

Abstract
The addition of HER2-targeted agents to neoadjuvant chemotherapy has dramatically improved pathological complete response (pCR) rates in early-stage, HER2-positive breast cancer. Nonetheless, up to 50% of patients have residual disease after treatment, while others are likely overtreated. Here, we performed multiplex spatial proteomic characterization of 122 samples from 57 HER2-positive breast tumors from the neoadjuvant TRIO-US B07 clinical trial sampled pre-treatment, after 14–21 d of HER2-targeted therapy and at surgery. We demonstrated that proteomic changes after a single cycle of HER2-targeted therapy aids the identification of tumors that ultimately undergo pCR, outperforming pre-treatment measures or transcriptomic changes. We further developed and validated a classifier that robustly predicted pCR using a single marker, CD45, measured on treatment, and showed that CD45-positive cell counts measured via conventional immunohistochemistry perform comparably. These results demonstrate robust biomarkers that can be used to enable the stratification of sensitive tumors early during neoadjuvant HER2-targeted therapy, with implications for tailoring subsequent therapy.
Human epidermal growth factor receptor 2 (HER2)-positive breast cancer accounts for 15–30% of invasive breast cancers and is associated with an aggressive phenotype1. The neoadjuvant setting allows for early assessment of treatment response and pCR is a strong surrogate for long-term survival in HER2-positive disease2–4. While the addition of HER2-targeted agents to neoadjuvant chemotherapy has dramatically improved pCR rates in early-stage HER2-positive breast cancer, 40–50% of patients have residual disease after treatment5,6. Conversely, HER2 inhibition with two targeted agents and without chemotherapy can result in pCR, suggesting that it may be possible to eliminate chemotherapy in a subset of patients7–10. Given the heterogeneity in response to HER2-targeted therapy5,11, identification of biomarkers of response beyond HER2 and estrogen receptor status is needed. Bulk gene expression profiling of pre-treatment samples has identified tumor (HER2-enriched intrinsic subtype, HER2 expression levels and estrogen receptor 1 expression levels10,12–15) and microenvironmental characteristics (increased immune infiltration13,15–18) that associate with pCR to HER2-targeted therapy. While pCR and long-term survival correlate strongly at an individual level2–4, tumor characteristics may correlate in opposite directions with pCR and recurrence-free survival (RFS) (for example, with the HER2-enriched intrinsic subtype correlating with higher pCR rate and shorter RFS), while immune characteristics may correlate in the same direction (that is, increased immune infiltration correlating with higher pCR rate and longer RFS)19.
Because tumor cells are profiled simultaneously with both colocalized and distant stroma and immune cells, bulk expression profiling is an imperfect tool for analyzing tumor and microenvironmental changes across treatment. In particular, it is difficult to assign observed changes to specific geographical or phenotypic cell populations within the complex tumor ecosystem, where malignant tumor cells interact with fibroblasts, endothelial cells and immune cells. Moreover, immune cells can be further divided into those that infiltrate the tumor core and those that are excluded20. As of yet, how the tumor and immune microenvironment change during therapy is poorly understood, necessitating multiplexed in situ profiling of longitudinal tissue samples. It is unknown whether such on-treatment in situ profiling might outperform pre-treatment bulk expression biomarkers in predicting HER2-targeted therapy response.
We used the GeoMx Digital Spatial Profiling (DSP, NanoString) technology to assay archival tissue from an initial discovery set of 28 patients with HER2-positive breast cancer enrolled on the neoadjuvant TRIO-US B07 clinical trial21, whose tumors were sampled pre-treatment, after 14–21 d of HER2-targeted therapy, consisting of lapatinib, trastuzumab or both (on-treatment), and at the time of surgery after completion of combination chemotherapy with HER2-targeted therapy (post-treatment). We subsequently validated our results in an independent set of 29 patients from the same trial. DSP enables geographical and phenotypic selection of tissue regions for multiplex proteomic characterization of cancer signaling pathways and the tumor-colocalized immune microenvironment22,23. In particular, we characterized spatial heterogeneity in untreated breast tumors as well as changes in cancer signaling pathways and microenvironmental composition in matched on-treatment biopsies and post-treatment surgical samples by profiling 40 tumor and immune proteins across multiple pan-cytokeratin (pan-CK)-enriched regions per sample. On-treatment protein expression changed dramatically in tumors that went on to achieve a pCR; a classifier based on these data outperformed models based on transcriptomic data, as well as established predictors such as PAM50 subtype10,14,24 and stromal lymphocytes (cellularity and tumor-infiltrating lymphocytes (TILs))17. This new spatial proteomic model robustly predicted treatment response in the validation cohort. Moreover, on-treatment CD45 protein expression alone predicted outcome with high accuracy—a finding that was reproduced with CD45 immunohistochemistry (IHC). Collectively, these results suggest new avenues to personalize therapy in early-stage HER2-positive breast cancer.
Results
Spatial proteomic analysis of HER2-positive breast tumors.
Participants in the TRIO-US B07 clinical trial (NCT00769470) in early-stage HER2-positive breast cancer received one cycle of neoadjuvant HER2-targeted therapy, including either trastuzumab, lapatinib or both agents, which was followed by six cycles of the assigned HER2-targeted therapy plus docetaxel and carboplatin given every three weeks21. Core biopsies were obtained pre-treatment and on-treatment after 14–21 d of HER2-targeted therapy; surgical excision specimens were obtained post-treatment (Fig. 1a). We defined a discovery cohort of 28 patients for whom formalin-fixed paraffin-embedded (FFPE) samples were available from all three time points (pre-treatment, on-treatment and at surgery). The cohort was balanced for both pCR and estrogen receptor status (Extended Data Fig. 1a,b) and was used for all exploratory analyses. We additionally defined an independent validation cohort of 29 patients from the same trial with matched pre-treatment and on-treatment FFPE samples to evaluate model performance.
Fig. 1 |. Spatial proteomic analysis of untreated HER2-positive breast tumors.
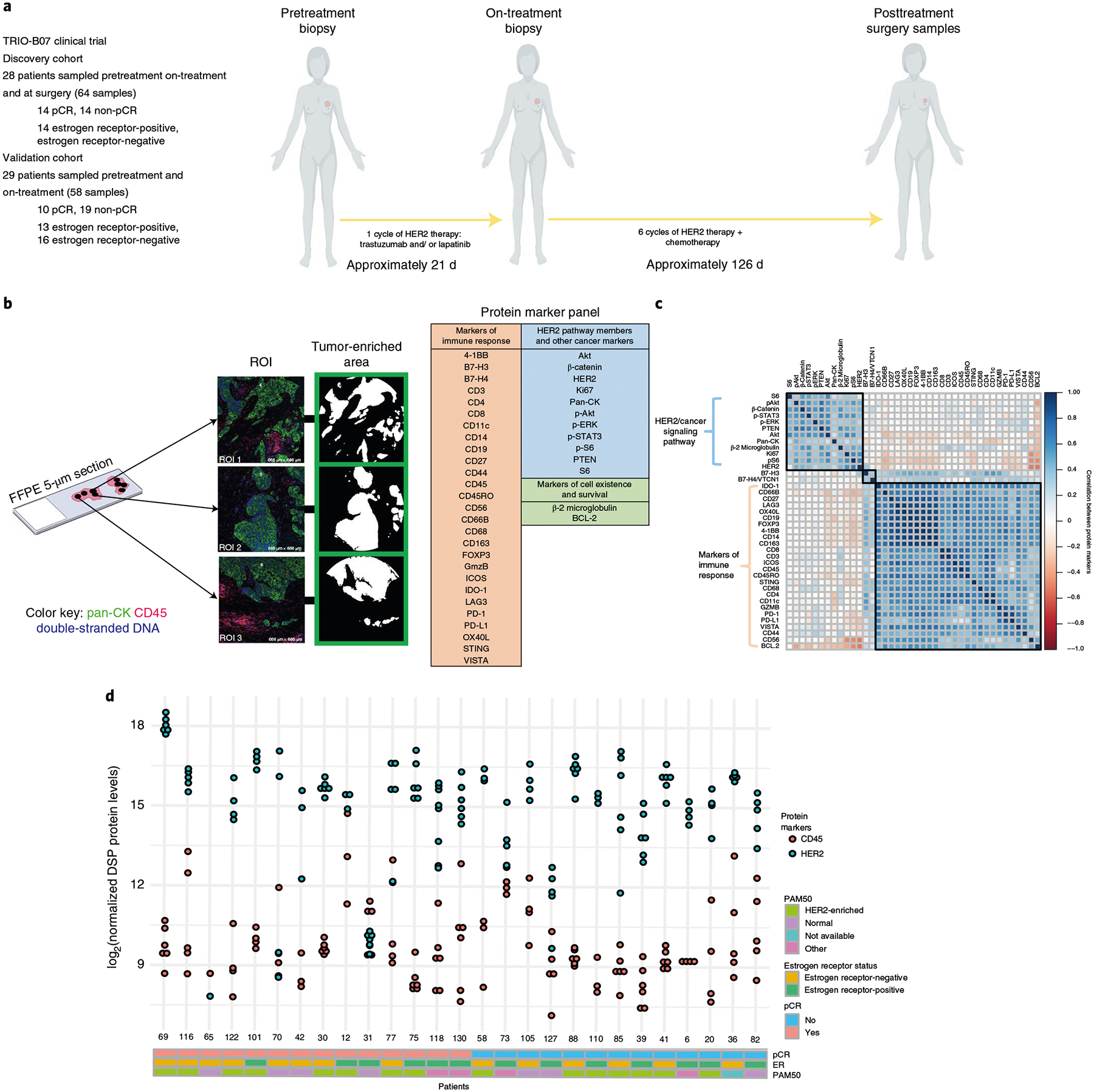
a, Schematic overview of the discovery and validation cohorts analyzed with the GeoMx DSP technology. Patients with invasive HER2-positive breast cancer enrolled on the TRIO-US B07 clinical trial were treated with one cycle of the assigned HER2-targeted therapy, followed by six cycles of the assigned HER2-targeted treatment plus chemotherapy (docetaxel and carboplatin). Tissue was obtained at three time points (pre-treatment, on-treatment and post-treatment/surgery). Base graphics for humans and tumors are from BioRender via a paid and licensed user account. b, Multiple ROIs per tissue sample were selected based on pan-CK enrichment and subjected to spatial proteomic profiling of 40 tumor and immune markers. Protein counts were measured within phenotypic regions corresponding to the pan-CK-enriched masks that included tumor cells and colocalized immune cells and separately for the inverted mask corresponding to pan-CK-negative regions. c, Pairwise correlation of pre-treatment protein marker expression across all ROIs in the discovery cohort. The black squares indicate probes in the same hierarchical cluster. d, Inter- and intratumor variability in HER2 and CD45 protein expression in n = 28 untreated HER2-positive breast tumors from the discovery cohort, where each point corresponds to an ROI. Clinical characteristics, including pCR status, estrogen receptor status and PAM50 subtype (based on gene expression profiling) are indicated.
DSP enables multiplex proteomic profiling of FFPE tissue sections (Extended Data Fig. 2a), where regions of interest (ROIs) can be selected based on both geographical and phenotypic characteristics (Extended Data Fig. 2b). We employed a pan-CK enrichment strategy to profile cancer cells and colocalized immune cells across an average of four regions per tissue specimen (Fig. 1b). Using CD45, pan-CK and double-stranded DNA immunofluorescence markers for visualization (Extended Data Fig. 1c), we selected spatially separated regions; a mask governing the ultraviolet (UV) illumination for protein quantitation was generated based on pan-CK immunofluorescence. In total, 40 tumor and immune proteins were profiled using DSP and proteins assessed using both DSP and orthogonal technologies showed strong concordance (Fig. 1b and Extended Data Fig. 2c–e). We further utilized paired pre-treatment and on-treatment bulk gene expression data from the same patients to infer PAM50 subtype and enable comparisons with the spatially resolved DSP data21.
In untreated tumors, the correlation among immune markers was striking, suggesting the coordinated action of multiple immune cell subpopulations (Fig. 1c). HER2 pathway members and other downstream cancer signaling markers were also highly correlated, while the correlation between tumor and immune markers was minimal for most marker pairs. Inter- and intratumor variability at the proteomic level was evident before treatment, including for HER2 and the pan-leukocyte marker CD45 (Fig. 1d). Averaging all ROIs per tumor to derive a composite score per marker, we found that tumors that achieved a pCR and those that did not had similar baseline HER2 levels (mean pCR cases, 14.50; mean non-pCR cases, 15.00) and baseline CD45 levels (mean pCR, 9.90; mean non-pCR, 9.64). Using a linear mixed-effects model with blocking by patient (Methods), we further found that individual DSP protein markers, including HER2 and CD45, did not significantly differ between pCR and non-pCR cases before treatment (unadjusted P > 0.10 for all markers).
Cancer signaling and immune changes after short-term therapy.
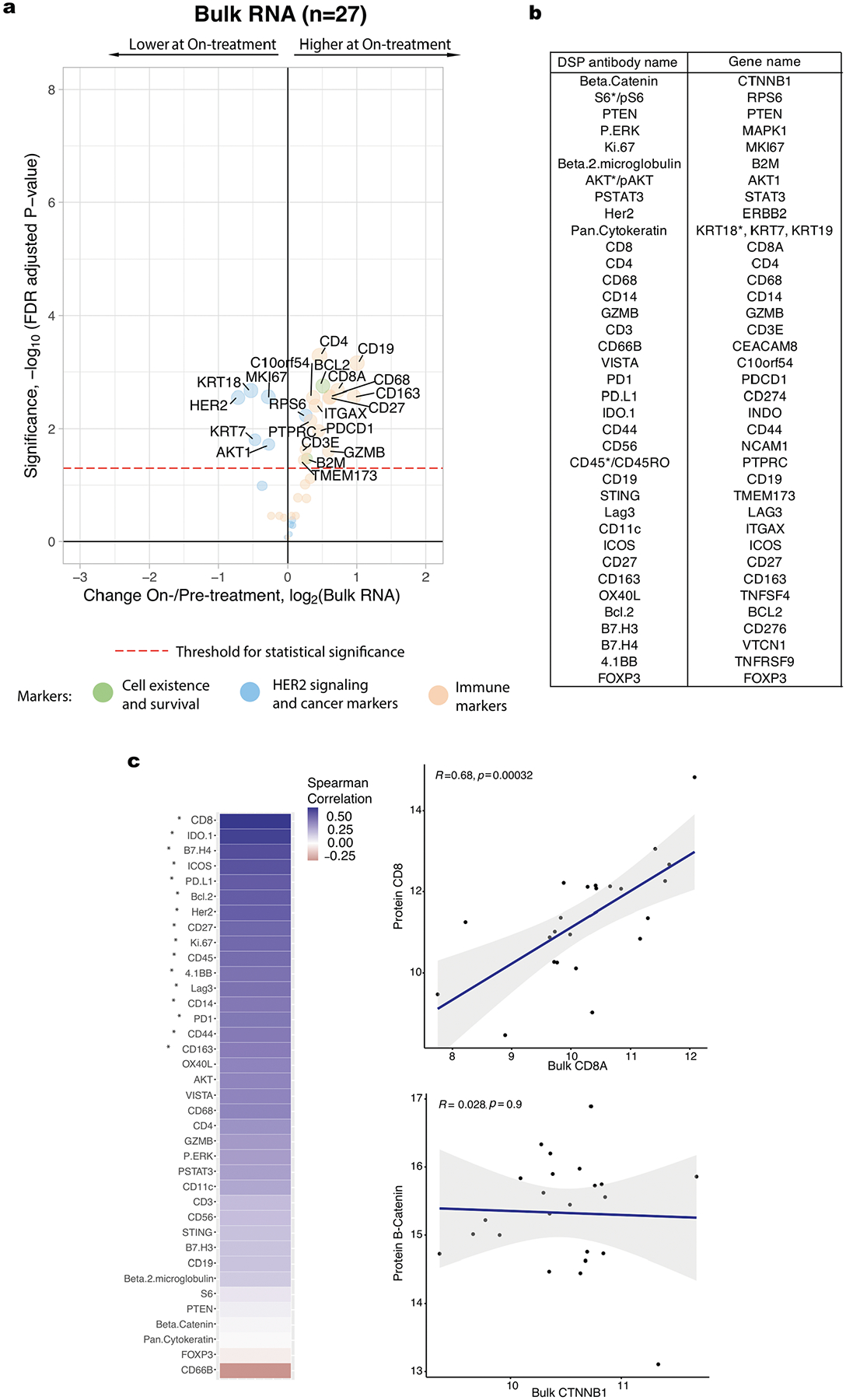
We used DSP to investigate treatment-related changes in breast tumor and immune markers during short-term HER2-targeted therapy by profiling on-treatment (after a single cycle of HER2-targeted therapy alone) biopsies in the discovery cohort. The protein markers that were most associated with pCR at the on-treatment time point were CD45 (unadjusted P = 0.0024) and CD56, a natural killer (NK) cell marker (unadjusted P = 0.0055) (Extended Data Fig. 1d,e). We quantified the fold change in protein levels on-treatment relative to pre-treatment using a linear mixed-effects model with blocking by patient and visualized the significance (false discovery rate (FDR)-adjusted P) of all markers relative to their fold change in volcano plots. These analyses revealed a dramatic reduction in HER2 and Ki67, accompanied by other downstream pathway members, including pAkt, Akt, pERK, S6 and pS6, with the phosphorylated proteins decreasing comparatively more (Fig. 2a). Immune markers—including CD45 and CD8, a marker of cytotoxic T cells—exhibited the greatest increase in expression with treatment. Of note, increased expression of CD8-positive T cells was similarly observed in the TRIO-US B07 transcriptomic data through cell type deconvolution21. More generally, the on-treatment versus pre-treatment bulk transcriptome data mirrored the changes seen at the protein level but the fold changes were attenuated (Extended Data Fig. 3). For example, using genes that corresponded with the DSP protein markers, we found that the expression of Erbb2, Akt1, Mki67 and breast cancer-associated keratin genes (Krt7, Krt18 and Krt19)25 decreased significantly with treatment, while immune markers increased (Extended Data Fig. 3). Despite the use of different analytes, measurements and tissue sections, the DSP protein and bulk RNA datasets consistently showed reduced HER2 signaling and breast cancer-associated markers, accompanied by increased immune cell infiltration during neoadjuvant treatment. Given that lapatinib was associated with lower pCR rates in the TRIO-US B07 trial and more generally26, we additionally assessed on-treatment changes in the trastuzumab-treated cases (arms 1 and 3, n = 23) and observed similar patterns as in the full discovery cohort (Extended Data Fig. 4a).
Fig. 2 |. Spatial proteomic analysis reveals changes in cancer signaling and immune infiltration after short-term HER2-targeted therapy.
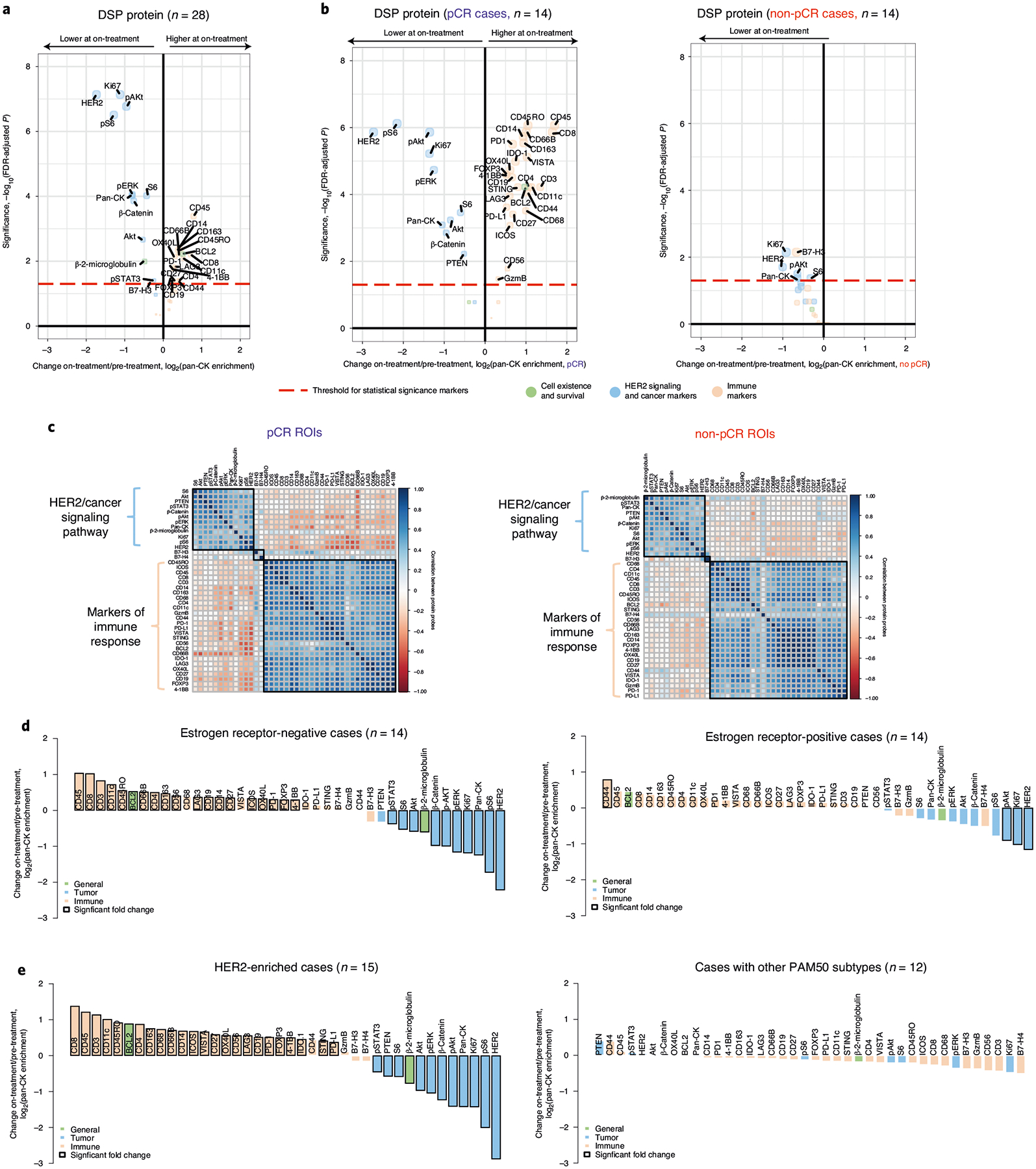
a, Volcano plot demonstrating treatment-associated changes based on comparison of pre-treatment versus on-treatment protein marker expression levels in pan-CK-enriched regions. Significance, −log10(FDr-adjusted P), is indicated along the y axis. The P value is determined based on a linear mixed-effects model (two-sided test) with blocking by patient and is derived from the t-value (a measure of the size of the difference relative to the variation in the sample data). n indicates the number of biologically independent cases used in the analysis. b, Volcano plots demonstrating treatment-associated changes in pCR versus non-pCR cases. n indicates the number of biologically independent cases used in each analysis. c, Pairwise correlation of protein markers in pCR versus non-pCR cases. The black squares demarcate hierarchical clusters. d, Waterfall plots illustrating treatment-associated changes (pre-treatment to on-treatment) in estrogen receptor-positive and estrogen receptor-negative cases based on protein expression. e, Waterfall plots illustrating treatment-associated changes in DSP protein expression (pre-treatment to on-treatment) in HER2-enriched and non-HER2-enriched cases (n = 7 normal-like, n = 2 luminal B, n = 2 basal, n = 1 luminal A). Analyses were performed in the discovery cohort.
We next examined how treatment-associated changes differed based on tumor sensitivity to HER2-targeted therapy, stratifying tumors based on achievement of pCR after neoadjuvant therapy (Fig. 2b). In the pCR cases, a multitude of immune markers increased with treatment, while, in the non-pCR cases, no significant treatment-associated immune changes were observed and the reduction in Ki67 and HER2 signaling was modest. These patterns can also be visualized via pairwise comparisons of protein marker correlations, which revealed a stronger negative correlation between the immune marker cluster and the cancer cell marker cluster in tumors that went on to achieve a pCR (mean fold change across all markers in pCR cases = −0.231, non-pCR cases = −0.075, two-sided Wilcoxon rank-sum test, P < 2.2 × 10−16) (Fig. 2c). Notably, no such differences between pCR and non-pCR cases were observed in the on-treatment bulk expression data from this same cohort21.
Since both estrogen receptor status8,24 and HER2-enriched subtype have been associated with response to neoadjuvant therapy10,14,18, we explored how protein marker expression changed with these covariates. Estrogen receptor-negative tumors exhibited more significant changes on-treatment (relative to pre-treatment) compared to estrogen receptor-positive tumors (mean absolute fold change estrogen receptor-negative cases = 0.59, mean estrogen receptor-positive cases = 0.36, two-sided Wilcoxon rank-sum test, P = 0.0045; Fig. 2d). However, when tumors were stratified by outcome, pCR cases exhibited more significant changes than non-pCR cases irrespective of estrogen receptor status (Extended Data Fig. 5a) and estrogen receptor status was not predictive of pCR in this cohort (P = 0.47). Similarly, tumors classified as HER2-enriched before treatment exhibited more significant changes in tumor and immune markers in the on-treatment biopsy relative to other subtypes (Fig. 2e and Extended Data Fig. 5b,c). For example, while CD8-positive T cells increased significantly with treatment in HER2-enriched cases, they decreased slightly in other cases. However, as in the full TRIO-US B07 transcriptomic cohort21, HER2-enriched subtype was not predictive of pCR (P = 0.87).
To assess the utility of multiregion sampling, we measured changes on-treatment versus pre-treatment using a single randomly selected region per tissue sample averaged across 100 simulations (Extended Data Fig. 5d). Consistent with the findings based on all tumor regions, CD45 and CD8 showed the greatest increase on-treatment, while HER2 and pS6 decreased most in the single region analysis. While the magnitude of marker fold change with treatment was greater for pCR cases than non-pCR cases (mean absolute fold change across all markers in pCR cases = 0.87 versus non-pCR cases = 0.33, two-sided Wilcoxon rank-sum test, P = 1.02 × 10−7), individual markers did not increase significantly with treatment in the single region analysis, reflecting increased variance.
We also examined treatment-associated changes in patients with residual tumor cells present at the time of surgery (non-pCR cases) to elucidate the biology associated with combined HER2-targeted therapy and chemotherapy. While the non-pCR cases showed limited changes at the on-treatment time point, by the time of surgery there was a substantial reduction in the HER2 and downstream Akt signaling pathway and a concomitant increase in immune markers in pan-CK-enriched regions (Extended Data Fig. 1f). Notably, HER2 decreased more significantly than its downstream pathway members, which may reflect compensatory pathway activation contributing to resistance9,27. While some immune markers increased significantly in non-pCR cases at surgery (n = 8), the fold change was diminished relative to pCR cases sampled on-treatment (mean fold change non-pCR post-treatment = 0.30, mean fold change pCR on-treatment = 0.85, two-sided Wilcoxon rank-sum test, P = 0.0021; Fig. 2b). Among the immune markers that increased at surgery in the non-pCR cases, CD56 was most significant and potentially related to the role of NK cells in identifying and killing chemotherapy-stressed tumor cells28. NK cells were similarly found to increase at the time of surgery in the TRIO-US B07 bulk expression data21.
Increased heterogeneity during HER2-targeted therapy.
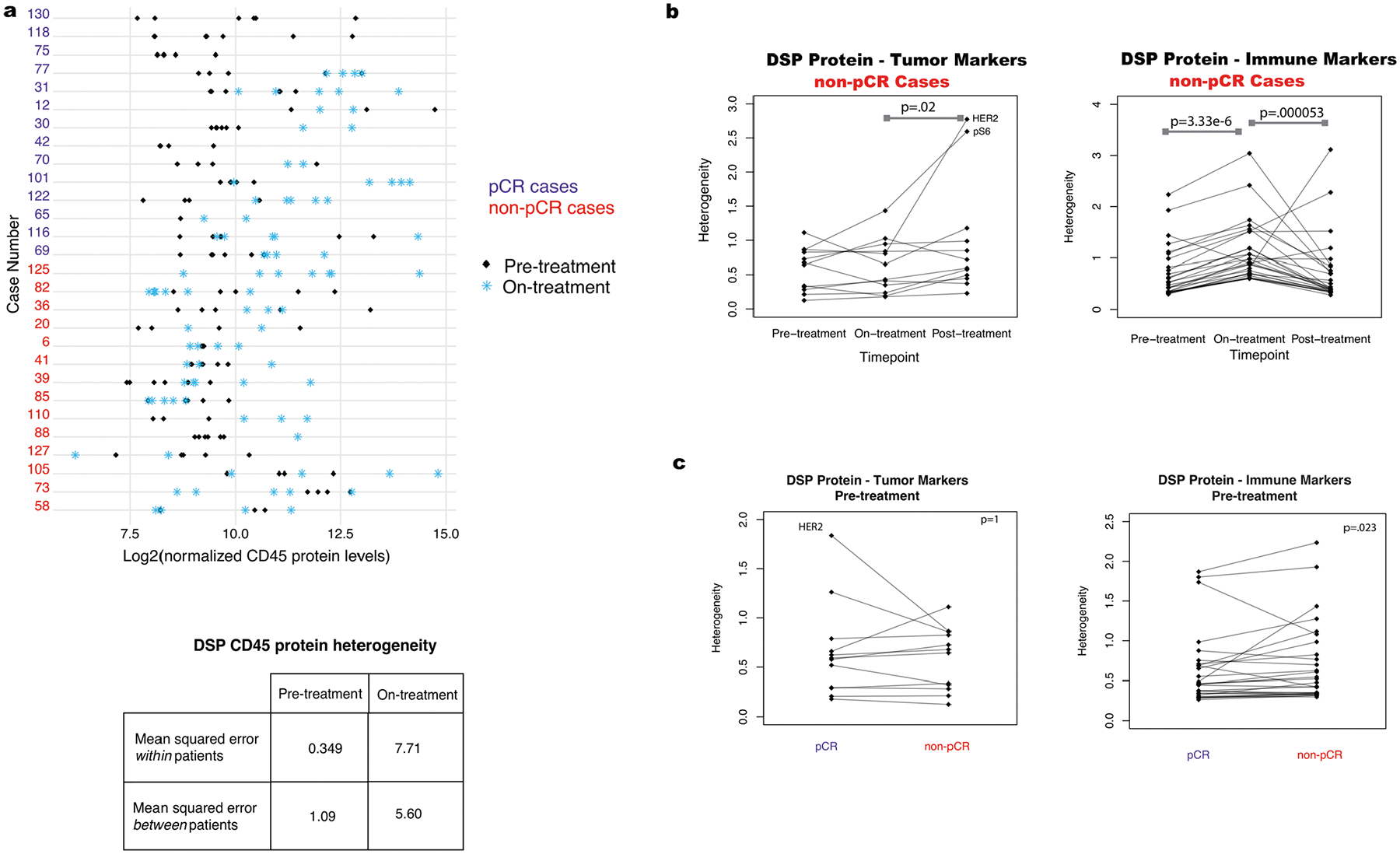
Given that genomic heterogeneity is a defining feature of HER2-positive breast cancer, we next evaluated the extent to which HER2 protein expression varied within different regions of a breast tumor biopsy through neoadjuvant treatment and between patients. As shown for two exemplary cases (Fig. 3a), HER2 protein levels across geographically disparate regions within each tissue sample exhibited relatively consistent HER2 protein levels before treatment in most cases (Fig. 3b). Far greater heterogeneity in HER2 protein expression was observed on-treatment both between regions and between patients (Fig. 3c). Similarly, CD45, a pan-leukocyte marker, showed increased heterogeneity between regions on-treatment and was higher in the pCR cases on-treatment as compared to the non-pCR cases (Extended Data Fig. 6a). Such regional heterogeneity may reflect pharmacokinetic differences due to vasculature, tissue architecture, immune infiltration or the biopsy itself, underscoring the importance of profiling multiple regions per sample on-treatment.
Fig. 3 |. Increased heterogeneity of tumor and immune markers during HER2-targeted therapy.
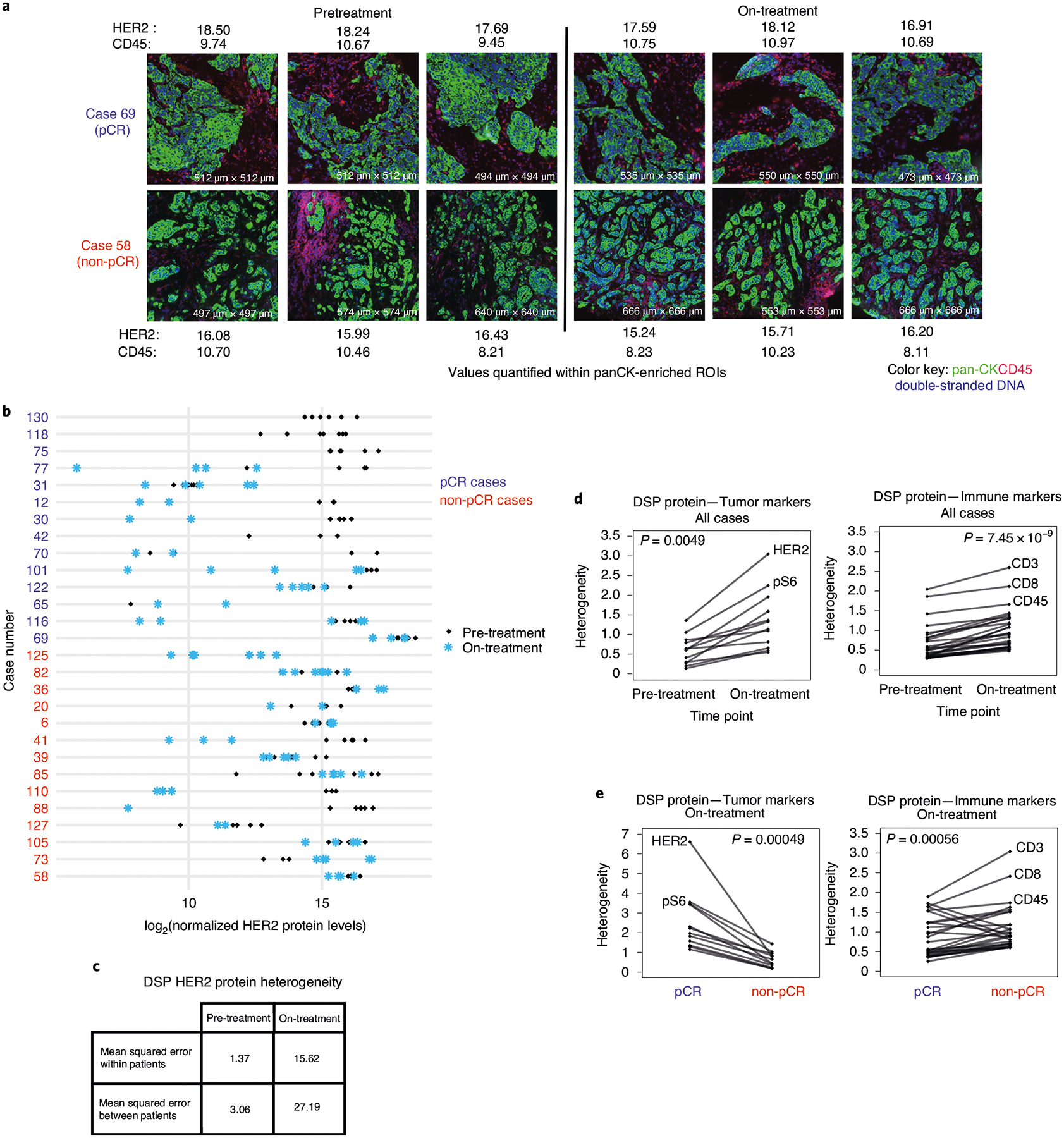
a, representative images of ROIs from two cases and quantification of HER2 and CD45 protein levels (log2-normalized) in pan-CK-enriched regions. b, Comparison of DSP HER2 protein levels pre-treatment and on-treatment for all regions profiled per case per time point (n = 28 patients). c, Comparison of the mean squared error in DSP HER2 protein expression pre-treatment versus on-treatment within and between patients. d, Pre-treatment versus on-treatment heterogeneity for each DSP tumor and immune marker. e, On-treatment heterogeneity in DSP protein markers for pCR and non-pCR cases. Heterogeneity was calculated as the mean squared error within patients based on ANOVA in c–e. P values are based on a two-sided paired Wilcoxon signed-rank test. Analyses are based on the discovery cohort (n = 28 patients).
We also investigated regional heterogeneity across all tumor and immune protein markers during treatment. For each marker and at each time point, regional heterogeneity across the cohort was computed as the within-patient mean squared error based on analysis of variance (ANOVA) (Methods). Across all markers, DSP protein heterogeneity increased significantly on-treatment relative to pre-treatment (Fig. 3d), similar to that noted for HER2. These changes were widespread, with heterogeneity being higher for all tumor and immune markers on-treatment compared to pre-treatment. The probes with the greatest heterogeneity included both tumor (HER2, pS6) and immune (CD3, CD8) markers. Among tumors that failed to achieve a pCR, we evaluated heterogeneity throughout the course of neoadjuvant therapy. Heterogeneity among tumor markers was not significantly different on-treatment and pre-treatment (two-sided Wilcoxon rank-sum test, P = 0.52) but increased at surgery (post-treatment), whereas immune marker heterogeneity increased on-treatment with a subsequent decrease at surgery (Extended Data Fig. 6b,c). Tumors that achieved a pCR exhibited higher protein heterogeneity among tumor markers (including HER2) on-treatment, whereas those that did not exhibited higher heterogeneity across immune markers (Fig. 3e). Higher immune marker heterogeneity on-treatment in the non-pCR cases may reflect a less consistent immune response with some regions experiencing a greater immune influx than others. We did not observe higher pre-treatment HER2 heterogeneity in the non-pCR cases compared to pCR cases, as recently suggested based on fluorescence in situ hybridization (FISH)29; comparable regional heterogeneity among tumor markers was noted in pCR and non-pCR cases (Extended Data Fig. 6b).
We further analyzed the DSP data to investigate the composition of immune cells in pan-CK-enriched regions (used in all other analyses) relative to the surrounding pan-CK-negative regions designed to capture the neighboring microenvironment (Extended Data Fig. 7). Before treatment, both T-cell (CD3, CD4, CD8) and macrophage (CD68) markers were more prevalent in the surrounding microenvironment, while CD56-positive NK cells and immunosuppressive markers (for example, B7-H4 (VCTN1), PD-L1, IDO) were higher in pan-CK-enriched regions (Extended Data Fig. 7b). Although we cannot rule out the possibility of nonspecific staining, these findings are consistent with T-cell exclusion, where IDO and PD-L1 are thought to impair intratumor proliferation of effector T cells30. A similar immune profile was observed during HER2-targeted therapy alone. However, post-treatment, in the non-pCR cases with tumor remaining, most immune markers were more prevalent in the pan-CK-enriched regions compared to the neighboring microenvironment, including CD8 and CD68 (Extended Data Fig. 7b). Both before treatment and on-treatment, immune cell localization was similar in patients who achieved a pCR and those who did not (Extended Data Fig. 7c), as well as for estrogen receptor-positive versus estrogen receptor-negative cases (Extended Data Fig. 7d).
As proof of principle that other multiplexed imaging technologies can similarly be used to profile pan-CK-enriched tumors, we also used multiplex IHC to profile tissue samples from a patient who achieved a pCR and one who did not. Pan-CK antibodies were to define mask regions and several markers that changed significantly with treatment based on DSP—namely HER2, CD45 and CD8—were quantified across the whole tissue section and within pan-CK-enriched regions (Extended Data Fig. 8a,b). As expected, changes in protein expression signals were muted when the whole tissue section was considered relative to pan-CK-enriched regions. These data further support the concept that pan-CK enrichment may be beneficial for defining tumor and colocalized immune changes in breast and other tumors.
The geospatial distribution of tumor and immune cells has been associated with relapse and survival in multiple tumor types31,32. Therefore, we investigated the relationship between treatment and the tumor microenvironment border using perimetric complexity, which is proportional to the perimeter of a region squared, divided by the area of the region33 (Methods and Extended Data Fig. 8c). Before treatment, no significant difference in perimetric complexity was observed between pCR and non-pCR cases (P = 0.299; Extended Data Fig. 8d). However, perimetric complexity decreased significantly on-treatment relative to pre-treatment (P = 1.32 × 10−6; Extended Data Fig. 8e). These data suggest that treatment may affect the geographical distribution of tumor cells as well as tumor cell content. Indeed, the proliferative marker Ki67 was highly correlated with perimetric complexity (Extended Data Fig. 8f). Thus, for highly proliferative tumors, the perimeter of the tumor microenvironment border may be relatively larger, allowing for increased cross talk with the surrounding microenvironment.
DSP of tumor biopsies reveals features associated with pCR.
Given the dramatic differences in treatment-associated changes in pCR cases compared to non-pCR cases (Fig. 2b), we next sought to evaluate whether DSP protein marker status before treatment or early on during the course of therapy could be used to predict pCR. We used an L2-regularized logistic regression to classify tumors by pCR status based on average DSP protein expression levels across multiple ROIs profiled pre-treatment and on-treatment, or the average marker expression both pre-treatment and on-treatment (denoted ‘on-treatment plus pre-treatment’) and evaluated model performance via nested cross-validation within the discovery cohort (Methods). Cases with tumor-enriched regions identified via DSP at both time points were utilized in this analysis (n = 23 cases; Extended Data Fig. 1b). A model based on on-treatment protein expression outperformed that based on pre-treatment protein expression (mean area under the receiver operating characteristic (AUROC) = 0.728 versus 0.614) and performed comparably to a model incorporating both on-treatment and pre-treatment protein expression levels (mean AUROC = 0.733) (Fig. 4a). A classifier trained using both immune and tumor markers outperformed a model using tumor markers alone (Fig. 4b).
Fig. 4 |. DSP of pan-CK-enriched paired pre- and on-treatment biopsies is associated with pathological complete response in the discovery cohort and outperforms established markers.
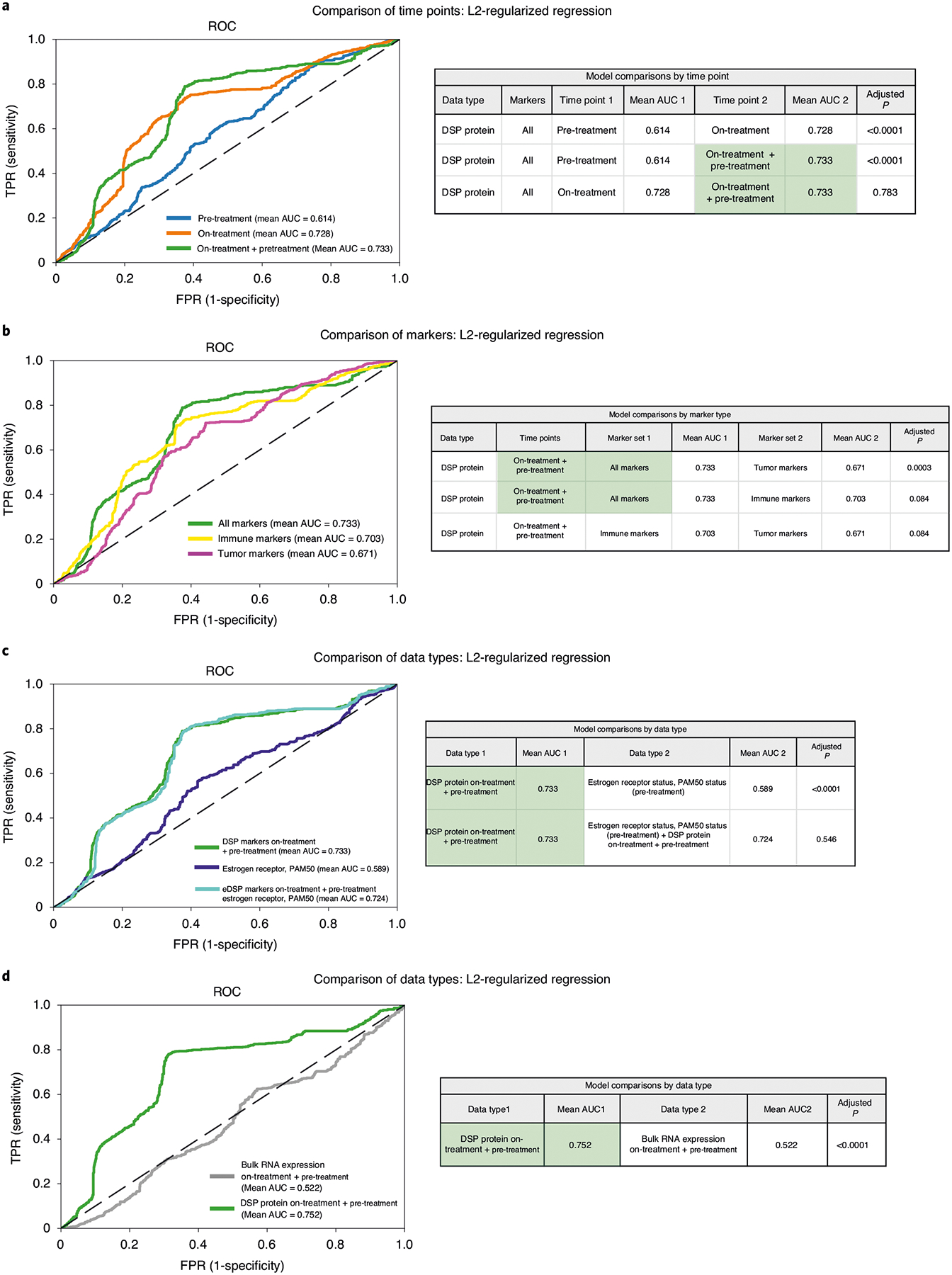
The AUROC performance of various models was compared using nested cross-validation (based on a two-sided t-test) with Holm–Bonferroni correction for multiple hypotheses in the discovery (training) cohort (n = 23 patients with paired data for both time points). a–c, ROC curves were generated using cases with DSP pan-CK-enriched data from both the pre-treatment and on-treatment time points (23 biologically independent cases were used in these analyses). a, ROC curves and statistical comparison of L2-regularized classifiers trained using DSP protein marker mean values (averaged across ROIs) pre-treatment, on-treatment and the combination of pre-treatment and on-treatment (‘on-treatment + pre-treatment’). AUC, area under the curve. b, ROC curves and statistical comparison of DSP protein on-treatment + pre-treatment L2-regularized classifiers trained using all marker, tumor marker and immune marker mean values. Cross-region mean marker values from both the pre-treatment and on-treatment time points were used in this analysis. c, ROC curves and statistical comparison of the on-treatment + pre-treatment DSP protein L2-regularized classifier to a model trained using estrogen receptor and PAM50 status. These two models were compared to a model that incorporates on-treatment + pre-treatment DSP protein data, estrogen receptor and PAM50 status. d, ROC and statistical comparison (based on a two-sided t-test) of on-treatment + pre-treatment L2-regularized classifiers trained using DSP protein marker mean values versus bulk RNA expression using RNA transcripts corresponding to the DSP protein markers. ROC curves were generated using cases with DSP pan-CK-enriched data and bulk expression data from both the pre-treatment and on-treatment time points (21 biologically independent cases were used in this analysis). In all panels, the chance (dashed) line is included for reference.
For the DSP protein on-treatment + pre-treatment classifier, we investigated the importance of multiregion sampling and heterogeneity by extending the model to incorporate both the mean marker expression across all regions and the s.e.m. for each marker between regions (Methods). This analysis was restricted to patients with at least 3 regions profiled at both time points (n = 16; Methods). We found that utilizing the mean immune values and the s.e.m. for tumor markers outperformed a model based on mean values for both tumor and immune markers (Extended Data Fig. 9a), suggesting value in capturing heterogeneity among tumor markers.
We next compared the performance of the DSP protein on-treatment + pre-treatment classifier with features previously associated with outcome (estrogen receptor status and PAM50 subtype), where models were again evaluated via cross-validation in the discovery cohort. Of note, a model based on estrogen receptor status and HER2-enriched PAM50 status performed poorly in this cohort (mean AUROC = 0.589); the addition of these two features or additional pathological features to the DSP protein on-treatment + pre-treatment dataset did not improve the AUROC (Fig. 4c and Extended Data Fig. 9b–d). Given the availability of bulk transcriptomic data for these cases, we also evaluated a model using paired on-treatment and pre-treatment bulk RNA expression data for the 37 markers that overlapped with the DSP protein panel. This model also performed significantly worse than that based on the DSP protein data (Fig. 4d; P < 0.0001 via cross-validation). This is not surprising since among the 37 overlapping DSP and bulk RNA expression markers, only 16 were positively correlated pre-treatment (Extended Data Fig. 3b,c). Various factors may contribute to the lack of strong correlation between protein and RNA expression levels, including pan-CK enrichment, RNA transience/degradation34 and post-translational regulation35, where protein expression is a more proximal readout of cellular phenotype.
DSP predicts pCR in an independent validation cohort.
In light of these promising findings, we further sought to evaluate the performance of the DSP protein on-treatment + pre-treatment classifier in an independent cohort (n = 29) of patients from the TRIO-US B07 clinical trial (Extended Data Fig. 9e). As with the discovery cohort, an average of four pan-CK-positive regions were profiled from each pre-treatment and on-treatment tumor tissue and the same panel of 40 protein antibodies was utilized. The change in markers on-treatment relative to pre-treatment mirrored that observed in the discovery cohort (Extended Data Fig. 9f): T-cell markers (CD3, CD4 and CD8) increased while the tumor markers HER2 and Ki67 showed the most significant reduction. Similar to the discovery cohort, in the validation cohort, we observed that on-treatment versus pre-treatment protein expression differences were more dramatic in tumors that ultimately underwent pCR (Extended Data Fig. 9g). AUROC performance of the L2-regularized logistic regression model, trained in the discovery cohort, was evaluated in the validation (test) cohort. The performance of the DSP protein on-treatment + pre-treatment model in predicting pCR was comparably high in the discovery (assessed via cross-validation, mean AUROC = 0.733) and validation (assessed via train-test, AUROC = 0.725) cohorts (Fig. 5a). In the on-treatment + pre-treatment classifier, which was trained in the discovery cohort and tested in the validation cohort, the marker with the largest L2-regularized coefficient was on-treatment CD45 protein levels. Other features with large coefficients included on-treatment markers that represent TIL and macrophage populations (CD44, CD66B) (Fig. 5b). On-treatment HER2 protein expression had a negative coefficient in the model, which is consistent with poor outcome being associated with high HER2 levels during treatment.
Fig. 5 |. Validation of the DSP-based spatial proteomic biomarker in an independent cohort.
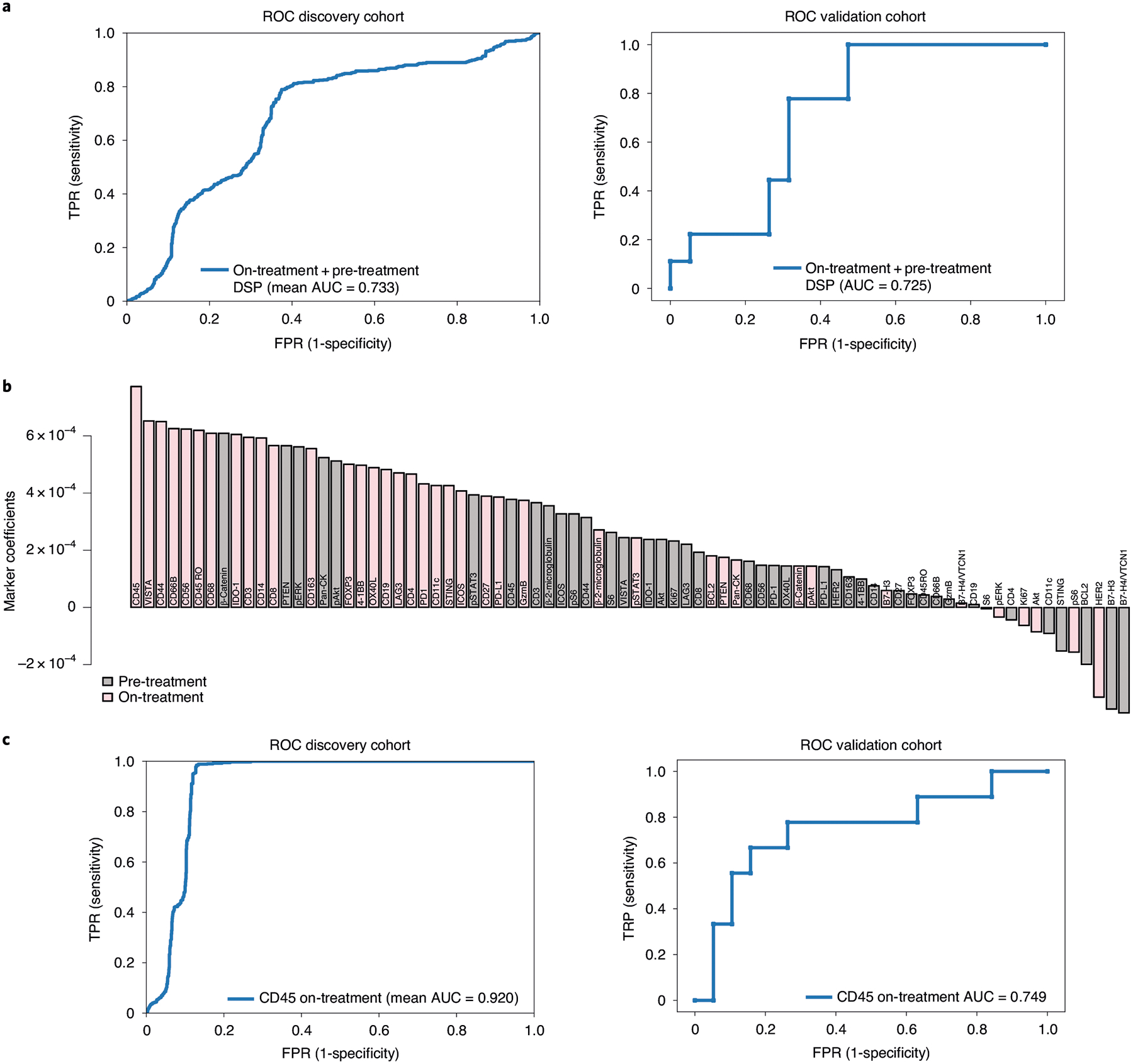
a, ROC curves for the on-treatment + pre-treatment DSP protein L2-regularized classifier in the discovery (training) cohort (n = 23 patients assessed via cross-validation) and the validation (test) cohort (n = 28 patients assessed via train-test) using the 40-plex DSP protein marker panel. b, Coefficients for each of the 40 markers in the L2-regularized on-treatment + pre-treatment DSP protein model, trained in the discovery cohort and tested in the validation cohort. c, ROC curves for on-treatment L2-regularized classifier in the discovery (training) cohort (n = 23, assessed via cross-validation) and the validation (test) cohort (n = 28, assessed via train-test) using CD45 from the DSP protein marker panel.
In light of the widespread use of trastuzumab in current neoadjuvant treatment paradigms36, we further assessed model performance for the best performing on-treatment + pre-treatment DSP protein model in the trastuzumab-containing cases (arms 1 and 3, n = 19). Again, similar model performance and marker coefficients were seen as in the full discovery cohort and in the subset of cases in the validation cohort who were treated with trastuzumab (Extended Data Fig. 4b–d). The validation of these findings in an independent cohort demonstrates the robustness of this approach and potential utility of multiplex spatial proteomic profiling to identify candidate biomarkers.
Given that CD45 on-treatment values had the greatest coefficient in the model (Fig. 5b), we assessed model performance using this single marker by training an L2-regularized logistic regression model in the discovery cohort and evaluating this model in the validation (test) cohort. The performance of the DSP CD45 on-treatment model was high in both the discovery (assessed via cross-validation, mean AUROC = 0.920) and validation (assessed via train-test, AUROC = 0.749) cohorts (Fig. 5c).
On-treatment CD45 IHC measurements are associated with pCR.
Building on the striking finding that a single immune marker (CD45) predicted pCR with high accuracy, we sought to investigate whether quantitative DSP measurements correlated with conventional IHC (Fig. 6a). Using 28 cases from a combination of the discovery and validation cohorts with additional tissue remaining (Extended Data Fig. 10a) we performed CD45 IHC and quantified the percentage of CD45-positive cells within tumor-enriched regions from the on-treatment time point (Methods). Pan-CK-enriched CD45 measurements from DSP correlated with the CD45 IHC measurements (r = 0.53) (Extended Data Fig. 10b) and with stromal TILs (r = 0.41). Using the subcohort with CD45 IHC data, performance was strong in both a model trained using on-treatment IHC CD45-positive cell percentage (mean AUROC = 0.871) and a model trained using on-treatment DSP CD45 (mean AUROC = 0.811, both models assessed using cross-validation; Fig. 6b). Cases that ultimately achieved a pCR tended to have higher CD45 staining (Fig. 6c) and higher CD45 DSP levels (Fig. 6d and Extended Data Fig. 10c) in tumor-enriched areas on treatment; this separation between pCR and non-pCR cases was greater for both CD45 measurements than for on-treatment stromal TILs, on-treatment tumor cellularity or pre-treatment HER2 FISH ratios (Fig. 6d and Extended Data Fig. 10c). In light of the significant separation between pCR and non-pCR cases using CD45 IHC and CD45 DSP levels, we next used each feature to establish thresholds to predict pCR; in the case of CD45 IHC, the selected threshold was 20% CD45-positive cells (Extended Data Fig. 10d). Positive predictive value is among the most relevant model performance parameters in early neoadjuvant treatment selection, where the goal is to de-escalate treatment for tumors that are highly likely to undergo pCR while not depriving others of potentially curative therapy. In the CD45 DSP validation set, the positive predictive value was 0.714, modestly lower than the CD45 DSP discovery set (0.750) and the CD45 IHC discovery set (0.818) (no validation set was available for CD45 IHC). Performance was similar when excluding the lapatinib-only arm (Extended Data Fig. 4e). Overall, these findings demonstrate the potential utility of profiling a single marker in an on-treatment biopsy to predict which patients will respond early during HER2-targeted therapy, such that subsequent therapy could be tailored accordingly.
Fig. 6 |. On-treatment IHC-based measurement of percentage CD45-positive cells in tumor-enriched regions is associated with pathological complete response.
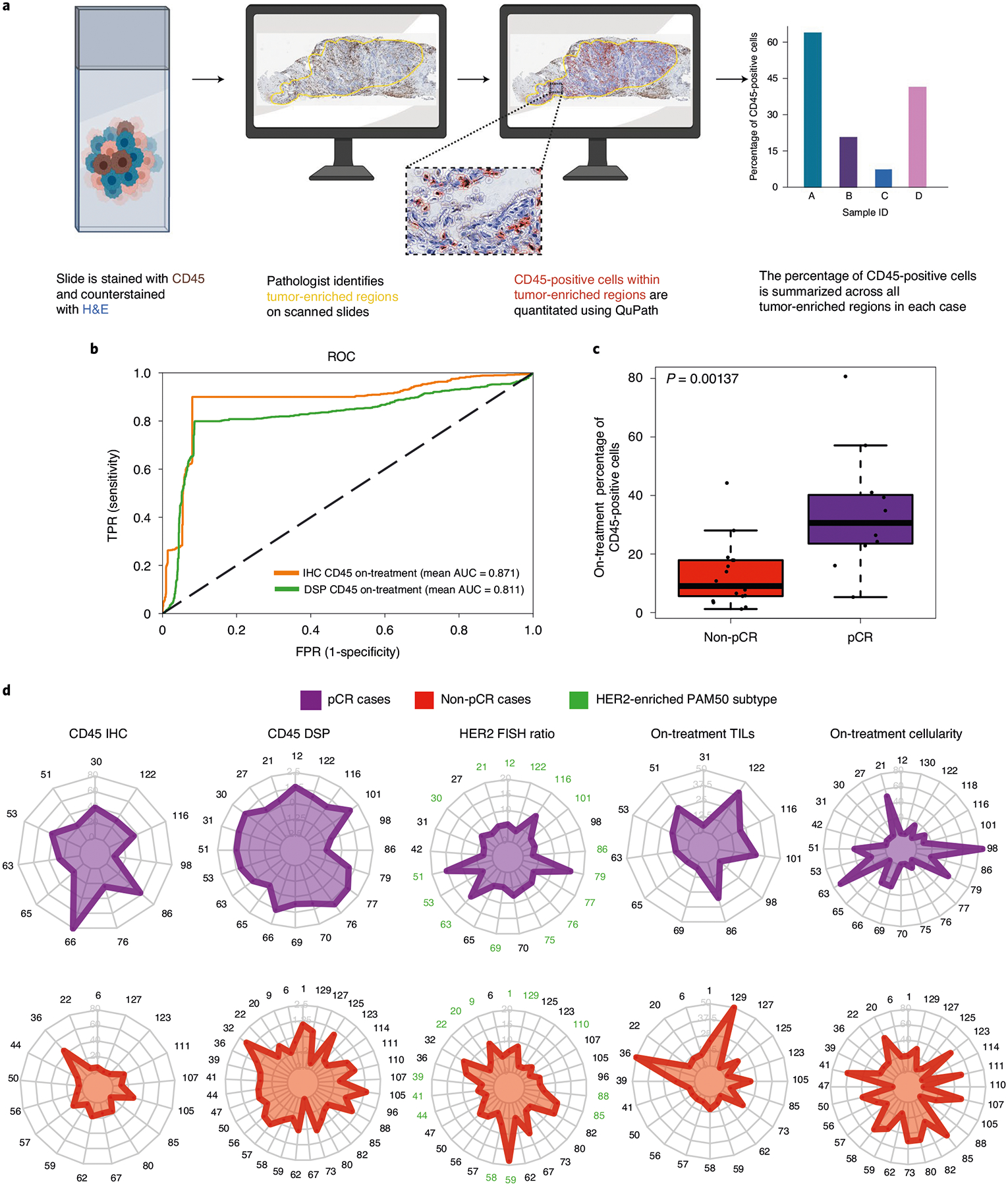
a, Schematic showing the process for quantitative assessment of CD45-positive cell counts from IHC. b, ROC curves for classifiers trained using either on-treatment DSP CD45 levels or on-treatment IHC-based CD45-positive percentage of cells. These models were evaluated on the subset of cases on which CD45 IHC was performed (n = 28 biologically independent tumors). c, Box plot showing IHC CD45 percentage positive within tumor-enriched regions stratified by pCR. Comparison as evaluated on the subset of cases on which CD45 IHC was performed (n = 28 biologically independent tumors). P values were derived using a two-sided Wilcoxon rank-sum test. For each box plot, the center is the median, the bounds of the box are the 25th and 75th percentile and the bounds of the whiskers extend to the most extreme data points that are no more than 1.5× the interquartile range from the bounds of the box. All individual data points, including maxima and minima, are overlaid on the boxplot. d, Radar plots showing on-treatment IHC CD45 percentage positive, on-treatment DSP CD45 levels, pre-treatment HER2 FISH ratios, on-treatment TIL score and on-treatment tumor cellularity across patients stratified by pCR status. All cases where a given feature was measured across the DSP discovery and validation cohorts are shown.
Discussion
Bulk genomic and transcriptomic profiling has been a mainstay of cancer biomarker discovery efforts in recent years. However, admixture among heterogeneous cellular populations complicates the analysis of such data, issues that are compounded when studying longitudinal samples, where the changing composition and localization of cell populations may reflect the biology of disease progression or mechanisms of treatment response. Indeed, efforts to establish validated biomarkers of response to HER2-targeted therapy based on bulk genomic and transcriptomic profiling have met with limited success to date in other trial cohorts and in TRIO-US B0710,12,21. We reasoned that in situ proteomic profiling of the tumor immune microenvironment through therapy would circumvent the limitations of dissociative techniques and improve our ability to uncover features associated with response to neoadjuvant HER2-targeted therapy. In this study, we used the DSP technology23 to simultaneously profile 40 tumor and immune markers en bloc on a single 5-μm section of archival tissue from breast tumors sampled before, during and after neoadjuvant HER2-targeted therapy in the TRIO-US B07 clinical trial. To enhance signal while accounting for intratumor heterogeneity, we employed a pan-CK masking strategy to enrich for tumor cells and colocalized immune cells across multiple regions, each comprised of approximately 300–600 cells per sample. Our results illustrate the feasibility and power of multiplex in situ proteomic analysis of archival tissue samples to provide proximal readouts of tumor and immune cell signaling through therapy. Many signaling proteins/phosphoproteins, including those profiled in this study, are considered protein network bottlenecks and integrate mutational and transcriptional changes37,38, making this a particularly powerful approach to characterize compensatory signaling and mechanisms of resistance.
DSP of longitudinal breast biopsies from this trial cohort uncovered changes associated with therapy, including markedly decreased HER2 and downstream Akt signaling on-treatment, which was accompanied by increased CD45 and CD8 expression, consistent with infiltrating leukocytes and cytotoxic T cells, respectively. By the time of surgery, after a full course of neoadjuvant therapy, the tumor immune composition changed considerably with increased CD56 expression in non-pCR cases, potentially reflecting NK cell-mediated killing of chemotherapy-stressed tumor cells28. While immune influx on-treatment was also observed in the bulk expression data from this cohort21, there it did not correlate with response, leaving open the question of whether the biopsy itself may have induced these changes. DSP, in contrast, revealed that changes in both tumor and immune markers on-treatment were more dramatic in tumors that went on to achieve a pCR and, critically, on-treatment protein expression robustly predicted response in an independent validation cohort. Neither pre-treatment protein expression, bulk pre-treatment and on-treatment gene expression data, nor established predictive features were predictive in this cohort, thus emphasizing the power of this quantitative multiplexed spatial proteomic biomarker. This work also illuminates study design considerations, including the value of pan-CK enrichment of tumor cells (or other markers to enrich for specific cell populations) and multiregion profiling to capture regional tumor heterogeneity and treatment-associated changes, concepts that should be broadly applicable to biomarker discovery in other epithelial tumor types.
Thus, our findings address a critical unmet clinical need given the considerable emphasis devoted to identifying subsets of the population in which therapy should be escalated, for example, by combining HER2-targeted agents, or safely de-escalated, for example, through shortening or omission of chemotherapy and its associated toxicities39,40. While many biomarkers have been considered to help guide personalized targeting of escalated versus de-escalated approaches in early-stage HER2-positive breast cancer—including imaging, circulating tumor DNA and pre-treatment immune scores or intrinsic subtype—there is currently no validated biomarker that can guide patient stratification. The increasing plethora of options for HER2-targeted therapy, including new highly effective but potentially toxic agents41,42, combined with great heterogeneity in response make HER-positive breast cancer the ideal setting for the development of optimally personalized therapy over the next decade. The next step toward clinical translation of the biomarker described in this study based on CD45 protein expression or cell counts will involve prospective validation in a neoadjuvant clinical trial that selects therapy based on biomarker status and assesses both pCR and RFS43.
Methods
Cohort selection.
The TRIO-US B07 clinical trial was a randomized, multicenter study that included 130 women with stage I–III unilateral, HER2-positive breast cancer21. The institutional review board at the University of California Los Angeles approved the TRIO-US B07 clinical trial (no. 08-10-035). Informed written consent was obtained from all participants. This covers consent from patients for their samples to be shared with other researchers. Patients were not compensated for their participation. The institutional review board at Stanford University approved the use of the TRIO-US B07 clinical trial specimens for correlative studies in the Curtis Lab (eProtocol no. 32180). Enrolled patients were randomly assigned to three treatment groups, dictating the type of targeted therapy namely trastuzumab, lapatinib or trastuzumab and lapatinib in combination. Breast tumor biopsies were obtained before treatment and after 14–21 d of the assigned HER2-targeted therapy (without chemotherapy), which was followed by 6 cycles of the assigned HER2-targeted treatment plus docetaxel and carboplatin given every 3 weeks and surgery. For each time point, core biopsies or surgical tissue sections were obtained and stored as either fresh-frozen or FFPE material. In total, 28 cases with FFPE samples available from all 3 time points (pre-treatment, on-treatment and at surgery) were selected for inclusion in the discovery cohort based on sample availability and quality, with balancing by pCR status and estrogen receptor status (Fig. 1a and Extended Data Fig. 1a). An additional 29 cases with FFPE samples available pre-treatment and on-treatment were selected for the validation cohort to assess performance of the classifier (Extended Data Fig. 9e). Of note, the validation cohort was used exclusively to evaluate model performance. All other analyses are based on the discovery cohort. Thus, across both cohorts, tissue was utilized from a total of 57 female patients who were between the ages of 28 and 70. Tumor cellularity was assessed by a subspecialty breast pathologist (G.R.B.) using tumor sections stained with hematoxylin and eosin (H&E). Samples with a cellularity of 0 were omitted from further analysis. For other tissue sections estimated to have a cellularity of 0, tumor cells were identified on the FFPE sections used to perform DSP (distinct from the H&E sections used for the pathology review), and these were included in the analysis (Extended Data Fig. 1b).
DSP data generation and analysis.
DSP (NanoString) was performed as described previously23. Briefly, tissue slides were stained with a multiplexed panel of protein antibodies that contained a photocleavable indexing oligonucleotide, enabling subsequent readouts (Extended Data Fig. 2a). ROIs were selected on a DSP prototype instrument and illuminated using UV light. Released indexing oligonucleotides from each ROI were collected and deposited into designated wells on a microtiter plate, allowing for well indexing of each ROI during nCounter (nCounter MAX system version 4.0.0.3) readout (direct protein hybridization). Custom masks were generated using an ImageJ (NIH) pipeline, as described previously44. For each tissue sample, counts for each marker were obtained from an average of 4 (range 1–7) pan-CK-enriched ROIs. Raw protein counts for each marker in each ROI were generated using nCounter45. The raw counts were External RNA Control Consortium-normalized (based on the geometric mean of the three positive control markers). Histone H3 was used as a housekeeping marker and ROIs with extreme histone H3 (more than three s.d. away from the mean) were filtered (<1% of ROIs). The geometric mean of two immunoglobulin G antibodies were used to calculate the background noise and we noted markers with a signal-to-noise ratio <3× (Extended Data Fig. 2f). Immune markers were normalized based on the ROI area to measure the total density of immune content in the region. Tumor markers were normalized using the housekeeping antibody (histone H3) to capture the status of the cancer signaling pathways on a per-cell basis. As further quality control, area normalization factors and housekeeping normalization factors were compared per ROI and ROIs were filtered with disparate normalization factors across the two methods. (This represented 6% of all ROIs.) All normalized counts were converted to log2 space for downstream analysis. The analyses carried out in this study are comparative in nature (for example, pre-treatment versus on-treatment, pCR versus non-pCR) and are robust to variations in normalization methods.
Bulk messenger RNA expression analysis.
Bulk mRNA expression analysis was performed as described elsewhere21. Briefly, RNA was extracted using the RNeasy Mini Kit (QIAGEN) and quantified by the NanoDrop One spectrophotometer (Thermo Fisher Scientific). RNA samples were labeled with cyanine 5-CTP or cyanine 3-CTP (PerkinElmer) using the Quick Amp Labeling Kit (Agilent Technologies). The gene expression microarray experiments were performed by comparing each baseline sample to samples taken after 14–21 d of HER2-targeted therapy (on-treatment). Each on-treatment sample was compared to the pre-treatment sample from the same patient. Limma v.3.28.21 (refs. 46,47) was used for background correction (‘normexp’), within-array normalization (‘loess’), between-array normalization and for averaging over replicate probes. For the downstream analyses, including batch correction and comparisons with the DSP cohort, the normalized counts were converted to log2space. Combat48 was used to remove potential batch affects associated with microarray run date. PAM50 status pre-treatment and on-treatment was inferred using absolute intrinsic molecular subtyping, an n-of-1 algorithm that is robust to variations in dataset composition49. This approach was utilized given the expected preponderance of HER2-enriched cases in this cohort.
Correlation analyses.
Plots showing the correlation between protein markers (Figs. 1c and 2c) were overlapped with hierarchical clustering in the form of black squares. The difference between the distribution of correlation values in pCR versus non-pCR cases was evaluated using a two-sided Wilcoxon two-sample t-test. For the correlation between DSP protein data and bulk RNA data, Spearman rank correlation was computed for pre-treatment samples using the average of all DSP ROIs (both pan-CK-enriched ROIs and surrounding microenvironment-enriched ROIs) per patient. For the correlation between DSP protein data and bulk RNA data, Spearman rank correlation was computed per region. For all other correlation analyses (Extended Data Figs. 2c,e and 10b), Pearson correlation was computed per tissue.
Comparative analyses.
For the comparative analyses (for example, pre-treatment versus on-treatment, pCR versus non-pCR, pan-CK-enriched versus pan-CK negative) of DSP protein data, where multiple regions were sampled per patient, we utilized a linear mixed-effects model with blocking by patient50. This model allows for marker levels to be compared in a patient-matched manner while controlling for differences in the number of ROIs profiled per patient. The coefficient of the fixed effect is the change attributable to that variable (x axis of volcano plots) and the P value used to calculate the FDRs (y axis of volcano plots) is based on the t-value (a measure of the size of the difference relative to the variation in the sample data). FDRs were computed using the Benjamini–Hochberg procedure51; an FDR-adjusted P of 0.05 was set as the significance threshold.
Region subsampling.
We sought to investigate the impact of utilizing a single randomly selected region per tissue sample, rather than multiple regions, when assessing on-treatment versus pre-treatment protein expression changes.
For these analyses (Extended Data Fig. 5d), we performed 100 iterations where a single region was selected from each tissue and computed fold changes and corresponding P values averaged over these 100 experiments. The number of random samplings was chosen empirically by raising the number of iterations beyond the number required to make the resulting output robust to further increases in the number of iterations used (P convergence).
L2-regularized logistic regression using molecular data.
Models and features.
L2-logistic regression using LIBLINEAR as a solver was used for the classification of pCR versus non-pCR cases. DSP marker values pre-treatment and on-treatment were averaged across all ROIs to derive a composite value for each marker for that time point. Five patients were excluded from the DSP models because data were available only at a single time point (Extended Data Fig. 1b). Mean DSP marker expression features were used in models comparing patient time points, tumor versus immune markers, DSP protein features versus established predictive features (estrogen receptor status and PAM50 classification) and DSP protein versus bulk RNA features (using RNA gene transcripts corresponding to DSP protein markers). To assess heterogeneity, the s.e.m. was calculated for marker values across all ROIs for tissues with at least three ROI to derive a composite value for each marker for that time point. These s.e.m. features were used in combination with mean expression features in models assessing the predictive value of heterogeneity. Additional single-feature models were compared in a subcohort with IHC data available (Extended Data Fig. 10a): CD45 DSP on-treatment with CD45 IHC on-treatment. The generation of CD45 IHC data is described below.
Model comparisons and evaluation of performance via internal cross-validation.
Model performance was evaluated and models compared using nested cross-validation using the Python package scikit-learn v.0.21.3 (ref. 52). Data were divided into N folds using stratified sampling (stratified cross-validation). The number of folds was chosen based on the number of cases in the non-pCR group (the class with fewer cases) such that the testing data would contain two cases from each class. Each model was trained using N−1 folds and scored using the mean AUROC on the remaining fold. This process was repeated iteratively holding out a different fold each time. The L2-penalization weight was chosen using stratified cross-validation within the N−1 training dataset, with the weight associated with the highest mean accuracy within this inner cross-validation selected for scoring. This nested cross-validation process was repeated 100 times using randomly generated folds. Model scores were then compared using an unpaired two-sided t-test with Holm–Bonferroni correction for multiple hypotheses. ROC curves were generated by averaging across the ROC curves from the 100 repeats of N-fold cross-validation, with each repeat containing a different random split of folds.
Evaluation of model performance in an independent validation cohort.
As described above, marker values pre-treatment and on-treatment were averaged across all ROIs to derive a composite value for each marker for that time point. Model selection was carried out using cross-validation as described above. The best performing model was selected and trained using the entire discovery cohort. Finally, model performance based on the AUROC was evaluated in the independent validation (test) cohort. For the models assessing CD45 IHC, cross-validation was used, as described above, to assess model performance.
Metrics of heterogeneity.
Marker heterogeneity was calculated as the mean squared error from the ANOVA done on a linear model with marker values as the dependent variable and patient identity as the independent variable. For these analyses, the dataset was subset to the particular time point or clinical outcome of interest.
Perimetric complexity.
Perimetric complexity was computed for the pan-CK-enriched binary masks for each ROI using ImageJ33. A linear mixed-effects model50 with blocking by patient was used to the compare the perimetric complexities of all the pan-CK-enriched regions pre-treatment and on-treatment regions and for cases that achieved a pCR versus those for cases that did not achieve a pCR.
Multiplex IHC analysis.
Unstained, paraffin-embedded sections were analyzed by multiplex IHC analysis used the following markers: pan-CK (AE1/AE2); CD8; CD45 leukocyte common antigen; and HER2 (29D8 CST). Using the Fred Hutchinson Cancer Research Center Experimental Histopathology core, stained samples were scanned, digitized as a series of square subimages (‘stamps’) and visualized using HALO. Pan-CK masking and tissue area masking was performed on each stamped tissue region using Fiji (ImageJ) v.2.0.0 (ref. 53). Briefly, the pan-CK channel was used to generate the masks (using the ImageJ tools Enhance Contrast, Threshold, Dilate, Fill Holes and Create Selection) for the pan-CK-positive region and the entire tissue region; CD8, CD45 and HER2 were quantified within each masked region (using the ImageJ Measure tool). A weighted average (with weights corresponding to each mask area) was used to calculate CD8, CD45 and HER2 levels across all the scanned subimages that comprised the tissue (either tissue mask or pan-CK mask area).
CD45 IHC analysis.
The concordance between CD45 DSP protein levels and CD45 counts based on IHC staining and digital quantification was evaluated in 28 on-treatment cases with remaining tissue. IHC was performed on the VENTANA BenchMark ULTRA instrument (VENTANA Medical Systems) according to the manufacturer’s protocols using primary antibody specific for CD45 (clone 2B11 + PD7/26, prediluted; VENTANA Medical Systems) and CC1 antigen retrieval. Slides were counterstained with H&E and then scanned with a Leica Aperio ScanScope (Leica Biosystems). Adjacent 5-μm paraffin sections were also cut for standard H&E staining. A board-certified pathologist specializing in breast cancer (G.R.B.) reviewed each corresponding H&E slide and circled the tumor-enriched regions on CD45-stained images, which were then analyzed with the QuPath image analysis software v.0.2.0-m9 (ref. 54). Pathologist-circled tumor-enriched regions were used for downstream analysis to mirror the pan-CK enrichment strategy employed in the DSP studies. Cells positive for CD45 were identified as having a mean cell 3,3′-diaminobenzidine optical density (by default settings). Proportions of CD45-positive cells were calculated as a cell-weighted average (based on the total number of cells) for tissues with multiple noncontiguous tumor-enriched regions.
Other pathological features.
Other pathological features that were assessed in this cohort using available tissue included cellularity, stromal TILs, HER2 FISH ratio, estrogen receptor status, HER2 IHC and Ki67 IHC, which were all assessed by board-certified pathologists. Ki67 scoring, estrogen receptor status, cellularity, TIL scoring and cellularity measurement for this cohort were described previously21.
Statistics and reproducibility.
The size of our cohort (n = 28 patients in the discovery cohort, n = 29 in the validation cohort, with multiple time points and regions per sample) was determined based on the availability of high-quality FFPE tissue from the TRIO-US B07 clinical trial. Our goal was to achieve the largest discovery cohort possible that was balanced with regard to estrogen receptor status and clinical outcome (pCR). All initial analyses and model development were performed within the discovery cohort exclusively. We subsequently assembled a validation cohort from an independent subcohort of samples from the same clinical trial. Samples were excluded from the DSP profiling and all analyses (Figs. 1–6 and all extended data) if no pan-CK-positive tumor regions were detected (Extended Data Fig. 1b). The criteria for inclusion were established at the start of the project; as such samples could not have been included with our planned study design. Due to the limiting nature of the biopsy specimens derived from independent donors, we did not perform technical replicates of DSP, IHC or perimetric complexity analyses. However, multiple regions per sample were profiled. Previous studies have demonstrated strong correlation among technical replicates using the DSP technology23. Randomization was not relevant for this study since we used tissue samples from a previously completed clinical trial (NCT00769470). The discovery cohort was balanced regarding estrogen receptor status and clinical outcome (pCR). Individuals involved in the pathological assessment, multiplex IHC staining and DSP data collection were blinded to the clinical characteristics of the cohort, including pCR outcome. For the individuals performing the data analysis, blinding was not necessary since the analysis did not involve any (even potentially) subjective measurements.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Discovery cohort description and preliminary proteomic analysis.
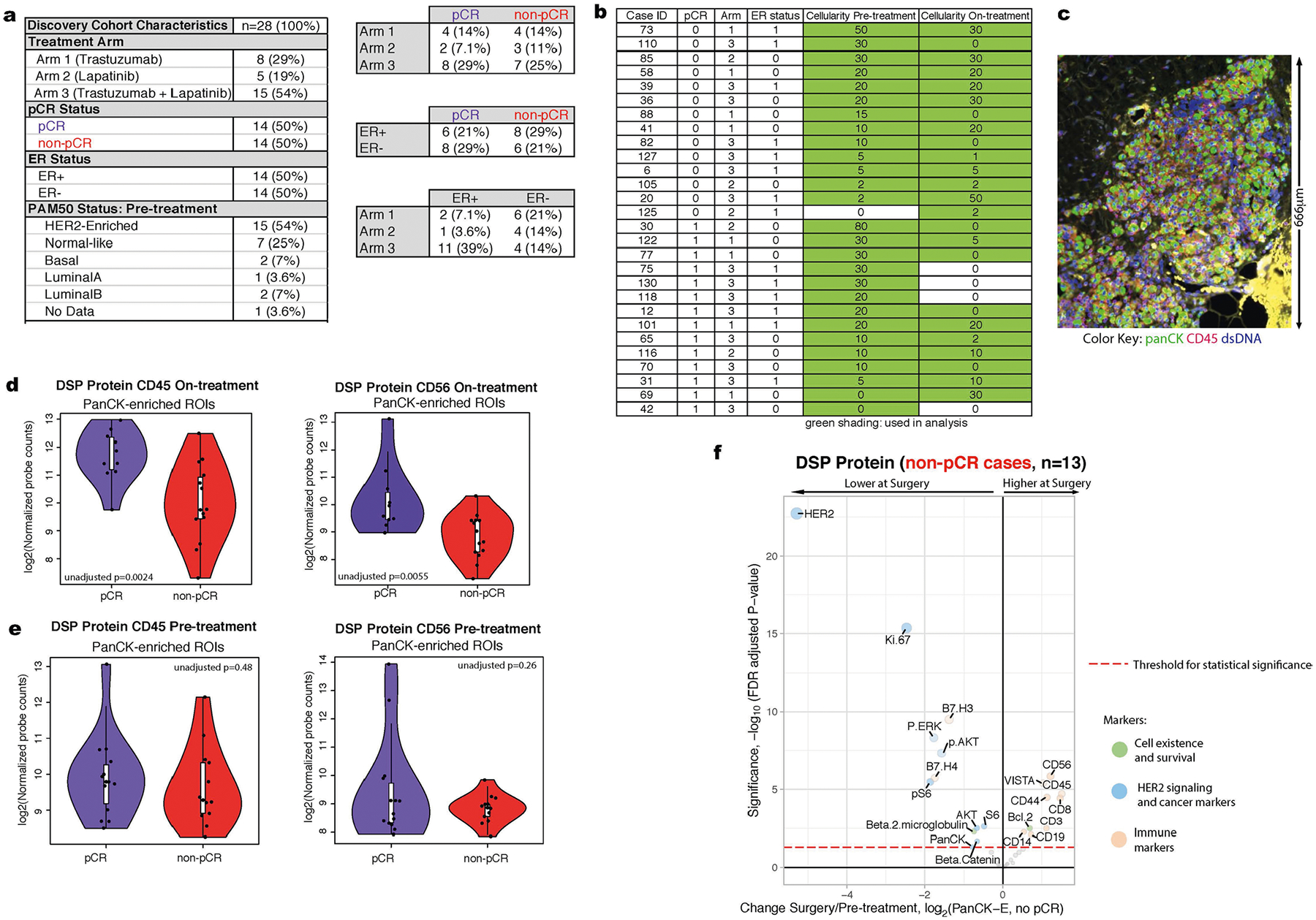
a. Summary of the clinical characteristics of the TRIO-US B07 DSP discovery cohort, including treatment arm, pathologic complete response (pCR), estrogen receptor (ER) status, and PAM50 status inferred based on pre-treatment bulk expression data. Two-way contingency tables compare the distribution of ER status, pCR status, and treatment arm. b. Pathology-estimated cellularity pre-treatment and on-treatment for the discovery cohort. Samples with green shading indicate those used for subsequent analysis. For the pCR column, 0=non-pCR, 1=pCR. For ER status column, 0=ER-negative, 1=Er-positive. c. An example region from case 30 sampled on-treatment. While cellularity was estimated to be 0 based on pathology review of a distinct tissue section, tumor regions were identified upon imaging the tissue section used in this analysis. d,e. CD45 values and CD56 values from the Digital Spatial Profiling (DSP) protein data on-treatment (d, n = 24 tumors with on-treatment data) and pre-treatment (e, n = 27 tumors with pre-treatment data) in the pCR cases versus the non-pCR cases. Each point represents the average probe values for all panCK-enriched ROIs for that case On-treatment. The p-value was derived using a linear mixed-effect model (two-sided test) over the multi-region data with blocking by patient. Adjustments were not made for multiple comparisons. For each violin plot, the white box represents the interquartile range and the black lines extending from the white box represent 1.5X the interquartile range. Analyses based on the discovery cohort. f. Volcano plot demonstrating treatment-associated changes from pre-treatment to surgery in tumors that did not undergo pCR using DSP protein expression levels in pancytokeratin-enriched (PanCK-E) regions from n = 28 tumors. Significance, −log10(FDR adjusted p-value), is indicated along the y-axis. The p-value is determined based on a linear mixed-effects model (two sided test) with blocking by patient and is derived from the t-value (a measure of the size of the difference relative to the variation in the sample data).
Extended Data Fig. 2 |. Digital Spatial Profiling (DSP) is used for multiplex protein quantification within tumor regions.
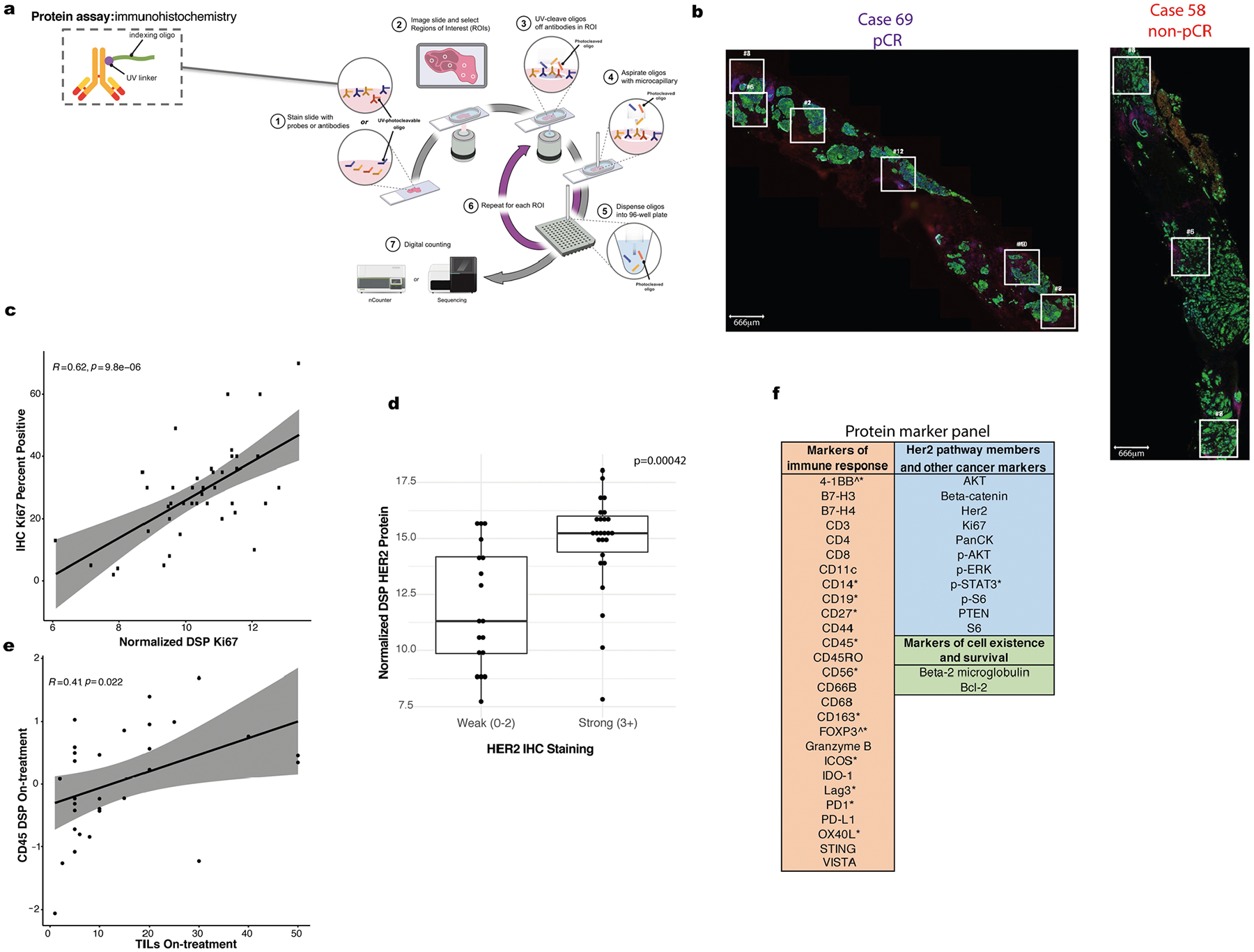
a Overview of NanoString DSP workflow adapted with permission from NanoString, Inc. b. Location of spatially separated ROIs within tissue specimens for a representative pCR (69) and non-pCR case (58). An average of 4 ROIs were profiled per tissue (range: 1–7). c. Correlation plot comparing Ki67 percent positive (evaluated using IHC) with normalized DSP Ki67 expression (averaged across ROIs) across n = 42 tumor biopsies (24 pre-treatment, 18 on-treatment). Pearson correlation coefficient and p-value are noted. Grey shading indicates 95% confidence interval (CI) of the correlation coefficient. d. Boxplot comparing normalized DSP Her2 expression (averaged across all ROIs in a given sample) between cases that exhibited strong (3+) IHC Her2 staining (using a distinct tissue slice from the same case and timepoint) or weaker (0–2) IHC Her2 staining. A total of n = 44 biologically independent tumor biopsies (23 pre-treatment, 21 on-treatment) with paired Her2 IHC and DSP data were utilized in this analysis. A two-sided Wilcoxon test was used to assess significance. For each boxplot, the center is the median, the bounds of the box indicate the 25th and 75th percentile, the bounds of the whiskers extend to the most extreme data points that are no more than 1.5x the interquartile range from the bounds of the box; all individual data points, including maxima and minima are overlaid. e. Correlation plot comparing normalized on-treatment CD45 DSP protein values with on-treatment stromal tumor infiltrating lymphocyte (TILs) score for all cases (n = 31) with both data types available. Grey shading indicates the 95% CI of the correlation coefficient. P value is assessed via a one-sided t-test (correlation coefficient not equal to 0). f. Markers with a signal to noise ratio (SNR) < 3 (Methods) in the discovery cohort indicated by a caret (^) and those with SNR < 3 in the validation cohort indicated with an asterisk (*).
Extended Data Fig. 3 |. Treatment-associated changes observed with bulk RNA data.
a. Volcano plot demonstrating treatment-associated changes based on comparison of pre-treatment versus on-treatment bulk RNA expression levels. RNA transcripts with corresponding Digital Spatial Profiling (DSP) protein markers were used in this analysis. Significance, −log10(FDR adjusted p-value), is indicated along the y-axis. Analyses based on the discovery cohort (n = 28 tumors). b. Pairing of protein antibodies and gene names used in comparative analyses between DSP and bulk expression data. Genes listed here were used to generate the bulk expression volcano plot shown in panel a. For direct comparisons yielding the correlation plots shown in panel c, the marker names indicated by the asterisk were used; specifically, for proteins with multiple form (for example AKT/pAKT, CD45/CD45RO), the unmodified form of the protein was used when available and the breast-cancer associated keratin gene with the highest mean expression level was used. c. Spearman correlation between DSP protein probes (averaged across all ROIs per case) and bulk RNA transcripts corresponding to these markers pre-treatment. Significantly correlated probes (with p-value < 0.05, correlation coefficient not equal to 0, assessed via a one-sided t-test) are indicated by an asterisk. Two exemplary correlation plots are shown, where each dot represents a single case. The grey shading indicates the 95% confidence interval of the correlation coefficient. Analyses based on the discovery cohort cases with paired DSP and bulk RNA data (n = 23 tumors).
Extended Data Fig. 4 |. Treatment-associated changes and model performance assessed in cases treated with trastuzumab.
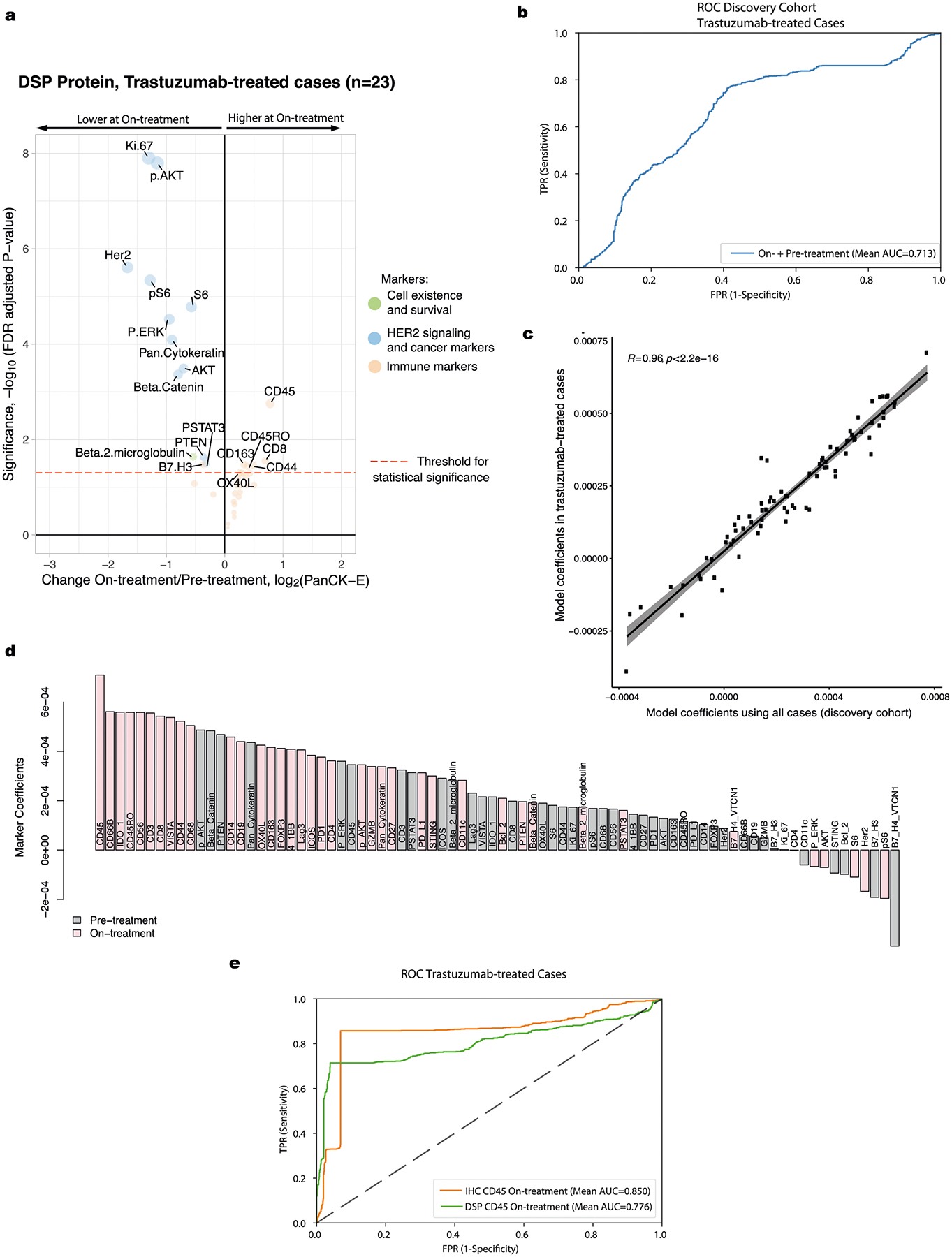
a. Volcano plot demonstrating treatment-associated changes based on comparison of pre-treatment versus on-treatment protein marker expression levels in pancytokeratin-enriched (PanCK-E) regions in the trastuzumab-treated cases (arms 1 and 3, n = 23 biologically independent tumors). Significance, −log10(FDR adjusted p-value), is indicated along the y-axis. Analyses based on the discovery cohort. b. Receiver operating characteristic (ROC) curves for On- plus Pre-treatment DSP protein L2-regularized classifier in the discovery cohort using the subset of cases with data available at both timepoints that were treated with trastuzumab or trastuzumab+lapatinib (n = 19 biologically independent tumors). Model performance was assessed via cross-validation using the 40 DSP protein markers profiled in both cohorts c. Correlation plot comparing the marker coefficients for the On- plus Pre-treatment DSP protein trained using all cases in the discovery cohort (n = 23 tumors with both timepoints versus using only those cases treated with trastuzumab (arms 1 and 3, n = 19 tumors with both timepoints). The grey shading indicates the 95% confidence interval of the correlation coefficient. P value is assessed via a one-sided t-test (correlation coefficient not equal to 0). d. Coefficients for each marker in the L2-regularized On- plus Pre-treatment DSP protein model, trained using only those cases treated with trastuzumab (arms 1 and 3, n = 19 tumors with both timepoints). e. OC curves for classifiers trained using either DSP on-treatment CD45 levels or IHC CD45 % positive. These models were evaluated on the subset of cases on which CD45 IHC was performed that were treated with trastuzumab or trastuzumab+lapatinib (n = 20 biologically independent tumors).
Extended Data Fig. 5 |. Waterfall plots illustrating treatment-associated changes in various data subsets.
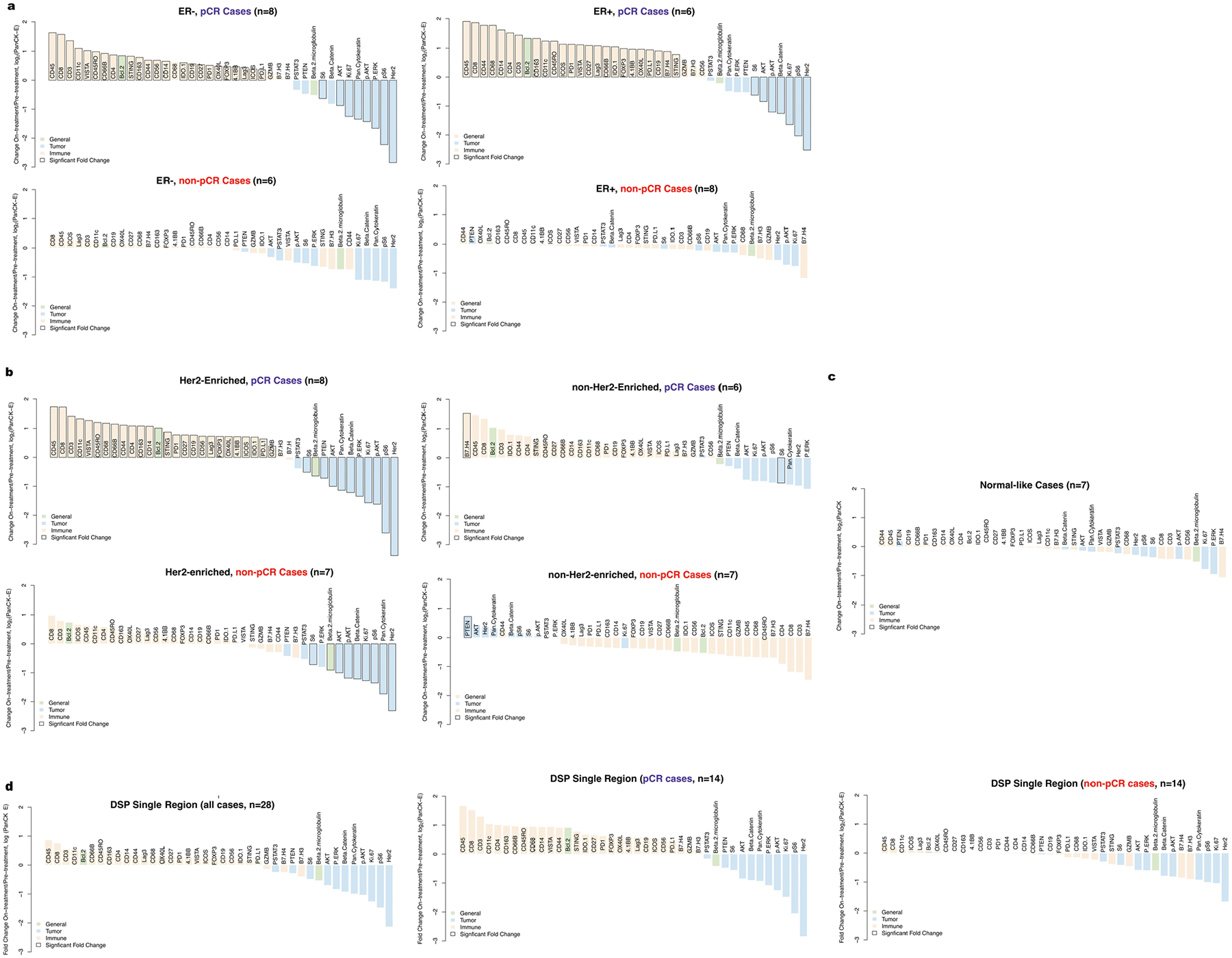
Waterfall plots show changes (pre-treatment to on-treatment) based on in pancytokeratin-enriched (PanCK-E) regions from DSP protein expression data. Analyses based on the discovery cohort (n = 28 tumors). a. Input data was stratified both by estrogen receptor (ER) status and pathologic complete response (pCR) outcome. b. Samples were stratified both by PAM50 status (Her2-Enriched or other) and pCR. c. Waterfall plots illustrating treatment-associated changes (pre-treatment to on-treatment) in the Normal-like cases in the discovery cohort. PAM50 status was determined using bulk expression data from the pre-treatment biopsies. d. Waterfall plots when only one region is used to profile each sample (averaged across 100 iterations of random samples of a single region per timepoint), rather than the 2–7 regions from each sample used in other analyses. The leftmost plot is for all patients, and the plots on the right are stratified by pCR status.
Extended Data Fig. 6 |. Regional heterogeneity profiled through treatment in tumor and immune markers.
Analyses based on the discovery cohort (n = 28 tumors). Heterogeneity was calculated as the mean squared error within patients based on analysis of variance. P-values are based on a two-sided Wilcoxon matched-pair signed rank test. a. Comparison of DSP CD45 protein levels pre-treatment and on-treatment for all regions profiled per case per timepoint. Also shown is a comparison of the mean squared error in DSP CD45 protein expression pre-treatment versus on-treatment within and between patients. b. Pre-, on-, and post-treatment heterogeneity for each DSP protein marker in non-pCR cases (patients with tumor cells present at surgery). c, Pre-treatment heterogeneity in DSP protein marker expression in pCR and non-pCR cases.
Extended Data Fig. 7 |. Comparison of immune markers in panCK-enriched and panCK-negative regions.
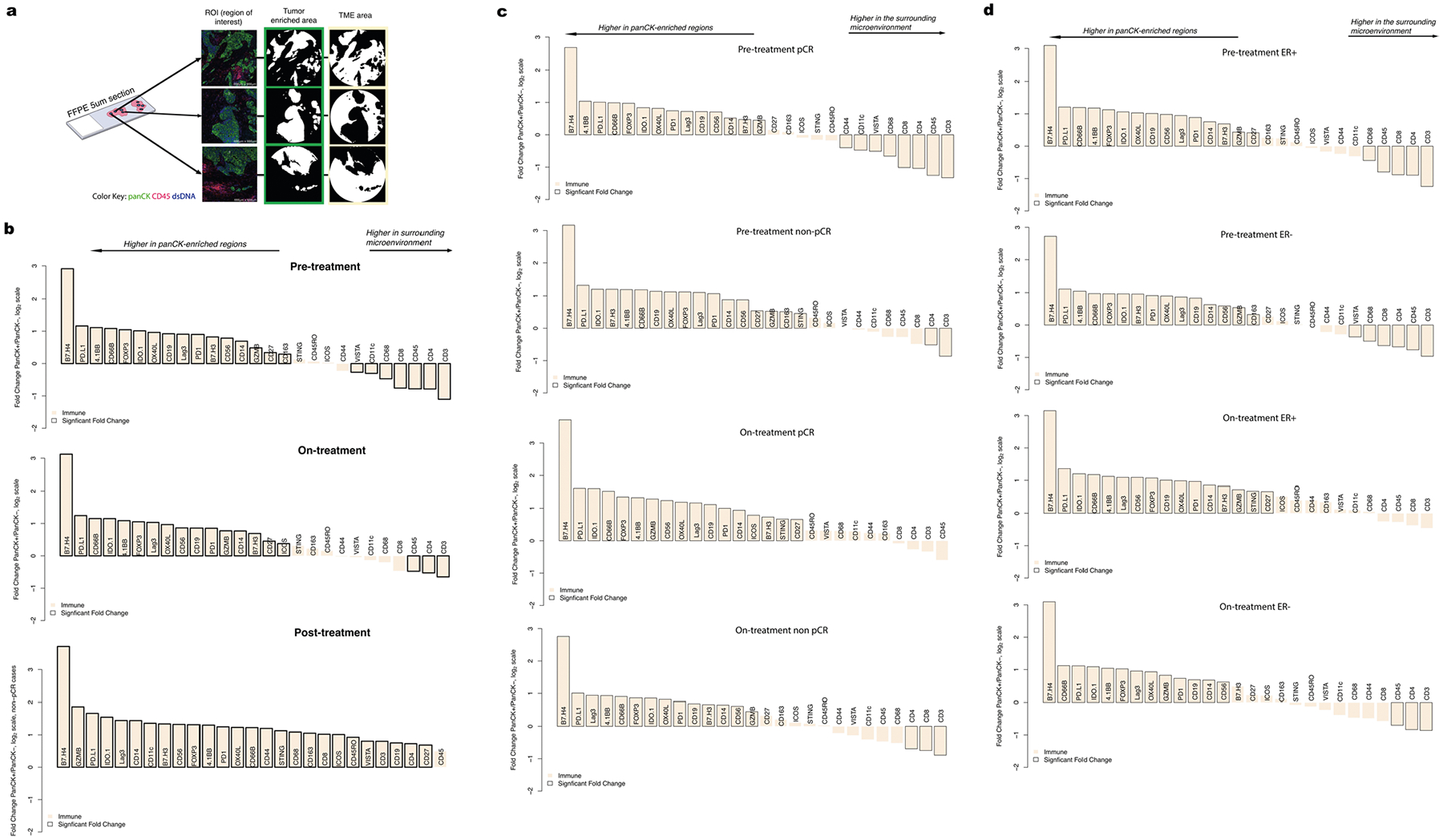
a. Digital Spatial Profiling (DSP) was performed on multiple regions of interest (ROIs) per tissue sample. Protein counts were measured within phenotypic regions corresponding to the PanCK-enriched (tumor-enriched) masks that include tumor cells and co-localized immune cells and separately for the inverted mask corresponding to panCK-negative (tumor microenvironment, TME) regions. b–d. Waterfall plots, generated using the DSP protein data, comparing immune marker expression between the panCK-enriched regions and the surrounding panCK-negative regions. Analyses based on the discovery cohort (n = 28 tumors). b. A comparison of all data pre-treatment on-treatment, and post-treatment. c. A comparison of pre-treatment and on-treatment timepoints, in pCR (n = 14) and non-pCR cases (n = 14). Pre-treatment, the correlation between immune marker fold-change values in the pCR and non-pCR cases was 0.98 indicating similar immune distribution across the panCK-enriched regions and surrounding microenvironment regardless of pCR outcome and this correlation remained high on-treatment (0.95). d. A comparison of pre-treatment and on-treatment timepoints, in ER-positive cases (n = 14), and ER-negative cases (n = 14).
Extended Data Fig. 8 |. Multiplex immunohistochemistry and perimetric complexity.
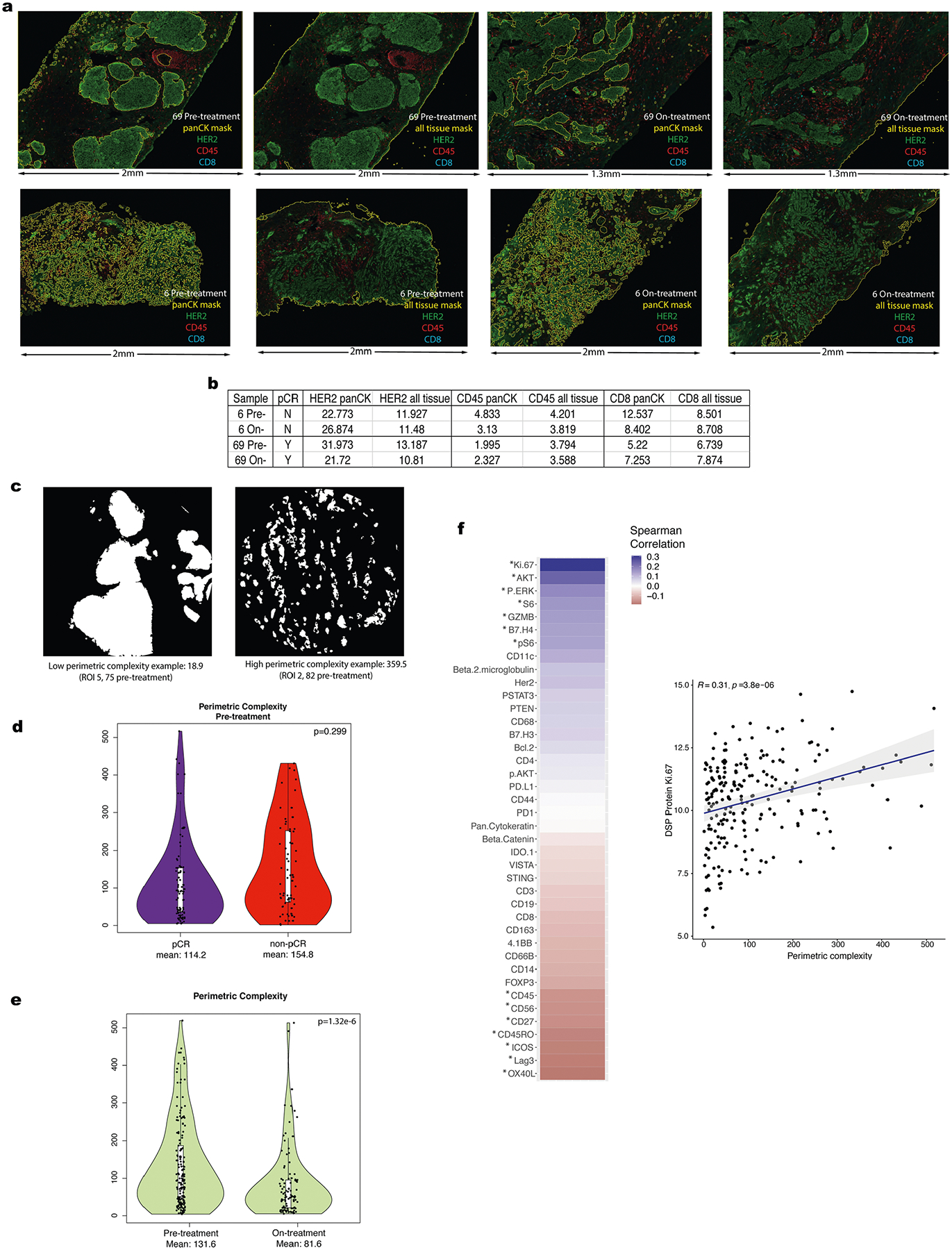
a. Multiplex immunohistochemistry (mIHC) images showing the distribution of HER2, CD45, and CD8 signal in representative tissue stamps pre-treatment and on-treatment. The panCK mIHC channel (not shown) was used to generate the panCK mask and the tissue mask (outlined in yellow). Due to the limiting nature of the biopsy specimens which derive from independent donors, we did not perform technical replicates of the mIHC pilot experiments. b. IHC marker expression levels for HER2, CD45, and CD8 were quantified for the whole tissue section (across all digitized sub-images) and within the panCK-enriched tumor regions (across all digitized sub-images). c. Illustration of panCK-enriched binary masks and perimetric complexity-based quantification of the tumor-microenvironment border. d. Comparison of perimetric complexity values pre-treatment between pCR cases and non-pCR cases. The p-value was derived using a linear mixed-effect model (two-sided test) over the multi-region data with blocking by patient (n = 28 patients). Adjustments were not made for multiple comparisons. For each violin plot, the white box represents the interquartile range and the black lines extending from the white box represent 1.5X the interquartile range. e. Comparison of pre-treatment versus on-treatment perimetric complexity values. PanCK-enriched ROIs were used to quantify perimetric complexity. P-values computed with a linear model, blocked by patient (n = 28 patients). Adjustments were not made for multiple comparisons. For each violin plot, the white box represents the interquartile range and the black lines extending from the white box represent 1.5X the interquartile range. f. Spearman correlation between the DSP protein expression values and perimetric complexity per region of interest (ROI) in the pre-treatment and on-treatment tissue specimens from the discovery cohort. Significantly correlated probes: p-value < .05 are denoted by an asterisk. P value is assessed via a one-sided t-test (correlation coefficient not equal to 0). Correlation plot for Ki-67, the marker with the highest correlation with perimetric complexity, where each dot represents an individual ROI. The grey shading indicates the 95% confidence interval of the correlation coefficient. Analyses are based on the discovery cohort (n = 28 patients).
Extended Data Fig. 9 |. Comparison of alternative models including non-DSP measures and model validation cohort.
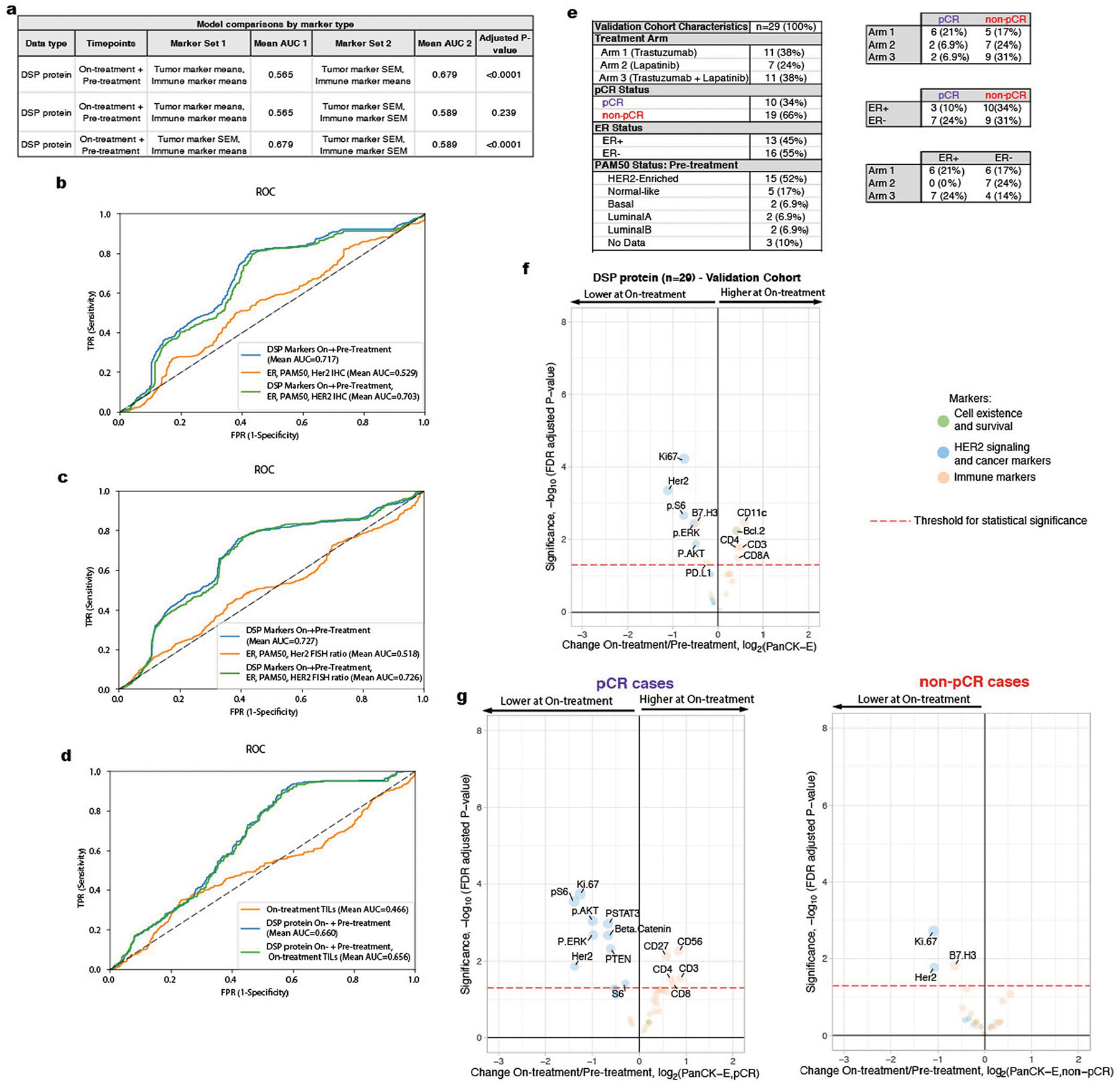
a. AUROC (Area Under the Receiver Operating Characteristics) performance (using nested cross-validation with Holm-Bonferroni correction for multiple hypotheses) comparing DSP protein on- plus pre-treatment L2-regularized classifiers trained using marker means versus marker standard error of the mean (SEM) for tumor markers and immune markers (n = 23 patients with paired data across both timepoints). b–d. Receiver operating characteristic (ROC) curves and AUROC quantification for the On- plus Pre-treatment DSP protein L2-regularized classifier using all 40 markers compared to other models: (b) model trained using estrogen receptor (ER) status, PAM50 status, and HER2 IHC (3 + vs other) (n = 19 patient) and model incorporating these three measures as well as On- plus Pre-treatment DSP; (c) model trained using Er status, PAM50 status, and pre-treatment HER2 FISH ratio (n = 21 patients) and model incorporating these three measures as well as On- plus Pre-treatment DSP; (d) model trained using on-treatment stromal tumor infiltrating lymphocytes (TILs) (n = 16 patients) and model incorporating TILs as well as On- plus Pre-treatment DSP. Model comparisons were performed in the discovery cohort. e. Summary of the clinical characteristics for the TRIO-US B07 clinical trial Digital Spatial Profiling (DSP) validation cohort used for model testing. Treatment arm, pathologic complete response (pCR), ER status, and PAM50 status inferred based on pre-treatment bulk expression data are included. Two-way contingency tables compare the distribution of ER status, pCR status, and treatment arm. f. Volcano plot demonstrating treatment-associated changes based on comparison of pre-treatment versus on-treatment protein marker expression levels in pancytokeratin-enriched (PanCK-E) regions in the validation cohort (n = 29 patients). g. Volcano plots demonstrating treatment-associated changes in pCR versus non-pCR cases in the PanCK-E regions in the validation cohort (n = 29 patients). Significance, −log10(FDR adjusted p-value), is indicated along the y-axis. The p-value was derived using a linear mixed-effect model (two-sided test) over the multi-region data with blocking by patient.
Extended Data Fig. 10 |. On-treatment immunohistochemistry for CD45 predicts pathologic complete response.
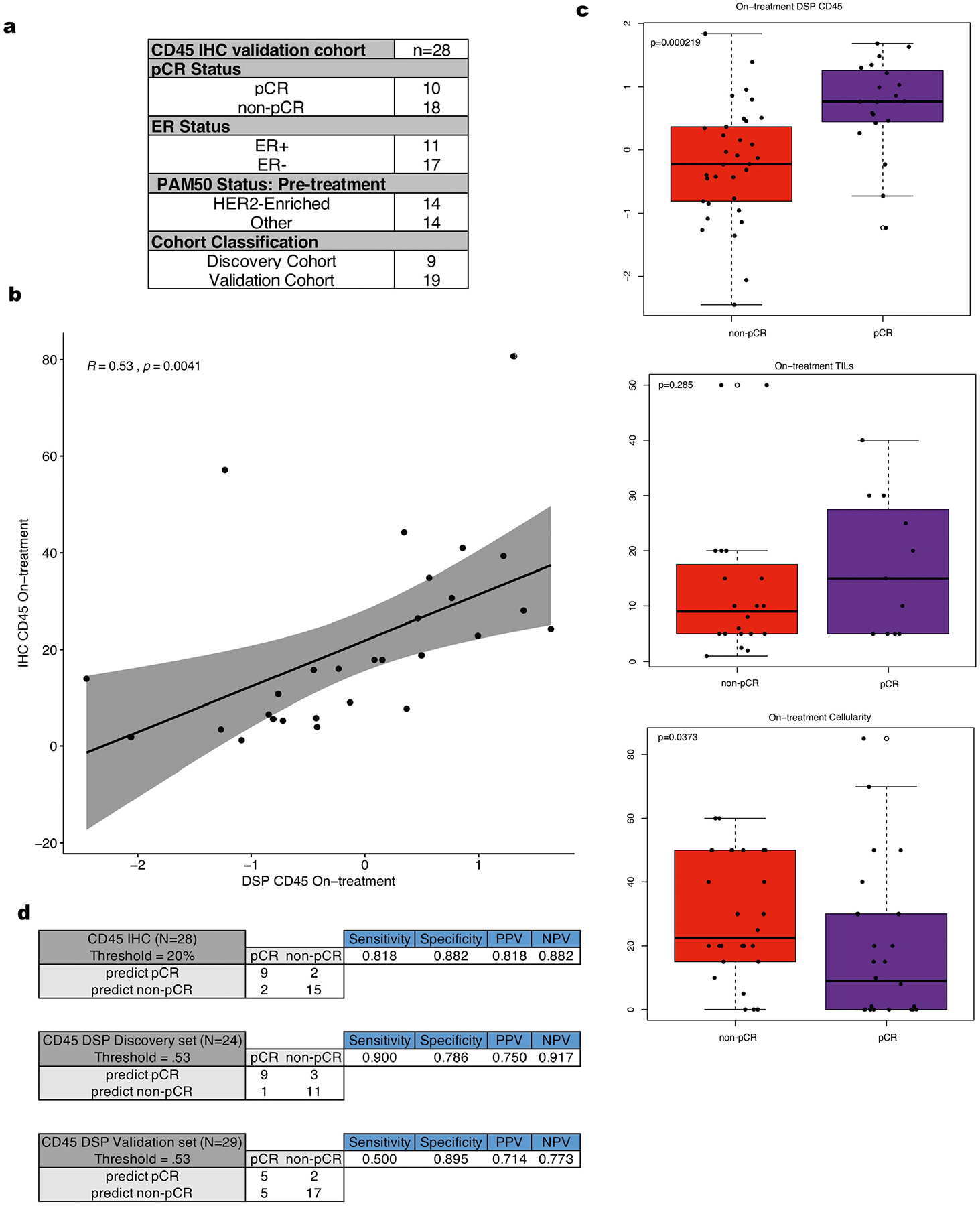
a. Summary of the clinical characteristics of cohort used to evaluate IHC CD45 percent positive cells within tumor enriched regions as a biomarker. These features include pathologic complete response (pCR), estrogen receptor (ER) status, and PAM50 status. b. Correlation plot comparing the normalized on-treatment CD45 DSP protein values with the paired on-treatment CD45 % positive derived from IHC. The grey shading indicates the 95% confidence interval of the correlation coefficient. P value is assessed via a one-sided t-test (correlation coefficient not equal to 0) using n = 28 tumors with paired data. c. Boxplots showing DSP on-treatment CD45 levels (n = 53 biologically independent tumors), on-treatment stromal tumor infiltrating lymphocyte (TILs) score (n = 31 biologically independent tumors), and on-treatment tumor cellularity (n = 47 biologically independent tumors) stratified by pCR utilizing all available data where these features were measured across the discovery and validation cohorts. P-values were derived using the two-sided Wilcoxon rank-sum test. For each boxplot, the colored box represents the interquartile range and the black lines extending from the box represent 1.5X the interquartile range. d. For both the CD45 on-treatment IHC data (n = 28 patients) and the normalized CD45 on-treatment DSP data (n = 24 patients in the discovery cohort, n = 29 patients in the validation cohort), thresholds were defined to predict pCR vs non-pCR cases. Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) are shown for such thresholds.
Acknowledgements
We thank NanoString for technical support. We thank members of the Curtis lab, especially Z. Hu, J. A. Seoane, A. Khan and J. Brugge for input on the HER2 and Ki67 IHC. This project was supported by awards to C.C. from the National Institutes of Health (NIH)/National Cancer Institute (NCI) (no. R01CA182514) and the Breast Cancer Research Foundation. C.C. is a Susan G. Komen Scholar. K.L.M. is supported by grant no. F30CA239313-02 from the NIH/NCI. J.L.C. was a Damon Runyon Physician-Scientist supported by the Damon Runyon Cancer Research Foundation (no. PST 11-17) and a Susan G. Komen Postdoctoral Fellowship Award (no. PDR17481769). M.F.P. was supported by the Breast Cancer Research Foundation and Tower Cancer Research Foundation and grant no. R01CA182514. D.J.S. and S.S. were also supported in part by grant no. R01CA182514. The multiplex IHC staining performed at the Fred Hutchinson Cancer Research Center experimental histopathology shared resources was supported by the Fred Hutchinson/University of Washington Cancer Consortium (no. P30 CA015704).
Competing interests
M.F.P. received research funding from Cepheid, Eli Lilly and Company, Zymeworks and Novartis; performed consulting/advisory board work for Biocartis, Eli Lilly and Company, Zymeworks, Novartis, Puma Biotechnology, Merck Millipore, AstraZeneca, CME Outfitters and Clinical Care Options; and has private equity in TORL Biotherapeutics. D.J.S. received research funding from Pfizer, Novartis, Syndax, Millenium Pharmaceuticals, Aileron Therapeutics, Bayer and Genentech; owned stock in BioMarin, Amgen, Seattle Genetics and Pfizer; served on the board of directors for BioMarin; and performed consulting/advisory board work for Eli Lilly and Company, Novartis, Bayer and Pfizer. S.A.H. received contracted research and medical writing assistance from Ambrx, Amgen, Arvinas, Bayer, Daiichi Sankyo, Genentech/Roche, GlaxoSmithKline, Immunomedics, Eli Lilly and Company, Macrogenics, Novartis, Pfizer, OBI Pharma, Pieris, Puma Biotechnology, Radius Health, Sanofi, Seattle Genetics and Dignitana. R.J. is currently employed by Tempus. M.K., Z.Z., M.H. and J.B. are all employees of NanoString and declare that they have competing interests. C.C. performed advisory board/consulting work for Genentech, GRAIL, Illumina and NanoString and reports GRAIL stockholdings. C.C. and K.L.M. are coinventors on a patent application filed by Stanford University relating to this manuscript. J.L.C., Z.M., E.K., G.R.B. and J.Z. declare no conflicts of interest.
Footnotes
Code availability
Code associated with the manuscript is available at https://github.com/cancersysbio/BreastCancerSpatialProteomics. R v.3.6.0 was used with base/standard packages and the following additional packages: vioplot_0.3.2; lmerTest_3.1-0; dplyr_0.8.3; ggrepel_0.8.1; tidyr_0.8.3; reshape_0.8.8; ggplot2_3.2.1; limma_3.28.21. Python v.3.6.8 was used with base/standard packages and the following additional packages: pandas_0.25.1; numpy_1.17.2; matplotlib_3.1.1; seaborn_0.9.0; statsmodels_0.10.1; scipy_1.3.1; pystan_2.19.1.1; arviz_0.6.1; joblib_0.13.2; scikit-learn_0.21.3.
Extended data is available for this paper at https://doi.org/10.1038/s43018-021-00190-z.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43018-021-00190-z.
Data availability
DSP protein counts, bulk RNA expression data, perimetric complexity data, CD45 IHC data and the clinical covariate data generated for this study are available through the Curtis Lab GitHub repository (https://github.com/cancersysbio/BreastCancerSpatialProteomics). Source data for Figs. 1–6 and Extended Data Figs. 1–10 are provided with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Slamon DJ et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Huober J et al. Survival outcomes of the NeoALTTO study (BIG 1–06): updated results of a randomised multicenter phase III neoadjuvant clinical trial in patients with HER2-positive primary breast cancer. Eur. J. Cancer 118, 169–177 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Broglio KR et al. Association of pathologic complete response to neoadjuvant therapy in HER2-positive breast cancer with long-term outcomes: a meta-analysis. JAMA Oncol 2, 751–760 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Cortazar P et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014). [DOI] [PubMed] [Google Scholar]
- 5.de Azambuja E et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): survival outcomes of a randomised, open-label, multicentre, phase 3 trial and their association with pathological complete response. Lancet Oncol 15, 1137–1146 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Gianni L et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol 15, 640–647 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Gianni L et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol 13, 25–32 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Rimawi MF et al. Multicenter phase II study of neoadjuvant lapatinib and trastuzumab with hormonal therapy and without chemotherapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer: TBCRC 006. J. Clin. Oncol 31, 1726–1731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veeraraghavan J et al. De-escalation of treatment in HER2-positive breast cancer: determinants of response and mechanisms of resistance. Breast 34, S19–S26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Llombart-Cussac A et al. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): an open-label, single-group, multicentre, phase 2 trial. Lancet Oncol 18, 545–554 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Gianni L et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet 375, 377–384 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Lesurf R et al. Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy: results from the ACOSOG Z1041 (Alliance) trial. Ann. Oncol 28, 1070–1077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fumagalli D et al. RNA sequencing to predict response to neoadjuvant anti-HER2 therapy: a secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol 3, 227–234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prat A et al. Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin. Cancer Res 20, 511–521 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Tanioka M et al. Integrated analysis of RNA and DNA from the phase III trial CALGB 40601 identifies predictors of response to trastuzumab-based neoadjuvant chemotherapy in HER2-positive breast cancer. Clin. Cancer Res 24, 5292–5304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loi S et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann. Oncol 25, 1544–1550 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Nuciforo P et al. A predictive model of pathologic response based on tumor cellularity and tumor-infiltrating lymphocytes (CelTIL) in HER2-positive breast cancer treated with chemo-free dual HER2 blockade. Ann. Oncol 29, 170–177 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Varadan V et al. Immune signatures following single dose trastuzumab predict pathologic response to preoperative trastuzumab and chemotherapy in HER2-positive early breast cancer. Clin. Cancer Res 22, 3249–3259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Martinez A et al. Survival, pathologic response, and genomics in CALGB 40601 (Alliance), a neoadjuvant phase III trial of paclitaxel-trastuzumab with or without lapatinib in HER2-positive breast cancer. J. Clin. Oncol 38, 4184–4193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binnewies M et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurvitz SA et al. Pathologic and molecular responses to neoadjuvant trastuzumab and/or lapatinib from a phase II randomized trial in HER2-positive breast cancer (TRIO-US B07). Nat. Commun 11, 5824 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toki MI et al. Validation of novel high-plex protein spatial profiling quantitation based on NanoString’s Digital Spatial Profiling (DSP) technology with quantitative fluorescence (QIF). Cancer Res 77, 3810 (2017). [Google Scholar]
- 23.Merritt CR et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol 38, 586–599 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Untch M et al. Neoadjuvant treatment with trastuzumab in HER2-positive breast cancer: results from the GeparQuattro study. J. Clin. Oncol 28, 2024–2031 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Shao M-M et al. Keratin expression in breast cancers. Virchows Arch 461, 313–322 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Valachis A, Nearchou A, Lind P & Mauri D Lapatinib, trastuzumab or the combination added to preoperative chemotherapy for breast cancer: a meta-analysis of randomized evidence. Breast Cancer Res. Treat 135, 655–662 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Pohlmann PR, Mayer IA & Mernaugh R Resistance to trastuzumab in breast cancer. Clin. Cancer Res 15, 7479–7491 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zingoni A et al. Natural killer cell response to chemotherapy-stressed cancer cells: role in tumor immunosurveillance. Front. Immunol 8, 1194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzger Filho O et al. HER2 heterogeneity as a predictor of response to neoadjuvant T-DM1 plus pertuzumab: results from a prospective clinical trial. J. Clin. Oncol 37, 502 (2019). [Google Scholar]
- 30.Joyce JA & Fearon DT T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015). [DOI] [PubMed] [Google Scholar]
- 31.AbdulJabbar K et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat. Med 26, 1054–1062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keren L et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watson AB Perimetric complexity of binary digital images: notes on calculation and relation to visual complexity. Mathematica 14 https://content.wolfram.com/uploads/sites/19/2012/02/Watson.pdf (2012). [Google Scholar]
- 34.Schwanhäusser B et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Truitt ML & Ruggero D New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 16, 288–304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B & Hurvitz S Long-term outcomes of neoadjuvant treatment of HER2-positive breast cancer. Clin. Adv. Hematol. Oncol 14, 520–530 (2016). [PubMed] [Google Scholar]
- 37.Sever R & Brugge JS Signal transduction in cancer. Cold Spring Harb. Perspect. Med 5, a006098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu H, Kim PM, Sprecher E, Trifonov V & Gerstein M The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput. Biol 3, e59 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pernas S & Tolaney SM HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol 11, 1758835919833519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goutsouliak K et al. Towards personalized treatment for early stage HER2-positive breast cancer. Nat. Rev. Clin. Oncol 17, 233–250 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Modi S et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med 382, 610–621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murthy RK et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N. Engl. J. Med 382, 597–609 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Javier Cortes GG et al. Chemotherapy (CT) de-escalation using an FDG-PET/CT (F-PET) and pathological response-adapted strategy in HER2+ early breast cancer (EBC): PHERGain Trial. J. Clin. Oncol 38, 503 (2020). [Google Scholar]
- 44.Amaria RN et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med 24, 1649–1654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malkov VA et al. Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounter Assay System. BMC Res. Notes 2, 80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritchie ME et al. A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Johnson WE, Li C & Rabinovic A Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Paquet ER, Lesurf R, Tofigh A, Dumeaux V & Hallett MT Detecting gene signature activation in breast cancer in an absolute, single-patient manner. Breast Cancer Res 19, 32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bates D, Mächler M, Bolker BM & Walker SC Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48 (2015). [Google Scholar]
- 51.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995). [Google Scholar]
- 52.Pedregosa F et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res 12, 2825–2830 (2011). [Google Scholar]
- 53.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bankhead P et al. QuPath: open source software for digital pathology image analysis. Sci. Rep 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
DSP protein counts, bulk RNA expression data, perimetric complexity data, CD45 IHC data and the clinical covariate data generated for this study are available through the Curtis Lab GitHub repository (https://github.com/cancersysbio/BreastCancerSpatialProteomics). Source data for Figs. 1–6 and Extended Data Figs. 1–10 are provided with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.