

Abstract
Fusidic acid (FA) is a potent steroidal antibiotic that has been used in Europe for more than 60 years to treat a variety of infections caused by Gram-positive pathogens. Despite its clinical success, FA requires significantly elevated dosing (3 g on the first day, 1.2 g on subsequent days) to minimize resistance, as FA displays a high resistance frequency, and a large shift in minimum inhibitory concentration is observed for resistant bacteria. Despite efforts to improve on these aspects, all previously constructed derivatives of FA have worse antibacterial activity against Gram-positive bacteria than the parent natural product. Here, we report the creation of a novel FA analogue that has equivalent potency against clinical isolates of Staphylococcus aureus (S. aureus) and Enterococcus faecium (E. faecium) as well as an improved resistance profile in vitro when compared to FA. Importantly, this new compound displays efficacy against an FA-resistant strain of S. aureus in a soft-tissue murine infection model. This work delineates the structural features of FA necessary for potent antibiotic activity and demonstrates that the resistance profile can be improved for this scaffold and target.
Keywords: antibiotics, fusidic acid analogues, resistance profile, in vivo efficacy, EF-G inhibitor
Graphical Abstract
Recent reports have confirmed that the worrisome rise of antibiotic-resistant bacterial infections is continuing.1,2 Some of the most problematic Gram-positive pathogens are methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE), and accordingly, these have been classified as serious threats by the Centers for Disease Control and Prevention (CDC).3 The emergence of bacteria resistant to vancomycin,4 a drug of last resort against Gram-positive infections, highlights the need to develop or re-engineer drugs that could be efficacious against these problematic pathogens.
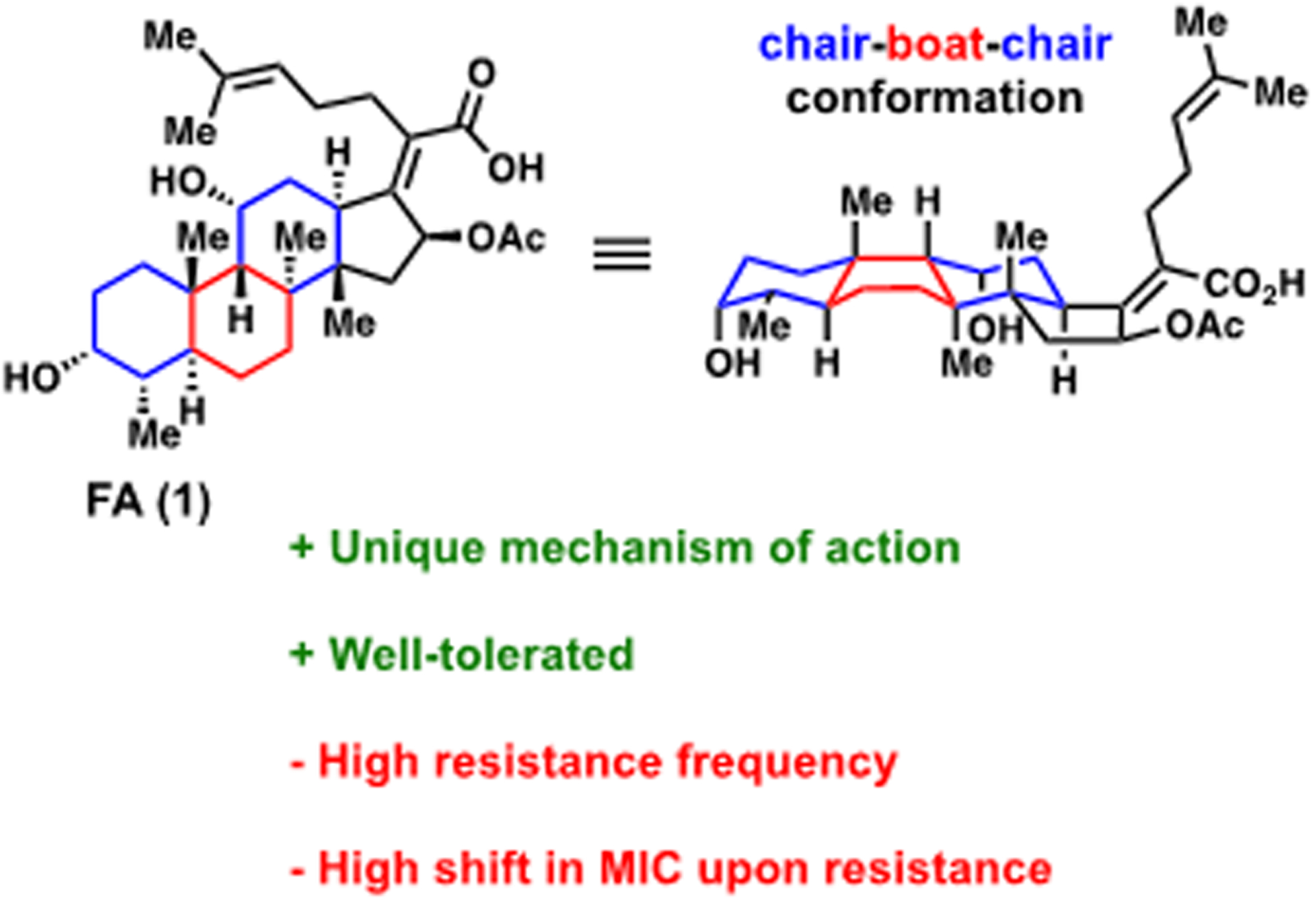
Fusidic acid (1, Figure 1) is a steroidal antibiotic that is produced by the fungus Fusidium coccineum.5 Its mechanism of action involves inhibition of protein synthesis by stabilizing the elongation factor G (EF-G) complex, resulting in the truncation of peptide elongation.6 EF-G being an unusual antibacterial target likely minimizes the cross-resistance of FA with other antibiotics.7 FA is a Gram-positive-only antibiotic that has been used since the 1960s in Europe to treat Gram-positive infections and has been particularly effective in treating MRSA.7,8 To date, FA is approved in 23 countries and is administered via oral, intravenous, and topical routes.7 In the United States, FA has achieved its primary and secondary efficacy end points in phase 3 clinical trials for the treatment of patients with acute bacterial skin and skin structure infections (ABSSSI).9 FA has also received Orphan Drug Designation from the Food and Drug Administration (FDA) for the treatment of prosthetic joint infections and gained Qualified Infectious Disease Product Designation under the Generating Antibiotic Incentives Now (GAIN) Act.10 Strikingly, an analysis conducted in 2011 showed that virtually all (99.7%) of S. aureus strains in the United States were susceptible to FA.7
Figure 1.
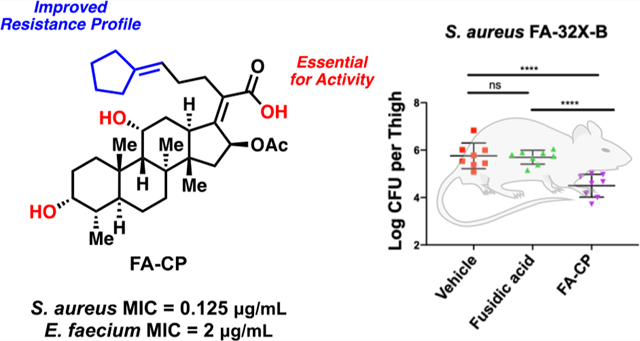
Structure of fusidic acid (FA), with some of its properties.
Despite the clinical success of FA, the high resistance frequency in the range of 1 × 10−6 to 1 × 10−8 at 2–16X the minimum inhibitory concentration (MIC) coupled with drastic MIC shifts upon resistance remain a major concern and are a hindrance to its wider usage.7 FA-resistant bacterial strains display a varied profile, illustrated by the heterogeneity in mutations observed in the laboratory and in the clinic.11 Some of the most prevalent mutations in the fusA gene (encoding EF-G) observed in the clinic are V90L, H457Q, L461K, and A655V.7 Especially concerning is the L461K mutation, observed clinically in S. aureus strains resistant to FA, which leads to MIC values of >256 μg/mL, a >2048-fold increase in MIC upon resistance.7 This large shift in MIC upon resistance observed from a single amino acid mutation is consistent with the hypothesis that single enzyme inhibitors are more prone to display elevated shifts in MIC upon resistance relative to antibiotics with multiple targets.12,13 In addition to mutations in the fusA gene, another source of clinical resistance for FA is the overexpression of the FusB family of proteins encoded by the fusB gene usually carried on a 21-kb plasmid (pUB101); FusB binds to EF-G and drives the dissociation of FA from its target.14 More recently, fusC and fusD genes have also been discovered in clinical isolates of S. aureus.15 The fusC gene has been associated with driving resistance in S. aureus and coagulase-negative staphylococci, while the fusD gene has been linked to conferring resistance among Staphylococcus saprophyticus.15
The aforementioned resistance challenges necessitate a front-loading dosing regimen, with 3 g of FA taken on the first day of treatment and 1.2 g taken daily thereafter, allowing for considerable drug plasma levels to be reached (Cmax of greater than 130 μg/mL in patients).7 Despite multiple efforts to construct FA analogues,16–32 none of these derivatives are as active as FA against Gram-positive pathogens;7 in addition, none of them improve on the resistance issues noted above. No FA derivatives have gained traction translationally, and only FA itself has moved forward to the clinic from this antibiotic class. While no FA derivatives with improved activity against Gram-positive bacteria have been reported, derivatives with improved activity relative to FA have been reported against the malaria parasite Plasmodium falciparum.21,26,30
Notable challenges in constructing FA variants through direct modification of this natural product include the presence of reactive functional groups such as the olefinic side chain, an α,β-unsaturated carboxylic acid, an acetate, and two sterically hindered alcohols (Figure 1). Furthermore, the unique chair–boat–chair core conformation of FA makes the total synthesis of this antibiotic challenging, and such routes are not yet suitable for the rapid generation of derivatives.24,25,33 Intrigued by the translational potential of FA, here we report the development of synthetic routes from FA that provide rapid access to novel variants with modified side chains, leading to the discovery of a novel FA analogue with an improved resistance profile.
RESULTS
Synthesis and Antibacterial Assessment of FA Derivatives Where the Carboxylic Acid and Alcohols Are Modified.
Key to this study was to first determine the important structural features necessary to retain the potent antibacterial activity of FA. Although FA derivatives have been reported in the literature,16–32 a systematic antimicrobial activity assessment of FA derivatives with MIC values against key Gram-positive pathogens does not exist. Thus, as a prelude to our work, we first synthesized several known FA analogues and assessed their antibacterial activity, with the specific goals of probing the importance of the carboxylic acid and alcohols.
The role of the carboxylic acid was investigated via construction of ester 2 and amide 3, using previously published routes (Figure 2A).26,27 Diketone 4 was synthesized previously using the Jones oxidation reaction.34 Herein, diketone 4 was synthesized using pyridinium chlorochromate (Figure 2A). Oxidation solely at C3 was effected using recently published conditions to provide compound 5 (Figure 2A).28 The monoketone at C11 was previously synthesized by treating FA with Kiliani’s reagent in a low yielding process.29 Herein, a five-step sequence was developed to generate the C11 ketone (Figure 2B). This route required the protection of the carboxylic acid to generate intermediate 6.21,23 The steric hindrance at the C11 position induced by 1,3-diaxial interactions was leveraged, and the C3 alcohol was selectively protected with TBS to generate 7. The C11 alcohol was then oxidized with pyridinium chlorochromate to generate 8. Removal of the TBS protecting group in compound 8 with aqueous hydrofluoric acid provided 9, and subsequent deprotection of the pivaloyloxymethyl protecting group under mild basic conditions afforded the target monoketone, compound 10 (Figure 2B). Key to this work was utilizing a suitable protecting group for the carboxylic acid that could be removed under mild basic conditions, as lactonization between the C16 acetyl and the C21 carboxylic acid occurs under strongly basic conditions.21,23 Accordingly, POM-Cl, used previously to protect the FA carboxylic acid,21,23 was utilized to protect the carboxylic acid as a pivaloyloxy-methyl ester based on its ability to be removed under mild conditions.21,23
Figure 2.
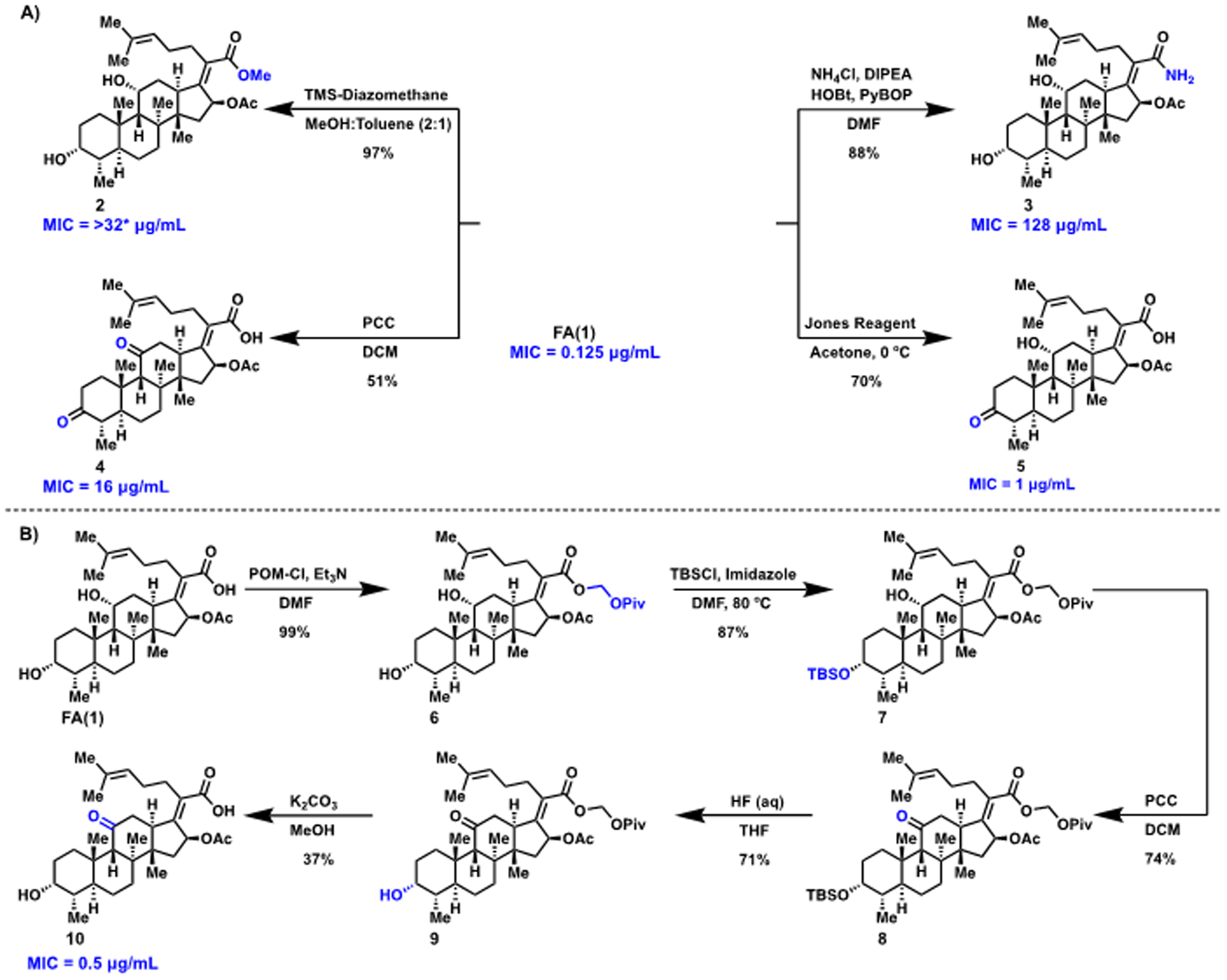
(A) Structure–activity relationship investigation of the carboxylic acid and alcohols of FA. (B) Synthesis of the C11 ketone of FA. MICs determined using CLSI guidelines against S. aureus ATCC 29213, n = 3. Asterisk indicates solubility limit.
Antibacterial activity of FA analogues 2–5 and 10 was assessed against S. aureus ATCC 29213 using standard MIC assays following Clinical and Laboratory Standards Institute (CLSI) guidelines.35 As shown in Figure 2, FA analogues 2–4 display significantly reduced antibacterial activity, reinforcing the importance of the free carboxylic acid and both of the alcohols. Oxidation of the secondary alcohols separately is more tolerated, with the monoketones at C3 (compound 5) and at C11 (compound 10) having MIC values only 4–8-fold worse than FA (Figure 2).
Synthesis and Antibacterial Evaluation of Side Chain Analogues.
As modifications to FA shown in Figure 2 were all deleterious to antibacterial activity, the focus shifted to modifications of the gem-dimethyl alkene side chain. This work was informed by the crystal structure of the ribosome trapped with EF-G in a post-translocational state with FA bound;36 this complex indicates that the side chain of FA is positioned in a large, hydrophobic pocket of EF-G.36
Several FA analogues with modifications to the side chain have been previously reported, such as derivatives where the C24 and C25 or the C17 and C20 double bonds have been reduced.17,22 Furthermore, truncation of the C26 and C27 methyl groups has also been reported as well as analogues where the C26 and C27 methyl groups were modified via microbial oxidation.37,38 Heteroatoms have also been introduced via ozonolysis of the C24 and C25 double bond.18 Aryl side chain analogues of FA have also been synthesized and used to conduct photolabeling studies.23 Notably, all of the previously reported side chain analogues of FA have reduced antibacterial activity relative to FA.
To provide access to FA derivatives with novel side chains, a modular synthetic route for rapid generation of a variety of compounds with modified side chains was envisioned. Aldehyde 11, generated previously by ozonolysis23 and herein via dihydroxylation followed by oxidative cleavage (Figure 3A), was selected as the pluripotent intermediate that could be transformed via olefinations, one-carbon homologations, and cross-metathesis reactions to the various analogues. Using 11, halogens were introduced via a one-carbon homologation reaction with carbon tetrachloride, carbon tetrabromide, and carbon tetraiodide, respectively, to generate intermediates 12–14 (Figure 3B). Subsequent deprotection with potassium carbonate in methanol afforded novel halogenated compounds 15–17 (Figure 3B). Construction of the analogous fluorinated compound was accomplished by treating aldehyde 11 with chlorodifluoroacetic acid and triphenylphosphine at elevated temperatures to generate 18 and deprotection to generate difluorinated derivative 19 (Figure 3C). Aldehyde 11 was also converted to olefin 20 and fully deprotected to generate 21 as previously reported (Figure 3D).37
Figure 3.
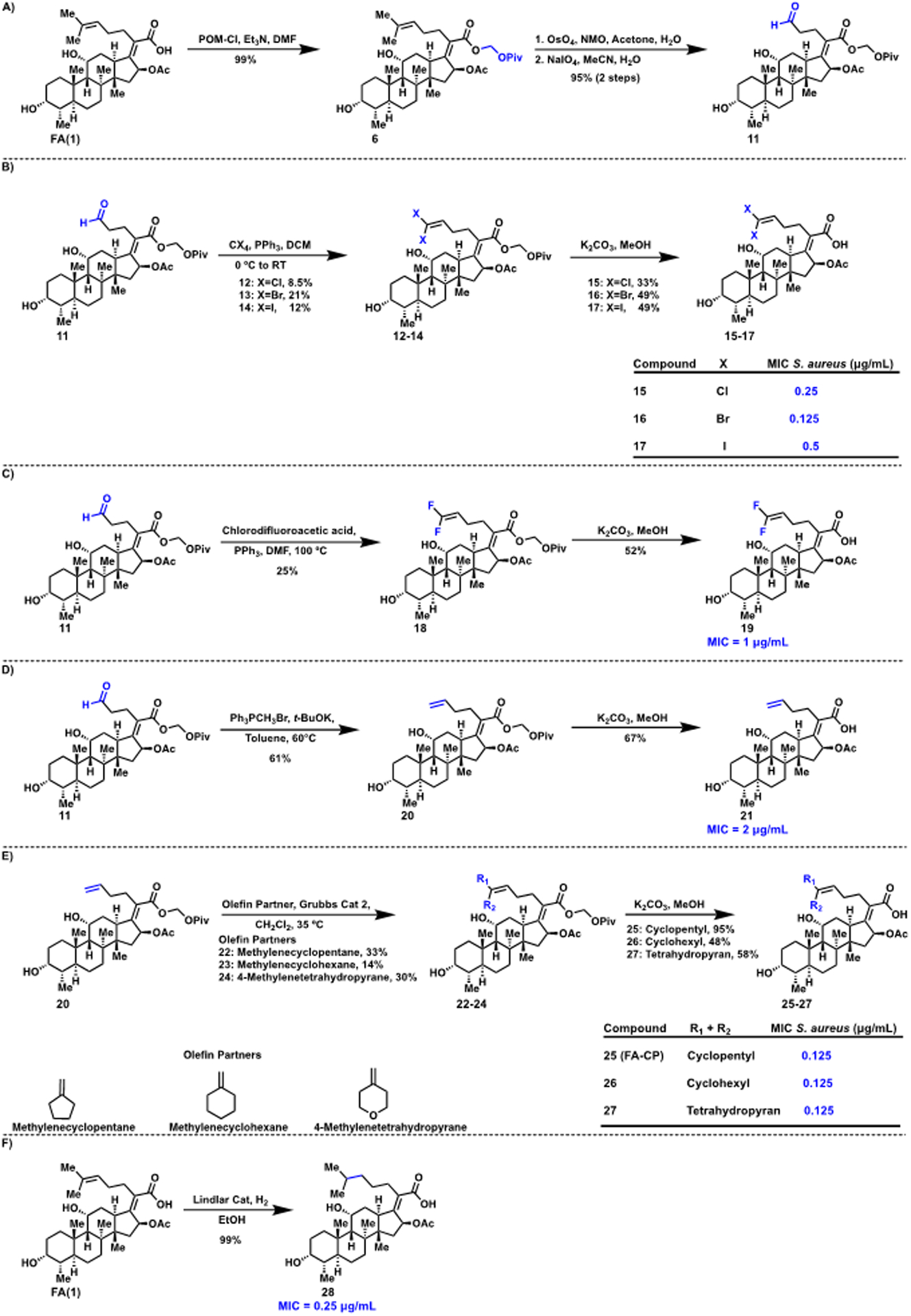
(A) Synthesis of aliphatic aldehyde derivative of FA. (B) Construction of halogenated side chain analogues of FA. (C) Generation of fluorinated side chain analogue of FA. (D) Synthesis of an FA analogue with a terminal olefin side chain. (E) Synthesis of cyclic side chain derivatives of FA. (F) Formation of saturated side chain derivative of FA. MICs determined using CLSI guidelines, against S. aureus ATCC 29213, n = 3.
Olefin 20 was used as another key point of diversity and divergence to generate compounds containing symmetrical side chains with the C24 and C25 double bond intact. Thus, to form cyclic derivatives, olefin 20 was coupled via a cross-metathesis reaction with methylenecyclopentane, methylenecyclohexane, and 4-methylenetetrahydropyran using Grubbs’ catalyst 2 to generate analogues 22–24. Deprotection generated novel cyclic side chain derivatives 25–27 (Figure 3E). Lastly, compound 28 with a fully saturated side chain was synthesized as previously reported using Lindlar’s catalyst (Figure 3F).22
With this collection of FA derivatives in hand, the MIC of each compound against S. aureus was determined (Figure 3). Compounds 15, 17, 19, 21, and 28 displayed modest decreases in potency relative to FA. Notably, analogues 16, 25 (hereafter referred to as FA-CP), 26, and 27 displayed equipotent activity to FA, suggesting the antibiotic potential of FA derivatives with modified side chains (Figure 3).
Evaluation of Resistance Frequency of Equipotent Analogues of FA.
The resistance frequency of the equipotent analogues (16, 25 (FA-CP), 26, and 27) was assessed in head-to-head experiments with FA. The large inoculum method was used in order to generate resistance mutants and derive the resistance frequency of FA and the equipotent analogues.39 The resistance frequency was determined at 2, 4, 8, 16, and 32X the MIC, which is 0.125 μg/mL for all compounds. The resistance frequency at the listed MICs was then compared head-to-head against FA, and statistical significance was then determined using one-way ANOVA with Tukey’s multiple comparisons. As shown in Figure 4A,B, compounds FA-CP and 26 displayed an improvement in resistance frequency relative to FA. FA-CP displayed a modest improvement in resistance frequency at 2 and 32X the MIC, while analogue 26 only displayed an improvement at 32X the MIC (Figure 4A,B). This is in contrast with analogues 16 and 27, which displayed a worse resistance frequency relative to FA (Figure 4C,D).
Figure 4.
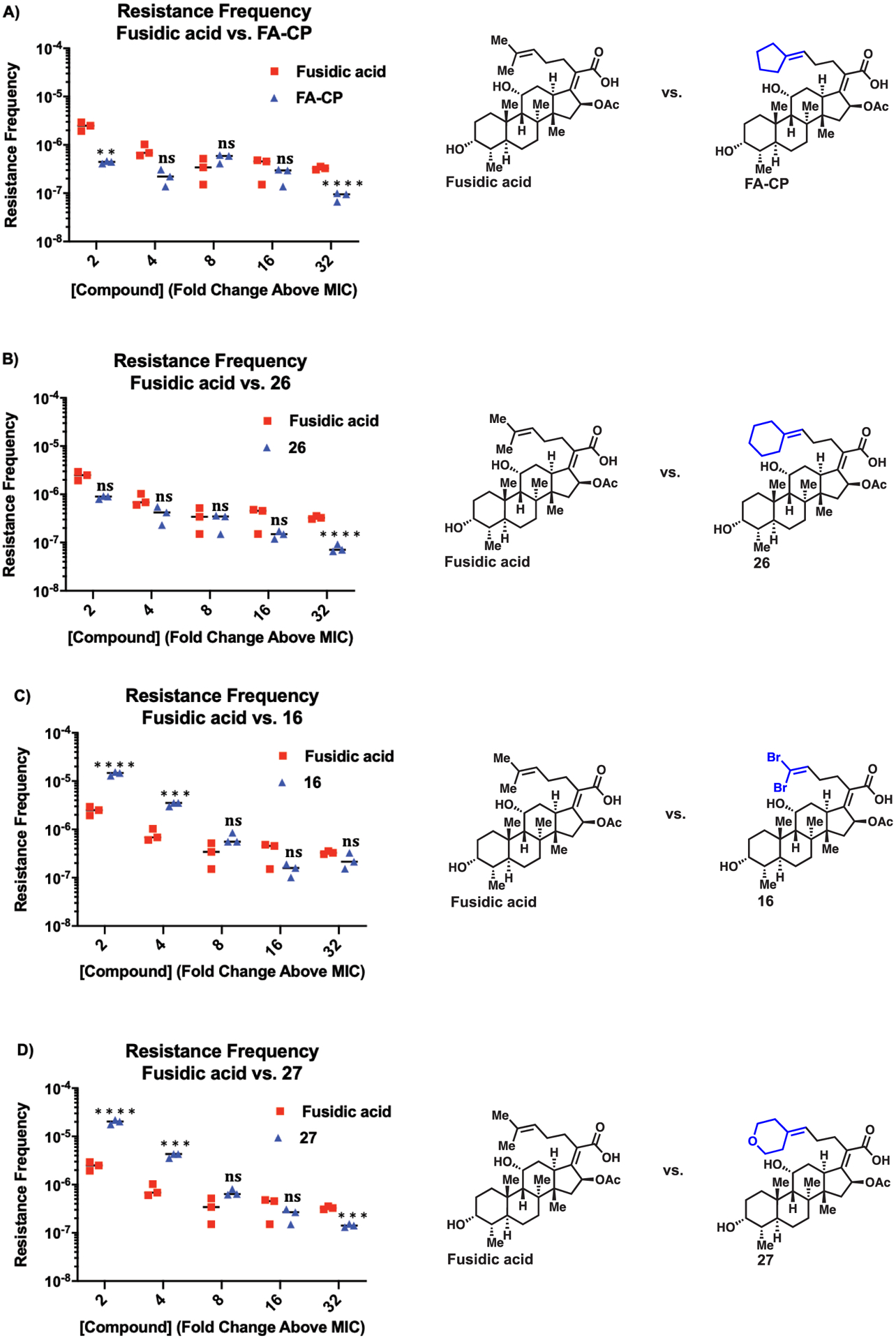
(A) Resistance frequency of FA-CP. (B) Resistance frequency of compound 26. (C) Resistance frequency of compound 16. (D) Resistance frequency of compound 27. Resistance frequency of FA and equipotent analogues was determined in S. aureus ATCC 29213. FA data is the same in all four experiments. Error is SEM, n = 3. NS = not significant, **p < 0.01, *** p < 0.001, and **** p < 0.0001.
Effect of Human Serum Binding, Assessment of Metabolic Stability, and Mammalian Cell Toxicity.
The translational potential of the novel derivatives was further investigated by assessing parameters important in antibacterial development, such as the effect of human serum binding, metabolic stability, and mammalian cell toxicity. FA is reported to be highly protein bound, with human plasma protein binding of 97%.40 Additionally, the MIC for FA when 50% human serum is included in the media shifts to 16–256X higher.40 In this work, the shift in MIC for S. aureus in 50% human serum was determined to be very similar for FA as compared to compounds 16, FA-CP, 26, and 27 (Table S1). In addition, the stability of the novel derivatives to mouse liver microsomes was similar to FA (Table S2). FA, 16, FA-CP, 26, and 27 exhibited low toxicity toward human foreskin fibroblast-1 cells (HFF-1), with all compounds giving IC50 values greater than 50 μM (Table S3).
Mode-of-Action of Novel FA Derivative.
FA-CP was selected for additional studies investigating the mode-of-action. For these studies, resistance mutants were generated at 32X the MIC of FA-CP and FA, and sequencing of the fusA gene (coding for EF-G, the target of FA) revealed mutations in fusA, suggesting that FA-CP engages the same target as FA. Specifically, for FA and FA-CP, the fusA gene was sequenced for 40 different resistant colonies generated at 32X the MIC, 20 colonies for each compound. As shown in Table 1, the resistance mutants observed for FA are in accord with those seen previously in analogous studies. Specifically, mutations F88L,11,41 T436I,11,41 H457N,41 and D434N11 have been identified in bacterial cell culture studies with S. aureus and FA. Additionally, H457Y11,41–44 has been observed in bacterial cell culture studies as well as clinical isolates of S. aureus that are resistant to FA. Consistent with the notion that FA-CP engages the same target as FA, single amino acid substitutions (H457Y, D434N, and F88L) within EF-G were found in the 20 different FA-CP colonies (Table 1).
Table 1.
Observed Mutations in the EF-G Protein (Encoded by fusA) and MIC Values for FA and FA-CP at 32X the MIC in S. aureus ATCC 29213
| strains resistant to FA | EF-G mutation | MIC for FA | strains resistant to FA-CP | EF-G mutation | MIC for FA-CP |
|---|---|---|---|---|---|
| FA-32X-1 | H457Y | 128 | FA-CP-32X-1 | H457Y | 32 |
| FA-32X-2 | T436I | 8 | FA-CP-32X-2 | H457Y | 64 |
| FA-32X-3 | H457Y | 128 | FA-CP-32X-3 | H457Y | 64 |
| FA-32X-4 | H457N | 256 | FA-CP-32X-4 | H457Y | 32 |
| FA-32X-5 | F88L | 256 | FA-CP-32X-5 | H457Y | 64 |
| FA-32X-6 | T436I | 16 | FA-CP-32X-6 | H457Y | 64 |
| FA-32X-7 | H457Y | 128 | FA-CP-32X-7 | H457Y | 64 |
| FA-32X-8 | H457Y | 128 | FA-CP-32X-8 | H457Y | 64 |
| FA-32X-9 | F88L | 256 | FA-CP-32X-9 | H457Y | 32 |
| FA-32X-10 | H457Y | 128 | FA-CP-32X-10 | D434N | 32 |
| FA-32X-11 | F88L | 256 | FA-CP-32X-11 | H457Y | 32 |
| FA-32X-12 | H457Y | 128 | FA-CP-32X-12 | D434N | 32 |
| FA-32X-13 | H457Y | 128 | FA-CP-32X-13 | H457Y | 64 |
| FA-32X-14 | H457Y | 128 | FA-CP-32X-14 | H457Y | 32 |
| FA-32X-15 | H457Y | 128 | FA-CP-32X-15 | H457Y | 64 |
| FA-32X-16 | F88L | 256 | FA-CP-32X-16 | F88L | 64 |
| FA-32X-17 | H457N | 256 | FA-CP-32X-17 | F88L | 64 |
| FA-32X-18 | D434N | 256 | FA-CP-32X-18 | H457Y | 64 |
| FA-32X-19 | H457Y | 128 | FA-CP-32X-19 | H457Y | 32 |
| FA-32X-20 | H457Y | 128 | FA-CP-32X-20 | H457Y | 32 |
The MICs of FA and FA-CP against these resistance mutants were also determined, and it was found that the mutants generated from FA-CP typically displayed a reduced shift in MIC upon resistance relative to the mutants generated from FA (Table 1). Specifically, the highest MIC observed for colonies generated from FA-CP was 64 μg/mL (fold increase from wild-type (WT) S. aureus: 512) (Table 1). In our in-house studies, the highest MIC observed for FA was 256 μg/mL (fold increase from WT S. aureus: 2048) (Table 1).
In a separate experiment, S. aureus strains resistant to FA were generated at 4, 8, 16, and 32X the MIC. Mutations found in the fusA gene of the resistant colonies are consistent with what has been observed in previous studies conducted with FA. Specifically, observed mutations in the fusA gene causing single amino acid substitutions, T385N,45 R464L,11 F88L,11,41 H457L,41 T436I,11,41 H457N,41 and D434N,11 have been identified in bacterial cell culture studies with S. aureus and FA. Furthermore, H457Y11,41–44 and P406L11,41,42,45 have been observed in bacterial cell culture studies as well as clinical isolates of S. aureus that are resistant to FA. FA-CP was evaluated against these 11 FA-resistant strains for antimicrobial activity. Although FA-CP displays cross-resistance with FA, expected as the target is conserved between the two compounds, FA-CP displays a 2–8-fold improvement in activity against these FA-resistant strains relative to FA (Table 2).
Table 2.
MIC Values for FA and FA-CP against 11 Different FA-Resistant Strains of S. aureus ATCC 29213a
| strains resistant to FA | EF-G mutation | MIC for FA | MIC for FA-CP |
|---|---|---|---|
| FA-4X-A | T385N | 8 | 2 |
| FA-4X-B | T385N | 8 | 2 |
| FA-8X-A | T385N | 8 | 2 |
| FA-8X-B | R464L | 16 | 8 |
| FA-16X-A | H457Y | 128 | 64 |
| FA-32X-A | F88L | 256 | 128 |
| FA-32X-B | P406L | 32 | 4 |
| FA-32X-C | H457L | 256 | 128 |
| FA-32X-D | T436I | 8 | 2 |
| FA-32X-E | H457N | 256 | 128 |
| FA-32X-F | D434N | 256 | 32 |
FA-resistant strains were generated at 4X (n = 2), 8X (n = 2), 16X (n = 1), or 32X (n = 6) the MIC as indicated. MICs determined using CLSI guidelines, n = 3.
Activity of FA-CP and FA against Multidrug-Resistant Clinical Isolates of S. aureus, E. faecium, and an S. aureus Strain Harboring the fusC Gene.
The activity of FA, FA-CP, and vancomycin was assessed against 29 different clinical isolates of S. aureus. FA and FA-CP displayed no cross-resistance with vancomycin and retained potent activity against the 29 different clinical isolates assessed (Figure 5A). Noteworthy is that within this collection of clinical isolates, there are strains with resistance to a wide variety of antibiotics, including cefoxitin, clindamycin, doxycycline, erythromycin, gentamicin, levofloxacin, oxacillin, penicillin, tetracycline, and trimethoprim/sulfamethoxazole (see Tables S4 and S5 for a full list of resistance profiles).
Figure 5.
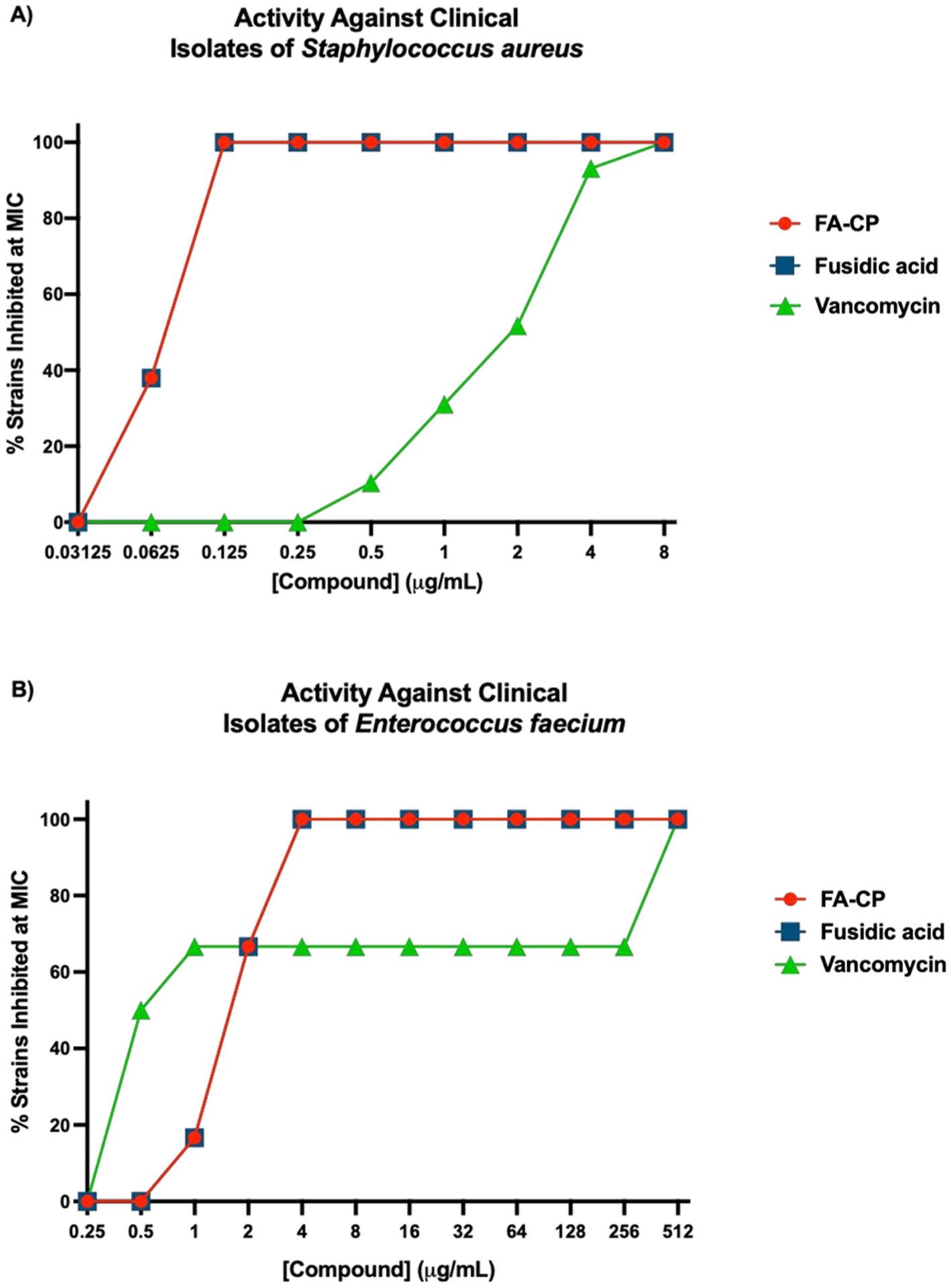
(A) Activity against 29 clinical isolates of S. aureus. (B) Activity against six different clinical isolates of E. faecium. MICs determined using CLSI guidelines, n = 3. See Tables S4–S6 for a list of resistance genes in clinical isolate panels. A full listing of this MIC data is in Tables S5 and S6.
The activity of FA and FA-CP was also assessed against a S. aureus strain that possesses the fusC gene, strain S. aureus ATCC BAA-1721.46 FA-CP displayed cross-resistance with FA, as both compounds displayed an MIC of 8 μg/mL against this strain.
The activity of FA, FA-CP, and vancomycin was also assessed against six different clinical isolates of E. faecium (Figure 5B). As a reference point, the MIC of FA and FA-CP against a sensitive strain of E. faecium ATCC 19434 is 2 μg/mL for both antibacterial agents. FA and FA-CP displayed no cross-resistance with vancomycin in the E. faecium clinical isolates. Excitingly, FA and FA-CP retained potent activity against VRE with high levels of vancomycin resistance (vancomycin MIC = 512 μg/mL). See Table S6 for a full list of resistance profiles.
Pharmacokinetic Studies and in vivo Efficacy of FA and FA-CP.
As a prelude to the mouse infection studies, the tolerability of FA (sodium fusidate) and FA-CP was assessed in mice at 50 mg/kg (intraperitoneal injection, once-a-day for 4 days), and both compounds were well tolerated by mice at this dose. Noteworthy is the rapid metabolism of FA in rodents, which is a major hurdle in the preclinical evaluation of FA analogues in mouse models.28,32 Head-to-head assessment of the pharmacokinetics of FA and FA-CP in mice revealed that these compounds have similar PK profiles (Figure 6A). The mouse and human PK profiles for FA are considerably different, as FA has a long half-life in humans of around 10–14 h,7,47 with predominant metabolism by the liver;48 the rapid clearance observed in mice could be driven by the formation of 3-epiFA, a metabolite that has not been observed in humans.28,32
Figure 6.
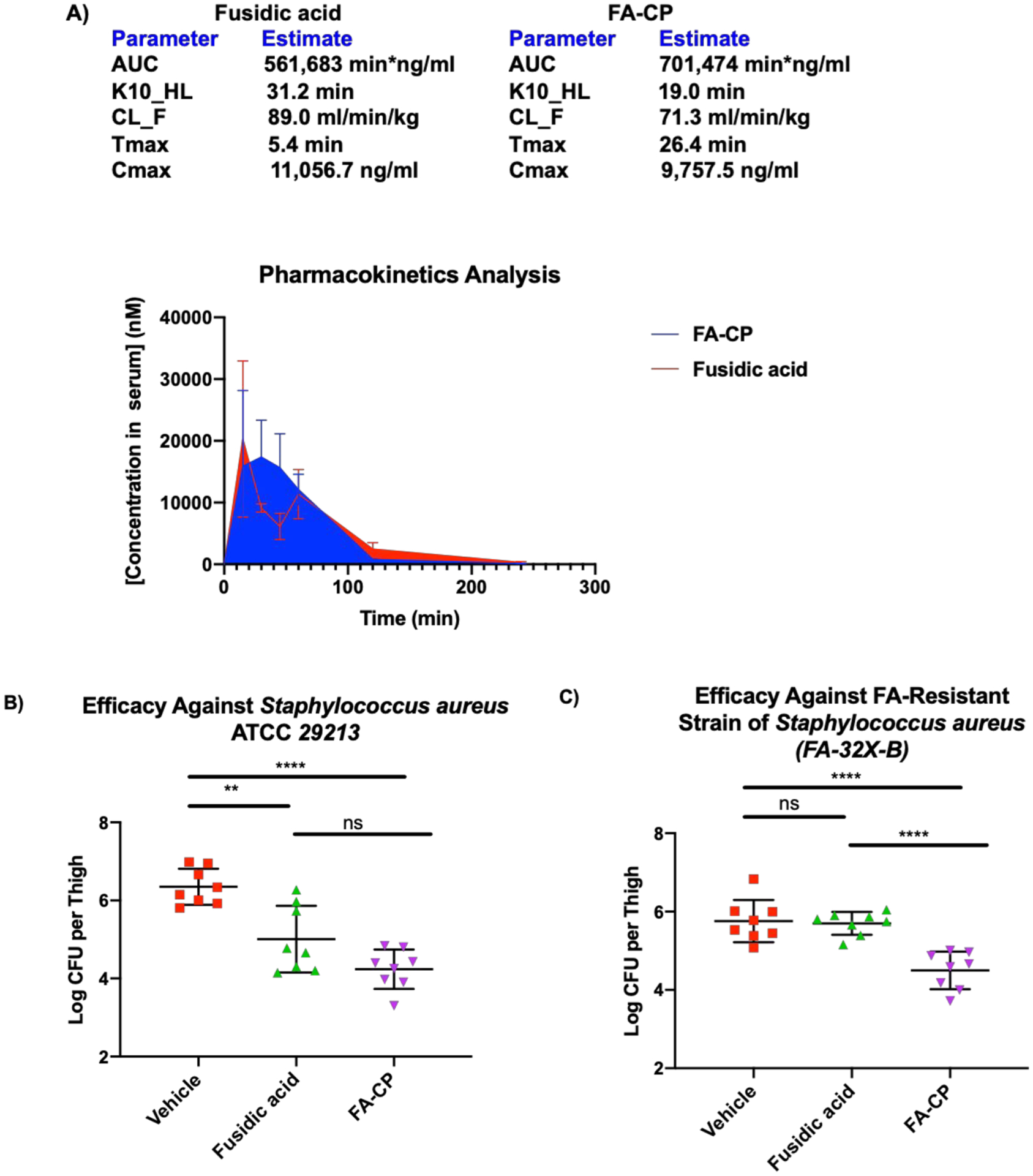
(A) Pharmacokinetic analysis of fusidic acid and FA-CP. C57BL/6 mice were treated with 50 mg/kg of fusidic acid or FA-CP via intraperitoneal injection, with three mice per time point (0, 15, 30, 45, 60, 120, and 240 min). After the stated time points, mice were sacrificed, and the serum concentrations of fusidic acid or FA-CP were determined by liquid chromatography–tandem mass spectrometry (LC–MS/MS). The data are illustrated as the mean, and error bars represent the standard error. The pharmacokinetic parameters illustrated above were calculated with a one-compartment model using a nonlinear regression program (Phoenix WinNonlin Version 8.1, Certara USA Inc., Princeton, NJ, USA). (B) S. aureus ATCC 29213 neutropenic thigh infection burden study. CD-1 mice were first rendered neutropenic with cyclophosphamide. Mice were injected on Day -4 to Day -2 with 150 mg/kg and on Day -1 with 100 mg/kg of cyclophosphamide, respectively. Acute thigh infections initiated in CD-1 mice with S. aureus ATCC 29213 (8.9 × 105 colony forming units (CFU) mouse−1 via intramuscular injection) were treated with vehicle, fusidic acid, and FA-CP (eight mice per group) at 1, 2, and 3 h postinfection (50 mg/kg via intraperitoneal injection), and the bacterial burden was evaluated at 24 h postinfection. (C) S. aureus (FA-32X-B) neutropenic thigh infection study. CD-1 mice were first rendered neutropenic with cyclophosphamide as described above. Acute thigh infections initiated in CD-1 mice with S. aureus (FA-32X-B) (1.08 × 106 CFU mouse−1 via intramuscular injection) were treated with vehicle, fusidic acid, and FA-CP (eight mice per group) at 1, 2, and 3 h postinfection (50 mg/kg via intraperitoneal injection), and the bacterial burden was evaluated at 8 h postinfection. Drugs were formulated with 5% DMSO, 10% Tween 20, 85% PBS immediately before injection. The data is shown as means ± s.d., and statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons. NS indicates not significant (P > 0.05), **P < 0.01, ****P < 0.0001.
Encouraged by the efficacy of FA-CP against the panel of clinical isolates, the lack of toxicity to mammalian cells, and the promising tolerability and pharmacokinetic data, FA-CP was compared head-to-head to FA in in vivo efficacy studies; the design of these efficacy studies was guided by mouse models previously conducted with FA.49 First, a sensitive strain of S. aureus was assessed in a neutropenic thigh infection burden model utilizing S. aureus ATCC 29213, in which FA-CP and FA display the same MIC of 0.125 μg/mL. After infection, three doses of FA-CP and FA were administered (50 mg/kg, intraperitoneal injection) 1, 2, and 3 h postinfection to separate groups of mice. Mice were sacrificed 24 h postinfection, and bacterial burden in the thigh muscle tissue homogenates was determined by serial dilution plating onto tryptic soy agar. Significant reductions in bacterial burden were observed with FA and FA-CP (Figure 6B).
The improved potency of FA-CP against FA-resistant strains motivated and guided the second neutropenic thigh infection burden model. An FA-resistant S. aureus strain, FA-32X-B, generated at 32X the MIC of FA, was used for this study. FA has an MIC of 32 μg/mL, while FA-CP displays an MIC of 4 μg/mL (Table 2). After infection, three doses of FA-CP and FA were administered (50 mg/kg, intraperitoneal injection) 1, 2, and 3 h postinfection to separate groups of mice. Mice were sacrificed 8 h postinfection, and bacterial burden in the thigh muscle tissue homogenates was determined by serial dilution plating onto tryptic soy agar. This resulted in reductions in bacterial burden with FA-CP and no efficacy with FA (Figure 6C). This study is, to our knowledge, the first mouse bacterial infection model in which a novel derivative of FA displays improved efficacy relative to FA.
DISCUSSION
Despite FA being approved in Europe since the 1960s to treat problematic S. aureus infections, there have been no follow-on drugs, and FA remains the only member of the fusidane class to be used clinically.7 The high resistance frequency, drastic shifts in MIC upon bacterial resistance to FA, and lack of effective derivatives have narrowed the development path for this antibiotic class.
A considerable challenge in the creation of new and effective derivatives of FA has been a clouded structure–activity relationship for this compound, due to heterogeneity in the reported biological data for derivatives, with many compounds evaluated against the malaria parasite Plasmodium falciparum21,26,30 and Mycobacterium species28,31,32 but not against Gram-positive bacteria. In this work, we have constructed and assessed both previously synthesized derivatives and novel derivatives, allowing for a clearer structure–activity relationship (SAR) to emerge. Most notably, while previous investigations suggested that changes in the hydrophobic side chain of FA diminished antibacterial activity,17,18,22,23,37,38 here we show that certain changes at this position are not only tolerable but lead to derivatives with favorable resistance profiles compared to FA.
Specifically, the reduced shift (relative to that observed for FA) in MIC upon resistance of S. aureus to FA-CP is an important feature of this compound. The highest MIC for the resistance mutants generated to FA-CP was 64 μg/mL, versus 256 μg/mL for resistance mutants generated to FA. In principle, this improvement could allow for administration of less FA-CP or a similar compound. Although FA is considered a safe antibiotic, gastrointestinal side effects have been observed in 30–58% of patients treated with FA, and up to 30% of patients have elevated bilirubin.47
Another interesting and potentially useful aspect of FA-CP is the ability of this compound to retain some activity against strains of S. aureus that are resistant to FA, including in a mouse model of infection. Specifically, FA-CP displayed in vivo efficacy against an FA-resistant S. aureus strain generated at 32X the MIC of FA (strain FA-32X-B). Our sequencing studies reveal that FA-32X-B possesses the mutation P406L in the EF-G protein. Noteworthy is the fact that this mutation has not only been observed in bacterial cell culture studies but also been identified in clinical isolates of S. aureus,11,42 highlighting the potential of FA-CP to retain efficacy in vivo against some clinical isolates of S. aureus that are resistant to FA.
Sequencing studies also revealed some interesting differences between the mutational profile of bacteria arising from FA versus FA-CP treatment. In our in-house studies, five different amino acid changes were observed in EF-G for FA-resistant S. aureus, while only three different amino acid mutations in EF-G were observed for FA-CP-resistant S. aureus. Mutations in EF-G have been classified into four different groups based on their role in resistance to FA: Group A (mutations affecting FA binding), Group B (mutations affecting EF-G ribosome interactions), Group C (mutations affecting EF-G conformation), and Group D (mutations affecting EF-G stability).50 Moreover, Group A mutations involve amino acids that are in direct contact with FA and with residues that can shape the drug pocket.50 Additionally, it has been postulated that Group C mutations are related to the interdomain orientation of EF-G, which can affect the FA-binding pocket along with the conformation dynamics and FA locking of EF-G.50 The mutations in FA-resistant strains found in our studies have been previously classified and belong to Group A and Group C, whereas all the mutations observed for FA-CP-resistant strains belong to Group A, indicating that these mutations are in regions that can affect FA binding.
While mutation to fusA, the gene encoding for EF-G, is a major mechanism of resistance for FA, it is important to note that other resistance mechanisms are operational in clinical isolates, specifically, the horizontal acquisition of plasmids harboring fusB or fusC.15 These resistance genes encode protective proteins that drive the dissociation of the EF-G ribosome complex from FA.51 Analogous to the shifts in MIC observed for FA upon mutations to the fusA gene, an increase in MIC has also been reported in clinical isolates that possess the fusB and/or fusC genes. For example, MIC ranges of 2–64 μg/mL in staphylococcal clinical isolates have been reported for clinical isolates that carry the fusC gene, and MIC ranges of 4–1024 μg/mL have been reported for strains that possess the fusB gene.15,44,46 Due to the clinical relevance of strains that have acquired the fusC gene, the MIC of FA-CP was assessed against a strain that possesses this gene. FA-CP displays the same MIC as FA against S. aureus ATCC BAA-1721, with an MIC of 8 μg/mL. While resistance driven by the fusB and fusC genes is worrisome, the primary mechanism of clinical resistance to FA is still resistance mediated by mutations in the fusA gene.7,42 Specifically, mutation L461K found in clinical isolates of S. aureus is of high concern due its drastic shift in MIC (MIC > 256 μg/mL) upon a single amino acid mutation.7 Perhaps even more problematic are clinical isolates of S. aureus that are resistant to FA and possess four different EF-G amino acid alterations within the same strain. Five such clinical isolates have been isolated, and they all showed the following alterations in EF-G: V90I, H457Q, L461K, and A655V.42 All of these strains have an MIC greater than 256 μg/mL, highlighting the prominent role that mutations to the fusA gene play in the development of resistance to FA in the clinic.42 It may be possible to design FA derivatives that retain strong binding to these mutant versions of EF-G, and FA derivatives with a further improved resistance profile can also be envisioned.
FA-CP is the first analogue to outperform FA in a mouse infection model, which more broadly suggests the potential for certain FA derivatives. The important improvement in the resistance profile observed for FA-CP has some precedence in antibacterial drug discovery, for example in the dihydrofolate reductase inhibitor iclaprim52 and leucyl-tRNA synthetase inhibitor DS86760016,53 with improvement in each case likely due to subtly different modes of target engagement relative to the parent antibiotics (trimethoprim and GSK2251052, respectively). With access to derivatives now facile and the ability to improve upon its resistance profile now demonstrated, novel antibiotics of the fusidane class can be developed for the treatment of a variety of bacterial infections. Finally, as demonstrated by its outstanding activity against permeability defect Escherichia coli,54 FA has potential against Gram-negative bacteria but is accumulation limited; the detailed SAR reported herein should assist in exploiting this untapped potential of FA to combat drug-resistant Gram-negative infections.
METHODS
Bacterial Strains.
S. aureus ATCC 29213, E. faecium ATCC 19434, and S. aureus ATCC BAA-1721 were obtained from the American Type Culture Collection (ATCC). Resistant strains of E. faecium were obtained from the CDC. Resistant strains of S. aureus were obtained from the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) and the CDC.
Antimicrobial Susceptibility Tests.
Susceptibility testing was performed in biological triplicate, using the microdilution broth method as outlined by CLSI. Bacteria were cultured with cation-adjusted Muller-Hinton broth (Sigma-Aldrich, Cat# 90922) in round-bottom 96-well plates (Corning, Cat# 3788). Human serum (ultrafiltrate, unspecified gender, 30K Dalton membrane filtered) was purchased from BioIVT (Hicksville, NY).
Cell Culture.
HFF-1 cells were obtained from ATCC. HFF-1 cells were grown in DMEM with 15% fetal bovine serum (Gemini Benchmark, Cat# 100–106), 100 μg/mL of penicillin, and 100 μg/mL of streptomycin. All cells were cultured at 37 °C in a 5% CO2 environment. Media was prepared by the University of Illinois School of Chemical Sciences Cell Media Facility.
Cell Viability.
Cells were harvested, seeded in a 96-well plate, and allowed to adhere. Cells were treated with investigational compounds in DMSO (1% final concentration). Cells were incubated for 72 h before viability was assessed by the Alamar Blue Assay. Raptinal (20 μM) was used as a dead control.
Mouse Liver Microsome Stability Assay.
A mixture of PBS (pH 7.4), NADPH regenerating system solution A (Corning Life Sciences), and NADPH regenerating system solution B (Corning Life Sciences) was incubated at 37 °C in a shaking incubator for 5 min. Next, compound was added in DMSO (final concentration 50 μM, 0.5% DMSO) before ice-cold mouse liver microsomes (Thermo Fisher, male CD-1 mice, pooled) were added (final protein concentration of 1 mg/mL). An aliquot was immediately removed, quenched with an equal volume of 100 μM internal standard, and centrifuged at 13 000 rcf for 3 min. The reactions were incubated at 37 °C in a shaking incubator for 3 h. A second aliquot was removed and quenched. Samples were analyzed with the 5500 QTRAP LC-MS/MS system (Sciex, Framingham, MA) in the Metabolomics Laboratory of Roy J. Carver Biotechnology Center, University of Illinois at Urbana–Champaign. Software Analyst 1.6.2 was used for data acquisition and analysis. The 1200 series HPLC system (Agilent Technologies, Santa Clara, CA) includes a degasser, an autosampler, and a binary pump. The LC separation was performed on an Agilent Sb-Aq column (4.6 × 50 mm, 5 μm) with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The flow rate was 0.3 mL/min. The linear gradient was as follows: 0–3 min, 100% A; 10–16 min, 5% A; 16.5–22 min, 100% A. The autosampler was set at 10 °C. The injection volume was 1 μL. Mass spectra were acquired under both positive (ion spray voltage +5500 V) and negative (ion spray voltage −4500 V) electrospray ionization (ESI). The source temperature was 450 °C. The curtain gas, ion source gas 1, and ion source gas 2 were 33, 65, and 60 psi, respectively. Multiple reaction monitoring (MRM) was used for quantitation.
Selection of Resistant Mutants.
Resistant mutants were selected via the large inoculum method. Briefly, S. aureus ATCC 29213 (1 × 109 CFU) was plated on 100 mm plates of LB agar containing 4, 2, 1, 0.5, and 0.25 μg/mL. Colonies were visible after incubation at 37 °C for 24 h. Resistant colonies were confirmed by streaking on selective media with the same concentration of fusidic acid and compounds 16, FA-CP (25), 26, 27.
Sequencing of fusA.
FusA was amplified by colony polymerase chain reaction (PCR). Colonies were picked and diluted in 100 μL of sterile H2O. PCR reactions are set up by combining MiFi Mix (Bioline, London, UK), 20 μM primer mix [fusA-F2, fusA_seq1, forward EF-G2, and reverse EF-G] (S. aureus ATCC 29213), template DNA, and H2O. Reactions were performed on a C1000 Thermal Cycler (Bio-Rad, Hercules, CA) with the following conditions: 5 min of denaturation at 95 °C, followed by 30 cycles of 20 s at 95 °C, 20 s at 50 °C, and either 1 min (fusA1) or 1.5 min (fusA2) at 72 °C. A 10 μL portion of the PCR reaction mixture was analyzed by agarose gel to confirm the product. PCR reactions were purified using GeneJET PCR Purification Kit (Thermo Scientific). PCR amplicons were submitted to the Core DNA Sequencing Facility at the University of Illinois at Urbana–Champaign for Sanger sequencing with the following primers to sequence the fusA [fusA2, fusA_seq1, forward EF-G2, forward EF-G3, forward EF-G4]. See the Supporting Information to find the sequence of the primers used in this study.
Mouse Maximum Tolerated Dose (MTD) of Sodium Fusidate and FA-CP (25).
The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Illinois at Urbana–Champaign (protocol number: 16144 and 19181). In these studies, 10- to 12-week-old female C57BL/6 mice purchased from Charles River were used. The maximum tolerated dose (MTD) of a single compound was determined first. Sodium fusidate and FA-CP (25) were formulated in 5% DMSO, 10% Tween 20, and 85% PBS. Sodium fusidate and FA-CP (25) were given by intraperitoneal (IP) injection. All the mice were monitored for signs of toxicity for 2 weeks. For multiple doses, the compound was given by daily IP injection for 4 consecutive days, and mice were monitored for signs of toxicity for 1 month. The MTD was the highest dosage with acceptable toxicity (e.g., < 20% weight loss). Sodium fusidate and FA-CP (25) were well tolerated as a single dose of 50 mg/kg. Further analysis showed that sodium fusidate and FA-CP (25) were well tolerated with daily dosing of 50 mg/kg for 4 consecutive days. The MTD of FA-CP (25) was used to inform the dosing schedule used in subsequent efficacy studies.
Pharmacokinetic Assessment of Sodium Fusidate and FA-CP (25).
The protocol was approved by the IACUC at the University of Illinois at Urbana–Champaign (protocol number: 16144, 19181). In these studies, 10- to 12-week-old female C57BL/6 mice purchased from Charles River were used. The compounds were formulated in 5% DMSO, 10% Tween 20, and 85% PBS. Mice were treated with sodium fusidate or FA-CP (25) (50 mg/kg) via IP injection with three mice per time point (15, 30, 45, 60, 120, and 240 min). At specific time points, mice were sacrificed, blood was collected and centrifuged, and the serum was frozen at −80 °C until analysis. The proteins in a 10 μL aliquot of serum were precipitated by the addition of 50 μL of acetonitrile with the addition of 10 μL of 1.6 μg/mL of internal standard (sodium fusidate was the internal standard when measuring FA-CP (25), and FA-CP (25) was the internal standard when measuring sodium fusidate). The sample was then vortexed and centrifuged to remove the proteins. Supernatants were analyzed with the QTRAP 5500 LC-MS/MS system (Sciex) in the Metabolomics Laboratory of the Roy J. Carver Biotechnology Center, University of Illinois at Urbana–Champaign. Software Analyst 1.6.2 was used for data acquisition and analysis. The 1200 Series HPLC System (Agilent Technologies) includes a degasser, an autosampler, and a binary pump. The LC separation was performed on an Agilent Zorbax SB-Aq column (4.6 × 50 mm; 5 μm) with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The flow rate was 0.3 mL/min. The linear gradient was as follows: 0–1 min, 95% A; 8–13 min, 0% A; 8.1–18.5 min, 95% A. The autosampler was set at 10 °C. The injection volume was 5 μL. Mass spectra were acquired under negative electrospray ionization with a voltage of −4500 V. The source temperature was 450 °C. The curtain gas, ion source gas 1, and ion source gas 2 were 32, 60, and 60 psi, respectively. MRM was used for quantitation: fusidic acid: m/z 515.3 → 393.3, FA-CP (25): m/z 541.4 → 437.2. The limit of quantitation of (S/N = 10) was 1 nM. Pharmacokinetic parameters were calculated with a one-compartment model using a nonlinear regression program (Phoenix WinNonlin Version 8.1; Certara USA).
Neutropenic Thigh Infection Burden Study with Sodium Fusidate and FA-CP (25).
Mouse studies were carried out in strict accordance with the recommendations in the guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the IACUC at the University of Illinois at Urbana–Champaign (protocol number: 17271). Briefly, seven-week-old male CD-1 mice (cohorts of eight) were rendered neutropenic by IP injection of cyclophosphamide (150 mg/kg on Day -4 to Day -2 and 100 mg/kg on Day -1). On Day -1, mice were anesthetized with a combination of xylazine/ketamine, and furs on the right hind thigh were removed by clipping with a pair of scissors followed by application of depilating gel (Veet Aloe Vera Legs & Body Hair Remover Gel Cream). After 24 h, mice were anesthetized with isoflurane, and infected with S. aureus ATCC 29213 or the S. aureus FA-resistant strain 32X-B at a concentration of ~1 × 106 CFUs (in 50 μL) by injection into the thigh muscle (bicep femoris) with a 25G 5/6″ needle. Infected mice were intraperitoneally treated with vehicle (85% PBS, 10% Tween, 5% DMSO), 50 mg/kg of sodium fusidate, or FA-CP (25) at 1, 2, and 3 h postinfection (hpi) individually in 100 μL of volume. Infected animals were monitored for myositis and lameness until euthanasia. At indicated times (24 hpi for the 50 mg/kg cohorts for the S. aureus ATCC 29213 infection model and 8 hpi for the 50 mg/kg cohorts for the S. aureus (FA-32X-B) infection model), mice were euthanized with CO2 asphyxiation from a compressed gas source followed by cervical dislocation. Infected thigh muscle tissues were harvested and homogenized with a Omni Soft Tissue Tip Homogenizer (OMNI International) in 2 mL of sterile PBS. Bacterial burden in the tissue homogenates was determined by serial dilution plating onto tryptic soy agar.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH (AI136773) and the University of Illinois at Urbana—Champaign. We thank L. Li (Metabolomics Center, Roy J. Carver Biotechnology Center, UIUC) for LC-MS/MS analysis and Professor Levent Dirikolu (School of Veterinary Medicine, Louisiana State University) for the help with PK parameter calculations. M.G.C. and A.G. are members of the NIH Chemistry-Biology Interface Training Grant (T32-GM136629). M.G.C. is also supported by the NSF Graduate Research Fellowship (NSF-GRFP). We are also grateful for the support services of the NMR and mass spectrometry facilities of the University of Illinois at Urbana-Champaign.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00869.
Tables S1–S6, MIC values for FA and equipotent analogues in S. aureus in 50% human serum, stability to mouse liver microsomes, IC50 in human foreskin fibroblast-1 cells, list of antibiotics and associated reference numbers with clinically relevant levels of resistance in S. aureus and E. faecium, antimicrobial assessment of FA, vancomycin, and FA-CP in panel of multidrug-resistant clinical isolates of S. aureus, and antimicrobial assessment of FA, vancomycin, and FA-CP in panel of multidrug-resistant clinical isolates of E. faecium. Synthetic procedures and characterization for core and side chain derivatives of FA (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.0c00869
The authors declare the following competing financial interest(s): The University of Illinois has filed patents on some of the compounds described in this manuscript.
Contributor Information
Martin Garcia Chavez, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States.
Alfredo Garcia, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States.
Hyang Yeon Lee, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States.
Gee W. Lau, Department of Pathobiology, College of Veterinary Medicine, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States;.
Erica N. Parker, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States
Kailey E. Komnick, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States
Paul J. Hergenrother, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States;.
REFERENCES
- (1).Cassini A, Högberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, Colomb-Cotinat M, Kretzschmar ME, Devleesschauwer B, Cecchini M, Ouakrim DA, Oliveira TC, Struelens MJ, Suetens C, Monnet DL, Strauss R, Mertens K, Struyf T, Catry B, Latour K, Ivanov IN, Dobreva EG, Tambic Andraševic A, Soprek S, Budimir A, Paphitou N,Žemlicková H, Schytte Olsen S, Wolff Sönksen U, Märtin P, Ivanova M, Lyytikäinen O, Jalava J, Coignard B, Eckmanns T, Abu Sin M, Haller S, Daikos GL, Gikas A, Tsiodras S, Kontopidou F, Tóth Á, Hajdu Á, Guólaugsson Ó, Kristinsson KG, Murchan S, Burns K, Pezzotti P, Gagliotti C, Dumpis U, Liuimiene A, Perrin M, Borg MA, de Greeff SC, Monen JCM, Koek MBG, Elstrøm P, Zabicka D, Deptula A, Hryniewicz W, Caniça M, Nogueira PJ, Fernandes PA, Manageiro V, Popescu GA, Serban RI, Schréterová E, Litvová S,Štefkovicová M, Kolman J, Klavs I, Korošec A, Aracil B, Asensio A, Pérez-Vázquez M, Billström H, Larsson S, Reilly JS, Johnson A, and Hopkins S (2019) Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect. Dis 19, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Theuretzbacher U, Outterson K, Engel A, and Karlén A (2020) The global preclinical antibacterial pipeline. Nat. Rev. Microbiol 18, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).US Department of Health and Human Services, CDC. (2019) Antibiotic Resistance Threats in the United States 2019, US Department of Health and Human Services, CDC, Atlanta, GA. [Google Scholar]
- (4).Markwart R, Willrich N, Haller S, Noll I, Koppe U, Werner G, Eckmanns T, and Reuss A (2019) The rise in vancomycin-resistant Enterococcus faecium in Germany: data from the German Antimicrobial Resistance Surveillance (ARS). Antimicrob. Resist. Infect. Control 8, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Godtfredsen WO, Jahnsen S, Lorck H, Roholt K, and Tybring L (1962) Fusidic Acid: a New Antibiotic. Nature 193, 987–987. [DOI] [PubMed] [Google Scholar]
- (6).Tanaka N, Kinoshita T, and Masukawa H (1968) Mechanism of protein synthesis inhibition by fusidic acid and related antibiotics. Biochem. Biophys. Res. Commun 30, 278–283. [DOI] [PubMed] [Google Scholar]
- (7).Fernandes P (2016) Fusidic Acid: A Bacterial Elongation Factor Inhibitor for the Oral Treatment of Acute and Chronic Staphylococcal Infections. Cold Spring Harbor Perspect. Med 6, a025437–a025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jones RN, Castanheira M, Rhomberg PR, Woosley LN, and Pfaller MA (2010) Performance of fusidic acid (CEM-102) susceptibility testing reagents: broth microdilution, disk diffusion, and Etest methods as applied to Staphylococcus aureus. J. Clin. Microbiol 48, 972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cempra. Cempra’s Fusidic Acid Achieves Primary Endpoint in Phase 3 Study of ABSSSI, http://ir.melinta.com/static-files/d621fa2f-d0d4-4fc3-833f-a64286cfb7ed (accessed November 11, 2020).
- (10).Melinta Therapeutics. Cempra, Inc. Receives U.S. Orphan Drug Designation for Taksta(TM) for Treatment of Prosthetic Joint Infections, http://ir.melinta.com/news-releases/news-release-details/cempra-inc-receives-us-orphan-drug-designation-takstatm (accessed November 11, 2020).
- (11).Nagaev I, Björkman J, Andersson DI, and Hughes D (2001) Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Mol. Microbiol 40, 433–439. [DOI] [PubMed] [Google Scholar]
- (12).Silver LL (2017) The Antibiotic Future. Top. Med. Chem 25, 31–67. [Google Scholar]
- (13).Silver LL (2016) Appropriate Targets for Antibacterial Drugs. Cold Spring Harbor Perspect. Med. 6, a030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tomlinson JH, Kalverda AP, and Calabrese AN (2020) Fusidic acid resistance through changes in the dynamics of the drug target. Proc. Natl. Acad. Sci. U. S. A 117, 25523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Abouelfetouh A, Kassem M, Naguib M, and El-Nakeeb M (2017) Investigation and Treatment of Fusidic Acid Resistance Among Methicillin-Resistant Staphylococcal Isolates from Egypt. Microb. Drug Resist 23, 8–17. [DOI] [PubMed] [Google Scholar]
- (16).Von Daehne W, Godtfredsen WO, and Rasmussen PR (1979) Structure-Activity Relationships in Fusidic Acid-Type Antibiotics. Adv. Appl. Microbiol 25, 95–146. [DOI] [PubMed] [Google Scholar]
- (17).Duvold T, Sørensen MD, Björkling F, Henriksen AS, and Rastrup-Andersen N (2001) Synthesis and Conformational Analysis of Fusidic Acid Side Chain Derivatives in Relation to Antibacterial Activity. J. Med. Chem 44, 3125–3131. [DOI] [PubMed] [Google Scholar]
- (18).Janssen G, and Vanderhaeghe H (1967) Modification of the Side Chain of Fusidie Acid (Ramycin). J. Med. Chem 10, 205–208. [DOI] [PubMed] [Google Scholar]
- (19).Bodley JW, and Godtfredsen WO (1972) Studies on translocation XI: Structure-function relationships of the fusidane-type antibiotics. Biochem. Biophys. Res. Commun 46, 871–877. [DOI] [PubMed] [Google Scholar]
- (20).Willie GR, Richman N, Godtfredsen WO, and Bodley JW (1975) Translocation. XV. Characteristics of and structural requirements for the interaction of 24,25-dihydrofusidic acid with ribosome. elongation factor G complexes. Biochemistry 14, 1713–1718. [DOI] [PubMed] [Google Scholar]
- (21).Kaur G, Pavadai E, Wittlin S, and Chibale K (2018) 3D-QSAR Modeling and Synthesis of New Fusidic Acid Derivatives as Antiplasmodial Agents. J. Chem. Inf. Model 58, 1553–1560. [DOI] [PubMed] [Google Scholar]
- (22).Wu P-P, He H, Hong WD, Wu T-R, Huang G-Y, Zhong Y-Y, Tu B-R, Gao M, Zhou J, Zhao S-Q, Li D-L, Xu X-T, Sheng Z-J, Ward SA, O’Neill PM, and Zhang K (2018) The biological evaluation of fusidic acid and its hydrogenation derivative as antimicrobial and anti-inflammatory agents. Infect. Drug Resist 11, 1945–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Riber D, Venkataramana M, Sanyal S, and Duvold T (2006) Synthesis and Biological Evaluation of Photoaffinity Labeled Fusidic Acid Analogues. J. Med. Chem 49, 1503–1505. [DOI] [PubMed] [Google Scholar]
- (24).Dauben WG, Kessel CR, Kishi M, Somei M, Tada M, and Guillerm D (1982) A formal total synthesis of fusidic acid. J. Am. Chem. Soc 104, 303–305. [Google Scholar]
- (25).Ireland RE, Giger R, and Kamata S (1977) Studies on the total synthesis of steroidal antibiotics. 3. Generation and correlation of tetracyclic derivatives from the degradation of fusidic acid and total synthesis. J. Org. Chem 42, 1276–1282. [DOI] [PubMed] [Google Scholar]
- (26).Kaur G, Singh K, Pavadai E, Njoroge M, Espinoza-Moraga M, De Kock C, Smith PJ, Wittlin S, and Chibale K (2015) Synthesis of fusidic acid bioisosteres as antiplasmodial agents and molecular docking studies in the binding site of elongation factor-G. MedChemComm 6, 2023–2028. [Google Scholar]
- (27).Hill CK, and Hartwig JF (2017) Site-selective oxidation, amination and epimerization reactions of complex polyols enabled by transfer hydrogenation. Nat. Chem 9, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Njoroge M, Kaur G, Espinoza-Moraga M, Wasuna A, Dziwornu GA, Seldon R, Taylor D, Okombo J, Warner DF, and Chibale K (2019) Semisynthetic Antimycobacterial C-3 Silicate and C-3/C-21 Ester Derivatives of Fusidic Acid: Pharmacological Evaluation and Stability Studies in Liver Microsomes, Rat Plasma, and Mycobacterium tuberculosis culture. ACS Infect. Dis 5, 1634–1644. [DOI] [PubMed] [Google Scholar]
- (29).Suchkova GS, Lokshin GB, Kuzovkov AD, and Chernyshev AI (1979) Oxidation of fusidic acid. Chem. Nat. Compd 15, 54–56. [Google Scholar]
- (30).Espinoza-Moraga M, Singh K, Njoroge M, Kaur G, Okombo J, De Kock C, Smith PJ, Wittlin S, and Chibale K (2017) Synthesis and biological characterisation of ester and amide derivatives of fusidic acid as antiplasmodial agents. Bioorg. Med. Chem. Lett 27, 658–661. [DOI] [PubMed] [Google Scholar]
- (31).Dziwornu GA, Kamunya S, Ntsabo T, and Chibale K (2019) Novel antimycobacterial C-21 amide derivatives of the antibiotic fusidic acid: synthesis, pharmacological evaluation and rationalization of media-dependent activity using molecular docking studies in the binding site of human serum albumin. MedChemComm 10, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Strydom N, Kaur G, Dziwornu GA, Okombo J, Wiesner L, and Chibale K (2020) Pharmacokinetics and Organ Distribution of C-3 Alkyl Esters as Potential Antimycobacterial Prodrugs of Fusidic Acid. ACS Infect. Dis 6, 459–466. [DOI] [PubMed] [Google Scholar]
- (33).Tanabe M, Yasuda DM, and Peters RH (1977) Partial synthesis of fusidic acid. Tetrahedron Lett 18, 1481–1484. [Google Scholar]
- (34).Krakower GW, Van Dine HA, Diassi PA, and Bacso I (1967) Transformations of fusidic acid. III. 17-Oxa-4.alpha.,8,-14-trimethyl-D-homo-18-norandrostanes. J. Org. Chem 32, 184–191. [DOI] [PubMed] [Google Scholar]
- (35).CLSI. (2018) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 11th ed., Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- (36).Gao Y-G, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, and Ramakrishnan V (2009) The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science 326, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhao SW, Panpan, Zhang K, Hong W, Jiang Z, Cui X, Cheng A, and Qin J Fusidic acid chemically modified compounds, and preparation method and application thereof, CN 105924488A, September. 7, 2016. [Google Scholar]
- (38).Ibrahim A-RS, Elokely KM, Ferreira D, and Ragab AE (2018) Microbial Oxidation of the Fusidic Acid Side Chain by Cunninghamella echinulata. Molecules 23, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Biedenbach DJ, Rhomberg PR, Mendes RE, and Jones RN (2010) Spectrum of activity, mutation rates, synergistic interactions, and the effects of pH and serum proteins for fusidic acid (CEM-102). Diagn. Microbiol. Infect. Dis 66, 301–307. [DOI] [PubMed] [Google Scholar]
- (40).Laue H, Valensise T, Seguin A, Hawser S, Lociuro S, and Islam K (2007) Effect of human plasma on the antimicrobial activity of iclaprim in vitro. J. Antimicrob. Chemother 60, 1388–1390. [DOI] [PubMed] [Google Scholar]
- (41).O’Neill AJ, Bostock JM, Morais Moita A, and Chopra I (2002) Antimicrobial activity and mechanisms of resistance to cephalosporin P1, an antibiotic related to fusidic acid. J. Antimicrob. Chemother 50, 839–848. [DOI] [PubMed] [Google Scholar]
- (42).Besier S, Ludwig A, Brade V, and Wichelhaus TA (2003) Molecular analysis of fusidic acid resistance in Staphylococcus aureus. Mol. Microbiol 47, 463–469. [DOI] [PubMed] [Google Scholar]
- (43).Chen W, He C, Yang H, Shu W, Cui Z, Tang R, Zhang C, and Liu Q (2020) Prevalence and molecular characterization of methicillin-resistant Staphylococcus aureus with mupirocin, fusidic acid and/or retapamulin resistance. BMC Microbiol 20, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chen C-M, Huang M, Chen H-F, Ke S-C, Li C-R, Wang J-H, and Wu L-T (2011) Fusidic acid resistance among clinical isolates of methicillin-resistant Staphylococcus aureus in a Taiwanese hospital. BMC Microbiol 11, 98–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Norström T, Lannergård J, and Hughes D (2007) Genetic and Phenotypic Identification of Fusidic Acid-Resistant Mutants with the Small-Colony-Variant Phenotype in Staphylococcus aureus. Antimicrob. Agents Chemother 51, 4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).O’Neill AJ, McLaws F, Kahlmeter G, Henriksen AS, and Chopra I (2007) Genetic basis of resistance to fusidic acid in staphylococci. Antimicrob. Agents Chemother 51, 1737–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Still JG, Clark K, Degenhardt TP, Scott D, Fernandes P, and Gutierrez MJ (2011) Pharmacokinetics and Safety of Single, Multiple, and Loading Doses of Fusidic Acid in Healthy Subjects. Clin. Infect. Dis 52, S504–S512. [DOI] [PubMed] [Google Scholar]
- (48).Godtfredsen WO, and Vangedal S (1966) On the metabolism of fusidic acid in man. Acta Chem. Scand 20, 1599–1607. [DOI] [PubMed] [Google Scholar]
- (49).Murphy TM, Little S, Wu R, Slee A, and Fernandes P Assessment of In Vivo Activity of CEM-102 (Fusidic Acid) in Murine Infection Models, http://melinta.com/wp-content/uploads/alltime/pdf/c/Assessment%20of%20In%20Vivo%20Activity%20of%20CEM-102%20in%20Murine.pdf (accessed November 11, 2020).
- (50).Chen Y, Koripella RK, Sanyal S, and Selmer M (2010) Staphylococcus aureus elongation factor G–structure and analysis of a target for fusidic acid. FEBS J 277, 3789–803. [DOI] [PubMed] [Google Scholar]
- (51).Cox G, Thompson GS, Jenkins HT, Peske F, Savelsbergh A, Rodnina MV, Wintermeyer W, Homans SW, Edwards TA, and O’Neill AJ (2012) Ribosome clearance by FusB-type proteins mediates resistance to the antibiotic fusidic acid. Proc. Natl. Acad. Sci. U. S. A 109, 2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Oefner C, Bandera M, Haldimann A, Laue H, Schulz H, Mukhija S, Parisi S, Weiss L, Lociuro S, and Dale GE (2009) Increased hydrophobic interactions of iclaprim with Staphylococcus aureus dihydrofolate reductase are responsible for the increase in affinity and antibacterial activity. J. Antimicrob. Chemother 63, 687–698. [DOI] [PubMed] [Google Scholar]
- (53).Purnapatre KP, Rao M, Pandya M, Khanna A, Chaira T, Bambal R, Upadhyay DJ, and Masuda N (2018) In Vitro and In Vivo Activities of DS86760016, a Novel Leucyl-tRNA Synthetase Inhibitor for Gram-Negative Pathogens. Antimicrob. Agents Chemother 62, e01987–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, and Shimer G (2001) Antibiotic Susceptibility Profiles of Escherichia coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob. Agents Chemother 45, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.