

Summary
Most cases of adult myeloid neoplasms are routinely assumed to be sporadic. Here, we describe an adult familial acute myeloid leukemia (AML) syndrome caused by germline mutations in the DEAD/H-Box helicase gene DDX41. DDX41 was also found to be affected by somatic mutations in sporadic cases of myeloid neoplasms as well as in a biallelic fashion in 50% of patients with germline DDX41 mutations. Moreover, corresponding deletions on 5q35.3 present in 6% of cases lead to haploinsufficient DDX41 expression. DDX41 lesions caused altered pre-mRNA splicing and RNA processing. DDX41 is exemplary of other RNA helicase genes also affected by somatic mutations, suggesting that they constitute a family of tumor suppressor genes.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of myeloid neoplasms characterized by cytopenia, morphologic dysplasia, cytogenetic abnormalities and propensity to progress to secondary acute myeloid leukemia (sAML). While closely related to primary forms of AML, MDS predominantly affects the elderly. Next-generation sequencing (NGS) in MDS led to the discovery of relevant somatic mutations and their combinations (Patel et al., 2012; Walter et al., 2013). The spectrum of affected genes overlaps with those seen in AML and the closely related myeloproliferative/myelodysplastic (MDS/MPN) syndromes.
Familial MDS has been rarely reported, usually in the context of early-onset disease and germline mutations. Patients with germline RUNX1 mutations, present with thrombocytopenia, and frequent progression towards MDS/AML (Owen et al., 2008). Similarly, germline CEBPA and GATA2 mutations have been associated with AML and early-onset MDS/AML (Owen et al., 2008; Hahn et al., 2011). Among patients with typical MDS, late presentation makes it difficult to distinguish hereditary factors from aging and cumulative environmental exposures (Pfeilstöcker et al., 2007; Sekeres, 2010). Nevertheless, in rare cases, a strong family history may suggest a genetic predisposition, illuminating the seemingly sporadic cases.
While investigating genetic causes of AML families affected by myeloid neoplasms we identified germline mutations in a DEAD/H-box helicase gene that induced late onset MDS/AML with a predisposition to acquisition of somatic DEAD/H-box mutations.
Results
Identification of myeloid leukemias with mutant familial DDX41 mutations
In the index family, father, son and paternal grandmother were affected by de novo AML, while sAML from antecedent MDS (Refractory anemia with excess blasts; RAEB) was diagnosed in a daughter. Age at disease onset ranged from 44–70 years. Using whole exome sequencing (WES) we found a recurrent germline mutation of DDX41 (c.419insGATG, p.D140fs) in the father, son and daughter. The prevalence of this germline minor allele in the general population is 1/12,518 (NHLBI GO Exome Sequencing Project; (ESP); https://esp.gs.washington.edu/). This alteration was not found in 200 internal controls. Subsequent analysis of acquired sequence alterations also revealed the concomitant presence of a canonical somatic mutation of DDX41 (c.G1574A, p.R525H) in the father and son (Figure 1A, Figure S1A). Germline and somatic DDX41 mutations were distinguished by analysis of buccal DNA in patients. In two other sons (55 and 56 years), the heterozygous mutation (c.419insGATG, p.D140fs) was detected with no apparent disease, but both of them have developed slight monocytosis. Blood smears from both showed the presence of immature monocytes (Figure S1B). They were younger than most of the patients with germline DDX41 mutations (Figure S1C). The canonical somatic mutation of DDX41 (c.G1574A, p.R525H) was not detected. In a second family, identical twin brothers both developed MDS (Refractory cytopenia with multilineage dysplasia; RCMD), and AML was observed in their father. Both were successfully treated with lenalidomide for transfusion-dependent anemia. Both twins showed a germline DDX41 variant (c.T1187C; p.I396T, Figure 1B, Figure S1D). As in the index family, they also showed the somatic DDX41 (p.R525H) mutation. The germline alteration p.I396T was not found in 200 internal controls, or in available databases (1092 controls; http://www.1000genomes.org/ and ESP). In the 3rd family, we identified a 67-year-old male patient (case 6) diagnosed with MDS-RAEBI who harbored both canonical germline and somatic DDX41 mutations, p.D140fs and p.R525H respectively. His brother also died from AML at the age of 58 years (Figure 1C, Figure S1E).
Figure 1: Germline and somatic mutations of three families with history of MDS and leukemia.
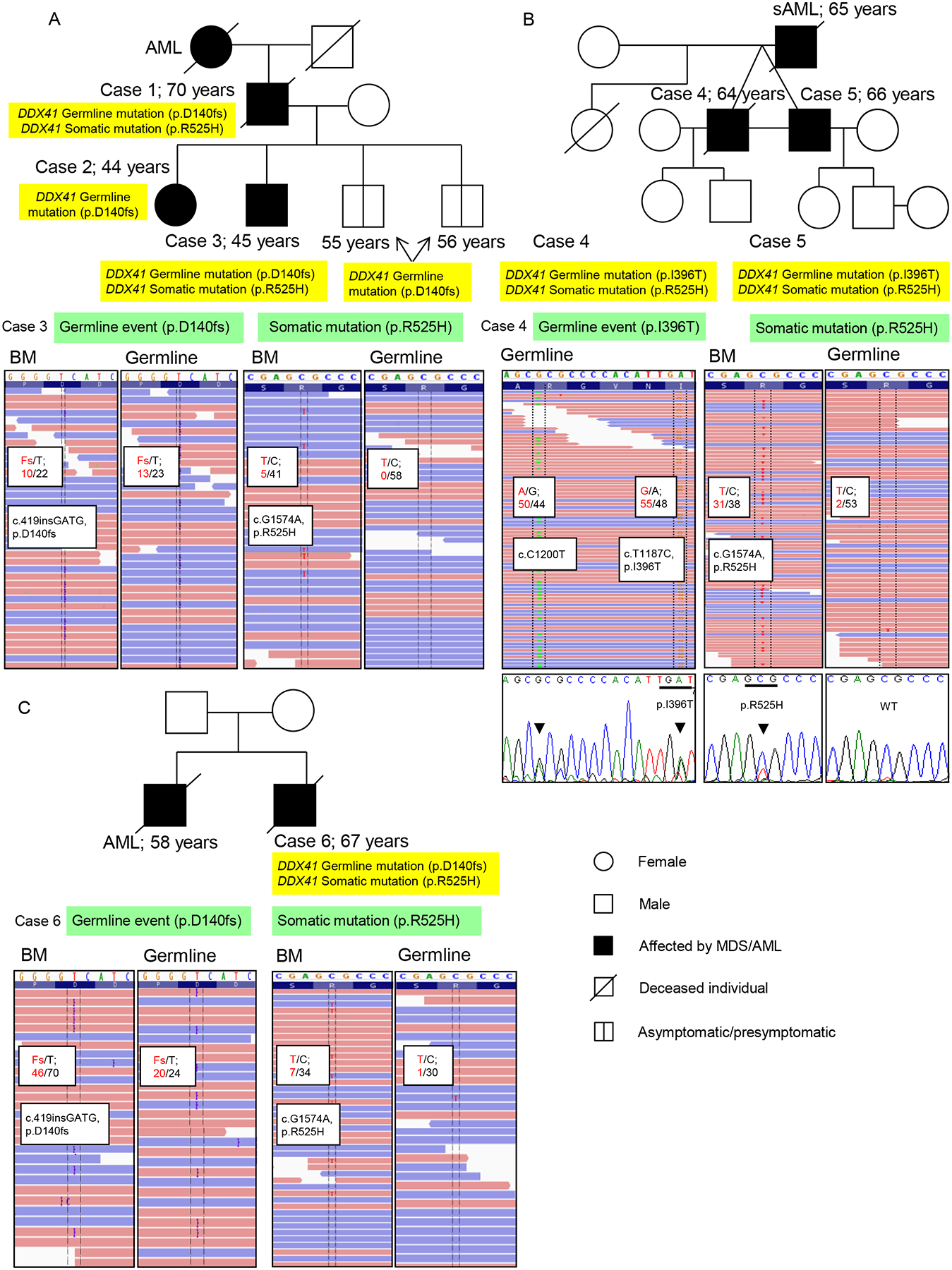
(A, B, C) Pedigrees of family 1 (A), family 2 (B), and family 3 (C) (upper panels). Age of diagnosis and detected DDX41 mutations are indicated. Lower panels show sequencing reads from whole exome sequencing (WES) with frequencies of detected mutations in bone marrow (BM) and germline samples. Confirmation of germline and somatic DDX41 mutations by Sanger sequencing is exemplarily shown for the family 2 (B). Arrows and bars indicated the specific nucleotide and predicted codon, respectively. Case number is annotated according to Table 1. Asymptomatic/presymptomatic carrier-clinically unaffected at this time but could later exhibit symptoms (Bennett et al., 2008)
See also Figure S1
In another leukemia family (Figure S2A), a 73-year-old male patient (case 7) was diagnosed with secondary AML, whereas his nephew (case 8) presented with AML at the age of 56. Both patients harbored the canonical germline DDX41 mutation. Paternal cousins of the index case were also afflicted by AML at the age of 79 and 89, but declined genetic testing. Through further search (Table 1) we identified additional 3 cases with advanced MDS and a strong family history of MDS/AML (families 5–7) who harbored DDX41 alterations, two of them showed both germline and somatic mutations of DDX41 (Figure S2B).
Table 1.
Characteristics of DDX41 mutants.
| Family | Cases | Age | Sex | Disease | Germline event | DDX41 somatic mutation | Cytogenetics |
|---|---|---|---|---|---|---|---|
| Family 1 | 1 | 70 | M | pAML | P.D140fs | P.R525H | Normal |
| Family 1 | 2 | 44 | F | sAML | P.D140fs | No | Normal |
| Family 1 | 3 | 45 | M | pAML | P.D140fs | P.R525H | Normal |
| Family 2 | 4 | 64 | M | RCMD | P.I396T | P.R525H | Normal |
| Family 2 | 5 | 66 | M | RCMD | P.I396T | P.R525H | Normal |
| Family 3 | 6 | 67 | M | RAEB-I | P.D140fs | P.R525H | Normal |
| Family 4 | 7 | 73 | M | sAML | P.D140fs | N/A | 46,XY,r(7)(p11q21)[7]/46,XY[8] |
| Family 4 | 8 | 56 | M | pAML | P.D140fs | N/A | 46,XY,del(20)(q11.21q13.33)[4]/46,XY[14] |
| Family 5 | 9 | 72 | M | RAEB-I | P.D140fs | P.R525H | Normal |
| Family 6 | 10 | 62 | M | RAEB-II | P.D140fs | No | Normal |
| Family 7 | 11 | 65 | M | RAEB-I | P.F183I | P.R525H | Normal |
| 12 | 85 | M | sAML | P.D140fs | P.R525H | 47,XY,+8[2]/46,XY[18] | |
| 13 | 74 | M | sAML | P.Q52fs | P.A225D | 44,XY,del(7)(q22),−16,−17,−18,−20, +2mar[2]/45,idem,+8[10]/46,XY[8] | |
| 14 | 58 | M | RAEB-I | P.D140fs | No | Normal | |
| 15 | 69 | M | CMML-1 | P.D140fs | No | N/A | |
| 16 | 88 | M | RAEB-I | P.D140fs | No | Normal | |
| 17 | 71 | M | pAML | P.D140fs | No | Normal | |
| 18 | 68 | M | sAML | P.D140fs | No | 46,XY,−7,+mar[2]/46,XY[19] | |
| 19 | 78 | M | RAEB-1 | P.M155I | No | 46,XY,del(20)(q11.2)[17] | |
| 20 | 64 | M | 5q-syndrome | No | P.R525H | 46,XY,del(5)(q12q33)[6] | |
| 21 | 68 | M | RAEB-II | No | P.R525H | Normal | |
| 22 | 63 | M | pAML | No | P.R525H | Normal | |
| 23 | 66 | M | RCMD | No | P.R525H | Normal | |
| 24 | 46 | M | RAEB-II | No | P.R525H | Normal | |
| 25 | 78 | M | RCMD | No | P.P321L | Normal | |
| 26 | 70 | F | 5q-syndrome | No | P.E247K | 46,XX,del(5)(q13q33) [20] | |
| 27 | 68 | M | pAML | No | Splice site (e11+1) | Normal |
DDX41 and other helicase defects in myeloid neoplasms
In addition to cases with a strong family history of MDS/AML (families 1–7) a cohort of 1,034 patients with MDS and sAML was subjected to a targeted screening by NGS (Table S1). We identified 6 additional patients with the germline c.419insGATG variant (case 12, 14–18, Figure S2C) and 2 patients with two different germline mutations of DDX41 (case 13, c.156_157insA; p.Q52fs and case 19, c.G465A; p.M155I). The former alteration was not found in public databases but the prevalence of the latter alteration in the general population is 5/13,006 (ESP). In total, DDX41 mutations were identified in 27 among 1,045 patients with myeloid neoplasms (Table 1). Simultaneously, we identified a total of 17 cases with somatic DDX41 mutations: 13 with recurrent missense mutations (p.R525H), 3 with non-recurrent missense mutations (c.C674A; p.A225D, c.C962T; p.P321L and c.G739A; p.E247K), and 1 with splice-site mutation (e11+1). The occurrence of somatic DDX41 mutations was closely linked to the presence of DDX41 germline mutations. About 50% of patients with germline DDX41 mutations also acquired the somatic mutation, while 0.8% patients with wild-type (WT) DDX41 acquired a somatic DDX41 mutation (p<.001, Figure S2D, S2E). The additional somatic mutations were always acquired in the remaining WT allele (Figure S2F).
Somatic mutations of DDX41 affected the ATP binding domain (Figure 2A). Because germline mutations were predominantly out-of-frame insertions and the co-occurrence with somatic DDX41 mutations suggested that defects of this gene may result in a loss of function. Consequently, deletion and mutations of this gene may be functionally equivalent. Deletions of the long arm of chr.5 involving the DDX41 locus (5q35.3) were present in 6% of all cases and 26% of the del(5q) cases (Figure 2A, Figure S3A) and resulted in decreased DDX41 mRNA levels (p=.0004; Figure 2B). Of note, the inactivating germline c.419insGATG mutation was not found among patients with del(5q35.3). Germline DDX41 mutations, per definition can be considered founder lesions. When we analyzed the clonal architecture of somatic DDX41 mutations we observed an average variant allelic frequency (VAF) of 25±10%. VAF in other important genes showed larger (SF3B1, TET2) and smaller (SMC3, NPM1) clone sizes (Figure 2C–D) in the corresponding cases. Whereas in some patients somatic DDX41 mutations are ancestral, they also appear to be present as subclones in others (Figure 2E).
Figure 2: DDX41 gene structure and clonal architecture in DDX41 mutants.
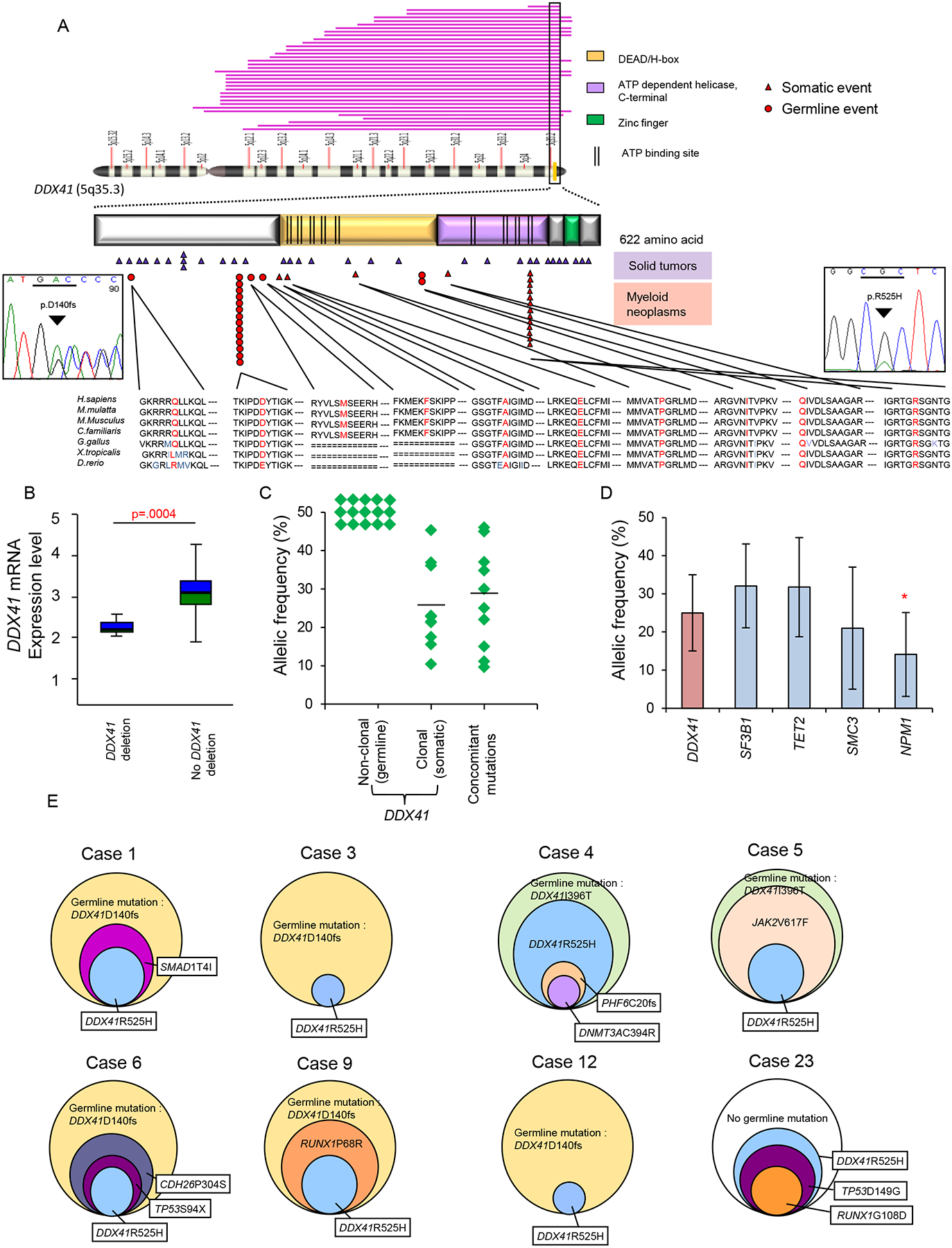
(A) DDX41 is located at the distal end of chromosome 5q, 5q35.3, and encodes a protein that contains three known domains and ATP binding sites, as illustrated. The pink bars visualize deletions of chromosome 5q in our MDS cohort that include the DDX41 locus. The red triangles indicate DDX41 mutations in patients with hematological malignancies from our cohort and TCGA. Red circles indicate the identified germline mutations of DDX41 (p.Q52fs, p.D140fs, p.M155I and p.I396T). The p.R525H mutation was detected in 13 out of 1,045 cases. Purple triangles show DDX41 mutations in non-hematological malignancies. Sanger sequencing confirming recurrent germline mutation (p.D140fs; left) and somatic mutation (p.R525H; right) of DDX41 are shown.
(B) DDX41 mRNA expression was analyzed by real-time RT-PCR in cases with deleted DDX41 locus compared with cases without deletion. Boxes represent 25–75 percentiles. A line inside a box represents median. Whiskers indicate maximum and minimum values.
(C) For cases with DDX41 mutations, variant allelic frequencies (VAFs) of DDX41 mutations (germline and somatic) and concomitant mutations of other genes (somatic) are shown. Mean values of VAFs were compared between somatic DDX41 and concomitant mutations (Mean±SD: 25±10% and 29±14%, respectively; p>.05).
(D) For cases in the whole examined cohort, VAFs of DDX41 mutations (Mean±SD: 25±10%) were compared to those of other genes affected by somatic mutations in myeloid neoplasms, including patients with DDX41 mutations. VAF is indicated as mean±SD. * indicates a p value of p=.004.
(E) Clonal architecture of 8 cases with DDX41 mutations. The percentages represent allelic frequencies with 50% set as the largest circle; case 1: 50% of DDX41-D140fs (germline mutation), 25% of SMAD1-T4I and 21% of DDX41-R525H; case 3: 50% of DDX41-D140fs (germline mutation) and 12% of DDX41-R525H; case4: 50% of DDX41-I396T (germline mutation), 42% of DDX41-R525H, 15% of PHF6-C20fs and 14% of DNMT3A-C394R; case5: 50% of DDX41-I396T (germline mutation), 46% of JAK2-V617F and 37% of DDX41-R525H; case 6: 50% of DDX41-D140fs (germline mutation), 37% of CDH26-P304S, 22% of TP53-S94X and 20% of DDX41-R525H; case 9: 50% of DDX41-D140fs (germline mutation), 30% of RUNX1-P68R and 25% of DDX41-R525H; case 12: 50% of DDX41-D140fs (germline mutation) and 11% of DDX41-R525H; case 23: 36% of DDX41-R525H, 34.8% of TP53-D149G and 11.1% of RUNX1-G108D. (Case number is annotated according to Table1)
See also Figure S3
Somatic mutations of DDX41 occur but are rare in non-hematologic malignancies (www.sanger.ac.uk). We observed 15 somatic mutations (n=342) in genes encoding other members of DEAD/H-box RNA helicase family. Defects of the helicase family were mutually exclusive (Figure S3B). We also identified 2 rare germline events: DHX29 c.G1627A (p.V543M) and c.G1561A (p.E521K), not found in ESP. In addition, deletions of DDX4 (5q11.2) and DHX58 (17q21.2) loci were identified in 14 and 13 cases, respectively (data not shown).
Clinical aspects of DDX41 mutations
DDX41 mutations and deletions occurred more frequently in patients with advanced MDS (19% in advanced MDS vs. 6% in low-risk MDS; p=.02; Figure 3A, B) and in AML (12% of primary; n=302 and 13% of secondary AML; n=154). Overall, patients with either DDX41 mutations or deletions had inferior overall survival (OS) (Figure 3C). Similar phenotypic associations (Figure 3B) (Boultwood et al., 2007) and effects on OS (Figure 3D) were observed in cases with decreased DDX41 expression.
Figure 3: Clinical impact of DDX41 deficiency in myeloid neoplasms.
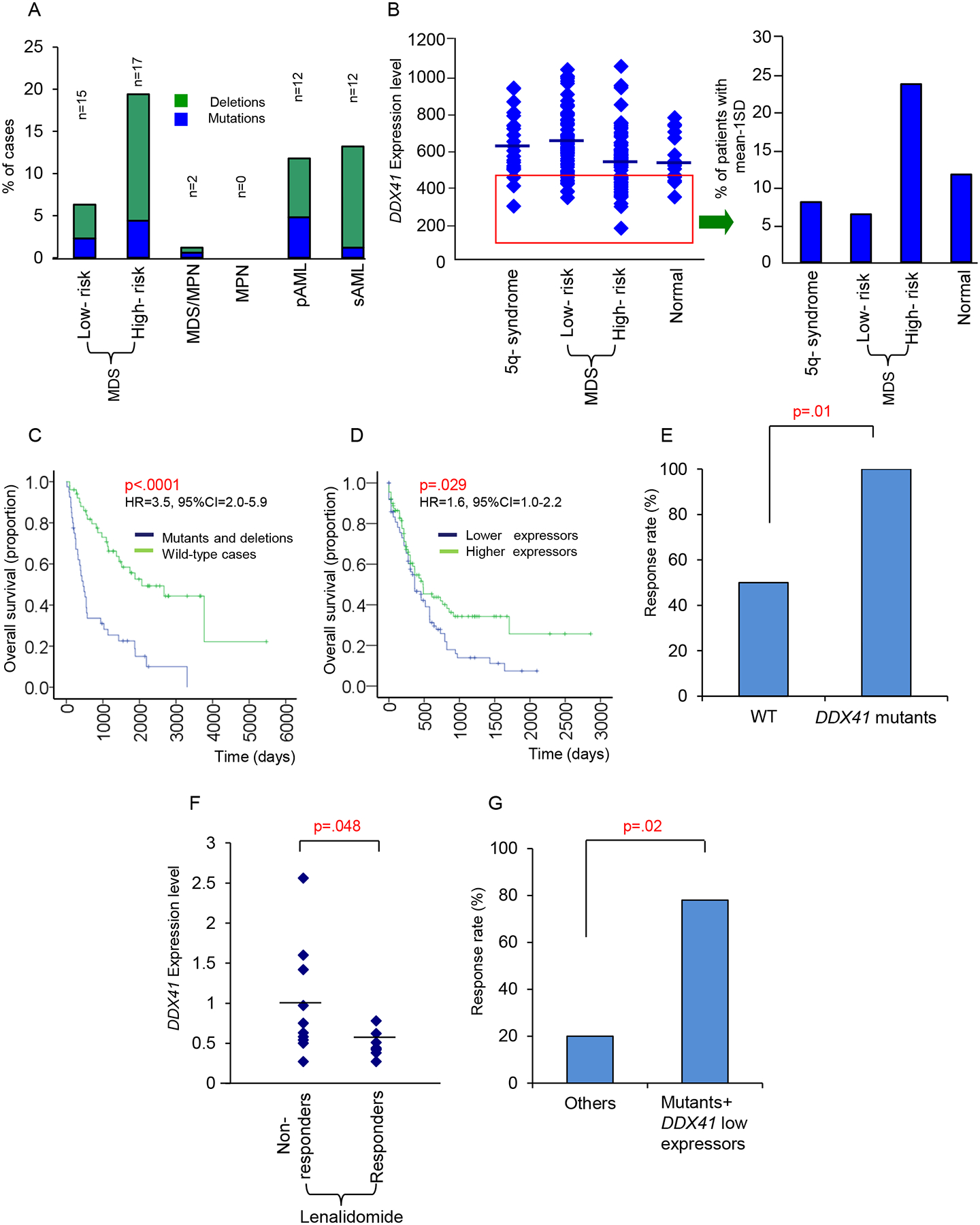
(A) Patients with somatic DDX41 defects (mutations and deletions) in different types of myeloid neoplasms. Indicated is the percentage of patients of each cohort with DDX41 deletions and mutations. The absolute number of patients with alterations is shown on the top of each bar.
(B) DDX41 mRNA levels in MDS patients with different subtypes. Reduced DDX41 expression was also demonstrated in various categories. Bars represent mean value.
(C) Overall survival analysis in patients with DDX41 mutations or deletions compared with wild-type cases (HR=3.5, 95%CI=2.0–5.9, p<.0001).
(D) Overall survival analysis in patients with low DDX41 mRNA expression compared with patients with higher expression (HR=1.6, 95%CI=1.0–2.2, p=.029). Cases with high and low DDX41 expression were dichotomized by the mean of relative mRNA transcription levels (mean=3.85 relative mRNA expression).
(E) Response rate to lenalidomide in patients with DDX41 mutants (n=8/8) compared with wild-type cases (n=55/103). p=.01.
(F) DDX41 mRNA expression in lenalidomide responders (n=9) compared with non-responders (n=10). Single square represent individual patients. Horizontal line indicates mean value. p=.048.
(G) Response rate to lenalidomide treatment of patients with DDX41 mutations and/or low DDX41 expression (n=7/9) compared with others (n=2/10).
We noted that both twins (Family 2) with germline and somatic DDX41 mutations responded well to lenalidomide, despite the absence of del(5q) (List et al., 2006) which led us to investigate the association between somatic DDX41 mutations and response to lenalidomide within a cohort of 111 patients with and without del(5q) treated with this drug (Table S2). Patients with DDX41 mutations responded better to lenalidomide treatment (Figure 3E). Furthermore, 19/111 cases were also analyzed for DDX41 expression by Taqman PCR (Figure 3F–G, Table S3). When we compared the expression of DDX41 between responders (n=9) and refractory cases (n=10), responders showed significantly lower DDX41 mRNA levels (Figure 3F). Patients with either low expression or DDX41 mutations (7/9) showed better response rate (78% vs. 20%) compared to others (Figure 3G).
Germline and somatic DDX41 mutations were associated with normal karyotype disease (70% vs. 47% without DDX41 mutations; p=.0045; Table S4). About 50% of DDX41 mutant cases did not harbor additional mutations (13 cases were sequenced by whole exome sequencing, whereas 14 cases were sequenced by deep-targeted re-sequencing). Nine of eleven cases with familial MDS/AML cases presented with normal karyotype and lacked typical AML-associated mutations. In addition to the predisposition for somatic DDX41 mutations, germline DDX41 mutations were associated with several other somatic mutations; among the 62 most frequently mutated genes, DDX41 lesions coincided with TP53, RUNX1 and LUC7L2 mutations (Figure S4).
Functional consequences of DDX41 lesions
DDX41 is expressed in CD14+, CD33+, and CD34+ myeloid cells (Figure S5A, S5B), consistent with a function in hematopoiesis. The DDX41 protein is highly conserved among species. The existing structure of the partial helicase domain of human DDX41 (PDB ID: 2P6N) and the structure of Drosophila Vasa (PDB ID: 2DB3) were used to generate a structural model (Figure S5C). Germline DDX41 frameshift mutations lead to a loss of function. Somatic DDX41 mutations are possibly hypomorphic, based on the location in the ATP binding domain. To model consequences of DDX41 deficiency, we used lentiviral shRNA delivery to knock down DDX41 in K562 cells and observed enhanced proliferation compared to mock transduced cells (Figure 4A–B). Similarly, CD34+ hematopoietic progenitor cells transduced with shRNA against DDX41 showed significantly enhanced colony formation (Figure 4C). When serial replating assays were used to assess the effects of DDX41 on retention of clonogenic capacity (Sontakke et al., 2014 and He et al., 2011), knockdown cells showed significantly increased replating efficiency consistent with retained clonogenic properties (Figure 4C). Anti-proliferative properties of DDX41 were also suggested by the results of cultures performed in the presence of various growth conditions. When we cultured CD34+ cells with knock down of DDX41, decreased levels of DDX41 resulted in a higher sensitivity to growth factor stimuli compared to control (Figure 4D–E). As an experiment for the tumor suppressor functions of DDX41, forced expression in U937 cells, which express low levels of endogenous DDX41, inhibited growth (Figure 4F). In primary MDS, we applied similar ectopic expression experiments for haploinsufficient DDX41 expression due to del(5q). We selected one representative sAML case in which deletion with haploinsufficient DDX41 expression was confirmed by SNP-A and quantitative RT-PCR, respectively. Forced expression of DDX41 in these DDX41-defective primary cells reduced colony formation (Figure S5D). These results further suggested that loss of DDX41 expression is associated with enhanced proliferative capacity in myeloid neoplasms. To validate the results of in vitro studies, we performed xenograft experiments with cell lines in which DDX41 was knocked down and demonstrated accelerated tumor growth compared to mock transduced cell (Figure 4G).
Figure 4: Biological consequences in DDX41 deficient cells.
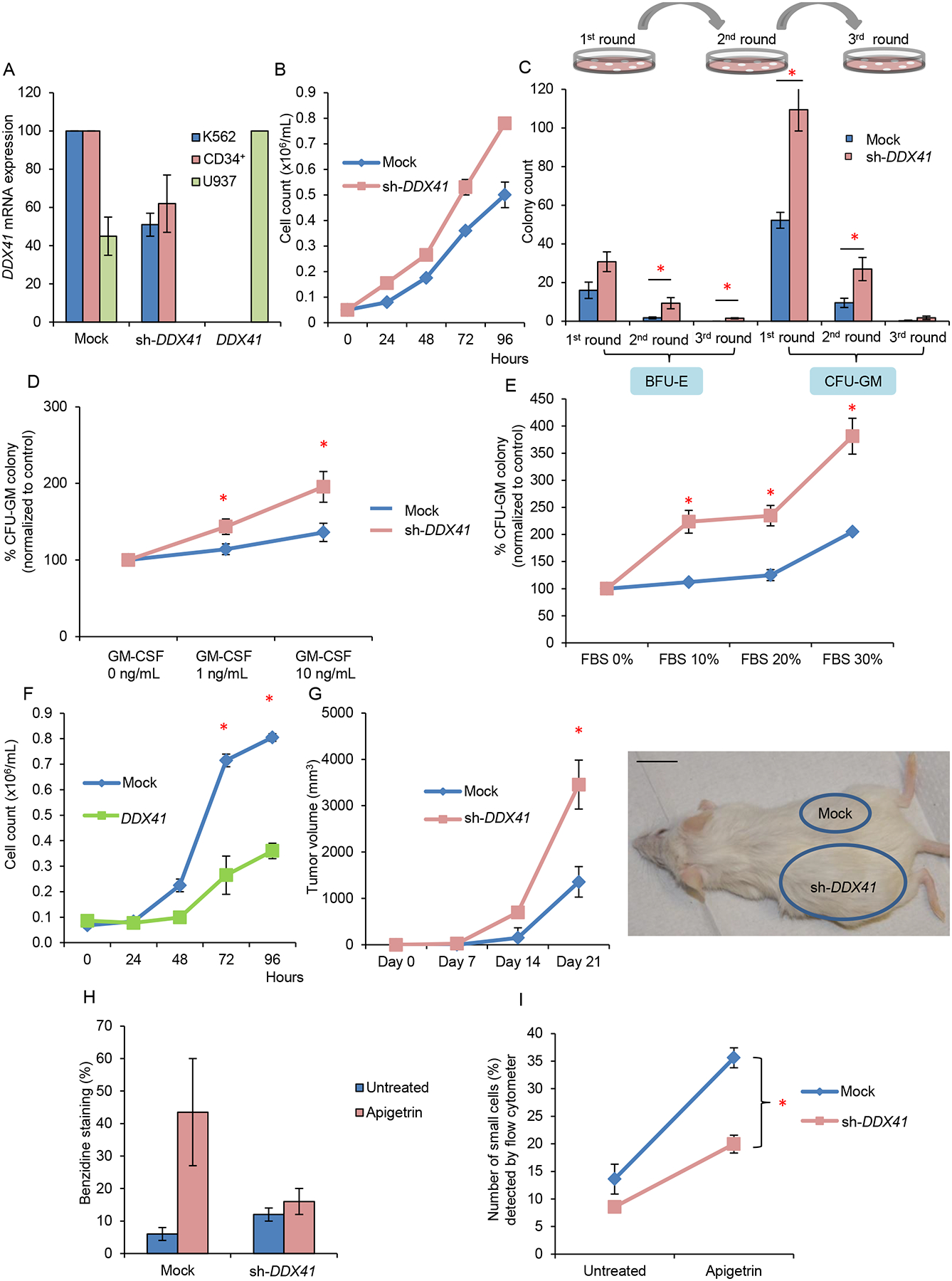
(A) Expression level of DDX41 in leukemic cell lines K562 and U937 and primary CD34+ cells as determined by normalization to GAPDH upon knockdown or overexpression of DDX41.
(B) Growth curves of K562 leukemic cells transduced with DDX41 knockdown construct (sh-DDX41) or mock-transduced. Doubling time of mock vs. sh-DDX41 = 29 hr. vs. 24 hr.
(C) Colony forming assay comparing DDX41 deficient with control primary CD34+ cells. Numbers of plating are indicated. BFU-E burst forming unit-erythroid; CFU-GM colony forming unit-granulocyte/macrophage.
(D) Percentage of CFU-GM colonies derived from plating of DDX41 deficient primary CD34+ cells in the absence or presence of different concentrations of GM-CSF in methylcellulose semisolid medium.
(E) Percentage of CFU-GM colonies derived from plating of DDX41 deficient CD34+ primary cells with various concentrations of FBS.
(F) Growth curves of U937 cells after lentiviral infection with a DDX41 expression construct compared to mock infected control cells. Doubling time of mock vs. DDX41 = 31 hr. vs. 36 hr.
(G) K562 cells with decreased DDX41 expression were injected into the left flank and those with mock transduction were injected into the right flank of NSG mice. n=3 for each experiment. A representative image and quantification of tumor volume are shown. Three series of independent experiments were performed. Scale bar: 1 inch.
(H) Hemoglobin detection of K562 cells with decreased DDX41 expression compared to those with mock transduction before and after exposure to apigetrin. Hemoglobin was measured by benzidine staining.
(I) Percentage of small cell population (mature erythroid cells) as detected by flow cytometry in DDX41 knockdown K562 cells compared with control cells after exposure to apigetrin.
Each bar/value represents the mean±SEM of 3 independent experiments performed in duplicates unless stated otherwise. * p<.05
See also Figure S5
In addition to increased proliferative capacity, we evaluated anti-differentiation and anti-apoptotic potential due to reduction of DDX41 function. DDX41 knockdown in K562 cells slightly impaired apigetrin-induced erythroid differentiation (Figure 4H–I; Figure S5E–F). As a rescue experiment, flow cytometry analysis showed higher expression of the CD11b and CD14 differentiation marker in U937 cells with forced expression of DDX41 (Figure S5G). In addition, up-modulation of DDX41 reversed the relative apoptotic resistance of U937 cells (Figure S5G). Using murine lin−sca 1+c-kit+ (LSK) cell model (Oakley et al., 2012), we also confirmed higher levels of c-kit and lower expression of Gr-1 upon DDX41 knockdown, illustrating defective differentiation in DDX41 deficient cells (Figure S5H).
Defective function of DDX41 may constitute a vulnerability of affected cells and provides a rationale for synthetic lethal intervention. When we tested a previously described helicase inhibitor, compound 8 (C14H15N3O) (Radi et al.,2011) in the lentiviral-induced knock down model in vitro, in the range of 1–10 μM, knockdown cells displayed increased susceptibility to C14H15N3O inhibition compared to mock transduced controls (Figure S5I–K).
The precise function of DDX41 is not yet known, but an involvement of RNA helicases in RNA splicing has been proposed (Cordin et al., 2012; Staley et al., 1998; Schwer et al., 2000). Mutations of other spliceosomal proteins are common in myeloid neoplasms (Maciejewski et al., 2012) and were mutually exclusive with DDX41 mutations in our cohort (Figure S6A). To further elucidate a role for DDX41 in the spliceosome, we expressed an epitope tagged version of WT DDX41 in HEK293 cells and performed a mass spectrometry analysis of proteins associated with DDX41 in an antibody pull-down followed by peptide sequencing. Spliceosomal proteins constituted the top functional group associated with DDX41 (Figure S6B) and many of them interact with DDX41 (Figure 5A; Table S5). Among many, PRPF8 and SF3B1 are exemplary spliceosomal proteins found in a complex with DDX41. Western blotting of native protein in primary extract as well as immunoprecipitates obtained with anti-DDX41 confirmed the findings of the mass spectrometry experiments (Figure S6C). Of note, mutations in DDX41 (R525H) altered the native DDX41 interactome especially for major components in U2 (SF3B1, SF3B2, SF3B3) and U5 (PRPF8, SNRNP200) spliceosome (Figure 5B). Western blots performed for SF3B1 and PRPF8 (Figure S6D) confirmed the differences between mutant and WT DDX41 immunoprecipitates. Notably, our analyses showed that these protein-protein associations persisted after nuclease digestion of RNA.
Figure 5: Protein Interactions of DDX41 and splicing factors.
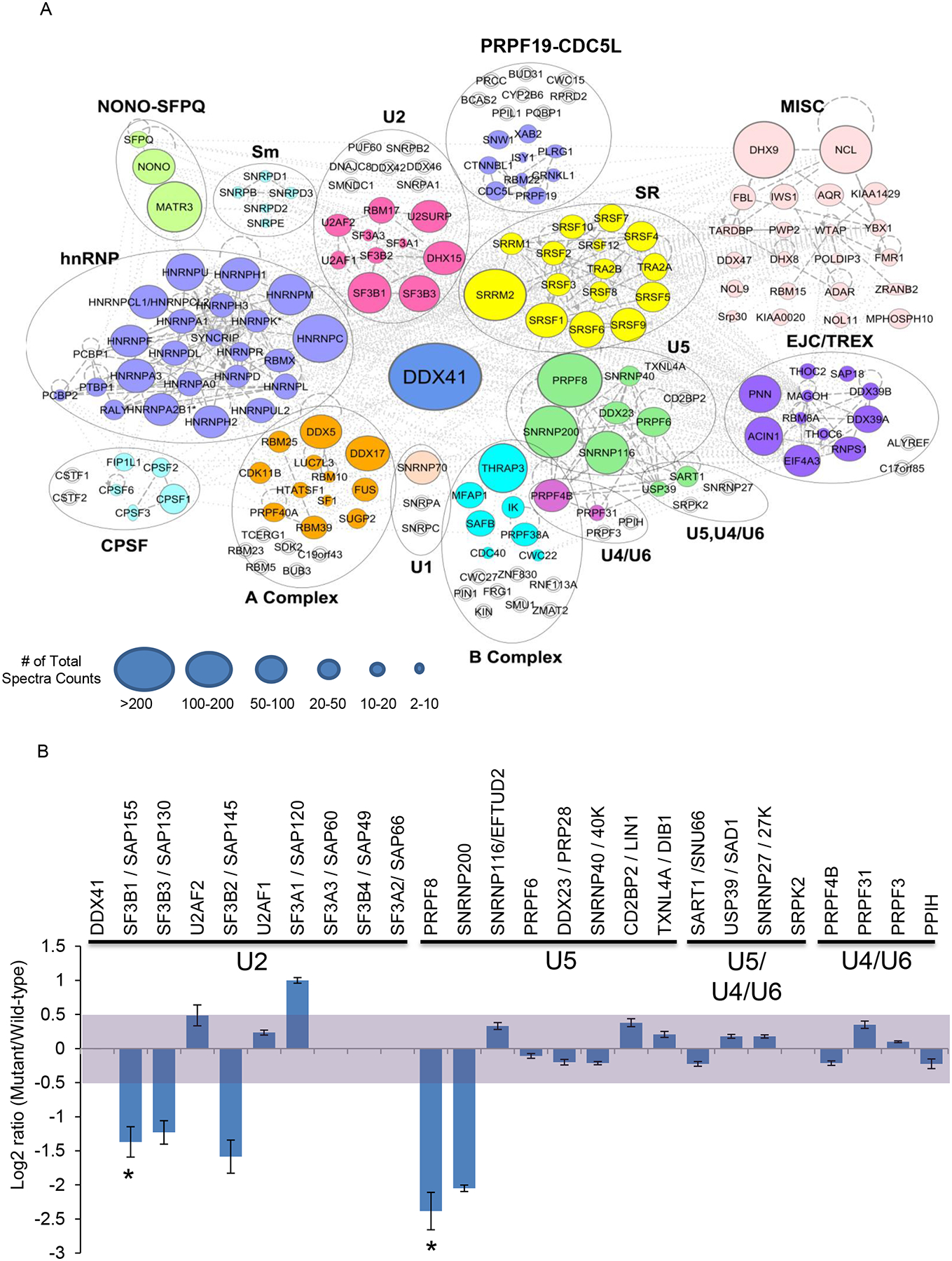
(A) DDX41 interactions with spliceosomal protein complexes are indicated. Spliceosomal proteins that coimmunoprecipitated with DDX41 were organized in colored functional protein complexes based on Ingenuity pathway analysis and published data (Hegele et al., 2012). Individual protein enrichment was presented as total spectral counts and displayed by different circle size. Increased circle size indicates higher number of total spectra counts for the protein. Total spectral count is a semi-quantitative method to predict abundance of a specific protein and is not used to compare with abundance of other proteins. Unfilled double ring symbols indicate proteins that were not identified in DDX41 co-immunoprecipitation experiments, but which have been linked to the spliceosome.
(B) Summary of cataloging and quantification of protein interactions with wild-type and mutant DDX41. Protein names and their associated spliceosomal complex are shown on top of bars. Protein abundance was normalized to DDX41 and presented as ratio of mutant to wild-type in log2 scale. Light purple shading indicates no significant difference in protein interaction between wild-type and mutant DDX41 (log2 scale between 0.5 and −0.5). Standard deviation (+/−) was calculated based on the three strongest peak intensities used in the calculation.
To investigate the possible impact of defective DDX41 on pre-mRNA splicing, deep whole RNA sequencing was investigated in deletion, mutant and WT cases (Przychodzen et al., 2013). The analysis involved 148,318 exons and comparison of their average usages between DDX41 defects (n=5) and controls (n=11) (Figure 6A, Figure S7A). DDX41 defects were associated with more avid exon skipping (excess of shorter mRNA missing an exon) and more exon retention (excess of longer mRNA incorporating an exon) in 61 and 95 genes, respectively. The top 10 most differentially misspliced exons (Figure 6B, top 40 gene list in Table S6) in functionally important genes were examined by reverse transcription polymerase chain reaction (RT-PCR). The difference of skipping ratio in ZMYM2 exon 3 can be used as an illustrative example (13% difference between DDX41 defect and wild-type; p=.019) (Figure 6C). The enhanced skipping of this exon, located in the 5’UTR was recapitulated by DDX41 knockdown in K562 and CD34+ cells. In contrast, overexpression of wild-type DDX41 in U937 cells led to decreased exon skipping of ZMYM2 in comparison to mock transduction (Figure 6D, Figure S7B–C). In addition to the changes in spliced isoform ratios, ZMYM2 mRNA was expressed at significantly lower levels in DDX41 low expressors (p<.001; Figure S7D). ZMYM2 encodes a zinc finger protein involved in a histone deacetylase complex (Bantscheff et al., 2011) and may constitute one of the downstream elements associated with DDX41 defects via its interaction with the LSD1-CoREST-HDAC1 co-repressor complex (Gocke et al., 2008). This complex is activated in various cancers and down-regulates transcription of tumor suppressor genes. When we analyzed RNA expression patterns (Boultwood et al., 2007) low ZMYM2 mRNA levels were associated with down-modulation of SMC3, RAD21, RUNX1, which were also significantly under-expressed in cases with low DDX41 (Figure S7E).
Figure 6: Deep whole RNA sequencing showed splicing defects in DDX41 deficient cells.
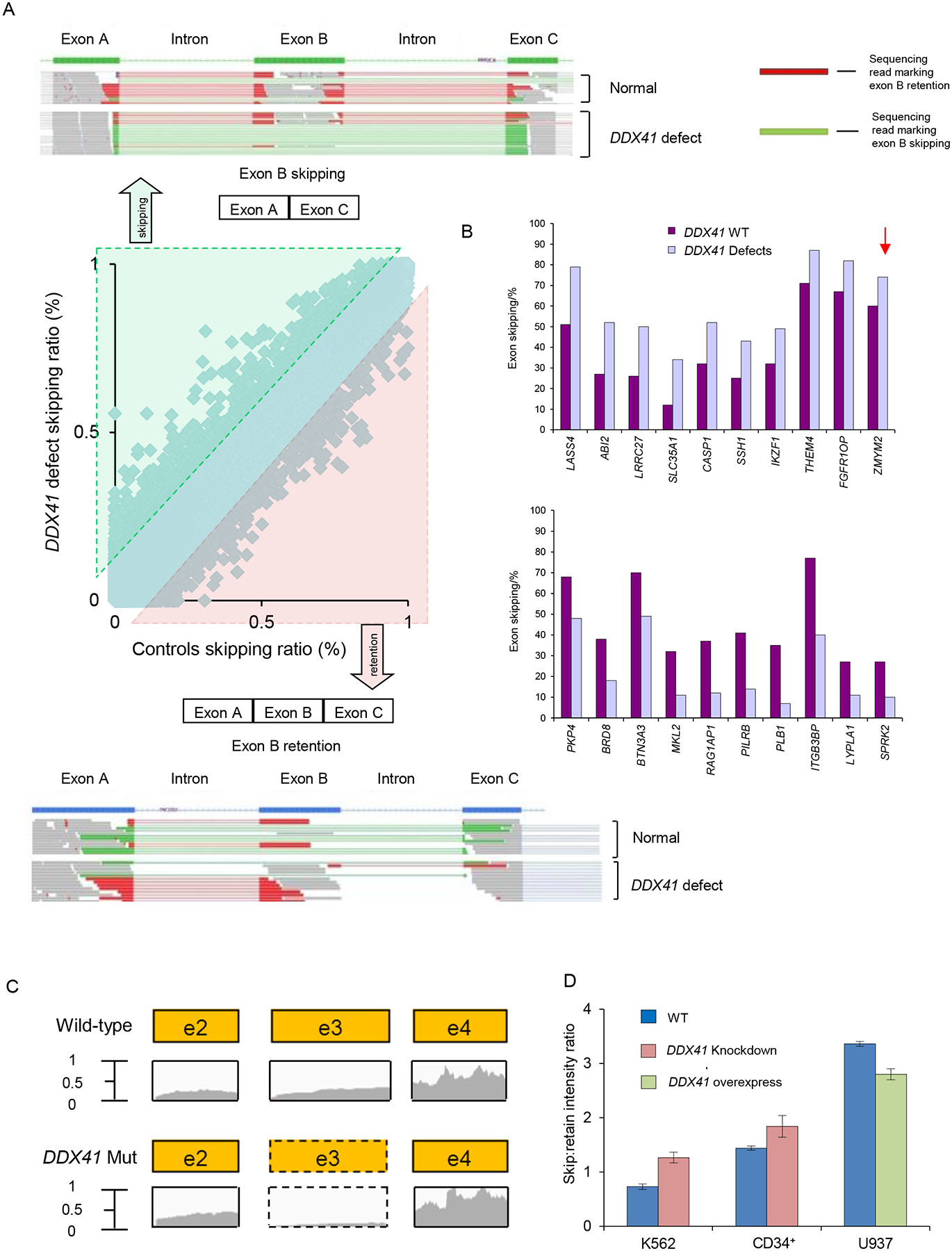
(A) Increased exon skipping (top) and retention (bottom) in patients with DDX41 defects are indicated by an excess of green reads and red reads, respectively. The center panel shows a scatter plot of exon skipping in RNA isolated from control cells versus RNA from DDX41 defective mutant cells. Lines show the 10% difference cutoff limit used to select the most frequently affected exons.
(B) Deep RNA sequencing was performed for blasts from patients with DDX41 mutations, deletions and wild-type to analyze altered splicing. The bar diagrams indicate the top 10 genes significantly more skipped in DDX41 defects (top) and in DDX41 wild-type (bottom). The arrow indicates the 13% difference of exon skipping in the ZMYM2 gene when comparing DDX41 defect and wild-type samples.
(C) Exon 3 of ZMYM2 was skipped in DDX41 deficient cells as demonstrated by the read counts from deep sequencing.
(D) RT-PCR was performed in K562 cells, CD34+ progenitors and U937 cells to evaluate ZMYM2 exon 3 skipping compared with controls. Depicted is the skip:retain intensity ratio for wild-type and DDX41 knockdown/overexpressing samples. Each bar represents the mean±SEM of 3 independent experiments.
Discussion
Several familial leukemia syndromes have been identified (Owen et al., 2008; Hahn et al., 2011; Liew et al., 2011). However, the incomplete penetrance and the increased frequency of myeloid disorders at older age may have hindered the identification of more inherited leukemia syndromes in older adults.
Germline DDX41 lesions define a hereditary MDS/AML syndrome which is characterized by long latency, advanced disease (high risk MDS/AML), normal karyotype and poor prognosis. Germline variants of DDX41 may convey a strong predisposition to MDS and subsequent AML and are likely to serve as a “first hit”, or an ancestral event. The canonical frameshift insertion in DDX41 might be more frequent than previously described germline RUNX1, CEBPA and GATA2 defects (Owen et al., 2008; Hahn et al., 2011).
In addition to germline DDX41 mutation, we report here in myeloid neoplasms canonical somatic mutations in this gene, often coinciding as a second hit with the germline mutations. By all the established and recognized criteria (Lawrence et al., 2014), somatic DDX41 mutations found by us are driver mutations (Makishima et al., 2014). Of note is that a somatic splice site DDX41 mutation was previously listed along other somatic mutations found in TCGA (Ding et al., 2012; Cancer Genome atlas Research Network 2013); TCGA cohort also contained the recurrent p.R525H mutation. However, the aforementioned studies did not further investigate somatic DDX41 mutations. The association between somatic and germline variants had not been elucidated. The frequency of the leukemogenic allele of DDX41 in the general population is very low and, due to late presentation, some healthy carriers may still anticipate disease. Mild abnormalities can be noted on careful evaluation of asymptomatic carriers. The germline DDX41 lesions strongly predispose to further somatic hits in the remaining healthy allele of this gene, suggesting that a higher level of haploinsufficiency created by a somatic missense mutation further enhances the clonal advantage. However, carriers of the inactivating variant germline c.419insGATG acquire somatic hypomorphic rather than inactivating mutations. Thus, total (bi-allelic) inactivation of the gene does not seem to be permissive, as we did not find any cases of germline DDX41 mutations followed by somatic deletion of its healthy allele.
The pro-leukemogenic properties of DDX41 lesions are supported by the presence of somatic mutations in this gene and the consistent lack of pathognomonic AML lesions, such as typical cytogenetic abnormalities, or primary AML-specific mutations. Germline alterations may constitute a predisposition factor for the acquisition of somatic mutations in the same genes, as is the case with JAK2, in which the rs10974944 polymorphism increases the risk for somatic JAK2 V617F mutations (Kilpivaara et al., 2009; Olcaydu et al., 2009). Similar to CEBPA and RUNX1 mutations, biallelic DDX41 mutations occur in germline DDX41 frame-shift mutations carriers as secondary somatic mutations. Notably, similar to the somatic CEBPA mutations, the somatic DDX41 mutations are hypomorphic, but the penetrance of CEBPA mutations is high, whereas it is currently unknown for DDX41. Index family 1 had 4 affected members in clear succession. Since the father did not develop disease until the age of 70, it is unclear whether all affected individuals will eventually develop MDS/AML. The long latency is also supported by the finding of the germline mutation in adult carrier family members that did not so far develop leukemia. However, they were younger than most of the patients with germline DDX41 mutations. In contrast to the RUNX1/CEBPA mutations (Owen et al., 2008), DDX41 mutations induce disease at age > 40 years. In germline GATA2 mutations carriers, only 16% remain asymptomatic by the age of 40 (Spinner et al., 2014). In our cohort, we found both germline and somatic mutations of DDX41. Germline mutations in familial leukemia syndromes and somatic mutations of the same gene in sporadic cases are a hallmark of key drivers in leukemogenesis such as CEBPA.
Similar to previously described spliceosomal mutations (Yoshida et al., 2011; Makishima et al., 2012), the precise mechanisms by which DDX41 lesions exert their pro-leukemogenic defects are unclear. A recent report on comprehensive gene expression and mutational profiles in medulloblastoma also showed frequently lower expression levels of DDX41 and frequent mutations of another RNA helicase DDX3X, which suggests that defective helicase functions might be related to common mechanisms for tumorigenesis (Kool et al., 2014). The physiological role of spliceosomal proteins is well characterized, but the functions of RNA helicases are far less well defined and might include a possible involvement in spliceosomal function, ribosome biogenesis, and translation initiation (Putnam et al., 2013). Both somatic and germline mutations indicate that DDX41 is a tumor suppressor gene and an important driver in myeloid malignancies. To that end DDX41 is exemplary of other DEAD/H-Box helicases that are also mutated in myeloid neoplasms. It is possible that RNA helicase mutations constitute a separate class of spliceosomal defects. Spliceosomal mutations induce splicing dysfunction (Przychodzen et al., 2013; Makishima et al., 2012) and our results indicate that DDX41 mutations also result in a specific missplicing pattern and altered expression of specific downstream genes.
DDX41 defects lead to a hereditary leukemia syndrome, and somatic lesions of this gene also occur in sporadic myeloid neoplasms. A significant proportion of del(5q) cases include the DDX41 locus, which leads to haploinsufficiency in a sizable proportion of patients with myeloid neoplasms. It is possible that DDX41 plays a role in the pathogenesis of del(5q), in particular in those cases with longer interstitial deletions, which, unlike the smaller defects, convey unfavorable prognosis (Jerez et al., 2012). Indeed, DDX41 defects were associated with advanced disease and poor prognosis.
The presence of DDX41 mutations or deletions was associated with responsiveness to lenalidomide. This finding might constitute a possible therapeutic intervention for otherwise poor risk disease, but further studies are necessary to determine the predictive value of DDX41 mutations, deletions or low expression for lenalidomide response.
Recent reports suggest the existence of pre-leukemic stem cells in MDS (Woll et al, 2014) and in AML (Shlush et al., 2014) These pre-leukemic stem cells contain a first hit that significantly enhances the likelihood of subsequent leukemia development. The germline DDX41 mutations induce MDS/leukemia with long latency but significant penetrance. Thus, DDX41 lesions might genuinely induce a pre-leukemic state that predisposes for leukemia.
In summary, we identified germline mutations in DDX41 that are associated with the development of hereditary MDS and AML. The strong family history and late onset suggest high penetrance with long disease latency. Germline DDX41 defects strongly predispose to somatic DDX41 mutations.
EXPERIMENTAL PROCEDURES
PATIENTS’ SAMPLES
Bone marrow aspirates or blood samples were collected from the 8 index cases (Family1-4), an additional 840 patients with myeloid neoplasms seen at Cleveland Clinic, University of Muenster and University of Chicago and 197 cases from TCGA database (n=1,045; Table S1). Informed consent for sample collection was obtained according to protocols approved by the Institutional Review Boards (Cleveland clinic, Ethik-Kommission der Ärztekammer Westfalen-Lippe und der medizinischen Fakultät der Westfälischen Wilhelms Universität Münster and University of Chicago) and in accordance with the Declaration of Helsinki. Diagnosis was confirmed according to 2008 WHO classification criteria. Tumor DNAs were extracted from patients’ bone marrow cells. For germline controls, DNA was obtained from paired CD3+ T cells or buccal swab. Index patients from family 1 were analyzed at diagnosis (and relapse). First complete remission samples were additionally used as surrogate for germline DNA. Germline DDX41 mutation was confirmed by Sanger sequencing in buccal swab DNA in index patients Case 2 and Case 3.
NGS STUDIES
Whole exome sequencing (WES) was performed as previously described (Yoshida et al., 2011). To detect allelic frequencies for mutations or SNPs, we applied deep next-generation multi-amplicon sequencing to targeted exons (Yoshida et al., 2011). The multi-amplicon panel included 62 genes. Libraries were generated according to standard procedures and paired-end sequenced (Supplemental experimental procedures).
CYTOGENETICS AND SNP ARRAYS
Technical details regarding sample procession for SNP array assays were previously described (Maciejewski et al., 2009, Gondek et al., 2008). The Gene Chip Mapping 250K Assay kit and the Genome-Wide Human SNP Array 6.0 (Affymetrix) were used. A stringent algorithm was applied for the identification of lesions using SNP arrays. Individuals with lesions identified by SNP array concordant with those identified in metaphase cytogenetics or typical lesions known to be recurrent required no further analysis. Changes reported in our internal or publicly available (Database of Genomic Variants; http://projects.tcag.ca/variation) copy number variation (CNV) databases were considered non-somatic and were excluded. Results were analyzed using CNAG (v3.0) (Nannya et al., 2005) or Genotyping Console (Affymetrix). All other lesions were confirmed as somatic or germline by analysis of CD3-sorted cells (Tiu et al., 2009).
QUANTITATIVE RT-PCR WITH TAQMAN PROBES
Total RNA was extracted from bone marrow mononuclear cells and cell lines. cDNA was synthesized from 500 ng total RNA using the SuperScript III First-Strand Synthesis System (Invitrogen). Quantitative gene expression levels were detected using real-time PCR with the ABI PRISM 7500 Fast Sequence Detection System and FAM dye labeled TaqMan MGB probes (Applied Biosystems). TaqMan assays were performed according to the manufacturer’s instructions. Primers and probes for all genes analyzed were purchased from Applied Biosystems gene expression assays products (DDX41: Hs00169602_m1; GAPDH: Hs99999905_m1). The expression level of target genes was normalized to the GAPDH mRNA.
WHOLE RNA SEQUENCING
We used publicly available RNAseq data from TCGA data portal for 97 patients (http://tcga-data.nci.nih.gov/tcga). We selected 3 cases which showed deletion of 5q including DDX41 locus, 1 case harbored DDX41 mutation (c.G1574A, p.R525H) and 1 case showed low expression of DDX41, for which deep RNAseq (Tarazona et al., 2011) data were available. We also selected 11 cases that were wild-type for any spliceosomal factor mutation (Supplemental experimental procedures).
CELL CULTURE, LENTIVIRAL-MEDIATED shRNA KNOCKDOWN AND LENTIVIRAL EXPRESSION VECTOR
HL60 (human promyelocytic cell line), U937 (human monocytic cell line) and K562 (human chronic myelocytic leukemia cell line) cells were cultured using Iscove’s Modified Dulbecco’s Medium + 10% Fetal Bovine Serum. The pLKO.1_DDX41-shRNA and the control non-target shRNA were purchased from Sigma-Aldrich (St.Louis, MO, USA). In brief, 293T cells were transfected with shRNA targeting DDX41 or non-target shRNA control plasmid together with packing plasmid pCMVΔ8.2 and envelope plasmid containing VSV-G. Viral supernatants were harvested at 48, 72 and 96 hours post transfection, and target cells were infected in the presence of 8 μg/mL polybrene for 24 hours, and selected with puromycin (2 μg/mL for K562 and 1 μg/mL for HL60). For CD34+ primary cells, we used 25 μg/mL of Retronectin® instead of polybrene. Lentiviral expression vector (pLX304, Clone ID: HsCD00442077; DNASU Plasmid Repository) was used to generate viral supernatants. U937 was transfected in the presence of 8 μg/mL polybrene for 24 hours, then selected with blasticidin (5 μg/mL).
HUMAN CD34+ COLONY ASSAYS
CD34+ cells were isolated from healthy bone marrow. Informed consent for sample collection was obtained according to the protocols and procedures approved by CCF Institutional review Board (IRB3952 and IRB5024) and in accordance with the Declaration of Helsinki. Approximately 5×104 sorted Human CD34+ cells from healthy donor were plated on methylcellulose according to the MethoCult technical manual (H4230; StemCell Technologies). Lentivirally infected human CD34+ cells were added to methylcellulose medium supplemented with 10 ng/mL human IL-3, 50 ng/mL SCF, 3 U/mL erythropoietin, 10 ng/mL GM-CSF and 20% FBS. The number of burst-forming unit erythroid (BFU-E) and colony-forming unit granulocyte/macrophage (CFU-GM) were accessed after 10- to 14-day culture at 37°C in humidified atmosphere with 5% CO2 as per the manufacturer’s instructions. For the assessment of sensitivity to stimuli, both DDX41-deficient and control CD34+ cells were plated in various amount of GM-CSF and FBS (0, 1, 10 ng/mL of GM-CSF and 0, 10, 20, 30% of FBS). Colonies were evaluated after 10- to 14-day culture. For serial methylcellulose replating assay, CD34+ cells were plated on methylcellulose with cytokines. After 10–14 days, the colony numbers were counted under microscope. The colonies were picked up, and cells were pooled and replated (104 cells/plate) onto secondary methylcellulose plates. Three rounds of replating were performed for each experiment (Sontakke et al., 2014 and He et al., 2011).
IN VIVO TUMOR XENOGRAFT
Tumor xenograft studies were performed in accordance with recommendations in Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and conducted under a protocol approved by Cleveland Clinic Institutional Animal Care and Use Committee. K562 cell line was transfected with lentiviruses carrying control shRNA, or DDX41-targeting shRNA. A total of 10×106 cells were diluted in PBS 100 μL and injected subcutaneously into the flank of 8 week-old NSG mice. Mock cells were injected in the right flank while sh-DDX41 cells were injected in the left flank. Tumor volumes were measured in two dimensions (length and width) using a Dial Caliper and calculated using the formula: tumor volume = (length × width2) x0.5. Tumor volume was measured every 7 days. Three independent experiments were performed in triplicates.
IMMUNOPRECIPITATION
V5 immunoprecipitation was performed with V5 tagged wild-type and mutant DDX41 (R525H) in HEK293 cells. Nuclear protein extracts (~10 mg of protein) were transferred to tubes with antibody-bound protein G beads and rocked gently at 4°C overnight. Nonspecifically bound proteins were removed with 5 washes of PBS containing 1% Nonidet P-40. Immunoprecipitation products were extracted from protein G beads using Laemmli sample buffer. Immunoprecipitates were analyzed by LC-MS/MS. Peak intensity based label-free comparison was employed to compare relative protein abundance.
STATISTICAL ANALYSIS
The Kaplan-Meier method was used to analyze overall survival (OS) by the log-rank test. Pairwise comparisons were performed by Wilcoxon test for continuous variables and by two-sided Fisher’s exact test for categorical variables. For multivariate analyses, a Cox proportional hazards model was employed. Variables considered for model inclusion were IPSS risk group, age, sex, and gene mutation status. The statistical significance of functional studies was evaluated using a 2-tailed t test. Significance was determined at a two-sided α level of .05, except for p values in multiple comparisons, in which Bonferroni correction was applied.
ACCESSION NUMBER
Whole-exome sequencing results have been deposited in the Sequence Read Archive (SRA; BioProject accession PRJNA275985).
The Gene Expression Omnibus accession number for SNP Arrays is GSE66668.
Supplementary Material
Significance.
We have identified a familial AML syndrome characterized by long latency and germline mutations in the gene coding for the DEAD-Box helicase DDX41 located on chr. 5q35. Recurrent somatic DDX41 mutations were identified in myeloid neoplasms; around 50% of cases in patients with germline mutations harbored somatic point mutations in the other allele. In addition to mutations, DDX41 locus was deleted in 26% of MDS cases with del(5q) and resulted in haploinsufficient expression. DDX41 defects led to loss of tumor suppressor function due to altered pre-mRNA splicing and RNA processing. Somatic mutations were also found in other RNA helicase genes, suggesting that they constitute a family of tumor suppressor genes in myeloid neoplasms.
ACKNOWNLEDGEMENT
Supported by grant from the MDS Foundation, young investigator grant award (C.P.), grant from National Institutes of Health (2K24HL077522, R01HL118281), Scott Hamilton CARES grant (H.M.), NIH grant R01CA143193 (Y.D.), grant from AA&MDS Int. Foundation (H.M.) and the project for the development of innovative research on cancer therapies (p-direct; S.O.). Supported by grants from the Deutsche Forschungsgemeinschaft (DFG), the Deutsche Krebshilfe and the Deutsche José-Carreras Stiftung (C.MT). We thank our patients for participating in this study. We are grateful to Peter Wieacker for genetic counseling of affected patients. The authors thank The Cancer Genome Atlas for access to the whole genome sequencing results described in the text. We thank George Rafidi and Maya Lewinsohn from University of Chicago for their contribution in DNA sequencing of the family members.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schleg J, Abraham Y, Becher I, Bergamini G,et al. (2011). Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol 29, 255–265. [DOI] [PubMed] [Google Scholar]
- Bennett RL, Freanc KS, Resta RG, Doyle DL (2008). Standardized human pedigree nomenclature: update and assessment of the recommendations of the national society of genetic counselors. J. Genet. Counsel 17, 424–33. [DOI] [PubMed] [Google Scholar]
- Boultwood J, Pellagatti A, Cattan H, Lawrie CH, Giagounidis A, Malcovati L, Della Porta MG, Jädersten M, Killick S, Fidler C, et al. (2007). Gene expression profiling of CD34+ cells in patients with the 5q- syndrome. Br. J. Haematol 139, 578–589. [DOI] [PubMed] [Google Scholar]
- Cancer Genome atlas Research Network. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordin O, Hahn D & Beggs JD (2012). Structure, function and regulation of spliceosomal RNA helicases. Curr. Opin. Cell Biol 24, 431–438. [DOI] [PubMed] [Google Scholar]
- Ding L. Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. (2012). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocke CB & Yu H (2008). ZNF198 stabilizes the LSD1-CoREST-HDAC1 complex on chromatin through its MYM-type Zinc Fingers. Plos One 22, e3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek LP, Tiu R, O’Keefe CL, Sekeres MA, Theil KS, Maciejewski JP (2008). Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 111, 1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, Babic M, Lin M, Carmagnac A, Lee YK, et al. (2011). Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet 43, 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Nguyen AT and Zhang Y (2011). KDM2B/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 117, 3869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburow A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, et al. (2012). Dynamic Protein-Protein Interaction Wiring of the Human Spliceosome. Molecular Cell 45, 567–580. [DOI] [PubMed] [Google Scholar]
- Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV, O’Keefe CL, Mohamedali AM, Batista D, Sekeres MA, et al. (2012). Topography, clinical and genomic correlates of 5q myeloid malignancies revisited. J. Clin. Oncol 30, 1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, Bass A, Marubayashi S, Heguy A, Garcia-Manero G, et al. (2009). A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat. Genet 41, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Jones DT, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, et al. (2014). Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer cell. 25, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanow P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew E & Owen C (2011). Familial myelodysplastic syndrome: a review of the literature. Haematologica 96, 1536–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, Powell B, Greenberg P, Thomas D, Stone R et al. (2006). Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med 355, 1456–1465. [DOI] [PubMed] [Google Scholar]
- Maciejewski JP & Padgett RA (2012). Defects in spliceosomal machinery: a new pathway of leukaemogenesis. Br. J. Haematol 158, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewski JP, Tiu RV, O’Keefe C (2009). Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br. J. Haematol 146, 479–488. [DOI] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. (2012). Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Yoshida K, LaFramboise T, Przychodzen BP, Ruffalo M, Gómez-Seguí I, Shiraishi Y, Sanada M, Nagata Y, Yusuke Sato Y, et al. (2014). In Analogy to AML, MDS Can be Sub-Classified By Ancestral Mutations. Blood 124, 823. [Google Scholar]
- Nannya Y. Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC et al. (2005). A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 65, 6071–6079. [DOI] [PubMed] [Google Scholar]
- Oakley K, Han Y, Vishwakarma BA, Chu S, Bhatia R, Gudmundsson KO, Keller J, Chen X, Vasko V, Jenkins NA, et al. (2012). Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood 119, 6099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcaydu D, Harutyunyan A, Jäger R, Berg T, Gisslinger B, Pabinger I, Gisslinger H, Kralovics R (2009). A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet 41: 450–454. [DOI] [PubMed] [Google Scholar]
- Owen C, Barnett M & Fitzgibbon J (2008). Familial myelodysplasia and acute myeloid leukemia-a review. Br. J. Haematol 140, 123–132. [DOI] [PubMed] [Google Scholar]
- Patel JP,Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Vlierberghe PV, Dolgalev I, Thomas S, Aminova O, et al. (2012). Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med 366, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeilstöcker M, Karlic H, Nösslinger T, Sperr W, Stauder R, Krieger O, Valent P (2007). Myelodysplastic syndromes, aging, and age : correlations, common mechanisms, and clinical implications. Leuk. Lymphoma 48, 1900–1909. [DOI] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H (2013). Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 122, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam AA & Jankowsky E (2013). DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 1829, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi M, Falchi F, Garbelli A, Samuele A, Bernardo V, Paolucci S, Baldanti F, Schenone S, Manetti F, Maga G, et al. (2012). Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: towards the next generation HIV-1 inhibitors. Bioorg. Med. Chem. Lett 22, 2094–2098. [DOI] [PubMed] [Google Scholar]
- Schwer B & Meszaros T (2000). RNA helicase dynamics in pre-mRNA splicing. EMBO J. 19, 6582–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekeres MA (2010). The epidemiology of myelodysplastic syndromes. Hematol. Oncol. Clin. N. Am 24, 287–294. [DOI] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. (2014) Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontakke P, Carretta M, Capala M, Schepers H, Shuringa JJ (2014). Ex Vivo Assays to Study Self-Renewal, Long-Term Expansion, and Leukemic Transformation of Genetically Modified Human Hematopoietic and Patient-Derived Leukemic Stem Cells. In Leukemia: methods and protocols, So CWE, ed. (New York, US: Springer-Verlag; ), pp. 195–210. [DOI] [PubMed] [Google Scholar]
- Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, Arthur DC, Gu W, Gould CM, Brewer CC, et al. (2014). GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics and immunity. Blood 123, 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP & Guthrie C (1998). Mechanical devices of the spliceosome: motors, clocks, springs and things. Cell 92, 315–326. [DOI] [PubMed] [Google Scholar]
- Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa, (2011). A Differential expression in RNA-seq: a matter of depth. Genome Res. 21, 2213–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiu RV, Gondek LP, O’Keefe CL, Huh J, Sekeres MA, Elson P, McDevitt MA, Wang XF, Levis MJ, Karp JE, et al. (2009). New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J. Clin. Oncol 27, 5219–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, Fulton R, et al. (2013). Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 27, 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll PS, Kjällquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, Erlandsson R, Ngara M, Anderson K, Deng Q, et al. (2014). Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer cell. 25, 794–808. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.