

Abstract
The breakdown of the blood‐brain barrier (BBB) is related to the occurrence and deterioration of neurological dysfunction in ischemic stroke, which leads to the extravasation of blood‐borne substances, resulting in vasogenic edema and increased mortality. However, a limited understanding of the molecular mechanisms that control the restrictive properties of the BBB hinders the manipulation of the BBB in disease and treatment. Here, we found that the glycocalyx (GCX) is a critical factor in the regulation of brain endothelial barrier integrity. First, endothelial GCX displayed a biphasic change pattern, of which the timescale matched well with the biphasic evolution of BBB permeability to tracers within the first week after t‐MCAO. Moreover, GCX destruction with hyaluronidase increased BBB permeability in healthy mice and aggravated BBB leakage in transient middle cerebral artery occlusion (t‐MCAO) mice. Surprisingly, ultrastructural observation showed that GCX destruction was accompanied by increased endothelial transcytosis at the ischemic BBB, while the tight junctions remained morphologically and functionally intact. Knockdown of caveolin1 (Cav1) suppressed endothelial transcytosis, leading to reduced BBB permeability, and brain edema. Lastly, a coimmunoprecipitation assay showed that GCX degradation enhanced the interaction between syndecan1 and Src by promoting the binding of phosphorylated syndecan1 to the Src SH2 domain, which led to rapid modulation of cytoskeletal proteins to promote caveolae‐mediated endocytosis. Overall, these findings demonstrate that the dynamic degradation and reconstruction of GCX may account for the biphasic changes in BBB permeability in ischemic stroke, and reveal an essential role of GCX in suppressing transcellular transport in brain endothelial cells to maintain BBB integrity. Targeting GCX may provide a novel strategy for managing BBB dysfunction and central nervous system drug delivery.
Keywords: blood‐brain barrier, brain edema, caveolin1, glycocalyx, ischemia stroke, transcytosis
Glycocalyx‐regulated transcellular transport across the BBB contributes significantly to the barrier function of the BBB in both normal and diseased conditions. GCX may be an essential target for manipulating BBB permeability to help with drug delivery to the brain or to protect against BBB injury under diseased conditions.

1. INTRODUCTION
The blood‐brain barrier (BBB) provides a controlled homeostatic microenvironment for the central nervous system (CNS) by strictly controlling the passage of materials between the blood and brain parenchyma (1). The integrity of the BBB is compromised by cerebral ischemia, which results in the unwanted entry of substances into the brain, leading to brain edema that critically worsens the clinical outcome (2). The BBB is mainly composed of vascular endothelial cells (ECs) encapsulated by the basement membrane and astrocytes. Enriched tight junctions (TJs) and a negligible rate of vesicular transcytosis, the two distinct features of brain capillary ECs, determine the restrictive properties of the BBB (3). However, the relative contribution of these two EC transport pathways to BBB integrity in ischemic injury remains unclear. A thorough study of these two processes may help us gain a deeper understanding of the alteration and regulation of the BBB and provide novel insights into therapeutic applications.
In animal stroke studies, BBB opening has been demonstrated to be biphasic (4, 5), with the first phase of enhanced permeability occurring 4–6 h after reperfusion and the second phase occurring 2–3 days (6) or 7 days (7, 8) after reperfusion. The disassembly and reassembly of endothelial TJs are considered to account for the featured dynamics of BBB disruption (4, 6). This hypothetical mechanism is supported by alterations in the protein/mRNA levels of TJs or by fluorescence imaging studies (9). However, it has never been convincingly proven at the ultrastructural level (10) and has been repeatedly questioned in recent years as accumulating experimental evidence shows that TJ morphology remains normal despite abundant vascular extravasation of tracers under stroke conditions (11, 12, 13, 14).
Recent advances have emphasized the importance of transcellular transport in limiting BBB permeability (11, 15, 16, 17, 18). Ultrastructural analysis has demonstrated that the significantly increased transcytosis after stroke, as evidenced by a rapid increase in endocytic vesicles, might be the initial response of ECs to ischemia (15). In addition, the uptake of albumin, horseradish peroxidase (HRP), and gold nanoparticles by EC vesicles is significantly increased (19, 20, 21). Another recent study highlighted the governing role of transcytosis in immature vessel leakage during the formation of the zebrafish BBB (16). These findings suggest that the traditionally neglected process of transcytosis may play a critical role in maintaining the integrity of the BBB. However, the mechanisms underlying the formation of EC vesicles and the regulation of transcytosis remain largely unknown.
The glycocalyx (GCX) is a carbohydrate‐rich, negatively charged gel‐like layer covering the surface of luminal ECs, which is vulnerable to be disrupted yet quickly reconstituted (22). Researches on the peripheral circulation system have shown that disruption of the GCX leads to increased permeability to plasma proteins and the exposure of EC adhesion molecules, resulting in local inflammation and edema (23). The endothelial cells that form the BBB are surrounded by the GCX, but the exact mechanism by which the GCX affects the BBB in the CNS remains unclear. As the brain capillary structure differs greatly from that in peripheral tissues because of its unique BBB barrier function and neurovascular coupling, the role of their GCX may also be far different from its counterpart in the peripheral circulation.
Using brain injury animal models of cardiac arrest (24) and status epilepticus (25), we have previously shown that the destruction of the GCX increases BBB permeability and aggravates brain edema, but the underlying mechanism remains unclear. In this study, we investigated the changes in the GCX and its contribution to ischemic BBB damage using a mouse stroke model of transient middle cerebral artery occlusion (t‐MCAO). We found that within the first week following t‐MCAO onset, the endothelial GCX displayed a characteristic change pattern, that is, degradation, reconstruction, and redegradation. Surprisingly, the time course of GCX change matched well with the biphasic pattern of BBB opening. At the same time, we observed a significant increase in the number of caveolae vesicles and an accelerated rate of transcytosis in the ECs of the ischemic BBB. Unexpectedly, the TJs of the ischemic BBB remained morphologically intact when observed under a transmission electron microscope (EM) for up to 7 days, which continuously halted the tracers to cross the gaps of the adjacent endothelial cells. More importantly, enzymatic degradation of the GCX not only aggravated t‐MCAO‐induced BBB leakage but also led to increased vesicular transcytosis and BBB leakage in healthy animals. Mechanistically, GCX degradation enhanced the interaction between syndecan1 and Src by promoting the binding of phosphorylated syndecan1 to the Src SH2 domain, thereby leading to rapid modulation of cytoskeletal proteins to promote caveolae‐mediated endocytosis.
2. MATERIALS AND METHODS
2.1. Animals
Male C57BL/6J background mice were obtained from the Experimental Animal Center of Southern Medical University and housed under a 12‐h light/dark cycle in a pathogen‐free facility with free access to water and standard chow before and after the operation. The study was approved by the Animal Care and Use Committee of Nanfang Hospital, Southern Medical University and was performed in accordance with the National Guidelines for Animal Experimentation (No.NFYY2018117).
2.2. Mouse ischemic stroke model
The 8‐ to 10‐week‐old mice were subjected to t‐MCAO using intraluminal filaments, as described previously (26). Ischemia was induced for 45 min, and reperfusion was restored.
2.3. Drug administration and study protocol
A total of 267 mice were used in this study (Figure S1), which was divided into three parts. In the first part, we investigated the dynamic evolution of the GCX and its effects on BBB breakdown and brain injury under t‐MCAO conditions. Of the 169 mice that underwent t‐MCAO, 8 mice displayed cerebral hemorrhage, 13 died before predefined time points and were thus excluded. First, the dynamic changes in BBB permeability and its ultrastructural components, including endothelial GCX, TJs, and vesicular activity, were detected at 1, 3, 6, 12 h, 1, 2, 5, and 7 days after reperfusion onset. Second, we examined the effect of the GCX on t‐MCAO‐induced brain injury. The mice subjected to t‐MCAO were randomly allocated to different groups receiving vehicle, hyaluronidase (HAase, 20 mg/kg, Type IV‐S, H3884, Sigma‐Aldrich, USA), and hydrocortisone (hc, 30 mg/kg, H3160, Sigma Aldrich, USA). HAase was intravenously injected immediately after reperfusion onset via the tail vein and was used to degrade the endothelial GCX, while hc was administered via intraperitoneal injection and was used to preserve the GCX (24). Mice in the control group received an equivalent volume of saline. In the second part, whether caveolin1 (Cav1) knockdown ameliorated BBB breakdown and the consequent formation of brain edema after ischemic stroke were examined, and in the third part, under physiological conditions, the contribution of the endothelial GCX to BBB properties and the potential molecular mechanisms were studied. In addition to HAase, heparinase (HPase, 1 unit/mouse, H8891; Sigma‐Aldrich, USA) that degrades the endothelial GCX (27) was used to exclude the nonspecific effects of HAase. Brain tissues were collected to detect BBB properties 6 h after intravenous injection of each enzyme. Moreover, Cav1‐knockdown mice were used to evaluate the regulatory effects of the GCX on caveolae vesicle‐mediated transcytosis.
2.4. Determination of GCX components (hyaluronan and syndecan1)
Following t‐MCAO, serum samples were collected at varying time points to assess the shedding of hyaluronan and syndecan1, the two degraded GCX components. The concentrations of hyaluronan and syndecan1were determined using ELISA kits from R&D Systems (Minneapolis, MN, USA) and Diaclone Research (Besancon, France), respectively.
2.5. Transmission EM and image analyses
EM imaging of the endothelial GCX was performed as previously described (24). Briefly, mice were anesthetized and perfused with 0.1 mol/L sodium cacodylate buffer containing 2% lanthanum nitrate, 2.5% glutaraldehyde, and 2% paraformaldehyde. The ischemic cortex tissue was harvested and diced. Three or four 1‐mm3 tissue blocks were immersed in 2.5% glutaraldehyde overnight for fixation, soaked overnight in sodium cacodylate buffer, and washed in an alkaline (0.03/L NaOH) sucrose (2%) solution. The samples were then dehydrated using a series of graded ethanol solutions and embedded in epoxy resin. Ultrathin sections stained with uranyl acetate and lead citrate were examined using an EM (H‐7500, Hitachi, Tokyo, Japan).
For the mice receiving HRP injection, HRP (1000 units/mouse, P8250; Sigma Aldrich, USA) was dissolved in saline and injected into the tail vein 1 h before anesthesia. Brain tissue was harvested and fixed as described above and then cut into 100 μm thick slices using a vibrating microtome (Vt1000s, Leica), followed by incubation with diaminobenzidine (DAB) solution. Ultrathin sections were cut, collected on copper grids, counterstained, and examined under EM.
All ultrastructural quantifications were performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Quantitative assessments of the endothelial GCX occupation area and the corresponding inner diameter of the capillary were performed on six capillary vessels randomly chosen from the EM images collected from each mouse. The thickness of the GCX was then defined as the ratio of the area of the endothelial GCX to the length of the capillary inside diameter.
Endothelial vesicles were manually counted in six capillary cross‐sectional profiles at each time point after t‐MCAO. Vesicle counts from six capillaries were averaged within each animal to obtain a single data point per animal. Data were expressed as number per plasma membrane length (μm) for “free” cytoplasmic vesicles or HRP‐filled vesicles.
2.6. Two‐photon in vivo imaging
Fluorescein isothiocyanate‐labeled WGA (FITC‐WGA) lectin (6.25 mg/kg body weight, L4895, Sigma‐Aldrich, USA) was used to tag the GCX in vivo via tail vein injection and circulated for 30 min before imaging. Shortly before the two‐photon imaging, 70 kDa dextran (Rhodamine B, 1% in sterile saline, Sigma‐Aldrich, USA) was administered via the tail vein to label the blood plasma. The cortex was imaged using a two‐photon microscope (FV1200MPE; Olympus, Tokyo, Japan) coupled to a femtosecond pulsed tunable Mai‐Tai eHPDS laser (Spectraphysics, Newport, USA) set to 800 nm, with two fluorescence detection channels and water immersion objectives.
2.7. BBB permeability assay
Evans blue (EB, 2% in saline, 4 ml/kg, Sigma‐Aldrich, USA) dye was injected via the tail vein and allowed to circulate for 1 h. Then, the mice were perfused with saline to remove the residual dye from the vessels until the drainage became colorless. The brains were removed, separated into hemispheres, and weighed. Then, the hemispheres were placed in methanamide (Macklin, Shanghai, China) and soaked at 60°C overnight. The EB content of each hemisphere was calculated based on optical density using a microplate reader (SpectraMax M5, Molecular Devices, Sunnyvale) and expressed as μg per gram of brain.
2.8. Determination of brain edema formation
At 24 h after t‐MCAO, brain samples were weighed and dried in an oven at 60°C for 7 days and weighed to determine the dry weight. Brain water content (%) was calculated as (wet weight‐dry weight)/wet weight × 100%.
Fifteen contiguous coronal images of each mouse were collected for cranial magnetic resonance imaging (MRI). Edema formation was visualized from T2‐weighted images and diffusion‐weighted images (DWI) and semiquantified as midline‐shift (MLS) and relative ADC value (rADC) on restructured ADC images, respectively (26). MLS was calculated using the following formula: MLS = (A−B)/2, where A and B represent the distance between the cortex outer border of the ipsilateral side and contralateral side and the middle of the third ventricle, respectively. rADC% was expressed as (ADCips/ADCcon) × 100%, where the corresponding ADC value was measured in the ipsilateral ischemic and contralateral symmetric regions. The above data were calculated by a researcher blinded to group allocation.
2.9. TTC staining
TTC staining was performed as described previously (26). Briefly, the mice were sacrificed 24 h after the onset of reperfusion. Seven consecutive coronal sections were cut from the frontal pole with a brain matrix and incubated with 1% 2, 3, 5‐triphenyltetrazolium chloride (TTC, Sigma‐Aldrich) at 37°C for 10 min. Corrected infarct volume (%) was analyzed using ImageJ software by a researcher blinded to group assignment and quantified as follows: corrected infarct volume (%) = [contralateral hemisphere volume − (ipsilateral hemisphere volume − infarct volume)]/contralateral hemisphere volume × 100%.
2.10. Immunostaining and confocal microscopy
Mice were anesthetized with isoflurane and then transcardially perfused with phosphate‐buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. The brains were removed, postfixed in 4% PFA for 1 h at room temperature, and submerged in 20% and 30% sucrose for gradient hydration. The brains were cryopreserved in OCT and sectioned using a cryostat (Leica CM1800, Leica Microsystems GmbH, Heidelberg, Germany). Following IgG staining, the sections were incubated with biotinylated anti‐mouse IgG antibody (1:200, Proteintech) overnight at 4°C and then color‐developed using DAB solution. If not stated otherwise, the images were acquired at room temperature using a Zeiss LSM880 confocal microscope (Carl Zeiss, Jena, Germany).
2.11. Western blotting
Brain tissues from 2 to 4 mm posterior to the frontal pole were solubilized and immunoblotted as previously reported (28). In brief, protein extracts were separated on sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) gels and then transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% bovine serum albumin in Tris‐buffered saline with 0.1% Tween‐20 (TBST), and then incubated overnight at 4°C with the following primary antibodies: anti‐Cav1 (1:1000, Abcam), anti‐phospho‐Cav1 (1:1000, Cell Signaling Technology), anti‐syndecan1 (1:600, Proteintech), anti‐PY102 (1:1000, Cell Signaling Technology), and anti‐GAPDH (1:10,000, Proteintech). After washing with TBST, the blots were incubated with HRP‐conjugated secondary antibodies (1:20,000; Abcam) and were then developed by incubation with Luminata Crescendo Western HRP substrate (Millipore). Protein bands were imaged, and their densities were quantified using ImageJ after normalization to GAPDH.
2.12. Knockdown of Cav1 with shRNA
For the Cav1 gene, four different shRNAs were tested. Their ability to silence gene expression was determined using reverse transcription‐polymerase chain reaction (RT‐PCR) and immunoblotting. The two most potent shRNAs, including 5′CCGGCGACGTGGTCAAGATTGACTTCTCGAGAAGTCAATCTTGACCACGTCGTTTTTT3′ (shRNA1) and 5′CCGGGCAAGATATTCAGCAACATCCCTCGAGGGATGTTGCTGAATATCTTGCTTTTTT3′ (shRNA2), were selected to knockdown the expression of Cav1. Adeno‐associated virus 9 (AAV9) vectors were prepared by Shengbo Biotechnology (Shanghai, China), which encodes the following shRNAs: AAV‐Control‐shRNA, AAV‐Cav1‐shRNA1, and AAV‐Cav1‐shRNA2. To test the efficiency of Cav1 shRNAs, six‐week‐old mice received intracerebroventricular injections of control virus (AAV9‐mCherry, 2 µl of 1013vg/ml) or AAV9‐mCherry‐Cav1. Three weeks after injection, the distribution of AAV9‐mCherry in brain sections was observed under a confocal microscope, and the knockdown was detected by RT‐PCR and western blot analysis.
2.13. GST pull‐down assay
GST fusion proteins, His‐tagged fusion proteins, and GST empty vector were transformed into Escherichia coli BL21 cells for culture. After washing glutathione Sepharose 4B beads (GE Healthcare) twice with 1 ml of lysis buffer, and GST or GST fusion proteins were incubated with His‐tagged fusion proteins overnight at 4°C with constant shaking. The following four groups were achieved: GST + His‐Src, GST‐syndecan1 + His‐Src, GST + His‐Src ∆SH2 , and GST‐syndecan1 + His‐Src ∆SH2 . After washing, the complexes were eluted with freshly reduced glutathione hormone for 10 min. Pull‐down products were separated on 10% SDS‐PAGE gels and then analyzed by western blotting using anti‐GST and anti‐His antibodies.
2.14. Coimmunoprecipitation assay
After centrifugation at 10,000 g for 5 min at 4°C, the supernatant was obtained and quantified. An aliquot containing 500 μg protein was immunoprecipitated with 1 μg rabbit anti‐syndecan1 (Proteintech) at 4°C overnight. The immune complexes were collected and incubated with 30 μl of protein A/G magnetic beads (Bimake, Houston, USA) for 1 h at room temperature, followed by magnetic separation. After washing, the beads were resuspended in 35 μl of 1× loading buffer and boiled for 5 min. The protein complexes were immunoblotted as described above using rabbit anti‐Src (Cell Signaling Technology) and rabbit anti‐syndecan1 (Proteintech). Whole tissue lysate prepared for IP was used as an input, and homophytic IgG was used as the negative control.
2.15. Statistical analysis
All data are expressed as the mean ± standard deviation (SD). Statistical differences among multiple groups were compared using one‐way analysis of variance followed by Tukey's multiple comparisons test. The differences between the two groups were analyzed using unpaired t‐tests. Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad, La Jolla, USA) and SPSS 20.0 (IBM, Armonk, United States). Statistical significance was set at p < 0.05.
3. RESULTS
3.1. Dynamic changes of endothelial GCX in brain capillary within the first week after t‐MCAO
Initial experiments aimed to characterize the dynamic changes in the GCX during stroke progression and its time course by measuring the serous levels of two soluble components of the GCX, hyaluronan, and syndecan1, and monitoring the morphological changes in the GCX under EM at different time points after reperfusion onset. As shown in Figure 1A, serum hyaluronan started to increase at 1 h after ischemic stroke and reached the first peak level at 6 h with an approximately significant 4‐fold increase compared to the sham control group (1926 ± 119.5 ng/ml vs. 546.8 ± 77.58 ng/ml, p < 0.05), then decreased to 654.2 ± 146.4 ng/ml at 1 day and peaked again at 7 days with 1459 ± 324.7 ng/ml. Similarly, serum syndeacn1 peaked at 6 h and 7 days with approximately 7‐fold (14.51 ± 3.96 ng/ml) and 10‐fold (24 ± 3.28 ng/ml) increase compared to that of the sham group (2.32 ± 0.27 ng/ml), respectively (Figure 1B).
FIGURE 1.
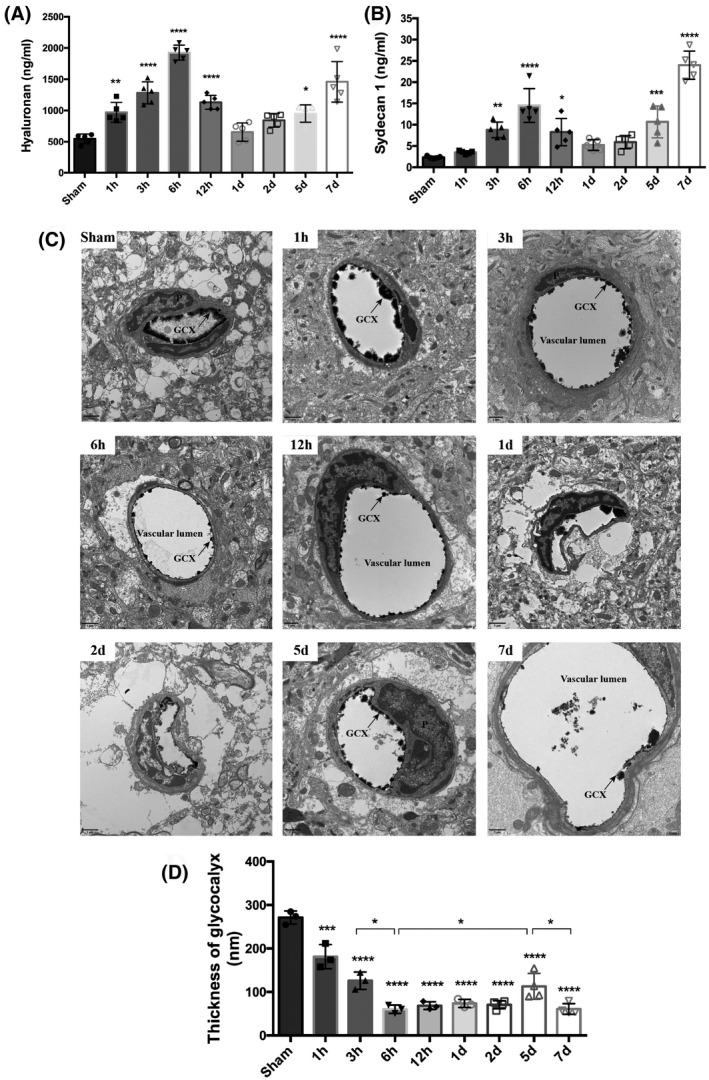
Evolutionary change pattern of endothelial GCX responded to ischemia‐reperfusion injury within the first week. Measurements of constitutional parts of the GCX, hyaluronan (A) and syndecan1 (B), in plasma at a series of discrete time points after t‐MCAO. (C) Transmission electron microscopic views of the cerebral capillaries endothelial GCX with lanthanum nitrate staining at different time points after t‐MCAO. Scale bar, 1 μm. The endothelial GCX densely covered the surface of vascular endothelium in sham group, while appeared to be sparse and thinner at different time points after ischemia, and obvious perivascular edema was visualized at 6 h, 1, 2 and 5 days. (D) Quantitative analysis of the thickness of endothelial GCX of cerebral capillaries. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 vs. sham group
Morphologically, we used EM to observe the changes in the GCX of brain capillaries in the ischemic cortex at 1, 3, 6, 12 h, 1, 2, 5, and 7 days after reperfusion. The overall view of the GCX is depicted in Figure 1C. The endothelial GCX densely covered the surface of the vascular endothelium in the sham‐operated group. At different time points after ischemia and reperfusion, the GCX appeared to be sparse and thinner, showing varying degrees of damage that progressively aggravated in a time‐dependent manner within the first 6 h. Quantitative analysis indicated that, compared with the sham group (with an average thickness of 271.1 ± 15.22 nm), the GCX thickness decreased significantly at 6 h and reached the lowest value (60.26 ± 9.61 nm), which was consistent with the first peak level of soluble serous GCX components. Next, although gradually increased from 2 to 5 days, the GCX thickness declined again at 7 days (60.91 ± 12.41 nm), consistent with the second peak level of the soluble serum GCX. Furthermore, apparent perivascular edema was visualized at 6 h, 1, 2, and 5 days after reperfusion.
These data clearly showed a biphasic change of the brain capillary GCX following t‐MCAO in which GCX degradation occurred within the first 6 h after reperfusion onset, followed by a brief reconstruction and then a second round of degradation at 7 days.
3.2. Biphasic pattern of BBB damage within the first week after t‐MCAO
We next studied the dynamics of BBB leakage during stroke progression through in vivo two‐photon microscopy, using two different tracers: lectin wheat germ agglutinin (WGA, 40 kDa) to visualize the GCX and rhodamine B‐labeled dextran (70 kDa) to mark the blood vessels. At around 4 h after reperfusion onset, the mice were placed under a two‐photon microscope to directly visualize BBB leakage in the same field. As shown, both 40 kDa WGA and 70 kDa dextran leakage exhibited a gradual increase around the ischemic microvessels from 4 h and 20 min to 5 h and 50 min after reperfusion onset within the ischemic cortical area, but no leakage of either tracer was observed from the contralateral side (Figure 2A, Movies [Link], [Link], [Link], [Link]). Notably, the diffuse increase in extravascular 40 kDa tracer leakage was strongest near 6 h.
FIGURE 2.
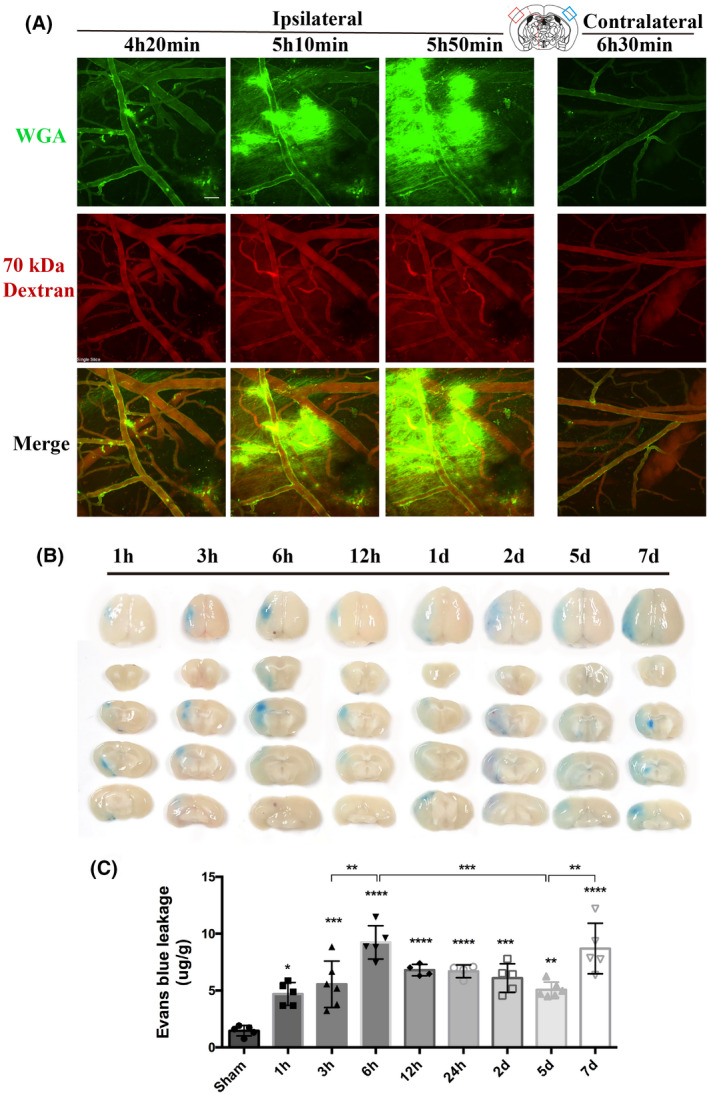
Dynamic evolution pattern of BBB permeability within the first week following cerebral ischemia/reperfusion onset. (A) Representative live two‐photon imaging within the cortical infarct region over time after t‐MCAO. The tracer leakage from blood vessels into the CNS parenchyma was visualized 30min after tail vein injection of lectin wheat germ agglutinin (WGA, green background) and 70 kDa dextran (red background). Scale bar, 50 μm. (B) Representative images showing EB extravasation over time after t‐ MCAO. A column of images represents different brain slices of the same mouse. (C) Quantitative analysis of EB. Differences between operated and sham‐operated controls were significant at the *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 levels
BBB leakage was further assessed at longer time points, up to 7 days, based on EB extravasation. Qualitative macroscopic evaluation showed varying intensities of the characteristic blue coloration, which was strongest at 6 h and 7 days after reperfusion (Figure 2B), while dye extravasation was not observed in the non‐ischemic hemisphere of all t‐MCAO mice. Quantitative analysis showed that, compared with sham mice, EB leakage was consistently detected in the ischemic hemispheric tissue of t‐MCAO mice from 1 h to 7 days after reperfusion onset, with two peaks of EB leakage that appeared at 6 h and 7 days after reperfusion onset, respectively (Figure 2C). These data indicated that BBB leakage occurred continuously within 7 d after reperfusion onset under our experimental conditions, with a biphasic pattern: the first increasing period from 1 to 6 h, followed by a decreasing period from 6 h to 5 d and a second increasing period from 5 to 7 days, consistent with previous MRI findings (7, 8).
These data, combined with our observations of evolutionary changes of the endothelial GCX, indicated that the dynamic changes in the GCX correlated well with the temporal evolution of BBB breakdown following t‐MCAO, implicating an essential role of GCX degradation in BBB disruption within the first week after ischemic stroke.
3.3. Dynamic changes in endothelial transcytosis and TJ ultrastructure within first week after t‐MCAO
Next, we investigated whether TJs or transcytosis of CNS endothelial cells contributes to the biphasic evolution of BBB permeability after t‐MCAO. Therefore, we first examined the morphological changes of TJs in the ischemic region under EM using lanthanum nitrate as an electron‐dense marker at a series of reperfusion time points following t‐MCAO. Notably, in the peripheral enterocytes, EM analysis revealed disrupted TJs with paracellular leakage of lanthanum nitrate, which penetrated between two adjacent enterocytes and filled the whole paracellular space (29). As shown in Figure 3, in sham‐operated mice, thick and intact GCX was observed on the surface of the endothelium of the brain capillary (dark arrow). Between two adjacent endothelial cells, electron‐dense “kissing points” (green arrows) formed by TJs were clearly observed. In the t‐MCAO group, the endothelial GCX became sparse and thin at 1, 3, 6, 12 h, 1, 2, 5, and 7 days after reperfusion onset. Surprisingly, electron‐dense TJs (green arrows) between adjacent ECs were clearly observed at each reperfusion time point. Moreover, the electron‐dense marker lanthanum remained intraluminally and was halted sharply at the “kissing points,” where the TJs were positioned. These results indicated that the TJs of the ischemic BBB at these time points remained structurally and functionally intact and thus unlikely to contribute to BBB leakage under our experimental conditions.
FIGURE 3.
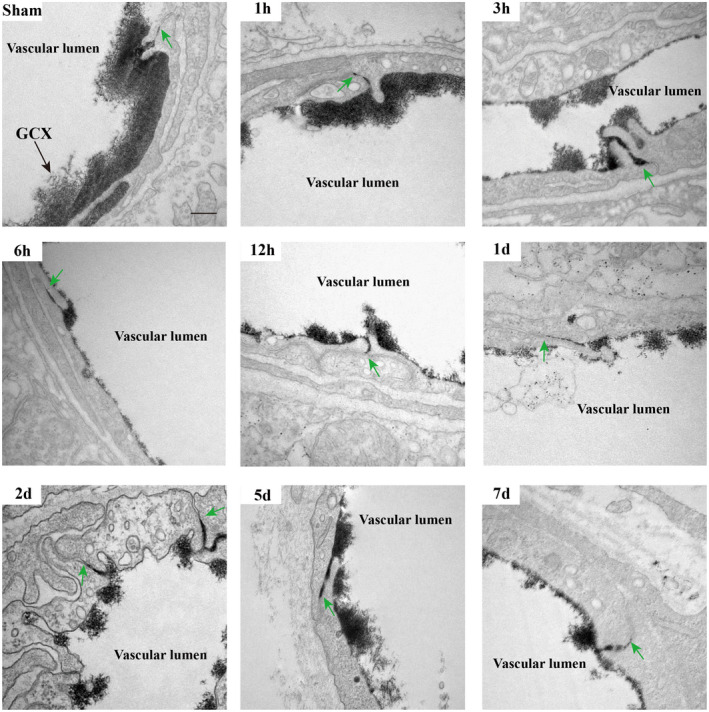
Continuous appearance of functional TJs structure throughout the period of BBB breakdown induced by t‐MCAO. At different time points after t‐MCAO, endothelial TJs showed no apparent ultrastructural defects and remained sealed, as assessed by electron‐dense lanthanum (black) diffused into intercellular clefts but stopped sharply at the junction (green arrows). Scale bar, 200 nm
We then observed vesicular transcytosis at these various time points following t‐MCAO. As shown in Figure 4A, the brain capillary ECs of the sham animals displayed very few lanthanum‐filled vesicles. In contrast, in t‐MCAO mice, a significantly increased number of free vesicles (blue arrows) and lanthanum‐containing vesicles (red arrows) were found in the ischemic brain capillary ECs at all selected reperfusion time points. We quantified the number of vesicles and found that t‐MCAO‐induced increase in vesicles also showed a biphasic pattern during the 7‐day reperfusion, with the two peaks occurring at 6 h and 7 days after reperfusion onset (Figure 4B). Taken together, our data provided evidence that the time course of vesicle change synchronized well with the change in BBB permeability, implying that vesicle‐mediated transcytosis might account for the increased BBB permeability under our experimental conditions.
FIGURE 4.
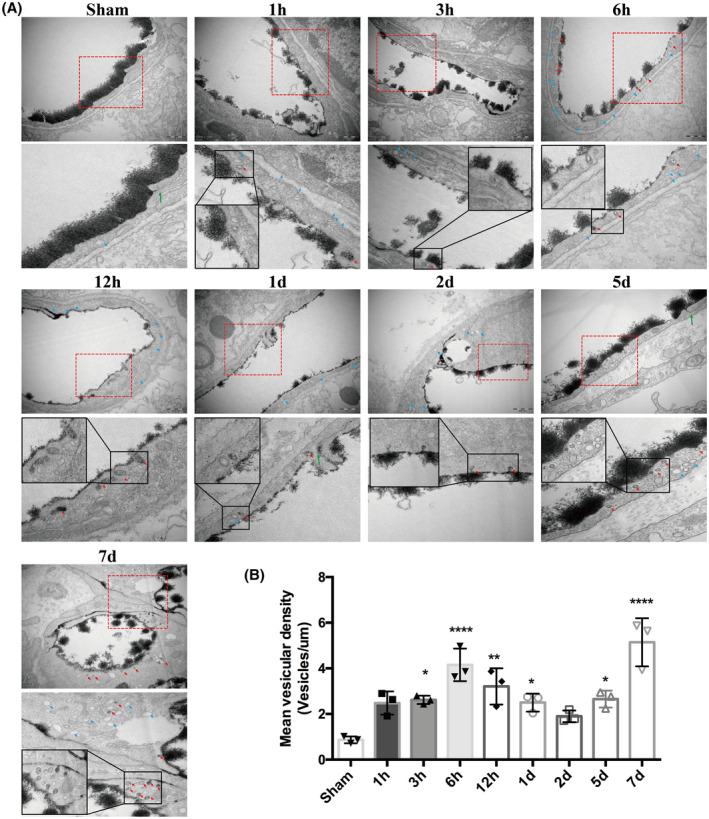
Active and abundant vesicular activity in cortical endothelium after ischemia and reperfusion. (A) Increased free vesicles (blue arrows) and lanthanum‐containing vesicles (red arrows) in endothelial cells appeared continuously during the whole course of BBB impairment. Sham endothelium displayed very few vesicles. Upper panel showed EM images (Scale bar, 500 nm) of individual capillary at different time points after reperfusion and the lower panel showed high magnification images of the areas boxed in upper panel, respectively. (B) Free vesicular density quantification. *p < 0.05, **p < 0.01 and ****p < 0.0001 vs. sham group
3.4. Extent of GCX degradation was positively correlated with endothelial transcytosis and BBB disruption following t‐MCAO
Based on the well‐matched timescale between GCX injury and BBB permeability changes in the ischemic brain, we speculated that the dynamic degradation and reconstruction of the GCX might account for the biphasic disruption of BBB permeability following t‐MCAO. To test this hypothesis, we pharmacologically potentiated or protected against degradation of the endothelial GCX in t‐MCAO mice using HAase or hydrocortisone, respectively, and assessed their impact on the t‐MCAO‐induced increase in BBB permeability.
Serum samples collected at 6 h after t‐MCAO were used to assess the shedding of hyaluronan and syndecan1, which reflected the extent of GCX degradation. Results showed that HAase significantly increased the shedding of hyaluronan to 1903 ± 118.1 ng/ml compared with the 6 h‐vehicle group (1277 ± 560.3 ng/ml, p < 0.05), while hydrocortisone decreased the amount of hyaluronan to 854.5 ± 296.2 ng/ml (Figure 5A). Similarly, HAase treatment induced an approximately 2‐fold increase in syndecan1 (45.26 ± 10.50 ng/ml, Figure 5B) compared with the 6 h‐vehicle group (19.79 ± 7.33 ng/ml, p < 0.05), while hydrocortisone remarkably lowered the release of syndecan1 to 7.72 ± 2.03 ng/ml (vs. 6 h group, p < 0.05).
FIGURE 5.
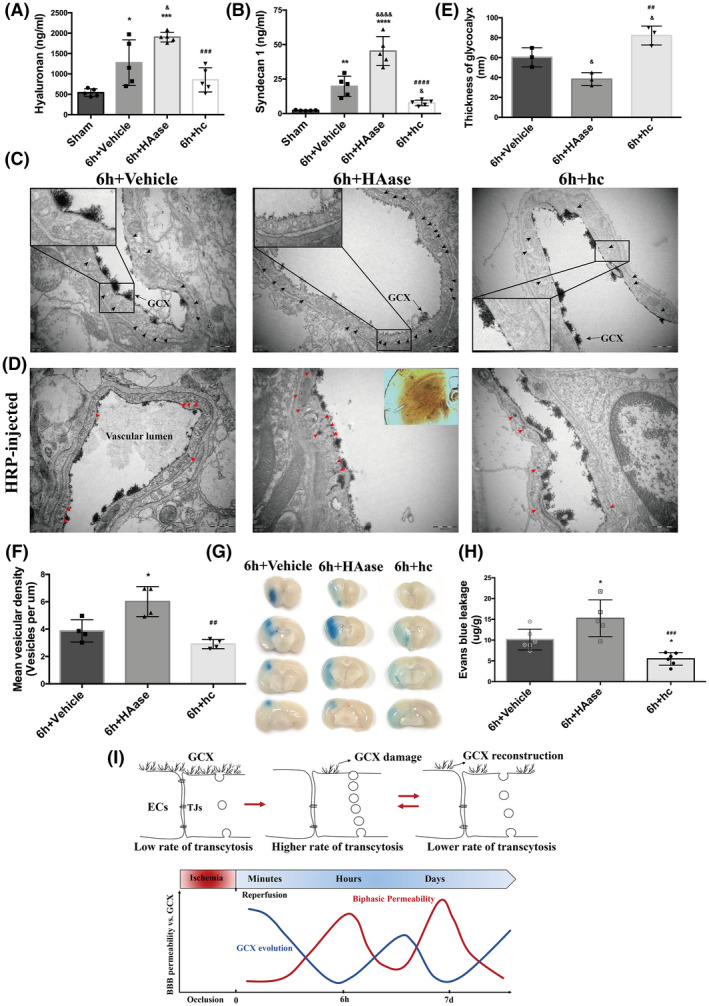
Further degradation of GCX activated more endothelial transcytosis and increased the BBB permeability after t‐MACO. (A, B) Serum levels of hyaluronan and syndecan1were measured in different treatment groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. the sham group; & p < 0.05, &&&& p < 0.0001 vs. the 6h‐vehicle group; ## p < 0.01, ### p < 0.001, #### p < 0.0001 vs. the 6h+HAase group. (C) Representative EM images at 6h after t‐MCAO revealed increased number of vesicles in HAase group. Scale bar, 500 nm. (D) Representative EM images of mice injected with HRP tracer illustrating increased HRP‐filled vesicles (red arrows). ECs of HAase‐treated mice at 6h after t‐MCAO possessed more HRP filled vesicles. Scale bar, 500 nm. (E) Quantification of lanthanum‐stained endothelial GCX of the cerebral capillaries in ischemic cortex at 6h after t‐MCAO. &p < 0.05 vs. the 6h‐vehicle group; ## p < 0.01 vs. the 6h+HAase group. (F) Quantification of endothelial vesicles. *p < 0.05 vs. the 6h group; ## p < 0.01 vs. the 6h+HAase group. (G, H) Representative images and quantitative analysis of EB extravasation from experimental groups. *p < 0.05 vs. the 6h‐vehicle group; ### p < 0.001 vs. the 6h+HAase group. (I) Schematic diagram of BBB biphasic events and GCX degradation and recovery associated with cerebral ischemia and reperfusion time‐course. Endothelial GCX displayed an evolutionary change pattern, whose timescale unexpectedly matched well to the biphasic evolution of BBB permeability to tracers, accompanying by increased number of endothelial vesicles and accelerated rates of transcytosis, during the first‐week mornitoring‐phase after t‐MCAO
Additionally, we observed ultrastructural changes in the GCX under EM. As depicted in Figure 5C, compared to the GCX coverage on the cerebral capillary luminal surface in the mice subjected to t‐MCAO with 6 h reperfusion, HAase treatment led to a further reduction of endothelial GCX coverage, while hydrocortisone treatment partly prevented the t‐MCAO‐induced GCX degradation. Quantitative analysis showed that HAase treatment reduced the average thickness of the cerebral capillary endothelial GCX from 60.26 ± 9.61 nm at 6 h after reperfusion onset to 38 ± 6.50 nm (p < 0.05), while in hydrocortisone‐treated animals, the GCX thickness was returned to 82.21 ± 9.49 nm (p < 0.05 vs. 6 h). Together, the above results demonstrated that the GCX structure was severely degraded after t‐MCAO, and this disruption could be exacerbated by HAase or protected by hydrocortisone.
Importantly, we observed that ECs in the HAase‐treated group displayed more intensive free vesicles (black arrows, Figure 5C), while the hydrocortisone‐treated group showed less. We further examined the transport function of these increased vesicles following the intravascular HRP injection. Results showed that degradation of the GCX by HAase exacerbated the t‐MCAO‐induced trafficking of HRP‐filled vesicles (red arrows, Figure 5D) in ECs of the brain capillary, while preservation of the GCX by hydrocortisone treatment significantly suppressed the vesicle‐mediated transcellular transportation induced by t‐MCAO. These data suggest that further degradation of the GCX activates more endothelial transcytosis after t‐MCAO.
To further establish a causal link between GCX degradation and BBB disruption following t‐MCAO, we assessed the effect of HAase and hydrocortisone on t‐MCAO‐induced EB extravasation in the ischemic brain tissue. As shown in Figure 5G, HAase treatment augmented EB leakage in t‐MCAO mice after 6 h of reperfusion, while hydrocortisone treatment reduced t‐MCAO‐induced EB leakage. Taken together, these data indicate that GCX degradation critically contributes to BBB disruption after t‐MCAO, possibly by promoting vesicle‐mediated transcellular transportation across the BBB.
3.5. GCX degradation aggravated infarct volume and brain edema after t‐MCAO
BBB destruction can cause vasogenic brain edema and exacerbate cerebral infarction (30). Thus, we investigated whether GCX degradation leads to increased infarct volume and brain edema following t‐MCAO. As stated above, we used HAase to potentiate or hydrocortisone to protect against t‐MCAO‐induced GCX degradation. Infarction volume and brain edema were assessed in t‐MCAO mice with 24 h reperfusion by TTC staining, MRI scanning, and brain water contents. As shown in Figure 6, TTC‐staining and MRI scanning (T2WI and ADC map) clearly showed that HAase treatment substantially increased t‐MCAO‐induced infarct volume, while hydrocortisone treatment significantly reduced infarction size. Similar effects were observed for HAase and hydrocortisone on t‐MCAO‐induced brain edema in which degradation of the GCX by HAase significantly aggravated MLS and increased brain water contents (85.95 ± 0.49%) compared with that of the t‐MCAO group (83.08 ± 0.31%) (p < 0.05), while preservation of the GCX by hydrocortisone significantly reduced MLS and decreased the brain water content to 81.79 ± 0.92% when compared with the t‐MCAO group (p < 0.05). These data indicate that the extent of GCX degradation in ischemic cerebral capillaries is closely associated with infarction size and brain edema severity following t‐MCAO.
FIGURE 6.
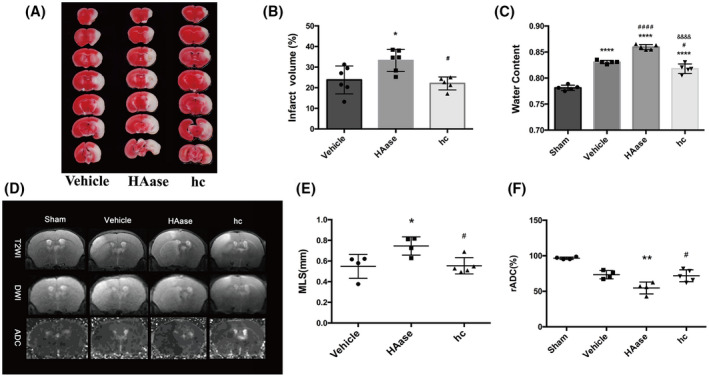
Further degradation of GCX aggravated brain edema after t‐MACO. (A, B) Representative images and quantification of TTC‐ stained brain slices from each group. *p < 0.05 vs. the 6h‐vehicle group; # p < 0.05 vs. the 6h+HAase group. (C) Quantitative analysis of brain parenchymal water content in different groups. ****p < 0.0001 vs. the sham group; # p < 0.05, #### p < 0.0001 vs. the 6h‐vehicle group; &&&& p < 0.0001 vs. the 6h‐HAase group. (D) Representative T2‐weighted images of the tenth slice from each mouse in different groups. ADC images were re‐structured on DWI. Normal signal presented in the left undamaged hemisphere. (E, F) Quantification of edema formation based on MRI images. *p < 0.05, **p < 0.01 vs. the vehicle group; #p < 0.05 vs. the 6h‐HAase group
3.6. GCX degradation triggered endothelial transcytosis and increased BBB permeability under physiological conditions
We then wondered whether GCX degradation would still increase BBB permeability through endothelial transcytosis under physiological conditions. First, we examined the impact of HAase or HPase on vesicular trafficking of cerebral capillary endothelial cells in healthy control mice using EM. Six hours after the administration of HAase or HPase, the mice were sacrificed for vesicular trafficking assessment. Surprisingly, as shown in Figure 7A, only a few vesicles were found in the cerebral capillary endothelial cells of control mice, while degradation of the GCX by HAase or HPase led to a remarkable increase in the number of free vesicles (black arrows) in cerebral capillary ECs. Increased vesicles containing shedding GCX components (black arrowheads) were also observed in ECs treated with HAase or HPase. These results indicate that brain capillary endothelial cells can rapidly respond to enzymatic degradation of the GCX by increasing the number of vesicles.
FIGURE 7.
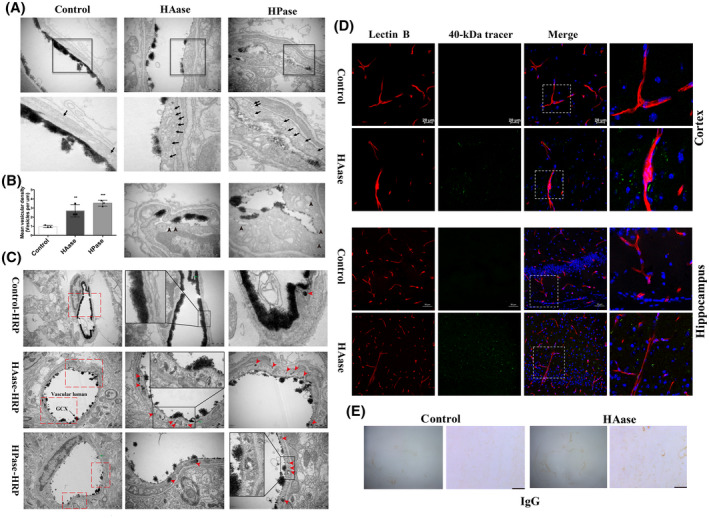
Endothelium displayed increased vesicular activity and evident transcytosis at 6h after enzymatic degradation of GCX under physiological condition. (A) Representative EM images were obtained after GCX was degraded by two enzymes (HAase and HPase) for 6 h. The lower panel showed high magnification images of the areas boxed in upper panel illustrating increased free vesicles (Black arrows). Scale bar, 500 nm. In addition, the EM images (the bottom panel, scale bar, 200 nm) in the HAase and HPase groups showed increased vesicles containing shedding GCX component (black arrowheads), indicating increased endothelial transcytosis. (B) Free vesicular density quantification revealed a significant increase in endothelial vesicle number after either HAase‐ or HPase‐induced GCX degradation. **p < 0.01, ***p < 0.001 vs. control group. (C) Increased transcytosis was evident in HRP‐injected HAase‐ or HPase‐treated mice. Right panel showed high magnification images of the areas boxed in left panel. Red arrows indicate single HRP‐filled vesicles (red arrows) either budding from the luminal membrane or traveling through the endothelial cytoplasm. ECs displayed functional TJs structure (green arrows). Scale bar, 1 μm on the left panel and 500 nm on the right panel. HAase, hyaluronidase; HPase, heparinase. (D) Histological analysis of 40 kDa FITC‐Dextran (green) leakage from isolectin‐B4 (IB4)‐ stained blood vessels (red) at 6 h after GCX degrasation induced by HAase. Upper and lower histological panels displayed BBB leakage in the cortex (Scale bar, 20 μm) and hippocampus (Scale bar, 50 μm), respectively. (E) IgG in the whole brain was visualized by peroxidase‐based immunohistochemistry. There was a macroscopically visible leakage of IgG in HAase group. Right histological panels displayed high magnifications. Scale bar, 100 μm
Second, we evaluated the transcellular transport function of these vesicles by intravenous administration of HRP to HAase or HPase‐treated mice. The mice were sacrificed to assess vesicle trafficking at 1 h after HRP injection. As shown in Figure 7C, ECs of either HAase‐ or HPase‐treated mice were scattered with HRP‐filled vesicles (red arrowheads) but few in control mice. These results demonstrate that enzymatic degradation of the GCX could trigger and enhance vesicle‐mediated endothelial transcytosis of the brain capillary under physiological conditions.
To further demonstrate whether GCX degradation‐induced transcytosis could increase substance transportation across the BBB, we intravenously injected 40 kDa FITC‐conjugated dextran into HAase‐treated mice and monitored its extravasation. We found that in the cortex of HAase‐treated mice, the 40 kDa dextran tracer leaked out of capillaries and was dotted in the nonvascular brain parenchyma (Figure 7D, upper panel). In contrast, the tracer was not detectable in the surrounding brain parenchyma of the control mice. Similarly, in the hippocampus, the tracer sporadically appeared in the brain parenchyma in the HAase‐treated mice but not in the control mice. Our observations of the BBB leakage caused by HAase were consistent with a previous study that HAase induced a significantly higher rate of plasma leakage of both low molecular weight sodium fluorescein and 155 kDa dextran into the surrounding tissue in the blood‐retinal barrier (31). In addition, we visualized IgG in the whole brain by peroxidase‐based immunohistochemistry, which revealed a macroscopically visible leakage of IgG in the HAase group (Figure 7E).
The above data demonstrate that enzymatic degradation of the GCX could promote transcellular transportation to increase BBB permeability in healthy mice, further supporting the GCX as an essential component in maintaining the normal barrier function of the BBB.
3.7. GCX degradation induced BBB disruption via caveolae‐mediated transcytosis
Under EM, the increased vesicles were identified as caveolae vesicles rather than clathrin‐coated vesicles because of the lack of electron‐dense coating. To distinguish between caveolin‐ and clathrin‐dependent mechanisms of GCX degradation‐induced transcytosis, we knocked down Cav1, a crucial membrane‐associated protein involved in caveolae formation, and observed subsequent changes in BBB permeability and brain edema after t‐MCAO. AAV9‐encoded Cav1‐shRNAs and AAV9‐control virus (red fluorescence labeling) were observed in the brain parenchyma under confocal microscopy (Figure 8A). In addition, Cav1 mRNA and protein levels were evidently decreased in brain homogenates of mice injected with AAV9‐Cav1‐shRNAs compared with AAV9‐control (Figure 8B–D), which indicated an effective knockdown of Cav1. Notably, the EB leakage and brain edema induced by t‐MCAO were remarkably lower in Cav1 knockdown mice than in control mice (Figure 8E–I), suggesting that caveolae‐mediated transcellular transport plays an essential role in BBB disruption and brain edema formation.
FIGURE 8.
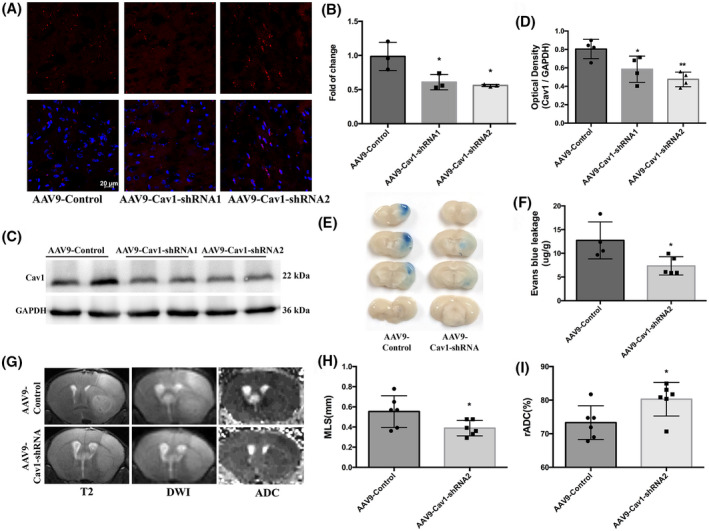
Cav1‐knockdown mice displayed reduced BBB permeability and lighter brain edema after t‐MCAO. (A) Six‐week‐old mice received intracerebroventricular injections of control virus (AAV9‐mCherry, two microliters of 1013 vg/ml) or one that decreased expression of Cav1(AAV9‐mCherry‐Cav1). Three weeks after injection, distribution of AAV9‐mCherry on brain sections were examined under confocal microscope. Red fluorescence was clearly seen in brain parenchyma. Comparison of mRNA levels (B) and protein levels (C, D) in cortical lysate from animals injected with either AAV9‐control or AAV9‐Cav1 showed significant decreases in Cav1 expression. *p < 0.05, **p < 0.0 vs. the AAV9‐control group. (E, F) Representative images and quantitative analysis of and EB extravasation at 6h after t‐MCAO between the AAV9‐control and AAV9‐Cav1 group. (G‐I) Representative T2‐weighted images and comparison of brain edema index in different groups. *p < 0.05, **p < 0.01 vs. the AAV9‐control group
To further determine the Cav1‐dependent mechanism of transcytosis induced by GCX degradation, we assessed 40 kDa FITC‐dextran extravasation in Cav1‐knockdown mice treated with HAase. Compared with control mice, vascular leakage caused by HAase was significantly reduced in Cav1‐knockdown mice (Figure 9A). Moreover, the vesicle‐transport function evaluated by EM showed that Cav1‐knockdown significantly reduced the number of HRP‐filled vesicles in HAase‐treated mice, compared with the control group (Figure 9B,C). These data, together with our observations of intact TJs under EM, suggest that GCX degradation‐induced BBB disruption is dependent on caveolae‐mediated transcytosis rather than TJs.
FIGURE 9.
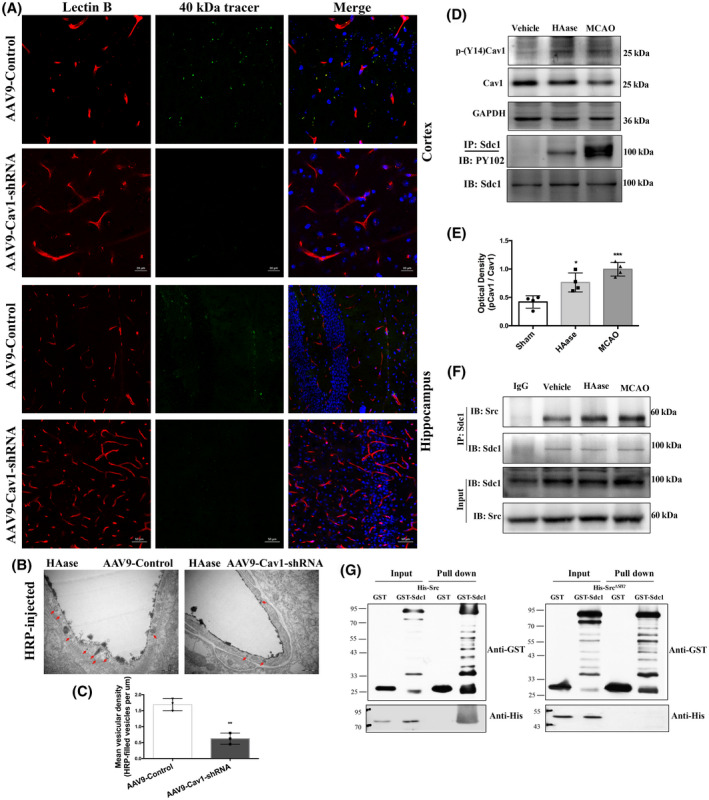
GCX degradation‐induced BBB disruption is Cav1‐dependent. (A) Histological analysis of 40 KD FITCDextran (green) leakage from isolectin‐B4 (IB4)‐stained blood vessels (red) in Cav1 knockdown mice at 6h after GCX degrasation induced by HAase. Upper and lower histological panels displayed BBB leakage in the cortex (Scale bar, 20 μm) and hippocampus (Scale bar, 50 μm), respectively. (B, C) EM evaluation of vesicles‐transport function showed that Cav1‐knockdown significantly reduced the the number of HRP‐filled vesicles in HAase treated mice, compared with the control group. **p < 0.01 vs. the AAV9‐control group. (D, E) Western blot results of p‐Cav1, p‐syndecan1. *p < 0.05 and ***p < 0.001vs. the sham group. (F) The association of syndecan1 and Src detected by immunoprecipitation at 6h after using HAase or t‐MCAO. (G) The direct interaction between syndecan1 and Src detected by GST pull down
3.8. Syndecan1/Src kinase complex is involved in caveolae‐mediated endocytosis promoted by GCX degradation
Caveolae‐mediated transcellular transport has been reported to play an important role in maintaining the integrity of the endothelial barrier in which the tyr14‐phosphorylation of Cav1 induced by Src contributes to caveolae vesicle formation (32, 33, 34). Therefore, we speculated that GCX degradation activated Src‐induced phosphorylation of Cav1, which in turn promoted caveolae‐mediated transcellular transport to increase BBB permeability under normal and stroke conditions. To test this hypothesis, we first examined the effects of GCX degradation induced by HAase or t‐MCAO on the phosphorylation level of Cav1, a substrate of Src kinase. Compared with the control mice, the levels of p‐Cav1 were significantly increased when the GCX was degraded by HAase or ischemic injury (Figure 9D). These results indicate that GCX destruction induces Src‐mediated Cav1 phosphorylation.
Tyrosine phosphorylation within the cytoplasmic domain of syndecan1 by Src kinase has been reported to mediate endocytosis via robust recruitment of cytoskeletal proteins (35). Syndecan1, a critical component of the GCX, is a transmembrane proteoglycan whose ectodomain shedding is accompanied by tyrosine phosphorylation within its cytoplasmic domain in the NMuMG cell model (36). Thus, we examined the tyrosine phosphorylation levels of syndecan1 (Figure 9D, last panel), which was detected in the HAase group and was enhanced after t‐MCAO. The results suggest that GCX degradation results in the shedding of syndecan1 and phosphorylation of the cytoplasmic domain of syndecan1.
Second, we conducted coimmunoprecipitation and GST pull‐down experiments to determine whether GCX degradation promoted the interaction between Src kinase and syndecan1. Our data showed that syndecan1 protein was associated with Src, and this association was enhanced when the GCX was disrupted by HAase (Figure 9F). Furthermore, we examined the structural domain of Src protein that mediated its association with syndecan1 by an in vitro binding assay using full‐length Src protein or Src protein with SH2 domain deletion. As shown in Figure 9G, His‐Src protein was efficiently precipitated by GST‐syndecan1 but not by GST alone, clearly supporting an interaction between syndecan1 and Src protein. Variants of Src carrying a deletion in the SH2 domain (Src ∆SH2 ) could not be precipitated, indicating that the binding sites of syndecan1 and Src were located in the SH2 domain. These data suggest that GCX degradation enhances the interaction between syndecan1 and Src by promoting competitive binding of tyrosine‐phosphorylated syndecan1 to the Src SH2 domain. This association might enhance the efficiency of Src‐mediated syndecan1 phosphorylation, leading to rapid modulation of cytoskeletal proteins and accelerated caveolae‐mediated endocytosis.
4. DISCUSSION
The mechanisms underlying BBB function and cerebrovascular integrity in physiological and pathological conditions are crucial but far from being fully understood. In this study, we found that in response to ischemia‐reperfusion injury within the first week following t‐MCAO onset, the endothelial GCX displayed a characteristic change pattern, that is, degradation, reconstruction, and redegradation, and surprisingly, the time course of changes in the GCX matched well with the biphasic pattern of BBB damage. In addition, we observed the continuous appearance of functional TJ structures between adjacent endothelial cells and active endothelial transcytosis in ischemic cortical capillaries under EM at a series of time points within the first week after t‐MCAO onset, suggesting that vesicle‐mediated transcytosis may account for the leakage of BBB observed at these time points. Moreover, under t‐MCAO conditions, extensive destruction of the GCX aggravated BBB permeability, in conjunction with changes in the corresponding caveolae vesicles. Lastly, we demonstrated that enzymatic degradation of the GCX in the normal BBB led to increased vesicular activity, evident transcytosis, and subsequent BBB leakage, which is Cav1‐dependent, and the activation of the syndecan1/Src complex may be involved in this process. Our results emphasize the significance of the low rate of transcytosis, which is closely related to the integrity of the endothelial GCX, to the BBB function, suggesting that the current understanding of BBB disruption in stroke must be reconsidered.
The endothelial GCX is a hot topic in the peripheral circulation system, and great progress has been made in determining its function and clinical relevance. The GCX, which covers the surface of vascular ECs, participates in a variety of physiological processes, such as microvascular permeability, inflammation, and endothelial mechanical conduction (23). Recently, increasing attention has been paid to the role of the vascular GCX in the CNS. At the level of single cortical capillaries, Kutuzov et al. (37) demonstrated that the GCX may serve as a barrier for large but not small molecules. We have previously shown that GCX degradation contributes to higher BBB permeability, deteriorated brain edema, and worse neurologic outcomes following cardiac arrest (24) or status epilepticus (25). These studies support the essential role of GCX in maintaining BBB integrity. In this study, we assessed the time course of GCX destruction in a mouse model of t‐MCAO and showed a biphasic change in the brain capillary GCX following t‐MCAO in which GCX degradation occurred within the first 6 h after reperfusion onset, followed by a brief reconstruction and then a second round of degradation starting at 7 days. In a previous study, Potter et al. (38) reported that it takes 5 to 7 days for the GCX to endogenously recover to its intrinsic thickness after acute degradation of the GCX in mouse cremaster muscle venules, indicating that an endogenous reconstructing mechanism repairs the disrupted GCX. In this study, we observed a transient reconstruction of the GCX from 2 d to 5 d after reperfusion onset. In a recent clinical study, multiple soluble components of the GCX have been reported to increase in the plasma of patients with mild ischemic stroke on day 7 (39), which is in line with our observations that increased GCX degradation is detected in the BBB of t‐MCAO mice at 7 days after reperfusion onset. Future studies are required to determine the exact mechanism underlying the second round of GCX degradation starting at 7 days. Multiple studies have reported the biphasic BBB opening after ischemic stroke. However, the time points for the second opening were quite inconsistent among these studies, including 72 h (5), 22 and 46 h (40), and 48 h (41) after reperfusion onset. These discrepancies may result from differences in the ischemia‐reperfusion regime, selected time points, monitoring duration, and intervals. Our data show a continuous post‐ischemic BBB leakage to EB from 1 h to 7 days after reperfusion onset with two peaks: the first appeared at 6 h and the second at 7 days, which is in line with previous MRI findings (7, 8).
Previous studies have emphasized the predominant role of the disassembly and reassembly of endothelial TJs in contributing to biphasic changes in ischemia‐induced BBB leakage (3, 11, 14, 16). However, no ultrastructural evidence of TJ breakdown has been provided in these studies. In the current study, we studied the changes in BBB TJ ultrastructure in the ischemic cortex at a series of time points (1, 3, 6, 12 h, 1, 2, 5, and 7 days) after t‐MCAO onset. We found a continuous appearance of functional TJ ultrastructure in the ipsilateral stroke cortex, characterized by an abrupt cessation of tracer at the “kissing points” where the TJs are positioned. These data clearly demonstrate that increased BBB leakage is not because of changes in TJs under our experimental conditions. Surprisingly, we consistently observed significantly increased free vesicles and lanthanum‐containing vesicles in the ECs of ischemic BBB at all the time points, indicating an increased rate of transcytosis. Notably, the t‐MCAO‐induced increase of vesicles in BBB ECs also showed a biphasic pattern during the 7‐day reperfusion, and similar to BBB leakage, the two peaks were also detected at 6 h and 7 days after reperfusion onset. These data indicate that the time course of vesicle change synchronizes well with the increase in BBB permeability, suggesting that under our experimental conditions, activation of previously neglected vesicle‐mediated transcytosis may be the primary cause of BBB permeability increase within the first week of ischemic stroke.
Consistent with our data, an early increase in endothelial caveolae vesicles at 6 h has also been reported to be responsible for the first peak of BBB permeability in a previous study (15). However, unlike our results, in that study, TJ abnormalities, including gaps or protrusions, were observed at later time points (48–58 h after reperfusion) and were proposed to account for the second increase in BBB permeability. This discrepancy may be because of the differences in stroke severity, monitoring time point selection, and TJ structure observation methods. The gaps or protrusions of a small segment of TJs detected under an EM may not mean a real abnormal function of the whole TJs.
In addition to increased transcytosis within the ECs of the ischemic BBB, we observed biphasic loss of the endothelial GCX, which covers the luminal surface of the capillary endothelium following t‐MCAO. Moreover, the dynamic loss of the GCX correlates well with the temporal evolution of BBB breakdown, implicating a significant role of GCX degradation in BBB disruption. In addition, there is good synchronization between the vibration (increase/decrease) of vesicle number and GCX degradation/reconstruction during the 7‐day reperfusion after t‐MCAO. These data strongly suggest that GCX degradation may promote transcellular transport and thus increase BBB permeability. Indeed, we obtained several additional lines of evidence to support our hypothesis.
First, under physiological conditions, enzymatic degradation of the GCX of the normal BBB induces evident endothelial transcytosis and tracer leakage across the BBB, indicating that decreased GCX coverage itself can promote transcellular transportation across the brain capillary endothelium to increase BBB permeability in healthy mice. Surprisingly, the thickness of the cerebral endothelial GCX is much higher than that of the cardiac and pulmonary capillary endothelial GCX (42), yet the number of caveolae vesicles in brain ECs was only one‐tenth of that in peripheral vascular ECs (10). These findings raise an interesting hypothesis that the thick endothelial GCX and low abundance of caveolae vesicles may be an intrinsic structural feature of the brain capillaries (i.e., the BBB) that maintains a very low transcellular transport rate of the BBB and brain homeostasis.
Second, under ischemic stroke conditions, our data show that pharmacologically enhancing or protecting against endothelial GCX degradation in t‐MCAO mice accordingly increases or decreases the number of functional HRP‐filled vesicles, leading to aggravated or alleviated BBB permeability, respectively. Therefore, our data emphasize that transcytosis is the key factor limiting BBB permeability under physiological conditions or stroke conditions in which the integrity of the GCX plays a key role.
From a clinical point of view, the finding that the GCX plays an essential role in the regulation of BBB permeability by regulating BBB transcytosis is of great significance. Brain edema caused by BBB disruption is the main cause of high mortality and disability in patients with various CNS diseases, especially stroke (2). However, current medical treatments focused on osmotic therapy, such as hypertonic saline and mannitol, and surgical interventions, such as decompressive craniectomy, are often ineffective or incompletely understood (43). Our findings suggest that the effective treatment of brain edema can start from the new perspective of BBB transcytosis and targeting the GCX may not only be a new strategy for the prevention and treatment of brain edema but also be helpful for BBB drug delivery in the treatment of some life‐threatening diseases.
It is generally accepted that vasogenic brain edema is directly caused by the leakage of vascular fluid from TJ rupture (2). However, within the first week after t‐MCAO, we detected edema formation but no ultrastructural evidence of TJ destruction. Is the endothelial transcytosis‐mediated BBB breakdown a previously unrecognized contributor to cerebral vasogenic edema? We found that Cav1 knockdown significantly reduced BBB permeability and brain edema induced by t‐MCAO, suggesting that caveolae‐dependent endothelial transcytosis plays an essential role in BBB disruption and secondary vasogenic brain edema. Notably, a recent study has shown that cerebrospinal fluid provides the earliest source of fluid that drives acute ischemic tissue swelling (44). On this basis, we propose that endothelial transcytosis plays a key role in vascular fluid influx, which directly results in brain edema in later stages when the BBB is open after stroke. This finding was further supported by the effect of the GCX degradation‐induced vesicular transcytosis on brain edema formation. Our data showed that aggravating GCX degradation with HAase or ameliorating GCX degradation with hydrocortisone augmented or reduced t‐MCAO‐induced brain edema, supporting a positive correlation between the extent of GCX degradation and brain edema severity. In addition to its association with brain edema, we also showed that the extent of GCX degradation in ischemic cerebral capillaries is closely associated with infarction size. Although we did not further explore how GCX degradation augments brain infarction following t‐MCAO, secondary injuries resulting from brain edema may be an important contribution, such as high intracranial pressure, worsened blood supply, and augmented inflammation.
Src is a nonreceptor protein tyrosine kinase that acts as a critical regulator of the actin cytoskeleton and endothelial permeability. It consists of a kinase domain, an Src‐homology 2 (SH2) domain, and a 3 (SH3) domain (45). Src SH2 domain can be competitively bound by tyrosine phosphorylation molecules, thus promoting various important cellular functions by stimulating Src kinase activation or enhancing downstream signal transduction (45, 46). Our data showed that GCX destruction induced Src‐mediated Cav1 phosphorylation, which contributed to caveolae vesicle formation. Besides, the tyrosine phosphorylation level of syndecan1 increased, and this change was accompanied by the shedding of its ectodomain shed, which was consistent with the results obtained from the in vitro cell model (36). Moreover, we have demonstrated that syndecan1 is intrinsically bound to the SH2 domain of Src, and interestingly, this interaction is enhanced when GCX is destroyed. In a previous study, Src‐mediated phosphorylation of syndecan1 has been shown to recruit cortactin and thus promote efficient actin‐dependent endocytosis (35). Moreover, this enhanced interaction between Src and syndecan1 enhances the efficiency of Cav1 trafficking and syndecan1 phosphorylation, leading to rapid modulation of cytoskeletal proteins and activation of caveolae‐mediated endocytosis. Given the multitude of potential signaling molecules simultaneously released after GCX degradation in vivo, future studies using in vitro cellular models are required to identify the specific signal transduction involved in this process.
There are several limitations to this study. First, Mfsd2a has been shown to regulate BBB integrity in Mfsd2a−/− mice (3, 11), which is accompanied by increased vesicle trafficking; however, the TJs appear to be intact, which is similar to what we observed for GCX damage in our t‐MCAO model. However, we did not examine the possibility that Mfsd2a may regulate CNS endothelial transcytosis by modulating GCX or that Mfsd2a mediates the effect of GCX on endothelial transcytosis. Second, although, consistent with our observation, emerging studies have found no TJ loss, we cannot completely exclude the possibility of TJs being open and closed instantaneously, and we may miss the time points to see these changes. Third, we did not design experiments to determine the specific mechanisms underlying GCX redegradation at 7 days after reperfusion onset.
In conclusion, this study is the first to reveal the essential role of the GCX in suppressing transcellular transport in CNS endothelial cells to maintain BBB integrity. Our data also emphasize that GCX‐regulated transcellular transport across the BBB contributes significantly to the barrier function of the BBB in both normal and diseased conditions. GCX may be an essential target for manipulating BBB permeability to help with drug delivery to the brain or to protect against BBB injury under diseased conditions.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
AUTHOR CONTRIBUTIONS
Juan Zhu and Suyue Pan designed the study. Juan Zhu, Zheqi Li, Kewei Liu, Yuan Chang, and Yuqin Peng performed the experiments and analyzed the data. Zhenzhou Lin and Yihua He bred the mice and performed the experiments. Zhong Ji, Yongming, Shengnan Wang, and Dongmei Wang provided valuable comments and reagents and edited the manuscript. Kaibin Huang and Juan Zhu analyzed the data and wrote the manuscript.
Supporting information
Fig S1 Experimental procedure and flow diagram of the study. (A) Experimental procedures, drug delivery, and measurements after t‐MCAO. (B) Flow diagram of the experiment. t‐MCAO, temporary middle cerebral artery occlusion; EM, Electron Microscope; EB, Evans blue; TTC, 2,3,5‐triphenyltetrazolium chloride; MRI, magnetic resonance imaging; GCX, glycocalyx; BBB, blood brain barrier; HAase, Hyaluronidase; hc, Hydrocortisone; HPase, heparinase; Cav1, caveolin1; HRP, horseradish peroxidase; WT, wild‐type mice; i.v., intravenous injection; i.p., intraperitoneal injection.
Video S1 Live two‐photon imaging within the cortical infarct region at 4 h and 20 min after t‐MCAO
Video S2 Live two‐photon imaging within the cortical infarct region at 5 h and 10 min after t‐MCAO
Video S3 Live two‐photon imaging within the cortical infarct region at 5 h and 50 min after t‐MCAO
Video S4 Live two‐photon imaging of the contralateral side after t‐MCAO.
ACKNOWLEDGMENTS
This work was supported by the National Key R&D Program of China (No. 2017YFC1307500), National Natural Science Foundation of China (No. 81871030 & 82072133), Natural Science Foundation of Guangdong Province (2019A1515011446), and Guangzhou Science and Technology Planning Project (Grant No. 201804010055).
Zhu J, Li Z, Ji Z, Wu Y, He Y, Liu K, et al. Glycocalyx is critical for blood‐brain barrier integrity by suppressing caveolin1‐dependent endothelial transcytosis following ischemic stroke. Brain Pathology. 2022;32:e13006. 10.1111/bpa.13006
Contributor Information
Kaibin Huang, Email: hkb@smu.edu.cn.
Suyue Pan, Email: pansuyue@smu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Daneman R, Prat A. The blood‐brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36(3):513–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andreone BJ, Chow BW, Tata A, Lacoste B, Ben‐Zvi A, Bullock K, et al. Blood‐brain barrier permeability is regulated by lipid transport‐dependent suppression of caveolae‐mediated transcytosis. Neuron. 2017;94(3):581–94.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sandoval KE, Witt KA. Blood‐brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32(2):200–19. [DOI] [PubMed] [Google Scholar]
- 5. Kuroiwa T, Ting P, Martinez H, Klatzo I. The biphasic opening of the blood‐brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 1985;68(2):122–9. [DOI] [PubMed] [Google Scholar]
- 6. Prakash R, Carmichael ST. Blood‐brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol. 2015;28(6):556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, et al. The blood–brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153(1):175–81. [DOI] [PubMed] [Google Scholar]
- 8. Lin C‐Y, Chang C, Cheung W‐M, Lin M‐H, Chen J‐J, Hsu CY, et al. Dynamic changes in vascular permeability, cerebral blood volume, vascular density, and size after transient focal cerebral ischemia in rats: evaluation with contrast‐enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2008;28(8):1491–501. [DOI] [PubMed] [Google Scholar]
- 9. Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, et al. Blood‐brain barrier dysfunction and recovery after ischemic stroke. Prog Neurogibol. 2018;163:144–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ayloo S, Gu C. Transcytosis at the blood‐brain barrier. Curr Opin Neurobiol. 2019;57:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ben‐Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature. 2014;509(7501):507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood‐brain barrier. Nature. 2010;468(7323):557–61. [DOI] [PubMed] [Google Scholar]
- 13. Reeson P, Tennant KA, Gerrow K, Wang J, Weiser Novak S, Thompson K, et al. Delayed inhibition of VEGF signaling after stroke attenuates blood‐brain barrier breakdown and improves functional recovery in a comorbidity‐dependent manner. J Neurosci. 2015;35(13):5128–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krueger M, Härtig W, Reichenbach A, Bechmann I, Michalski D. Blood‐brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS One. 2013;8(2):e56419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knowland D, Arac A, Sekiguchi K, Hsu M, Lutz S, Perrino J, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood‐brain barrier breakdown in stroke. Neuron. 2014;82(3):603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Brown NM, Megason SG, Gu C. Suppression of transcytosis regulates zebrafish blood‐brain barrier function. Elife 2019;8:e47326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haley MJ, Lawrence CB. The blood‐brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab. 2017;37(2):456–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood‐brain barrier breakdown in the peri‐infarct zone in young adult and aged mice. J Cereb Blood Flow Metab. 2016;36(2):413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ito U, Ohno K, Yamaguchi T, Takei H, Tomita H, Inaba Y, et al. Effect of hypertension on blood‐brain barrier. Change after restoration of blood flow in post‐ischemic gerbil brains. An electronmicroscopic study. 1980;Stroke 11(6):606–11. [DOI] [PubMed] [Google Scholar]
- 20. De Bock M, Van Haver V, Vandenbroucke RE, Decrock E, Wang N, Leybaert L, et al. Into rather unexplored terrain‐transcellular transport across the blood‐brain barrier. Glia. 2016;64(7):1097–123. [DOI] [PubMed] [Google Scholar]
- 21. Cipolla MJ, Crete R, Vitullo L, Rix RD. Transcellular transport as a mechanism of blood‐brain barrier disruption during stroke. Front Biosci. 2004;9:777–85. [DOI] [PubMed] [Google Scholar]
- 22. Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, oude Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salmon AH, Satchell SC Endothelial glycocalyx dysfunction in disease: albuminuria and increased microvascular permeability. J Pathol. 2012;226(4):562–74. [DOI] [PubMed] [Google Scholar]
- 24. Zhu J, Li X, Yin J, Hu Y, Gu Y, Pan S, et al. Glycocalyx degradation leads to blood‐brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J Cereb Blood Flow Metab. 2018;38(11):1979–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li X, Zhu J, Liu K, Hu Y, Huang K, Pan S. Heparin ameliorates cerebral edema and improves outcomes following status epilepticus by protecting endothelial glycocalyx in mice. Exp Neurol. 2020;330:113320. [DOI] [PubMed] [Google Scholar]
- 26. Wang X, Chang Y, He Y, Lyu C, Li H, Zhu J, et al. Glimepiride and glibenclamide have comparable efficacy in treating acute ischemic stroke in mice. Neuropharmacology 2020;162:107845. [DOI] [PubMed] [Google Scholar]
- 27. Gao L, Lipowsky HH. Composition of the endothelial glycocalyx and its relation to its thickness and diffusion of small solutes. Microvasc Res. 2010;80(3):394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu J, Liu K, Huang K, Gu Y, Hu Y, Pan S, et al. Metformin improves neurologic outcome via amp‐activated protein kinase‐mediated autophagy activation in a rat model of cardiac arrest and resuscitation. J Am Heart Assoc. 2018;7(12):e008389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schellekens DHSM, Hundscheid IHR, Leenarts CAJI, Grootjans J, Lenaerts K, Buurman WA, et al. Human small intestine is capable of restoring barrier function after short ischemic periods. World J Gastroenterol. 2017;23(48):8452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liebeskind DS, Jüttler E, Shapovalov Y, Yegin A, Landen J, Jauch EC, et al. Cerebral edema associated with large hemispheric infarction. Stroke. 2019;50(9):2619–25. [DOI] [PubMed] [Google Scholar]
- 31. Leskova W, Pickett H, Eshaq RS, Shrestha B, Pattillo CB, Harris NR, et al. Effect of diabetes and hyaluronidase on the retinal endothelial glycocalyx in mice. Exp Eye Res. 2019;179:125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sverdlov M, Shajahan AN, Minshall RD. Tyrosine phosphorylation‐dependence of caveolae‐mediated endocytosis. J Cell Mol Med. 2007;11(6):1239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joshi B, Bastiani M, Strugnell SS, Boscher C, Parton RG, Nabi IR, et al. Phosphocaveolin‐1 is a mechanotransducer that induces caveola biogenesis via Egr1 transcriptional regulation. J Cell Biol. 2012;199(3):425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zimnicka AM, Husain YS, Shajahan AN, Sverdlov M, Chaga O, Chen Z, et al. Src‐dependent phosphorylation of caveolin‐1 Tyr‐14 promotes swelling and release of caveolae. Mol Biol Cell. 2016;27(13):2090–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen K, Williams KJ. Molecular mediators for raft‐dependent endocytosis of syndecan‐1, a highly conserved, multifunctional receptor. J Biol Chem. 2013;288(20):13988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reiland J, Ott VL, Lebakken CS, Yeaman C, McCARTHY J, Rapraeger AC, et al. Pervanadate activation of intracellular kinases leads to tyrosine phosphorylation and shedding of syndecan‐1. Biochem J. 1996;319 (Pt 1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kutuzov N, Flyvbjerg H, Lauritzen M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood–brain barrier. Proc Natl Acad Sci U S A. 2018;115(40):E9429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Potter DR, Jiang J, Damiano ER. The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ Res. 2009;104(11):1318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DellaValle B, Hasseldam H, Johansen FF, Iversen HK, Rungby J, Hempel C, et al. Multiple soluble components of the glycocalyx are increased in patient plasma after ischemic stroke. Stroke. 2019;50(10):2948–51. [DOI] [PubMed] [Google Scholar]
- 40. Huang ZG, Xue D, Karbalai H, Buchan AM, Huang ZG, Xue D, et al. Biphasic opening of the blood‐brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci. 1999;26(4):298–304. [DOI] [PubMed] [Google Scholar]
- 41. Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood‐brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739(1–2):88–96. [DOI] [PubMed] [Google Scholar]
- 42. Ando Y, Okada H, Takemura G, Suzuki K, Takada C, Tomita H, et al. Brain‐specific ultrastructure of capillary endothelial glycocalyx and its possible contribution for blood brain barrier. Sci Rep. 2018;8(1):17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thrane AS, Rangroo Thrane V, Nedergaard M. Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014;37(11):620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mestre H, Du T, Sweeney AM, Liu G, Samson AJ, Peng W, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020;367(6483):eaax7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roskoski R Jr. Src protein‐tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9–25. [DOI] [PubMed] [Google Scholar]
- 46. Shah NH, Amacher JF, Nocka LM, Kuriyan J. The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Crit Rev Biochem Mol Biol. 2018;53(5):535–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1 Experimental procedure and flow diagram of the study. (A) Experimental procedures, drug delivery, and measurements after t‐MCAO. (B) Flow diagram of the experiment. t‐MCAO, temporary middle cerebral artery occlusion; EM, Electron Microscope; EB, Evans blue; TTC, 2,3,5‐triphenyltetrazolium chloride; MRI, magnetic resonance imaging; GCX, glycocalyx; BBB, blood brain barrier; HAase, Hyaluronidase; hc, Hydrocortisone; HPase, heparinase; Cav1, caveolin1; HRP, horseradish peroxidase; WT, wild‐type mice; i.v., intravenous injection; i.p., intraperitoneal injection.
Video S1 Live two‐photon imaging within the cortical infarct region at 4 h and 20 min after t‐MCAO
Video S2 Live two‐photon imaging within the cortical infarct region at 5 h and 10 min after t‐MCAO
Video S3 Live two‐photon imaging within the cortical infarct region at 5 h and 50 min after t‐MCAO
Video S4 Live two‐photon imaging of the contralateral side after t‐MCAO.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.