation in untreated mice expressed more CSPG4 than the same BT168-NS cells cultured in vitro (99% versus 49%) (Fig. 5A). In contrast, in vivo up-regulation was not observed for other known CAR-T target antigens such as HER-2 and IL-13Rα2 (fig. S10). To dissect the dynamic of CSPG4 expression, we sorted BT168-NS grown in vitro to obtain CSPG4H (81.2 ± 3.2%) and CSPG4L (1.4 ± 0.2%) cells (Fig. 5B), respectively. When sorted cells were cultured in vitro for 3 weeks, we found that CSPG4 expression remained high in CSPG4H cells and low in CSPG4L cells (Fig. 5C). Tumors removed by day 10 after inoculation from mice implanted with CSPG4L cells showed CSPG4 up-regulation (63.5 ± 5%), whereas tumors derived from CSPG4H cells maintained high CSPG4 expression (75.2 ± 2.2%) (Fig. 5D). Notably, CSPG4H and CSPG4L cells showed similar growth kinetics when cultured in vitro (Fig. 5E) and similar tumorigenicity in immunodeficient mice, causing death by day 75 upon cell inoculation (Fig. 5F). These results argue against a selective advantage growth of CSPG4H cells versus CSPG4L cells in vivo.
Fig. 5. GBM-NS up-regulate CSPG4 in vivo.
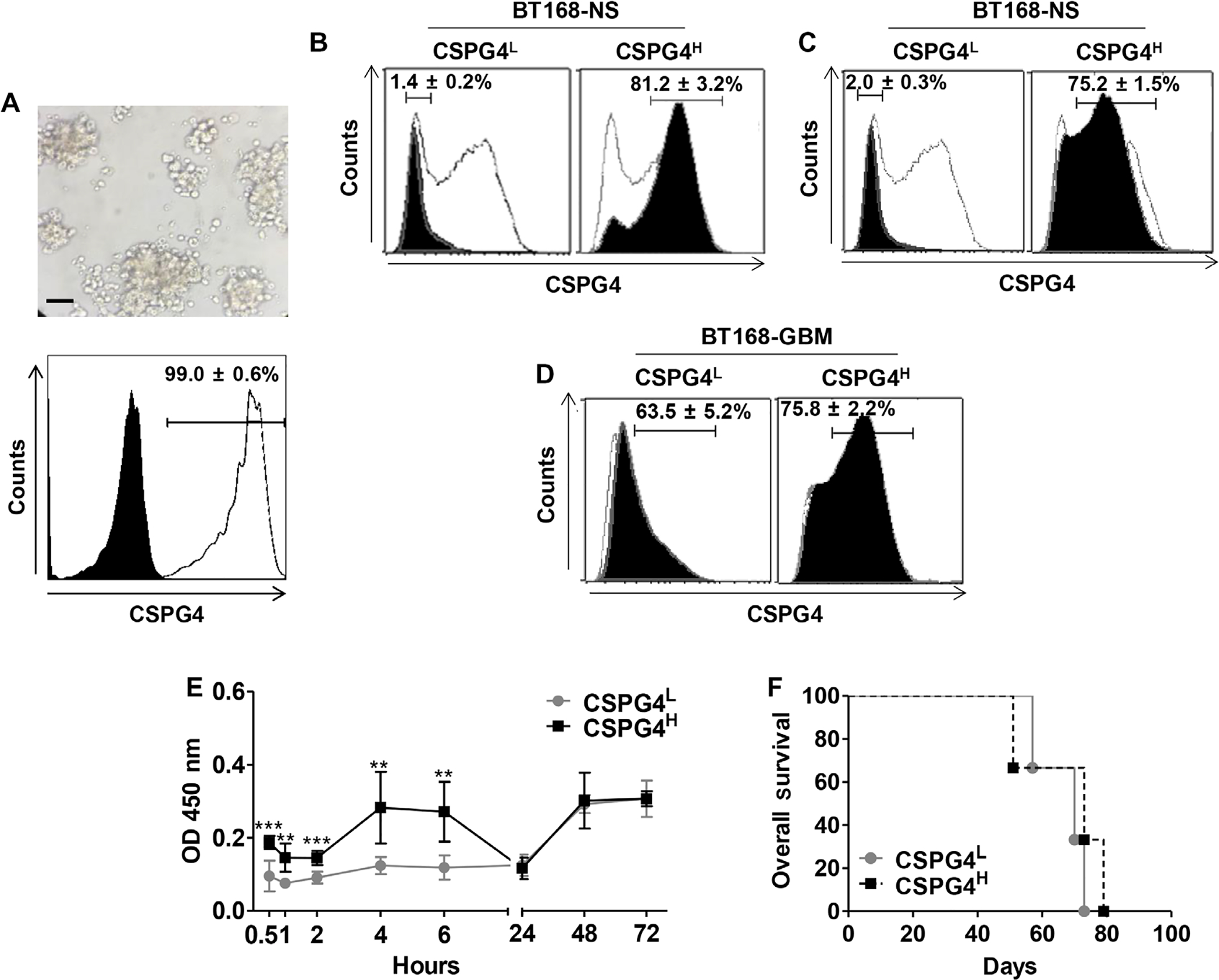
(A) Xenograft gliomas originating from the GBM-NS CSPG4ML (49%, BT168-NS) were explanted from engrafted mice and depleted of contaminant murine cells. Representative in vitro image shows GBM-NS 12 hours after isolation and murine cell depletion (upper panel) (scale bar, 150 μm). CSPG4 expression was assessed by flow cytometry (white histogram) compared to isotype control (black histogram). (B) BT168-NS were selected to obtain CSPG4L and CSPG4H cells using immunomagnetic sorting. Black histograms indicate the expression of CSPG4 in CSPG4L and CSPG4H cells as compared to unselected BT168-NS (white histograms). Data are representative of three different experiments. (C) Expression of CSPG4 (black histograms) in BT168-NS CSPG4L and BT168-NS CSPG4H versus unselected BT168-NS (white histograms) after 3 weeks in culture. (D) Gliomas originating from BT168-NS CSPG4L and CSPG4H cells were removed from engrafted mice. Upon depletion of contaminant murine cells, the expression of CSPG4 was measured by flow cytometry. Black histograms indicate the expression of CSPG4 in BT168 gliomas from CSPG4L and CSPG4H cells, whereas white histograms represent CSPG4 expression in CSPG4L and CSPG4H cells before in vivo implantation. (E) Kinetics of in vitro growth of CSPG4L and CSPG4H as measured by WST assay. (F) Kaplan-Meier survival curves of nude mice implanted intracranially with either BT168-NS CSPG4L or BT168-NS CSPG4H.
TNFα released from tumor-associated microglia induces CSPG4 expression in GBM-NS
Resident microglia and recruited inflammatory cells are critical components of the GBM microenvironment and contribute in inducing tumor cell proliferation and infiltration, at least in part via tumor necrosis factor–α (TNFα) and IL-6 (32). We observed that BT168-NS xenografts, removed 1 week after tumor implantation, were infiltrated by murine CD45lowCD11bhighLy6C+ cells, which represented ~10% of the cellularity upon tumor dissociation (Fig. 6A). When murine and human cells from engrafted BT168-NS were separated, we detected high murine TNFα concentration in the murine fraction of the xenograft but relatively low murine IL-6 concentration (Fig. 6B). BT168-NS cultured ex vivo in the presence of the murine fraction isolated from the xenograft showed CSPG4 up-regulation (from 49% to 68 ± 1.3%) (P < 0.01) (Fig. 6C). Considering the high homology between murine and human TNFα and previous evidence that human and murine TNFα are cross-reactive (33), we hypothesized that the CSPG4 up-regulation in engrafted tumors was mediated by TNFα released by murine microglia. We confirmed that human recombinant TNFα (hTNFα) up-regulated CSPG4 when added in vitro to either unselected BT168-NS or selected BT168-NS CSPG4L cells (from 49% to 71 ± 3% and from 1% to 36 ± 1%, respectively) (P < 0.01 and P < 0.005) (Fig. 6, C and D). A similar pattern of CSPG4 expression was obtained in another CSPG4ML GBM-NS (BT462-NS) upon incubation with hTNFα (CSPG4 from 42.5 ± 0.2% to 72.1 ± 0.8%) (Fig. 6E). We further characterized the hTNFα-dependent CSPG4 up-regulation in GBM-NS by assessing nuclear factor κB (NF-κB) activation and found that hTNFα up-regulated p65 phosphorylation in BT168-NS. In contrast, blocking TNFα with infliximab prevented the expression of both NF-κB p65 and CSPG4 (Fig. 6, F and G). To evaluate whether CSPG4 up-regulation in GBM-NS in response to murine microglia is clinically relevant, we assessed the content of the microglia, identified as Iba1+ cells, in GBM specimens. We observed that GBM expressing higher amounts of CSPG4 showed more microglia content (P = 0.04) (Fig. 7, A to C) and that the microglia express TNFα (Fig. 7D).
Fig. 6. TNFα derived from murine microglia influences the expression of CSPG4 in GBM-NS in vitro and in vivo.
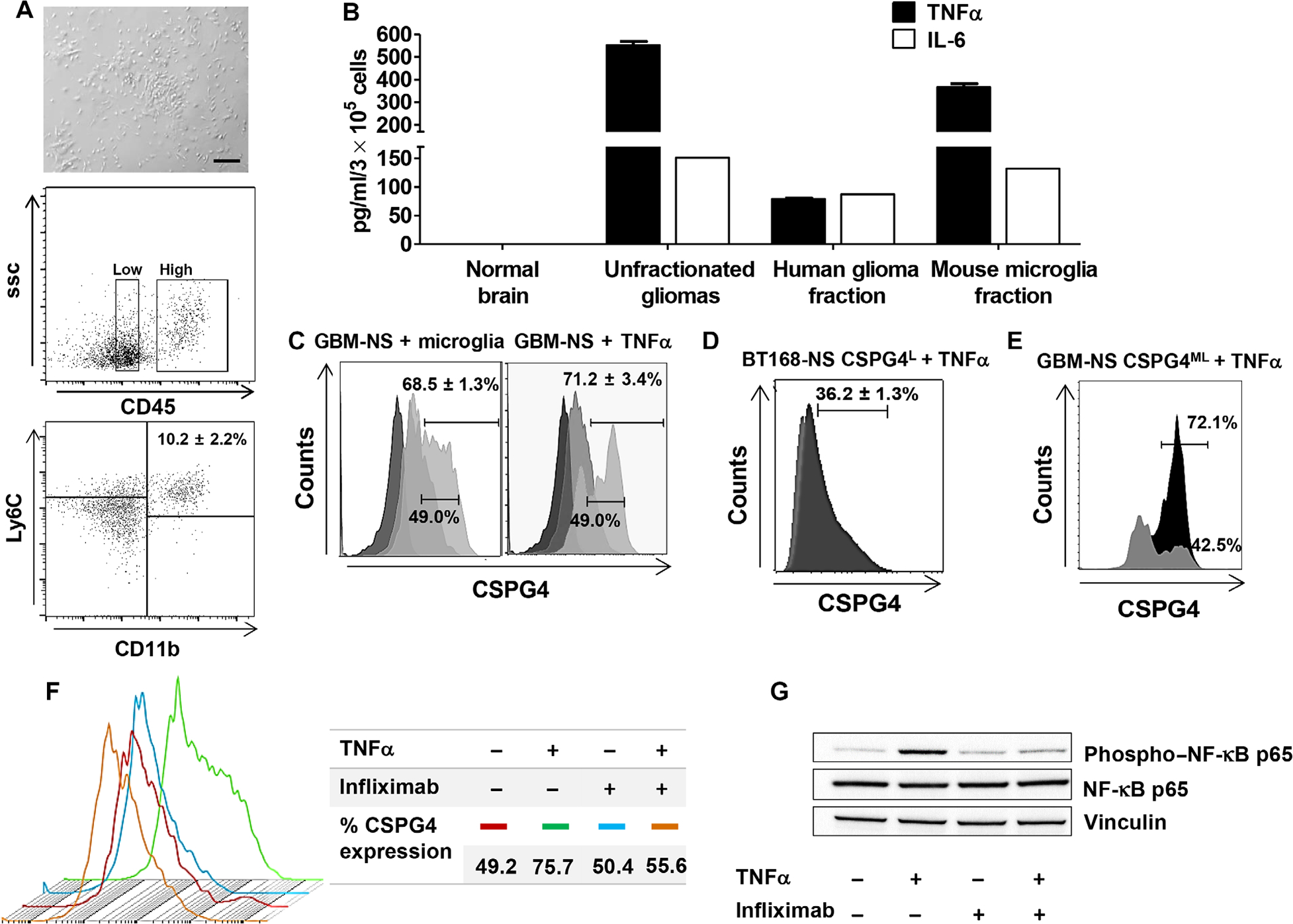
(A) Gliomas derived from GBM-NS in vivo were removed from engrafted nude mice. The murine fraction of the removed tumors was isolated and plated in culture (upper panel) (scale bar, 200 μm). The lower panels show murine CD45lowCD11bhighLy6C+ cells removed from the glioma, which correspond to murine-derived microglia. (B) Lysates from the unfractionated glioma, selected human glioma fraction, selected murine microglia fraction, and normal brain were analyzed for the presence of murine TNFα and IL-6 by specific ELISA. (C) BT168-NS were cultured in vitro in the presence of either the murine fraction isolated from the xenograft GBM or hTNFα. CSPG4 expression was measured by flow cytometry 24 hours after incubation. Light gray, gray, and dark gray histograms indicate CSPG4 expression after hTNFα stimulation, no stimulation, and isotype control, respectively. (D) BT168-NS CSPG4L were stimulated with hTNFα in vitro. CSPG4 expression was measured by flow cytometry. Light gray and dark gray histograms indicate CSPG4 expression in the absence or presence of hTNFα, respectively. (E) Up-regulation of CSPG4 by GBM-NS CSPG4ML (BT462-NS) upon hTNFα treatment. Light gray and black histograms indicate CSPG4 expression without and with hTNFα, respectively. (F) CSPG4 expression in BT168-NS treated with or without hTNFα and with or without TNFα-blocking Ab (infliximab, 50 μg/ml). (G) Immunoblot of BT168-NS treated with or without hTNFα and with or without TNFα-blocking Ab (infliximab, 50 μg/ml). hTNFα activates NF-κB in BT168-NS after 24 hours, as measured by the increase of phospho–NF-κB p65. Vinculin is used as loading control.
Fig. 7. CSPG4-high GBMs show more microglia than CSPG4-low GBMs and express TNFα.
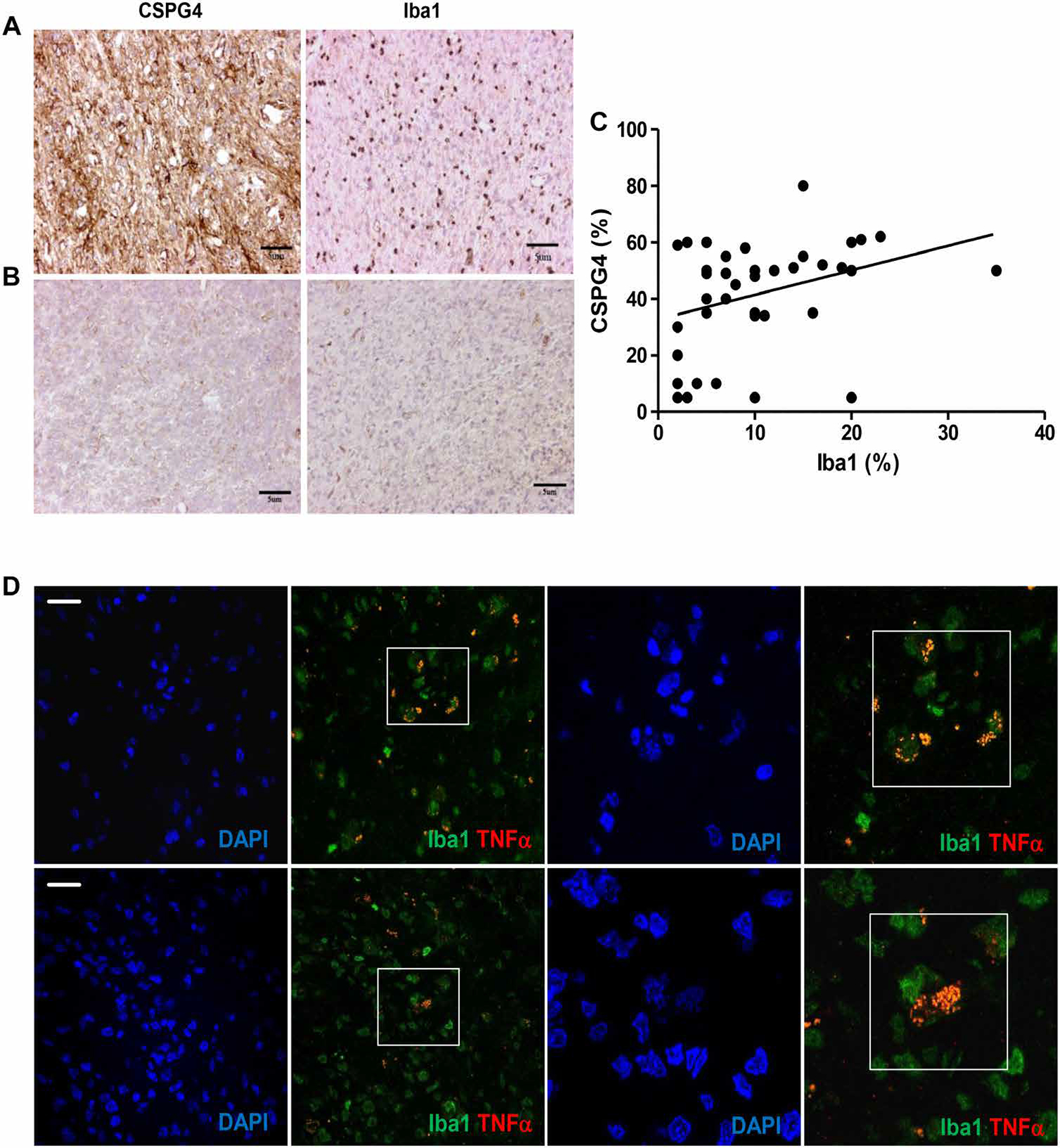
(A) GBM expressing high amounts of CSPG4 (BT168) shows a high number of Iba1+ cells identified as microglia. (B) The only GBM negative for CSPG4 expression (BT274) in our cohort shows a total lack of Iba1+ cells. (C) Correlation of the percentage of CSPG4-positive tumor cells and Iba1-positive cells in the tumor mass (linear regression, P = 0.04). The cutoff for Iba1 expression was set at 10%. (D) Representative GBM specimen evaluated by immunofluorescence shows the presence of Iba1+ cells (green) coexpressing TNFα (merge). Two different areas are shown at two magnifications (scale bars, 25 μm). Nuclei are visualized by DAPI staining.
DISCUSSION
The high intra- and intertumoral heterogeneity of cell surface expression of most of the identified TAAs in GBM is a major obstacle for developing effective immunotherapy. Here, we demonstrate that CSPG4 is highly expressed in 67% of GBM specimens with limited intratumoral heterogeneity. Furthermore, CSPG4 expression is constitutive in a broad array of GBM-NS and induced by TNFα released from the microglia surrounding the tumors. Because of its high constitutive and inducible expression, CSPG4 is effectively targeted by CSPG4.CAR-Ts that promote control of tumor growth in clinically relevant GBM xenograft models.
Because of their surface expression in GBM and not in normal brain parenchyma, IL-13Rα2, HER2, EGFRvIII, and EphA2 antigens are suitable targets for CAR-Ts (6, 7, 10, 29, 34). CAR-Ts targeting IL-13Rα2, HER2, and EGFRvIII have been tested in clinical trials. Three patients with recurrent GBM received autologous T cells expressing a first-generation IL-13Rα2–specific CAR. Multiple doses of CAR-Ts delivered intratumorally via a reservoir implanted at the time of surgery were well tolerated and promoted transient anti-glioma responses (35). More recently, T cells expressing a second-generation IL-13Rα2–specific CAR encoding the 4–1BB endodomain were infused either intracavity or intraventricularly in seven patients. Sustained tumor regression was observed in one patient (36). In another phase 1 study, autologous virus-specific T cells expressing a second-generation HER2-specific CAR (encoding the CD28 endodomain) were infused intravenously in 17 patients with recurrent GBM. Treatment was well tolerated, without on-target toxicity toward normal tissues, and promoted disease stabilization in seven patients (37). Finally, 10 patients with recurrent GBM were safely infused intravenously with autologous EGFRvIII-specific CAR-Ts. Although T cells were detectable in regions of active GBM, no measurable antitumor activity was documented, and EGFRvIII antigen decrease was observed in five of seven evaluable patients (15).
These clinical data not only support the potential of achieving clinical benefits using CAR-Ts in GBM but also highlight that the heterogeneous intratumor expression of the targeted antigens remains a critical issue, because outgrowth of tumor cells with low expression of a targeted antigen was observed (38). Tumor escape secondary to antigen loss was also reported in patients with GBM vaccinated to target the EGFRvIII antigen (14).
Our results indicate that CSPG4 is an attractive target for immunotherapy in GBM. We found that CSPG4 is highly expressed in 67% of the GBMs analyzed, showing limited intratumoral heterogeneity, expression in tumor-associated vessels, and lack of expression in the normal brain parenchyma. CSPG4 overexpression in GBM correlates with a more aggressive clinical outcome, as reported for other types of CSPG4-expressing malignancies (39–41). Finally, CSPG4 is also a particularly attractive target for immunotherapy because of its critical role in promoting tumor progression (42), causing chemo- and radiotherapy resistance (43), inducing angiogenesis (44, 45), and, as reported here, being consistently expressed or induced in GBM-NS.
Because of their capacity to self-renew and differentiate into cell types present within the tumor of origin in immunodeficient mice, while simultaneously maintaining the molecular and histopathological characteristics of the parental tumors, GBM-NS are highly relevant tumor cells to study the efficacy of potential therapies in GBM (16, 17). In addition, we and others have reported that patients with GBM that can generate GBM-NS with high replicative potentials in vitro have poorer clinical outcomes (17, 46). IL-13Rα2 and HER2 can also be expressed by GBM-NS generated in vitro and by CD133+ cells selected from primary GBM tissues, respectively (10, 29, 47). Our cohort of GBM-NS includes a panel of varied GBM-NS that reflect GBM with different molecular characteristics. Our data highlight the higher expression of CSPG4 in GBM-NS, as compared to IL-13Rα2 or HER2, and have confirmed the colocalization of CSPG4 and nestin in primary GBM samples. The latter is a recognized marker of aggressiveness in GBM.
Relevant to the use of targeted immunotherapy in GBM is our observation that CSPG4, but not IL-13Rα2 or HER2, expression is up-regulated in vivo in response to TNFα. The source of TNFα is the surrounding tumor-associated microglia. Microglia are well known to shape the complex microenvironment of GBM (48). The interactions between GBM and microglia enhance the invasion properties of GBM through the secretion of EGF and CSF-1 (colony-stimulating factor 1), as demonstrated in the GL261 murine glioma model (49). Microglia differ from monocytes/macrophages by having a lower expression of CD45 (50) and Ly6C (51). In pathological conditions, microglia release large amount of TNFα involved in sensitizing cancer cells to apoptosis (52). Notably, CSPG4 protects tumor cells from TNFα-induced apoptosis (43), exerting many biological functions through the NF-κB signaling pathway (52). In addition, NF-κB promotes the up-regulation of many proteoglycans in GBM (53). Our findings support this correlation because, by blocking TNFα, we prevented NF-κB activation and CSPG4 expression in GBM-NS. Resident microglia and recruited inflammatory cells are important components of the GBM microenvironment actively involved in promoting tumor expansion (54). Our experimental observation of CSPG4 up-regulation in GBM-NS in response to murine microglia is corroborated by the clinical observation that the microglia, identified using the specific marker Iba1 (55), are more abundant in GBM specimens with high expression of CSPG4 and coexpression of TNFα.
High constitutive and TNFα-induced CSPG4 expression in GBM-NS contributes to the marked antitumor activity of CSPG4.CAR-Ts encoding the 4–1BB endodomain, which we have observed in our xenotransplant models. 4–1BB–mediated costimulation was superior to either CD28 or the combination of CD28 and 4–1BB costimulation, promoting better intratumor persistence of CSPG4.CAR-Ts, a feature consistent with the reported prolonged persistence of CD19-specific CAR-Ts encoding 4–1BB in patients with lymphoid malignancies (56, 57). Sixty percent of mice treated with CSPG4.CAR-Ts remained tumor-free up to 180 days, and tumor recurrence was not due to antigen loss by tumor cells. Although 4–1BB costimulation provided better intratumor persistence for CSPG4.CAR-Ts than other costimulation, CSPG4.CAR-Ts were only detectable for 8 to 10 days after intratumor inoculation, which is likely insufficient to consistently eliminate all tumor cells. We observed that GBM-NS up-regulate PD-L1 in response to IFN-γ in vitro and in vivo when IFN-γ is released by CSPG4.CAR-Ts. This suggests that, even if our mouse model does not assess the potential immunosuppressive role of the GBM tumor microenvironment, the PD-1/PD-L1 axis may contribute to limiting the efficacy of CAR-Ts in GBM. This observation also indicates that although recent TCGA (The Cancer Genome Atlas) data show that IFN-related genes are expressed in low amounts in GBM compared to other tumors (58), upon encounter with T cells, releasing IFN-γ GBM can up-regulate PD-L1. Our observation in the mouse model is supported by recent clinical evidence showing expression of inhibitory molecules in GBM after the infusion of EGFRvIII-specific CAR-Ts (15).
We elected to use intratumor delivery of CAR-Ts to treat GBM because this approach has been safely used in patients (35, 36), overcomes the interference of the blood-brain barrier to T cell trafficking, and mitigates potential “on-target off-tumor” toxicities in the event that the antigen is expressed in low amounts in normal organs. However, the intratumor/intracavity delivery of CAR-Ts does not recapitulate the spatiotemporal biodistribution of T cells, which usually reach tumors via vasculature, and can at least partially explain the short-term persistence of CAR-Ts in our and other GBM models (59). Nevertheless, for effective clinical application, the limited persistence of intratumorally inoculated CAR-Ts can be compensated by the administration of multiple doses of CAR-Ts (36, 60) or by developing implantable scaffolds or biomaterials that promote the gradual release of CAR-Ts and/or the local delivery of growth factors to sustain their survival.
In conclusion, the high constitutive and TNFα-induced CSPG4 expression in GBM allows the application of CAR-Ts to a high proportion of patients and reduces the risk of tumor escape due to the heterogeneous antigen expression. A combination of CAR-Ts targeting multiple antigens likely remains a prerequisite for successful tumor eradication of GBM in all patients (61), but our data suggest that targeting CSPG4 as a single antigen is beneficial and that CSPG4 should be included in future combinations aimed at targeting multiple antigens in GBM.
MATERIALS AND METHODS
Study design
The aim of the study was to test the expression of CSPG4 in GBM and the activity of CAR-Ts targeting CSPG4 in GBM models. We first characterized a cohort of newly diagnosed GBM patients in one institution for CSPG4 expression by immunohistochemistry. All the analyzed samples were also molecularly characterized as part of the routine diagnostic workup. The sample size allowed us to demonstrate statistical significance. Our strategy was to use GBM-NS as GBM tumor models both in vitro and in vivo. All GBM-NS were generated in the same laboratory from GBM samples that were also analyzed for the expression of CSPG4 by immunohistochemistry. For the in vitro experiments, blood obtained from the blood bank was used to generate the CAR-Ts. We selected to use nude mice for the in vivo experiments because these mice allow the engraftment of human cells. We selected to use the intracranial injection of CAR-Ts after tumor implantation because this approach is clinically feasible. Tumor engraftment, eradication, and recurrence were followed by bioluminescence imaging. In all in vivo experiments, to ensure similar tumor sizes in all treatment groups, mice were randomized after imaging analysis, performed the day before CAR-T injection. To ensure statistical power, experimental groups were composed of 10 animals each. For each experiment, the number of mice, statistical analyses, and numbers of experimental replicates are described in the figure legends. Investigators were not blinded during follow-up of mice and evaluation of the in vivo experiments.
GBM specimens and cell lines
GBM specimens were obtained from the Department of Neurosurgery at the Istituto Neurologico Carlo Besta and diagnosed according to World Health Organization criteria (62). The protocol was approved by the local institutional review board, and all patients provided their informed consent. GBM-NS were derived from GBM specimens as previously described (17, 63) and cultured in standard medium containing B27 supplement (Thermo Fisher Scientific) and the mitogenic factors EGF and bFGF (basic fibroblast growth factor) (PeproTech) (17). All GBM-NS were confirmed mycoplasma-free by the PromoKine PCR Test Kit. Murine microglia isolated from tumor xenografts were cultured in DMEM supplemented with 10% serum and replaced with GBM-NS medium 3 days before plating them in coculture experiments.
Generation of the CSPG4-specific CAR and transduction of T lymphocytes
The CSPG4.CAR was generated using the anti-CSPG4 mAb 763.74 (56). The CAR cassette includes the CD8α hinge and transmembrane domains (30). As costimulatory endodomains, we used CD28, 4–1BB, or the combination of CD28 and 4–1BB. As control, we used a CAR specific for the CD19 antigen (CD19.CAR). Transient retroviral supernatant was generated by cotransfection of 293T cells with the RD114 envelope (RDF plasmid), the MoMLV gag-pol (PegPam3-e plasmid), and the retroviral vector, as previously described (26). For the generation of CAR-Ts, peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood of healthy donors (Gulf Coast Regional Blood Center) using Ficoll-Paque (Amersham Biosciences). PBMCs were activated with plate-bound CD3 (Miltenyi Biotec) and CD28 (BD Biosciences Pharmingen) mAbs, transduced with the retroviral supernatant by day 3 of culture, and then expanded in complete medium containing 45% RPMI 1640 (Hyclone) and 45% Click’s medium (Irvine Scientific) supplemented with 10% fetal bovine serum (Hyclone), penicillin (100 IU/ml), streptomycin (100 mg/ml), and 2 mM GlutaMAX (Gibco). Cells were supplemented with IL-7 and IL-15 (PeproTech) twice a week for 2 weeks (64).
Coculture assay and cytotoxicity assay
For the coculture experiments, GBM-NS were plated at 5 × 105 cells per well in 24-well plates with CSPG4.CAR-Ts or cTs (either nontransduced T cells or T cells expressing the CD19.CAR) at E:T ratios of 1:5 or 2:5 in GBM-NS medium without serum and in the presence of B27 supplements. CSPG4.CAR-Ts or cTs were maintained in GBM-NS medium for 4 to 5 days before plating the cocultures. Coculture supernatants were collected after 24 hours to measure IFN-γ and IL-2 using specific enzyme-linked immunosorbent assay (ELISA) (R&D Systems). After 3 to 4 days of coculture, GBM-NS and T cells were collected, and residual tumor cells and T cells were measured by flow cytometry based on CSPG4 and CD3 expression, respectively. The cytotoxic activity of CSPG4.CAR-Ts and cTs was determined in a standard 51Cr release assay using different E:T ratios (40:1, 20:1, 10:1, and 5:1) (25).
CFSE assay
CSPG4.CAR-Ts and cTs were labeled with 1.5 μM CFSE (Invitrogen) and plated with GBM-NS at an E:T ratio of 1:5 or 2:5. CFSE dilution was measured by gating on CD3+ T cells using flow cytometry by day 7 of coculture. CSPG4.CAR-Ts alone were used as a negative control.
Flow cytometry
CD3, CD4, CD8, CD45, and PD-L1 anti-human mAbs (BD Biosciences) were used to identify T cells, and the CSPG4 mAb (Miltenyi Biotec) was used to label GBM-NS. CD45, CD11b, and Ly6C anti-mouse mAbs (Miltenyi Biotec) were used to identify murine microglia in xenograft GBM models. CAR expression on the cell surface of T cells was detected using protein L-based staining (GenScrip) (65). Acquisitions were performed using a Canto II flow cytometer or a MACSQuant (Miltenyi Biotec) flow cytometer, and data were analyzed using the FlowJo software (version 9.0) or FlowLogic software (version 7.2, Miltenyi Biotec).
Xenograft mouse models
Animal studies were performed in accordance with the Lineberger Cancer Center Animal Studies Core Facility at the University of North Carolina at Chapel Hill and following directives of Istituto Neurologico C. Besta in Milan in accordance to the Italian Principle of Laboratory Animal Care (D. Lgs. 26/2014) and European Communities Council Directives (86/609/EEC and 2010/63/UE). Antitumor activity of CSPG4.CAR-Ts was evaluated using nude mice purchased from the University of North Carolina Animal Studies Core Facility and engrafted with GBM-NS. Five- to 6-week-old mice were injected intracranially with 0.1 × 106 GBM-NS in 2 μl of phosphate-buffered saline (PBS). The coordinates, with respect to the bregma, were 0.7 mm post, 3 mm left lateral, 3.5 mm deep, and within the nucleus caudatum. Upon tumor engraftment, 2 × 106 CD19.CAR-Ts (cTs) or CSPG4.CAR-Ts were injected intratumorally in 5 μl of PBS using the same tumor coordinates. For survival studies, mice were monitored three times a week and euthanized when signs of discomfort appeared in accordance with the institutional guidelines. Brains were collected and used to perform cell isolation on fresh samples or fixed in 4% paraformaldehyde for histological studies.
Measuring GBM-NS growth and CAR-T persistence in vivo
Tumor growth was measured by in vivo bioluminescence using the Xenogen-IVIS Imaging System. GBM-NS were transduced with a retroviral vector encoding the GFP/FFluc gene. CAR-T persistence was also measured by transducing them with an enhanced GFP/FFluc gene, permitting detection of less than 10 cells (30). GFP expression by transduced cells was evaluated by flow cytometry, whereas expression of FFluc was detected using D-luciferin. For imaging purposes, mice were injected intraperitoneally with D-luciferin (150 mg/kg). Signal intensity was measured after drawing a region of interest over the brain region and expressed as total flux (photons per second).
Magnetic sorting and mouse cell depletion
CSPG4H and CSPG4L BT168-NS cells were obtained by magnetic sorting using the Anti-PE MicroBead Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Briefly, dissociated BT168-NS were labeled with anti–NG2 Ab–phycoerythrin (PE) (Miltenyi Biotec) and were subsequently incubated with anti-PE microbeads. The purity of enriched cells was verified using anti–CSPG4 Ab–PE (Miltenyi Biotec) with the MACSQuant Analyzer. Xenograft tumors were explanted and dissociated using the human Tumor Dissociation Kit (Miltenyi Biotec) in combination with GentleMACS (Miltenyi Biotec). The cell suspension was labeled with Mouse Cell Depletion Cocktail (Miltenyi Biotec) and incubated for 15 min at 2° to 8°C. Magnetic sorting was performed using LS columns according to the manufacturer’s instructions to separate human GBM-NS and tumor-infiltrating murine cells. Murine TNFα and IL-6 cytokines secreted by the cells included in the positive fraction were measured using a mouse-specific ELISA assay (PeproTech).
Cell proliferation assay
CSPG4H and CSPG4L BT168-NS cells were seeded at 5 × 103 cells per well in 96-well plates. Cell proliferation was measured at different time points using the cell proliferation reagent WST-1 (Roche Applied Science), according to the manufacturer’s instructions. Six replicates per point were analyzed. The absorbance was determined at 450 nm.
IFN-γ stimulation and PD-L1 blocking
GBM-NS were seeded at 2 × 105 cells in six-well plates and incubated with or without hIFN-γ (0.5 ng/ml; PeproTech). After 24 hours, GBM-NS were manually dissociated and evaluated for the expression of PD-L1 (anti-human PD-L1 Ab, BD Biosciences) by flow cytometry. In other experiments, GBM-NS were plated at 2 × 105 cells per well in 24-well plates, cocultured with CSPG4.CAR-Ts or cTs at an E:T ratio of 1:5, and incubated with isotype control or αPD-L1 Ab (10 μg/ml, eBioscience). After 3 days of coculture, GBM-NS and T cells were collected, and residual GBM-NS and T cells were quantified by flow cytometry based on CSPG4 and CD3 expression, respectively. Coculture supernatants were collected after 24 hours to measure IFN-γ using specific ELISA (R&D Systems).
TNFα stimulation and blocking
GBM-NS were seeded at 2 × 105 cells in six-well plates and exposed to hTNFα (0.5 ng/ml; PeproTech) for 24 hours. In specific experiments, TNFα was blocked by adding infliximab (50 μg/ml; Schering-Plough) (66). GBM-NS were also cocultured in the presence of murine microglia isolated from glioma xenografts. After incubation with either soluble TNFα or murine microglia, GBM-NS were collected and evaluated for CSPG4 expression by flow cytometry.
Western blot
Proteins from cell cultures were extracted in lysis buffer [50 nM tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, 2× phenylmethylsulfonyl fluoride, leupeptin (10 μg/ml), aprotinin (10 μg/ml), 1 mM Na3VO4, 1 mM NaF], and protein concentrations were assessed using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were resolved on a 4 to 12% SDS–polyacrylamide gel electrophoresis gel and electroblotted onto nitrocellulose membranes. Nonspecific protein-binding sites on membranes were blocked by incubating the membranes for 3 hours in 7% bovine serum albumin (BSA) in TBS-T (tris-buffered saline with Tween 20) before incubating them with primary Abs overnight at 4°C. The following primary Abs were used: anti–NF-κB p65 (1:1000 dilution, Cell Signaling Technology), anti–phospho–NF-κB p65 (Ser536) (1:1000 dilution, Cell Signaling Technology), and anti-vinculin (1:10,000 dilution, Santa Cruz Biotechnology). As secondary Abs, we used anti-mouse and anti-rabbit horseradish peroxidase (HRP)–conjugated mAbs (1:3000 dilution, Bethyl Laboratories). The reactions were developed using the enhanced chemiluminescence kit (ECL, Amersham, GE Healthcare).
Immunohistochemistry and immunofluorescence
GBM specimens were sliced into 2-μm-thick sections, deparaffinized in xylene, rehydrated through decreasing concentrations (100%, 95%, 90%, 80%, and 70%) of ethyl alcohol, and then rinsed in distilled water. Antigen retrieval was performed in 90°C solution of 0.001 M EDTA buffer (pH 8.0) for 20 min. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide in distilled water. Slides were treated with 1% BSA (Santa Cruz Biotechnology) and 5% normal goat serum in PBS containing 0.05% Triton X-100 (Sigma-Aldrich) and incubated in a closed humid chamber with the anti-CSPG4 rabbit polyclonal Ab (1:200 dilution, Abcam) for 1 hour at room temperature. Staining was detected using the EnVision+ System-HRP–labeled polymer anti-rabbit Ab for 1 hour at room temperature and then the chromogen DAB/substrate reagent (Dako). Slides were counterstained with hematoxylin (Sigma-Aldrich), dehydrated, and mounted. The expression of CSPG4 was evaluated for intensity of reactivity. A double immunofluorescence staining with the anti-nestin (1:50 dilution, R&D Systems) or anti–α-SMA (1:50 dilution, Sigma-Aldrich) Ab and the same anti-CSPG4 Ab (1:50 dilution) was performed. The Alexa Fluor 488 goat anti-rabbit Ab (1:100 dilution, Thermo Fisher Scientific) was used for detecting CSPG4, and the Cy3-conjugated AffiniPure goat anti-mouse Ab (1:100 dilution, Jackson ImmunoResearch) was used to detect nestin or α-SMA. Rabbit anti-Iba1 Ab (1:1000 dilution, Wako Chemicals) was used to detect human microglia in the tumor mass. A double immunofluorescence staining with anti-TNFα (1:200 dilution, R&D Systems) and the same anti-Iba1 Ab (1:500 dilution) was performed. Cell nuclei were counterstained with DAPI (Sigma-Aldrich), and the slides were mounted with a PBS/glycerol solution. Quantitative analyses of CSPG4 and Iba1 expression were blindly performed by three independent investigators on three to five independent fields per tumor by counting the number of cells in the photographed fields using the 40× objective of a Leica DM-LB microscope.
Statistical analysis
In vitro data are presented as means ± SD, and a paired Student’s t test (two-tailed) and ANOVA were used to determine the statistical significance of the difference. The in vivo data are presented as means ± SEM, and Kaplan-Meier curves were used to evaluate the significance of the difference between CAR-treated and control-treated groups.
Supplementary Material
Fig. S1. Normal brain vessels do not express CSPG4.
Fig. S2. CSPG4.CAR is expressed in T cells.
Fig. S3. CSPG4.CAR-Ts prolonged the survival of mice injected with BT275-NS and BT462-NS.
Fig. S4. CSPG4.CAR-Ts controlled tumor growth in the U87-MG xenograft model.
Fig. S5. CSPG4.CAR encoding either CD28 or CD28/4–1BB showed lower antitumor activity than CSPG4.CAR encoding 4–1BB.
Fig. S6. CSPG4.CAR encoding either CD28 or CD28/4–1BB costimulatory endodomains showed limited antitumor activity in vivo.
Fig. S7. CSPG4.CAR-Ts encoding either CD28 or CD28 and 4–1BB persisted for less than 5 days.
Fig. S8. GBM-NS–derived gliomas recurring in vivo retained CSPG4 expression.
Fig. S9. GBM-NS up-regulated PD-L1 in response to IFN-γ.
Fig. S10. HER-2 and IL-13Rα2 expression is not up-regulated in GBM-NS in xenograft models.
Table S1. GBM characterization based on CSPG4 expression and molecular subtypes.
Acknowledgments:
This work is dedicated to the memory of Viola Kraus. We thank D. Lecis (Department of Experimental Oncology and Molecular Medicine, AmadeoLab—Fondazione IRCCS Istituto Nazionale dei Tumori) for providing reagents, help, and advice with TNFα-blocking experiment.
Funding:
This work was supported by University Cancer Research Fund at University of North Carolina, Lineberger Comprehensive Cancer Center. S.P. was supported by “Il Fondo di Gio Onlus.”
Footnotes
Competing interests: G.D. is an inventor on patent application (WO2015080981A1) held and submitted by Baylor College of Medicine that covers the CSPG4.CAR. All other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/10/430/eaao2731/DC1
Data and materials availability:
GBM-NS are available from G.F. under a material transfer agreement with Istituto Neurologico Besta, Milan, Italy.
REFERENCES AND NOTES
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff R-O; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group, Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group, Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Sadelain M, Brentjens R, Rivière I, The basic principles of chimeric antigen receptor design. Cancer Discov. 3, 388–398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dotti G, Gottschalk S, Savoldo B, Brenner MK, Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 257, 107–126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heimberger AB, Suki D, Yang D, Shi W, Aldape K, The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 3, 38 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan C-T, Song H, Zhang W, Fine HA, Rosenberg SA, Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 23, 1043–1053 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, Fraietta JA, Kloss CC, Posey AD Jr., Engels B, Singh R, Ezell T, Idamakanti N, Ramones MH, Li N, Zhou L, Plesa G, Seykora JT, Okada H, June CH, Brogdon JL, Maus MV, Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 7, 275ra22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffman KT, Hu M, Carles-Kinch K, Tice D, Donacki N, Munyon K, Kifle G, Woods R, Langermann S, Kiener PA, Kinch MS, Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 63, 7907–7912 (2003). [PubMed] [Google Scholar]
- 10.Chow KKH, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, Rainusso N, Wu M-F, Liu H, Kew Y, Grossman RG, Powell S, Lee D, Ahmed N, Gottschalk S, T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 21, 629–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, Sahin A, Carter BS, Brem H, Junghans RP, Sampath P, Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor–modified T cells. Clin. Cancer Res. 18, 5949–5960 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suryadevara CM, Verla T, Sanchez-Perez L, Reap EA, Choi BD, Fecci PE, Sampson JH, Immunotherapy for malignant glioma. Surg. Neurol. Int. 6, S68–S77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weller M, Butowski N, Tran D, Recht L, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust S, Iwamoto F, Drappatz J, O’Rourke D, Wong M, Finocchiaro G, Perry J, Wick W, He Y, Davis T, Stupp R, Sampson J, ATIM-03. Act IV: An international, double-blind, phase 3 trial of rindopepimut in newly diagnosed, EGFRvIII-expressing glioblastoma. Neuro Oncol. 18, vi17–vi18 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon II JE, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling RJ, Shi W, Vredenburgh JJ, Bigner DD, Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 28, 4722–4729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot J-M, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV, A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Bacco F, Casanova E, Medico E, Pellegatta S, Orzan F, Albano R, Luraghi P, Reato G, D’Ambrosio A, Porrati P, Patanè M, Maderna E, Pollo B, Comoglio PM, Finocchiaro G, Boccaccio C, The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 72, 4537–4550 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Finocchiaro G, Pellegatta S, Immunotherapy with dendritic cells loaded with glioblastoma stem cells: From preclinical to clinical studies. Cancer Immunol. Immunother. 65, 101–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, Abate-Daga D, Rosati SF, Fine HA, Ferrone S, Rosenberg SA, Morgan RA, Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells., J. Immunother. Cancer 2, 25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, Ferrone CR, Favoino E, Koya Y, Campoli MR, McCarthy JB, DeLeo AB, Ferrone S, CSPG4 in cancer: Multiple roles. Curr. Mol. Med. 10, 419–429 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Hafner C, Breiteneder H, Ferrone S, Thallinger C, Wagner S, Schmidt WM, Jasinska J, Kundi M, Wolff K, Zielinski CC, Scheiner O, Wiedermann U, Pehamberger H, Suppression of human melanoma tumor growth in SCID mice by a human high molecular weight-melanoma associated antigen (HMW-MAA) specific monoclonal antibody. Int. J. Cancer 114, 426–432 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Pluschke G, Vanek M, Evans A, Dittmar T, Schmid P, Itin P, Filardo EJ, Reisfeld RA, Molecular cloning of a human melanoma-associated chondroitin sulfate proteoglycan. Proc. Natl. Acad. Sci. U.S.A. 93, 9710–9715 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivera Z, Ferrone S, Wang X, Jube S, Yang H, Pass HI, Kanodia S, Gaudino G, Carbone M, CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin. Cancer Res. 18, 5352–5363 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stallcup WB, The NG2 proteoglycan: Past insights and future prospects. J. Neurocytol. 31, 423–435 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Geldres C, Savoldo B, Dotti G, Chimeric antigen receptors for cancer immunotherapy. Methods Mol. Biol. 1393, 75–86 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, Del Vecchio M, Creighton CJ, Ittmann M, Ferrone S, Dotti G, T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. 20, 962–971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svendsen A, Verhoeff JJC, Immervoll H, Brøgger JC, Kmiecik J, Poli A, Netland IA, Prestegarden L, Planagumà J, Torsvik A, Kjersem AB, Sakariassen PØ, Heggdal JI, Van Furth WR, Bjerkvig R, Lund-Johansen M, Enger PØ, Felsberg J, Brons NHC, Tronstad KJ, Waha A, Chekenya M, Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 122, 495–510 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lama G, Mangiola A, Proietti G, Colabianchi A, Angelucci C, D’Alessio A, De Bonis P, Geloso MC, Lauriola L, Binda E, Biamonte F, Giuffrida MG, Vescovi A, Sica G, Progenitor/stem cell markers in brain adjacent to glioblastoma: GD3 ganglioside and NG2 proteoglycan expression. J. Neuropathol. Exp. Neurol. 75, 134–147 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Chinnaiyan P, Wang M, Rojiani AM, Tofilon PJ, Chakravarti A, Ang KK, Zhang H-Z, Hammond E, Curran W Jr., Mehta MP, The prognostic value of nestin expression in newly diagnosed glioblastoma: Report from the Radiation Therapy Oncology Group. Radiat. Oncol. 3, 32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, Grossman RG, Heslop HE, Gottschalk S, HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 16, 474–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D, Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18, 676–684 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Rabinovich BA, Ye Y, Etto T, Chen JQ, Levitsky HI, Overwijk WW, Cooper LJN, Gelovani J, Hwu P, Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 14342–14346 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sliwa M, Markovic D, Gabrusiewicz K, Synowitz M, Glass R, Zawadzka M, Wesolowska A, Kettenmann H, Kaminska B, The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain 130, 476–489 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Marmenout A, Fransen L, Tavernier J, Van der Heyden J, Tizard R, Kawashima E, Shaw A, Johnson M-J, Semon D, Müller R, Molecular cloning and expression of human tumor necrosis factor and comparison with mouse tumor necrosis factor. Eur. J. Biochem. 152, 515–522 (1985). [DOI] [PubMed] [Google Scholar]
- 34.Choi BD, Suryadevara CM, Gedeon PC, Herndon JE II, Sanchez-Perez L, Bigner DD, Sampson JH, Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J. Clin. Neurosci. 21, 189–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang W-C, Naranjo A, Starr R, Wagner J, Wright C, Zhai Y, Bading JR, Ressler JA, Portnow J, D’Apuzzo M, Forman SJ, Jensen MC, Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 21, 4062–4072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, Harshbarger TL, D’Apuzzo M, Ressler JA, Jensen MC, Barish ME, Chen M, Portnow J, Forman SJ, Badie B, Regression of glioblastoma after chimeric antigen receptor T-Cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, Ghazi A, Gerken C, Yi Z, Ashoori A, Wu M-F, Liu H, Rooney C, Dotti G, Gee A, Su J, Kew Y, Baskin D, Zhang YJ, New P, Grilley B, Stojakovic M, Hicks J, Powell SZ, Brenner MK, Heslop HE, Grossman R, Wels WS, Gottschalk S, HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 3, 1094–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, Fisher WE, Heslop HE, Rooney CM, Brenner MK, Leen AM, Vera JF, Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. 22, 623–633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farnedi A, Rossi S, Bertani N, Gulli M, Silini EM, Mucignat MT, Poli T, Sesenna E, Lanfranco D, Montebugnoli L, Leonardi E, Marchetti C, Cocchi R, Ambrosini-Spaltro A, Foschini MP, Perris R, Proteoglycan-based diversification of disease outcome in head and neck cancer patients identifies NG2/CSPG4 and syndecan-2 as unique relapse and overall survival predicting factors. BMC Cancer 15, 352 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warta R, Herold-Mende C, Chaisaingmongkol J, Popanda O, Mock A, Mogler C, Osswald F, Herpel E, Küstner S, Eckstein V, Plass C, Plinkert P, Schmezer P, Dyckhoff G, Reduced promoter methylation and increased expression of CSPG4 negatively influences survival of HNSCC patients. Int. J. Cancer 135, 2727–2734 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Hsu NC, Nien P-Y, Yokoyama KK, Chu P-Y, Hou M-F, High chondroitin sulfate proteoglycan 4 expression correlates with poor outcome in patients with breast cancer. Biochem. Biophys. Res. Commun. 441, 514–518 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Al-Mayhani MTF, Grenfell R, Narita M, Piccirillo S, Kenney-Herbert E, Fawcett JW, Collins VP, Ichimura K, Watts C, NG2 expression in glioblastoma identifies an actively proliferating population with an aggressive molecular signature. Neuro Oncol. 13, 830–845 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chekenya M, Krakstad C, Svendsen A, Netland IA, Staalesen V, Tysnes BB, Selheim F, Wang J, Sakariassen PØ, Sandal T, Lønning PE, Flatmark T, Enger PØ, Bjerkvig R, Sioud M, Stallcup WB, The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene 27, 5182–5194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Svendsen A, Kmiecik J, Immervoll H, Skaftnesmo KO, Planagumà J, Reed RK, Bjerkvig R, Miletic H, Enger PØ, Rygh CB, Chekenya M, Targeting the NG2/CSPG4 proteoglycan retards tumour growth and angiogenesis in preclinical models of GBM and melanoma. PLOS ONE 6, e23062 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yotsumoto F, You W-K, Cejudo-Martin P, Kucharova K, Sakimura K, Stallcup WB, NG2 proteoglycan-dependent recruitment of tumor macrophages promotes pericyte-endothelial cell interactions required for brain tumor vascularization. Oncoimmunology 4, e1001204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pallini R, Ricci-Vitiani L, Banna GL, Signore M, Lombardi D, Todaro M, Stassi G, Martini M, Maira G, Larocca LM, De Maria R, Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 8205–8212 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, Barish ME, Forman SJ, Jensen MC, Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine–redirected T Cells. Clin. Cancer Res. 18, 2199–2209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quail DF, Joyce JA, The microenvironmental landscape of brain tumors. Cancer Cell 31, 326–341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE, Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 18, 519–527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kushchayev SV, Sankar T, Eggink LL, Kushchayeva YS, Wiener PC, Hoober JK, Eschbacher J, Liu R, Shi FD, Abdelwahab MG, Scheck AC, Preul MC, Monocyte galactose/N-acetylgalactosamine-specific C-type lectin receptor stimulant immunotherapy of an experimental glioma. Part 1: Stimulatory effects on blood monocytes and monocyte-derived cells of the brain. Cancer Manag. Res. 4, 309–323 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJC, Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 205, 2319–2337 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Lin Y, Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 29, 1275–1288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wade A, Robinson AE, Engler JR, Petritsch C, James CD, Phillips JJ, Proteoglycans and their roles in brain cancer. FEBS J. 280, 2399–2417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hambardzumyan D, Gutmann DH, Kettenmann H, The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korzhevskii DE, Kirik OV, Brain microglia and microglial markers. Neurosci. Behav. Physiol. 46, 284–290 (2016). [Google Scholar]
- 56.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M, CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA, Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG, Genomic analysis of immune cell infiltrates across 11 tumor types. J. Natl. Cancer Inst. 108, djw144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krenciute G, Krebs S, Torres D, Wu M-F, Liu H, Dotti G, Li X-N, Lesniak MS, Balyasnikova IV, Gottschalk S, Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Rα2-positive glioma. Mol. Ther. 24, 354–363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown CE, Starr R, Martinez C, Aguilar B, D’Apuzzo M, Todorov I, Shih C-C, Badie B, Hudecek M, Riddell SR, Jensen MC, Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 69, 8886–8893 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KKH, Brawley VS, Byrd TT, Krebs S, Gottschalk S, Wels WS, Baker ML, Dotti G, Mamonkin M, Brenner MK, Orange JS, Ahmed N, Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P, The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Behnan J, Stangeland B, Langella T, Finocchiaro G, Murrell W, Brinchmann JE, Ultrasonic surgical aspirate is a reliable source for culturing glioblastoma stem cells. Sci. Rep. 6, 32788 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, Liu H, Creighton CJ, Gee AP, Heslop HE, Rooney CM, Savoldo B, Dotti G, Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng Z, Chinnasamy N, Morgan RA, Protein L: A novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J. Transl. Med. 10, 29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lecis D, De Cesare M, Perego P, Conti A, Corna E, Drago C, Seneci P, Walczak H, Colombo MP, Delia D, Sangaletti S, Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 4, e920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Normal brain vessels do not express CSPG4.
Fig. S2. CSPG4.CAR is expressed in T cells.
Fig. S3. CSPG4.CAR-Ts prolonged the survival of mice injected with BT275-NS and BT462-NS.
Fig. S4. CSPG4.CAR-Ts controlled tumor growth in the U87-MG xenograft model.
Fig. S5. CSPG4.CAR encoding either CD28 or CD28/4–1BB showed lower antitumor activity than CSPG4.CAR encoding 4–1BB.
Fig. S6. CSPG4.CAR encoding either CD28 or CD28/4–1BB costimulatory endodomains showed limited antitumor activity in vivo.
Fig. S7. CSPG4.CAR-Ts encoding either CD28 or CD28 and 4–1BB persisted for less than 5 days.
Fig. S8. GBM-NS–derived gliomas recurring in vivo retained CSPG4 expression.
Fig. S9. GBM-NS up-regulated PD-L1 in response to IFN-γ.
Fig. S10. HER-2 and IL-13Rα2 expression is not up-regulated in GBM-NS in xenograft models.
Table S1. GBM characterization based on CSPG4 expression and molecular subtypes.
Data Availability Statement
GBM-NS are available from G.F. under a material transfer agreement with Istituto Neurologico Besta, Milan, Italy.