

SUMMARY
BACKGROUND.
Seronegative coeliac disease is poorly defined.
AIMS.
To study clinical phenotypes and long-term outcomes of seronegative coeliac disease in a multicentre cohort over 20 years.
METHODS.
Seronegative coeliac disease was diagnosed in HLA-DQ2/DQ8 positive patients with villous atrophy (VA), negative IgA endomysial (EmA), tissue transglutaminase (tTG) and deamidated-gliadin antibodies (DGP), clinical and histological response to gluten-free diet (GFD),and no alternative causes for VA. In patients with IgA deficiency, coeliac disease was diagnosed through VA, positive IgG EmA/tTG/DGP and clinical/histological response to a GFD (coeliac disease+IgAd). Patients with seropositive coeliac disease served as controls.
RESULTS.
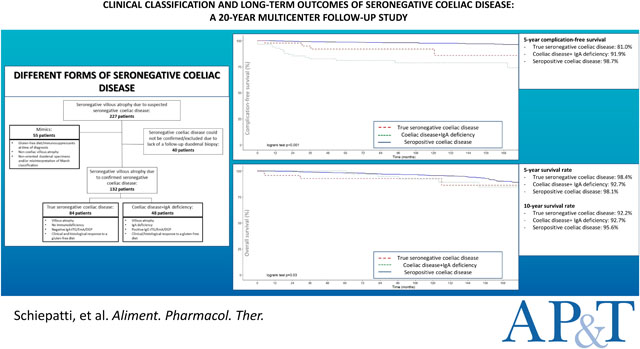
Of 227 patients previously diagnosed with seronegative coeliac disease, true seronegative coeliac disease was confirmed in 84, coeliac disease+IgAd in 48 and excluded in 55. Lack of follow-up duodenal biopsy precluded diagnosing seronegative coeliac disease in 40 patients. 2084 seropositive coeliac patients served as controls. True seronegative coeliac disease had more severe symptoms at diagnosis, higher risk of complications (HR 10.87, 95%CI 6.11–19.33,p<0.001) and mortality (HR 2.18, 95%CI 1.12–4.26,p<0.01) than seropositive coeliac disease (no differences between true seronegative coeliac disease and coeliac disease+IgAd). On multivariate analysis, age at diagnosis, lack of clinical response to a GFD, true seronegative coeliac disease, coeliac disease+IgAd and classical presentation predicted complications. Age at diagnosis, complications and absence of clinical response to a GFD predicted mortality.
CONCLUSIONS.
Seronegative coeliac disease has a more aggressive disease phenotype than seropositive coeliac disease. These data argue against over-reliance on serology for the diagnosis of coeliac disease, and support a strict clinical and histologic follow-up in seronegative coeliac disease.
Keywords: coeliac disease, seronegative villous atrophy, endomysial antibodies, tissue transglutaminase, IgA deficiency, mortality
Graphical Abstract
INTRODUCTION
Coeliac disease is a chronic immune-mediated enteropathy affecting nearly 1% of the population worldwide and characterized by villous atrophy (VA) and positive IgA tissue transglutaminase (tTG)/endomysial antibodies (EmA), which normalise upon a gluten-free diet (GFD) [1–3].
A minority of coeliac patients present with negative specific serology and are considered as affected by seronegative coeliac disease [4–6]. Although in Western Countries seronegative coeliac disease is the most common aetiology among patients with VA and negative coeliac serology (seronegative villous atrophy-SNVA) [4–12], there are still uncertainties concerning the serological and histological criteria for the definition and diagnosis of seronegative coeliac disease. This is due to the different study designs and diagnostic criteria adopted, endpoints and limited sample sizes of the populations under investigation in previous studies [4, 10, 11, 13–24]. Sensitivity and specificity of serological markers for coeliac disease have improved over the last few years [25], thus making it more difficult to define the real prevalence of this condition. Indeed, debate exists on whether to consider positive coeliac IgG based serology in the context of IgA deficiency as seronegative coeliac disease [9, 10], or instead as a conventional form of coeliac disease associated with IgA deficiency [4–6, 11]. Finally, negative coeliac serology was reported to occur in patients already on immunosuppressive therapies or a GFD prior to serological testing [4, 5, 15, 20], early stage disease [14, 19], late stage disease (with possible refractory coeliac disease or lymphoma) [21], dermatitis herpetiformis [26], and seldom in first-degree relatives of coeliac patients [27]. Therefore, the epidemiology, clinical features and natural history of seronegative coeliac disease are still poorly defined.
The objective of the present study was to provide a comprehensive overview on the clinical spectrum and long-term outcomes of a large cohort of patients affected by different forms of seronegative coeliac disease, evaluated over a 20-year period in three referral centres. Moreover, we sought to compare their features to those of patients diagnosed with conventional seropositive coeliac disease in the same timeframe, who served as controls.
PATIENTS AND METHODS
Study design and setting
This was a multicentre combined retrospective-prospective longitudinal study aiming to evaluate the clinical spectrum and natural history of patients with seronegative coeliac disease followed-up over 20 years (2000–2020) in three major referral centres (Royal Hallamshire Hospital, Sheffield, UK; Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; Beth Israel Deaconess Medical Center, Boston, USA).
Study population
The study population includes adult patients (≥18 years old) evaluated over 20 years in these three centres for an initial diagnosis of seronegative coeliac disease. Patients in whom a diagnosis of seronegative coeliac disease or coeliac disease and IgA deficiency was confirmed, were included in the study. Patients in whom seronegative coeliac disease was not confirmed after thorough re-evaluation were excluded from the analysis and considered separately.
The control group consisted of patients diagnosed with conventional seropositive coeliac disease over the same time-span.
Diagnostic criteria for different forms of seronegative coeliac disease
The flow chart in Figure 1 shows the diagnostic criteria for enrolment in each study group. Past medical history of all patients with suspected seronegative coeliac disease was carefully reviewed by either clinical letters and patients’ notes. Initial histology was re-evaluated by an expert gastrointestinal histopathologist [5, 6].
Figure 1. Flow chart summarising the enrolment criteria for each study group (number of patients in each group is also provided).
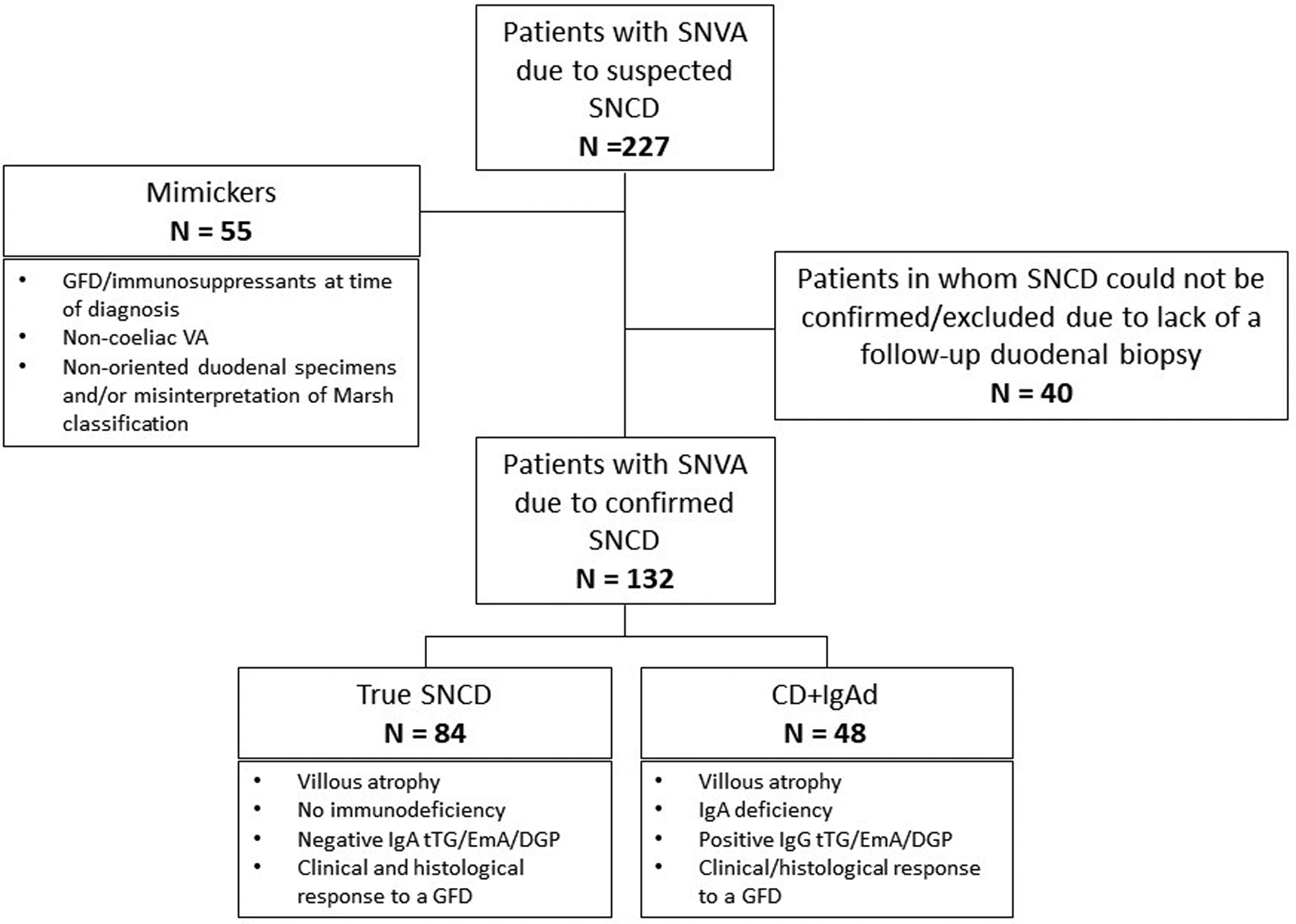
SNVA: seronegative villous atrophy; VA: villous atrophy; SNCD: seronegative coeliac disease; GFD: gluten-free diet; CD: coeliac disease; tTG: tissue transglutaminase antibodies; EmA: endomysial antibodies; DGP: deamidated gliadin antibodies
* These patients had confirmed villous atrophy, DQ2/DQ8 positive HLA typing and a satisfactory clinical response to a gluten-free diet.
Patients without evidence of VA after revision of their initial histology, those in whom investigations leading to initial diagnosis were performed while already on a GFD and/or on immunosuppressive therapy, those with negative HLA-DQ2 and DQ8, and those in whom VA was due to other non-coeliac enteropathies were defined “mimics”, excluded from the main study cohort and considered separately.
In HLA-DQ2/-DQ8 patients with normal immunoglobulins levels, VA, negative IgA EmA, IgA tTG and IgA DGP, while consuming gluten, and no alternative causes for VA, a diagnosis of “true seronegative coeliac disease” was made on the basis of both a clinical and histological response to a GFD. This means that patients with suspected seronegative coeliac disease who did not undergo a follow-up duodenal biopsy confirming histological response to a GFD were excluded. We specify that, before diagnosing true seronegative coeliac disease all the causes of SNVA unrelated to gluten ingestion were thoroughly investigated, and excluded by means of an algorithmic approach [4–7, 9, 10]. These included autoimmune enteropathy with positive enterocyte antibodies, common variable immunodeficiency, giardiasis and other parasitic infections, Whipple’s disease, HIV enteropathy, tuberculosis, small intestinal bacterial overgrowth, tropical sprue, lymphoproliferative disorders affecting the small bowel, graft versus host disease, drug-related enteropathies (ARB2s particularly olmesartan, chronic use of NSAIDs, radio/chemotherapy, methotrexate, azathioprine), peptic duodenitis (± H. Pylori)) [5–7, 9, 10].
For patients affected by total IgA deficiency (serum IgA< 0.08 g/dL) and VA, positive class IgG tTG/EmA/DGP supported the diagnosis of coeliac disease associated to IgA deficiency, together with a clinical/histological response to a GFD. Past medical history of dermatitis herpetiformis and family history of coeliac disease, if present, were supportive elements for the diagnosis of seronegative coeliac disease.
Diagnostic criteria for conventional seropositive coeliac disease and complications of coeliac disease
Diagnosis of conventional seropositive coeliac disease was made in accordance with major international guidelines on the basis of VA and positive IgA tTG/EmA/DGP while on a GCD. Patients affected by potential coeliac disease were excluded from the study [1].
Diagnosis of complications of coeliac disease was made as previously described [28, 29]. In particular, persistence of malabsorption symptoms and VA despite a strict GFD for at least 12 months in the absence of lymphoma, malignancies and non-coeliac enteropathies allowed the diagnosis of refractory coeliac disease [1, 28–30]. GFD adherence was assessed by means of dietary interview with a dietician. Distinction between type 1 and type 2 refractory coeliac disease was made on the basis of presence of an aberrant immune-phenotype of intraepithelial lymphocytes assessed by means of flow cytometry, immunohistochemistry and molecular genetics for γ-TCR clonality [1,28–30]. Diagnosis of malignancies (primary lymphomas and carcinomas of the small bowel) were based on histopathology [28–30].
Prevalence study
The prevalence of true seronegative coeliac disease and coeliac disease associated to IgA deficiency among all coeliac patients was calculated taking into account only individuals directly diagnosed by each centre over the period 2000–2020. These patients were defined as “incident cases”. On the contrary, patients referred to our tertiary centre for confirmation of the diagnosis of coeliac disease, or for a suspicion of complicated/refractory coeliac disease, or to obtain certificates entitling them to seasonal prescriptions for gluten-free products were excluded from this part of the study, as to avoid a selection bias. These patients were defined as ‘referred patients’.
Clinical phenotype study
Age at diagnosis, gender, presenting symptoms at diagnosis of coeliac disease, family history of coeliac disease, past medical history and HLA typing were collected and compared between all patients affected by true seronegative coeliac disease, coeliac disease associated to IgA deficiency and seropositive coeliac disease.
Follow-up and mortality study
Data on clinical response to a GFD, results of follow-up duodenal biopsy, date of last outpatient clinic access/phone contact, date of onset and type of any complications, and date and cause of death (where available) were recorded for all patients until February 2020.
Clinical response to a GFD was defined as improvement or disappearance of symptoms and biochemical abnormalities present at baseline. Histological response to a GFD was defined as histological improvement of VA on follow-up biopsy after at least 12 months of dietetic treatment.
For patients in the British cohort, data about development of complications, date of death and cause of death were obtained from a large and regularly updated local multi-dataset at the Royal Hallamshire Hospital, Sheffield, UK. Data for the Italian cohort were obtained from patients’ clinical notes, as previously done [28]. We specify that all the Italian patients who have not attended the clinic in the last six months before the study began, were contacted over the phone to ascertain whether they were still alive. For patients who were not reached on the phone, the Local Council Services were contacted, as previously done [28]. For the American cohort data were obtained from a large registry of the Coeliac Center, Beth Israel Deaconess Medical Center, Boston, USA.
Histology
Duodenal histology was graded according to the Marsh-Oberhuber classification [31] or the Corazza-Villanacci classification [32] on haematoxylin and eosin stained slides from the second duodenal portion. Severity of villous atrophy was graded on the samples showing the worst degree of villous blunting. Immunohistochemistry for CD3 and CD8 lymphocyte markers was performed on formalin-fixed, paraffin-embedded duodenal specimens.
Polymerase chain reaction-based analysis for γ-TCR gene rearrangement was performed on DNA extracted from formalin-fixed paraffin embedded duodenal specimens, in accordance to standard Euroclonality/BIOMED-2 protocol.
Presence of aberrant intraepithelial lymphocytes, the hallmark of type 2 refractory coeliac disease, was defined as >50% of CD3+ CD8- intraepithelial lymphocytes on traditional immunohistochemistry or >20% CD3-CD8-CD103+CD7+ cytoplasmatic CD3+ intraepithelial T-lymphocytes by means of flow cytometry [33].
Coeliac serology
EmA were detected by means of indirect immunofluorescence on monkey oesophagus/jejunum slides (INOVA Diagnostics, San Diego, USA was used for patients in the Italian cohort; The Binding Site, Birmingham, UK was used for patients in the British cohort). tTG and DGP were tested by using ELISA kits (EliA Celikey IgA and Celikey IgG, EliA Gliadin DP IgA and EliA Gliadin DP IgG; Phadi AB, Uppsala, Sweden were used for the Italian patients. Aesku Diagnostics, Wendelsheims, Germany was used for the British patients. INOVA Diagnostics, San Diego, USA was used for patients in the American cohort). Class IgG antibodies were tested only in patients affected by IgA deficiency.
Statistical analysis
Data were summarized as counts and percentage if categorical and as mean and standard deviation or median and interquartile range (25th–75th percentiles) if continuous. Categorical variables were compared between groups using Fisher’s exact test. Continuous variables were compared using Welch’s test, the Kruskal-Wallis test or Wilcoxon’s signed-rank test. The prevalence of SNCD and CD+IgAd deficiency were calculated only on ‘incident cases’ and reported together with their 95% exact binomial confidence interval (95% CI). Mortality rate and complications rate per 100 person years and 95% CI were computed. Survival and complication-free survival were described by means of Kaplan-Meier curves and compared between groups with the log-rank test. Hazard ratios and 95% CI were estimated from a Cox model. A multivariable Cox model stratified by Center of diagnosis was used to evaluate predictors of complication and mortality. Response to treatment was included as a time-dependent covariate. The Harrell’s c statistic was computed (95%CI) to assess discrimination using the testing/validation sample strategy. A two-sided p-value <0.05 was considered statistically significant. Bonferroni correction was applied for post-hoc comparisons. Stata 16.0 (StataCorp., College Station, Texas, USA) was used for computation.
Ethic approval
The study protocol of the leading centre was approved by the Yorkshire and Humber Research Ethics committee and registered with the local research and development department of Sheffield Teaching Hospital NHS Foundation Trust (REC reference 19/YH/0095). The protocol was also approved by the Ethics committee of the Fondazione IRCCS Policlinico San Matteo, Pavia, Italy and by the Ethic Committee of Beth Israel Deaconess Medical Center, Boston, USA (protocol number 2019P000927). The study protocol conforms to the Declaration of Helsinki, 6th revision, 2008.
RESULTS
Between January 2000 and July 2020, 227 patients with an initial diagnosis of seronegative coeliac disease were reinvestigated and 2084 were diagnosed with seropositive coeliac disease. True seronegative coeliac disease was confirmed in 84 patients (37%) and coeliac disease associated to IgA deficiency in 48 (21%), whereas in 55 patients (24%) seronegative coeliac disease was completely excluded (mimics group). In 40 patients (18%) with compatible HLA-DQ2/-DQ8, no alternative causes for VA and good clinical response to a GFD, seronegative coeliac disease could be neither confirmed nor excluded because of lack of a follow-up biopsy showing histological response to a GFD. These patients, were therefore excluded from the analysis (Figure 1).
Prevalence
Overall, 59 patients with true seronegative coeliac disease, 39 with coeliac disease associated to IgA deficiency and 2084 with seropositive coeliac disease were directly diagnosed in the three centres (‘incident cases’). Prevalence of true seronegative coeliac disease was 2.70% (95% CI 2.06%−3.47%), and prevalence of coeliac disease associated to IgA deficiency was 1.79% (95% CI 1.27%−2.44%), without substantial differences among the centres (Table 1).
Table 1.
Prevalence of seronegative coeliac disease and coeliac disease associated to IgA deficiency in coeliac patients according to centre of diagnosis.
| CENTRE | Total patients n = 2216 |
SPCD n= 2084 |
SNCD n= 84 |
Prevalence* (95% CI) |
CD+IgAd n= 48 |
Prevalence (95% CI) |
|---|---|---|---|---|---|---|
| ALL CENTRES-incident | 2182 | 2084 | 59 | 2.70% (2.06%−3.47%) |
39 | 1.79% (1.27%−2.44%) |
| ALL CENTRES-referred | 34 | 0 | 25 | - | 9 | - |
| Italian cohort-incident | 252 | 244 | 5 | 1.98% (0.65%−4.57%) |
3 | 1.19% (0.25%−3.44%) |
| Italian cohort-referred | 6 | 0 | 4 | 2 | ||
| British cohort-incident | 1098 | 1050 | 36 | 3.28% (2.31%−4.51%) |
12 | 1.09% (0.57%−1.90%) |
| British cohort-referred | 16 | 0 | 12 | - | 4 | - |
| American cohort-incident | 832 | 790 | 18 | 2.16% (1.29%−3.40%) |
24 | 2.88% (1.86%−4.26%) |
| American cohort-referred | 12 | 0 | 9 | - | 3 | - |
prevalence is calculated over the total population
SNCD: seronegative coeliac disease; SPCD: conventional seropositive coeliac disease; CD+ IgAd: coeliac disease+IgA deficiency; incident: patients directly diagnosed in the centre; referred: patients diagnosed elsewhere and then referred to our centres
Clinical phenotype
Baseline clinical and laboratory features of patients affected by true seronegative coeliac disease, coeliac disease associated to IgA deficiency and seropositive coeliac disease are summarized in Table 2. True seronegative coeliac disease patients were older at diagnosis (50±17 vs 42±16 years, p<0.001) and presented more frequently with intestinal and extra-intestinal features of severe malabsorption such as weight loss (p< 0.001), diarrhoea (p<0.001), and osteoporosis (p<0.01) than seropositive patients. No differences were found in the clinical phenotype between coeliac disease associated to IgA deficiency and true seronegative coeliac disease. Overall results on the clinical phenotype did not differ significantly between the centres (data not shown).
Table 2.
Baseline clinical and laboratory features of patients affected by seronegative coeliac disease, coeliac disease associated to IgA deficiency and conventional seropositive coeliac disease
| SNCD n=84 |
CD+IgAd n=48 |
SPCD n=2084 |
Overall p-value | p-value SNCD vs. SPCD* |
p-value SNCD vs. CD+IgAd* |
p-value SPCD vs. CD+IgAd* |
|
|---|---|---|---|---|---|---|---|
| Age at diagnosis (mean±SD) | 50±17 | 46±17 | 42±16 | <0.001 | <0.001 | 0.49 | 0.35 |
| Gender, Females | 60 (71.4%) | 26 (54.2%) | 1470 (70.5%) | 0.055 | - | - | - |
| Weight loss | 35 (41.7%) | 17 (35.4%) | 327 (15.7%) | <0.001 | <0.001 | 1.00 | <0.01 |
| Vomiting | 16 (19.0%) | 6 (12.5%) | 106 (5.1%) | <0.001 | <0.001 | 1.00 | 0.11 |
| Dyspepsia | 28 (33.3%) | 10 (20.8%) | 358 (17.2%) | <0.01 | <0.01 | 0.48 | 1.00 |
| Diarrhoea | 59 (70.2%) | 28 (58.3%) | 797 (38.3%) | <0.001 | <0.001 | 0.56 | 0.02 |
| Abdominal pain | 34 (40.5%) | 11 (22.9%) | 711 (34.1%) | 0.12 | - | - | - |
| Osteoporosis § | 17 (23.3%) | 6 (17.1%) | 125 (7.2%) | <0.001 | <0.001 | 1.00 | 0.12 |
| IDA | 23 (27.4%) | 18 (38.3%) | 685 (33.0%) | 0.41 | - | - | - |
| Low B12 anaemia | 7 (8.3%) | 2 (4.3%) | 112 (5.4%) | 0.44 | - | - | - |
| Folate deficiency anaemia | 5 (6.0%) | 0 | 76 (3.7%) | 0.24 | - | - | - |
| DH | 4 (4.8%) | 1 (2.1%) | 67 (3.2%) | 0.66 | - | - | - |
| Family history CD | 4 (4.8%) | 8 (17.0%) | 326 (15.7%) | 0.01 | 0.014 | 0.082 | 1.00 |
| Autoimmunity | 17 (20.2%) | 13 (27.1%) | 456 (21.9%) | 0.62 | - | - | - |
| Hypothyroidism | 9 (10.7%) | 6 (12.5%) | 249 (12.0%) | 0.93 | - | - | - |
| Hyperthyroidism | 1 (1.2%) | 0 | 34 (1.6%) | 1.00 | - | - | - |
| Type I diabetes | 3 (3.6%) | 4 (8.3%) | 107 (5.1%) | 0.44 | - | - | - |
| Addison’s disease | 0 | 0 | 3 (0.1%) | 1.00 | - | - | - |
| Rheumatoid arthritis | 5 (6.0%) | 0 | 21 (1.0%) | <0.01 | <0.01 | 0.47 | 1.00 |
| Systemic sclerosis | 2 (2.4%) | 0 | 1 (0.0%) | <0.01 | 0.01 | 1.00 | 1.00 |
| SLE/Sjogren syndrome | 3 (3.6%) | 1 (2.1%) | 16 (0.8%) | 0.03 | 0.10 | 1.00 | 0.97 |
| HLA typing § | 0.01 | 0.02 | 1.00 | 0.40 | |||
| DQ2 +ve | 59 (85.5%) | 18 (81.8%) | 532 (86.6%) | ||||
| DQ8 +ve | 7 (10.1%) | 2 (9.1%) | 24 (3.9%) | ||||
| DQ2/DQ8 +ve | 1 (1.4%) | 1 (4.5%) | 52 (8.5%) | ||||
| **DQ2 and DQ8 −ve | 2 (2.9%) | 1 (4.5%) | 6 (1.0%) |
post-hoc comparison p-values adjusted according to Bonferroni;
patients with negative HLA-DQ2/DQ8 typing were HLA-DQ7.5 positive and showed clinical and histological response to a gluten-free diet.
data not available for all patients, patients with missing data were not included in the analysis; percentages calculated based on only patients with available data, CD: coeliac disease; SNCD: seronegative coeliac disease; SPCD: seropositive coeliac disease; CD+IgAd def: coeliac disease associate to IgA deficiency; IDA: iron deficiency anaemia; DH: dermatitis herpetiformis; SLE: systemic lupus erythematosus; HLA: Human Leukocyte Antigen; SD: standard deviation,
Statistical analysis: proportions were compared with Fisher’s exact test, continuous variables were compared among groups with the Kruskal-Wallis test
Mimics group
Eighteen patients (33%) in the mimics group were on a GFD or immunosuppressive therapy at time of their first diagnosis of VA, but then developed positive IgA EmA/tTG/DGP after gluten challenge, or when immunosuppressants were stopped. Therefore, a diagnosis of conventional seropositive coeliac disease was made. In 14 patients (25%), VA was confirmed, being due to non-coeliac enteropathies including type 1 idiopathic villous atrophy (n=9), collagenous sprue (n=2), Crohn’s disease, autoimmune enteropathy with positive anti-enterocyte antibodies and tropical sprue (n=1 each) [5, 6, 9, 34]. Finally, the remaining 23 patients (42%) had no confirmed evidence of VA, upon revision of their initial slides, because of either poor orientation of duodenal specimens, or a misinterpretation of Marsh classification (i.e. patients with correctly oriented specimens and no evidence of VA). Seventeen of these 23 patients had normal duodenal biopsies and negative coeliac serology after a gluten-challenge, and their final diagnoses included irritable bowel syndrome, bile-acid diarrhoea and small-intestinal bacterial overgrowth. The remaining six patients did not repeat gastroscopy (4 had negative HLA-DQ2 and DQ8, one refused further investigations and one resumed normal diet without experiencing symptoms). Patients in the ‘mimics group’ differ from true seronegative coeliac disease in terms of presenting characteristics, being younger (mean age at diagnosis 41±14 years vs. 50±17 years, p<0.01) and less likely to have classical features of malabsorption (53% vs. 70%, p=0.02).
Long-term outcomes and mortality
Patients were followed-up for a median of 79 months (25th-75th, 37–134). Follow-up duodenal biopsies were performed in 84/84 (100%) true seronegative coeliac disease patients, 20/48 (42%) patients with coeliac disease associated to IgA deficiency and 620/2084 (30%) seropositive coeliac disease patients.
Table 3 summarizes the long-term outcomes in patients with true seronegative coeliac disease, coeliac disease associated to IgA deficiency and seropositive coeliac disease. A total of 58 patients developed a complication after a median of 32 months (IQR 17–72) since diagnosis of coeliac disease (33 in the British cohort, 6 in the Italian cohort and 19 in the American cohort) and 96 died (74 in the British cohort, 8 in the Italian cohort and 14 in the American cohort).
Table 3.
Long-term outcomes of patients affected by seronegative coeliac disease, coeliac disease associated to IgA deficiency and conventional seropositive coeliac disease.
| SNCD n=84 |
CD+IgAd n=48 |
SPCD n=2084 |
Overall p-value |
p-value SNCD vs. SPCD* |
p-value SNCD vs. CD+IgAd* |
p-value SPCD vs. CD+IgAd* |
|
|---|---|---|---|---|---|---|---|
| Strict GFD adherence± | 74 (88.1%) | 16 (80%) | 516 (83.2%) | 0.48 | - | - | - |
| COMPLICATIONS | 17 (20.2%) | 4 (8.3%) | 37 (1.8%) | <0.001 | <0.001 | 0.26 | 0.04 |
| Age at complication (median, IQR) | 65 (54–70) | 65 (56–72) | 56 (48–68) | 0.39 | - | - | - |
| Months to complication (median, IQR) | 18 (10–32) | 34 (24–58) | 46 (24–85) | 0.02 | 0.02 | 1.00 | 1.00 |
| RCD1 | 12 (14.3%) | 3 (6.2%) | 17 (0.8%) | <0.001 | <0.001 | 0.50 | 0.03 |
| RCD2 | 3 (3.6%) | 0 | 6 (0.3%) | <0.01 | <0.01 | 0.85 | 1.00 |
| UJI | 1 (1.2%) | 0 | 0 | 0.052 | - | - | - |
| EATL | 0 | 1 (2.1%) | 4 (0.2%) | 0.11 | - | - | - |
| BCL | 0 | 0 | 4 (0.2%) | 1.00 | - | - | - |
| SBC | 0 | 0 | 2 (0.1%) | 1.00 | - | - | - |
| Oesophageal cancer | 0 | 0 | 3 (0.1%) | 1.00 | - | - | - |
| Collagenous sprue | 1 (1.2%) | 0 | 0 | 0.052 | - | - | - |
| Celiac crisis | 0 | 0 | 1 (0.0%) | 1.00 | - | - | - |
| MORTALITY | 10 (11.9%) | 4 (8.3%) | 82 (4.0%) | <0.01 | <0.01 | 1.00 | 0.38 |
| Age at death (median, IQR) | 72 (66–77) | 73 (63–82) | 74 (65–80) | 0.89 | - | - | - |
| Months to death (median, IQR) | 137 (86–214) | 17 (5–54) | 84 (40–144) | 0.01 | 0.08 | 0.11 | 0.17 |
| CD Complications | 3 (3.5%) | 2 (4.2%) | 11 (0.5%) | <0.01 | 0.045 | 1.00 | 0.10 |
| Cardiovascular disease | 0 | 0 | 11 (0.5%) | 1.00 | - | - | - |
| Respiratory disease | 1 (1.2%) | 0 | 11 (0.5%) | 0.52 | - | - | - |
| Cancer | 5 (5.9%) | 0 | 15 (0.7%) | <0.01 | <0.01 | 0.47 | 1.00 |
| Infectious | 2 (2.4%) | 1 (2.1%) | 6 (0.3%) | 0.01 | 0.11 | 1.00 | 0.44 |
| Other | 0 | 0 | 9 (0.4%) | 1.00 | - | - | - |
| Unknown | 3 (3.6%) | 1 (2.1%) | 21 (1%) | 0.06 | - | - | - |
CD: coeliac disease; SNCD: seronegative coeliac disease; SPCD: seropositive coeliac disease; CD+IgAd: coeliac disease associate to IgA deficiency; GFD: gluten-free diet; RCD1; refractory celiac disease type 1; RCD2: refractory celiac disease type 2; UJI: ulcerative jejunitis; EATL: enteropathy associated T-cell lymphoma; SCL; B-cell lymphomas; SBC: small bowel carcinoma.
post-hoc comparison p-values adjusted according to Bonferroni
gluten-free diet adherence was evaluated in patients with follow-up duodenal biopsy and dietary evaluation at time of follow-up duodenal biopsy
Statistical analysis: proportions were compared with Fisher’s exact test, continuous variables were compared among groups with the Kruskal-Wallis test
A significantly higher number of complications occurred in true seronegative coeliac disease compared to coeliac disease associated to IgA deficiency and seropositive coeliac disease (20.2% vs. 8.3% vs. 1.8%, p<0.001). Median time between diagnosis of coeliac disease and onset of complications was 32 months (IQR 17–72) overall. Time to complication differed significantly among the three groups, being shorter in true seronegative coeliac disease than in coeliac disease associated to IgA deficiency and seropositive coeliac disease (p=0.02). Refractory coeliac disease was the most common complication arising in patients with seronegative coeliac disease (p<0.01), being type 1 refractory coeliac disease the most represented. Additionally, a significantly higher number of patients died in the true seronegative coeliac disease group than in coeliac disease associated to IgA deficiency and seropositive coeliac disease (11.9% vs. 8.3% vs. 4.0%, p<0.01), complications of coeliac disease and cancer unrelated to coeliac disease being the main causes of death (p<0.01). Median time between diagnosis and death was 85 months (IQR 42–150) overall. Time between diagnosis and death differed significantly among true seronegative coeliac disease, coeliac disease associated to IgA deficiency and seropositive coeliac disease, p=0.01. True seronegative coeliac disease appeared to have the longest time between diagnosis and death (median 138 months, IQR 86–214), although post-hoc pairwise analysis corrected by Bonferroni did not reach statistical significance between groups (true seronegative coeliac disease vs seropositive, p=0.08). Overall mortality was higher in the British cohort (HR 2.92, 95%CI 1.65–5.17, p<0.001) than in the American and the Italian cohorts. Centre of diagnosis did not influence development of complications.
Rates of complications, mortality rates and hazards ratios are summarized in Table 4. Kaplan-Meier curves in Figure 2 show complication-free survival in true seronegative coeliac disease, coeliac disease associated to IgA deficiency and seropositive coeliac disease. 5-year complication-free survival was 81.0% in true seronegative coeliac disease, 91.9% in coeliac disease associated to IgA deficiency, and 98.7% in seropositive coeliac disease (Log-rank test, p<0.001). Figure 3 shows overall survival in the three groups. 5-year survival rate was 98.4% in true seronegative coeliac disease, 92.7% in coeliac disease associated to IgA deficiency and 98.1% in seropositive coeliac disease. 10-year survival rate was 92.2% in true seronegative coeliac disease, 92.7% in coeliac disease associated to IgA deficiency and 95.6% in seropositive coeliac disease (Log-rank test, p=0.03).
Table 4.
Complication rate, mortality rate and hazard ratios in seronegative coeliac disease and conventional seropositive CD, stratified by center
| COMPLICATIONS | |||||
|---|---|---|---|---|---|
| Patients, n | Incidence per 100 person-year (95% CI) |
Hazard ratio (95% CI) |
*p-value | ||
| All SNCD ** | 21 | 2.07 (1.28–3.16) | Vs SPCD 8.92 (5.22–15.25) | <0.001 | |
| True SNCD | 17 | 2.48 (1.44–3.97) | Vs SPCD 10.87 (6.11–19.33) | <0.001 | |
| CD+IgAd | 4 | 1.21 (0.33–3.10) | Vs SPCD 5.04 (1.79–14.20) | <0.01 | |
| Vs SNCD 0.44 (0.15–1.32) | 0.42 | ||||
| SPCD | 37 | 0.24 (0.17–0.34) | 1 | - | |
| MORTALITY | |||||
| All SNCD ** | 14 | 1.27 (0.69–2.13) | Vs SPCD 2.22 (1.23–3.99) | <0.01 | |
| True SNCD | 10 | 1.32 (0.63–2.42) | Vs SPCD 2.18 (1.12–4.26) | 0.07 | |
| CD+IgAd | 4 | 1.17 (0.32–2.99) | Vs SPCD 2.32 (0.82–6.56) | 0.34 | |
| Vs SNCD 1.14 (0.33–3.92) | 1.00 | ||||
| SPCD | 82 | 0.54 (0.43–0.67) | 1 | - | |
SNCD: seronegative coeliac disease; SPCD: seropositive coeliac disease; CD+IgAd: coeliac disease associated to IgA deficiency; vs.:versus;
post-hoc comparison p values adjusted according to Bonferroni
this group included patients with true seronegative coeliac disease and coeliac disease associated to IgA deficiency together
Figure 2. Kaplan-Meier complication-free survival in seronegative coeliac disease, coeliac disease associated to IgA deficiency and conventional seropositive coeliac disease.
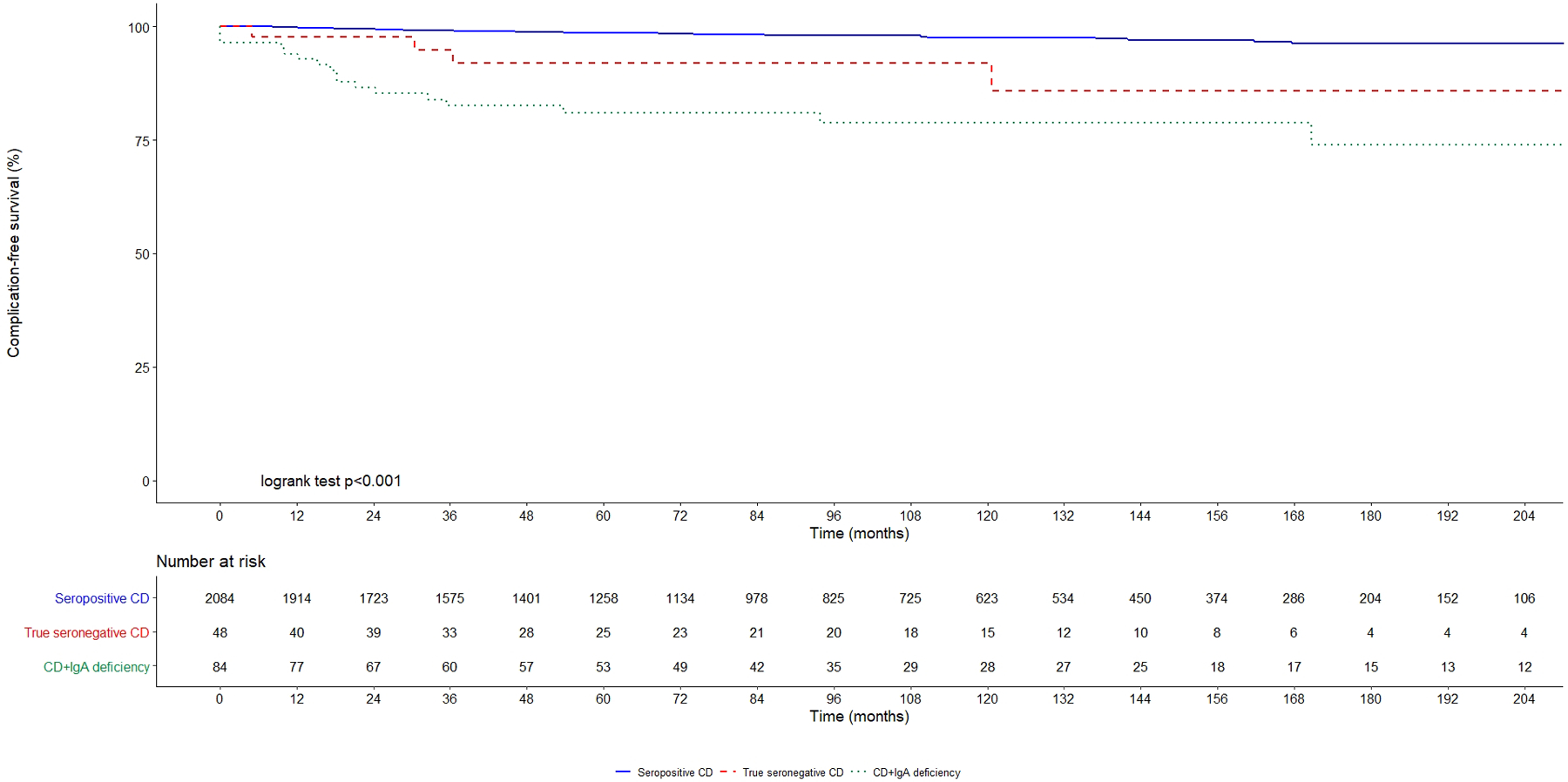
SPCD: seropositive coeliac disease (continuous blue line); CD+IgAd: coeliac disease associated to IgA deficiency (dotted green line); SNCD: seronegative coeliac disease (dashed red line).
Figure 3. Survival in seronegative coeliac disease, coeliac disease associated to IgA deficiency and conventional seropositive coeliac disease.
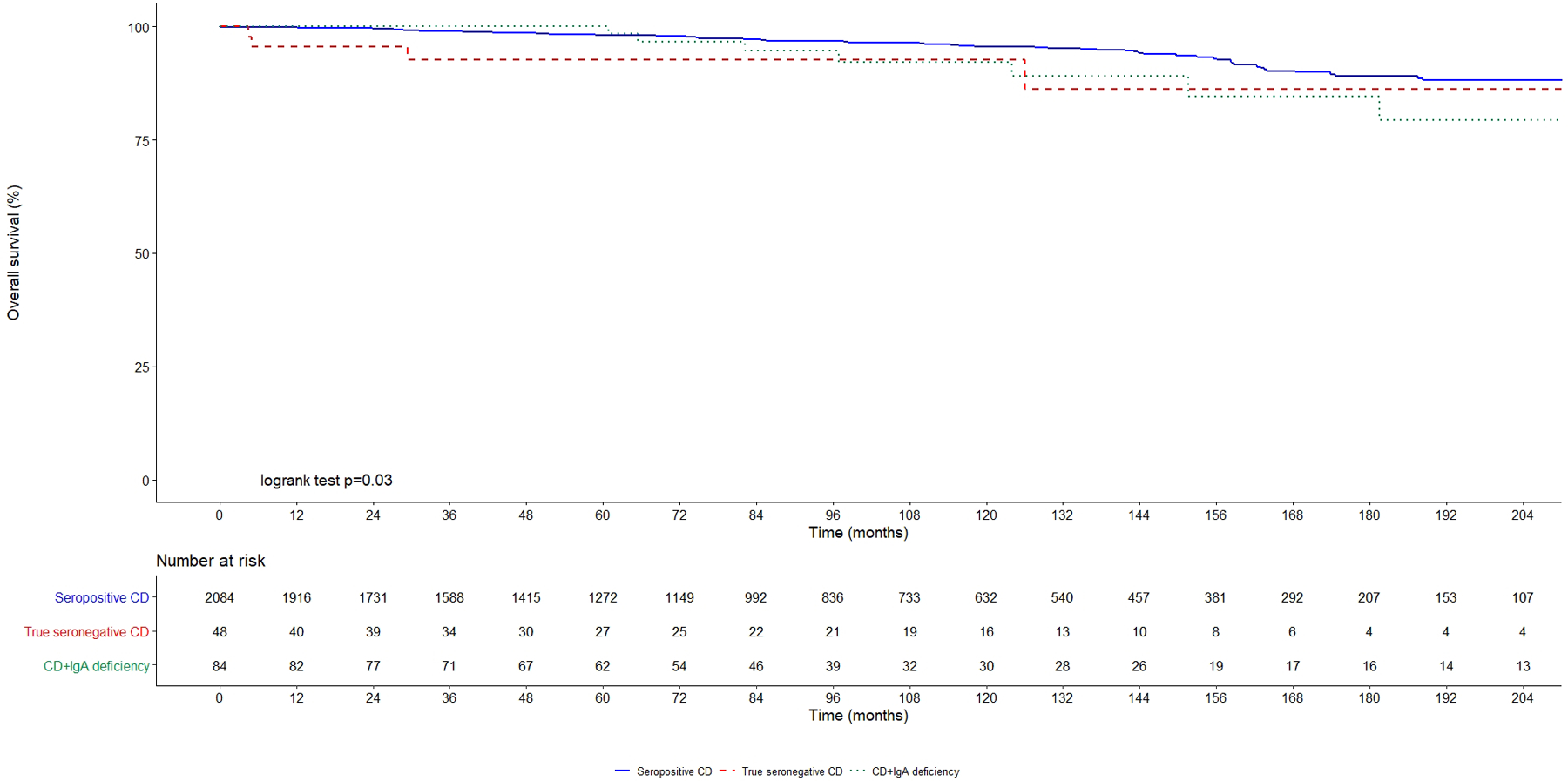
SPCD: seropositive coeliac disease (continuous blue line); CD+IgAd: coeliac disease associated to IgA deficiency (dotted green line); SNCD: seronegative coeliac disease (dashed red line).
Finally, Table 5 shows multivariable Cox models for complication-free survival and overall survival stratified by centre. Age at diagnosis of coeliac disease (p<0.001), lack of clinical response to a GFD (p<0.01), true seronegative coeliac disease (p<0.01), coeliac disease associated to IgA deficiency (p<0.01) and classical clinical presentation (p<0.01) were predictors of complications. Harrell’s c statistic was 0.87 (95%CI 0.82–0.92).
Table 5.
Multivariable Cox models for development of complications and mortality stratified by centre
| COMPLICATIONS | |||
|---|---|---|---|
| Factor | Hazard ratio (95% CI) |
*p-value | |
| Age at diagnosis (years) | 1.04 (1.02–1.06) | <0.001 | |
| True SNCD | 7.78 (4.23–14.31) | <0.001 | |
| CD+IgAd | 5.25 (1.80–15.34) | <0.01 | |
| SPCD | 1 | - | |
| Male | 1.39 (0.80–2.42) | 0.25 | |
| Female | 1 | - | |
| Classical presentation | 1 | - | |
| Non-classical presentation | 0.32 (0.16–0.63) | <0.01 | |
| Silent presentation | 0.16 (0.04–0.69) | 0.01 | |
| Clinical response to a GFD | 0.12 (0.06–0.25) | <0.001 | |
| MORTALITY | |||
| Age at diagnosis (years) | 1.10 (1.08–1.12) | <0.001 | |
| True SNCD | 1.04 (0.48–2.26) | 0.93 | |
| CD+IgAd | 1.38 (0.46–4.12) | 0.56 | |
| SPCD | 1 | - | |
| Male | 1.48 (0.97–2.27) | 0.07 | |
| Female | 1 | - | |
| Classical presentation | 1 | - | |
| Non-classical presentation | 0.98 (0.61–1.57) | 0.94 | |
| Silent presentation | 1.04 (0.51–2.15) | 0.91 | |
| Clinical response to a GFD | 0.51 (0.32–0.81) | <0.01 | |
| Development of complications | 3.80 (2.10–6.86) | <0.001 | |
SNCD: seronegative coeliac disease; SPCD: seropositive coeliac disease; CD+IgAd: coeliac disease associated to IgA deficiency; vs.:versus;
this group included patients with true seronegative coeliac disease and coeliac disease associated to IgA deficiency together
Age at diagnosis (p<0.01), lack of clinical response to a GFD (p<0.01) and development of complications (p<0.01) significantly predicted mortality. Harrell’s c statistic was 0.88 (95%CI 0.84–0.91).
DISCUSSION
This study is the largest to date on patients affected by seronegative coeliac disease, providing a real world overview on the clinical spectrum and long-term outcomes of this condition. Coeliac patients presenting with negative IgA coeliac serology because of true seronegative coeliac disease or IgA deficiency show a distinctive clinical phenotype and a more aggressive disease course than conventional seropositive coeliac disease.
Seronegative coeliac disease and coeliac disease associated to IgA deficiency are rare among coeliac patients, accounting for no more than 3% and 2% of all coeliac cases, respectively. Data reported in the literature about the prevalence of seronegative coeliac disease vary widely and are difficult to compare because of the design of the studies and the different diagnostic criteria adopted throughout the years [4,10,11,13–21]. The first papers primarily aiming to evaluate the diagnostic accuracy of EmA and tTg based assays in patients with biopsy-proven coeliac disease suggested a prevalence of seronegative coeliac disease varying between 6–39% [13–21]. More recent data showed a prevalence of seronegative coeliac disease around 2% among all the coeliac patients [10,11], and the proportion of patients with seronegative coeliac disease in our study cohort approximates at the lower end of these previous reports. Although, interestingly, our prevalence results reflect the sensitivity of contemporary coeliac specific serology [4–6,25], it is undoubtedly difficult to ascertain the true prevalence of seronegative celiac disease is in a population-based setting. Only two papers provided data on the frequency of seronegative coeliac disease in a more general setting, which was that of patients referred to endoscopy [37,38]. Of course, also in this scenario prevalence figures are likely to overestimate the real prevalence of seronegative coeliac disease.
Seronegative coeliac patients were older at diagnosis, presented with a more severe clinical phenotype and were burdened by a higher risk of complications than conventional seropositive coeliac disease. Multivariable analysis confirmed that seronegative coeliac disease, coeliac disease associated to IgA deficiency, age at diagnosis, classical pattern at diagnosis, and lack of clinical response to a GFD [28] were major predictors of complications. Multivariable Cox model stratified by centre did not identify seronegative coeliac disease and coeliac disease associated to IgA deficiency as independent predictors of mortality, with age at diagnosis, lack of clinical response to a GFD and development of complications being the most important ones. Two explanations are likely to be considered. The first one is the limited number of deaths in our cohort, possibly affecting the results of the multivariable model, although it would be very difficult to find a larger sample size with such a diagnostic accuracy over two decades. The second explanation may be that the increased risk of complications in seronegative coeliac disease is predominantly given by patients who developed type 1 refractory coeliac disease. This condition has the better prognosis among the different complications of coeliac disease [28,30], thus possibly justifying the higher risk of complications without a synchronous increase in mortality rates.
Also coeliac disease associated to IgA deficiency diagnosed in adulthood was characterised by a more severe disease phenotype than seropositive coeliac disease, which was overall similar to that of true seronegative coeliac disease. This result is the first of its kind to be reported, since the majority of studies on coeliac disease associated to IgA deficiency were conducted in paediatric settings and did not specifically compare clinical features and long-term outcomes between conventional seropositive coeliac disease and coeliac disease associated to IgA deficiency [39–41]. Only a Swedish population-based cohort study reported an increased mortality in patients affected by IgA deficiency [42].
Seronegative coeliac disease was misdiagnosed in up to 25% of patients who received an initial diagnosis of seronegative coeliac disease. Poor orientation of duodenal biopsies and/or misinterpretation of the Marsh classification were the major causes of diagnostic errors, leading to a diagnosis of seronegative coeliac disease in patients who, instead, had a normal duodenal architecture. The second most common diagnostic pitfall occurred when patients were investigated while on concomitant immunosuppressive therapies, or after a GFD had already been started. However, when patients withdrew immunosuppressants or were started again on a gluten-containing diet, their serological response was restored, and a diagnosis of conventional seropositive coeliac disease was made. The third type of diagnostic error occurred when non-coeliac enteropathies with VA were labelled as seronegative coeliac disease. In our series, this was the case for autoimmune enteropathy, tropical sprue and forms of idiopathic villous atrophy [5, 6, 9, 34, 35]. This is particularly relevant because of the higher mortality observed in non-coeliac SNVA compared to coeliac patients [9, 10, 36].
This study has some limitations. First, follow-up duodenal biopsies were not available in 40 patients with positive HLA-DQ2/-DQ8, no alternative causes for VA and satisfactory clinical response to a GFD. Although their clinical characteristics were similar to patients affected by true seronegative coeliac disease in whom histological recovery upon a GFD was assessed, they could not be considered for the final analysis in order to preserve a rigorous methodological approach. Secondly, a consensus on the diagnostic criteria for seronegative coeliac disease and the differential diagnosis with other non-coeliac enteropathies is currently missing [3–5]. The possibility of applying the method of the “three duodenal biopsies” for the diagnosis of seronegative coeliac disease still needs to be evaluated. While the rationale behind the three-biopsy method would be that of differentiating seronegative coeliac disease from patients with another form of self-limited enteropathy [9,34,43] that could spontaneously heal while being placed on a gluten-free diet, this should be carefully balanced against the potential risk of triggering relapse of severe malabsorption symptoms and developing complications. Moreover, all our patients were suffering from chronic diarrhoea (lasting more than 4 weeks), the waiting lists for endoscopy were at least one month in the three centres, and they were tested for stool cultures and parasites. This makes unlikely the possibility of villous atrophy being due to an acute gastroenteritis, as previously reported [43].
Thirdly, due to the retrospective collection of data, HLA was not available for all the patients and the results for coeliac serology was obtained through different diagnostic kits. Lastly, although two of the centres developed two scores to evaluate GFD adherence [44, 45], for the purpose of this study only dietetic interview was considered. However, these discrepancies reflect the real world clinical practice in three major centres and show how the concept and management of coeliac disease has evolved throughout the years. Supporting the generalizability of our results is the fact that prevalence, clinical features and risk of complications in seronegative coeliac disease and coeliac disease associated to IgA deficiency did not differ significantly between the centres despite potential differences in practice patterns. Trends in mortality of seronegative coeliac disease, and coeliac disease associated to IgA deficiency were similar among centres, although the British cohort had a higher overall mortality, likely influenced by the different methods for assessing mortality among the three centres.
Conclusions
Seronegative coeliac disease is a complex clinical entity characterised by a more severe clinical phenotype and disease course than conventional coeliac disease, which includes both true seronegative coeliac disease and coeliac disease associated to IgA deficiency. Our data would argue against over-reliance on coeliac serology for the diagnosis of coeliac disease and strongly support the need of assessing histological response to a GFD in patients with seronegative coeliac disease. Future perspectives should focus on early detection, and the optimization of the diagnostic criteria and follow-up strategies for these patients, including development of biomarkers for the disease.
AKNOWLEDGEMENTS
FUNDING
Dr. A Schiepatti has received a grant from Collegio Ghislieri, Pavia (Assegno di ricerca annuale per giovani ricercatori, year 2018).
Dr Jocelyn Silvester received financial support from the National Institute Of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Number K23DK119584. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Dr Amelie Therrien was supported by the DG Kinnear Award from the Association Quebecoise des Gastroenterologues du Quebec, and Phase 2 Award from the Fonds de Recherche Sante Quebec.
Role of the founding source:
The founding sources had no role in any of the following: study design; collection, analysis, and interpretation of data; writing of the report; decision to submit the paper for publication.
Declaration of interests:
Dr. Daniel Leffler receives salary support from Takeda Pharmaceuticals. Jocelyn A Silvester has served on an advisory board for Takeda Pharmaceuticals and has received research funding from Biomedal S.L., Cour Pharma and Glutenostics. Prof. David Surendran Sanders has received an educational grant from Dr Schӓr, a gluten-free manufacturer
REFERENCES
- 1.Ludvigsson JF, Bai JC, Biagi F et al. British Society of Gastroenterology. Diagnosis and management of adult celiac disease: guidelines from the British Society of Gastroenterology. Gut. 2014;63:1210–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lebwohl B, Sanders DS, Green PHR. Celiac disease. Lancet 2018;391:70–81 [DOI] [PubMed] [Google Scholar]
- 3.Singh P, Arora A, Strand TA et al. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2018;16:823–836 [DOI] [PubMed] [Google Scholar]
- 4.Schiepatti A, Sanders DS, Biagi F. Seronegative celiac disease: clearing the diagnostic dilemma. Curr Opin Gastroenterol 2018;34:154–158. [DOI] [PubMed] [Google Scholar]
- 5.Schiepatti A, Sanders DS, Zuffada M et al. Overview in the clinical management of patients with seronegative villous atrophy. Eur J Gastroenterol Hepatol. 2019;31:409–417 [DOI] [PubMed] [Google Scholar]
- 6.Leonard MM, Lebwohl B, Rubio-Tapia A et al. AGA Clinical Practice Update on the Evaluation and Management of Seronegative Enteropathies. Gastroenterology 2020. :S0016–5085(20)35220–3. [DOI] [PubMed] [Google Scholar]
- 7.Pallav K, Leffler DA, Tariq S et al. Nonceliac enteropathy: the differential diagnosis of villous atrophy in contemporary clinical practice. Aliment Pharmacol Ther 2012;35:380–90. [DOI] [PubMed] [Google Scholar]
- 8.DeGaetani M, Tennyson CA, Lebwohl B et al. Villous atrophy and negative celiac serology: a diagnostic and therapeutic dilemma. Am J Gastroenterol 2013;108:647–53. [DOI] [PubMed] [Google Scholar]
- 9.Aziz I, Peerally MF, Barnes JH et al. The clinical and phenotypical assessment of seronegative villous atrophy; a prospective UK center experience evaluating 200 adult cases over a 15-year period (2000–2015). Gut 2017;66:1563–1572 [DOI] [PubMed] [Google Scholar]
- 10.Schiepatti A, Biagi F, Fraternale G et al. Mortality and differential diagnoses of villous atrophy without celiac antibodies. Eur J Gastroenterol Hepatol 2017;29:572–576. [DOI] [PubMed] [Google Scholar]
- 11.Volta U, Caio G, Boschetti E et al. Seronegative celiac disease: Shedding light on an obscure clinical entity. Dig Liver Dis 2016;48:1018–22. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Bañares F, Crespo L, Núñez C et al. Gamma delta+ intraepithelial lymphocytes and celiac lymphogram in a diagnostic approach to celiac disease in patients with seronegative villous atrophy. Aliment Pharmacol Ther 2020;51:699–705. [DOI] [PubMed] [Google Scholar]
- 13.Rostami K, Kerckhaert J, von Blomberg BM et al. SAT and serology in adult celiacs, seronegative celiac disease seems a reality. Neth J Med 1998;53:15–9. [DOI] [PubMed] [Google Scholar]
- 14.Rostami K, Kerckhaert J, Tiemessen R et al. Sensitivity of antiendomysium and antigliadin antibodies in untreated celiac disease: disappointing in clinical practice. Am J Gastroenterol 1999;94:888–94. [DOI] [PubMed] [Google Scholar]
- 15.Dickey W, Hughes DF, McMillan SA. Reliance on serum endomysial antibody testing underestimates the true prevalence of celiac disease by one fifth. Scand J Gastroenterol 2000;35:181–3. [DOI] [PubMed] [Google Scholar]
- 16.Dahele A, Kingstone K, Bode J et al. Anti-endomysial antibody negative celiac disease: does additional serological testing help? Dig Dis Sci 2001;46:214–21. [DOI] [PubMed] [Google Scholar]
- 17.Dahele AV, Aldhous MC, Humphreys K et al. Serum IgA tissue transglutaminase antibodies in celiac disease and other gastrointestinal diseases. QJM 2001;94:195–205. [DOI] [PubMed] [Google Scholar]
- 18.Clemente MG, Musu MP, Frau F et al. Antitissue transglutaminase antibodies outside celiac disease. J Pediatr Gastroenterol Nutr 2002;34:31–4. [DOI] [PubMed] [Google Scholar]
- 19.Abrams JA, Diamond B, Rotterdam H et al. Seronegative celiac disease: increased prevalence with lesser degrees of villous atrophy. Dig Dis Sci 2004;49:546–50. [DOI] [PubMed] [Google Scholar]
- 20.Collin P, Kaukinen K, Vogelsang H et al. Antiendomysial and antihuman recombinant tissue transglutaminase antibodies in the diagnosis of celiac disease: a biopsy-proven European multicenter study. Eur J Gastroenterol Hepatol 2005;17:85–91. [DOI] [PubMed] [Google Scholar]
- 21.Salmi TT, Collin P, Korponay-Szabó IR et al. Endomysial antibody-negative celiac disease: clinical characteristics and intestinal autoantibody deposits. Gut 2006;55:1746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dore MP, Pes GM, Dettori I et al. Clinical and genetic profile of patients with seronegative celiac disease: the natural history and response to gluten-free diet. BMJ Open Gastroenterol 2017;4:e000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farina MH, Kumar Mandhwani R et al. Clinicopathological Study of Seronegative Celiac Disease in Adults in Pakistan: A Pilot Study. Middle East J Dig Dis 2017;9:94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ríos León R, Crespo Pérez L, Rodríguez de Santiago E et al. Genetic and flow cytometry analysis of seronegative celiac disease: a cohort study. Scand J Gastroenterol 2019;54:563–570. [DOI] [PubMed] [Google Scholar]
- 25.Leffler DA, Schuppan D. Update on serologic testing in celiac disease. Am J Gastroenterol 2010;105:2520–4. [DOI] [PubMed] [Google Scholar]
- 26.Reunala T, Salmi TT, Hervonen K et al. Dermatitis Herpetiformis: A Common Extraintestinal Manifestation of Celiac Disease. Nutrients 2018;1:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esteve M, Rosinach M, Fernández-Bañares F et al. Spectrum of gluten-sensitive enteropathy in first-degree relatives of patients with celiac disease: clinical relevance of lymphocytic enteritis. Gut 2006;55:1739–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biagi F, Schiepatti A, Maiorano G et al. Risk of complications in celiac patients depends on age at diagnosis and type of clinical presentation. Dig Liver Dis 2018;50:549–552. [DOI] [PubMed] [Google Scholar]
- 29.Cellier C, Delabesse E, Helmer C et al. Refractory sprue, celiac disease, and enteropathy-associated T-cell lymphoma. French Celiac Disease Study Group. Lancet 2000;356:203–8. [DOI] [PubMed] [Google Scholar]
- 30.Malamut G, Afchain P, Verkarre V et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology 2009;136:81–90. [DOI] [PubMed] [Google Scholar]
- 31.Oberhuber G, Granditsch G, Vogelsang H. The histopathology of celiac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol Hepatol 1999;11:1185–94. [DOI] [PubMed] [Google Scholar]
- 32.Corazza GR, Villanacci V, Zambelli C et al. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin Gastroenterol Hepatol 2007;5:838–43. [DOI] [PubMed] [Google Scholar]
- 33.van Wanrooij RL, Müller DM, Neefjes-Borst EA et al. Optimal strategies to identify aberrant intra-epithelial lymphocytes in refractory celiac disease. J Clin Immunol 2014;34:828–35 [DOI] [PubMed] [Google Scholar]
- 34.Schiepatti A, Sanders DS, Aziz I et al. Clinical phenotype and mortality in patients with idiopathic small bowel villous atrophy: a dual-center international study. Eur J Gastroenterol Hepatol 2020;32:938–949. [DOI] [PubMed] [Google Scholar]
- 35.Corazza GR, Biagi F, Volta U et al. Autoimmune enteropathy and villous atrophy in adults. Lancet 1997;350:106–9. [DOI] [PubMed] [Google Scholar]
- 36.Pensieri MV, Pulvirenti F, Schiepatti A et al. The high mortality of patients with common variable immunodeficiency and small bowel villous atrophy. Scand J Gastroenterol 2019;54:164–168. [DOI] [PubMed] [Google Scholar]
- 37.Hopper AD, Cross SS, Hurlstone DP et al. Pre-endoscopy serological testing for coeliac disease: evaluation of a clinical decision tool. BMJ 2007;334:729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mooney PD, Wong SH, Johnston AJ et al. Increased Detection of Celiac Disease With Measurement of Deamidated Gliadin Peptide Antibody Before Endoscopy. Clin Gastroenterol Hepatol 2015;13:1278–1284. [DOI] [PubMed] [Google Scholar]
- 39.Collin P, Mäki M, Keyriläinen O et al. Selective IgA deficiency and celiac disease. Scand J Gastroenterol 1992;27:367–71 [DOI] [PubMed] [Google Scholar]
- 40.Cataldo F, Marino V, Ventura A et al. Prevalence and clinical features of selective immunoglobulin A deficiency in celiac disease: an Italian multicenter study. Italian Society of Paediatric Gastroenterology and Hepatology (SIGEP) and “Club del Tenue” Working Groups on Celiac Disease. Gut 1998;42:362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cataldo F, Lio D, Marino V et al. IgG(1) antiendomysium and IgG antitissue transglutaminase (anti-tTG) antibodies in celiac patients with selective IgA deficiency. Gut 2000;47:366–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ludvigsson JF, Neovius M, Hammarström L. IgA deficiency and mortality: a population-based cohort study. J Clin Immunol 2013;33:1317–24. [DOI] [PubMed] [Google Scholar]
- 43.Goldstein NS. Non-gluten sensitivity-related small bowel villous flattening with increased intraepithelial lymphocytes: not all that flattens is celiac sprue. Am J Clin Pathol 2004;121:546–50. [DOI] [PubMed] [Google Scholar]
- 44.Biagi F, Andrealli A, Bianchi PI, Marchese A, Klersy C, Corazza GR. A gluten-free diet score to evaluate dietary compliance in patients with celiac disease. Br J Nutr 2009;102:882–7. [DOI] [PubMed] [Google Scholar]
- 45.Leffler DA, Dennis M, Edwards George JB et al. A simple validated gluten-free diet adherence survey for adults with celiac disease. Clin Gastroenterol Hepatol 2009;7:530–6 [DOI] [PubMed] [Google Scholar]