

Abstract
The striatonigral and olivopontocerebellar systems are known to be vulnerable in multiple system atrophy (MSA), showing neuronal loss, astrogliosis, and alpha‐synuclein‐immunoreactive inclusions. MSA patients who displayed abundant neuronal cytoplasmic inclusions (NCIs) in the regions other than the striatonigral or olivopontocerebellar system have occasionally been diagnosed with variants of MSA. In this study, we report clinical and pathologic findings of MSA patients characterized by prominent pathologic involvement of the hippocampus. We assessed 146 consecutively autopsied MSA patients. Semi‐quantitative analysis of anti‐alpha‐synuclein immunohistochemistry revealed that 12 of 146 patients (8.2%) had severe NCIs in two or more of the following areas: the hippocampal granule cells, cornu ammonis areas, parahippocampal gyrus, and amygdala. In contrast, the remaining 134 patients did not show severe NCIs in any of these regions. Patients with severe hippocampal involvement showed a higher representation of women (nine women/three men; Fisher's exact test, p = 0.0324), longer disease duration (13.1 ± 5.9 years; Mann–Whitney U‐test, p = 0.000157), higher prevalence of cognitive impairment (four patients; Fisher's exact test, p = 0.0222), and lower brain weight (1070.3 ± 168.6 g; Mann–Whitney U‐test, p = 0.00911) than other patients. The hippocampal granule cells and cornu ammonis area 1/subiculum almost always showed severe NCIs. The NCIs appeared to be ring‐shaped or neurofibrillary tangle‐like, fibrous configurations. Three of 12 patients also had dense, round‐shaped NCIs that were morphologically similar to pick bodies. The patients with Pick body‐like inclusions showed more severe atrophy of the medial temporal lobes and broader spreading of NCIs than those without. Immunohistochemistry for hyperphosphorylated tau and phosphorylated TDP‐43 revealed minimal aggregations in the hippocampus of the hippocampal MSA patients. Our observations suggest a pathological variant of MSA that is characterized by severe involvement of hippocampal neurons. This phenotype may reinforce the importance of neuronal alpha‐synucleinopathy in the pathogenesis of MSA.
Keywords: alpha‐synuclein, dementia, hippocampus, multiple system atrophy, neuronal inclusions
This study describes a clinicopathological phenotype of the hippocampal multiple system atrophy (MSA) characterized by abundant alpha‐synuclein‐immunopositive neuronal cytoplasmic inclusions and neuronal loss in the hippocampus with concomitant cognitive impairment in the patients. The phenotype may be a pathological variant of MSA and reinforces the importance of neuronal alpha‐synucleinopathy in the pathogenesis of MSA.
![]()
1. INTRODUCTION
Multiple system atrophy (MSA) is a sporadic, adult‐onset, and progressive neurodegenerative disorder, which manifests as variable combinations of dysautonomia, cerebellar ataxia, parkinsonism, and pyramidal signs (1, 2). The neuropathological diagnosis of MSA is made in the presence of alpha‐synuclein‐immunoreactive glial cytoplasmic inclusions (GCIs) in association with prominent neuronal loss and astrogliosis in the striatonigral (SN) or the olivopontocerebellar (OPC) systems (3, 4). GCIs have been considered to play important roles in the pathogenesis of MSA; it has been reported that GCIs are broadly spread throughout the central nervous system of MSA patients during the early phase of the disease, and the density of the GCIs correlates with the severities of neuronal loss (5, 6). Neuronal aggregations of alpha‐synuclein, neuronal cytoplasmic inclusions (NCIs), and intranuclear inclusions (NNIs) are also neuropathological findings in MSA (7). However, the burden of neuronal inclusions is usually less than that of the GCIs (8).
The SN and OPC systems are known to be vulnerable regions in MSA. However, MSA patients who display abundant neuronal inclusions in the regions other than SN or OPC system have occasionally been reported as pathologic variants (9, 10, 11, 12, 13, 14, 15). These patients sometimes present with cortical symptoms, including frontotemporal dementia (FTD), corticobasal syndrome (CBS), or progressive aphasia (PA), which are not presented in classical cases of MSA (9).
In this study, we identified 12 patients with severe involvement of the hippocampus from 194 consecutively autopsied patients with MSA. These patients showed not only pathologic findings of olivopontocerebellar atrophy (OPCA) or striatonigral degeneration (SND) but also extremely abundant neuronal inclusions, neuronal loss, and atrophy of the medial temporal lobes. We compared the clinical and neuropathologic findings of these patients to those of the patients with classical MSA.
2. MATERIALS AND METHODS
2.1. Patients
We reviewed the clinical and neuropathologic findings of 194 pathologically confirmed patients with MSA, who were consecutively autopsied between 1978 and 2018 at the Institute for Medical Science of Aging, Aichi Medical University. Clinical diagnosis of MSA was made by neurological experts of the Aichi Medical University, Nagoya University, or affiliated hospitals. The clinical data were obtained from medical records and clinicopathological conferences involving the neurological physicians. The ethnicity of all autopsied patients was east Asians. Of the MSA patients, 48 patients were excluded because of insufficient clinical information (n = 13), insufficient tissue material (n = 18), and comorbid pathological changes in the limbic areas (severe ischemic change, n = 14; intracerebral hemorrhage, n = 1; glioblastoma, n = 1; and limbic encephalitis, n = 1). The remaining 146 patients were included in the study.
2.2. Clinical findings
Assessments of clinical findings were retrospective. Disease onset was defined as awareness of either motor or autonomic symptoms that are described in the second consensus criteria of MSA (16). Disease duration was set as years from disease onset to death or to the induction of artificial ventilation. Clinical phenotypes were categorized as MSA with predominant cerebellar ataxia (MSA‐C) and MSA with predominant parkinsonism (MSA‐P). The presence of dementia was defined based on the description of cognitive impairment, including amnesia, behavioral disorders, and aphasia, in the clinical records before the patients acquired a bedridden state. The scores of a Japanese version of the Mini‐Mental State Examination (MMSE) or Revised Hasegawa Dementia Scale (HDS‐R) were also obtained from the medical records. The cut‐off values were set at 23 for MMSE and 21 for HDS‐R (17, 18). The remaining patients were classified as having no cognitive impairment.
2.3. Tissue preparation and immunohistochemical procedures
The tissues were fixed in 20% formalin for at least 2 weeks (19). The right hemisphere was cryopreserved. At first, we coronally cut the left hemisphere at a thickness of 8 mm using a brain knife and standardized slicer. The brainstem was transversely cut, and the cerebellum was sagittally cut at a thickness of 5 mm. Next, the regions of interest were trimmed out for embedding. The regions are systematic in our institute as follows: the primary motor cortex, the isocortex of the frontal, temporal, and parietal cortices, the subcortical white matter of the frontal and temporal lobes, anterior cingulate gyrus, insular cortex, hippocampus, amygdala, the basal ganglia, midbrain, pons, medulla oblongata, cerebellum, spinal cord, and sympathetic ganglion. In addition, whole hemispherical sections without trimming have occasionally been embedded into the paraffin as is. Sections of 9 μm thickness were prepared for hematoxylin‐eosin (H&E), Klüver‐Barrera, and Gallyas‐Braak staining, and those of 4.5 μm thickness were prepared for immunohistochemistry analysis. Primary antibodies used for immunohistochemistry were as follows: anti‐alpha‐synuclein (polyclonal rabbit, 1:1000; Sigma Aldrich, St. Louis, MO), anti‐phosphorylated alpha‐synuclein (pSyn#64, monoclonal mouse, 1:1000; Wako Pure Chemical Industries, Osaka, Japan), anti‐beta‐amyloid (clone 6F/3D; monoclonal mouse, 1:100; Dako, Glostrup, Denmark), anti‐phosphorylated tau (clone AT‐8; monoclonal mouse, 1:1000; Innogenetics, Ghent, Belgium), and anti‐phosphorylated transactive response DNA‐binding protein 43 kDa (pTDP‐43, polyclonal rabbit, 1:5000; Cosmobio, Tokyo, Japan). A standard avidin/biotin technique was employed, and diaminobenzidine was used as a chromogen.
2.4. Neuropathological assessment
Neuropathological assessments were conducted by two neurologists (T.A. and M.Y.). We made pathological diagnosis of MSA based on the presence of alpha‐synuclein‐immunopositivity and positive Gallyas‐Braak staining for GCIs (3). Pathological sub‐classification, as OPCA, SND, or OPCA‐SND‐mixed, was made on the basis of an established three‐grade pathological scale (20, 21). The presence of concomitant Lewy bodies in the substantia nigra, locus coeruleus, or dorsal nucleus of the vagus was assessed using H&E staining (7). Comorbid pathologic changes, including the presence of neurofibrillary tangles (NFTs) (22), amyloid plaques (23), argyrophilic grains (24), and TDP‐43 proteinopathy (25) were evaluated according to established criteria.
2.5. Semi‐quantitative evaluation of pathologic findings
Severities of NCIs and GCIs were semi‐quantitatively scored using anti‐alpha‐synuclein immunohistochemistry. The grades of NCIs were set as follows: grade 0 (none): no NCIs, grade 1+ (mild): 1–9 NCIs, and grade 2+ (severe): 10 or more NCIs (Figure 1). GCIs were scored as follows: grade 0 (none): no GCIs, grade 1+ (mild): 1–19 GCIs, and grade 2+ (severe): 20 or more GCIs. The counts were averaged over five visual fields using a 20× objective. Further, we semi‐quantitatively assessed neuronal loss and astrogliosis using H&E staining as follows: grade 0: normal appearance, grade 1+: definite astrogliosis that is associated with subtle neuronal loss, and grade 2+: apparent neuronal loss and astrogliosis.
FIGURE 1.
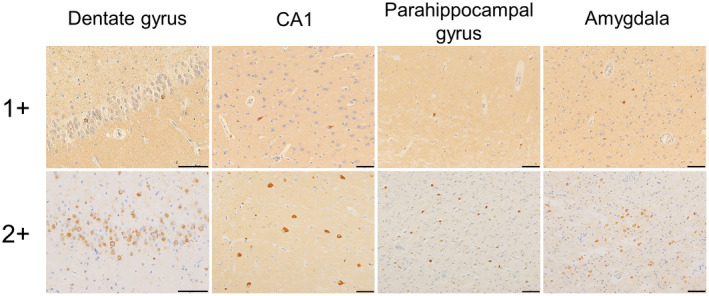
Semiquantitative assessment of NCIs. The scores are as follows: 0 (no NCIs), 1+ (mild to moderate: 1–9 NCIs in a visual field using a ×20 objective), and 2+ (severe, >10 NCIs in the same setting). All scale bars are 50 μm. NCIs, neuronal cytoplasmic inclusions
2.6. Electron microscopy
Formalin‐fixed tissues from the hippocampus of patients 4 and 10 were processed for electron microscopy. Ultrathin sections were cut, fixed, and stained with uranyl acetate and lead citrate and examined using a JEM‐1400 electron microscope (JEOL, Tokyo, Japan).
2.7. Genetic analysis
Frozen brains were available in 59 patients out of the 146 patients that were included in the study. Genomic DNA was extracted from frozen autopsied brain tissues using DNAzol (Thermo Fisher Scientific) according to the manufacturer's instructions. We evaluated variants in APOE, COQ2, GBA, and SNCA using Sanger sequencing as previously described (26, 27). Copy numbers of SNCA gene were evaluated using TaqMan Copy Number Assay (Applied Biosystems, Foster City, CA). If ambiguous results were obtained, we further analyzed the copy number using a custom‐designed array for array comparative genomic hybridization analysis.
2.8. Statistical analyses
Data are expressed as mean ± standard deviation (SD) or range. The Fisher's exact test was used for comparing categorical variables, and the Mann–Whitney U test was used for continuous and ordinal variables. The significance level was set at a p‐value of 0.05. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is based on R and R commander (The R Foundation for Statistical Computing, Vienna, Austria) (28).
3. RESULTS
3.1. Identification of hippocampal MSA patients
Twelve of the 146 patients with MSA (8.2%) showed severe (2+) alpha‐synuclein‐immunopositive NCIs in two or more of the following areas: the hippocampal granule cells, cornu ammonis area (CA) 1/subiculum, CA2‐CA4, parahippocampal gyrus, and amygdala. We classified the 12 patients as the ‘hippocampal MSA’ patients. The other 134 patients (91.8%) did not demonstrate severe (2+) NCIs in any of these regions and were classified as the ‘classical MSA’ patients in this study.
3.2. Clinical findings of hippocampal MSA patients
Tables 1 and 2 summarize the clinical findings of the included patients. A higher proportion of women was observed in the hippocampal MSA group (nine women/three men) than in the classical MSA group (55 women/79 men) (Fisher's exact test, p = 0.0324). Duration of disease was significantly longer in the hippocampal MSA patients (13.1 ± 5.9 years, range: 2–25) than that in the classical MSA patients (6.9 ± 3.6 years, range: 1–19) (Mann–Whitney U‐test, p = 0.000157). Only one patient of the hippocampal MSA (Patient 8) underwent artificial ventilation 14 years after disease onset and died a year later. Cognitive impairment was more prevalent in the hippocampal MSA group than in the classical MSA group (Fisher's exact test, p = 0.0222). Scores of cognitive examinations in the hippocampal MSA patients with cognitive deficits were as follows: HDS‐R was scored as 13 of 30 at 3 years after onset in Patient 2; HDS‐R was scored as 19 of 30 at 7 years after onset in Patient 6; MMSE was scored as 13 of 30 and HDS‐R was as 8 of 30 at 11 years after onset in Patient 7; and HDS‐R was scored as 5 of 30 at 5 years after onset in Patient 9 (Table S1). No patients of the hippocampal MSA presented with FTD, CBS, or PA. Persecutory delusion in Patient 3 and visual hallucination in Patient 6 were noted. There were no significant differences in the proportions of MSA‐C and MSA‐P between the hippocampal MSA and classical MSA groups.
TABLE 1.
Comparison between the hippocampal MSA and classical MSA
| Hippocampal MSA | Classical MSA | p‐value | |
|---|---|---|---|
| n (women/men) | 12 (9/3) | 134 (55/79) | 0.0324 c |
| Age at onset, years, mean ± SD (range) | 56.6 ± 9.3 (44–74) | 60.4 ± 9.3 (33–84) | 0.160 d |
| Duration of illness, years, mean ± SD (range) | 13.1 ± 5.9 (2–25) | 6.9 ± 3.6 (1–19) | 0.000157 d |
| Cognitive impairment, n (%) | 4 (33.3%) | 11 (8.2%) | 0.0222 c |
| Clinical phenotypes of MSA | |||
| MSA‐C, n (%) | 5 (41.7%) | 69 (51.5%) | 0.560 c |
| MSA‐P, n (%) | 7 (58.3%) | 61 (45.5%) | 0.548 c |
| Unclassified a , n (%) | 0 (0%) | 4 (3.0%) | 1.00 c |
| Brain weight, g, mean ± SD (range) | 1070.3 ± 168.6 (700–1390) |
1200.0 ± 160.0 (700–1650) (n = 117) |
0.00911 d |
| Pathologic subtypes of MSA | |||
| OPCA dominant, n (%) | 3 (25.0%) | 36 (26,9%) | 1.00 c |
| SND dominant, n (%) | 1 (8.3%) | 43 (32.1%) | 0.108 c |
| OPCA‐SND mixed, n (%) b | 8 (66.7%) | 55 (41.0%) | 0.127 c |
| Lewy body in brainstem, n (%) | 1 (8.3%) | 8 (6.0%) | 0.548 c |
| Braak's NFT stage, mean ± SD (range) | 1.6 ± 0.8 (1–3) | 1.4 ± 0.6 (1–4) | 0.449 d |
| Thal's amyloid phase, mean ± SD (range) | 0.8 ± 1.0 (0–3) | 0.9 ± 1.2 (0–5) | 0.938 d |
| Saito's argyrophilic grain stage, mean ± SD (range) | 0.0 ± 0.0 (0–0) | 0.1 ± 0.4 (0–2) | 0.333 d |
Abbreviations: MSA, multiple system atrophy; MSA‐C, multiple system atrophy with predominant cerebellar ataxia; MSA‐P, multiple system atrophy with predominant parkinsonism; NFT, Neurofibrillary tangle; OPCA, olivopontocerebellar atrophy; SD, standard deviation; SND, striatonigral degeneration.
The unclassified MSA refers to patients that could not be classified as either MSA‐P or MSA‐C based on clinical symptoms.
The OPC and the SN systems are equally affected.
Fisher's exact test.
Mann–Whitney U test.
TABLE 2.
Clinical and pathological findings of the hippocampal MSA patients
| Patients | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| Sex | F | M | M | F | F | F | F | F | M | F | F | F |
| Age at onset, years | 74 | 61 | 58 | 69 | 49 | 60 | 63 | 44 | 51 | 50 | 55 | 45 |
| Duration, years | 2 | 8 | 9 | 10 | 10 | 14 | 14 | 14 | 14 | 18 | 19 | 25 |
| Clinical phenotype | P | C | P | C | C | P | C | P | P | C | P | P |
| Cognitive impairment | − | + | − | − | − | + | + | − | + | − | − | − |
| Brain weight, g | 1120 | 1390 | 1200 | 1130 | 1110 | 1110 | 1065 | 920 | 1115 | 920 | 1064 | 700 |
| Jellinger's pathologic gradings | ||||||||||||
| OPCA | I | III | III | III | III | III | III | III | III | III | III | III |
| SND | III | I | III | III | II | III | I | III | III | III | III | III |
| Pick body‐like inclusion | − | − | − | − | − | − | + | − | − | + | − | + |
| Lewy body in brainstem | − | − | − | − | − | + | − | − | − | − | − | − |
| Braak's NFT Stage | III | II | I | II | I | III | I | I | I | I | II | I |
| Thal's amyloid phase | 3 | 1 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 1 | 1 |
| LATE stage | 0 | 0 | 0 | 0 a | 0 | 0 | 0 | 0 | 0 | 2 | 0 a | 1 |
Abbreviations: C, multiple system atrophy with predominant cerebellar ataxia; LATE, limbic‐predominant age‐related TDP‐43 encephalopathy; NCIs, neuronal cytoplasmic inclusions; NFT, neurofibrillary tangle; OPCA, olivopontocerebellar atrophy; P, multiple system atrophy with predominant parkinsonism; SND, striatonigral degeneration
There are a few granular TDP‐43 aggregations in CA1.
3.3. Neuropathologic findings of hippocampal MSA patients
The average brain weight of the hippocampal MSA patients was 1070.3 ± 168.6 g, which was significantly lower than that of the classical MSA patients (1200.0 ± 160.0 g) (Mann–Whitney U‐test, p = 0.00911). The medial temporal lobes were often atrophied in the hippocampal MSA patients, and H&E staining exhibited neuronal loss and astrogliosis in the CA1, subiculum, parahippocampal gyrus, or amygdala (Figures 2A–D and 3A–C, Figure S1, and Table S2). A semi‐quantitative analysis revealed that neuronal loss and astrogliosis of the hippocampus were significantly more severe in the hippocampal MSA patients than in the classical MSA patients (Table 3). Gallyas‐Braak staining and anti‐alpha‐synuclein immunohistochemistry revealed abundant NCIs with ring‐shaped or NFT‐like fibrous configurations in the hippocampus (Figure 3D,E). The hippocampal granule cells and CA1/subiculum were the most likely to show NCIs (Figure 4 and Table S2). The hippocampal granule cells and the CA1/subiculum almost always showed more than 10 NCIs when averaged over five visual fields using the 20 × objective, whereas these regions never had more than 5 NCIs in the classical MSA patients. Alpha‐synuclein‐immunopositive, thick neurites that indicated intra‐axonal aggregation of alpha‐synuclein was occasionally observed in the neuropil of the hippocampus (Figure 3F,G). Three of the hippocampal MSA patients (Patients 7, 10, and 12) showed dense, rounded NCIs, which were morphologically similar to Pick bodies (Figure 3B,H). Although ring‐shaped or NFT‐like NCIs were occasionally observed among the classical MSA patients, these Pick body‐like inclusions were never observed. Atrophy of the medial temporal lobes was much severe, and the spread of NCIs was more extensive in the patients with Pick body‐like inclusions than that in patients without (Figures 3I and 4, and Table S2). Electron microscopy of Patients 4 and 10 revealed that the NCIs in the dentate nucleus were composed of randomly orientated granule‐coated filaments (Figure 3K).
FIGURE 2.
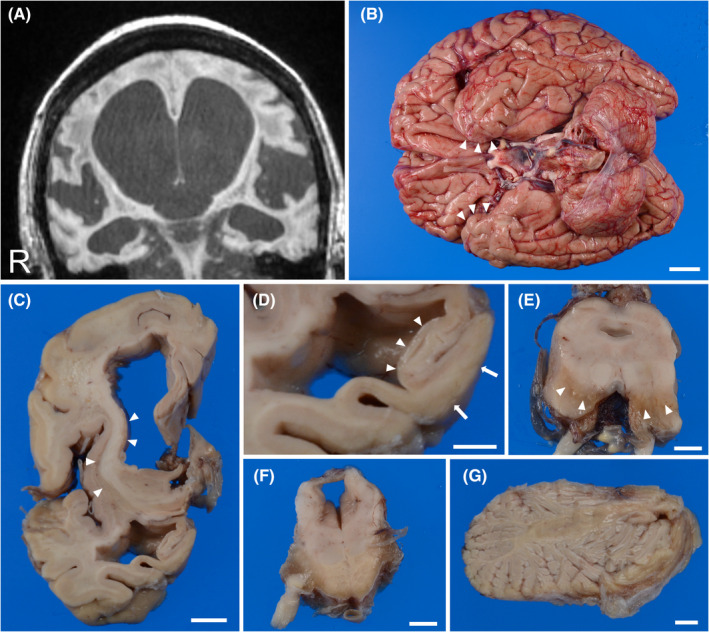
Radiologic and macroscopic findings of a hippocampal MSA patient. (A) T1‐weighted MRI 14 years after the disease onset reveals severe atrophy of the medial temporal lobe and dilatation of the lateral ventricle. (B) A bottom view of the brain demonstrates atrophy of the medial temporal lobes (arrowheads), the brain stem, and the cerebellum. (C) A coronal section of the cerebrum shows prominent atrophy of the striatum (arrowheads) and the white matter of the frontal lobe and a dilatation of the lateral ventricle. (D) A higher magnification of the former panel shows severe atrophy of the CA region (arrowheads), subiculum, and parahippocampal gyrus (arrows). (E–G) Discoloration of the substantia nigra (E, arrowheads), an atrophy of the basilar pons (F), and an atrophy of the cerebellar white matter (G) are also observed. A–G: Patient 10. Scale bars: B; 2 cm, C; 1 cm, D–G; 5 mm. CA, cornu ammonis area; MSA, Multiple system atrophy; MRI, magnetic resonance imaging
FIGURE 3.
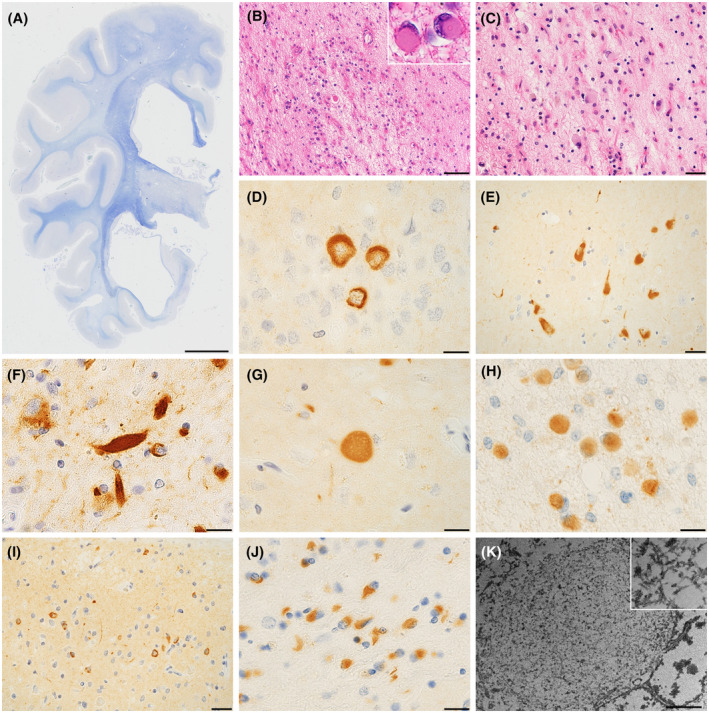
Microscopic findings of the hippocampal MSA patients. (A) A coronal section shows prominent atrophy of the medial temporal lobe. (B–C) The dentate gyrus (B) and CA1 (C) demonstrates severe neuronal loss, astrogliosis, and Pick body‐like inclusions (B, insert). (D–E) Anti‐alpha‐synuclein immunohistochemistry reveals ring‐shaped neuronal cytoplasmic inclusions (NCIs) in the dentate gyrus (D) and NFT‐like NCIs in the pyramidal neurons of the subiculum (E). (F–G) Alpha‐synuclein‐immunopositive thick neurites (F) or spheroids (G) are observed in the neuropils of the hippocampus. (H) Pick body‐like inclusions are alpha‐synuclein‐immunoreactive. (I) The cingulate gyrus shows abundant NCIs. (J) The putamen displays alpha‐synuclein immunopositive glial cytoplasmic inclusions. (K) The NCIs in the hippocampus consists of randomly orientated granule‐coated fibrillary structures. A–C, H: Patient 12. D–E, G: Patient 4. F, I, K: Patient 10. J: Patient 5. A: Klüver‐Barrera staining, B–C: H&E staining, D–E and G–H: alpha‐synuclein immunohistochemistry, F: phosphorylated alpha‐synuclein immunohistochemistry. K: electron microscopy. Scale bars: A; 1 cm, B, I; 50 μm, C, E; 20 μm, D, F‐H, J; 10 μm, K; 2 μm. CA, cornu ammonis area; H&E, hematoxylin‐eosin; MSA, Multiple system atrophy; NCIs, neuronal cytoplasmic inclusions
TABLE 3.
Comparison of neuronal loss and astrogliosis between the hippocampal MSA and classical MSA
| Hippocampal MSA (n = 12) | Classical MSA (n = 134) | p‐value | |
|---|---|---|---|
| Hippocampus and limbic system | |||
| Dentate gyrus, mean ± SD (range) | 0.5 ± 0.8 (0–2) (n = 11) a | 0.0 ± 0.0 (0–0) (n = 133) a | 2.31 × 10−12 c |
| CA1/subiculum, mean ± SD (range) | 1.1 ± 0.8 (0–2) (n = 11) a | 0.0 ± 0.0 (0–0) (n = 130) a | 1.54 × 10−23 c |
| CA2–4, mean ± SD (range) | 0.7 ± 0.8 (0–2) (n = 11) a | 0.0 ± 0.1 (0–1) (n = 131) a | 2.3 × 10−15 c |
| Parahippocampal gyrus, mean ± SD (range) | 1.0 ± 0.8 (0–2) (n = 11) a | 0.0 ± 0.2 (0–2) (n = 132) a | 3.73 × 10−17 c |
| Amygdala, mean ± SD (range) | 1.2 ± 0.6 (0–2) (n = 11) a | 0.2 ± 0.4 (0–1) (n = 131) a | 5.31 × 10−10 c |
| Cingulate gyrus, mean ± SD (range) | 0.8 ± 0.7 (0–2) | 0.1 ± 0.2 (0–1) (n = 119) b | 3.2 × 10−11 c |
| Insular cortex, mean ± SD (range) | 0.7 ± 0.8 (0–2) | 0.0 ± 0.2 (0–1) (n = 132) a | 5.95 × 10−9 c |
| Cerebral cortices | |||
| Prefrontal cortices, mean ± SD (range) | 0.7 ± 0.5 (0–1) | 0.1 ± 0.3 (0–2) (n = 127) b | 1.78 × 10−11 c |
| Temporal cortices, mean ± SD (range) | 0.4 ± 0.5 (0–1) | 0.0 ± 0.2 (0–2) (n = 130) b | 1.15 × 10−9 c |
A few patients are excluded because of comorbid ischemic changes.
A few patients are excluded because of comorbid ischemic changes or insufficient tissue material.
Mann–Whitney U test.
FIGURE 4.
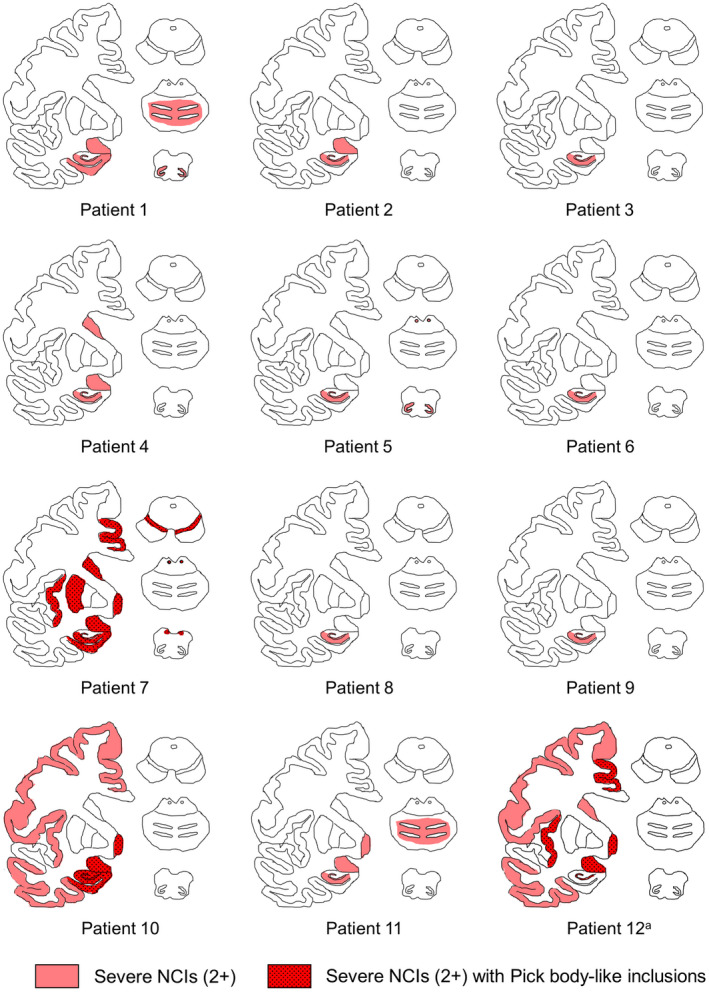
Distribution of neuronal cytoplasmic inclusions (NCIs) of the hippocampal MSA patients. Regions with severe (2+) NCIs are shown in red, and those with Pick body‐like inclusions are highlighted with a dotted pattern. The dentate gyrus and CA1/subiculum are vulnerable in all patients. Patients with Pick body‐like inclusions tend to show broad spreading of NCIs. aNote that only few neurons are retained in the hippocampus of Patient 12, and hence, they are not illustrated in red; however, neuronal inclusions are prevalent among the retained neurons. CA, cornu ammonis area; MSA, multiple system atrophy; NCIs, neuronal cytoplasmic inclusions
A subset of the hippocampal MSA patients showed astrogliosis and alpha‐synuclein‐immunopositive NCIs in the cerebral cortices (Figure 4 and Table S2). Semi‐quantitative analyses revealed that neuronal loss and astrogliosis in the cerebral cortices were significantly more severe in the hippocampal MSA patients than in the classical MSA patients (Table 3). The proportions in neuropathologic subtypes of OPCA and SND did not differ between two groups (Figures 2C,E–G and 3J, Table 1, Tables S2 and S3). In the hippocampal MSA, two patients (Patients 4 and 7) in the SN system and three patients (Patients 1, 5, and 11) in the OPC system showed abundant NCIs (Figure 4 and Table S2). In the classical MSA, three patients in the SN system and five patients in the OPC system revealed abundant NCIs.
Concomitant Lewy bodies were observed in the substantia nigra, locus coeruleus, and dorsal nucleus of the vagus in Patient 6. Deposits of hyperphosphorylated tau and beta‐amyloid were generally mild in the hippocampus and limbic areas of the hippocampal MSA patients (Figure 5 and Table 2). Four of the hippocampal MSA patients showed a few granular TDP‐43 aggregations in the hippocampus (Figure 5 and Table 2). The limbic‐predominant age‐related TDP‐43 encephalopathy stage of Patient 10 corresponded to stage 2, while Patient 12 corresponded to stage 1 (25). The other two patients with the hippocampal MSA (Patients 4 and 11) had occasional granular TDP‐43 aggregations that were restricted in CA1. Concomitant Lewy body pathology, Braak's NFT stages, Thal's amyloid phases, and Saito's argyrophilic grain stages did not differ between the hippocampal MSA and classical MSA groups (Table 1) (22, 23, 24).
FIGURE 5.
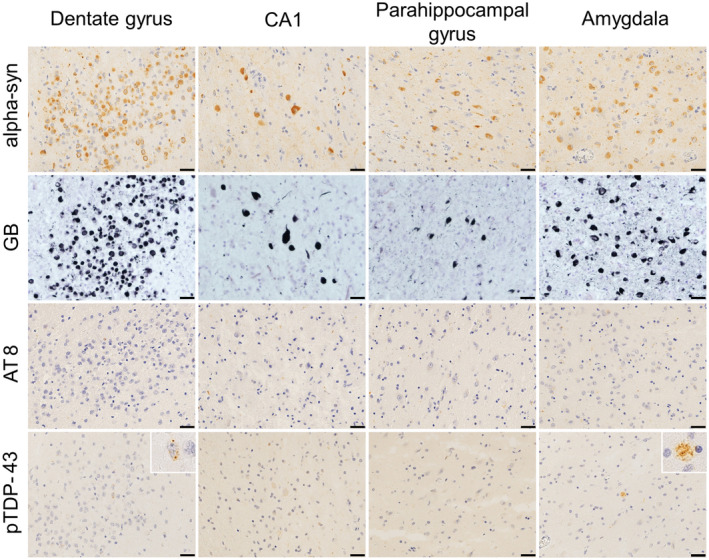
Immunohistochemical properties of neuronal cytoplasmic inclusions (NCIs) in a hippocampal MSA patient. Photomicrographs from Patient 10 are shown. NCIs in the dentate gyrus, CA1, parahippocampal gyrus, and amygdala are labeled using anti‐alpha‐synuclein immunohistochemistry (top row) and Gallyas−Braak staining (second row); however, staining is not observed following anti‐hyperphosphorylated tau (third row). A few granular TDP‐43 aggregations are observed in the dentate gyrus and amygdala using anti‐phosphorylated TDP‐43 immunohistochemistry (bottom row, insert). All scale bars are 20 μm. CA, cornu ammonis area; GB, Gallyas−Braak staining; MSA, Multiple system atrophy; NCIs, neuronal cytoplasmic inclusions; TDP‐43, transactive response DNA‐binding protein 43 kDa
3.4. Genetic findings
We conducted a mutational analysis of APOE, COQ2, GBA, and SNCA to identify variants located on these genes in the autopsied brains, where frozen tissues were available for DNA extraction in seven of the 12 hippocampal MSA patients (Patients 3–6 and 9–11) and 52 of the 134 classical MSA patients. The identified variants for COQ2 and GBA are listed in Table S4. Specifically, we identified five heterozygous carriers of the V393A variant of the COQ2 gene (9.6% carrier frequency) in the classical MSA group but none in the hippocampal MSA group. The most common APOE genotype identified was ε3/ε3 (85.7% in the hippocampal MSA, 77% in the classical MSA, Table S5). There were no significant differences in the allele frequencies of V393A between the hippocampal MSA and classical MSA (Fisher's exact test, p = 1.0). There were no significant differences in the distribution of APOE genotypes between the hippocampal and classical MSA, either (Fisher's exact test, p = 1.0). There were neither mutations nor copy number variations of the SNCA gene found in any of samples.
4. DISCUSSION
Our study has expanded the knowledge of MSA variants that are characterized by extremely abundant NCIs of alpha‐synuclein (7, 9). The hippocampal granule cells and CA1/subiculum always and most prominently displayed NCIs in the hippocampal MSA patients. The burden of NCIs and the severity of neuronal loss in the hippocampus of the hippocampal MSA patients were far greater than those of the other MSA patients. The NCIs of the hippocampus were labeled with anti‐alpha‐synuclein immunohistochemistry and silver staining and ultra‐structurally consisted of granule‐coated filaments, and these properties were characteristic of MSA (29). These neuropathological findings differed from those of Lewy bodies (9, 30). Although comorbidity of Lewy body pathology has been reported in a subset of MSA patients (31), the NCIs we found are not a comorbid Lewy bodies.
The hippocampal MSA group showed longer clinical duration than the classical MSA group in our study or previously reported patients (32, 33). However, the severe hippocampal lesions may not appear simply because of long‐term survival. A subset of the hippocampal MSA patients was associated with short clinical durations, whereas some patients of the classical MSA group showed long clinical durations (Figure S2). These clinical and pathologic results indicate that the hippocampal MSA is a pathological variant of MSA and not a pathologic finding of an advanced disease phase.
Another finding of interest was that a subset of the hippocampal MSA patients showed dense NCIs in the frontotemporal lobes. Aoki et al. have recently reported a pathological variant of MSA that showed abundant NCIs of alpha‐synuclein in the frontotemporal cortices, namely FTLD associated with alpha‐synuclein (FTLD‐synuclein) (9). Importantly, a subset of the previously reported patients with FTLD‐synuclein also had NCIs in the CA and dentate gyrus (9, 13). The distributions of cerebral lesion seem to be overlapped each other between the hippocampal MSA and FTLD‐synuclein. The hippocampal MSA and FTLD‐synuclein could be included in a continuous spectrum of cortical pathology.
The hippocampal MSA patients had a significantly higher frequency of cognitive impairment than the classical MSA patients, and the difference was independent of aging‐related or comorbid pathologic changes. The marked neuronal inclusions may facilitate neuronal dysfunctions in the hippocampi of these patients. Recent studies have also shown that the burden of alpha‐synuclein‐immunopositive inclusions within the hippocampal neurons is correlated with the occurrence of cognitive impairment among MSA patients (34, 35). By contrast, the FTLD‐synuclein was reported to correlate with the clinical presentation of FTD, CBS, or PA (9). These symptoms were not observed in our hippocampal MSA patients, which generates large differences in clinical findings between the hippocampal MSA and FTLD‐synuclein. The fact indicates that the distributions of NCIs in the cerebral gray matter absolutely impacts socio‐cognitive impairments among MSA patients.
The significance of alpha‐synuclein‐immunoreactive neuronal inclusions to the pathophysiology of MSA remains to be elucidated. Neuropathologic observations and studies in animal models have suggested that the formation of GCIs is the primary lesion in MSA, and neuronal inclusion is a secondary phenomenon that arises from the oligomyelin‐axon‐neuron complex mechanism (6, 36, 37). However, recent pathologic studies have revealed that intra‐neuronal aggregation of alpha‐synuclein is broadly observed even in patients who die during the early disease phase (4, 7). Neuronal inclusions were not only found within the vulnerable regions of MSA (38), but also out of the SN and OPC networks (7, 35). The fact that alpha‐synuclein is aggregated early and broadly within the neurons suggests that neuronal and glial inclusions synergistically accelerate the degenerative process of MSA (4, 7). Our observations support this hypothesis.
Although clinical characteristics of the hippocampal MSA remain to be further clarified, cognitive impairment during an early disease phase might be indicative of this variant. In our patient series, cognitive impairment was significantly more prevalent in the hippocampal MSA patients than in the classical MSA patients. In addition, the hippocampal MSA could be considered when the progression of hippocampal atrophy is observed over time in imaging studies. Pathologic changes of the hippocampus were generally minimal among the classical MSA of our patient series; hence, cognitive assessments and radiological observation of the hippocampus may yield reasonable clues to identify the hippocampal MSA.
Mutational analysis of APOE, COQ2, GBA, and SNCA did not detect any variants significantly associated with the hippocampal MSA, suggesting variations in these genes unlikely underlie the hippocampal MSA. Further analysis of a larger sample size is warranted to make a more accurate conclusion.
The study contains several limitations because of the retrospective design. First, the included patients were not consistently or longitudinally evaluated using standardized scales of cognitive functions. Although our results indicated that symptomatic dementia was correlated with the hippocampal MSA phenotype, cognitive impairments might be underestimated in both hippocampal MSA and classical MSA patients. Second, the sample size of the hippocampal MSA group was small; hence, it was hard to classify dementia syndromes or to make comparisons between distributions of NCIs and neurological manifestations.
In conclusion, we have reported 12 MSA patients that showed abundant NCIs, neuronal loss, and regional atrophy in the hippocampus. The phenotype may be a pathological variant of MSA and reinforces the importance of neuronal alpha‐synucleinopathy in the pathogenesis of MSA.
CONFLICT OF INTEREST
The authors report no competing interests.
AUTHOR CONTRIBUTIONS
Takashi Ando: Conceptualization, Methodology, Investigation, Writing – Original Draft. Yuichi Riku: Methodology, Investigation, Writing – Review & Editing. Akio Akagi: Investigation. Hiroaki Miyahara: Investigation. Mitsuaki Hirano: Investigation. Toshimasa Ikeda: Investigation. Hiroyuki Yabata: Investigation. Ryuichi Koizumi: Investigation. Chisato Oba: Investigation. Saori Morozumi: Investigation. Keizo Yasui: Investigation. Atsuko Goto: Investigation. Taiji Katayama: Investigation. Satoko Sakakibara: Investigation. Ikuko Aiba: Investigation. Motoko Sakai: Investigation. Masaaki Konagaya: Investigation. Keiko Mori: Investigation. Yasuhiro Ito: Investigation. Hiroyuki Yuasa: Investigation. Masayo Nomura: Investigation. Kristine Joyce L. Porto: Investigation. Jun Mitsui: Investigation, Writing ‐ Review & Editing. Shoji Tsuji: Investigation, Writing – Review & Editing. Maya Mimuro: Investigation. Yoshio Hashizume: Investigation. Masahisa Katsuno: Writing – Review & Editing, Supervision. Yasushi Iwasaki: Investigation, Writing – Review & Editing, Supervision. Mari Yoshida: Conceptualization, Methodology, Investigation, Writing – Review & Editing, Supervision.
ETHICAL APPROVAL
Written informed consent was obtained from the patients’ relatives before autopsies. All human studies were approved by the research ethical committee of the Aichi Medical University.
Supporting information
FIGURE S1 Chronological changes of T2‐weighted MRI
FIGURE S2 Pathologic findings of a classical MSA patient who had survived for 19 years after disease onset
TABLE S1 Summary of cognitive impairment in the hippocampal MSA patients
TABLE S2 Semi‑quantitative scores of neuronal loss and neuronal cytoplasmic inclusions in the hippocampal MSA patients
TABLE S3 Semi‑quantitative scores of glial cytoplasmic inclusions in the hippocampal MSA patients
TABLE S4 Distribution of identified COQ2 and GBA variants among the hippocampal MSA and classical MSA
TABLE S5 Distribution of APOE genotypes among the hippocampal MSA and classical MSA
ACKNOWLEDGMENTS
We would like to thank all patients and their families, and clinicians who referred patients to our institute. We also wish to express our gratitude to Kenji Kohtani, Toshiaki Mizuno, Chizuko Sano, and Chieko Uno for their technical support during pathologic analyses.
Ando T, Riku Y, Akagi A, Miyahara H, Hirano M, Ikeda T, et al. Multiple system atrophy variant with severe hippocampal pathology. Brain Pathology. 2022;32:e13002. 10.1111/bpa.13002
Funding information
This work was partly supported by AMED under Grant Numbers JP20ek0109392, JP20ek0109391, Intramural Research Grant (30‐8) for Neurological and Psychiatric Disorders of NCNP (M.Y.), and JSPS KAKENHI Grant Number JP20K16586 (Y.R.). This work was supported by Grants‐in‐Aid from the Research Committee of CNS Degenerative Diseases, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour, and Welfare Sciences Research Grants, the Ministry of Health, Labour, and Welfare, Japan (Y.I.)
DATA AVAILABILITY STATEMENT
The raw data that support the findings of this study are available upon reasonable request from the corresponding author.
REFERENCES
- 1. Fanciulli A, Wenning GK. Multiple‐system atrophy. N Engl J Med. 2015;372:249–63. [DOI] [PubMed] [Google Scholar]
- 2. Koga S, Dickson DW. Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry. 2018;89:175–84. [DOI] [PubMed] [Google Scholar]
- 3. Trojanowski JQ, Revesz T, Neuropathology Working Group on MSA . Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33:615–20. [DOI] [PubMed] [Google Scholar]
- 4. Yoshida M. Multiple system atrophy: alpha‐synuclein and neuronal degeneration. Neuropathology. 2007;27:484–93. [DOI] [PubMed] [Google Scholar]
- 5. Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain. 2004;127:2657–71. [DOI] [PubMed] [Google Scholar]
- 6. Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol. 2008;64:239–46. [DOI] [PubMed] [Google Scholar]
- 7. Cykowski MD, Coon EA, Powell SZ, Jenkins SM, Benarroch EE, Low PA, et al. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain. 2015;138:2293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jellinger KA. Multiple system atrophy: an oligodendroglioneural synucleinopathy. J Alzheimers Dis. 2018;62:1141–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha‐synuclein. Acta Neuropathol. 2015;130:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Homma T, Mochizuki Y, Komori T, Isozaki E. Frequent globular neuronal cytoplasmic inclusions in the medial temporal region as a possible characteristic feature in multiple system atrophy with dementia. Neuropathology. 2016;36:421–31. [DOI] [PubMed] [Google Scholar]
- 11. Horoupian DS, Dickson DW. Striatonigral degeneration, olivopontocerebellar atrophy and “atypical” Pick disease. Acta Neuropathol. 1991;81:287–95. [DOI] [PubMed] [Google Scholar]
- 12. Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, et al. Co‐localization of alpha‐synuclein and phosphorylated tan in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration. Acta Neuropathol. 2001;101:285–93. [DOI] [PubMed] [Google Scholar]
- 13. Rohan Z, Rahimi J, Weis S, Kapas I, Auff E, Mitrovic N, et al. Screening for alpha‐synuclein immunoreactive neuronal inclusions in the hippocampus allows identification of atypical MSA (FTLD‐synuclein). Acta Neuropathol. 2015;130:299–301. [DOI] [PubMed] [Google Scholar]
- 14. Saito M, Hara M, Ebashi M, Morita A, Okada K, Homma T, et al. Perirhinal accumulation of neuronal alpha‐synuclein in a multiple system atrophy patient with dementia. Neuropathology. 2017;37:431–40. [DOI] [PubMed] [Google Scholar]
- 15. Shibuya K, Nagatomo H, Iwabuchi K, Inoue M, Yagishita S, Itoh Y. Asymmetrical temporal lobe atrophy with massive neuronal inclusions in multiple system atrophy. J Neurol Sci. 2006;179:50–8. [DOI] [PubMed] [Google Scholar]
- 16. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ideno Y, Takayama M, Hayashi K, Takagi H, Sugai Y. Evaluation of a Japanese version of the Mini‐Mental State Examination in elderly persons. Geriatr Gerontol Int. 2012;12:310–6. [DOI] [PubMed] [Google Scholar]
- 18. Jeong JW, Kim KW, Lee DY, Lee SB, Park JH, Choi EA, et al. A normative study of the Revised Hasegawa Dementia Scale: comparison of demographic influences between the Revised Hasegawa Dementia Scale and the Mini‐Mental Status Examination. Dement Geriatr Cogn Disord. 2007;24:288–93. [DOI] [PubMed] [Google Scholar]
- 19. Riku Y, Watanabe H, Mimuro M, Iwasaki Y, Ito M, Katsuno M, et al. Non‐motor multiple system atrophy associated with sudden death: pathological observations of autonomic nuclei. J Neurol. 2017;264:2249–57. [DOI] [PubMed] [Google Scholar]
- 20. Jellinger KA, Seppi K, Wenning GK. Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord. 2005;20:S29–36. [DOI] [PubMed] [Google Scholar]
- 21. Lee YH, Ando T, Lee JJ, Baek MS, Lyoo CH, Kim SJ, et al. Later‐onset multiple system atrophy: a multicenter Asian study. Mov Disord. 2020;35:1692–3. [DOI] [PubMed] [Google Scholar]
- 22. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. [DOI] [PubMed] [Google Scholar]
- 24. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, et al. Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Expe Neurol. 2004;63:911–8. [DOI] [PubMed] [Google Scholar]
- 25. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitsui J, Matsukawa T, Ishiura H, Fukuda Y, Ichikawa Y, Date H, et al. Mutations in COQ2 in familial and sporadic multiple‐system atrophy. N Engl J Med. 2013;369:233–44. [DOI] [PubMed] [Google Scholar]
- 27. Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Dürr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015;2:417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanda Y. Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arima K, Murayama S, Mukoyama M, Inose T. Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 1. Neuronal cytoplasmic inclusions. Acta Neuropathol. 1992;83:453–60. [DOI] [PubMed] [Google Scholar]
- 30. Uchihara T, Nakamura A, Mochizuki Y, Hayashi M, Orimo S, Isozaki E, et al. Silver stainings distinguish Lewy bodies and glial cytoplasmic inclusions: comparison between Gallyas‐Braak and Campbell‐Switzer methods. Acta Neuropathol. 2005;110:255–60. [DOI] [PubMed] [Google Scholar]
- 31. Koga S, Li F, Zhao N, Roemer SF, Ferman TJ, Wernick AI, et al. Clinicopathologic and genetic features of multiple system atrophy with Lewy body disease. Brain Pathol. 2020;30:766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy—an analysis of 230 Japanese patients. Brain. 2002;125:1070–83. [DOI] [PubMed] [Google Scholar]
- 33. Wenning GK, Tison F, BenShlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord. 1997;12:133–47. [DOI] [PubMed] [Google Scholar]
- 34. Koga S, Parks A, Uitti RJ, van Gerpen JA, Cheshire WP, Wszolek ZK, et al. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord. 2017;32:405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miki Y, Foti SC, Hansen D, Strand KM, Asi YT, Tsushima E, et al. Hippocampal alpha‐synuclein pathology correlates with memory impairment in multiple system atrophy. Brain. 2020;143:1798–810. [DOI] [PubMed] [Google Scholar]
- 36. Lantos PL. The definition of multiple system atrophy: a review of recent developments. J Neuropathol Exp Neurol. 1998;57:1099–111. [DOI] [PubMed] [Google Scholar]
- 37. Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, et al. Mouse model of multiple system atrophy alpha‐synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–59. [DOI] [PubMed] [Google Scholar]
- 38. Nishie M, Mori F, Yoshimoto M, Takahashi H, Wakabayashi K. A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol Appl Neurobiol. 2004;30:546–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Chronological changes of T2‐weighted MRI
FIGURE S2 Pathologic findings of a classical MSA patient who had survived for 19 years after disease onset
TABLE S1 Summary of cognitive impairment in the hippocampal MSA patients
TABLE S2 Semi‑quantitative scores of neuronal loss and neuronal cytoplasmic inclusions in the hippocampal MSA patients
TABLE S3 Semi‑quantitative scores of glial cytoplasmic inclusions in the hippocampal MSA patients
TABLE S4 Distribution of identified COQ2 and GBA variants among the hippocampal MSA and classical MSA
TABLE S5 Distribution of APOE genotypes among the hippocampal MSA and classical MSA
Data Availability Statement
The raw data that support the findings of this study are available upon reasonable request from the corresponding author.